

Translate this page into:
Natural products-isoxazole hybrids: A review of developments in medicinal chemistry
⁎Corresponding author. luantian@symc.edu.cn (Tian Luan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
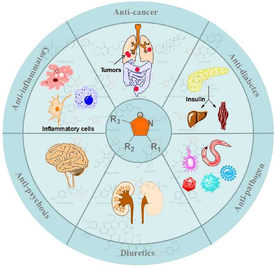
Five-membered heterocycles containing nitrogen, oxygen atoms, especially isoxazoles, are widely present in many natural products and drugs and have relevant medical and pharmaceutical applications. Due to their rich biological activity, the development of novel isoxazole derivatives has been the focus of researchers and scientists. It is worth noting that a large number of isoxazoles have been used frequently as clinical drugs to treat various diseases, showing great development value and wide medicinal potential. In this review, the research progress of isoxazole natural products in anti-bacterial, anti-tuberculosis, anti-virus, anti-tumor, anti-inflammation, anti-diabetes, anti-parasite, anti-psychosis and other biological activities was reviewed. Finally, the research and development prospects of isoxazole natural products were prospected. This review will provide reference for the design of natural isoxazole products with stronger activity and less toxicity.
Keywords
Isoxazole
Natural products
Pharmacological activity
Structural modifications

1 Introduction
N-containing heterocyclic compounds are a class of important heterocyclic compounds, that are widely used in the search for new bioactive compounds and play an important role in the fields of medicinal chemistry and organic chemistry (Sysak and Obminska-Mrukowicz, 2017). Azoles are a class of 5-membered nitrogen-containing heterocyclic compounds with one or more heteroatoms, such as nitrogen, oxygen, or sulfur (El-Garhy, 2015) (Fig. 1).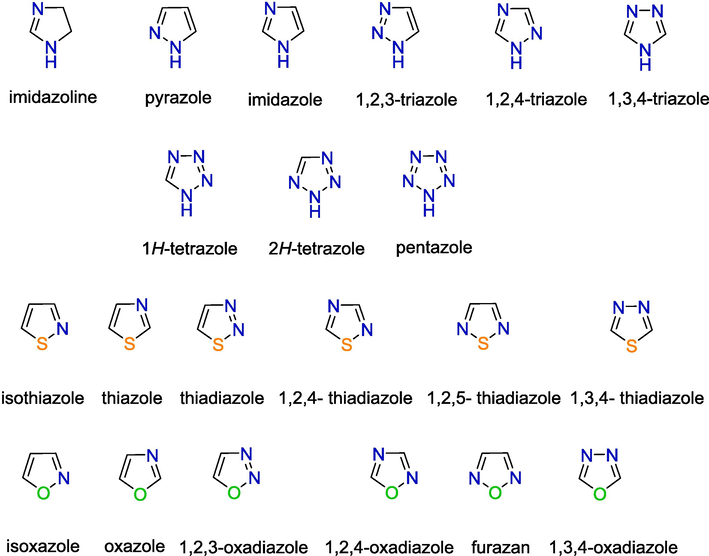
Various types of azoles.
Isoxazole is a class of heterocyclic compounds widely used in drug discovery research. The structural characteristics of isoxazole make it possible for a variety of noncovalent interactions, especially hydrogen bonds (hydrogen bond receptors N and O), π-π stack (unsaturated five-membered ring), and hydrophilic interactions (cLogP = 0.334 at physiological pH = 7.4) (Zhu et al., 2018). The introduction of isoxazole can improve efficacy, reduce toxicity, and improve pharmacokinetic characteristics (Sysak and Obminska-Mrukowicz, 2017; Anand and Singh, 2014; Barmade et al., 2016). Isoxazoles can interact with a wide range of protein targets, so isoxazoles have a wide range of biological activities, including anti-cancer, anti-bacterial, anti-fungal, anti-viral, anti-microbial, anti-tuberculosis, anti-inflammatory, etc. (Zhu et al., 2018). Currently, there are dozens of drugs available on the market that contain isoxazole fragments. These drugs can alleviate and eliminate the effects of various diseases, thereby safeguarding human health. (Fig. 2 and Table 1) (Thakur et al., 2022).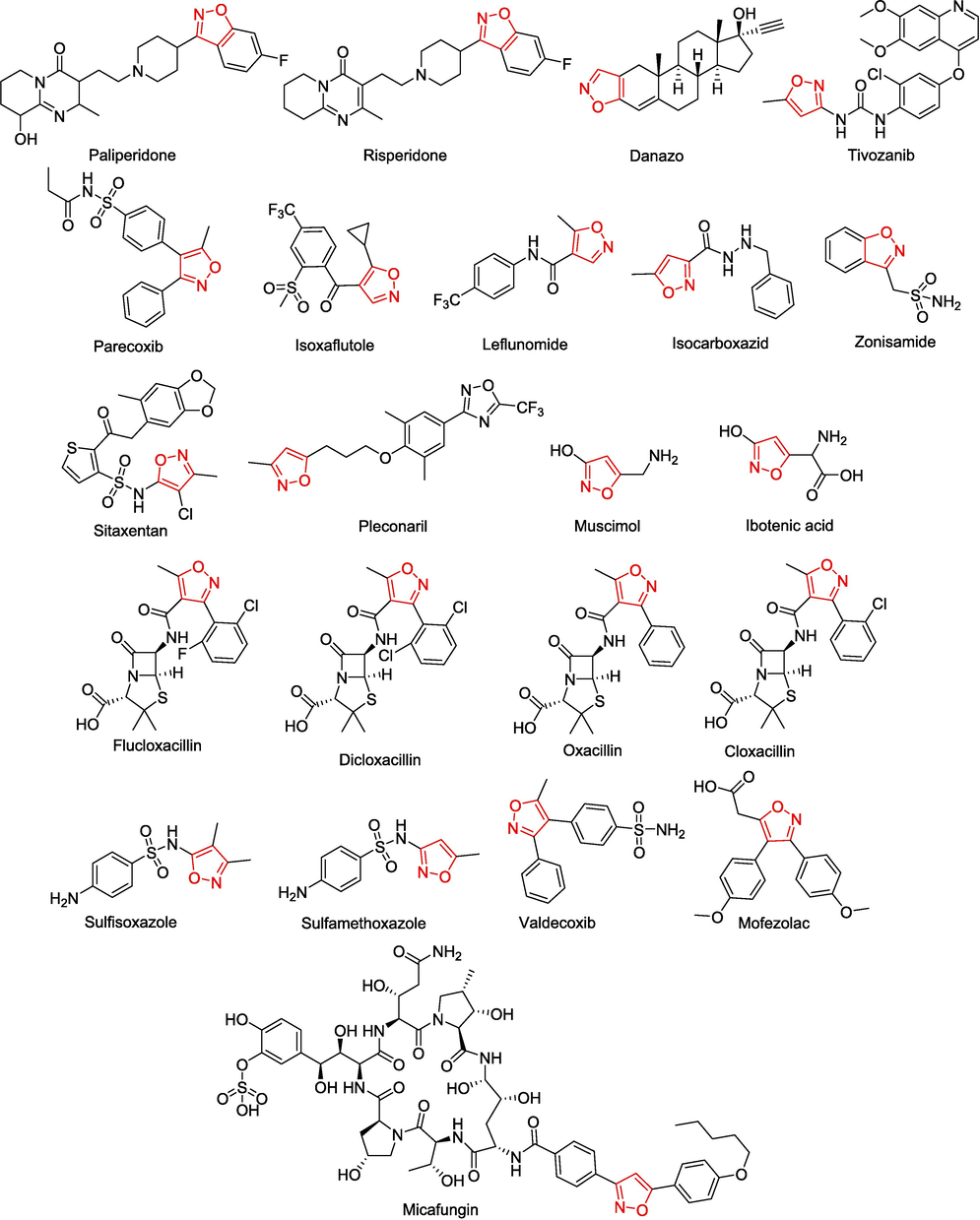
Isoxazole-based commercially available drugs.
Drug
Pharmacological mechanism
Treatment/Purpose
Ref.
Paliperidone
Dopamine type 2 receptor blockade
Schizophrenia
(Dolder et al., 2008)
Risperidone
Serotonin 5-HT2 receptor blockade
Schizophrenia
(Megens et al., 1994)
Danazol
Gonadotropin inhibitor
Endometriosis
(Andrews and Wentz, 1975)
Tivozanib
VEGF-TKI with high selectivity for VEGF receptors 1–3
Renal cell carcinoma
(Salgia et al., 2020)
Parecoxib
COX-2 inhibitor
Anti-inflammatory
(Urdaneta et al., 2009)
Isoxaflutole
Hydroxyphenylpyruvate dioxygenase (HPPD) inhibitor
Herbicidal
(Pallett et al., 1997; Viviani et al., 1998)
Leflunomide
Interfere with the metabolism of pyrimidine nucleotides
Rheumatoid arthritis
(Greene et al., 1995)
Isocarboxazid
The monoamine oxidase inhibitor
Treatment-resistant depression
(Larsen et al., 2015)
Zonisamide
It is likely to involve blockade of both sodium and T-type calcium channels
Anti-epileptic
(Mimaki, 1998)
Sitaxentan
Highly selective endothelin (ET) A receptor antagonist
Pulmonary arterial hypertension
(Scott, 2007)
Pleconaril
Pleconaril integrates into the capsid of picornaviruses, including enteroviruses and rhinoviruses, preventing the virus from attaching to cellular receptors and uncoating to release RNA into the cell
Enterovirus infections
(Rotbart et al., 2001)
Muscimol
GABAA-agonist
Anticonvulsant
(Allen et al., 2008; Collins, 1980)
Ibotenic acid
Aspartic acid receptor agonists
Hallucinogens
(Stebelska, 2013; Krogsgaard-Larsen et al., 1980)
Flucloxacillin
β-lactamase inhibitor
Anti-bacterial
(Lima et al., 2020)
Dicloxacillin
β-lactamase inhibitor
Anti-bacterial
(Lima et al., 2020)
Oxacillin
β-lactamase inhibitor
Anti-bacterial
(Lima et al., 2020)
Cloxacillin
β-lactamase inhibitor
Anti-bacterial
(Lima et al., 2020)
Sulfisoxazole
Endothelin receptor antagonist
Anti-bacterial
(Syed et al., 2006; Uchino et al., 2008)
Sulfamethoxazole
Competitive antagonists of para-aminobenzoic acid (PABA)
Anti-bacterial
(Yang et al., 2017; Brain et al., 2008)
Valdecoxib
COX-2 inhibitor
Anti-inflammatory
(Talley et al., 2000; Nussmeier et al., 2005)
Mofezolac
COX-1 inhibitor
Anti-inflammatory
(Pati et al., 2019; Cingolani et al., 2017)
Micafungin
Inhibiting the production of 1,3-β-D-glucan
Anti-fungal
(Wasmann et al., 2018)
Natural products extracted from microorganisms, plants, and animals have significant structural diversity and biological properties, and these natural products are an important source of drug discovery (Huang et al., 2022; Chopra and Dhingra, 2021). However, most natural products have low pharmacological activity or too many adverse reactions to be used directly in the clinical treatment of diseases (Wang et al., 2023). Structural modification can improve the physical and chemical properties and biological activities of natural products, reduce adverse reactions, and improve selectivity. Furthermore, structural modifications may also exhibit biological activities completely different from those of their parent compounds (Yao et al., 2017). Isoxazole plays an important role in natural products and has significant biological activity. More and more studies have reported the modification of natural products with isoxazole (Farooq and Ngaini, 2022).
Some reviews have been conducted on the research of some isoxazole derivatives as drugs (Zhang et al., 2011; Ohnmacht and Neidle, 2014; Defossa and Wagner, 2014), but there is no systematic report on the development status of isoxazole in natural product derivatives. Given this, the research progress of isoxazole derivatives in the field of natural product modification in anti-microbial, anti-fungal, anti-viral, anti-tuberculosis, anti-cancer, anti-inflammatory, hypoglycemic, anti-parasitic, anti-obesity, anti-psychosis, anti-oxidation and other aspects was reviewed. The research direction of isoxazole-natural products in medicinal chemistry was explored.
2 Hybridization of natural product and isoxazole
To facilitate the classification and management of the literature, all relevant literature is divided into eleven categories according to the introduction framework: cinnamamide and cinnamic acids, chalcones, styrene and polyphenols, coumarins, lignin, quinones, flavonoids, steroids, terpenes, alkaloids, carbohydrate, and other natural components.
2.1 Cinnamamide and cinnamic acid-isoxazole hybridization
Shultz et al. (Shultz et al., 2011) designed and synthesized N-hydroxy-(4-oxime)-cinnamide derivatives containing indole. Among them, compound 1 containing isoxazole showed certain inhibitory activity against HDAC-1 with an IC50 value of 17 nM. The synthesis route of compound 1 is depicted in Fig. 3. Methyl 4-formylcinnamate was reacted with (2-(3,5-dimethylisoxazol-4-yl)-1H-indol-3-yl)methanamine via reductive amination to generate substituted methyl 4-aminomethyl cinnamates, which were then converted to the corresponding hydroxamates using aqueous hydroxylamine in methanolic sodium methoxide. Histone deacetylases (HDACs) are enzymes involved in chromatin remodeling that play a key role in epigenetic regulation of gene expression. HDAC catalyzes the removal of acetyl groups from core histones and other lysine residues that control cell functions such as proliferation, migration, differentiation, and cell death. Therefore, inhibition of HDAC is a practical treatment for the discovery of new targeted anticancer drugs (Bolden et al., 2006). Giannini et al. (Giannini et al., 2009) designed a series of compounds with a N-hydroxy-(4-oxime)-cinnamide scaffold. The cytotoxic activity of the compounds against the NB4, H460 and HCT116 cell lines and their inhibitory activity against class I, II, and IV HDacs were evaluated. Compound 2, which contains isoxazole, exhibited significant inhibitory activity against HCT116 and NCI-H460 cell lines with IC50 values of 7.0 and 4.0 μM, respectively. Compound 2 also demonstrated good inhibitory activity against HDAC6 and HDAC8 with IC50 values of 0.175 and 0.127 μM, respectively.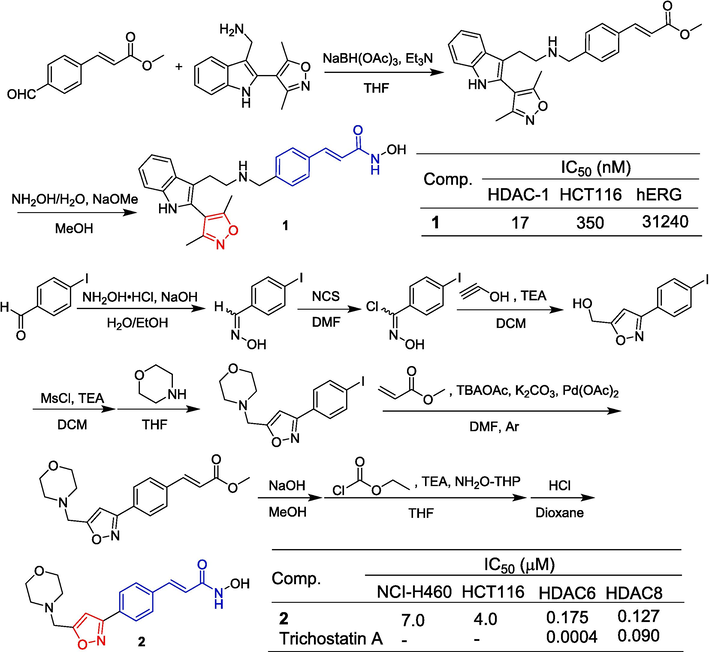
Cinnamamide-isoxazole derivatives 1–2.
Botulinum neurotoxin (BoNTs) is the most toxic protein known to man, and exposure to it can cause flaccid paralysis. Because these proteins are extremely potent, they have been studied as possible bioterrorism weapons. Smith et al. (Smith et al., 2012) reported a series of 2,4-dichloro cinnamyl hydroxamate derivatives and tested their inhibition of the BoNT/A light chain metalloprotease. Compound 3, which contains isoxazole, exhibited a certain inhibitory effect on BoNT/A with an IC50 value of 1.16 μM.
EP3 antagonists have the potential to reduce the risk of atherosclerotic thrombosis without increasing the risk of bleeding. Singh et al. (Singh et al., 2010) reported on the pharmacological effects of a series of EP3 receptor antagonists. In the absence of serum protein, compound 4 containing isoxazole showed some binding capacity to hEP3 with an IC50 value of 14.5 μM.
Shultz et al. (Shultz et al., 2011) described the design, synthesis, and biological evaluation of a novel isoindolyl hydroxate. Compound 5 containing isoxazole inhibited HDAC1 with an IC50 of 0.17 μM. The proliferation of HCT116 cells was inhibited with an IC50 of 2.2 μM. The synthesis route of the compound is shown in Fig. 4. Starting with 5-Bromoisoindoline-1,3-dione as the starting material, it was converted to tert-butyl carbamate through reduction and Boc protection. A subsequent Heck reaction with methyl acrylate was followed by removal of the Boc protecting group. The isoxazole fragment was then introduced through a nucleophilic substitution reaction. Finally, compound 5 was obtained by treating the methyl esters with hydroxylamine in the presence of sodium methoxide.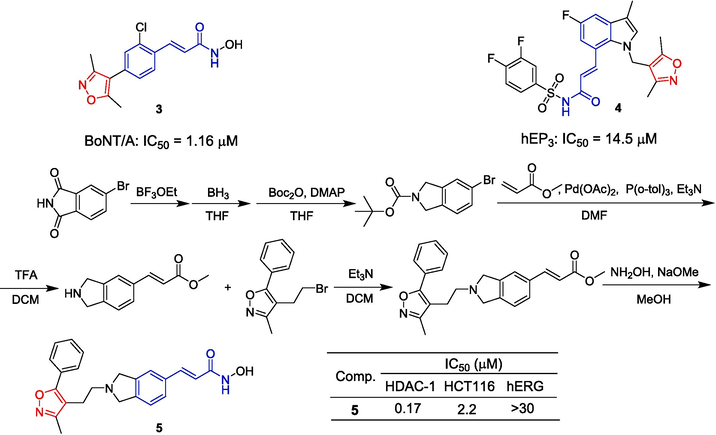
Cinnamamide-isoxazole derivatives 3–5.
Sunduru et al. (Sunduru et al., 2015) designed and synthesized a series of salicylhydrazide sulfonamides and investigated the in vitro replication activity of these compounds against Chlamydia trachomatis (C. trachomatis) and Chlamydia pneumoniae (C. pneumoniae) in HeLa cells. Compound 6 showed an inhibitory effect on C. trachomatis at a concentration of 50 μM with an inhibitory rate of 33.4 %. Compound 7 showed inhibitory effects on C. trachomatis and C. pneumoniae at a concentration of 50 μM, and inhibitory rates were 28.0 % and 22.4 %. Furthermore, compound 7 inhibits T3S in Yersinia pseudotuberculosis (Y. pseudotuberculosis) in a dose-dependent manner.
Marwaha et al. (Marwaha et al., 2014) found that acylated sulfonamides are novel growth inhibitors of the sexually transmitted pathogen C. trachomatis. Compounds 8–12 showed significant inhibitory effects on C. trachomatis with MIC values of 12 μM. These compounds showed strong inhibitory effects on C. trachomatis and C. pneumoniae with an IC50 ranging from 4.2 to 7.7 μM. There was no significant toxicity to host cells (HeLa: IC50 > 50 μM).
Caffeic acid sulfonamide derivatives were synthesized by coupling sulfonamide compounds to the caffeic acid main chain. Free radical scavenging ability was determined by the DPPH method. CASMZ (13) has a certain ability to clear DPPH, and the IC50 value is 42.2 μM, close to the activity of caffeic acid (IC50 value 40.9 μM). The pretreatment of cells with CASMZ (13) inhibited the viability of H2O2-induced cells, decreased ROS and MDA production, increased antioxidant enzyme activity, and further up-regulated the expression of Nrf2 and its target genes (Peng et al., 2020) (Fig. 5).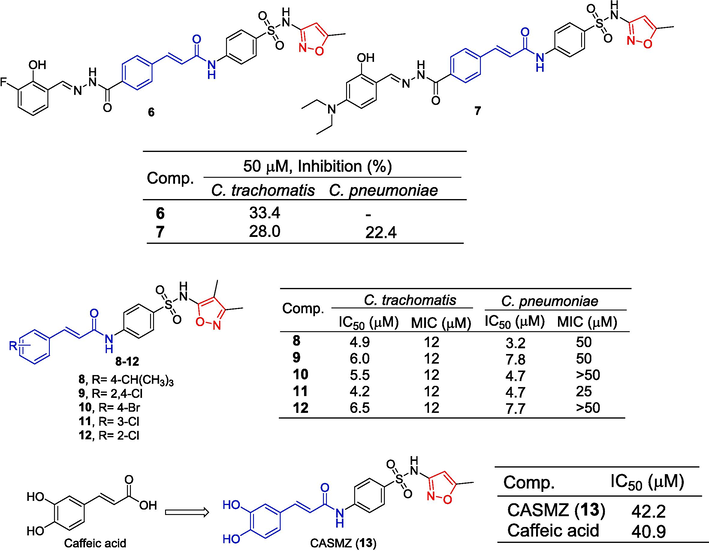
Cinnamamide-isoxazole derivatives 6–13.
Darwish et al. (Darwish et al., 2014) synthesized a variety of heterocyclic compounds containing sulfonamides and evaluated the antibacterial and anti-fungal activities of the compounds newly synthesized in vitro. Compounds 14 and 15 containing cinnamic acid showed significant inhibitory activity against a variety of bacteria and fungi. The diameters of the inhibited region of 14 and 15 on Bacillus subtilis (RCMB-010067) are 18.20 cm and 18.30 cm. The diameters of the inhibition zone of Geotricum candidum (RCMB-05097) are 18.30 and 19.00 cm.
Mohamed et al. (Mohamed et al., 2021) designed and synthesized a series of heterocyclic cyanoacrylamide and tested the anti-tumor activity of these compounds against four cell lines (A549, MCF-7, HepG-2, and Wi38). Compounds 16 and 17 containing isoxazole exhibited varying degrees of inhibitory activity against A549, MCF-7, HepG-2, and WI38 cell lines with an IC50 ranging from 187.1 to 1057 µg/mL. Compound 16 had the most obvious inhibitory effect on A549 cells with an IC50 value of 187.1 µg/mL. Compounds 16 and 17 increased the expression of apoptotic genes (BAX and P53) and decreased the expression of anti-apoptotic genes (Bcl2 and CDK4). Compounds 16 and 17 also increased caspase-3, -8, and -9 activity. Flow cytometry showed that compound 6 induced cell cycle arrest in the S phase and compound 17 induced cell cycle arrest in the G2/M phase. Compounds 16 and 17 caused significant DNA breaks in HepG-2 cells compared to control cells. Therefore, compounds 16 and 17 inhibit liver cancer growth by inducing endogenous and exogenous pathways of the apoptosis process (Fig. 6).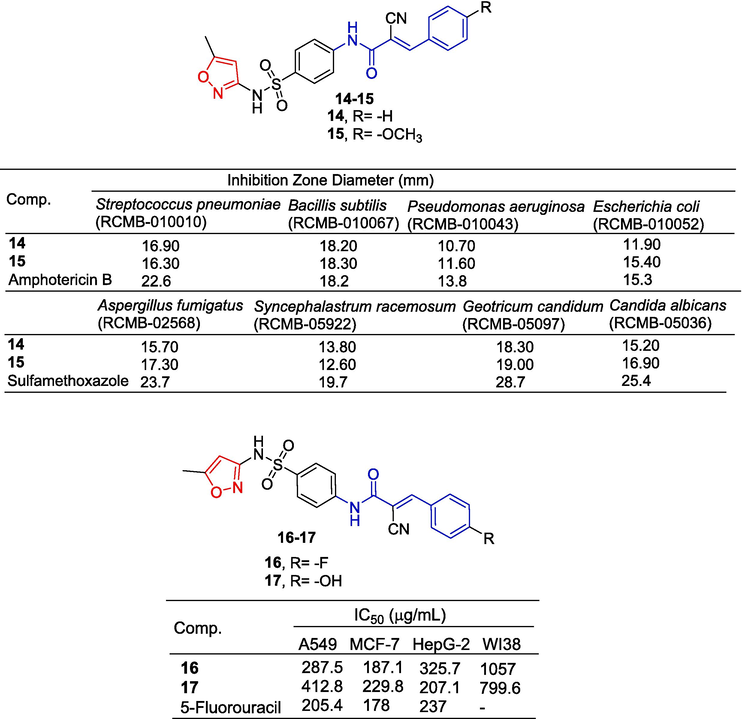
Cinnamamide-isoxazole derivatives 14–17.
Kamal et al. (Kamal et al., 2015) synthesized isoxazole-linked aryl cinnamic acid conjugates. Inhibition of these compounds on the growth of multiple human cancer cell lines (HeLa, DU-145, A549, and MDA-MB231) was evaluated. Among them, 18–20 showed good cytotoxicity in HeLa and MD-MB231 cells, with IC50 values between 2.3 and 3.3 µM. The preliminary structure–activity relationship indicated that the activity could be enhanced by introducing electron-donating groups at both the meta and para-positions of the phenyl group.
Kumar et al. (Kumar et al., 2017) synthesized a series of arylisoxazole-oxyindole derivatives and evaluated the anti-proliferative activity of these compounds against A549, HeLa, MCF-7, and DU-145 cell lines. Ethyl 2,4-dioxo-4-(substituted phenyl)butanoates were obtained by reacting 1-(3,4,5-trimethoxyphenyl)ethan-1-one with diethyl oxalate in the presence of sodium ethanolate in ethanol. Further cyclization was achieved by NH2OH•HCl in ethanol to produce ethyl 3-substituted phenylisoxazole-5-carboxylate. The carboxylates were then reduced to (3-substituted phenylisoxazol-5-yl)methanol using LiAlH4. The resulting compounds were selectively oxidized to 3-arylisoxazole-5-carbaldehyde by IBX in DMSO. Finally, the target compounds were synthesized through the Knoevenagel reaction between equimolar mixtures of 3-arylisoxazole-5-carbaldehyde and oxindoles (Fig. 7). Compounds 21–23 have excellent to moderate cytotoxicity, with IC50 values ranging from 0.82 to 2.59 µM. Compounds 22 and 23 showed the strongest inhibitory effects on A549 cells, with IC50 values of 0.91 and 0.82 µM, respectively.
Cinnamamide and cinnamic acid-isoxazole derivatives 18–24.
Cheng et al. (Cheng et al., 2006) reported the design and synthesis of a series of Malonyl-Coa decarboxylase (MCD) inhibitors. Compound 24 contains a C3-conjugated ethyl acrylate isoxazole fragment, showing strong inhibitory activity against MCD with an IC50 value of 3.16 µM.
To obtain new PDHK inhibitors, Meng et al. (Meng et al., 2014) designed and synthesized a series of diarylisoxazole HSP90 inhibitors using the homology of the ATP binding bag between HSP90 and PDHK. The starting material was reduced using LiAlH4 conditions to obtain the corresponding alcohol, which was subsequently oxidized with activated MnO2 to yield the aldehyde. Following this, the Horner-Wadsworth-Emmons reaction was employed to generate an α,β-unsaturated ester. Hydrolysis of this ester with aqueous sodium hydroxide resulted in the formation of an acid. Subsequent amidation with various substituted piperazines and debenzylation using boron trichloride provided compounds 25–28. These compounds were further demethylated utilizing boron tribromide to produce phenolic compounds 29–31 (Fig. 8). The 3-linked acrylamide compounds 25, 26, and 29 of isoxazole showed significant inhibitory activity against PDHK1, with IC50 values ranging from 0.491 to 0.604 µM. Compounds 27, 28, 30, and 31 showed significant inhibitory activity against HSP90 with IC50 values ranging from 0.491 to 0.604 µM. Furthermore, compound 29 has greater cellular activity and can effectively regulate the metabolic profile of cancer cells and inhibit cancer cell proliferation, which can be demonstrated by increasing oxidative phosphorylation, reducing glycolysis, and related oxidative stress. The preliminary structure–activity relationship suggests that, for PDHK1, the introduction of substituting phenyl groups on piperazine demonstrates superior inhibitory activity compared to heterocyclic groups. Conversely, for HSP90, the introduction of heterocyclic groups on piperazine exhibits enhanced inhibitory activity relative to substituting phenyl groups.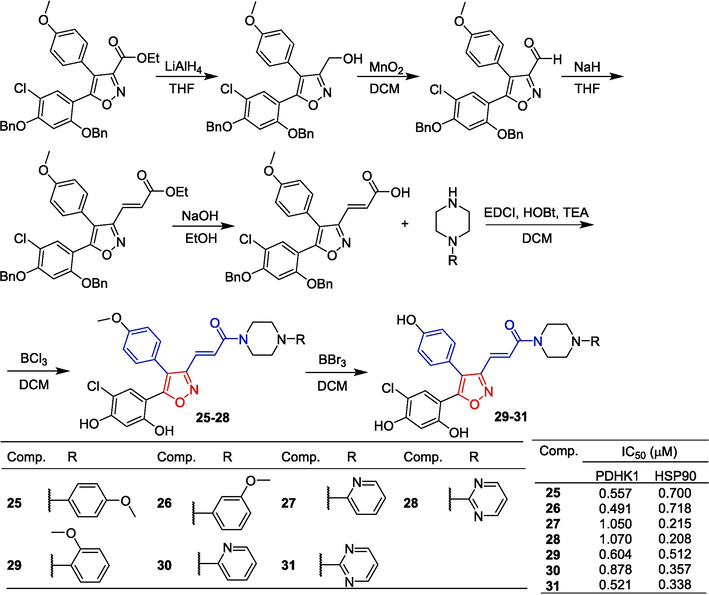
Cinnamamide-isoxazole derivatives 25–31.
