

Translate this page into:
Low-loading of oxidized platinum nanoparticles into mesoporous titanium dioxide for effective and durable hydrogen evolution in acidic media
⁎Corresponding author. mghanem@ksu.edu.sa (Mohamed A. Ghanem)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Low-loading of oxidized platinum nanoparticles (0.1–0.5 wt%) was incorporated into mesoporous titanium dioxide support (Ptx/meso-TiO2) via evaporation self-assembly (ESA) approach followed by a two-step calcination processes. The physicochemical characterizations showed that the oxidized Ptx/meso-TiO2 catalysts exhibit high surface area around 200 m2/g and Pt nanoparticles having an average size of 3.0 nm are uniformly incorporated into the mesoporous TiO2 matrix with the existence of Pt(II) and Pt(IV) oxidation states. The Ptx/meso-TiO2 electrocatalysts showed an enhanced electrocatalytic activity with hydrogen evolution onset potential at −10 mV vs. RHE, Tafel slope of −110 mV/dec, small charge transfer resistance, and mass activity that reaches up to 25.7 A/mgPt at −300 mV vs. RHE. The hydrogen evolution mass activity of Ptx/meso-TiO2 electrocatalysts is significantly more efficient than the commercial Pt/C catalysts and Pt nanoparticles supported on nanostructured carbon substrates. Moreover, the Ptx/meso-TiO2 electrocatalysts exhibit excellent durability for a 24-hour electrolysis in acid solution with a further current activation during the prolonged electrolysis. The enhanced mass activity and durability are attributed to the substrate mesoporosity, uniform distribution and strong bonding between the oxidized Pt nanoparticles and the TiO2 substrate. These results demonstrate the promise of the mesoporous TiO2 substrate modified with low loading of platinum nanoparticles for energy conversion technologies.
Keywords
Oxidized Pt nanoparticles
Mesoporous TiO2
Electrocatalyst
Hydrogen evolution

1 Introduction
Hydrogen is considered as the most promising fuel for the future renewable and clean energy sources (Crabtree et al., 2004; Jiao et al., 2016). The production of hydrogen by water electrolysis would be more favourable clean alternative path (Gong et al., 2016) to the current steam-methane reforming process if the electrochemical water splitting made economical and employed in the large-scale commercial application (Merki and Hu, 2011; Morales-Guio et al., 2014; Subbaraman et al., 2011). The noble metal platinum (Pt) is the commonly used catalyst for water electro-splitting because it exhibits an excellent electrocatalytic activity for hydrogen evolution reaction (HER) (Yan et al., 2016; Yin et al., 2015). However, Pt is an expensive and scarce metal which heavily impedes its utilization in large-scale commercial applications (Yu et al., 2012). As such, it is highly recommended to design and develop active, stable and inexpensive electrocatalysts for water splitting to produce high purity and large-scale of hydrogen gas. Significant research efforts have been devoted to producing cost-effective metal-based or nonmetal HER catalysts, for instance wide range of sulfide-based materials (Zheng et al., 2015; Chen et al., 2015; Voiry et al., 2013; Gao et al., 2015), metal chalcogenides (Zhuo et al., 2013; Jirkovsky et al., 2016; Li et al., 2016; Yu et al., 2016), metal carbides (MCs) (Liao et al., 2014; Harnisch et al., 2009), phosphosulphide (Cabán-Acevedo et al., 2015), and oxides (Wu et al., 2015, Xu et al., 2016), have been explored to improve the kinetics and activity of electrochemical HER. In addition WS2/graphene (Zhou et al., 2016a), ternary molybdenum sulfoselenide and nickel diselenide foam (Zhou et al., 2016b), sulfoselenide/nickel diselenide hybrid materials (Zhou et al., 2017) are found to be effective and stable electrocatalyst for the HER in acidic environment. To overcome the challenges associated with the platinum-based HER catalysts availability and to improve the cost-effectiveness of H2 production by water electrolysis, it is very important to develop high-performance electrocatalysts and significantly reduce the Pt loading to increase its mass activity and the Pt utilization efficiency (Stephens and Chorkendorff, 2011). Recently, many efforts have been done to enhance the mass activity and then HER performance of Pt including, the combination of Pt with metal hydroxide (Subbaraman et al., 2011; Yin et al., 2015; Wang et al., 2015a,b), nano-structuring (Chen and Kucernak, 2004), making alloys with the non-noble metals (Cao et al., 2014; Wang et al., 2017a,b), supporting sub-nanometer size Pt nanoparticles (NPs) (Esposito et al., 2010). More recently, a great number of studies have focused on the reduces the size of Pt nanoparticles to cluster size or even single atoms which reducing the noble metal usage and is found to be a highly effective for enhancing the Pt utilization thus decreasing the cost of the electrocatalysts (Yang et al., 2013). Qiao et al. (2011) demonstrated that the dispersion of single Pt atoms on iron oxide surface exhibited a higher catalytic activity for CO oxidation compared with the corresponding Pt nanoparticles. (Sun et al., 2013) reported the single atoms catalysts for methanol oxidation activity up to 10 times greater than the commercial Pt/C catalysts. In a relevant study by (Cheng et al., 2017a,b; Li et al., 2017) highly active sub-nanometer size of oxidized Pt clusters supported on TiO2 have demonstrated a significantly enhanced catalytic activity and durability for hydrogen evolution reaction that was attributed to the stabilization of Pt higher oxidation states and strong interaction with the TiO2 substrate.
Carbon-based materials with different morphologies and porosity are frequently used as a catalyst support in fuel cells and catalysis applications (Merino-Jimenez et al., 2016; Zhang et al., 2010; Huang and Wang, 2014). However, carbon materials support may not be robust enough for long-term HER durability under highly oxidizing conditions (Zhai et al., 2007; Hogarth and Ralph, 2002) due to the corrosion and collapse during the long-term operation. Consequently, this leads to separation and agglomeration of supported Pt nanoparticles from the carbon support which causes catalyst degradation and loss of the electrochemical activity (Huang et al., 2009). Therefore, searching for high stable catalyst support with withstanding physical and chemical stability in a harsh environment is very demanding. Titanium dioxide (TiO2) as a catalyst support has attracted great interest in recent years due to its inherent stability in the electrochemical environment and commercial availability (Bauer et al., 2010). The TiO2 based materials have shown an excellent corrosion resistance and strong metal support interaction, which can enhance the catalysts activities and utilization (Wang et al., 2010; Shaddad et al., 2013). However, titanium dioxide is a semiconductor with very high electrical resistivity (Tang et al.,1994) which limits its practical application in electrochemical applications (Wang et al., 2010; Zhou et al., 2013). Mesoporous TiO2 is regarded as a type of excellent catalytic support owing to their high thermal stability, low cost, tunable mesostructure and porosity, and large surface area (Fattakhova-Rohlfing et al., 2014) which facilitate the charge transport during the electrochemical reactions. Furthermore, a well-defined mesoporous system can also enhance the thermal (Comotti et al., 2006) and electrochemical stability of supported Pt nanoparticles by encapsulation into pore confinement (Galeano et al., 2012). Therefore an improved synthesis procedure for Pt nanoparticle modified mesoporous TiO2 would provide a high surface area and accessibility to the supported Pt nanoparticles via interconnected mesopores and enable excellent electronic conductivity. Recently our group reported the synthesis of low loading (3.28 wt%) and highly dispersed platinum nanoparticles (3–5 nm in diameter) supported on silica substrate as a catalyst for hydrogen production (Khdary and Ghanem, 2014, Khdary and Ghanem, 2016). Even though silica substrate is non-conducting, the Pt nanoparticles incorporated silica substrate displayed good performance for hydrogen production in acidic media with the mass activity of 11.9 A/gPt at an overpotential of −250 mV vs. SCE. In this work, very low loading of Pt nanoparticles (0.1–0.5 wt%) was incorporated onto mesoporous TiO2 (Ptx/meso-TiO2) via evaporation-induced self-assembly (EISA) of surfactant template followed by a two-step calcination process. Compared with the traditional method, this method was simple, convenient and it was easier to obtain very low loading of uniform size and high utilization of Pt nanoparticles on the mesoporous TiO2 substrate. The Ptx/meso-TiO2 catalysts with a platinum loading of 0.1–0.5 wt% exhibited higher mass H2 evolution activities than Pt supported on bulk TiO2 (Pt/bulk-TiO2) and commercial Pt/C catalysts evidenced by the significant shift in the hydrogen evolution onset potential, smaller Tafel slope and charge transfer resistance.
2 Experimental
2.1 Materials and chemicals
Poly(ethylene oxide)-block-poly (propylene oxide)-block-poly-(ethylene oxide) triblock copolymer Pluronic® F-127 (Mw = 12 600, PEO106PPO70PEO106), tetrabutyl orthotitanate (TBOT, >98.0%), ethanol (C2H5OH, >99.9%) and (H2PtCl6·6H2O ≥ 37.5%) were purchased from Sigma-Aldrich. Hydrochloric acid (37 wt%), nitric acid (HNO3), potassium hydroxide (KOH, >96.0%), (acetic acid 99%) and sulfuric acid (H2SO4, 95–98%) were obtained from BDH. Carbon Paper GDL-24BC was used as working electrode. Deionized water used in all experiments was obtained from a Milli-Q Ultrapure Water purification system with a resistivity of 18.2 MΩ cm and all chemicals were used as received without additional purification.
2.2 Preparation of ordered mesoporous Pt/meso-TiO2 catalysts
The ordered mesoporous Pt/meso-TiO2 catalysts were synthesized by adapting the previously reported procedure (Wang et al., 2015a,b) using a Ti precursor of tetrabutyl orthotitanate (TBOT) and the triblock copolymer Pluronic® F-127 (PEO106PPO70PEO106) surfactant as a template. Typically, Pluronic® F-127 (2.0 g) was first dissolved in highly pure ethanol (C2H5OH, 20 mL) and 2.4 g of concentrated HCl (37%). The mixture was vigorously stirred for 30 min at 40 °C to form a transparent solution. Then different weight percentages (0.1, 0.3, 0.5, 1.0, 2.0 and 3.0 wt%) of H2PtCl6·6H2O were added into the transparent template solution and mixed by vigorous stirring at 40 °C to obtain a clear yellow homogenous solution. Meanwhile, the titanium precursor solution was prepared by dissolving TBOT (3.4 g) in acetic acid (2.4 g) and the two solutions were mixed and stirred at 40 °C for 2.0 h. After that, the obtained homogenous golden solution was transferred to Petri dishes and left for 40 min at room temperature to evaporate the solvents, followed by solidifying the samples at 100 °C for 24 h to form the inorganic–polymer hybrids and completely remove the solvents. The golden samples were collected and heated with a ramp of 1.0 °C min−1 to 350 °C in N2 and held for 3.0 h to remove the template, resulting in the carbon-containing amorphous Ptx/meso-TiO2 catalyst powders. Crystallization of the amorphous Ptx/meso-TiO2 powders was carried out with a ramp of 1.0 °C min−1 to 400 °C in air for another 3.0 h. For the control experiment, the plain mesoporous TiO2 support (meso-TiO2) was prepared using similar above procedure but without adding H2PtCl6·6H2O In addition, platinum nanoparticles supported on bulk TiO2 (Pt0.5/bulk-TiO2) was also prepared via a similar route as used for Ptx/meso-TiO2 catalyst except for the absence of the triblock copolymer Pluronic® F-127 template. The obtained catalysts were denoted as (Ptx/meso-TiO2), where x represents the loading weight percentage of platinum.
2.3 Characterizations
Powder X-ray diffraction (XRD) pattern data were measured using a Rigaku Mini Flex 600 irradiation X-ray diffractometer using Cu Kα radiation (40 kV, 15 mA). The characterization of high-resolution scanning electron microscope (HRSEM) and transmission electron microscopy (TEM) were performed by a Hitachi S4800 (Japan) operated at 1.0 kV and 10 mA and a JEM-2100F microscope (Japan) operating at 200 kV respectively. For the TEM measurements, the catalyst powder was suspended in ethanol then a suspension drop was applied on a holey carbon film Cu grid and left to dry in an air atmosphere. X-ray photoemission spectroscopy (XPS) analyses were performed using JPS-9200, JEOL Photoelectron spectrometer while A NOVA 2200e analyzer (Quntachrome, Japan) was used to measure the specific Brunauer-Emmett-Teller surface area and prior to the measurements; the samples were degassed in vacuum at 180 °C for at least 6.0 h. The specific surface areas of the samples were calculated using the Brunauer-Emmett-Teller (BET) method, and the pore size, pore volume distributions were calculated by the Barrett-Joyner-Halenda (BJH) method by using the adsorption branch. ICP-MS analysis was performed using ThermoScientific iCAP6300. The platinum supported onto meso-TiO2 catalysts was dissolved by a mixture of concentrated nitric of (HNO3, ∼65%) and sulfuric acid (H2SO4, 95–98%) with a ratio of 3:1. The mixture was digested in a microwave oven for 20 min. A series of Pt solutions was obtained by diluting the 50 ppm Pt standard solution: 0, 0.25, 0.5, 0.75, and 1 mL standard solutions of 50 ppm were added to 25 mL volumetric flasks to produce calibration solutions with Pt concentrations of 0, 0.5, 1, 1.5 and 2 ppm, respectively. To prepare solution samples for Pt determination, the Ptx/meso-TiO2 catalysts (20 mg) were heated in 4.0 mL of acid mixture in an oil bath maintained at ∼100 °C. Next, the acid mixture was transferred to a 50 mL volumetric flask and deionized water was finally added to complete the volume of 50 mL, developing transparent solutions for ICP-MS (Thermo Scientific; UK, Model ICAP 6000) measurement of Pt content.
2.4 Electrochemical measurements
Electrochemical measurements were executed at room temperature of 21 °C with a potentiostat (BioLogic SAS, model) in a standard three-electrode setup, with the Ptx/meso-TiO2 catalyst deposited on carbon paper substrate as a working electrode, a Pt mesh and a saturated calomel electrode (SCE) as the counter and reference electrode respectively. A glass body filled with H2SO4 fitted to SCE reference electrode to prevent the contamination of the H2SO4 test solution with chloride ion. In all measurements, the potential was with referred to the RHE using the equation ERHE = ESCE + 0.243 + 0.059 pH and all polarization curves here are measured without IR compensation. The working electrode was fabricated by depositing the Ptx/meso-TiO2 catalyst thin film on carbon paper (SIGRACET®, grade GDL-24BC, geometric area 1 × 1 cm2) by electrophoretic deposition without added Nafion® solution. The deposition solution was prepared by dispersing 15 mg of the Ptx/meso-TiO2 powder in 1.0 mL of iodine- acetone solution (40 mg iodine dissolved in 15 mL of acetone) using ultrasonic probe for 20 min to get uniform catalyst dispersion. Then two carbon paper electrodes with (geometric area 1 × 1 cm2) each was immersed in parallel at ca. 1 cm distance in the deposition solution and then, +10 V of bias was applied between the two electrodes for 4 min with the catalyst particles coated on the negative electrode. After this process, the working electrodes were rinsed with deionized water, dried in air and then calcined at 350 °C for 30 min under a flow of N2 gas to improve of the catalyst with the carbon paper. The average weight of the deposited Ptx/meso-TiO2 catalyst on the carbon paper was about 1.1 mg in each case. The control experiment was performed by depositing about 1.1 mg/cm2 of 10 wt% Pt/C catalyst (PK catalyst, Fuel Cell store, USA) using the same electrophoretic deposition procedure reported above. The electrochemical impedance spectroscopy (EIS) measurements were carried out using the same cell configuration at the chosen potential from 100 kHz to 200 mHz with an amplitude of 10 mV. All the electrochemical measurements were performed in sulfuric acid solution that was purged with N2 prior to measurements and at 20 °C under normal atmosphere pressure.