Pfizer (formerly Agouron) has developed AG-7088, a peptide-aldehyde that targets the 3C protease of human rhinovirus (HRV) with the potential to treat the common cold (Witherell, 2000; McKinlay, 2001). Studies have shown that AG7088 is effective against EV6 and EV9, with EC50 of 43 nM and 15 nM, respectively (Binford et al., 2005). Based on the crystal structure of the RV 3C protease complex with AG7088, Kuo et al. (Kuo et al., 2008) designed and synthesized a series of AG7088 analogs and further tested their antiviral activity. The starting material was condensed with benzyloxycarbonyl (Cbz)-protected Phe to form the corresponding amide intermediate. Subsequently, derivative 32 was synthesized through the addition of 3-(5-methylisoxazol-3-yl)acryloyl chloride, followed by reduction and oxidation. The derivative 32 containing isoxazole showed a certain inhibitory effect on EV71 3C protease, with EC50 of 0.98 μM and no obvious toxicity (CC50 > 25 μM) (Fig. 9).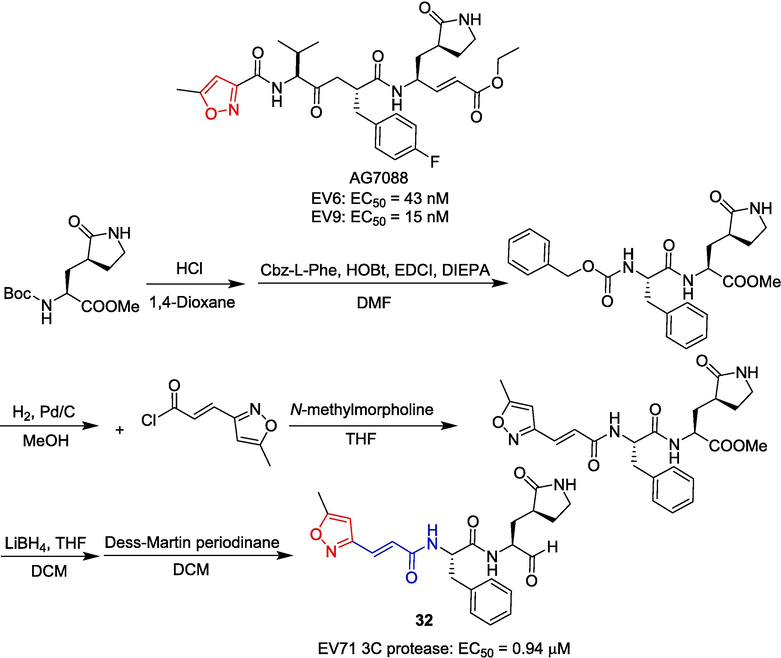
Cinnamamide-isoxazole derivative 32.
Summary: Cinnamamide and cinnamic acid-isoxazole hybridization natural products mainly have anti-tumor, anti-pathogen, and antioxidant activities. Among the anti-tumor activities, compound 1 showed the strongest inhibitory effect on the HCT116 cell line with an IC50 value of 0.35 μM, and compound 23 showed the strongest inhibitory effects on A549 and HeLa cells, with IC50 values of 0.82 and 0.91 µM, respectively. Therefore, the cinnamamide-linked isoxazole derivatives have the value of further research in the field of anti-tumor. Cinnamamide-isoxazole hybrid sulfonamide structure (compounds 8–12) has the best anti-chlamydia activity, and preliminary structure–activity relationship showed that introducing the electron-donating group was superior to the electron-withdrawing group in the para-position of the phenyl group of cinnamamide.
2.2 Chalcone-isoxazole hybridization
Niu et al. (Niu et al., 2016) designed and synthesized a series of chalcone derivatives containing isoxazole groups and evaluated their activity on melanin synthesis in mushroom tyrosinase and mouse B16 cells. 33–37 activity was stronger and EC50 was between 1.3–8.1 μM, which was significantly better than 8-methoxypsoralan (EC50 = 14.8 μM). The activity of these compounds with different substitutions on the benzene ring was found to be in the following order: 4-Cl > 3,4-F > 3,4-Cl > 4-F > 4-Cl, 3-F. In B16 cells, all compounds exhibited higher melanin-producing activity compared to 8-methoxypsoralan, with compound 34 (463 %) being three times more potent than the control 8-methoxypsoralan (115 %) (Fig. 10).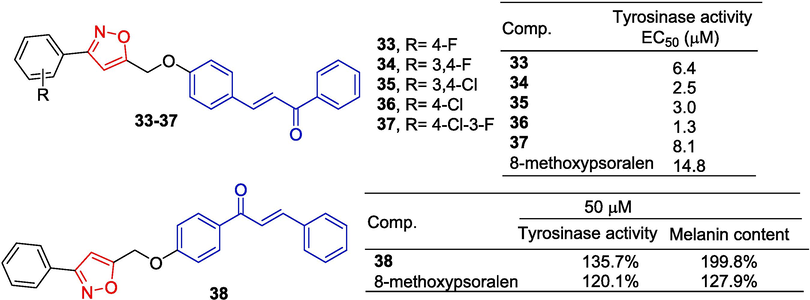
Chalcone-isoxazole derivatives 33–38.
Li et al. (Yin et al., 2017) reported a new isoxazole chalcone derivative 38. Pharmacological experiments showed that compound 38 (50 μM, 135.7 %) had a stronger inhibitory effect on tyrosinase in mouse B16 melanoma cells than 8-methoxy psoralen (50 μM, 120.1 %). Compound 38 can promote melanin synthesis in B16 cells and its activity (50 μM, 199.8 %) is better than that of 8-methoxypsoralen (50 μM, 127.9 %). Western blot experiments have shown that compound 38 promotes melanin production through Akt and GSK3β signaling pathways and has the potential to be developed as a drug for the treatment of vitiligo.
Sahoo et al. (Sahoo et al., 2021) designed and synthesized a series of 5-phenyl-3-isoxazolecarboxylic acid methyl ester-chalcone hybrids and evaluated their antimicrobial activity. As shown in Fig. 11, a series of chalcone acids were synthesized through aldol condensation of 4-formyl benzoic acid and various substituted acetophenones in the presence of potassium hydroxide as a base in methanol. The synthesized chalcone acids were then reacted with methyl 5-(3-aminophenyl)isoxazole-3-carboxylate using DMAP as a base and EDCI to form the target hybrid products, which are isoxazole methyl ester-chalcone derivatives. Compounds 39–42 showed strong in vitro activity against Mycobacterium tuberculosis H37Rv (Mtb H37Rv) with a MIC of 0.12 μg/mL. Cell viability tests in Vero cells did not show cytotoxicity of these compounds (CC50 > 10 μg/mL, SI > 320). Compound 41 has the strongest anti-drug activity against drug-resistant Mtb H37Rv with a MIC of 0.03–0.5 μg/mL.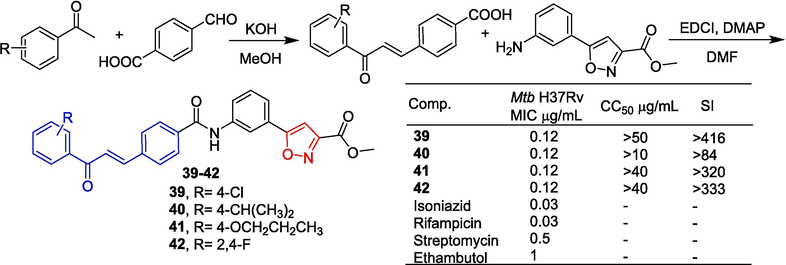
Chalcone-isoxazole derivatives 39–42.
Sunitha et al. (Sunitha et al., 2018) synthesized a series of diisooxazol-like chalcones and tested their anti-bacterial and anti-fungal activities. Compounds 43–46 were highly active at concentrations of 75 and 100 µg/mL against eight bacteria [Micrococcus luteus (M. luteus), Methicillin-resistant Staphylococcus aureus (S. aureus), Bacillus subtilis (B. subtilis), Bacillus cereus (B. cereus), Pseudomonas aeruginosa (P. aeruginosa), Klebsiella pneumoniae (K. pneumoniae), Escherichia coli (E. coli), Proteus vulgaris (P. vulgaris], three fungi (Microsporum canis, Microsporum gypseum, Epidermophyton floccosum) (Fig. 12). The structure–activity relationship demonstrates that R1 is the electron-withdrawing group, while R2 is the electron-donating group, which is beneficial to the activity..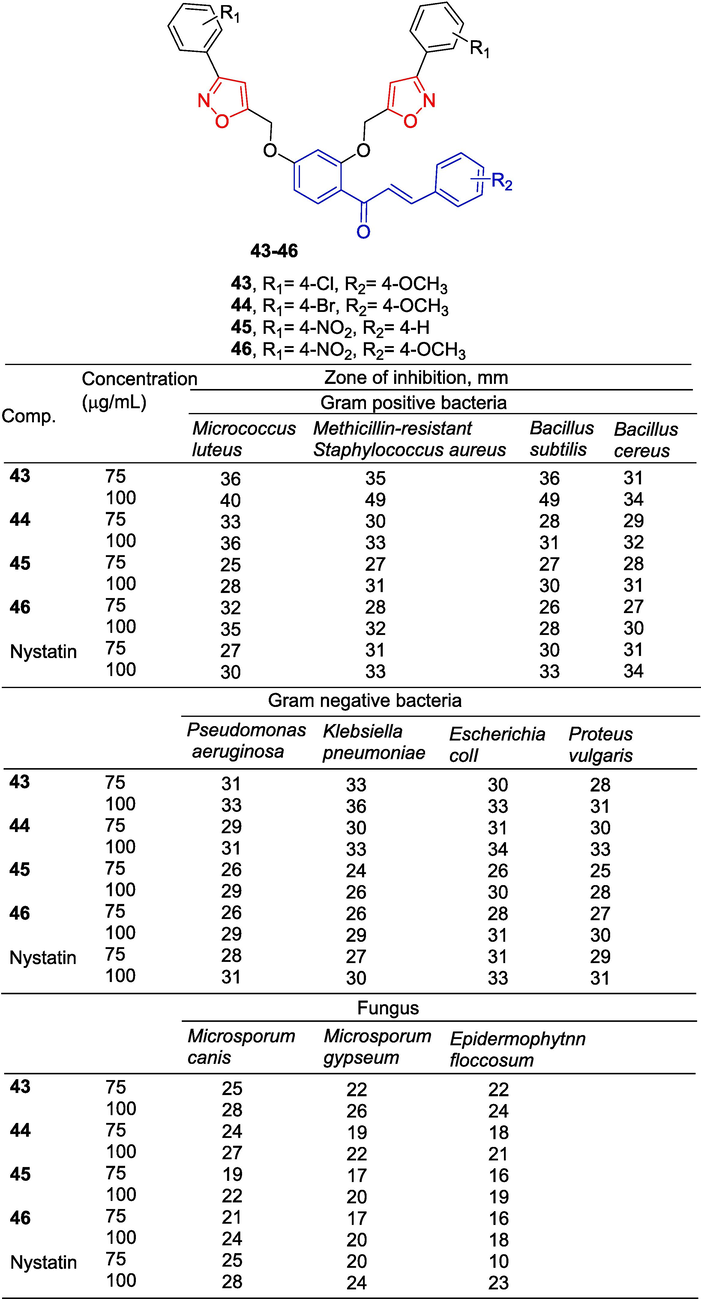
Chalcone-isoxazole derivatives 43–46.
Sunitha et al. (Sunitha et al., 2018) synthesized a series of monoisooxazol-like chalcones and tested their antibacterial activities. Compounds 47–50 were highly active at concentrations of 75 and 100 µg/mL against M.s luteus; S. aureus, B. subtilis, B. cereus, P. aeruginosa, K. pneumoniae, E. coli, and P. vulgaris (Fig. 13). The structure–activity relationship demonstrates that compounds with strong electron-withdrawing substituents on the phenyl ring attached to isoxazole enhance the antibacterial activity of the compounds.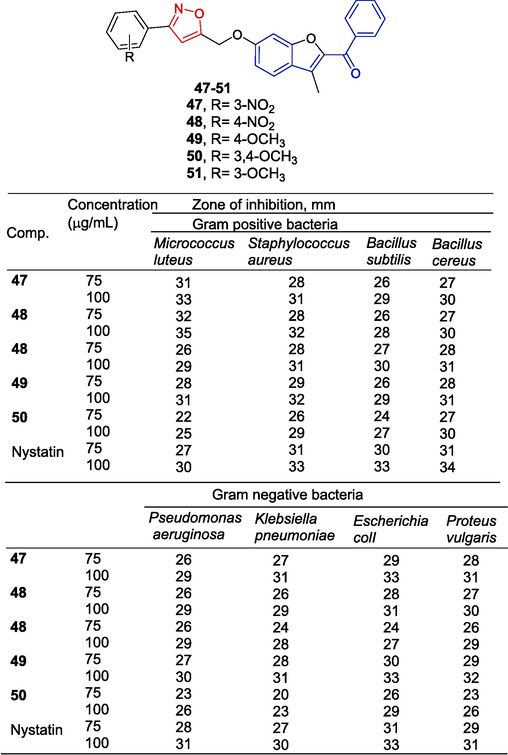
Chalcone-isoxazole derivatives 47–50.
Wan et al. (Wan et al., 2014) synthesized a series of isoxazoly-chalcone and evaluated the anti-tumor activity of H1792, H157, A549, and Calu-1 cancer cells in vitro. The IC50 values of compounds 51 and 52 against H1792, H157, A549, and Calu-1 cells were 1.35–19.63 μM. Among them, compounds 51 and 52 induce apoptosis of A549 cells through an exogenous pathway mediated by the death receptor 5 (DR5).
Shaik et al. (Shaik et al., 2020) designed and synthesized chalcone containing an isoxazole ring and determined its anti-bacterial, anti-oxidant, and anti-cancer activities. Compounds 53–55 exhibited potent antibacterial activity with MIC values ranging from 1 to 16 μg/mL, and the introduction of more electron-donating groups onto the phenyl moiety was found to enhance their activity. The compounds 56–58 and 54 exhibited potent antioxidant activity, with IC50 values ranging from 5 to 8 μg/mL. Compounds 55 and 59–60 demonstrated potent anti-tumor activity against DU-145 cells, with an IC50 range of 5–8 μg/mL (Fig. 14).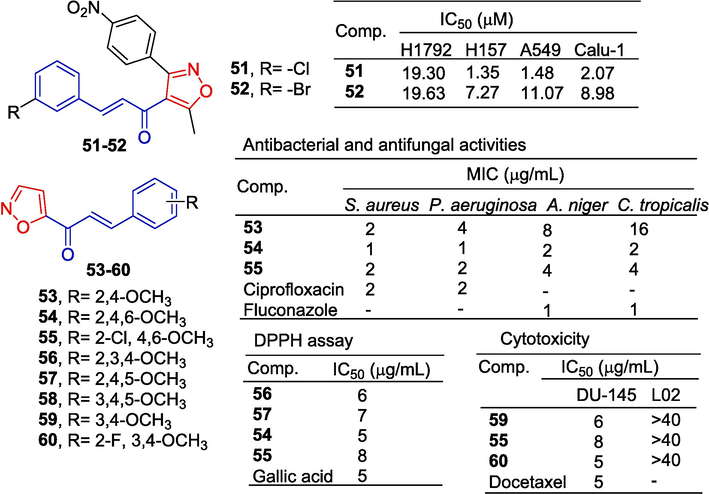
Chalcone-isoxazole derivatives 51–60.
Summary: Chalcone-isoxazole hybridization natural products mainly have anti-pathogen and anti-tumor activities. Compounds 39–42 have strong anti-tuberculosis activity and low toxicity in vitro. The structure–activity relationship demonstrates that nonpolar groups, such as halogens and alkyls, exhibit potent inhibitory capacity. Among them, compound 41 is valuable for further research in the development of anti-tuberculosis drugs. In terms of anti-tumor activity, these compounds did not reach the nanomolar level of IC50 values for cancer cells.
2.3 Styrene and polyphenols-isoxazole hybridization
Rodrigues et al. (Rodrigues et al., 2021) synthesized a derivative 61 containing isoxazole using curcumin as the skeleton (Fig. 15). Compound 61 showed strong inhibitory activity against the MCF-7 cell line, with IC50 = 3.97 μM, exceeding that of curcumin (IC50 = 21.89 μM). Compound 61 also showed significant inhibitory activity against the SKBR3 cell line with an IC50 value of 4.00 μM. However, it did not exceed curcumin (IC50 = 2.11 μM) (Amolins et al., 2009).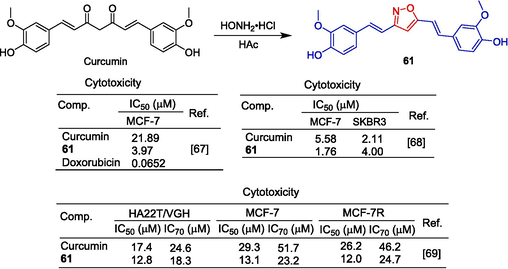
Styrene and polyphenol-isoxazole derivative 61.
According to another study, compound 61 showed strong inhibitory effects on HA22T/VGH, MCF-7, and MCF-7R cell lines with IC50 values of 12.8, 13.1, and 12.0 μM, respectively, surpassing the activity of curcumin (HA22T/VGH: IC50 = 17.4 μM; MCF-7: IC50 = 29.3 μM; MCF-7R: IC50 = 26.2 μM) (Simoni et al., 2008).
Actin and tubulin are contractile proteins and established targets for the development of anticancer drugs. Replacing the diketone portion of curcumin with isoxazole increased the stability of curcumin. Compound 61 retained actin polymerization inhibitory activity similar to that of curcumin. Additionally, compound 61 exhibited significant antioxidant activity, demonstrating a notable inhibition rate on DPPH with an EC50 value of 8 µM, which was higher than that of curcumin (EC50 = 40 µM) (Dhar et al., 2015; Chakraborti et al., 2013; Sherin and Rajasekharan, 2015). Kumboonma et al. (Kumboonma et al., 2019) found that compound 61 showed some inhibition activity of HDAC, with an inhibition rate of 54 % at 100 μM, which was lower than curcumin (62 %).
Curcumin binds to amyloid β peptide (Aβ) and inhibits or regulates the metabolism of amyloid precursor protein (APP). Compound 61 showed inhibitory activity on Aβ secretion, reducing the cleavage of Aβ38, Aβ40 and Aβ42 sites to varying degrees, with IC50 values of 6.1, 7.4 and 6.8 μM, respectively. It is a potent gamma-secretase inhibitor with an affinity for Aβ42 aggregates (IC50 = 138 nM), but with no significant activity against tau aggregates (Narlawar et al., 2008).
The curcumin IC50 on chloroquine-sensitive (CQ-S) and chloroquine-resistant (CQ-R) Plasmodium falciparum (P. falciparum) growth was 3.25 μM (MIC = 13.2 μM) and 4.21 μM (MIC = 14.4 μM). Mishra et al. (Mishra et al., 2008) synthesized a series of curcumin derivatives and evaluated their ability to inhibit the growth of P. falciparum. The IC50 of CQ-S P. falciparum and CQ-R P. falciparum were 8.44 and 7.92 μM, respectively, and the activity of derivative 61 was weaker than that of curcumin.
Mechanisms by which calcium/calmodulin-dependent protein kinase II (CaMKII) is involved in higher-order brain functions, such as learning and memory. Mayadevi et al. (Mayadevi et al., 2012) found that curcumin and its analogues inhibit Ca2+-dependent and independent kinase activity of CaMKII. The Ca2+-dependent activity of CaMKII was inhibited by compound 61 with an IC50 value of 11.37 μM. In autophosphorylation, CaMKII autophosphorylation, Ca2+ dependent, and Ca2+ independent activities were inhibited by compound 61 with IC50 values of 24.44 and 0.68 μM, respectively. Compound 61 inhibited the autophosphorylation of CaMKII in PSD and cytoplasm (IC50 = 22.79 μM), as well as Ca2+ dependent (IC50 = 22.87 μM) and Ca2 + independent activity (IC50 = 18.88 μM). Therefore, compound 61 is a potent CaMKII inhibitor.
Ahmed et al. (Ahmed et al., 2018) synthesized curcumin derivatives containing isoxazole and evaluated their anti-inflammatory and anti-nociceptive activities in experimental animal models. In vivo anti-inflammatory studies showed that the inhibition rate of compound 61 in induced edema was 66.1 %, which was lower than curcumin (71.6 % inhibition) and diclofenac sodium (84.4 % inhibition). Compound 61 has a relatively strong preventive effect on the onset of the torsional body, and its anti-nociceptive activity is 61 %. This was better than curcumin (58.1 % inhibition) but less than diclofenac sodium (67.1 % inhibition). The results of the in vitro COX-2 inhibition experiments showed that the inhibitory effect of compound 61 was stronger (49.3 % inhibition) than that of curcumin (19.1 % inhibition).
The ketoenol of curcumin is replaced by isoxazole to give compound 61. Compound 61 showed a strong scavenging ability for DPPH free radicals with an IC50 value of 10.71 μM. The activity was slightly higher than that of curcumin (IC50 = 11.06 μM). The inhibitory activities of the compounds in COX-1 and COX-2 were tested at 100 μM, and the compounds 61 and curcumin were equivalent to the COX-1 enzyme (compound 61: 80.8 %; curcumin: 80.5 %). The inhibitory activity of compound 61 on the COX-2 enzyme was significantly increased (compound 61: 58.1 %; curcumin: 35.0 %) and the COX-2/COX-1 ratio (0.72) was significantly increased (Selvam et al., 2005) (Fig. 16).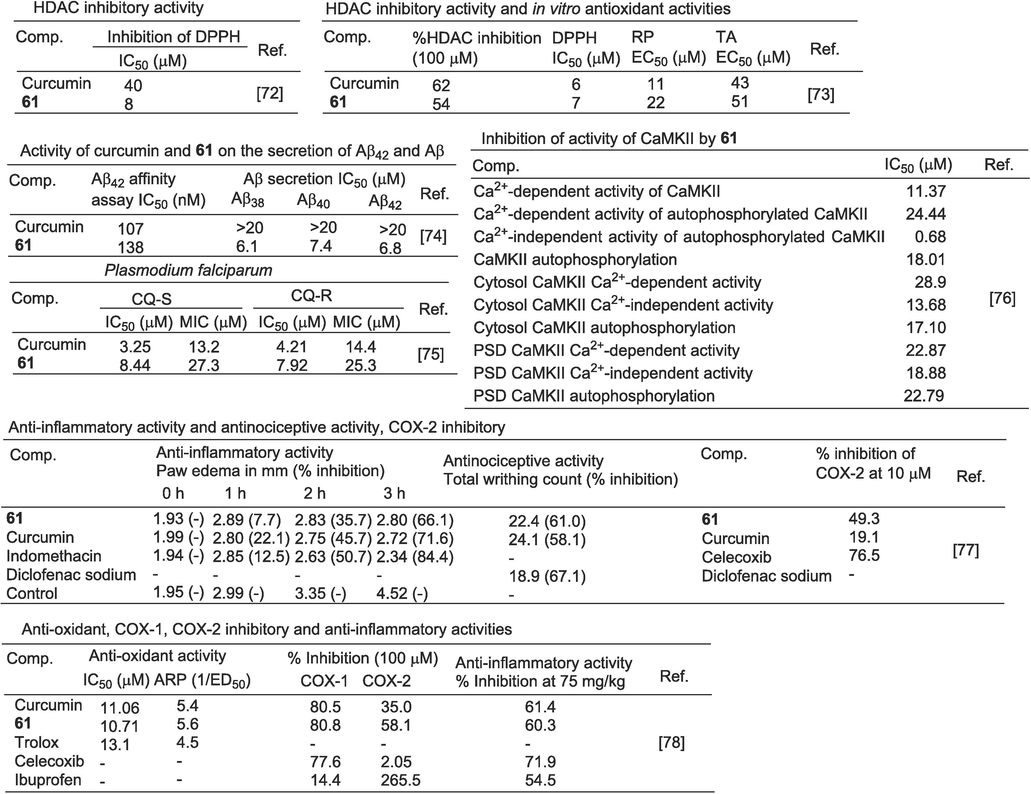
Multiple pharmacological activities of derivative 61.
The IC50 of derivative 62 against the MCF-7 and MCF-7R cell lines was 13.1 and 12.0 μM, respectively, surpassing the activity of curcumin (MCF-7: IC50 = 29.3 μM; MCF-7R: IC50 = 26.2 μM). The derivative 62 is similar to curcumin and is not cleared from cells by the P-gp protein (Poma et al., 2007).
Changtam et al. (Changtam et al., 2010) synthesized a series of curcumin derivatives and evaluated their activity against Mtb H37Rv. The synthesis method is similar to compound 61, and among them, compound 64 exhibits the highest activity with an MIC of 0.09 μg/mL. It is 1131 times more active than curcumin and about 18 times more active than the standard drugs kanamycin and isoniazid, respectively. The compound 64 also has strong activity against multidrug-resistant (MDR) M. tuberculosis isolated from patients (MIC = 0.195–3.125 μg/mL). Compounds 63, 65, and 66 also exhibited significant inhibitory activity against M. tuberculosis H37Ra, with a MIC of 0.39, 0.78, and 0.39 μg/mL, respectively. Compounds 63, 65, and 66 also showed high activity against clinical isolates of MDR-TB with a MIC ranging from 0.39 to 12.5 μg/mL.
The protein kinase C (PKC) family of serine/threonine kinases is an attractive drug target because it is involved in the regulation of various cellular functions, including cell growth, differentiation, metabolism, and apoptosis. To develop curcumin derivatives as effective PKC modulators, Das et al. (Das et al., 2011) found that the EC50 of curcumin derivatives 61 and 67 for protein fluorescence quenching varied in the range of 3.16–25.23 μM. These derivatives bind to PKChC1B to a higher degree than PKCdC1B and PKCeC1B.
Taka et al. (Taka et al., 2014) reported that a series of curcumin derivatives improved telomerase activity in vitro TRAP assays. Compound 68 increased telomerase activity by about 3 times at 60 μM (Fig. 17).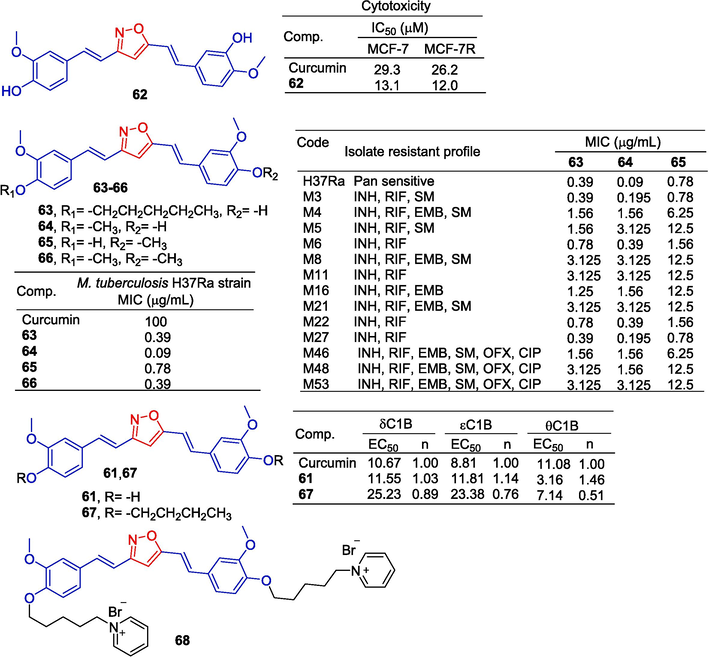
Styrene and polyphenols-isoxazole derivatives 62–68.