3 Results and discussion
3.1 Synthesis and characterizations of the mesoporous Ptx/TiO2 catalysts
Fig. 1 shows optical images of the produced mesoporous TiO2 modified with different wt% of Pt nanoparticles after the two steps annealing. Clearly, the bare mesoporous TiO2 has a white colour (Fig. 1a) which is gradually turned to dark brown colour as the Pt loading increases from 0.1 to 3.0 wt% (Fig. 1b–e). This confirms the precipitation of Pt catalyst onto the mesoporous TiO2 support.
Optical images of the produced mesoporous TiO2 modified with different wt% of Pt catalyst after the two steps annealing, (a) pure meso-TiO2 (b) Pt0.1/meso-TiO2, (c) Pt0.3/meso-TiO2, (d) Pt1.0/meso-TiO2 (e) Pt3.0/meso-TiO2.
The crystal structure of the obtained Ptx/meso-TiO2 catalysts was investigated by XRD and shown in Fig. 2. It is observable that for all the Ptx/meso-TiO2 catalysts the mesoporous TiO2 support exhibit the characteristic diffraction peaks for anatase phase. As shown in Fig. 2, the main diffraction peaks at 2θ of 25.5, 37.8, 48.2, 54.2 and 62.9°, can be assigned to the (1 0 1), (0 0 4), (2 0 0), (1 0 5) and (2 0 4) reflections of TiO2 in the anatase phase respectively (JCPDS, 21-1272). The characteristic diffraction peaks for Pt metal were absence in case of Pt loading less than 1.0 wt%. However when the Pt loading reaches 1.0 wt% and above, a faint diffraction peaks at the 2θ values 47.6°, 54.6°, and 69.9° are observed which can assign to the Pt (1 1 1), Pt (2 0 0) and Pt (2 2 0) diffraction plane respectively of face-centred cubic Pt phase (JCPDS No.01-1190).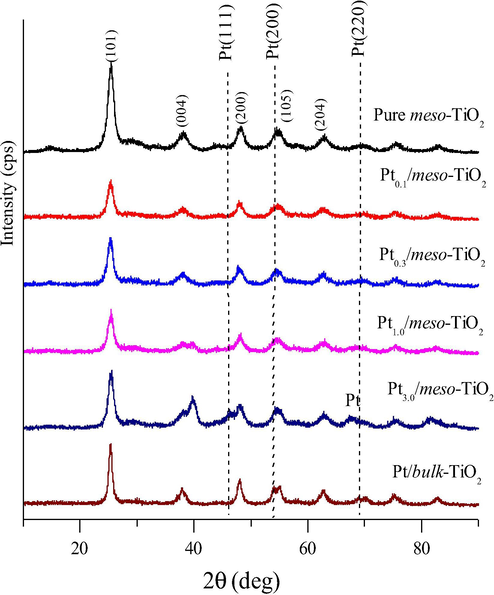
Wide-angle XRD patterns of the mesoporous TiO2 modified with different loading of Pt catalyst after calcination at 350 °C in N2 and at 400 °C in an air atmosphere for 3 h each.
The average crystallite size of pure mesoporous TiO2 was 6.9 nm while for Ptx/meso-TiO2 was reduced to 5.3 nm, as calculated from the peak width of the anatase (1 0 1) reflection using Scherrer’s equation (Lang and Willson, 1978). However, the crystallinity of Pt nanoparticles can’t be concluded because the weakness or absence of its diffraction peaks.
The mesoporosity and surface area of the mesoporous TiO2 and Ptx/meso-TiO2 catalysts were further measured by the nitrogen adsorption and desorption isotherm. Fig. 3 shows the N2 adsorption-desorption isotherms of the pure meso-TiO2 and Ptx/meso-TiO2 catalysts and the corresponding pore size distribution curves obtained by the Barrett-Joyner-Halenda (BJH) method from the adsorption branch. The N2 adsorption-desorption isotherms (Fig. 3a), clearly show that the pure meso-TiO2 and Ptx/meso-TiO2 catalysts exhibit a typical type of IV isotherm with a hysteresis loop, and capillary condensation at relative pressures P/Po between 0.45 and 0.8 which proves the presence of the mesoporous structures (Kruk and Jaroniec, 2001). The corresponding BET surface areas, specific pore volume, and pore size of these mesoporous catalysts are summarized in Table 1. We can find no significant change in the surface area, pore volumes or the pore size upon the incorporation of Pt nanoparticles into the meso-TiO2 framework. This indicates that the incorporation of H2PtCl6 hardly affects the evaporation-induced self-assembly (EISA) process of the titanium precursor and Pluronic® F-127 surfactant mixture.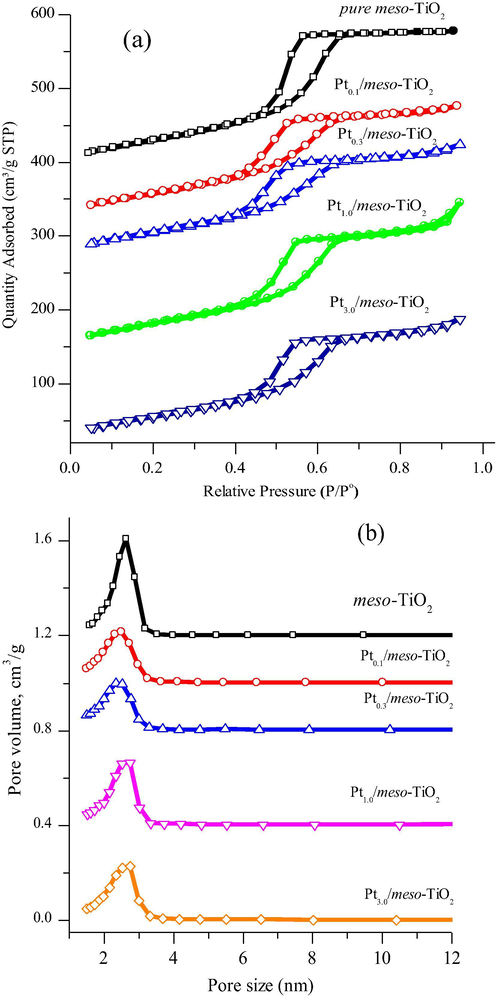
(a) N2 adsorption–desorption isotherms and (b) pore size distribution of the pure meso-TiO2 and Ptx/meso-TiO2 catalysts after calcination at 350 °C in N2 and at 400 °C in an air atmosphere for 3 h each. The curves of both figures are offsite for clarity.