Compound 69 showed the highest anti-fungal activity against C. albicans (MIC80 (alone) = 32 μg/mL, MIC80 (in combination) = 1 μg/mL) (CCM: MIC80 (alone) > 64 μg/mL, MIC80 (in combination) = 8 μg/mL. It showed strong synergistic antibacterial activity against C. krusei when combined with FLC (MIC80 = 0.5 μg/mL). It also showed strong synergistic antibacterial activity against C. tropicalis (MIC80 = 1 μg/mL). Furthermore, compound 69 showed strong antibacterial activity against C. krusei when used alone (MIC80 = 1 μg/mL). For C. tropicalis, compound 69 also showed the strongest synergistic anti-fungal activity (MIC80 = 1 μg/mL) (Fig. 18) (Dong et al., 2021).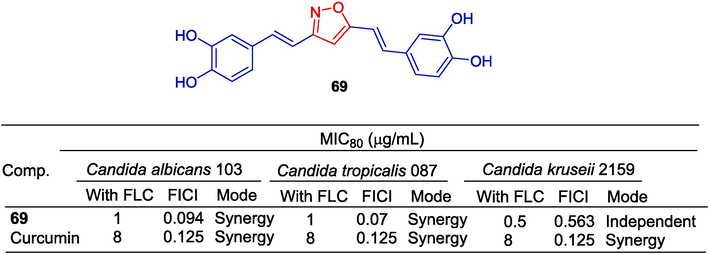
Styrene and polyphenol-isoxazole derivative 69.
Changtam et al. (Lal et al., 2013) synthesized a series of curcumin derivatives and evaluated their anti-bacterial and anti-tumor activities. Curcumin derivative 70 containing isoxazole: S. aureus (ATCC 11632), B. cereus (MTCC 7350), S. typhi (ATCC 23564), P. aeruginosa (ATCC 15499), and E. coli (ATCC 35218) showed significant inhibitory activity with MIC values of 20 μM. The derivative 70 showed moderate inhibitory activity against A. niger (MTCC 1108) with a MIC value of 40 μM. The IC50 values of derivative 70 against HepG-2, QG-56, and HCT116 cell lines were 25, 50, and 25 μM, respectively. Cytotoxicity was stronger than curcumin (HepG-2: IC50 = 50 µM; QG-56: IC50 = 100 µM; HCT116: IC50 = 50 µM).
Ahmed et al. (Ahmed et al., 2018) used curcumin scaffolds to synthesize sulfonamides, among which compound 71 containing isoxazole had the highest inhibitory activity against carbonic anhydrase isoenzyme I (human), with an IC50 value of 2.11 µM. The activity was similar to that of Acetazolamide (IC50 = 2.17 µM). Additionally, compound 71 showed good inhibitory activity against bCAII with an IC50 of 0.87 µM, the activity was better than Acetazolamide (IC50 = 0.94 µM). The kinetic study of compound 71 on the bCAII enzyme showed that the Ki value was 0.71 µM (Fig. 19).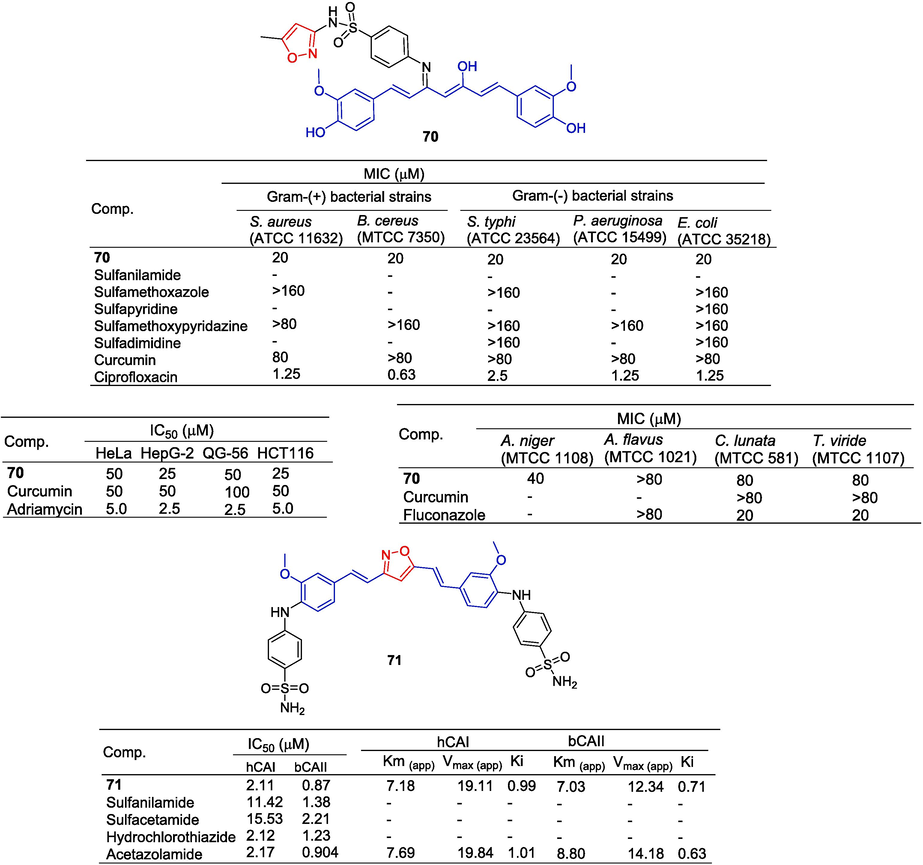
Styrene and polyphenols-isoxazole derivatives 70–71.
Balaji et al. (Balaji et al., 2019) synthesized several isoxazole-Hispolon derivatives. Among them, derivative 72 showed the best anti-tuberculosis activity against Mtb H37Rv (MIC = 1.6 µg/mL).
Reddy et al. (Reddy et al., 2020) synthesized a series of isoxazole derivative methyl β-orsellinate and evaluated its in vitro anti-proliferative activity against four human cancer cells and the normal cell line HEK-293 T(embryonic kidney). Compound 75 exhibits excellent anti-proliferative activity against the MCF-7 cell line with an IC50 of 7.9 µM, 5 times that of methyl β-orsellinate (IC50 = 46.63 µM). Compounds 73 and 75 showed inhibitory effects on MIAPACA cells with IC50 values of 9.862 and 9.442 μM. Compound 74 showed an inhibitory effect on IMR-32 cells with an IC50 value of 9.792 μM. Flow cytometry showed that compound 75 induced apoptosis of MCF-7 cells and kept the cell cycle in the G2/M phase.
The gallic acid derivative JEZTC (76) containing an isoxazole structure has anti-arthritic and cartilage protective effects. JEZTC (76) can significantly inhibit the expression of MMP and intracellular ROS, and significantly increase the expression of the metalloproteinase-1 (TIMP-1) gene in tissue. Additionally, there was less cartilage degradation in vivo in the JEZTC (76) group compared to the PBS group. The results also showed that JEZTC (76) activated the NF-κB pathway by regulating the MAPK and PI3K/Akt signaling pathways, resulting in the down-regulation of MMP, thus affecting osteoarthritis. The chondroprotective effect of JEZTC (76) may be related to its ability to inhibit chondrocyte apoptosis by reducing ROS production (Lu et al., 2018).
Mahapatra et al. (Mahapatra et al., 2022) synthesized a series of N-heteroaryl substituted gallamide derivatives. The antibacterial activity of three strains (S. aureus; E. coli, and Streptococcus pyogenes) was evaluated with compounds. JEZTC (76) was found to exhibit more inhibitory action against all three bacterial strains with the highest inhibition zone at 24, 24, and 25 mm; and had produced MIC at concentrations of 6.25 μg/mL with S. aureus, E.coli, and S. pyogenes (Fig. 20).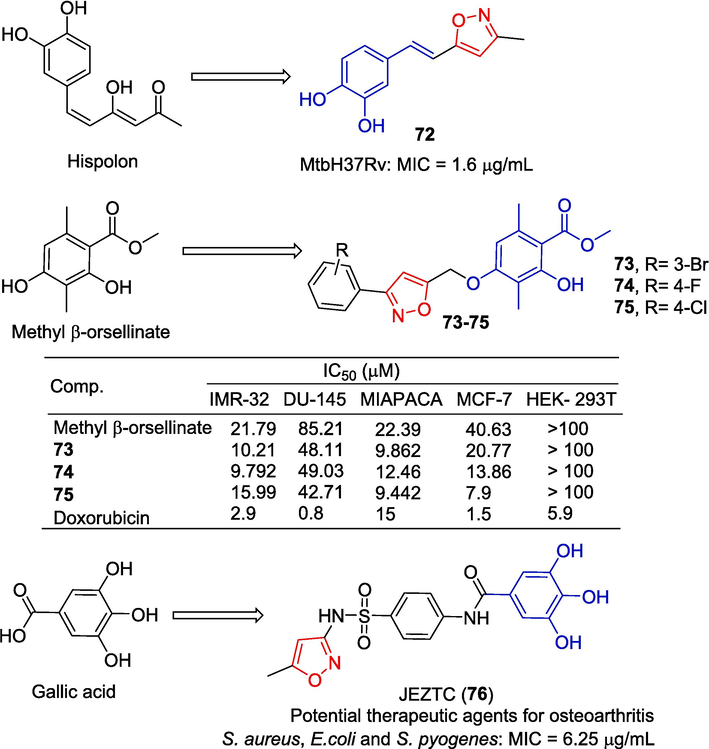
Polyphenols-isoxazole derivatives 72–76.
He et al. (He et al., 2016) synthesized a variety of stilbene derivatives containing isoxazole and determined the inhibitory activities of protein tyrosine phosphatase 1B (PTP1B) and TCPTP by colorimetry. Among them, compound 77 has the best inhibitory activity with IC50 values of 0.91 and 5.19 µM, respectively, which is stronger than that of the lead compound lithocholic acid (IC50 = 12.54 µM). Compound 78 showed remarkable activity and best selectivity (TCPTP/PTP1B = 20.7). The structure–activity relationship reveals that the introduction of electron-withdrawing groups, such as halogens, onto phenyl moieties can significantly enhance the inhibitory potency against PTP1B.
Pyrczak-Felczykowska et al. (Pyrczak-Felczykowska et al., 2019) synthesized a series of usnic acid derivatives and evaluated the anti-proliferation capacity of various cancer cells. Compounds 79 and 80 inhibit the survival of all cancer cell lines tested in a dose- and time-dependent manner. After 48 h of action, their IC50 value was approximately 3 μM for MCF-7 and PC-3 cells, and approximately 1 μM for HeLa cells. Both compounds 79 and 80 can induce G0/G1 blockade and decrease the proportion of HeLa cells in the S phase and the G2/M phase. Compounds 79 and 80 reduce the clonal potential of cancer cells, induce cell cycle arrest in the G0/G1 phase, and apoptosis of MCF-7 cells. In addition, they induced a large amount of cytoplasmic vacuolation through kinetoprotein-dependent endocytosis (Fig. 21).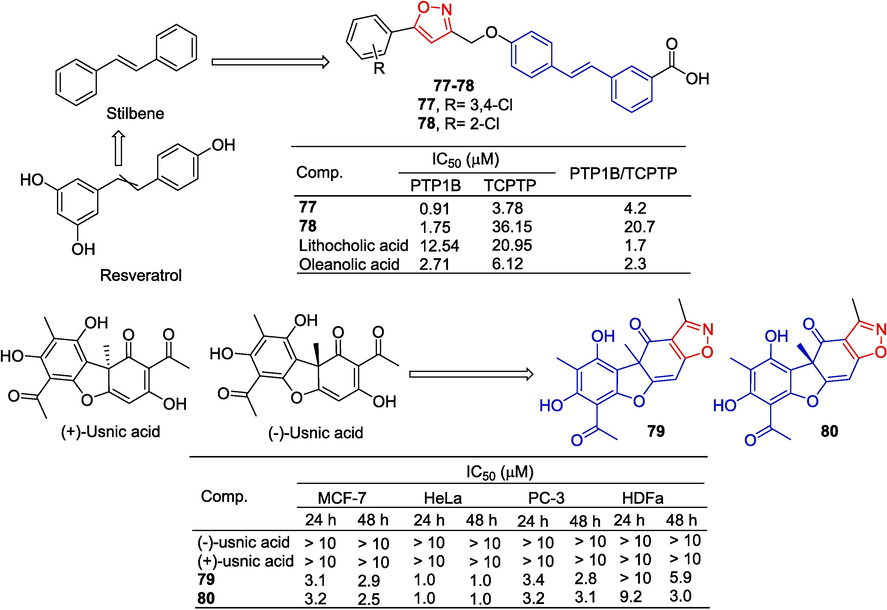
Styrene and polyphenols-isoxazole derivatives 77–80.
Summary: Curcumin, Resveratrol, Gallic acid, and Usnein are all polyphenols. The compounds summarized in this section combine isoxazole in the above natural products and their derived structures. These compounds have a variety of pharmacological activities by binding to different targets, including anti-tumor, anti-pathogen, antioxidant, anti-inflammatory, anti-arthritic, anti-nociceptive, and diuretic activities. Since curcumin and resveratrol have various pharmacological activities, these pharmacological activities are often improved after the hybridization of isoxazole. Similarly, the anti-tumor activity of gallic acid and usnein was significantly improved after the hybridization of isoxazole.
2.4 Flavone-isoxazole resveratrol
Wogonin (5, 7-dihydroxy-8-methoxy-flavonoids) has anticancer activity, and many results have shown that wogonin has strong anti-tumor activity in vivo and in vitro (Li-Weber, 2009; Baumann et al., 2008; Polier et al., 2011). Bian et al. (Bian et al., 2017) introduced isoxazole at site 7 of wogonin to obtain derivative 81 and tested its anti-tumor activity. Compound 81 showed some inhibitory activity in HepG-2 cells with an IC50 value of 21.66 μM. The activity was lower than wogonin (IC50 = 19.0 μM) and 5-fluorouracil (IC50 value 17.2 μM).
Rao et al. (Rao et al., 2018) designed and synthesized a series of heterocyclic flavono-isoxazole compounds and determined their anti-proliferative activity against MCF-7 by sulforhodamine B assay (SRB) and turbidimetry. Compound 82 showed a certain inhibitory effect on MCF-7 cells with an IC50 value of 34.2 μM. Compound 82 also showed moderate antibacterial activity, with an inhibitory rate of 41.7 % at a concentration of 30 µM (Fig. 22).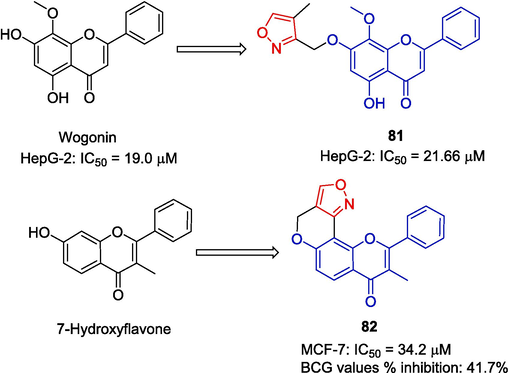
Flavono-isoxazole derivatives 81–82.
The flavonoid hydnocarpin, derived from the seeds of Hydnocarpus wightiana Blume, was chemically linked to isoxazole by Arya et al. (Arya et al., 2019) in order to synthesize a series of derivatives. These derivatives were subsequently evaluated for their anti-proliferative activity against A375, A549, and WI-38 cells. During the process of synthetic modification, the primary hydroxyl group at the C-9′ position of Hydnocarpin was initially transformed into its corresponding aldehyde via Moffatt oxidation. Subsequently, a one-pot [3 + 2] cycloaddition reaction was employed for synthesizing hydnocarpin-isoxazole derivatives. Compounds 83–86 exhibited significant inhibitory effects on A375 and A549 cells with IC50 values ranging from 0.65 to 7.5 μM (Fig. 23).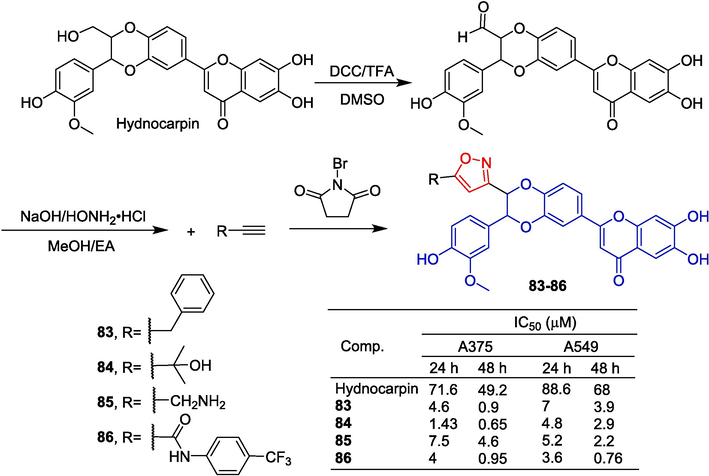
Flavono-isoxazole derivatives 83–86.
3-O-[(E)-4-(4-cyanophenyl)-2-oxobut-3-en-1-yl] kaempferol is a lead compound with anti-diabetic and anti-obesity activities. Nie et al. (Nie et al., 2020) reported a novel isoxazole derivative 87 based on this lead compound, which improves glucose consumption in insulin-resistant (IR) HepG-2 cells at the nanomolar level (EC50 = 0.8 nM). The methylation of kaempferol was achieved by subjecting it to dimethyl sulfate. For demethylation, anhydrous aluminum bromide in acetonitrile was employed as the chosen condition, resulting in the formation of the corresponding 3-hydroxyflavone. Subsequently, compound 87 underwent a nucleophilic substitution reaction followed by cyclization with chloro-benzaldoxime to yield isoxazole derivative (Fig. 24). Compound 87 significantly improved the level of AMPK phosphorylation (AMP-activated protein kinase) in HepG-2 cells. The levels of PEPCK (phosphoenolpyruvate carboxykinase) and G6Pase (glucose 6-phosphatase) were decreased. The molecular mechanism of 87 may be related to the activation of the AMPK/PEPCK/G6Pase pathway.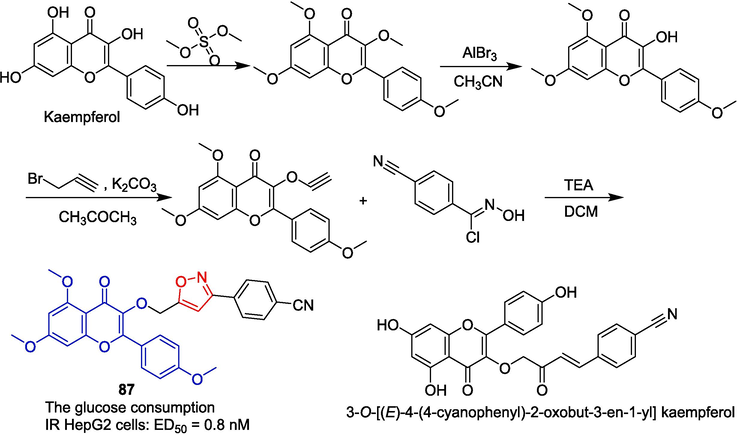
Flavono-isoxazole derivative 87.
Inhibition of alpha-amylase is considered a therapeutic strategy for the treatment of glucose absorption disorders. Flavonoids, isoxazoles, and their halogenated derivatives have become the focus of pharmaceutical chemistry research because of their high biological activity. Saidi et al. (Saidi et al., 2022, 1247) designed and synthesized a series of novel halogenated flavonoid isoxazoles and evaluated their inhibitory potential against α-amylase. The α-amylase inhibitory activity of compound 88 (IC50 = 16.2 μM) was the highest in vitro, which was comparable to that of acarbose (IC50 = 15.7 μM). Compounds 89–91 were slightly less active than 88, with IC50 values of 17.33, 17.58, and 18.36 μM, respectively. The above results suggest that the incorporation of electron-withdrawing groups in the para-position of the benzene ring confers enhanced activity.
Rongmao et al. (Qiu et al., 2018) designed and synthesized a series of isoflavone analogs and evaluated their lipid-lowering activities. Most of the compounds significantly reduced lipid accumulation in 3 T3-L1 adipocytes, and four compounds 92–95 had stronger inhibitory effects than GW4064. Compound 95 showed agonistic activity against FXR in cell-based luciferase reporting tests. At the same time, the expression of the FXR, SHP, and BSEP genes was up-regulated and the mRNA expression of the lipogenic gene SREBP-1c was down-regulated on 95. The safety of 95 in the HepG-2 cytotoxicity test also exceeds GW4064 (Fig. 25).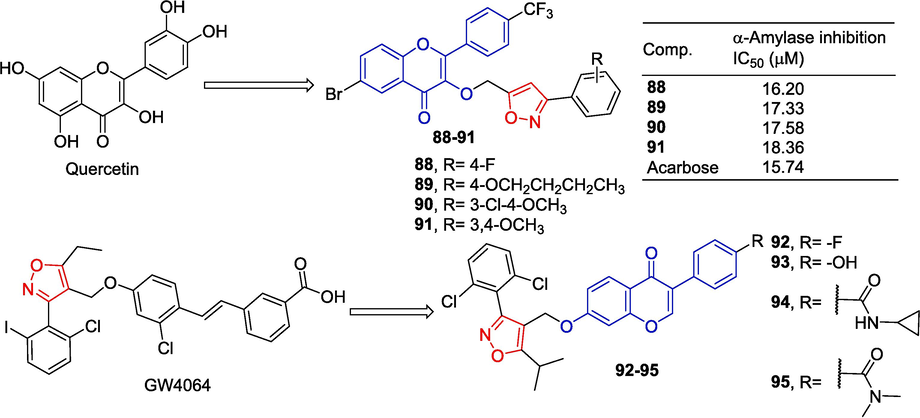
Flavono-isoxazole derivatives 88–95.
Asha Bhanu et al. (Asha Bhanu et al., 2020) designed and synthesized 7-hydroxyflavone derivatives and evaluated their anti-bacterial activity (Fig. 26). Compounds 96–98 showed strong antibacterial activity, exceeding or approaching that of standard drugs (Streptomycin and Cycloheximide).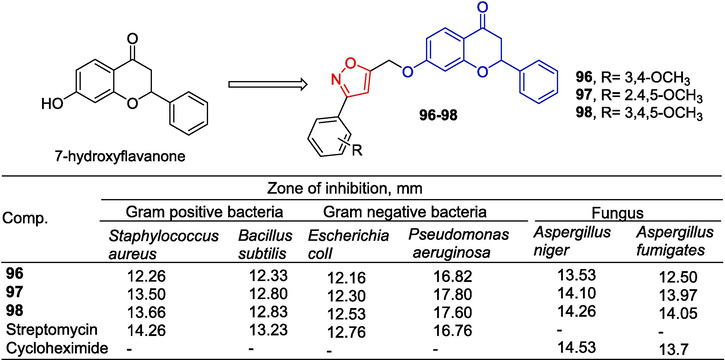
Flavono-isoxazole derivatives 96–98.
Gu et al. (Gu et al., 2017) designed and synthesized a series of dihydroflavone derivatives and evaluated their anti-psychotic activities in vitro and in vivo. In brief, dihydroflavones with various substitutions were first synthesized, followed by the acquisition of the target compounds through a three-step nucleophilic substitution reaction (Fig. 27). The activity of the D2 receptor/G protein α16a cotransfected into HEK293 cells was significantly reduced by compounds 99–101, exhibiting IC50 values ranging from 0.0513 to 0.116 μM. Moreover, compounds 99–101 effectively suppressed lipopolysaccharide/interferon-gamma-induced excess nitric oxide production by BV-2 microglia. In mice, the increase in motor activity induced by MK-801 (an antagonist of NMDA receptors) was reversed by gavage of 99–101. Reduce the overactivity of climbing behavior induced by apomorphine (a dopamine receptor agonist).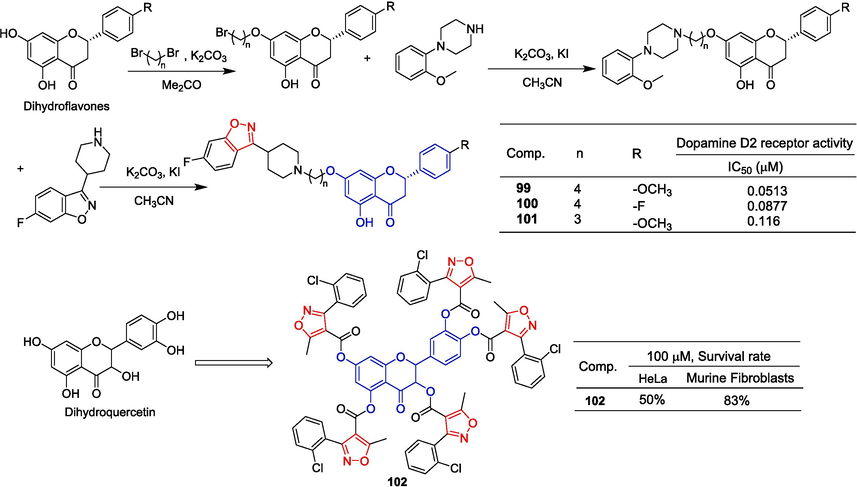
Flavono-isoxazole derivatives 99–102.
Nifantev et al. (Nifantev et al., 2015) synthesized dihydroquercetin aryl derivatives and determined the cytotoxicity of these compounds in HeLa cells and murine fibroblasts. The derivative 102 containing isoxazole showed weak HeLa cytotoxicity, with a survival rate of 50 % at 100 μM. At the same concentration, the survival rate of murine fibroblasts was 83 %.
Badadhe et al. (Badadhe et al., 2013) designed and synthesized a series of novel isoxazol pigment compounds and evaluated their antibacterial activities. Pharmacological experiments showed that compounds 103–105 had moderate anti-bacterial activity against Gram-positive bacteria.
Dengale et al. (Dengale et al., 2022) designed and synthesized a series of 2-(4,5,6,7-tetrahydrobenzo[c]isoxazol-3-yl)-4H-chromen-4-ones. Compounds 106–108 exhibited moderate anti-inflammatory activity compared to the standard drug diclofenac sodium and showed antioxidant activity comparable to the standard drug ascorbic acid. In terms of anti-inflammatory activity, the preliminary structure–activity relationship revealed that the presence of an electron-withdrawing group (R1) was conducive to enhanced activity, while the presence of an electron-donating group (R2) was detrimental to activity.
Diabetic retinopathy (DR) is one of the leading causes of blindness. Rho-associated coiled coils containing serine/threonine protein kinases (ROCKs) are considered potential targets for DR therapy. Zhao et al. designed and synthesized a new class of ROCK inhibitor 4H-chromen-4-one derivative. Compound 109 containing isoxazole can reduce ROCK I and ROCK II activity with IC50 values of 0.068 and 0.005 μM, respectively (Zhao et al., 2019) (Fig. 28).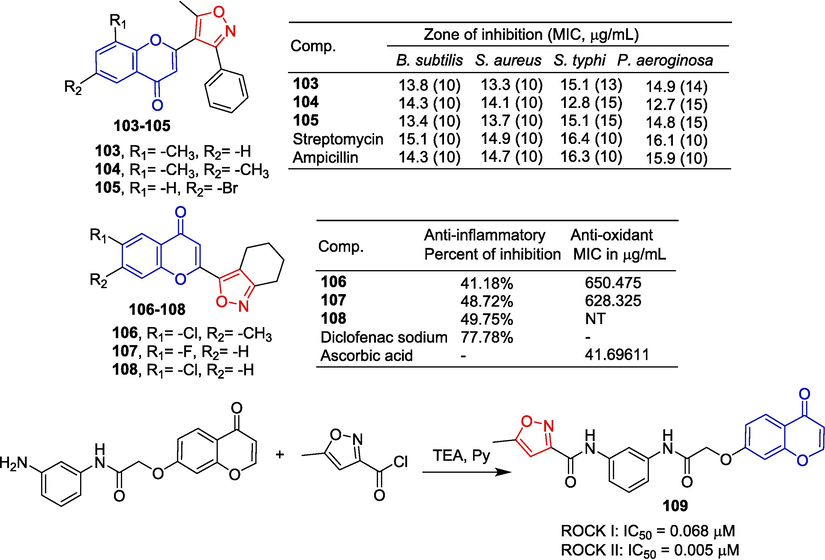
Chromone-isoxazole derivatives 103–109.
Abu-Bakr et al. (Abu-Bakr et al., 2019) synthesized a series of furo[3,2-g]chromones derivatives and screened for their inhibitory capacity in three human cancer cell lines, HFT-116, MCF-7, and HepG-2. Compound 110 showed a strong inhibitory effect on the HepG-2 cell line with an IC50 of 7.9 μg/mL. Compounds 111 and 112 showed strong inhibitory effects on the HCT116 cell line with an IC50 of 4.7 and 7.8 μg/mL, respectively.
Kaushik et al. (Kaushik et al., 2019) designed and synthesized a series of isoxazolylchromones, compound 113 showed obvious cytotoxicity in MCF-7 cells with an IC50 value of 31.25 μg/mL. The ERα selectivity of the ligand was confirmed by ERα silencing experiments. Deactivation of compound 113 showed that ERα luciferase reporter gene expression was negatively regulated and ERβGFP was induced in treated cells. The cell cycle showed an increase in sub-G0/G1 populations in the compound 113 treatment group. Compound 113 also induces cell death through apoptosis. Similar to tamoxifen, 113-induced cell death is mediated by an increase in ROS. Compared to tamoxifen-induced autophagy, compound 113 induced mitochondrial transmembrane potential loss and caspase activation without signs of autophagy.
Awadallah et al. (Awadallah et al., 2015) synthesized a series of isoxazole-containing pigment compounds and evaluated their cytotoxic activity against MCF-7 and A-549 cells. Compounds 114–116 showed weak inhibitory activity against the off-target cytoplasmic isomers hCA I and hCA II, but showed significant inhibitory activity against tumor-associated hCA IX and hCA XII. Compound 114 showed high inhibitory effects on MCF-7 and A-549 cell lines with IC50 of 25.80 and 57.23 µM, respectively (Fig. 29).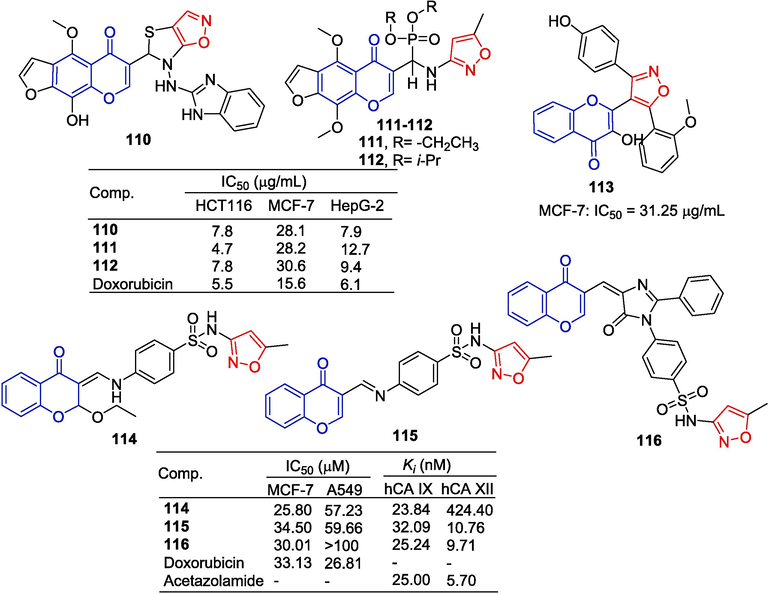
Chromone-isoxazole derivatives 110–116.
Summary: Flavone-isoxazole hybridization natural products mainly have anti-tumor, anti-psychotic, and lipid-lowering activities, and these compounds can also be used to treat diabetic retinopathy. Compound 87 exhibits promising potential as a valuable candidate in the quest to develop novel anti-diabetic drugs. Compounds 99–101 possess potential antipsychotic effects and may be beneficial in the treatment of schizophrenia. In terms of anti-tumor activity, most of the hybrid isoxazole derivatives showed no significant improvement in activity compared to the lead compounds. However, hydnocarpin derivatives (83–86), showed significantly improved anti-tumor activity. To some extent, the structure of isoxazole is an important pharmacophore for the anti-tumor effect of these hydnocarpin derivatives. Among these derivatives, compound 84 showed the strongest inhibitory effect on A375 cells with an IC50 of 0.65 μM, which is valuable for further study.
2.5 Coumarin-isoxazole hybridization
Shi et al. (Shi et al., 2017) designed and synthesized a series of coumarin-isoxazole compounds, and measured their anti-cancer activity against HCT116, Hun7, and SW620 cells in vitro by the MTT assay. Compound 118 showed inhibitory effects on HCT116, Hun7, and SW620 cell lines with IC50 values of 10.3, 12.1 and 10.5 μM, respectively. The IC50 values of compound 117 for these three cell lines were 9.21, 8.76, and 9.83 μM, respectively. Compounds 118 and 117 were less toxic to normal cells (HFL-1: The IC50 values were 90.9 and 74.2 μM, respectively).
Shakeel-u-Rehman et al. (Shakeel et al., 2014) designed and synthesized a series of 6-hydroxycoumarin isoxazoles. Compounds 119 and 120 showed the strongest inhibitory activity against PC-3 cells, with IC50 values of 8.2 and 13.6 μM, respectively.
The isoxazole-coumarin derivative 121 showed certain inhibitory activity against the HepG-2 cell line with an IC50 value of 15.3 µM (Gomha et al., 2015).
Krishna et al. (Krishna et al., 2017) synthesized isoxazole fused coumarin analogs and tested their anti-tumor activity in vitro. Compounds 122 and 123 showed moderate inhibitory effects on the Colo-205 cell line with IC50 values of 28.5 and 30.1 μM, respectively (Fig. 30).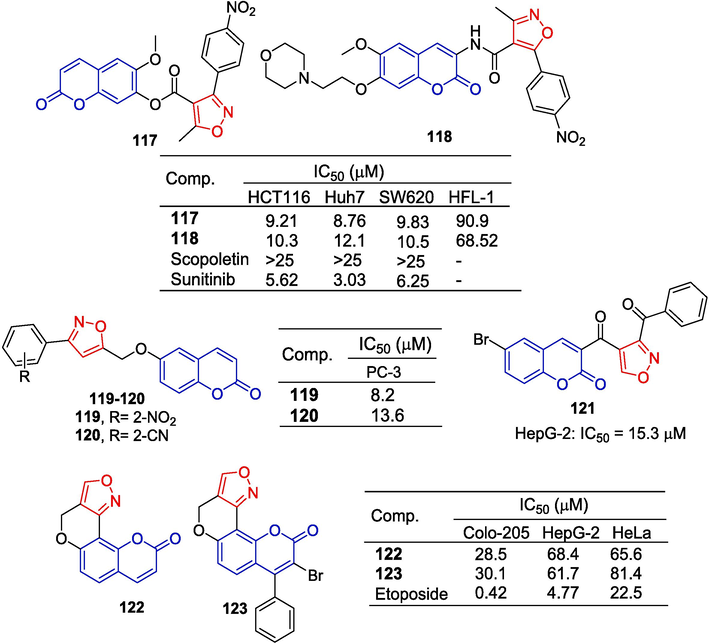
Coumarin-isoxazole derivatives 117–123.
Ghorab et al. (Ghorab et al., 2016) designed and synthesized a series of sulfanilyl coumarin derivatives. The anti-tumor activity of all compounds against the breast cancer cell line (T47D) was evaluated in vitro. Compound 124 containing isoxazole showed a moderate inhibitory effect on the T47D cell line with an IC50 of 68.4 μM.
Wang et al. (Wang et al., 2013) reported that a series of coumarin-containing sulfa compounds inhibited two human carbonic anhydrases. The starting diverse substituted malonic acid mono phenol esters were synthesized under solvent-free conditions using Meldrum’s acid and various substituted phenols, followed by cyclization with Eaton's reagent to yield the corresponding 4-hydroxycoumarin. The different substituted 3-formyl-4-chlorocoumarins were obtained through Vilsmeiere-Haack reactions. Finally, compounds 125–127 were prepared by reacting with sulfamethoxazole in ethanol (Fig. 31). These three compounds, which contain isoxazole, exhibited significant inhibitory effects on hCA II and hCA IX as well as on B16-F10 and MCF-7 cell lines. Compound 125 had the strongest inhibitory effect on the MCF-7 cell line and hCA II, with IC50 values of 0.012 and 0.026 μM. Compound 126 had the strongest inhibitory effect on hCA IX with an IC50 of 0.043 μM. As hCA II inhibitors, the substituted moieties on the benzene ring of the coumarin exhibit an active gradient of −H > −tBu > –CH3. However, for hCA IX, it is −tBu > –CH3 > -H.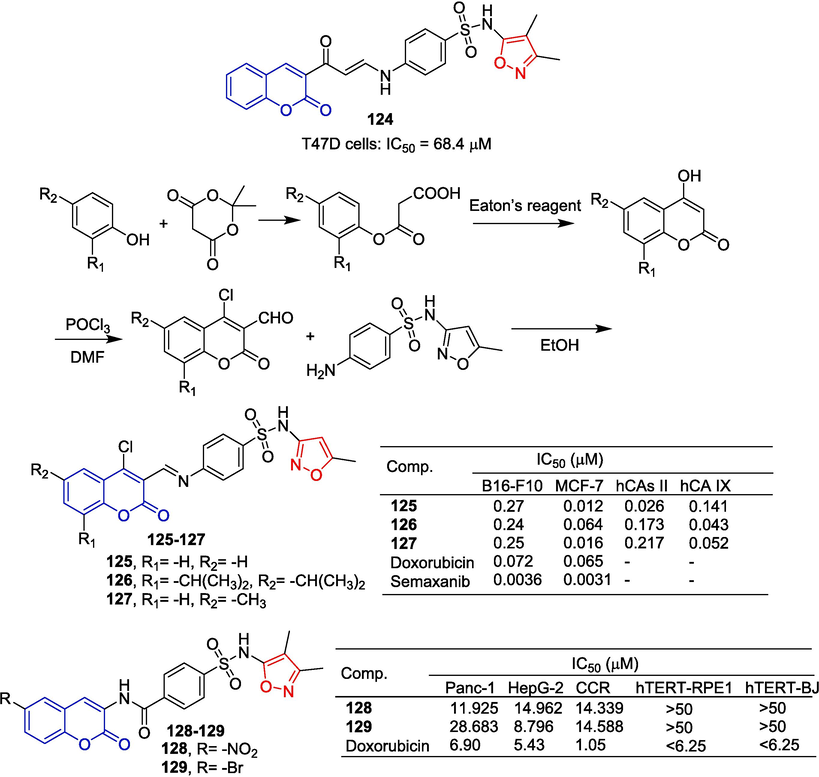
Coumarin-isoxazole derivatives 124–129.
Nasr et al. (Nasr et al., 2018) synthesized a series of derivatives of coumarin hydrazone and evaluated the anti-tumor activity of Panc-1, HepG-2, and CCR cell lines in vitro. Compound 128 containing isoxazole had the strongest inhibitory effect on Panc-1 cells with an IC50 value of 11.925 μM. Compound 129 had the strongest inhibitory effect on HepG-2 cells with an IC50 of 8.796 μM.
Chen et al. (Chen et al., 2013) synthesized novel anti-psychotic coumarin derivatives with potent dopamine D2, D3, and serotonin 5-HT1A, 5-HT2A receptor properties. The starting 7-hydroxycoumarin intermediates were obtained via the Pechmann reaction, and the target compounds were subsequently prepared through a two-step nucleophilic substitution reaction (Fig. 32). The isoxazole-piperidine derivative 130 has a high affinity for the D2, 5-HT1A, and 5-HT2A receptors (Ki of 4.9, 3.5, and 8.4 nM, respectively). The derivative 130 has a higher affinity for the 5-HT1A receptor than risperidone (Ki = 180 nM). The titer of compound 132 against the D2 and 5-HT2A receptors exceeds 130 (D2, Ki = 4.0 nM; 5-HT2A, Ki = 0.3 nM), and the affinity of the 5-HT1A receptor did not change. The derivatives 133 and 134 have the highest affinity for the 5-HT2A receptor (133: Ki = 0.3 nM; 134: Ki = 0.9 nM). Compounds 131, 133, and 134 exhibit high affinity for the D2 receptor with Ki of 3.9, 2.6, and 5.3 nM, respectively. Compound 133 has a higher affinity for the D2 receptor than compound 130 (Ki = 4.9 nM) and risperidone (Ki = 3.7 nM). Furthermore, compound 133 has a low affinity for the 5-HT2C and H1 receptors (which reduce the risk of obesity associated with chronic treatment) and the hERG channels (which reduce the incidence of point twisting). In animal models, compound 133 inhibited diamorphine-induced climbing behavior at the highest dose, hyperactivity and conditioned avoidance responses induced by MK-801, and no significant narcolepsy. Furthermore, in trials measuring prolactin secretion and weight gain, there were fewer preclinical adverse events at compound 133 compared to risperidone. The structure–activity relationship is as follows: the substitution at the 4-position (R1) with methyl as the favored substituent; the substitution at the 3-position (R2) with either unsubstituted or substituted (methyl) being preferred; and the substitution at the 8-position (R3) with an electron-withdrawing group (chloro) as the favored substituent.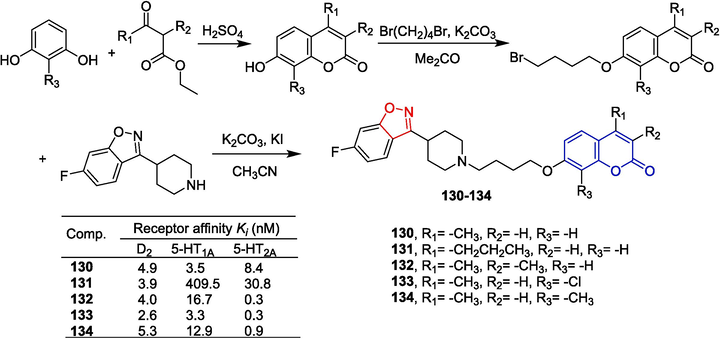
Coumarin-isoxazole derivatives 130–134.
Coumarin derivatives combine the properties of dopamine receptors D2, D3, and serotonin 5-HT1A, 5-HT2A. Compounds 135 and 136 were synthesized by the same research team using a similar synthetic approach to compounds 130–134. Compounds 135 and 136 have high affinity for dopamine D2, D3, and 5-HT1A, 5-HT2A receptors (Ki between 2.2–18.9 nM). Compound 135 has a low affinity for H1 receptors (reducing the risk of obesity under chronic treatment). In animal models, compound 135 inhibited apomorphine-induced climbing and hyperactivity induced by MK-801 at the highest dose without significant paralysis (Chen et al., 2014).
Park et al. (Park et al., 2016) synthesized a series of decursinol derivatives using the ka-reaction method and evaluated their biological activities. Compound 137 containing isoxazole has inhibitory activity in acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) with IC50 values of 61.91 and 14.64 μM, respectively.
Filoviruses, including Ebolavirus (EBOV), Marburgvirus (MARV) and Cuevavirus cause hemorrhagic fevers in humans with a mortality rate of up to 90 %. Gao et al. (Gao et al., 2020) designed and synthesized a piperidine-coumarin derivative and evaluated its anti-viral activity. Compound 138 containing isoxazole showed inhibitory activity against EBOV and MARV (EBOV: IC50 = 5.2 μM; MARV: IC50 = 3.2 μM). Compound 138 did not exhibit significant toxicity to host cells (A549: IC50 = 36.9 μM) (Fig. 33).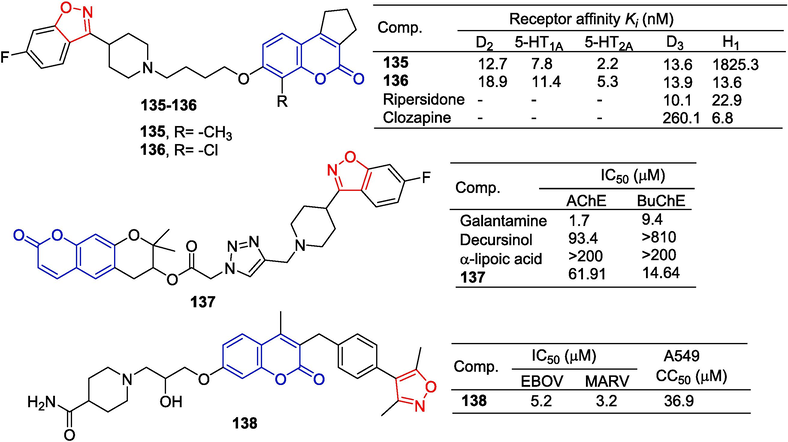
Coumarin-isoxazole derivatives 130–138.
Derivative 139 exhibited moderate anti-tumor activity against the Ehrlich ascite cells (EAC) model in vitro, with an IC50 of 52.65 μg/mL (W.S. Hamama, M.E. Ibrahim, H.H. Zoorob, Synthesis, DFT Study, 2017). Derivative 140 was synthesized by reacting 4-hydroxycoumarin with 5-amino-3-methylisoxazole in glacial acetic acid and exhibited significant anti-tumor activity; with an IC50 of 0.21 μM (Ibrahim et al., 2016).
Suresh et al. (Suresh et al., 2017) synthesized dihydro-6H-chromeno[4,3-b]isoxazolo[4,5-e] pyridine heterocycles. The anti-diabetic activity of the compounds on type 2 diabetes mellitus was evaluated. Compound 141 showed the strongest inhibitory effect on α-amylase activity with an IC50 value of 56.043 μg/mL.
In a rat model of carrageenan-induced metatarsal edema, the anti-inflammatory activity of the compound was evaluated at 100 mg/kg of body weight. The isoxazole-containing derivative 142 showed moderate anti-inflammatory activity, with a 42.87 % inhibition of edema formation compared to the control agent ibuprofen (100 mg/kg) (Dixit et al., 2013) (Fig. 34).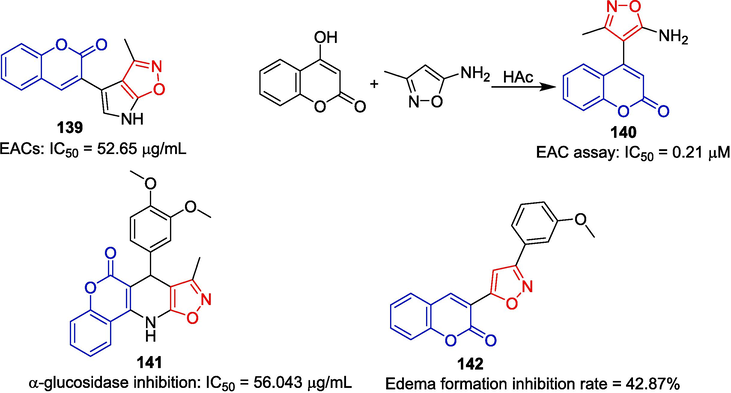
Coumarin-isoxazole derivatives 139–142.
Summary: Coumarin-isoxazole hybridization natural products mainly have anti-tumor, anti-inflammatory, anti-viral, anti-diabetic, and anti-psychotic activities. Carbonic anhydrase is a target for the development of anti-tumor chemotherapeutics, in terms of anti-tumor activity, compound 125 showed the strongest inhibitory activity against MCF-7 cells while inhibiting carbonic anhydrase, with IC50 values of 12 nM. In addition, the compound did not exhibit cytotoxic activity against macrophages. The results indicate that sulfonamides containing coumarin-isoxazole moieties have the potential to be potent and novel anti-tumor agents. Compared to the compounds in the previous sections, coumarin-isoxazole derivatives also have anti-psychotic activity by inhibiting central dopamine and serotonin receptors. Among them, compound 133 may constitute a new class of drugs for the treatment of schizophrenia.
2.6 Quinonoids-isoxazole hybridization
Vegfrecine has shown effective in vitro inhibitory activity against VEGFR-1 and VEGFR-2 tyrosine kinases, so vegfrecine becomes a VEGF receptor tyrosine kinase inhibitor. Adachi et al. (Adachi et al., 2021) synthesized a derivative of vegfrecine containing isoxazole rings. Compound 143 showed moderate inhibitory activity against VEGFR-1 tyrosine kinase (IC50 = 0.65 µM) and slightly lower inhibitory activity against VEGFR-2 tyrosine kinase (IC50 = 7.1 µM). The synthesis of the intermediate was carried out through oxidative amination of 2,5-dihydroxybenzamide in the presence of 2-((tert-butyldimethylsilyl)oxy)aniline. Subsequent ammonolysis of the intermediate yielded the 1-carbamoyl-2-aminoquinone derivative, which was then subjected to oxidative cyclization between the 1-carbamoyl group and 2-amino group using lead acetate to form the isoxazole ring-fused quinone. The TBS group of the isoxazole quinone derivative was deprotected to provide compound 143 (Fig. 35).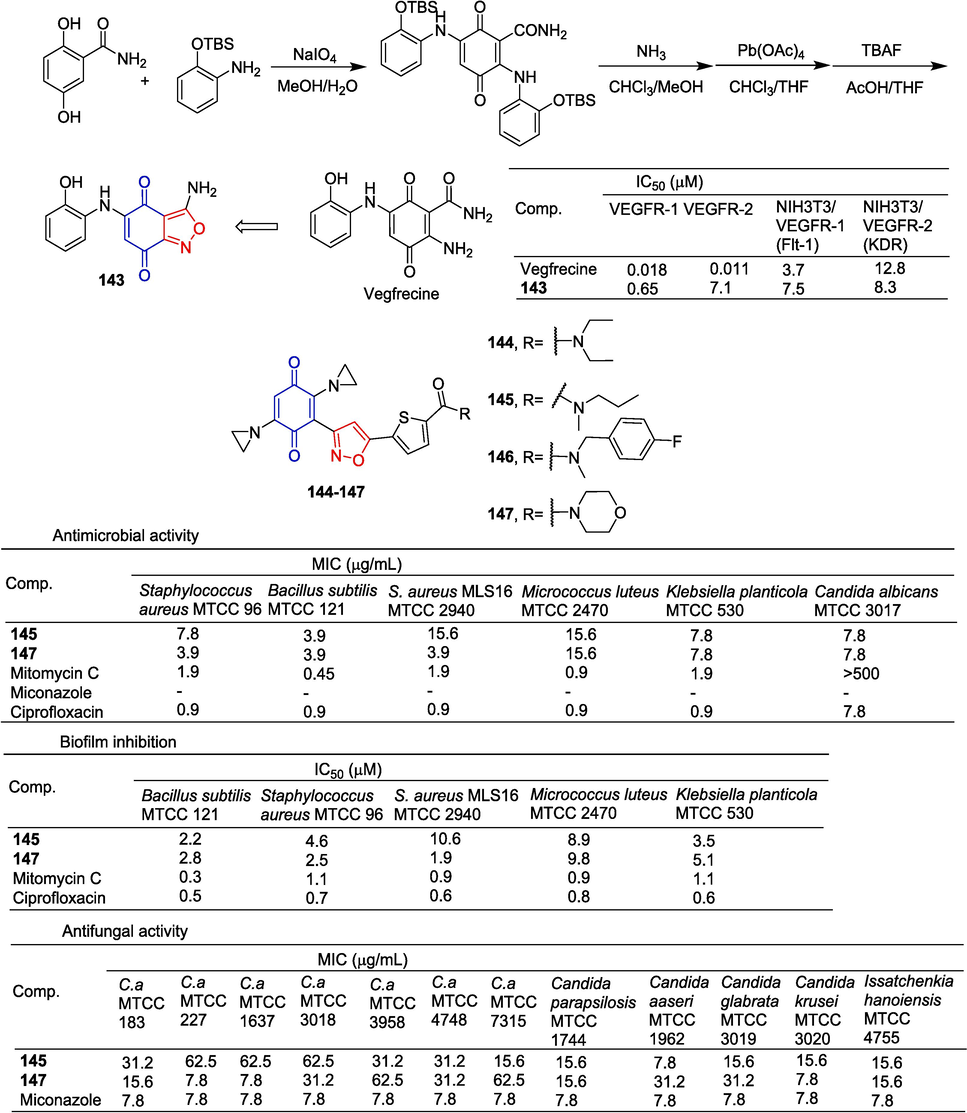
Benzoquinone-isoxazole derivatives 143–147.
Swapnaja et al. (Swapnaja et al., 2016) introduced quinone into the isoxazole structure and obtained a series of compounds with good activity against the tested strains. Compounds 145 and 147 have good bacteriostatic activity. Compound 145 can inhibit S. aureus MTCC 96, MIC values of B. subtilis MTCC 121 and Klebsiella planticola MTCC 530 were 7.8, 3.9, and 7.8 mg/mL, and IC50 values were 4.6, 2.2, and 3.5 μM, respectively. Compound 147 is associated with S. aureus MTCC 96, S. aureus MLS-16 MTCC 2940, MIC values of B. subtilis MTCC 121, and Klebsiella planticola MTCC 530 were 3.9, 3.9, 3.9, and 7.8 mg/mL, and IC50 values were 1.9, 2.5, 2.8, and 5.1 μM, respectively.
Li et al. (Li et al., 2019) synthesized a series of sampangine derivatives containing isoxazole. 6, 7-Dihydrobenzo[b]thiophen-4(5H)-one was used as the starting material to obtain the intermediate (5-bromobenzo[b]thiophene-4,7-dione) via three steps. Then, the target compound was prepared by reacting the intermediate with propionaldehyde oxime through a [1 + 2] cycloaddition reaction in the presence of NaClO. Compound 148 has strong anti-fungal activity against Cryptococcus neoformans (C. neoformans) (MIC80 = 0.031 μg/mL) higher than that of fluconazole (MIC80 = 2 µg/mL) and voriconazole (MIC80 = 0.12 µg/mL). Furthermore, compound 148 had a strong inhibitory effect on the important virulence factors (biofilm, melanin, and urease) of C. neoformans. The mechanism study showed that compound 148 could induce apoptosis of C. neoformans cells and keep the cell cycle in the G1/S phase. For the C3 substituents of isoxazole, alkyl substitutions were more favorable for anti-cryptococcal activity than heterocyclic and substituted phenyl derivatives. Among these, ethyl substitution yielded the best results (Fig. 36).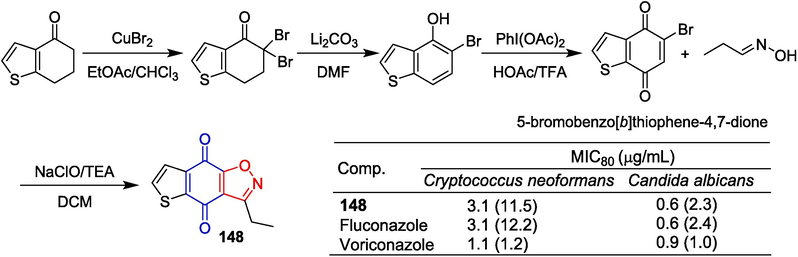
Benzoquinone-isoxazole derivative 148.
Santos et al. (Santos et al., 2010) synthesized a series of naphtho[2,3-d]isoxazole-4,9-diones by reacting 2,3-dichloro-1,4-naphthoquinone with nitromethyl derivatives in the presence of a base. Furthermore, they conducted an evaluation on the antifungal activity exhibited by these compounds. The MIC50 values of compound 149 for Candida parapsilosis PYCC 2545 and Candida glabrata PYCC 2418T were as low as 0.2 μg/mL. The MIC50 values of compound 150 for C. albicans PYCC 3436 and C. parapsilosis PYCC 2545 were 0.2 and 0.3 μg/mL.
Rajanarendar et al. (Rajanarendar et al., 2013) synthesized a series of isoxazole-indole-triones and evaluated the anti-inflammatory and analgesic activities of the resulting compounds. Compounds 151–153 exhibit significant anti-inflammatory activity by reducing the volume of foot PAWS produced by carrageenan. Compounds 151–153 significantly reduced the number of twists in mice and showed significant analgesic activity.
Uysal et al. (Uysal et al., 2021) designed and synthesized a series of novel 4-aminobenzenesulfonamide/carboxamide derivatives containing naphthoquinone pharmacophore, and evaluated their proteasomal inhibitory and anti-proliferative activities against the MCF-7. Compound 154 containing isoxazole showed significant inhibitory activity against the MCF-7 cell line with IC50 of 10.15 μM. Compound 154 showed some inhibitory activity in ChT-L (β5), C-L (β1), and T-L (β2) of proteasome at a concentration of 10 μM, with inhibition rates of 18.89 %, 8.39 %, and 6.55 %.