Catalyst
Pt (wt%)
Pt wt% ± 0.04 (ICP analysis)
SBET ± 5 (m2/g)a
Pore volume ± 0.01 (cm3/g)b
Pore size ± 0.02(nm)c
Pure meso-TiO2
0.0
00
218
0.33
2.65
Pt0.1/meso-TiO2
0.1
0.089
209
0.26
2.50
Pt0.3/meso-TiO2
0.3
0.27
203
0.26
2.30
Pt0.5/meso-TiO2
0.5
0.48
199
0.30
2.50
Pt1.0/meso-TiO2
1.0
0.86
221
0.34
2.80
Pt2.0/meso-TiO2
2.0
1.85
204
0.30
2.73
Pt3.0/meso-TiO2
3.0
2.8
201
0.28
2.74
Pt0.5/bulk-TiO2
0.5
0.56
29
0.022
–
The surface morphology of the Pt0.5/meso-TiO2 catalyst is characterized by high-resolution SEM and shown in Fig. 4a. Evidently, the SEM image shows that 2-dimensional hexagonal mesoporous channels are homogeneously formed throughout the catalyst film and extended over micrometre domains which confirms the formation of the mesoporous TiO2 structure after annealing and surfactant removal. The Pt0.5/meso-TiO2 catalyst surface analysis was examined by high-resolution XPS and Fig. 4b-d shows the Pt 4f, O 1s and Ti 2p core-level spectrum, respectively. The Pt 4f core level spectrum in Fig. 4b can be fitted distinguishably to Pt (0), Pt(II) and Pt (IV) oxidation states. The Pt(0) is clearly spitted into the 4f7/2 and 4f5/2 components at 69.98 and 73.95 eV respectively and slightly shifted to lower binding energy compared to Pt/C catalyst because the change in the Pt electronic structure induced by the interaction with TiO2 (Lewera et al., 2011; Cheng et al., 2017a,b). On the other hand, the 4f7/2 and 4f5/2 components of the Pt(II) and Pt(IV) oxidation states at peaks at 76.65 and 79.96 eV cannot be deconvoluted due to overlapping. The appearance of the strong signals for Pt (II) and Pt (IV) oxidized species in the Pt 4f core spectra confirms the existing of highly oxidized Pt surface and Pt-O-Ti coordination which produce strongly interacted Pt/TiO2 catalyst (Lewera et al., 2011; Cheng et al., 2017a,b). The core spectra of O 1s orbital of Pt0.5/meso-TiO2 catalyst is shown in Fig. 4c which can be fitted with three peaks. The main peak at 530.7 eV can be assign to Ti-O bond in anatase phase and the peak at about 532.3 can originate from Ti-O or Pt-O bonds while the peak at 535.8 eV is related to the adsorbed water (Cheng et al., 2017a,b). Fig. 4d shows the core spectra of the Ti 2p orbital of TiO2 substrate in Pt0.5/meso-TiO2 catalyst and the fitting shows that the two main peaks centered at 459.1 and 464.8 eV correspond to the Ti 2p3/2 and Ti 2p1/2 orbitals of the Ti(IV) oxidation state of TiO2, respectively, with the spin–orbit splitting equals 5.7 eV which perfectly matches with the literature (Cheng et al., 2017a,b).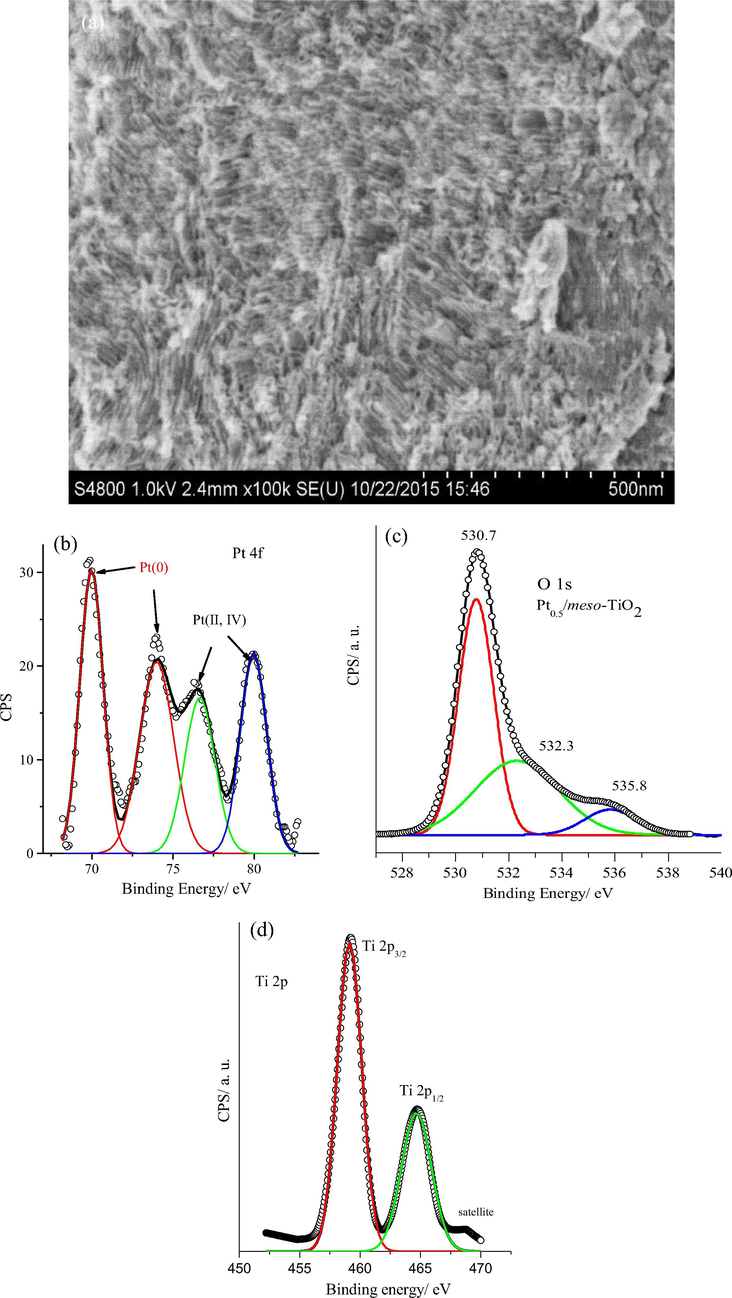
(a) High-resolution SEM of the Pt0.5/meso-TiO2 catalyst; XPS surface analysis of the XPS Pt0.5/meso-TiO2 catalyst (b) Pt 4f, (c) O 1s and (d) Ti 2p core level.
The fine mesostructures of the pure meso-TiO2 and Ptx/meso-TiO2 catalysts were characterized by TEM which allowed a precise determination of the size and distribution of the Pt nanoparticles and provides local structural information about the Pt species supported on the mesoporous TiO2. Fig. 5 shows typical TEM images of the pure meso-TiO2 (Fig. 5a) and Pt0.3/meso-TiO2 (Fig. 5b) catalysts. It can be clearly observed in Fig. 5a the hexagonal mesoporous channels and pores of TiO2 substrate with an average pore size of 3.0 nm which is consistent with the BET measurements shown above as well as with the reported literature (Fattakhova-Rohlfing et al., 2014). The TEM image in Fig. 5b obviously demonstrates the Pt nanoparticles (black spots) are well dispersed within the mesoporous TiO2 and they are not aggregate into larger particles and are roughly in a spherical shape with an average particle size of 3.0 nm as confirmed by the inset particle size distribution histogram. The high-resolution TEM image shown in Fig. 5c, clearly demonstrates the Pt nanoparticles are entirely crystalline because of the existence of lattice fringes through the entire nanoparticles. Moreover, the TEM image gives direct evidence that the Pt nanoparticles are effectively integrated within the mesoporous TiO2 framework. The inter-planar distance of the lattice fringes is d = 0.353 and 0.20 nm which matches with the (1 0 1) and (2 0 0) crystallographic plans of anatase-TiO2 and Pt centred cubic phase, respectively. Moreover, lattice fringes of TiO2 and Pt nanoparticles are overlapped together confirming the existence of strong interaction of the oxidized Pt nanoparticles with the mesoporous TiO2 substrate. An increase in chloroplatinic acid content apparently causes disruption for self-assembly process which produces a Pt nanoparticle supported on the disordered mesoporous TiO2 structure as shown in the TEM image in Fig. 5d of Pt3.0/meso-TiO2 catalyst. However, the surface area of Pt3.0/meso-TiO2 is still maintained (201 m2/g, Table 1 above).
TEM images of two-dimensional hexagonal mesoporous (a) pure meso-TiO2, (b) Pt0.3/meso-TiO2, the inset shows the Pt particle size distribution histogram, (c) high-resolution TEM image of the Pt0.3/meso-TiO2 showing the corresponding crystal lattice parameters, (the insets shows the SAED patterns for the Pt nanoparticles) and (d) TEM image of the Pt3.0/meso-TiO2 catalyst.