Lawrence et al. (Lawrence et al., 2010) synthesized isoxazole-containing compounds 155 and 156 and tested their inhibition of T-L and peptidyl glutamyl peptide hydrolase (PGPH). Compounds 155 and 156 significantly inhibited the activity of three proteasomes (CT-L, T-L, and PGPH), and the combined IC50 values of 155 for CT-L and T-L were 3.9 and 9.25 μM. The IC50 value of compound 156 for PGPH was 16.7 μM (Fig. 37).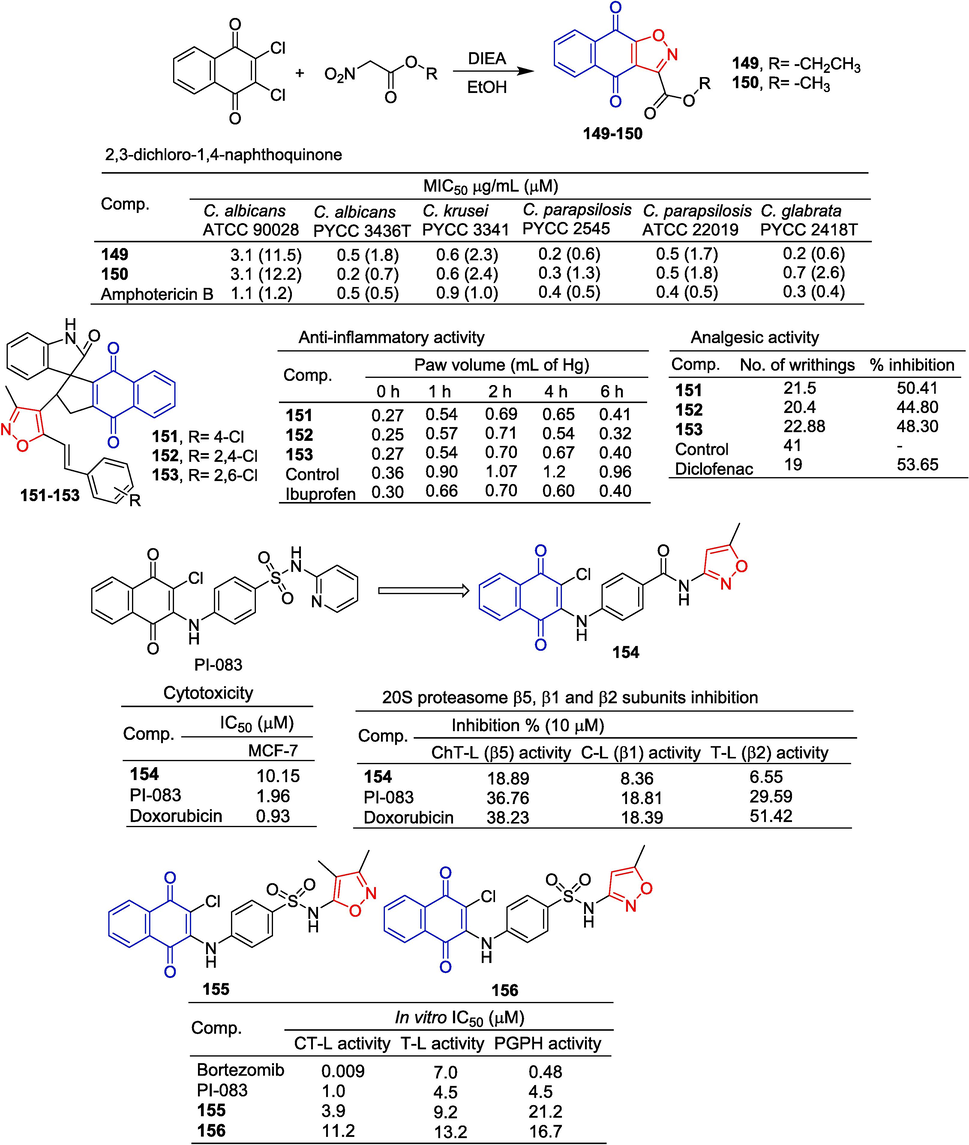
Naphthoquinone-isoxazole derivatives 149–156.
Yang et al. (Yang et al., 2011) synthesized a series of rhein derivatives and determined their cytotoxicity to HeLa and MOLT4 cell lines by the MTT assay. Rhein derivative 157 containing isoxazole showed inhibitory effects on HeLa and MOLT4 cell lines with IC50 values of 16 and 14 μM. More than emodin activity (HeLa: IC50 > 100 μM; MOLT4: IC50 = 37 μM).
Lee et al. (Lee et al., 2012) synthesized a series of 1,2-diaminoquinone derivatives and evaluated drug-induced cytotoxicity by SRB assay. The IC50 value of compound 158 containing isoxazole against the PC-3 cell line was 3.78 μM (Fig. 38).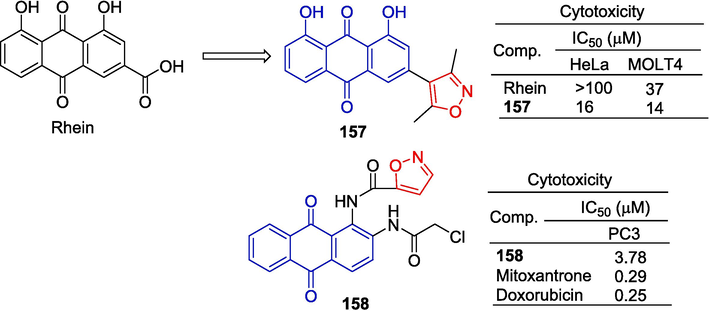
Anthraquinone-isoxazole derivatives 157–158.
Tyrosyl-DNA phosphodiesterase 2 (TDP2) is an enzyme that specifically repairs DNA damage mediated by topoisomerase II (TOP2). Yu et al. (Yu et al., 2018) report the synthesis of furoquinolinedione structure as a potential selective TDP2 inhibitor. Pharmacological experiments showed that compounds 159 and 160 were selective TDP2 inhibitors. The IC50 of the recombinant TDP2 and TDP2 whole cell extract (WCE) was 1.9 and 2.1 μM, respectively.
Yang et al. (Yang et al., 2021) synthesized a series of isoxazolinequinolinedione derivatives based on the TDP2 inhibitors of furanoquinolinedione and isoxazolinedione, and conducted enzyme inhibition tests. Enzymatic analysis showed that compounds 161–170 exhibited high TDP2 inhibitory activity with IC50 values ranging from 0.46 to 5.4 μM. The most potent compound 169, showed the strongest inhibitory activity with an IC50 of 0.46 μM (Fig. 39).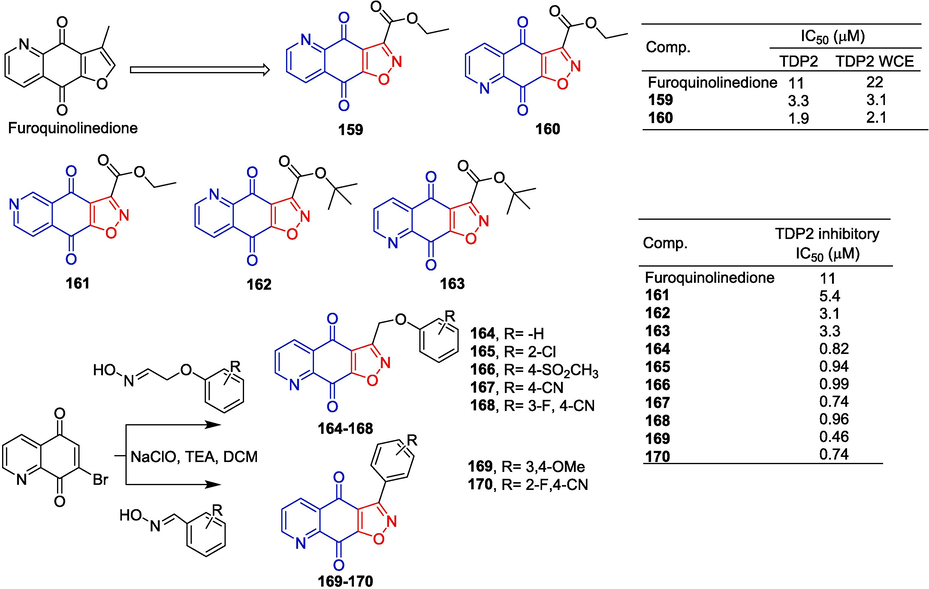
Naphthoquinone-isoxazole derivatives 159–170.
Combrink et al. (Combrink et al., 2007) designed and synthesized a series of 3-morpholino rifamycins and evaluated their antimicrobial activity. Compounds 171 and 172 containing isoxazole showed significant inhibitory activity against the M. smegmatis strain 43756Km1 with MIC values of 0.12 and 0.5 μM. It also showed significant inhibitory activity in M. smegmatis DSM 43756 with MIC values of 0.12 and 0.5 μM. Compound 172 pairs of wild-type E. coli (DH5) pCTF104 (Arr-2+), E. coli wild-type (DH5α) pCTF104 (Arr-2-), E. coli-CGSC# 5163 lpxC101 'leaky' strain (Arr-2+), and E. coli-CGSC# 5163 lpxC101 'leaky' strain (Arr-2-) showed significant inhibitory activity, MIC values are 4, 4, 2 and 2 μM. Compound 171 for the E. coli-CGSC# 5163 lpxC101 'leaky' strain (Arr-2+), and the E. coli-CGSC# 5163 lpxC101 'leaky' strain (Arr-2-) showed significant inhibitory activity with MIC values of 2 and 1 μM.
Bargiotti et al. (Bargiotti et al., 2012) synthesized a series of 3-arylnaphthalene (El-Garhy, 2015; Zhu et al., 2018)isoxazoles. Compounds were synthesized by reacting various naphthoquinones with in situ generated nitrile oxides, obtained through the treatment of corresponding oximes with triethylamine and aqueous NaClO in dichloromethane. These compounds were subsequently evaluated for their binding affinity to heat shock protein 90 and their antiproliferative activity against multiple human tumor cell lines (NCI-H460, A431, and STO). Compounds 173–184 have strong binding to Hsp90, with IC50 ranging from 0.031 to 0.77 μM. Compound 185, which contains a polar hydrophilic compound, exhibits the strongest affinity for Hsp90 with IC50 < 0.005 μM. Complexes 182 and 185 down-regulated the Hsp90 client proteins EGFR, Akt, Cdk4, Raf-1, survivin, and up-regulated Hsp70. Furthermore, the combination of 173–185 showed significant inhibitory effects on a variety of cancer cells (Fig. 40).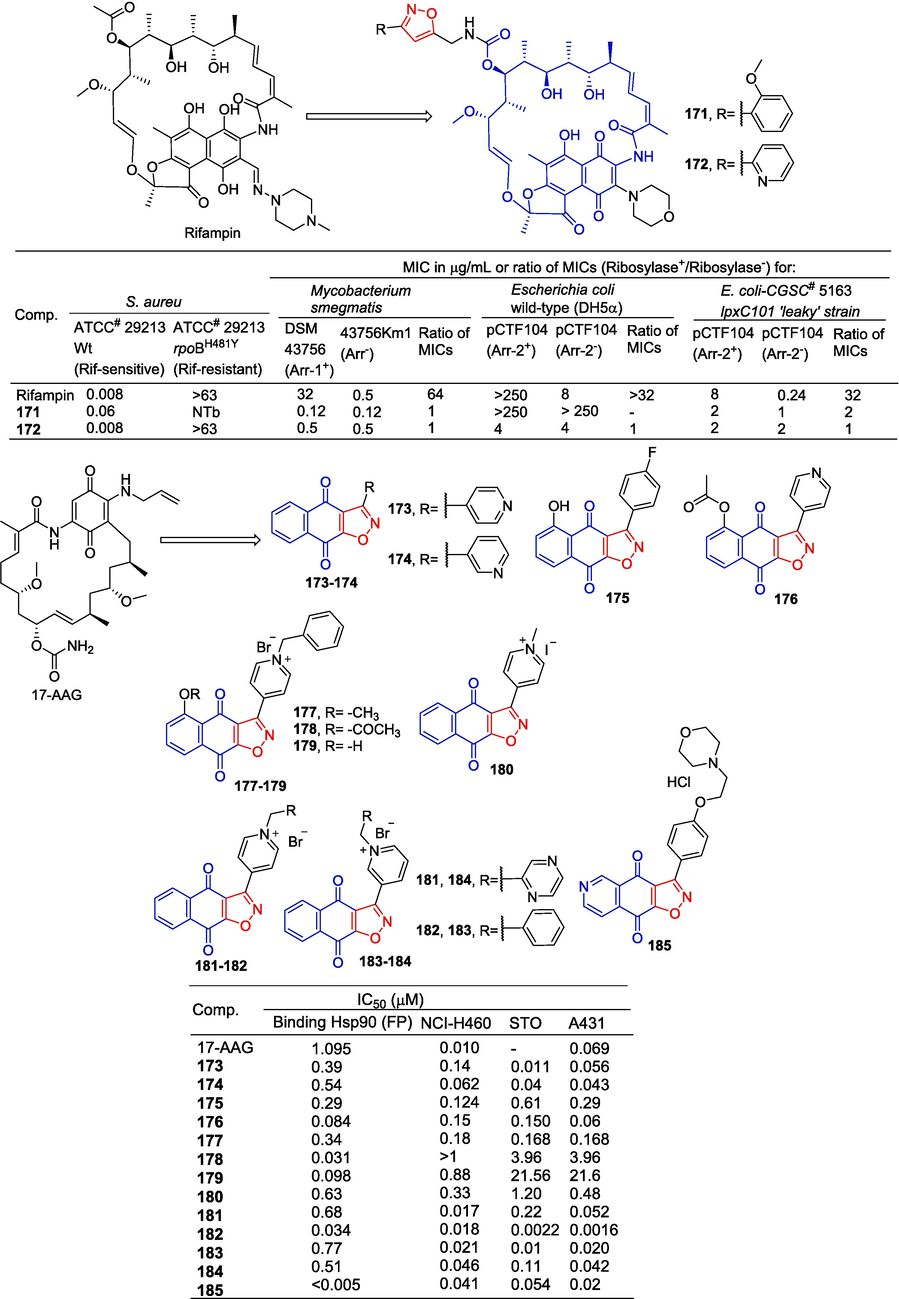
Naphthoquinone-isoxazole derivatives 171–185.
Summary: In this section, the hybrid natural products of benzoquinone-isoxazole, naphthoquinone-isoxazole, and anthraquinone-isoxazole were reviewed. These compounds mainly have anti-tumor, anti-inflammatory, and antimicrobial activities. The biological activities of benzoquinone and anthraquinone natural products after hybridization with isoxazole were not significantly improved compared to their lead compounds or positive control drugs.
TDP2 has the potential to be a new target for the development of anti-viral drugs. Among the naphthoquinone-isoxazole derivatives, furanoquinolinedione deserves to be developed. After replacing furan with isoxazole as bioisosteres, the inhibitory activity of these compounds (159–170) against TDP2 was significantly improved. Similarly, compounds 173–185 showed strong anti-tumor activity in vitro as a novel series of Hsp90 inhibitors. The inhibitory effect of 182 on the A431 cell line was the most obvious, with an IC50 value of 1.6 nM.
2.7 Lignin-isoxazole hybridization
Multidrug resistance (MDR) of anti-tumor drugs is a major obstacle to successful tumor treatment. Finding new drugs with anti-MDR activity is an effective way to overcome tumor resistance. Zhang et al. (Zhang et al., 2016) designed and synthesized a series of aromatic heterocyclic ester compounds of podophyllotoxin to evaluate their anticancer activity against two human chronic myeloid leukemia cell lines K562 and K562/ADR. Compound 186 with isoxazole showed significant inhibitory activity in K562 and K562/ADR cells with IC50 values of 0.052 and 0.025 μM, respectively. Anti-tumor activity was better than doxorubicin (K562: IC50 = 18.779 μM; K562/ADR: IC50 = 0.220 μM). The resistance factor (RF) value of compound 186 was 2.080, which was also better than doxorubicin (RF = 85.359).
Kamal et al. (Kamal et al., 2013) designed and synthesized a series of different hetero-aromatic linked 4β-aminopodophyllotoxin derivatives and evaluated their anti-tumor activity. Among them, the isoxazole-linked compound 187–195 showed inhibitory effects on A549, HT-29, ACHN, B-16, and HeLa cell lines, with IC50 values ranging from 5.26 to 38.4 μM. Among them, 187–189 showed obvious anti-proliferation activity against HeLa cell lines with IC50 values of 5.9, 7.5, and 5.26 μM.
Yang et al. (Yang et al., 2021) designed and synthesized a series of podophyllotoxin derivatives containing isoxazole and evaluated their insecticidal activity. Compounds 196–198 showed significant growth inhibition effects on Mythimna separata (M. separata), with final fatality rates of 65.5 %, 69.0 %, and 62.1 %, respectively. Toosendanin and podophyllotoxin were superior to positive controls (34.5 %). Podophyllotoxin has acaricidal activity that is close to zero for Tetranychus cinnabarinus (T. cinnabarinus), while its isoxazole derivative has acaricidal activity higher than podophyllotoxin. The MRS values of 197–200 at 72 h were 35.4 %, 38.0 %, 41.1 %, and 32.8 %, respectively. Therefore, compounds 199 and 200 showed the strongest insecticidal activity against Mythimna separata Walker and Tetranychus cinnabarinus Boisduval (Fig. 41).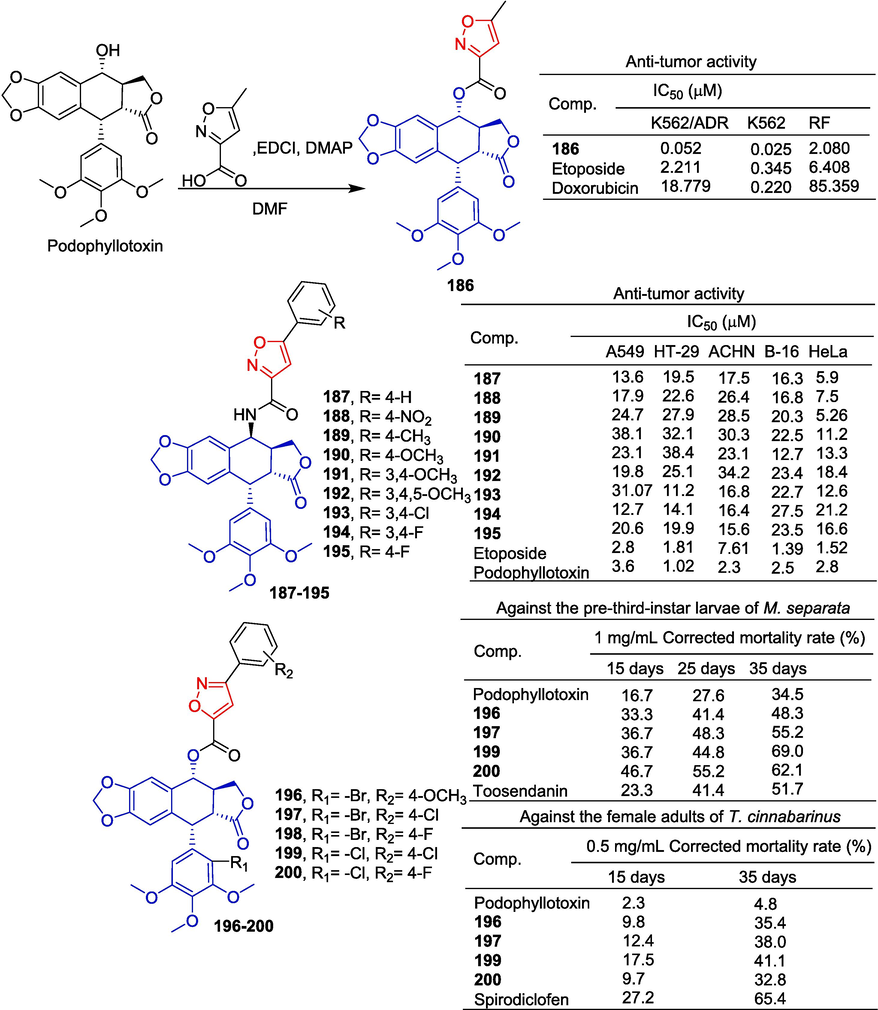
Lignin-isoxazole derivatives 186–200.
Summary: There are few natural products of lignin-isoxazole hybridization. Only podophyllotoxin derivatives have been reported, which have anti-tumor and insecticidal activities. The introduction of 5-methylisoxazole to the hydroxyl group of podophyllotoxin can significantly improve its anti-tumor activity in vitro, but the introduction of 5-phenylisoxazole can reduce its anti-tumor activity. Interestingly, the introduction of 5-phenylisoxazole increased the insecticidal activity of podophyllotoxin.
2.8 Steroidal-isoxazole hybridization
Jiang et al. (Jiang et al., 2013) designed and synthesized a series of 17β-estradiol derivatives that are linked to a large side chain at the C-7α position and determined their antagonistic activity of the endoplasmic reticulum by in vitro bioassays. Isoxazole-containing derivative 201 showed certain inhibition of ERα transfer activation activity in luciferase reporting experiments (ERα: IC50 = 481.8 nM, RBA = 1.2 %; and ERβ: IC50 = 331.1 nM, RBA = 2.6 %), and blocked the interaction between ERα and co-activators. Similarly, derivative 201 inhibited the growth of ERα-positive human breast cancer cells (Fig. 42).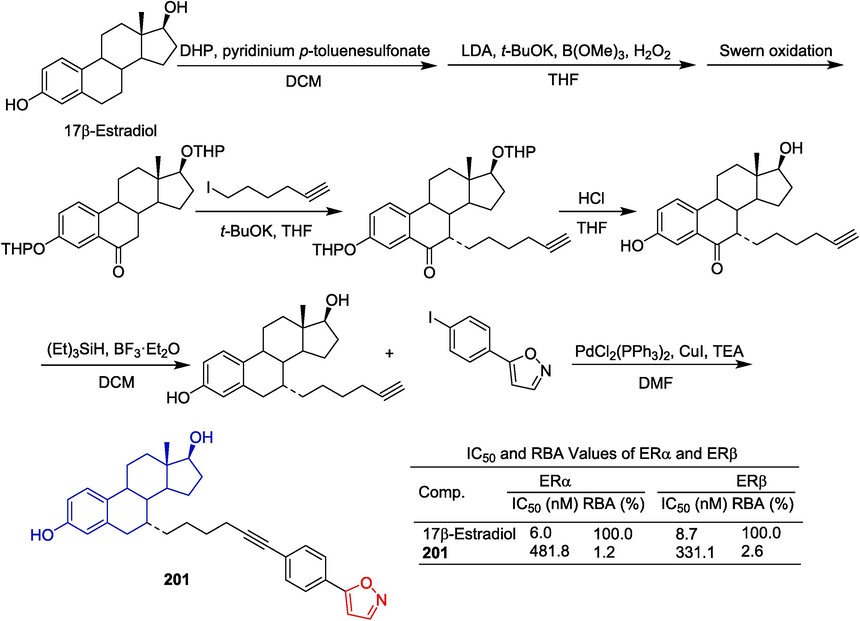
Steroidal-isoxazole derivative 201.
Baranovsky et al. (Baranovsky et al., 2019) designed and synthesized a steroid derivative containing isoxazole at site C-14. Compound 202 showed a certain inhibitory effect on the T-47D cell line, and the cell survival rate decreased to 49 % at a concentration of 50 μM.
A series of isoxazolyl steroid derivatives synthesized by Baranovsky et al. (Baranovsky et al., 2021). Compounds 203–205 were found to have strong binding ligands to human CYP51 in the micromolar range, with Kdapp of 1.50, 1.06, and 1.98 μM, respectively.
Kovacs et al. (Kovacs et al., 2012) prepared novel 17-exo-isoxazolyl derivatives by a “click” chemical method. The derivatives were screened in vitro to detect their anti-proliferative activity against three human cancer cell lines (HeLa, MCF-7, and A2780). Compound 206 showed a certain inhibitory effect on HeLa, MCF-7, and A2780 at a concentration of 10 μM, with inhibition rates of 68 %, 63 %, and 46 %, respectively (Fig. 43).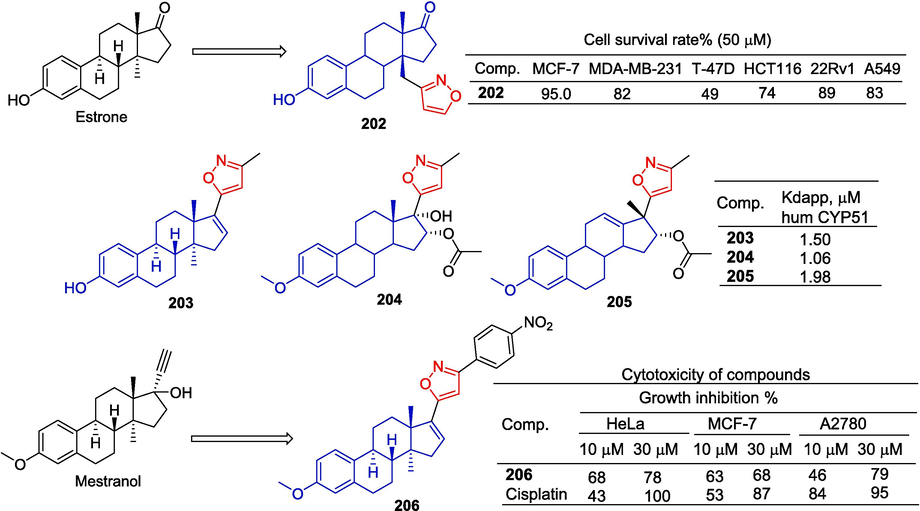
Steroidal-isoxazole derivatives 202–206.
Mohareb et al. (Mohareb et al., 2013) introduced various heterocycles in the C-17 position of the pregnenolone skeleton to obtain a series of derivatives and evaluated the cytotoxicity of some cancer cells. Compound 207, which introduced isoxazole, had the strongest killing effect on NUGC and DLDI cell lines with IC50 values of 44 and 60 nM, respectively, and there was no toxicity against normal fibroblast cells. The synthesis route is depicted in Fig. 44. Firstly, the reaction between pregnenolone and 3-oxo-3-phenylpropanenitrile in the presence of ammonium acetate yielded the Knoevenagel condensation intermediate. Then, this intermediate was reacted with elemental sulfur and a catalytic amount of triethylamine to afford the thiophene derivative. Finally, the compound 207 was obtained by reacting the thiophene derivative with hydroxylamine hydrochloride in 1,4-dioxane containing sodium acetate.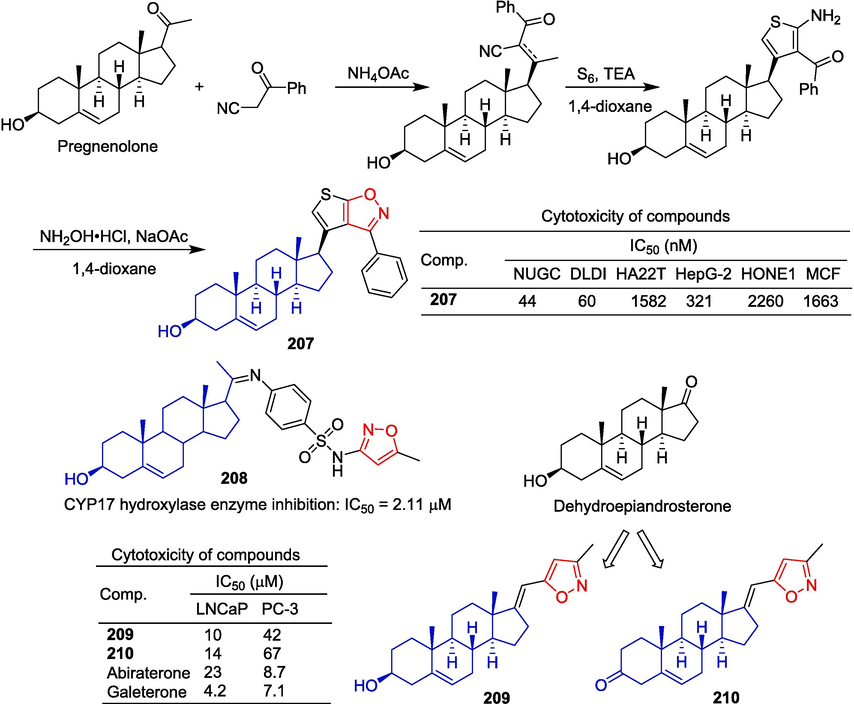
Steroidal-isoxazole derivatives 207–210.
Al-Masoudi et al. (Al-Masoudi et al., 2015) synthesized a series of pregnenolone analogues having aryl-imino groups at C-20 and evaluated the inhibitory activity of human CYP17 hydroxylase expressed in E. coli. Compound 208 was the most potent in the series with an IC50 of 2.11 μM. However, the activity was inferior to that of abiraterone acetate (IC50 = 0.072 μM).
Dalidovich et al. (Dalidovich et al., 2019) prepared isoxazole derivatives of [17(20)E]-21-norpregnene. The ability of the synthesized compound to inhibit the growth of prostate cancer cells was investigated. Among them, compounds 209 and 210 showed high inhibitory effects on LNCaP and PC-3 cell lines, with IC50 values ranging from 10–67 μM. The results also indicated that oxidation of the C3 hydroxyl group did not enhance the antiproliferative activity of prostate cancer cells.
Tsai et al. (Tsai et al., 2017) designed and synthesized a series of steroid derivatives with C-21 hydrophilic substituents. Isoxazol-containing derivative 211 showed a weak inhibitory effect on PC-3 cells with an IC50 of 369 μM.
Ajdukoviic et al. (Ajdukovic et al., 2015) evaluated the potential anti-tumor activity of different a-modified 17α-picolyl and 17(E)-picolinylidene androstane derivatives. The results of the 48 h in vitro experiment showed that compound 213 had the strongest inhibitory effect on PC-3 (IC50 = 6.6 µM).