3.2 Electrocatalytic activity of Ptx/meso-TiO2 catalysts
The electrocatalytic performance of the as-prepared Ptx/meso-TiO2 catalysts for hydrogen evolution reaction (HER) was evaluated in 0.5 M H2SO4 electrolyte using a typical three-electrode system at room temperature. For the purpose of comparison, blank carbon paper (CP), Pt0.5/bulk-TiO2 and a commercial 10 wt% Pt/C were also examined and by depositing them on CP electrode by electrophoretic deposited at catalyst average loading of 1.1 mg. Fig. 6a shows the linear sweep voltammetry (LSV) without IR compensation of the Ptx/meso-TiO2 catalysts loaded on CP electrode in 0.5 M H2SO4 at a scan rate of 10 mVs−1. Fig. 6b shows a comparison of LSV for Pt0.5/meso-TiO2 with Pt0.5/bulk-TiO2 and commercial 10 wt% Pt/C electrodes in 0.5 M H2SO4 at a scan rate of 10 mVs−1. As shown in Fig. 6a and 6b, the Ptx/meso-TiO2 catalysts exhibit electrocatalytic behaviour similar to that of commercial Pt/C catalyst with a Pt0.5/meso-TiO2 electrode having the most comparable current density to the commercial 10 wt% Pt/C electrode. Moreover, as shown in Fig. 6b and despite a similar platinum loading of 0.5 wt%, the Pt0.5/meso-TiO2 catalyst (green line) shows enhanced hydrogen evolution current enhancement in evaluating against Pt0.5/bulk-TiO2 catalyst (red line). The Pt0.5/meso-TiO2 exhibits significantly lower onset potential (measured graphically at 1.0 mA cm−2) and much higher electrolysis current for hydrogen evolution reaction. Presumably, this can attribute to the open porous structure and high surface area of the mesoporous TiO2 substrate (199 m2/g) in case of Pt0.5/meso-TiO2 catalyst. Fig. 6c and Table 2 show the electrochemical parameters of hydrogen evolution at the Ptx/meso-TiO2 catalysts and in comparison with Pt0.5/bulk-TiO2 and commercial 10 wt% Pt/C electrodes. Obviously and as shown in Fig. 6c, the hydrogen evolution onset potential (at 1.0 mA cm−2) was gradually decreased from −70 mV in case of pure meso-TiO2 to around −10 mV vs. RHE as the Pt content increases to 0.5 wt% indicating more favour hydrogen evolution reaction (HER). However, further increases in Pt content from 0.5 to 3.0 wt% leads to a decrease in current density and negative shift in the hydrogen evolution onset potential as shown in Fig. S1 (Supporting Information, SI). Apparently, as shown in Fig. 5d the disordered nature of the TiO2 support could be the reason behind the drop in electrochemical activity of Pt3.0/meso-TiO2 catalyst due to limitation in mass transport. Fig. 6d, Table 2 and Fig. S2 (SI) show the Tafel plot and Tafel slope for the studied Ptx/meso-TiO2 catalysts in comparison to commercial Pt/C catalyst which was obtained from the linear sweep voltammograms at low overpotential and a scan rate of 10.0 mV s−1. The Tafel slope in case of Pt0.5/bulk-TiO2 equals −180 mV/dec while in case of the mesoporous TiO2 substrate it is considerably decreased to −144 mV/dec for Pt0.1/meso-TiO2 and to an average of −110 mV/dec for Pt0.3/meso-TiO2, Pt0.5/meso-TiO2, and Pt1.0/meso-TiO2 catalysts. Evidently, the incorporation of a very small amount of Pt nanoparticles into mesoporous TiO2 substrate significantly enhanced the hydrogen evolution rate. The Tafel slope value of −110 mV/dec is close to the theoretical value of −118 mV/dec which indicates that HER follow the Volmer mechanism (Bard and Faulkner, 2000; Conway and Tilak, 2002). In a comparison of the catalysts apparent activity, it found that Pt0.5/meso-TiO2 is the most effective catalyst and its activity is enormously higher than the Pt0.5/bulk-TiO2 as well as very close to the activity commercial Pt/C catalyst as can be seen from Fig. 6b and Table 2. This proves that meso-structuring of TiO2 substrate significantly enhanced the hydrogen evolution activity due to enhancing mass transport through the open porous network. In evaluation of the catalyst mass activity as shown in Table 2 the Pt0.1/meso-TiO2 catalyst shows the most active hydrogen evolution catalyst with mass activity of 25.7 A mgPt, at −0.300 V vs RHE which is more than thirty times higher than the commercial Pt/C (0.75 A/mgPt) and the bulk Pt0.5/bulk-TiO2 (0.69 A/cm2 mgPt) catalysts respectively (Table 2). These results demonstrated that modifying the mesoporous TiO2 substrate with a trace amount of Pt nanoparticles tremendously enhance the catalytic activity of HER reaction even though the actual total Pt content within the mesoporous TiO2 is about 0.09, 0.27, and 0.48 wt%, for Pt0.1/meso-TiO2, Pt0.3/meso-TiO2 and Pt0.5/meso-TiO2 catalyst respectively as quantitatively determined by ICP analysis (Table 1).
(a) Linear sweep voltammetry (LSV) of the Ptx/meso-TiO2 and pure meso-TiO2 catalysts loaded on CP electrode in 0.5 M H2SO4 at a scan rate of 10 mV s−1, (b) comparison for linear sweep voltammetry of Pt0.5/meso-TiO2, Pt0.5/bulk-TiO2 and commercial 10 wt% Pt/C catalysts, (c) plot for the hydrogen evolution onset-potential against platinum loading for the Ptx/meso-TiO2 catalysts with different platinum loading, (d) Tafel plot at low polarization for hydrogen evolution reaction at Ptx/meso-TiO2 catalysts, the LSV was IR uncompensated.
Catalyst
Onset potential ± 5 mV vs. RHE (at 1.0 mA cm−2)
η, mV vs. RHE at 10 mA cm−2
Tafel slope, mV/dec
Pt loading ± 0.01, μg/cm2
Specific activity @ −300 mV (mA cm−2)
Mass activity @ −300 mV (A mgPt)
Pure meso-TiO2
−70
−238
–
16.0
–
Pt0.1/meso-TiO2
−26
−200
144
0.98
25.5
25.7
Pt0.3/meso-TiO2
−21
−186
107
2.86
33.0
11.52
Pt0.5/meso-TiO2
−10
−120
118
5.28
68.0
12.88
Pt1.0/meso-TiO2
−17
−137
106
9.45
52.0
5.50
Pt2.0/meso-TiO2
−40
−165
–
16.5
46.2
2.80
Pt3.0/meso-TiO2
−68
−190
–
29.76
37.8
1.27
Pt0.5/bulk-TiO2
−165
−400
180
6.08
4.2
0.69
Pt/C- 10 wt%
− 5
−55
82
110
82.4
0.75
Durability and stability of an electrocatalyst in a harsh environment is an important benchmark test for good electrocatalysts. Fig. 7a shows the linear sweep voltammograms (LSV) for Pt0.5/meso-TiO2 catalyst at 10 mV s−1 in different concentration of the sulfuric acid solution. Obviously, as the concentration of sulfuric acid increases, the hydrogen evolution apparent activity at Pt0.5/meso-TiO2 increases along with the onset potential drifted to more positive potential. Fig. 7b shows the plot obtained for the onset potential and the current density at 0.330 V vs. RHE with the H2SO4 concentration logarithm. Clearly the results show that the hydrogen evolution onset potential is linearly shift to more positive potential as function of the logarithm of the H2SO4 concentration. To evaluate the stability of the best performance catalyst of Pt0.5/meso-TiO2 for hydrogen evolution in the harsh acidic environment, a long-term cycling test and an extended electrolysis at fixed current in 2.0 M H2SO4 were executed. Fig. 7c shows the cyclic voltammetry curves of the Pt0.5/meso-TiO2 catalyst after 1000 cycles. The Pt0.5/meso-TiO2 catalyst appears to be stable 2.0 M H2SO4 and the apparent activity slightly increased after such long cycling. Moreover, as revealed in Fig. 7d, the chronopotentiometry diagram shows the Pt0.5/meso-TiO2 catalyst exhibits excellent long-term electrolysis with a trivial potential drift for a period of 24 h in 2.0 M H2SO4 environment at −20 mA cm−2 and −40 mA cm−2, respectively.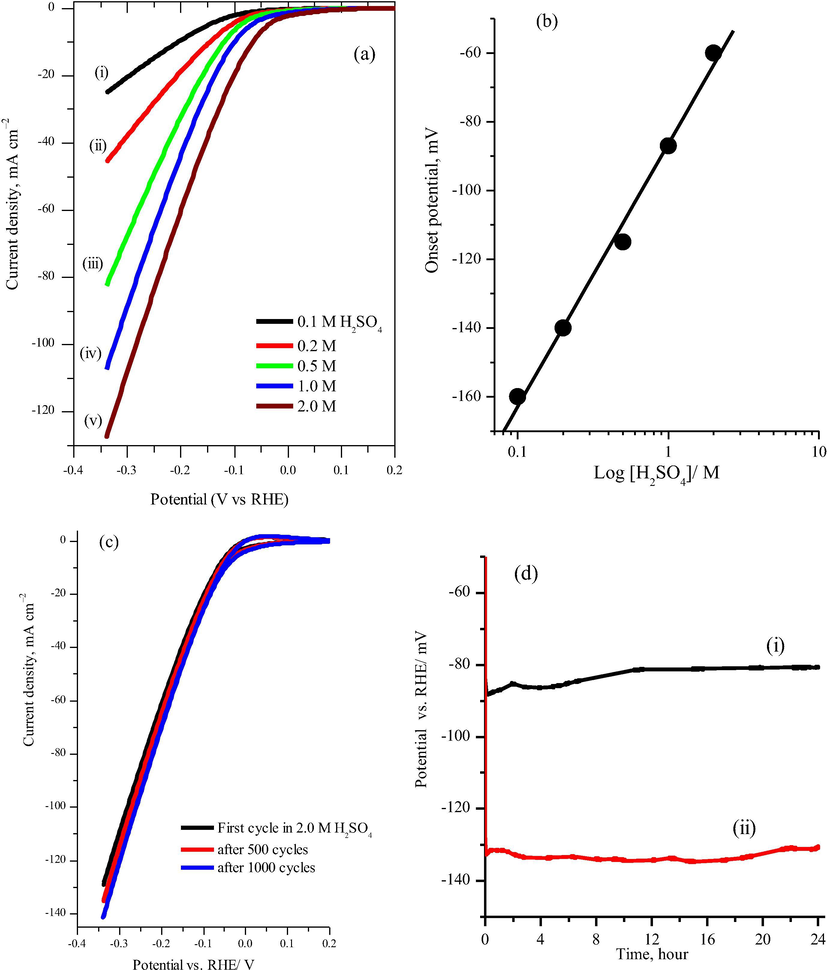
(a) Linear sweep voltammetry for Pt0.5/meso-TiO2 electrode at scan rate of 10 mV s−1 in different concentration of H2SO4, (b) plot of the onset potential and current density at 0.330 V vs. RHE against the logarithm of the concentration, (c) long term cyclic voltammetry (1000 cycles) in 2.0 H2SO4 at scan rate of 10 mV s−1, for clarity, only cycle number 1, 500 and 1000 are shown, (d) the chronopotentiometric curves of the Pt0.5/meso-TiO2 electrode for a continuous electrolysis for a 24 h in 2.0 H2SO4 solution at applied current of (i) −20 mA cm−2 and (ii) −40 mA cm−2 respectively, the LSV was IR uncompensated.
This long-term stability of Pt0.5/meso-TiO2 catalyst can be attributed to the strong interaction between the oxidized Pt nanoparticles with TiO2 substrate and the existence of the Pt(II) and Pt(IV) oxidation states as evidence from TEM and XPS characterizations. These results demonstrate that Ptx/meso-TiO2 catalysts with x in the range of 0.1–0.5 wt% is highly active and extremely stable for HER in acidic media. Therefore using the mesoporous TiO2 as a catalyst substrate can significantly enhance the Pt nanoparticle utilization and durability in comparison to the Pt/C counterparts for the HER (Chen and Kucernak, 2004; Huang and Wang, 2014) alongside the benefit of low platinum loading less cost.
In order to explore the charge-transfer resistance during HER process at Ptx/meso-TiO2 and Pt0.5/bulk-TiO2 electrodes, electrochemical impedance spectroscopy (EIS) was utilized to investigate the reaction kinetics and the electrode/electrolyte interfaces resistance. Fig. 8a shows the Nyquist plots of Pt0.5/meso-TiO2 and Pt0.5/bulk-TiO2 electrodes at overpotential of -200 mV in 0.5 M H2SO4 solution. The impedance spectra of Pt0.5/meso-TiO2 samples were fitted by the so-called 2CPE model comprising two constant phase elements (CPEs), i.e. two R||CPE circuits connected in series: Rpor||CPEpor related to the pore response and Rct||CPEdl related to the HER kinetics. The equivalent circuit model is shown in the inset of Fig. 8a. The equivalent circuit elements include a resistor (Rs), representing the electrolyte resistance which stays almost constant. According to the literature, (Hoshikawa et al., 2006; Sun et al., 2012) the arcs in the higher frequency region characterize the redox reaction at Pt/electrolyte interface whereas the arcs at lower frequency region are credited to the TiO2/electrolyte interface. The impedance of CPEdl is by Eq. (1) (Brug et al., 1984).
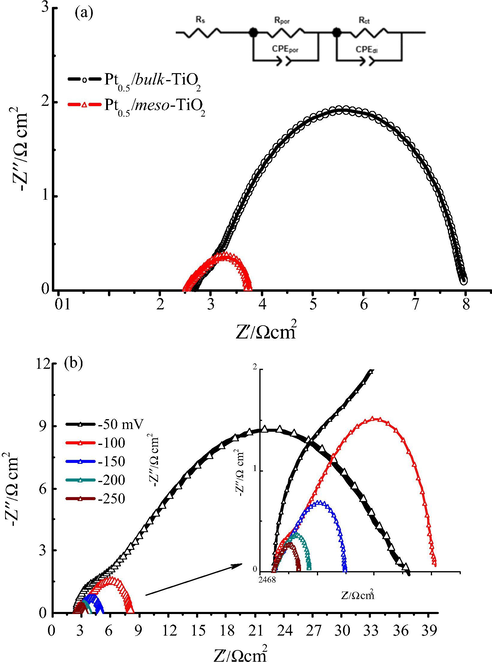
(a) Nyquist plots with an equivalent circuit for Pt0.5/meso-TiO2 and Pt0.5/bulk-TiO2 electrodes in 0.5 M H2SO4 solution at − 200 mV vs. RHE and the inset is the electrical equivalent circuit, (b) Nyquist plots of the Pt0.5/meso-TiO2 electrode at different cathodic overpotential, the inset is zoom in of the higher overpotential zone.