Qiu et al. (Qiu et al., 2018) designed and synthesized a series of isoxazole-chenodeoxycholic acid hybrids and evaluated the lipid-lowering ability of the compounds using a 3T3-L1 adipocyte model. Compound 214 showed a significant lipid-lowering effect, reducing fat accumulation by 30.5 % at 10 µM. Pharmacological mechanism studies have shown that 214 blocks lipid accumulation by activating the FXR-SHP signaling pathway and effectively down-regulates the expression of the key lipogenesis regulator SREBP-1c (Fig. 45).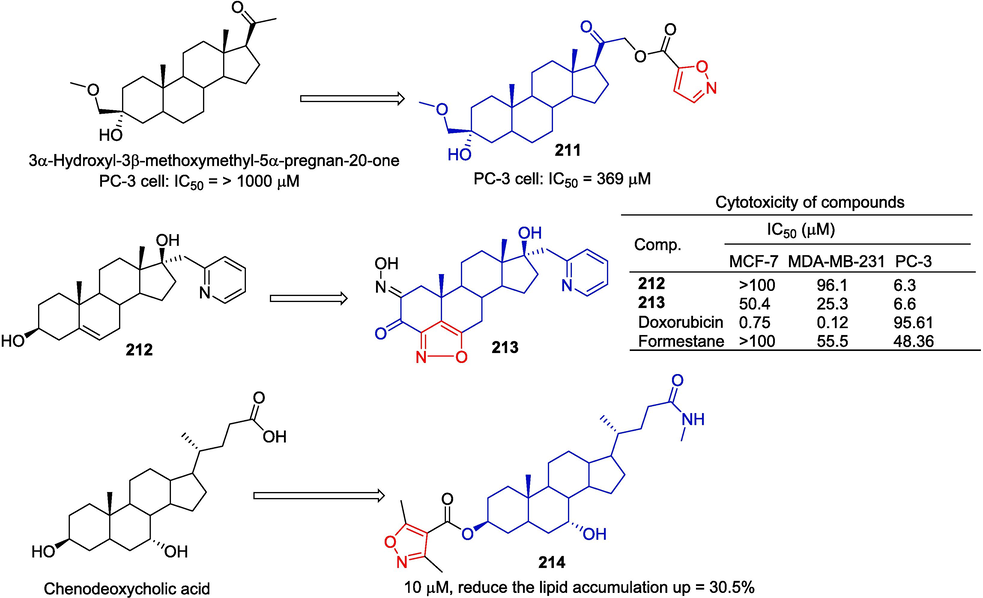
Steroidal-isoxazole derivatives 211 and 213–214.
Obticholic acid is a Farnesoid X receptor (FXR) agonist that has entered a Phase III clinical trial in patients with nonalcoholic steatohepatitis (NASH). Xiao et al. obtained a series of derivatives based on obeticholic acid modified at C24, in hopes of developing FXR agonists with higher selectivity. The derivative 215 (EC50 = 0.03 μM, efficacy = 121 %) linked to C3-OH isoxazole shows a strong affinity for FXR (Xiao et al., 2017). The synthesis route is depicted in Fig. 46. The starting material, obticholic acid, was subjected to oxidation, condensation, decarboxylation, and cyclization reactions to yield compound 215.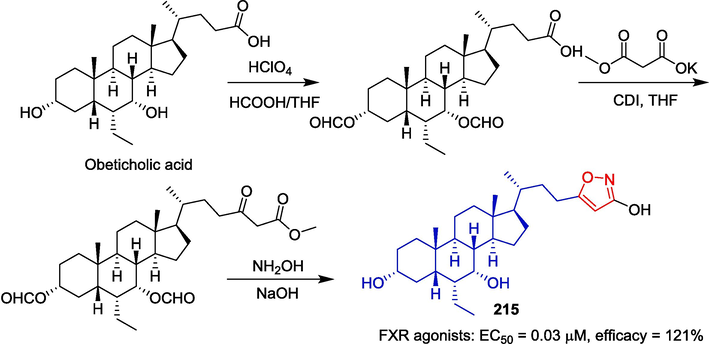
Steroidal-isoxazole derivative 215.
Figueroa-Valverde et al. (Figueroa-Valverde et al., 2011) synthesized the pregnenolone-danazol-ethylenediamine conjugate, and the antibacterial activity against S. aureus and Vibrio cholerae was tested. The bioactivity results showed that compound 216 inhibited the growth of both bacteria in a dose-dependent manner, with MIC values of 1.23 μM for S. aureus and V. cholerae.
Wang et al. (Wang et al., 2012) designed and synthesized a series of caudatin derivatives and detected their anti-hepatitis B virus (HBV) activity in HepG-2.2.15 cells. Compound 217 containing isoxazole showed inhibitory activity against HBV DNA replication with IC50 of 16.97 μM. Additionally, compound 217 inhibited HBsAg secretion to some extent (IC50 = 201.23 μM).
Yildiz et al. (Erdagi and Yildiz, 2022) modified diosgenin to obtain a series of derivatives and evaluated the cytotoxicity of these compounds in MCF-7 and A549. Compound 218 containing isoxazole showed stronger anti-tumor activity against MCF-7 and A549 cell lines with IC50 values of 9.15 µM and 14.92 µM, respectively, than diosgenin (IC50 values of 26.91 and 36.21 µM, respectively) (Fig. 47).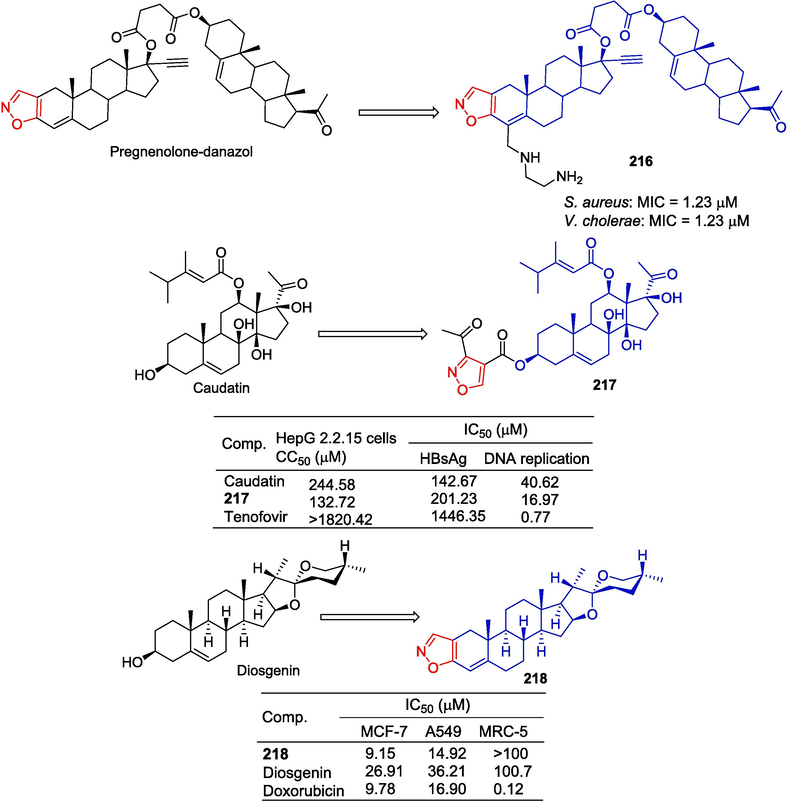
Steroidal-isoxazole derivatives 216–218.
Smirnova et al. (Smirnova et al., 2020) designed and synthesized a series of dipterocarpus alatus derivatives and evaluated their cholinesterase inhibitory activity and cytotoxicity. Compound 219 containing isoxazole showed some cytotoxicity to the MCF-7 cell line (EC50 = 16.2 µM), and slightly inhibited AChE (15.87 %). There was no inhibitory effect on butyrylcholinesterase.
20(R)-25-methoxyl-dammarane-3β,12β,20-triol (AD-1) is a newly discovered ginsenoside that induces G0/G1 cell cycle arrest, apoptosis, and ROS production. Ma et al. (Ma et al., 2021) obtained a series of AD-1 derivatives by introducing heterocyclic rings at the positions C-2 and C-3. The IC50 values of the 220 isoxazole-containing derivative for the RM-1 and HCT116 cell lines were 14.38 and 19.55 µM. Compound 220 did not show obvious cytotoxicity to normal GES-1 cells (IC50 > 100 μM).
To develop more effective anti-myocardial ischemia/reperfusion (MI/R) injury drugs, Wu et al. (Wu et al., 2020) designed and synthesized a series of heterocyclic panaxatriol fusion derivatives, and evaluated the cardiovascular activity of heterocyclic derivatives of panaxatriol. Compound 221 increased cell viability to approximately 88 % and 89.2 % at 10 µM. In vivo, captopril (20 mg/kg) and panaxatriol (40 mg/kg) reduced the size of the infarct by 34.0 % and 30.6 %, respectively. At a dose of 40 mg /kg, compound 221 reduced the size of the infarct to 20.5 %. Therefore, compound 221 can significantly reduce the size of the myocardial infarction, reduce the leakage of circulating cardiac troponin I (cTnI), and alleviate myocardial tissue injury (Fig. 48).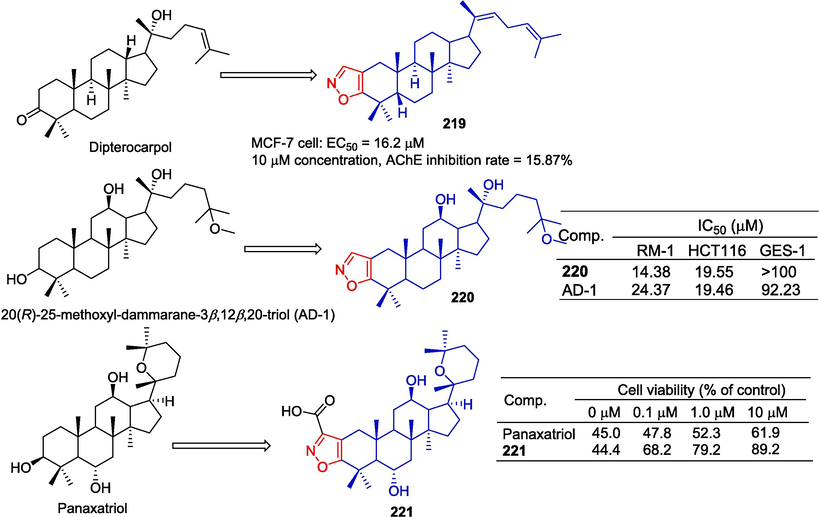
Steroidal-isoxazole derivatives 219–221.
Xu et al. (Xu et al., 2021) designed and synthesized a series of cholesterol oxime ether derivatives containing isoxazole, and determined the agricultural activity of these compounds against A. citricola, M. separata, and P. xylostella. The control effect was tested in the greenhouse. Compounds 223–225 and 231–232 showed significant inhibitory effects on M. separata, with mortality rates (MRs) ranging from 48.1 % to 66.6 % after 35 days at a concentration of 1 mg/mL. Among them, compound 224 has the highest MRs against M. separata, reaching 66.6 %. The third instar larvae of P. xylostella were treated with 20 μg/larva compounds 222–224, 226, and 228–229 for 48 h. These compounds showed good insecticidal activity, with MRs ranging from 26.6 % to 42.2 %. Among them, compound 224 had the highest MR against P. xylostella, reaching 42.2 %. The insecticidal activity of A. citricola treated with 0.04 μg/larva for 48 h showed that the MR of compounds 223, 224, 227, and 230 ranged from 38.0 % to 55.2 %. Among them, compound 230 had the highest MRs against A. citricola, reaching 55.2 %. The 48 h LD50 value of compound 230 for A. citricola was 0.041 μg/nymph, while the cholesterol 48 h LD50 value was 0.237 μg/nymph. Therefore, the aphid-killing activity of compound 230 is 5.8 times that of cholesterol. The control effect of compound 230 on A. citricola was tested in a greenhouse. The results showed that the control effects of 230 at 3, 5, and 7 days after treatment were 40.3 %, 54.5 %, and 59.8 %, respectively, while the corresponding cholesterol results were 9.5, 14.5 and 18.1 %, respectively. The results showed that the prophylactic effect of compound 230 was 3.3–4.2 times that of cholesterol (Fig. 49). Structure-activity relationships suggest that the C3 hydroxyl group and the C7 position of cholesterol are two important sites for modification.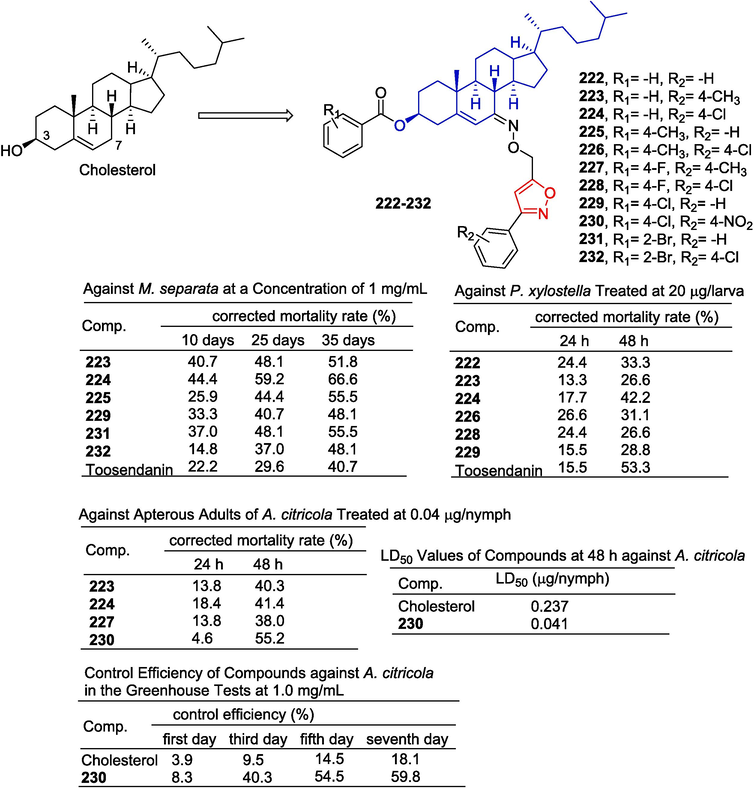
Steroidal-isoxazole derivatives 222–232.
Summary: In this section, the biological activities of various terpenoid natural products after hybridization with isoxazole molecules were reviewed, including estradiol, estrone, mestranol, pregnenolone, dehydroepiandrosterone, chenodeoxycholic acid, obeticholic acid, caudatin, diosgenin, dipterocarpol, panaxatriol, and cholesterol. Their semi-synthetic derivatives mainly have anti-tumor, antibacterial, anti-viral, lipid-lowering, anti-myocardial ischemia/reperfusion injury, and even agricultural activities. In anti-tumor activity, compound 207 showed good selectivity between tumor cells and normal cells. Compound 214 is valuable for further study in the treatment of hyperlipidemia. In addition, compared with panaxatriol, compound 221 can greatly improve the protective activity towards cardiomyocytes. The preliminary structure–activity relationship study suggested that fusing isoxazole can improve the activity of the parent compound.
2.9 Terpene-isoxazole hybridization
You et al. (You et al., 2003) designed and synthesized modified betulinic acid derivatives with A rings and evaluated the anti-tumor activity of three cancer cells. The derivative 233 containing isoxazole showed significant inhibitory effects on SK-MEL-2, A549, and B16-F10 cell lines with IC50 values of 1.15, 2.70, and 1.57 μM, respectively. On the activity of betulinic acid (SK-MEL-2: IC50 = 7.62 μM; A549: IC50 = 7.70 μM; B16-F10: IC50 = 4.98 μM). The findings also demonstrated that the introduction of isoxazole into the A ring of betulinic acid resulted in unfavorable anti-tumor activity.
Minassi et al. (Minassi et al., 2018) designed and synthesized betulate-isoxazole derivatives. Among them, compound 234 was found to be an inhibitor of HIF prolyl hydrolases (PHDs) with good biological activity (EC50 = 2.6 µM).
Xu et al. (Xu et al., 2012) designed and synthesized a series of betulinic acid derivatives by introducing different fused heterocycles into the A ring, and evaluated their inhibitory effects on the formation of osteoclasts induced by RANKL. At a concentration of 0.5 μM, the inhibition rate of compound 235 containing isoxazole was 66.8 %. The activity was much higher than that of betulinic acid (IC50 ≈ 20 μM).
Galaiko et al. (Galaiko et al., 2014) obtained a derivative from the methyl ester of betulonic acid and tested the cytotoxicity of compound 236 in RD TE32, A549 and MS cells. Compound 236 showed an obvious inhibitory effect on MS cells with an IC50 value of 16.36 μM.
Gundoju et al. (Gundoju et al., 2019) modified the C28 site of betulinic acid to obtain a series of derivatives and evaluated their inhibitory effects on α-glucosidase in RAW 264.7 cells. Compound 237 linked to isoxazole showed strong α-glucosidase inhibitory activity with an IC50 value of 10.8 μM. However, inhibitory activity was not as good as that of betulinic acid (IC50 = 7.8 μM).
Luginina et al. (Luginina et al., 2021) designed and synthesized betulinic acid-isoxazole conjugates and tested the anti-tumor activity of these compounds in vitro. Compounds 238 and 241 showed significant inhibitory effects on the A549 cell line with GI50 values of 11.05 and 11.51 μM, respectively. Compounds 239 and 240 showed significant inhibitory effects on the MCF-7 cell line with GI50 values of 11.47 and 12.49 μM, respectively. These compounds did not show significant cytotoxicity to immortalized human fibroblasts hTERT (GI50 > 100 μM) (Fig. 50).
Pentacyclic triterpenoids-isoxazole derivatives 233–241.
He et al. (He et al., 2016) synthesized a series of derivatives by introducing 5-substituted phenyl-3-isoxazole at position C3 of betulin. The PTP1B inhibitory activity of the target compounds was significantly improved than that of betulin. The IC50 of the compounds at 243 was 0.98 μM, which was 12 times that of betulin. The selectivity of the compound to the homologous enzyme TCPTP reached 3.95 times. The inhibitory activity of compound 242 on PTP1B was slightly weaker than that of 243, with IC50 of 1.62 μM. However, the selectivity of the homologous enzyme TCPTP was the highest, and the selectivity coefficient reached 5.25 times. The synthesis route is depicted in Fig. 51. As a protective group, 3,4-dihydro-2H-pyran selectively protected the hydroxyl group at C28 of betulin, followed by reaction with various substituted 3-(chloromethyl)-5-phenylisoxazoles. Finally, the protective group was removed in a methanol solution containing p-toluenesulfonic acid to obtain the target compounds.
Pentacyclic triterpenoids-isoxazole derivatives 242–245.
Koohang et al. (Koohang et al., 2009) reported that compound 244 showed a significant inhibitory effect on SK-MEL-2 and DAOY cell lines with an IC50 of 8 μM. The IC50 values of compound 245 for these two cancer cells were 8 and 7 μM, respectively.
Zhang et al. (Zhang et al., 2015) designed and synthesized 23-hydroxybetulinic acid (HBA) derivatives substituted with isoxazole and evaluated their anti-tumor activity. Compounds 246 and 247 showed inhibitory effects on various cancer cells, especially on the SF-763 cell line with IC50 values of 6.08 and 6.94 μM. More than 3-hydroxybetulinic acid activity (IC50 = 43.40 μM).
Grishko et al. (Grishko et al., 2017) reported a method of synthesis of novel ring-A fused heterocyclic derivatives. Derivatives 248 containing isoxazole showed significant inhibitory effects on RD-TE32, A549, MS, HEp-2, and HCT116 cell lines with IC50 values of 14.77, 7.85, 14.00, 22.07, and 19.01 μM, respectively (Fig. 52).
Pentacyclic triterpenoids-isoxazole derivatives 246–248.
Liu et al. (Liu et al., 2016) designed and synthesized a series of heterocyclic substituted derivatives of bevirimat as HIV-1 inhibitors. Cell culture efficacy of compound 249 against wild-type (wt) virus was weaker than that of bevirimat (EC50 = 10 nM). However, in the presence of human serum, the EC50 value at 249 was 0.295 μM, reflecting a much-reduced degree of serum-binding (7.5 times at 12 h versus 97.4 times for bevirimat). The synthesis route is depicted in Fig. 53. Betulin was used as the starting material, and the C28 primary hydroxyl group was protected using benzoic anhydride to form the corresponding ester. The triflate intermediate was prepared, followed by Suzuki coupling with aryl carboxylic acid. Finally, the benzoyl protecting group at the C28 position was removed, and the aryl ester was hydrolyzed.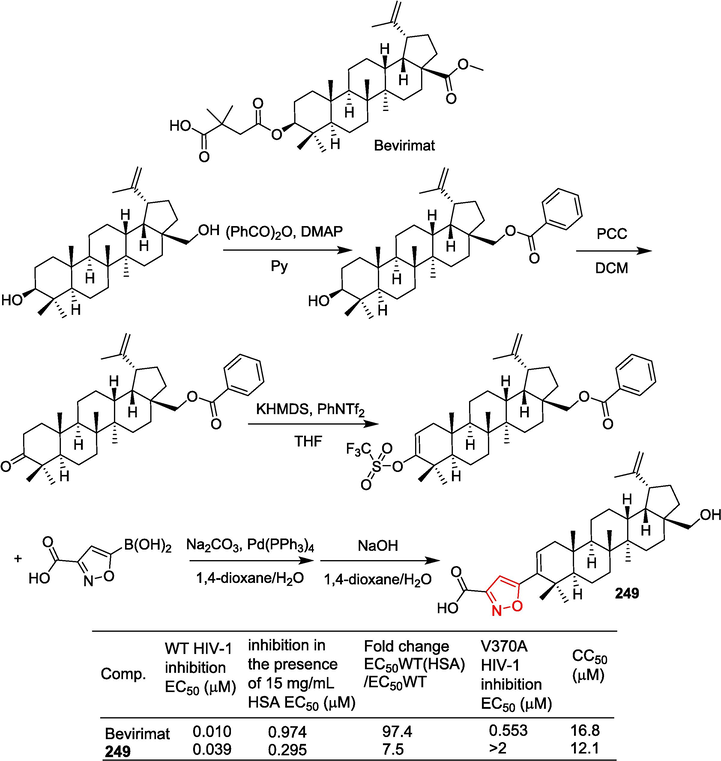
Pentacyclic triterpenoid-isoxazole derivative 249.
Wu et al. (Wu et al., 2016) synthesized oleanolic acid derivatives containing isoxazole and evaluated their anti-bone resorption activities. The results showed that the isoxazole derivative 250 inhibited the osteoclast formation of rank-induced RAW264.7 cells at a concentration of 2 μM with an inhibitory rate of 78.5 %. The inhibitory rate of oleanolic acid was more than 11.4 %.
Chouaib et al. (Chouaib et al., 2016) reported the synthesis of oleanolic acid derivatives coupled with isoxazole and evaluated their anti-inflammatory activity. In vitro anti-inflammatory tests showed that compounds 251–252 had a greater inhibitory effect on the production of pro-inflammatory IL-1β cytokines in LPS-stimulated human peripheral blood mononuclear cells (PBMC) than oleanolic acid. The compounds 251–252 produced 55 % and 50 % IL-1β (/control) at a concentration of 100 μM, respectively, which was less than 88 % of oleanolic acid. They also reported the synthesis of the oleanol-isoxazole conjugate and evaluated the anti-proliferation of the cancer cell lines EMT-6 and SW480. The results of the in vitro cytotoxicity test showed that 251–253 had stronger anti-proliferative activity, and the survival rate of EMT-6 cells decreased to 40 %, 44 %, and 32 %. The survival rate of SW480 cells decreased to 71 %, 61 %, and 57 %. The introduction of heterocyclic rings at the C3 position of isoxazole indicates, to some extent, that it exhibits superior anti-tumor activity compared to substituted phenyl.
Compound 254 has a strong inhibitory effect on leukemia cancer cells and inhibits the proliferation of K562 cells in a dose-dependent manner. After K562 cells were exposed to the compound 254 for 72 h, the IC50 value reached 39.252 μM. Compound 253 induces apoptosis and arrest of the G1 phase cell cycle, possibly due to decreased levels of pAkt (Pan et al., 2015).
Compound 255 has an additional benzene ring based on 254, which has inhibitory activity on MCF-7 cells with an IC50 value of 88.2 μM (Mallavadhani et al., 2014).
Wang et al. (Wang et al., 2017) modified oleanolic acid at position C3 to search for new interacting agents of FXR. The 256 isoxazole-modified oleanolic acid derivative showed an antagonistic effect on FXR with an IC50 value of 19.41 μM. The inhibitory effect did not exceed oleanolic acid (IC50 = 13.69 μM) (Fig. 54).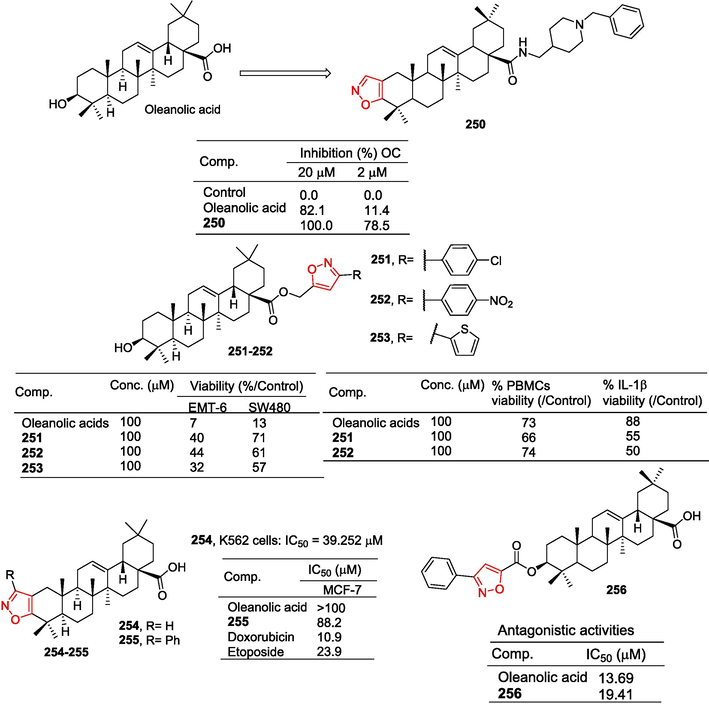
Pentacyclic triterpenoids-isoxazole derivatives 250–256.
You et al. (Gao et al., 2010) designed and synthesized A-ring modified glycyrrhetinic acid derivatives. All of these derivatives improved the anti-proliferation effect of human HL-60 leukemia cells. The derivative 257 containing isoxazole showed a significant inhibitory effect on HL-60 cells with an IC50 value of 1.7 μM. It exceeded the activity of glycyrrhetinic acid (IC50 > 40 μM).
Galaiko et al. (Galaiko et al., 2014) synthesis triterpene A-condensed isoxazoles and tested the cytotoxicity of compound 258 against RTE32, A549, and MS cells. Compound 258 showed the strongest inhibitory effect on MS cells with an IC50 value of 23.18 μM.
Olanipekun et al. (Olanipekun et al., 2023) designed and synthesized a series of isoxazole-arjunolic acid derivatives and evaluated their inhibitory potentials for tyrosinase and α-glucosidase. Compound 259 had the strongest inhibitory effect on tyrosinase (IC50 = 14.3 μM), about three times that of the standard drug kojic acid (IC50 = 41.5 μM). Compound 260 had strong inhibition of α-glucosidase (IC50 = 14.5 μM), and the value of IC50 was comparable to that of standard acarbose (IC50 = 10.4 μM).