Catalyst
Rs, Ω cm2
Rpor, Ω cm2
Ypor, m Ω cm−2sα
αpor
Rct, Ω cm2
Ydl, m Ω cm−2sα
αdl
Cdl, mF cm−2
R(total), Ω
Rf
Pt0.5/meso-TiO2
2.515
0.456
10.08
0.6078
0.786
8.97
0.846
12.14
3.757
607
Pt0.5/bulk-TiO2
2.656
0.768
3.63
0.6172
4.541
1.652
0.88
1.897
7.965
95
The Rct of Pt0.5/meso-TiO2 is equals 0.786 Ω cm2, which is much lower than its corresponding bulk electrodes (4.54 Ω cm2), signifying rapid charge transport during the HER process. One can observe that the Rtotal for Pt0.5/meso-TiO2 is smaller than for Pt0.5/bulk-TiO2 electrodes. In addition, the smallest Rtotal is obtained from Pt0.5/meso-TiO2, which should present improved charge transportation and higher electrochemical performance. It can also be evidenced that the absolute Cdl values of Pt0.5/meso-TiO2 electrodes obtained from the EIS analysis is about six times higher than Pt0.5/bulk-TiO2 electrodes indicating significantly higher electroactive surface area. The Nyquist plots of the Pt0.5/meso-TiO2 catalyst at different overpotential are shown in Fig. 8b with the corresponding electrochemical impedance parameters at different overpotential are reported in Table 4. It can be seen that the semicircle diameter and the Rpor and Rct values are significantly decreased as the negative overpotential shifted from −50 mV to −250 mV vs. RHE, which suggested that the HER process is significantly improved at the more negative potential that consistent with linear sweep voltammetry. As expected for the HER on Pt0.5/meso-TiO2 electrodes, Cdl decreased with increasing cathodic potential owing to the blocking effect of evolved hydrogen bubbles at higher current densities (see Table 4). Generally, as shown by the EIS analysis in Table 3, the electrical properties of the mesoporous TiO2 catalyst have been improved because the charge transfer resistance significantly decreased from 4.541 to 0.786 Ω cm2 as well as the electrode total resistance has been decreased, consequently, the Pt0.5/meso-TiO2 electrode exhibit more kinetically favourable hydrogen evolution reaction due to better electronic conductivity and diffusion than the Pt0.5/bulk-TiO2 electrode in consistent with those of cyclic voltammetry results.
−E/mV
Rs, Ω cm2
Rpor, Ω cm2
Ypor, m Ω cm−2 sα
αpor
Rct, Ω cm2
Ydl, m Ω cm−2 sα
αdl
Cdl, mF cm−2
R(total), Ω
50
2.348
32.77
4.078
0.611
3.5
0.285
0.713
0.314
38.62
100
2.432
4.207
5.376
0.760
1.54
4.25
0.527
14.87
8.179
150
2.506
0.883
9.187
0.534
1.626
6.76
0.818
10.15
5.015
200
2.515
0.456
10.08
0.608
0.786
8.97
0.846
12.14
3.757
250
2.5
0.585
9.983
0.850
0.294
7.95
0.683
11.17
3.379
Finally, to validate the HER activity of our Ptx/meso-TiO2 catalysts, Table S1 reports the electrocatalytic activity parameters of our Ptx/meso-TiO2 catalysts against the most efficient Pt nanoparticles supported onto carbon and oxides substrates previously reported in the literature. Under similar conditions, the Ptx/meso-TiO2 catalysts exhibited HER activities that are comparable to the values obtained for example for dendrimer Pt nanoparticles supported on carbon nanotubes and carbon nano-horns electrocatalysts (Devadas and Imae, 2016). In addition, the mass activity of our Ptx/meso-TiO2 catalysts is significantly higher than that reported for Pt nanoparticles loaded onto the pure TiO2 substrate and that modified with nitrogen-doped reduced graphene oxide (Roy et al., 2015) as shown in Table S1 (SI). This clearly documented that the hydrogen evolution mass activity of Ptx/meso-TiO2 catalysts is superior to the most of other Pt-based HER electrocatalysts.
4 Conclusion
In summary, we demonstrated the successful preparation of low loading of oxidized Pt nanoparticles (0.1–0.5 wt%) onto a mesoporous TiO2 substrate using evaporation-induced self-assembly (EISA) approach followed by a two-step calcination process. The XRD, BET, XPS and TEM analysis confirmed the formation of highly disperse oxidized Pt nanoparticles with an average diameter of 3.0 nm that are strongly bonded to the highly ordered mesoporous TiO2 framework. An enhanced electrocatalytic performance has been recorded at the Ptx/meso-TiO2 electrocatalysts with hydrogen evolution onset potential of −10 mV vs. RHE, Tafel slope of −110 mV/dec, small charge transfer resistance, and mass activity reaches up to 25.7 A/mgPt at −300 mV vs. RHE. It was suggested that such activity performance results from the strong bonding and the accessibility of oxidized Pt nanoparticles with the TiO2 substrate. Furthermore, the long-term stability is harsh acidic condition indicated the Ptx/meso-TiO2 catalysts are promising catalysts for the hydrogen evolution reaction in acidic solution.
Acknowledgement
This work was funded by The National Plan for Science, Technology and Innovation (MAARIFAH) King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia, Award number AT34-203.
References
- Electrochemical Methods: Fundamentals and Applications (2nd ed.). Wiley; 2000.
- Pt nanoparticles deposited on TiO2 based nanofibers: Electrochemical stability and oxygen reduction activity. J. Power Sources. 2010;195:3105-3110.
- [Google Scholar]
- The analysis of electrode impedances complicated by the presence of a constant phase element. J. Electroanal. Chem.. 1984;176:275-295.
- [Google Scholar]
- Efficient hydrogen evolution catalysis using ternary pyrite-type cobalt phosphosulphide. Nat. Mater.. 2015;14:1245-1251.
- [Google Scholar]
- Highly catalytic active PtNiCu nanochains for hydrogen evolution reaction. Nano Energy. 2014;9:301-308.
- [Google Scholar]
- Molybdenum sulfide clusters nitrogen-doped graphene hybrid hydrogel film as an efficient three dimensional hydrogen evolution electrocatalyst. Nano Energy. 2015;11:11-18.
- [Google Scholar]
- Electrocatalysis under conditions of high mass transport: investigation of hydrogen oxidation on single submicron Pt particles supported on carbon. J. Phys. Chem. B. 2004;108:13984-13994.
- [Google Scholar]
- Highly active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction. Energy Environ. Sci.. 2017;10:2450-2458.
- [Google Scholar]
- Highly active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction. Energy Environ. Sci.. 2017;10:2450-2458.
- [Google Scholar]
- Support effect in high activity gold catalysts for CO oxidation. J. Am. Chem. Soc.. 2006;128:917-924.
- [Google Scholar]
- Interfacial processes involving electrocatalytic evolution and oxidation of H2, and the role of chemisorbed H. Electrochim. Acta. 2002;47:3571-3594.
- [Google Scholar]
- Hydrogen evolution reaction efficiency by low loading of platinum nanoparticles protected by dendrimers on carbon materials. Electrochem. Commun.. 2016;72:135-213.
- [Google Scholar]
- Low-cost hydrogen-evolution catalysts based on monolayer platinum on tungsten monocarbide substrates. Angew. Chem. Int. Ed.. 2010;49:9859-9862.
- [Google Scholar]
- Toward highly stable electrocatalysts via nanoparticle pore confinement. J. Am. Chem. Soc.. 2012;134:20457-20465.
- [Google Scholar]
- An efficient molybdenum disulfide/cobalt diselenide hybrid catalyst for electrochemical hydrogen generation. Nat. Commun.. 2015;6:5982.
- [Google Scholar]
- A mini review on nickel-based electrocatalysts for alkaline hydrogen evolution reaction. Nano Res.. 2016;9:28-46.
- [Google Scholar]
- Harnisch, F., Sievers, G., Schröder, U., 2009. Tungsten carbide as electrocatalyst for the hydrogen evolution reaction in pH neutral electrolyte solutions. Appl. Catal., B, 89, pp. 455–458.
- Catalysis for low temperature fuel cells Part III: challenges for the methanol fuel cell. Platinum Met. Rev.. 2002;46:146-164.
- [Google Scholar]
- Effects of electrolyte in dye-sensitized solar cells and evaluation by impedance spectroscopy. Electrochim. Acta. 2006;51:5286-5294.
- [Google Scholar]
- Recent progress on carbon-based support materials for electrocatalysts of direct methanol fuel cells. J. Mater. Chem. A. 2014;2:6266-6291.
- [Google Scholar]
- Development of a titanium dioxide-supported platinum catalyst with ultrahigh stability for polymer electrolyte membrane fuel cell applications. J. Am. Chem. Soc.. 2009;131:13898-13899.