Compound 261, a derivative of maslinic acid containing isoxazole, has a higher anti-carcinogenic effect than crataegic acid. In the breast cancer cell line EMT-6, the survival rate (percentage of live cells/control) of 261 and crataegic acid at 10 μM was 5 % and 100 %, respectively. In colon cancer SW480, survival rates (percentage of live cells/controls) for 261 and cratonic acid at 10 μM were 9 % and 123 %, respectively (Chouaib et al., 2016). Carboxyl-linked isoxazole compounds 261–263 of maslinic acid were found to have anti-inflammatory activity. The derivative 261 was the most anti-inflammatory (100 μM: IL-1β production rate = 4 %). The derivative 262–263 also has anti-inflammatory effects (IL-1β production rate = 33 % and 44 %, 30 μM) (Fig. 55).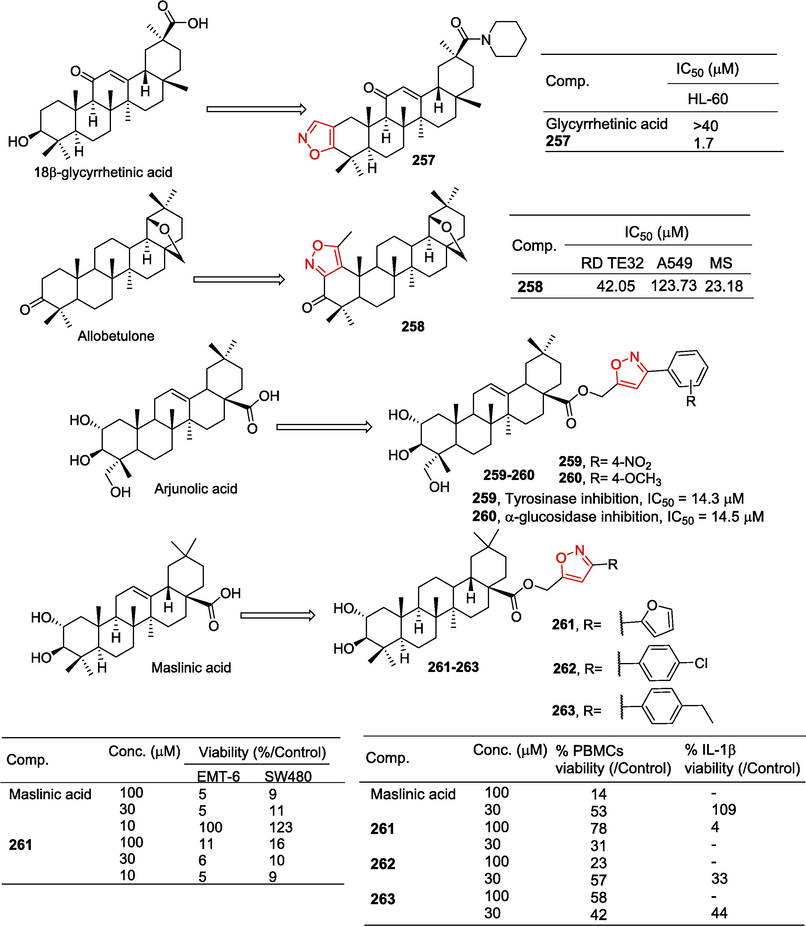
Pentacyclic triterpenoids-isoxazole derivatives 257–263.
Foskolin is a diterpene isolated from the roots of Coleus forskohlii. Burra et al. (Burra et al., 2017) performed a 1,3-dipolar cycloaddition reaction to introduce an isoxazole fragment onto the C1-OH of forskolin. The anti-cancer activity of these compounds was evaluated against MCF-7 and BT-474 cell lines (Fig. 56). The IC50 value of compounds 264–272 against MCF-7 cells was 0.5 μM. The IC50 value of compound 267 against BT-474 cells was 0.5 μM. The activity of both compounds was higher than that of forskolin (MCF-7: IC50 = 63.3 μM; BT-474: IC50 > 100 μM).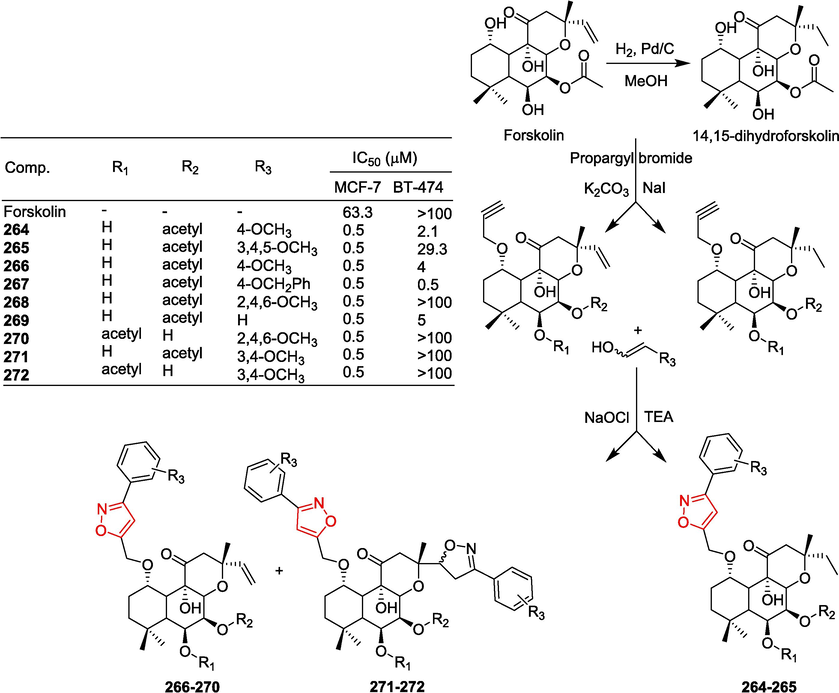
Terpene-isoxazole derivatives 264–272.
Mokenapelli et al. (Mokenapelli et al., 2021) synthesized a series of isoxazole-containing derivatives using andrographolide as the skeleton and evaluated the cytotoxicity of HCT15, HeLa, and DU145 cell lines. Compounds 273 and 274 showed significant cytotoxic activity against DU145 cell lines with IC50 values of 48.60 and 42.98 µM (Fig. 57).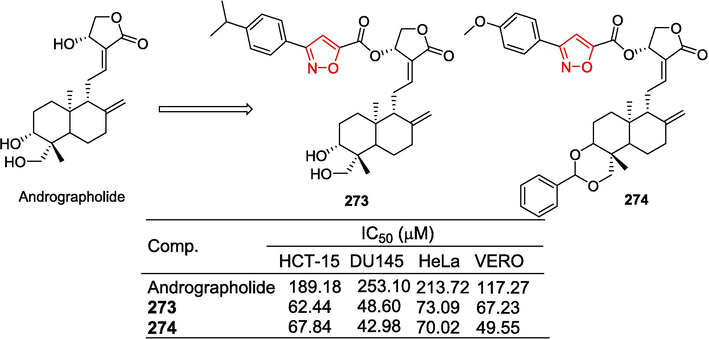
Terpene-isoxazole derivatives 273–274.
Zhang et al. (Zhang et al., 2018) studied the in vitro anti-S. aureus activity of N-sulfonaminoethyloxime derivatives of dehydroabietic acid. The derivative 275 showed the highest inhibitory activity against the S. aureus Newman strain and multidrug-resistant strains (NRS-1, NRS-70, NRS-100, NRS-108, and NRS-271), with a MIC of 1.56–3.13 μg/mL.
Alwaseem et al. (Alwaseem et al., 2018) modified C9 and C14 of parthenolide using P450-mediated chemical enzymes to obtain a series of parthenolide derivatives. Derivative 276 showed a significant inhibitory effect on the HCT116 cell line with an IC50 value of 8.3 μM. Anticancer activity was significantly higher than parthenolide (IC50 = 18.0 µM).
Modification of parthenolide C9 or C14 sites by Tyagi et al. (Tyagi et al., 2016) could effectively enhance the anti-leukemia activity of parthenolide. Derivatives 276 and 277 significantly inhibited M9-ENL1 cells with IC50 values of 2.6 and 13.7 μM, respectively.
Chen et al. (Chen et al., 2019) reported an anti-thrombotic derivative based on isosteviol with anticoagulant and anti-platelet activities. Compound 278 selectively inhibited FXa (Ki = 9.786 μM) containing an isoxazole structure against a panel of serine proteases, and it has certain anticoagulant activity (Fig. 58).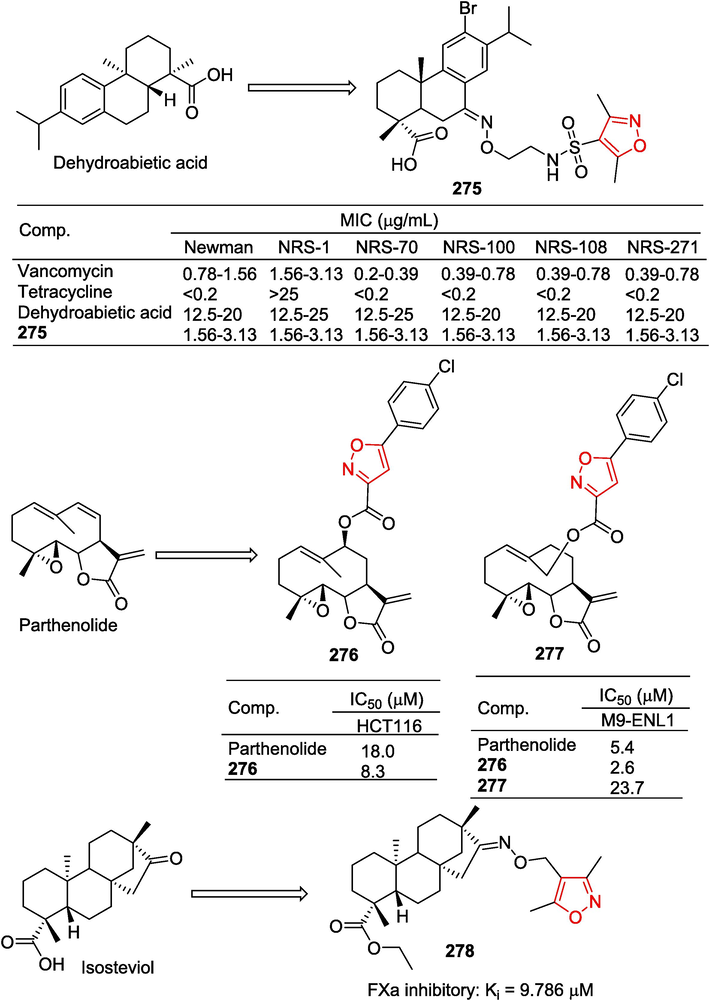
Terpene-isoxazole derivatives 275–278.
Summary: Terpene-isoxazole hybridization natural products mainly have anti-tumor, anti-inflammatory, anti-HIV, antibacterial, and anti-bone resorption activities. Moreover, PTP1B is recognized as a target for the therapy of diabetes. The inhibitory activity of compounds 242 and 243 on PTP1B was significantly better than that of the parent compound, and the preliminary structural-activity relationship showed that the introduction of electron-withdrawing groups to the meta position of the benzene ring or electron-donating groups to the para position of the benzene ring was beneficial to the activity.
Pentacyclic triterpenoids exist widely in many plants and have many pharmacological activities, including anti-tumor activity. Unfortunately, the anti-tumor activity of these pentacyclic triterpenoids-isoxazole derivatives was not satisfactory, the IC50 values of these derivatives on cancer cells did not reach the nanomolar level. Interestingly, forskolin, a labdane diterpene natural product, its C1-isoxazole derivatives (264–272) exhibited significant anti-cancer activity against the MCF-7 breast cancer cells with IC50 < 1 µM. Furthermore, the preliminary structure–activity relationship showed that the introduction of electron-donating groups to the benzene ring at the C3 position of isoxazole was beneficial to the activity.
2.10 Alkaloid-isoxazole hybridization
Camptothecin prevents the breakdown of the topoisomerase-I (Topo I)/DNA cleavable complex. Derivatives 279 and 280 showed significant inhibitory activity against A549, MCF-7, MDA-MBq231, HepG-2, KB, BEL-7402, MGC80-3, and SK-N-SH cell lines, with IC50 values ranging from 0.064–10.54 μM. Compound 279 had the strongest inhibitory effect on the HepG-2 cell line with an IC50 value of 0.064 μM (Pan et al., 2018). The synthetic routes for these two compounds are illustrated in Fig. 59. 10-Hydroxycamptothecin was used as the starting material, and the Duff reaction was first employed to introduce the aldehyde group at the C9 position. Subsequently, cyclization produced derivative 279 containing an isoxazole fragment. The compound 280 was synthesized by introducing N-succinylglycine to compound 279 using a one-pot method.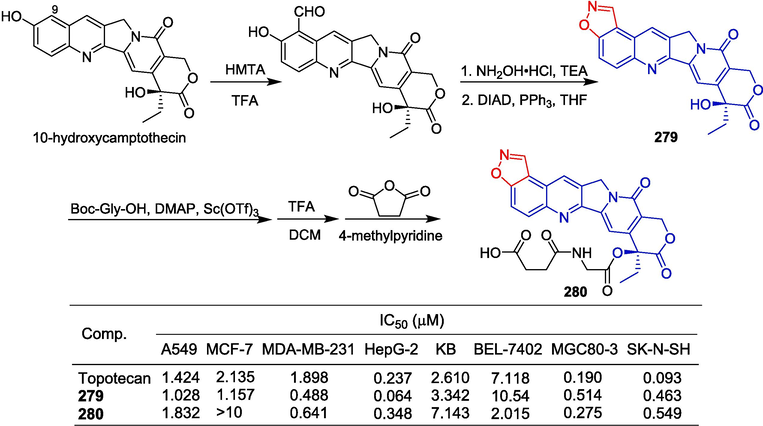
Alkaloid-isoxazole derivatives 279–280.
Zhu et al. (Zhu et al., 2020) developed a series of HSP90 inhibitor SN38 couplings at the 20-OH and 10-OH sites of ester and carbamate linkage of SN38. Among them, compounds 281 and 282 exhibit good HSP90α affinity with IC50 values of 72 and 70 nM. Compounds 281 and 282 showed significant inhibitory effects on A549, HCT116, MIA-PACA-2, Capan-1, and HL7702 cell lines with IC50 values ranging from 22–7893 nM. Furthermore, compound 282 showed excellent anti-tumor activity and low toxicity in HCT116 and Capan-1 xenograft tumor models. The pharmacokinetic analysis of the HCT116 and Capan-1 xenografts further confirmed that treatment with compound 282 led to effective cutting of tumor sites and prolonged exposure to SN38. The initial structure–activity relationship analysis suggested that the elongation of the carbon chain conferred enhanced antitumor activity. The synthetic routes for these two compounds are illustrated in Fig. 60.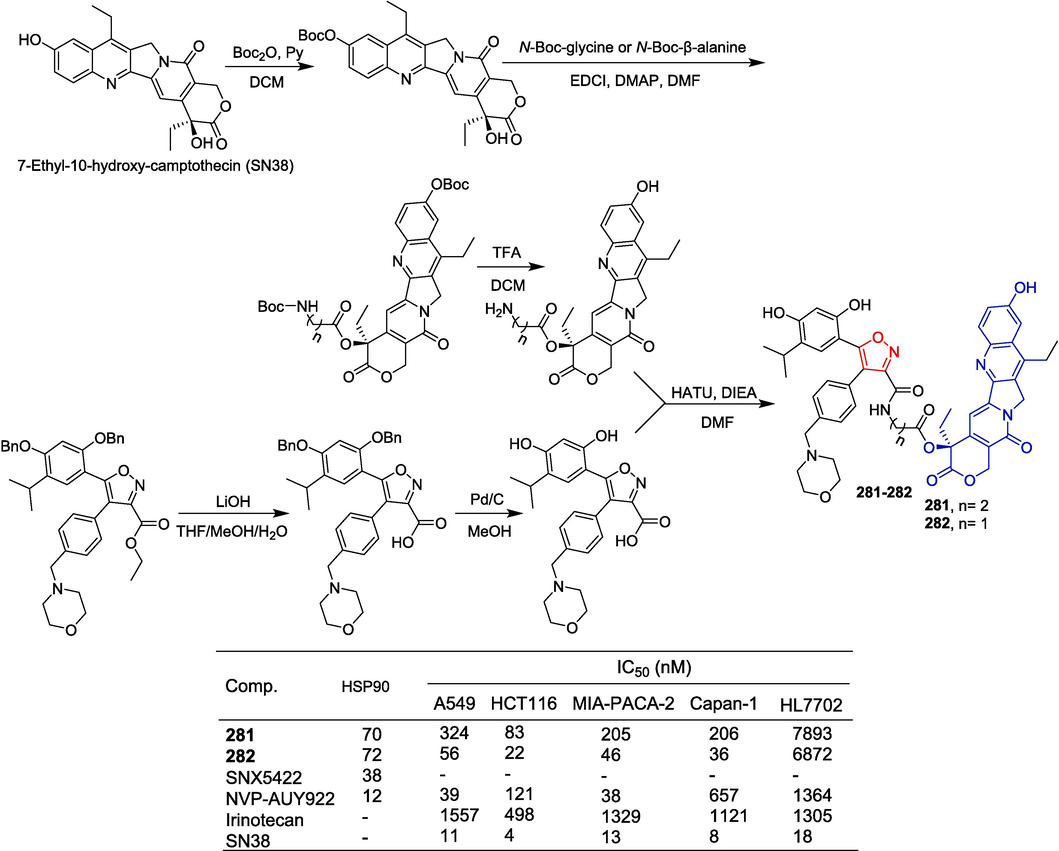
Alkaloid-isoxazole derivatives 281–282.
Compounds 283 and 284 were designed and synthesized by the same research group using similar synthesis methods as compounds 281 and 282 (Fig. 61) (Cao et al., 2023). In vitro experiments showed that these exhibited acceptable stability in PBS and plasma, with significant HSP90 binding affinity and strong cytotoxic capacity. Compounds 283 and 284 have a strong binding affinity for HSP90 with IC50 values of 70 and 72 nM. It showed significant inhibitory effects on A549, BGC823, HCT116, PSN-1, Capan-1, and MCF-7 cell lines, with IC50 values ranging from 8 to 302 nM.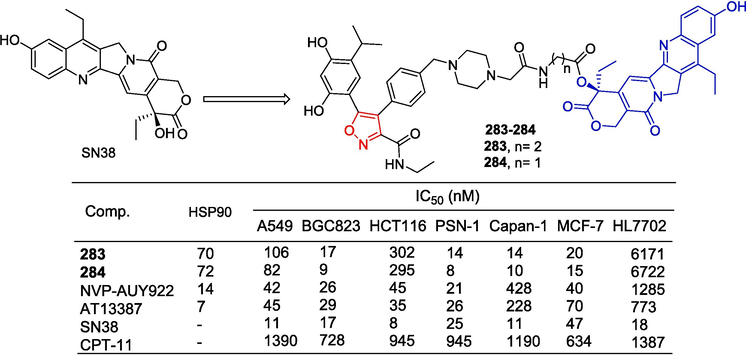
Alkaloid-isoxazole derivatives 283–284.
Liu et al. (Liu et al., 2019) reported a new conjugate of phosphatidylserine-targeted zinc (II) dipicolylamine drug and evaluated the in vitro cytotoxicity of these compounds against the COLO-205 cell line and the normal fibroblast Detroit 551. Cytotoxic compound 285 inhibited COLO-205 cells at 547 nM. Compound 285 is less toxic to normal cells (Detroit 551: IC50 = 7168 nM). As shown in Fig. 62, reductive amination of starting material with methyl 4-formylbenzoate and NaBH4. Then, the ester intermediate was treated with an acyl chloride containing an isoxazole group under basic conditions in CH2Cl2. Subsequently, the SN38-conjugated intermediate was obtained through hydrolysis and esterification. Incubation with 2 equivalences of zinc nitrate with SN38-conjugated intermediate then afforded the compound 285.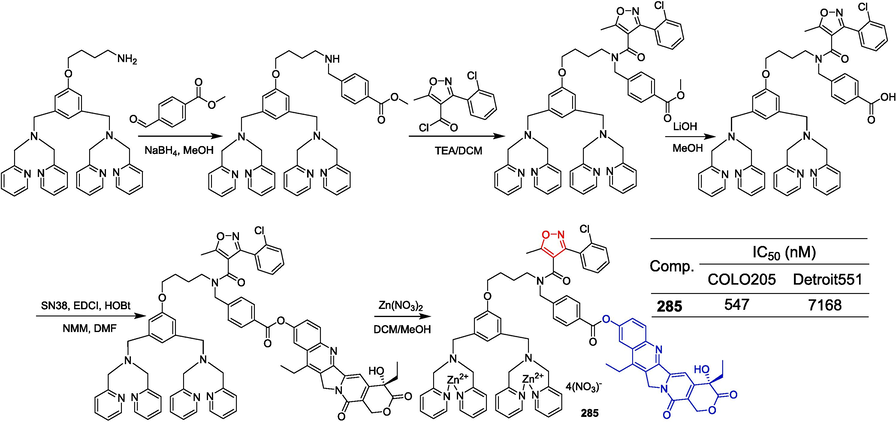
Alkaloid-isoxazole derivative 285.
Filali et al. (Filali et al., 2016) obtained a novel derivative of isoxazole by linking Harmine and evaluated its anti-proliferative activity against OCVAR-3, MCF-7, and HCT116 cell lines by MTT assay. Of the synthesized derivatives, compounds 286 and 287 showed the best activity, with IC50 values ranging from 5.0–73.08 µM for the three cancer cells. The above results suggest that the introduction of electron-donating groups at the para-position of the phenyl group may lead to a reduction in antiproliferative activity. Compounds 286 and 287 showed antiacetylcholinesterase activity with IC50 values of 22.9 and 17.9 µM.
Ravinaik et al. (Ravinaik et al., 2019) reported the synthesis of linked amide derivatives based on β-carboline and the evaluation of their anti-tumor activity. The synthetic approach to the targeted compounds 288–289 began with the cyclization of 2-aminobenzenethiol and 4-acetylbenzaldehyde in the presence of ZnO nanoparticles, resulting in the formation of 1-{4-(benzo[d]thiazol-2-yl)phenyl}ethanone. Subsequent reaction with 9H-pyrido[3,4-b]indole-1-carbaldehyde through Claisen-Schmidt condensation led to an α,β-unsaturated chalcone intermediate. The chalcone intermediate was then reacted with hydroxylamine hydrochloride and pyridine, leading to the formation of an isoxazole intermediate. Finally, coupling reactions between the isoxazole intermediate and various substituted benzoyl chlorides in the presence of Cs2CO3 afforded the target compounds 288–289. Compounds 288 and 289 showed significant inhibitory effects on MCF-7, A549, Colo-205, and A2780 cell lines with IC50 values ranging from 0.10–0.77 μM, respectively.
Wu et al. (Wu et al., 2007) reported carboline-based MK2 inhibitors. Compound 290 containing isoxazole inhibited MK2; IC50 as low as 1.50 μM, as measured in a DELFIA assay (Fig. 63).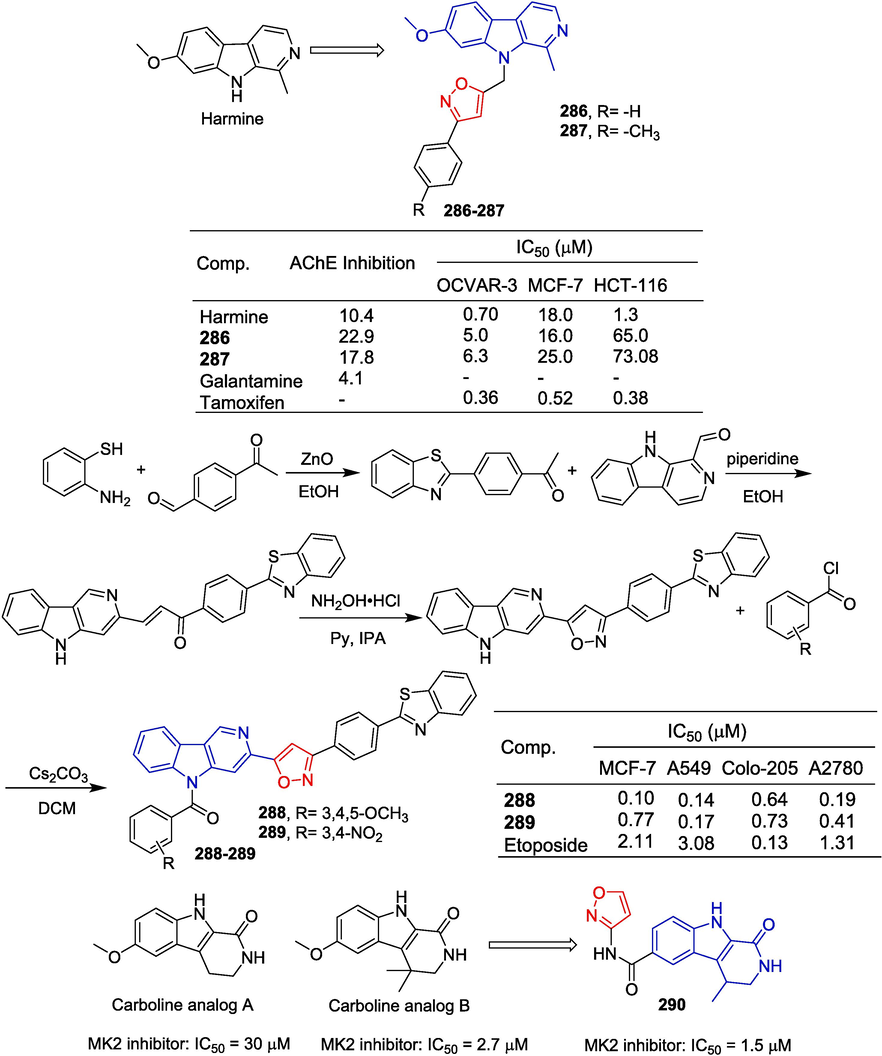
Alkaloid-isoxazole derivatives 286–290.
Indirubin has been shown to inhibit cyclin-dependent kinase (CDK) activity, leading to cell cycle arrest (Hoessel et al., 1999; Damiens et al., 2001). Furthermore, long-term animal studies have shown that indirubin does not have the toxicity of conventional anticancer drugs and does not affect bone marrow or hematopoietic stem cell production (Moon et al., 2006). Tang et al. (Chiou et al., 2015) synthesized a series of derivatives of 3-ylideneoxindole acetamides. Compounds 291–295 showed significant inhibitory activity against NCI-H460, MCF-7, Hep3B, KB, SF-268, and MKN-48 cell lines with IC50 ranging from 2.2–8.6 µM. Compound 295 inhibits cells in the G1 phase, increasing the number of cells in the sub-G1 phase, suggesting that compound 295 exerts its anti-tumor activity through the apoptosis-inducing pathway. Furthermore, compound 295 causes up-regulation of the cell cycle regulator cyclin D1, which is maintained at high levels. Compound 295 in CT26-xenografted BALB/c mice treated by the i.p. route showed a reduction in tumor volume comparable to cisplatin.
Bisindole alkaloids, which are metabolites of deep-sea sponges, have been widely concerned for their antiviral, antibacterial, anti-inflammatory, and anti-tumor activities (Shin et al., 1999; Casapullo et al., 2000; Bao et al., 2005; Gul and Hamann, 2005). Diana et al. (Diana et al., 2010) synthesized a series of bisindolyl isoxazole derivatives and evaluated them in a variety of human tumor cell lines. Among them, compound 296 had a strong inhibitory effect on the GXF 251L, A549, LXFA 629L, PANC1, and UXF 1138L cell lines with IC50 value < 10 µM (Fig. 64).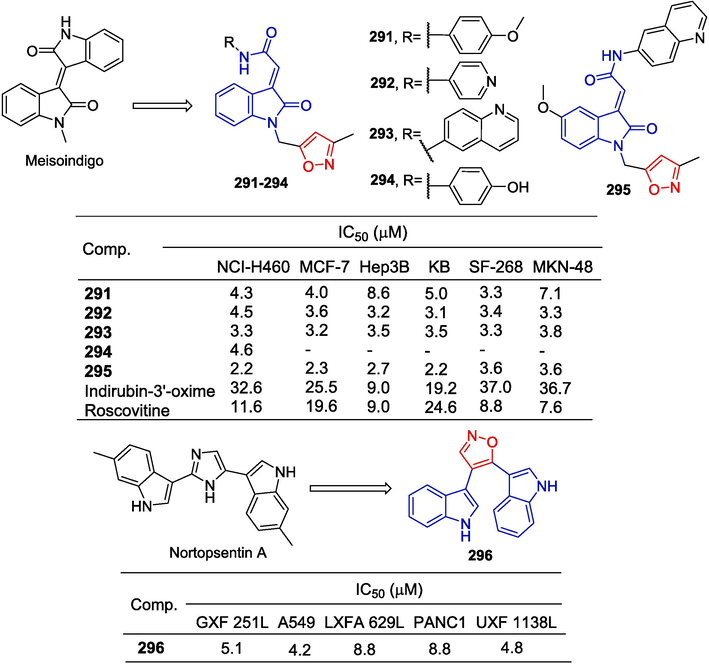
Alkaloid-isoxazole derivatives 291–296.