- [Google Scholar]
- Activity origin and catalyst design principles for electrocatalytic hydrogen evolution on heteroatom-doped graphene. Nat. Energy. 2016;1:16130.
- [Google Scholar]
- Design of active and stable Co-Mo-Sx chalcogels as pH-universal catalysts for the hydrogen evolution reaction. Nat. Mater.. 2016;2016(15):197-203.
- [Google Scholar]
- Service life test of the NiSn coatings as cathodes for hydrogen evolution in industrial chlor-alkali electrolysis. Int. J. Hydrogen Energy. 2014;39:8947-8958.
- [Google Scholar]
- Highly dispersed platinum nanoparticles supported on silica as catalyst for hydrogen production. RSC Adv.. 2014;4:50114.
- [Google Scholar]
- Khdary, N.H., Ghanem, M.A., 2016. Method for synthesizing platinum nanoparticles incorporated on silica. US 9243338 B2.
- Gas adsorption characterization of ordered organic−inorganic nanocomposite. Chem. Mater.. 2001;13:3169-3183.
- [Google Scholar]
- Sherrer after sixty years: A survey and some new results in determination of crystallite size. J. Appl. Cryst.. 1978;11:10-13.
- [Google Scholar]
- Metal support interactions between nanosized Pt and metal oxides (WO3 and TiO2) studied using X-ray photoelectron spectroscopy. J. Phys. Chem. C. 2011;115:20153-20159.
- [Google Scholar]
- Activating and optimizing MoS2 basal planes for hydrogen evolution through the formation of strained sulphur vacancies. Nat. Mater.. 2016;15:48-53.
- [Google Scholar]
- Highly active, stable oxidized platinum clusters as electrocatalysts for the hydrogen evolution reaction. Energy Environ. Sci.. 2017;2017(10):2450-2458.
- [Google Scholar]
- A nanoporous molybdenum carbide nanowire as an electrocatalyst for hydrogen evolution reaction. Energy Environ. Sci.. 2014;7:387-392.
- [Google Scholar]
- Merino-Jimenez, I., Santoro, C., Rojas-Carbonell, S., Greenman, J., Ieropoulos, I., Atanassov, P., 2016. Carbon-based air-breathing cathodes for microbial fuel cells. Catalysts 6 (2016) 127. Catalysts, 6, 127.
- Recent developments of molybdenum and tungsten sulfides as hydrogen evolution catalysts. Energy Environ. Sci.. 2011;4:3878-3888.
- [Google Scholar]
- Nanostructured hydrotreating catalysts for electrochemical hydrogen evolution. Chem. Soc. Rev.. 2014;43:6555-6569.
- [Google Scholar]
- Nitrogen doped reduced graphene oxide based Pt–TiO2 nanocomposites for enhanced hydrogen evolution. J. Phys. Chem. C. 2015;119:19117-19125.
- [Google Scholar]
- Chemical deposition and electrocatalytic activity of platinum nanoparticles supported on TiO2 nanotubes. Int. J. Electrochem. Sci.. 2013;8:2468-2478.
- [Google Scholar]
- Minimizing the use of platinum in hydrogen-evolving electrodes. Angew. Chem. Int. Ed.. 2011;50:1476-1477.
- [Google Scholar]
- Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)₂-Pt interfaces. Science. 2011;334:1256.
- [Google Scholar]
- Single-atom catalysis using Pt/graphene achieved through atomic layer deposition. Sci. Rep.. 2013;3:1775.
- [Google Scholar]
- High efficiency dye-sensitized solar cells based on a Bi-layered photoanode made of TiO2 nanocrystallites and microspheres with high thermal stability. J. Phys. Chem. C. 2012;116(22):11859-11866.
- [Google Scholar]
- Electrical and optical properties of TiO2 anatase thin films. J. Appl. Phys.. 1994;1994(754):2042.
- [Google Scholar]
- Enhanced catalytic activity in strained chemically exfoliated WS2 nanosheets for hydrogen evolution. Nat. Mater.. 2013;12:850-855.
- [Google Scholar]
- Highly Stable and CO-Tolerant Pt/Ti0.7W0.3O2 electrocatalyst for proton-exchange membrane fuel cells. J. Am. Chem. Soc.. 2010;132:10218-10220.
- [Google Scholar]
- Designed synthesis of size-controlled Pt-Cu alloy nanoparticles encapsulated in carbon nanofibers and their high efficient electrocatalytic activity toward hydrogen evolution reaction. Adv. Mater. Interfaces 2017 10.1002/admi.201700005
- [Google Scholar]
- Optimizing the Volmer step by single-layer nickel hydroxide nanosheets in hydrogen evolution reaction of platinum. ACS Catal.. 2015;5(2015):3801-3806.
- [Google Scholar]
- In situ synthesis of ordered mesoporous Co-doped TiO2 and its enhanced photocatalytic activity and selectivity for the reduction of CO2. J. mater. Chem. A. 2015;3(2015):9491-9501.
- [Google Scholar]
- Monodisperse Pt nanoparticles anchored on N-doped black TiO2 as, high performance bifunctional electrocatalyst. J. Alloys Comp.. 2017;701:669-675.
- [Google Scholar]
- Metallic WO2–Carbon mesoporous nanowires as highly efficient electrocatalysts for hydrogen evolution reaction. J. Am. Chem. Soc.. 2015;137:6983-6986.
- [Google Scholar]
- A Perovskite electrocatalyst for efficient hydrogen evolution reaction. Adv. Mater.. 2016;28:6442-6448.
- [Google Scholar]
- MoSe2 nanosheets grown on polydopamine-derived porous fibers: a high-performance catalyst for hydrogen evolution reaction. Adv. Mater. Interfaces. 2016;4:1600825.
- [Google Scholar]
- Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res.. 2013;46:1740-1748.
- [Google Scholar]
- Ultrathin platinum nanowires grown on single-layered nickel hydroxide with high hydrogen evolution activity. Nat. Commun.. 2015;6:6430.
- [Google Scholar]
- Non-syngas direct steam reforming of methanol to hydrogen and carbon dioxide at low temperature. Nat. Commun.. 2012;3 1230 1230
- [Google Scholar]
- General Formation of M-MoS3 (M = Co, Ni) hollow htructures with enhanced electrocatalytic activity for hydrogen evolution. Adv. Mater.. 2016;28:92-97.
- [Google Scholar]
- The stability of Pt/C catalyst in H3PO4/PBI PEMFC during high temperature life test. J. Power Sources. 2007;164:126-133.
- [Google Scholar]
- Carbon nanotube architectures as catalyst supports for proton exchange membrane fuel cells. Energy Environ. Sci.. 2010;3:1286-1293.
- [Google Scholar]
- Advancing the electrochemistry of the hydrogen-evolution reaction through combining experiment and theory. Angew. Chem. Inter. Ed.. 2015;54(2015):52-65.
- [Google Scholar]
- Outstanding hydrogen evolution reaction catalyzed by porous nickel diselenide electrocatalysts. Energy Environ. Sci.. 2017;2017(10):1487-1492.
- [Google Scholar]
- Highly active and durable self-standing WS2/graphene hybrid catalysts for the hydrogen evolution reaction. J. Mater. Chem. A. 2016;4:9472-9476.
- [Google Scholar]
- Efficient hydrogen evolution by ternary molybdenum sulfoselenide particles on self-standing porous nickel diselenide foam. Nat. Commun.. 2016;7:12765.
- [Google Scholar]
- A review of hollow Pt-based nanocatalysts applied in proton exchange membrane fuel cells. J. Power Sources. 2013;232:310-322.
- [Google Scholar]
- Hierarchical nanosheet-based MoS2 nanotubes fabricated by an anion-exchange reaction of MoO3–amine hybrid nanowires. Angew. Chem. Int. Ed.. 2013;52:8602-8606.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.arabjc.2018.04.010.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