Staurosporine, an alkaloid, is a potent but non-selective inhibitor of GSK-3β. Ye et al. (Ye et al., 2013) synthesized a series of 3-benzoisoxazol-4-indolyl maleimide and evaluated their GSK-3β inhibitory activity. The reaction of indole with oxalyl chloride in diethyl ether, followed by treatment with sodium methoxide, afforded methyl 2-(1H-indol-3-yl)-2-oxoacetate. Subsequent N-alkylation reactions were performed using various alkyl chlorides. Finally, the target compounds were synthesized through a condensation reaction with 2-(benzo[d]isoxazol-3-yl)acetamide. Compounds 297–299 showed significant inhibitory effects on GSK-3β with IC50 values ranging from 0.73 to 22.1 μM. In cell function experiments, compounds 297–299 significantly reduced Aβ-induced Tau hyperphosphorylation by inhibiting GSK-3β (Fig. 65).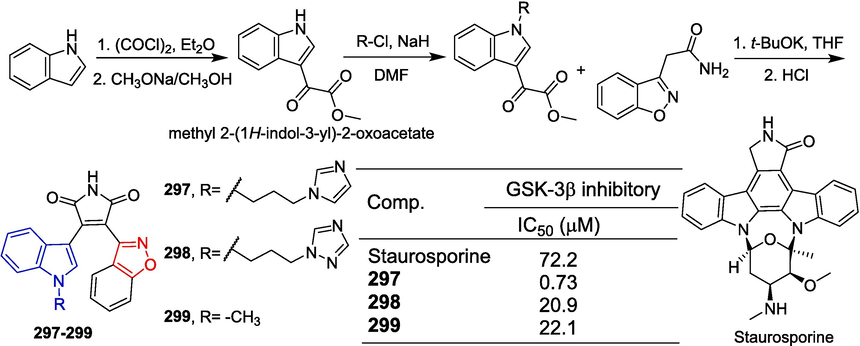
Alkaloid-isoxazole derivatives 297–299.
Li et al. (Li et al., 2017) synthesized a series of 12 N-substituted derivatives using matrine as the lead compound and evaluated their activity against the Coxsackie B3 virus (CVB3). The isoxazole-linked derivative 300 showed a significant inhibitory effect on the CVB3 virus with an IC50 value of 6.87 μM. There was no toxicity to normal Vero cells (IC50 > 556.3 μM).
Norwood et al. (Norwood et al., 2021) synthesized a series of vincamine compounds. Compound 301 showed strong inhibition of chloroquine-resistant P. falciparum Dd2, EC50 = 2.14 μM. For wild-type P. falciparum 3D7 cells, EC50 = 0.97 μM; HepG-2 cell: EC50 > 25 μM.
Lukesh et al. (Lukesh et al., 2017) synthesized vinblastine-amides derivatives and tested their anti-tumor activity. As depicted in Fig. 66, the azide intermediate was available directly in a single step from commercially available vindoline and catharanthine by enlisting first their Fe(III)-promoted coupling. Subsequent in situ Fe(III)-mediated free radical hydrogen atom transfer hydroazidation of anhydrovinblastine. The target compound was then obtained through a reduction reaction followed by a condensation reaction. Isoxazol-linked derivative 302 showed significant inhibitory effects on L1210, HCT116, and HCT116/VM46 cell lines with IC50 values of 4.5, 7.0, and 90 nM, respectively. Activity exceeding or approaching that of vinblastine (L1210: IC50 = 6.0 nM; HCT116: IC50 = 6.8 nM; HCT116/VM46: IC50 = 600 nM).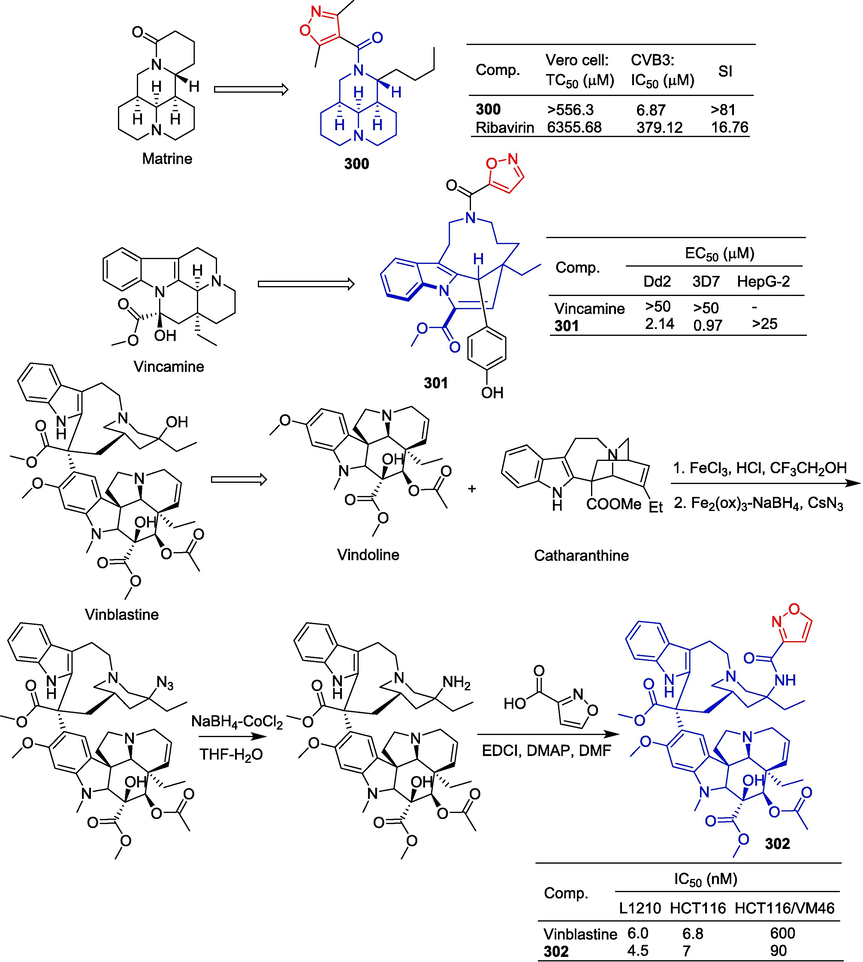
Alkaloid-isoxazole derivatives 300–302.
Summary: Alkaloid-isoxazole hybridization natural products mainly have anti-tumor, anti-viral, and anti-parasitic activities. Among these derivatives, camptothecin hybrid isoxazole derivatives showed the strongest anti-tumor activity in vitro, compound 284 was the most sensitive to the PSN-1 cell line with an IC50 value of 8 nM. Preliminary structure–activity relationships indicate that shortening carbon chains increases anti-tumor activity and reduces toxicity to normal cells (NL7702). Compound 300, a matrine hybrid isoxazole derivative, showed significantly better anti-viral activity than ribavirin. Besides, compound 301 significantly enhanced the anti-parasitic activity compared to its parent compound vincamine.
2.11 Carbohydrate-isoxazole hybridization
Zhang et al. (Zhang et al., 2015) designed and synthesized 23-hydroxybetulinic acid (HBA) derivatives substituted with isoxazole and evaluated their anti-tumor activity. The glycosylated derivative 303 showed strong inhibitory effects on HL-60, BEL-7402, SF-763, HeLa, and B16 cell lines with IC50 ranging from 15.10–26.71 μM (Fig. 67).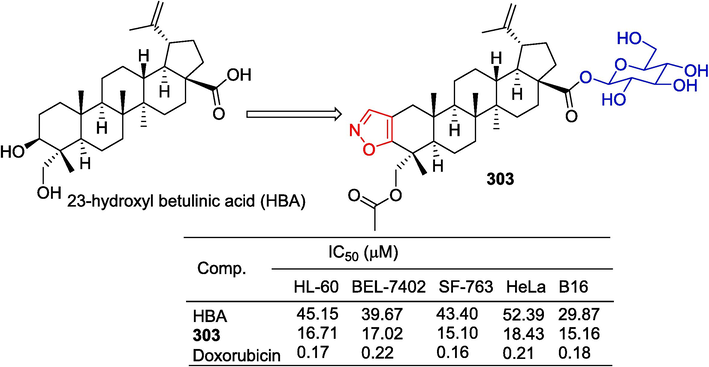
Carbohydrate-isoxazole derivative 303.
Neuroinflammation is an early stage of several neurological and neurodegenerative diseases. Constitutive microglia COX-1 is one of the pro-inflammatory factors in neuroinflammation. Mofezolac (a highly selective COX-1 inhibitor) is linked to galactose via an ester or amide bond to obtain a substance that can cross the blood–brain barrier (BBB) and control the inflammatory response of the central nervous system. Compound 304 is a selective COX-1 inhibitor (COX-1: IC50 = 0.27 μM; COX-2: IC50 = 3.1 μM, SI = 11.5). Compound 305 is a COX-2 inhibitor (COX-1: IC50 = 0.40 μM; COX-2: IC50 = 0.27 μM). Compound 304 is chemically and metabolically stable, able to cross the monolayer of Caco-2 cells (similar to the blood–brain barrier), and its transport is detected mediated by GLUT-1. Additionally, compound 304 inhibited PGE2 release more effectively than mofezolac in LPS-stimulated mouse BV2 microglia cell lines (Perrone et al., 2017) (Fig. 68). The esterification of mofezolac with 1,2:3,4-di-O-isopropylidene-α-D-galactopyranose (DIPG) was initially conducted in the presence of EDC and DMAP to yield the corresponding intermediate. Subsequently, deprotection of the intermediate using trifluoroacetic acid resulted in the synthesis of compound 304. Compound 305 was synthesized utilizing 11-aminoundecanoic acid as a linker following the same procedure as compound 304.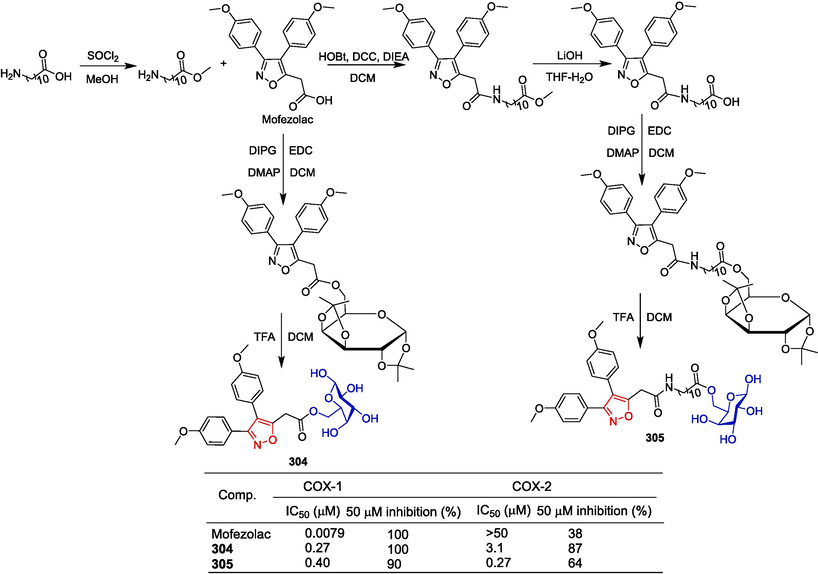
Carbohydrate-isoxazole derivatives 304–305.
Thiamethoxazole (HYM) is a broad-spectrum fungicide. When HYM is absorbed by plants, it is rapidly converted into two glucoside metabolites, which have less anti-fungal activity than HYM. To maintain strong anti-fungal activity in vitro and in vivo, HYM performs glycosylation with amino sugars, simulating plant glycosylation. The anti-fungal activity of glycosides 306 and 307 was the highest. N-acetylglucosamine had an obvious synergistic effect with HYM. Furthermore, glucoside 307 can significantly promote plant growth and induce an increase in plant defense enzyme activity. In addition, electron microscopy and proteomic results showed that glycoside 307 had a unique anti-fungal mechanism compared to HYM (Gao et al., 2022) (Fig. 69).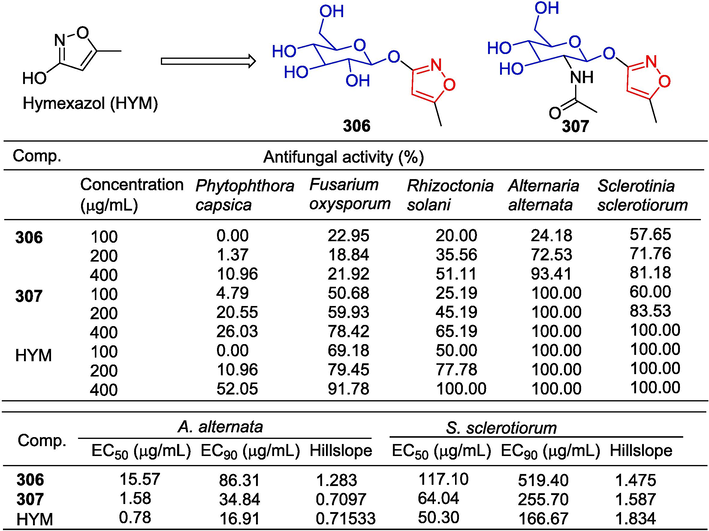
Carbohydrate-isoxazole derivatives 306–307.
Giguere et al. (Giguère et al., 2006) synthesized galactosides containing isoxazole and discovered specific galactin-1 and −3 inhibitors. Compounds 308 and 309 showed inhibitory effects on galectin-1 with inhibitory properties of 2.5 and 1.25 mM.
Hajlaoui et al. (Hajlaoui et al., 2017) designed and synthesized new glycosubstituted isoxazole and screened these compounds for antibacterial activity against the Mtb H37Rv strain. Compounds 310–313 showed potent anti-tuberculosis activity with a MIC of 3.125 μg/mL. Compounds 314–317 showed moderate inhibitory activity with a MIC of 12.5 μg/mL.
Gueron et al. (Gueron et al., 2014) synthesized a series of D-nuclear furanoside derivatives containing isoxazole and tested their anti-tumor activity. Compound 318 can inhibit PC3 cell growth, and the inhibition rate is 34.14 % at 10 μM concentration. After treatment with compound 318 for 24 h, PC3 cells remained in the G0/G1 phase (54.5 %). Under the treatment of compound 318, the increase in the population of G0/G1 cells was mainly at the expense of S phase cells, while the decrease in the population of G2/M cells was minimal.
Marchiori et al. (da Rosa et al., 2017) reported the synthesis of O-3-triazole-linked galactosyl arylsulfonamides as a potential inhibitor of Trypanosoma Cruzi cell invasion. The infection index of compound 319 was reduced (∼20), and in corning Epic label-free assays, compound 319 had a high binding affinity to galactin-3 (EC50 = 17.2 µM).
Chen et al. (Chen et al., 2001) describe the synthesis of compound 320 and its ability to inhibit αGal binding to human anti-Gal antibodies. With two known αGal trisaccharide standards, Galα1-3Galβ1-4GlcNAcβ-Allyl (IC50 = 0.03 mM) and Galα1-3Galβ1-4Glcβ-NHAc (IC50 = 0.07 mM) were compared. Compound 320 showed an anti-Gal binding affinity comparable to the two αGal standards, with an IC50 value of 0.08 mM (Fig. 70).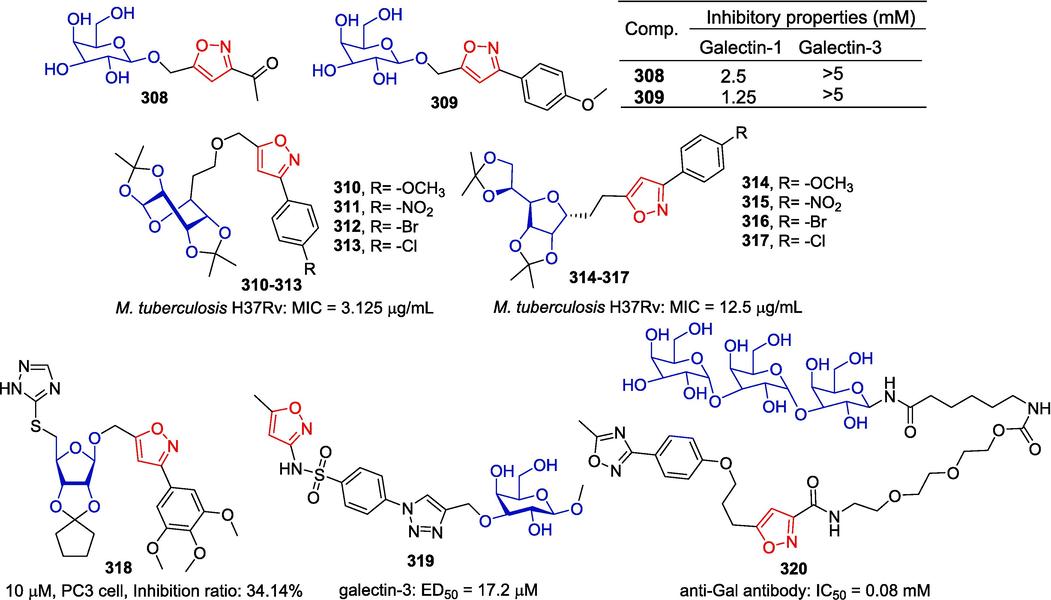
Carbohydrate-isoxazole derivatives 308–320.
Lopopolo et al. (Lopopolo et al., 2012) reported a series of potent serine protease factor Xa (fXa) inhibitors containing O-glucoside. Compounds 321–323 showed good anti-coagulant activity in vitro with Ki values of 37, 0.2, and 0.06 nM, respectively (Fig. 71).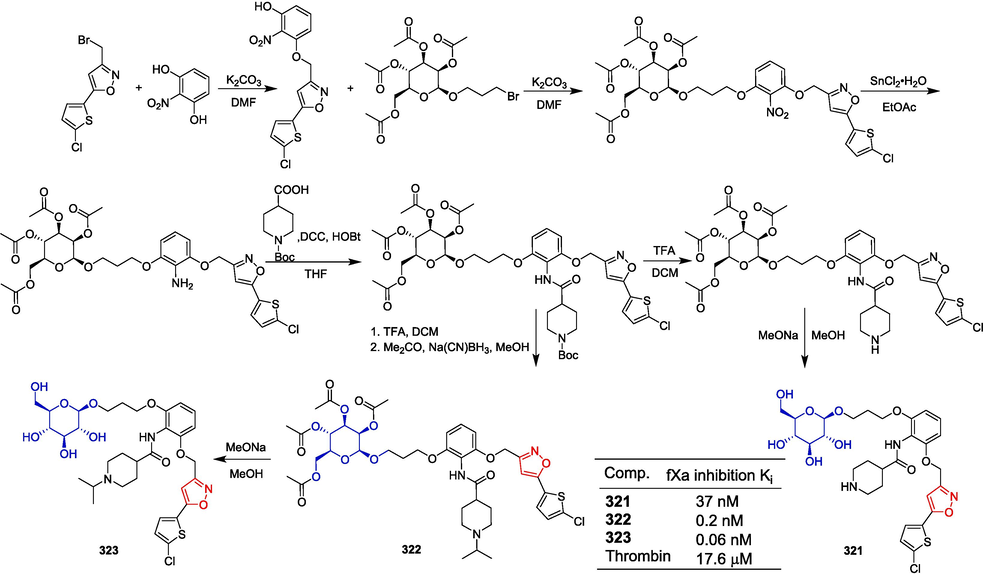
Carbohydrate-isoxazole derivatives 321–323.
Pérez-Balderas et al. (Pérez-Balderas et al., 2005) designed and synthesized a series of isoxazole-glycoconjugates and evaluated the binding affinity of these conjugates with concanavalin A using an enzyme-linked lectin essay (ELLA). Conjugates 324–328 showed inhibitory effects on HRP-labeled concanavalin A binding with IC50 ranging from 0.067 to 0.96 mM (Fig. 72).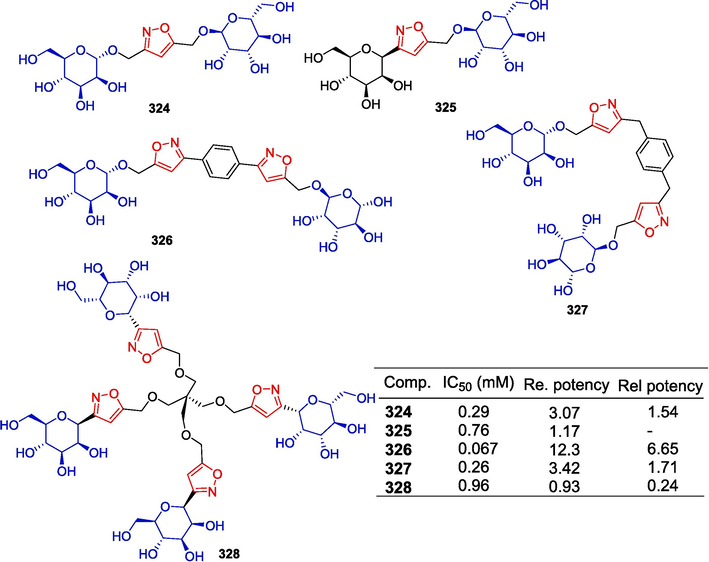
Carbohydrate-isoxazole derivatives 324–328.
Summary: Natural products often have the shortcomings of low bioavailability and poor water solubility. Therefore, the introduction of carbohydrates into their structures can often improve their physical properties. Carbohydrate-isoxazole hybridization natural products mainly have anti-tumor, anti-inflammatory, anti-fungal, anti-coagulant, and anti-parasitic activities. To develop anti-coagulant drugs with better safety, fXa inhibitors have emerged as attractive options for venous thromboembolism treatment. Compound 323 achieved in vitro excellent inhibition potency against fXa, with a Ki value of 60 pM. The other compounds in this section do not exhibit particularly desirable pharmacological activity. For instance, mofezolac is an anti-inflammatory drug bearing isoxazole moiety, unfortunately, the introduction of carbohydrate fragments into its structure did not increase its inhibitory activity against COX-1.
2.12 Other natural products-isoxazole hybridization
Ousidi et al. (Ousidi et al., 2023) synthesized the corresponding thiazolidin-4-one from natural (R)-camphor, followed by N-alkylation with propargyl bromide prior to subjecting the resulting product to 1,3-dipolar cycloaddition reactions with a series of nitrile oxides. Subsequently, in vitro cytotoxicity evaluation was performed on the four synthetic products containing camphor and isoxazole fragments. Unfortunately, their activity against three human cancer cells did not surpass that of the positive control drug Doxorubicin (Fig. 73). The preliminary structure–activity relationship revealed that introducing an electron-withdrawing group in the para-position of the phenyl group conferred beneficial effects on activity. Additionally, compound 311 exhibited significant inhibition potential against BCl-2 protein as confirmed by molecular docking studies. Furthermore, through hydrogen bonding interactions with ARG109 and SER105 residues, the nitrogen atom of isoxazole acted as a hydrogen bond acceptor indicating its crucial role.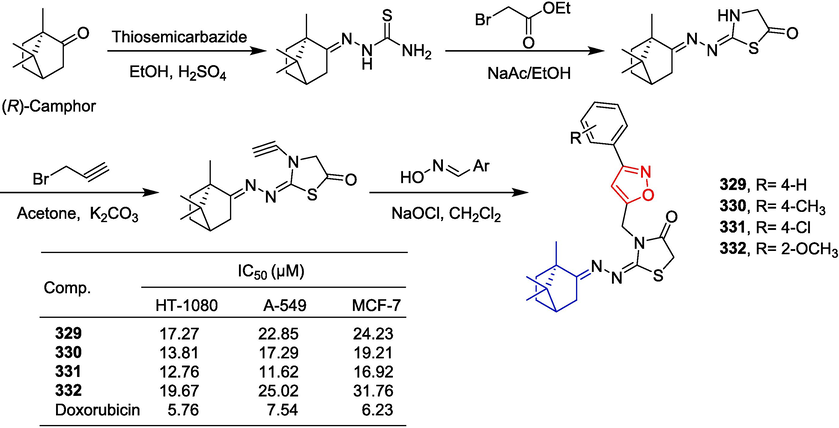
Camphor-isoxazole derivatives 329–332.
β-thalassemia can be treated with fetal hemoglobin (HbF) inducers. Currently, hydroxyurea (HU) is the only approved drug capable of inducing HbF, and studies have found that HU treatment is associated with a down-regulation of the heat shock protein (HSP) HSP90AA1 gene. However, the side effects of HU treatment limit enthusiasm for its use. Radicicol is a natural antibiotic, a resorcylic acid lactone known as a natural inhibitor of HSP90 and therefore a potential inducer of HbF in erythroid cells from β-thalassemia patients (Fig. 74). Additionally, it has been discovered that the resorcinolic portion plays a crucial role as a pharmacophore. However, radicicol presents several challenges for therapeutic application including poor solubility, significant liver toxicity, intrinsic chemical instability or loss of activity in vivo. Therefore, to develop a novel inducer for HbF, the researchers introduced isoxazole fragments for structural modification. Compound 333 exhibited remarkable induction activity towards HbF and thus represents a strong candidate for ameliorating the phenotype associated with β-thalassemia (Ilaria et al., 2021; Zuccato et al., 2024).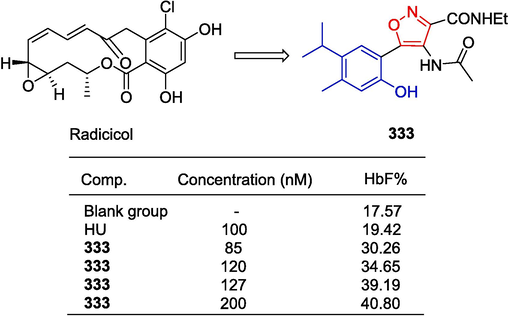
Radicicol-isoxazole derivative 333.
The synthesis route of a series of nucleoside-isoxazole hybrid molecules, as illustrated in Fig. 75, was reported by Kim et al. (Kim et al., 2003). The amino groups at both the N1 and N3 positions were protected by starting with 5-substituted uracil before selective deprotection of the amino acid at the N3 position through stirring in a weak base solution was performed. Subsequently, Mitsunobu and cyclization reactions were employed to synthesize the target compounds. Evaluation against poliovirus (Cox.B3) and vesicular stomatitis virus (VSV) revealed that compounds 334–336 exhibited superior antiviral activity compared to ribavirin. Among them, compound 336 demonstrated the highest activity with significantly higher selectivity index than ribavirin, indicating its ideal antiviral efficacy and low toxicity towards host cells.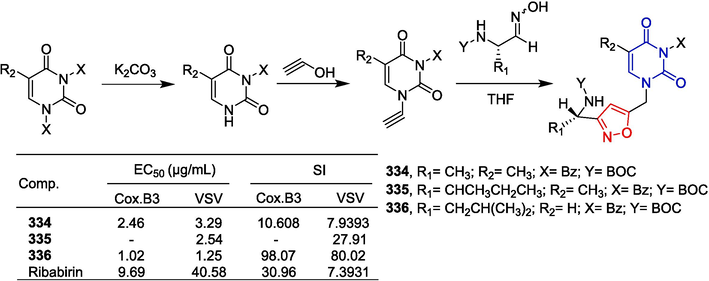
Uracil-isoxazole derivatives 334–336.
3 Conclusions and perspectives
Natural products encompass all metabolites derived from animals, plants, and microorganisms. Over the past century, structurally diverse natural products have made significant contributions to drug research, including antibiotics, immunosuppressants, anti-tumor agents, cholesterol-lowering drugs, and other clinically utilized medications, most of which are closely associated with natural products. In comparison to synthetic compounds, natural products exhibit greater diversity in terms of structural scaffold and stereochemistry while also possessing a wider range of molecular weight and lipid-water partition coefficient. Furthermore, natural products can actively participate in regulating various aspects of biological processes. These distinctive characteristics and advantages position natural products at the forefront of drug research.
Optimizing the structure of lead compounds is the main task for the development of new drugs, and the structure combination principle has become a strategy for the optimization of lead compounds. With natural products as lead compounds, isoxazole is introduced into different positions of their structure through structural modification may obtain active molecules with high efficiency and low toxicity. As previously mentioned, natural products-isoxazole hybrids have made great progress in medicinal chemistry, but there are still some problems and directions for further development: (i) To obtain both more active compounds and novel chemical structures, researchers can hybridize three or more of the above fragments with the same pharmacological activity. For example, combining two or more natural products of chalcone, flavone, coumarin and naphthoquinone with isoxazole may obtain more powerful anti-tumor agents. (ii) The key to the development of new drugs is the discovery of drug targets. Even though most natural product-isoxazole derivatives have good pharmacological activity, the targets of these compounds are still unclear, making it difficult to enter clinical trials. Computer-aided drug design can be used to find the target of natural product-isoxazole derivatives. At the same time, by extracting pharmacophore features and combining them with scaffold hopping, novel inhibitors for various targets can be obtained to solve the structural disadvantages of natural products such as large molecular weight. (iii) Most natural products have the disadvantages of poor water solubility and low bioavailability. Given this, researchers can use rational drug design strategies or novel drug delivery systems to improve the water solubility, absorption, distribution, metabolism, excretion, and toxicity of these natural product-isoxazole derivatives. Moreover, substituted imidazole, triazole, pyrazole, thiazole, amide, etc. can be used as bioisosteres of isoxazole. If isoxazole is changed into other bioisosteres, more effective bioactive molecules may be developed. (iv) Although many natural product-isoxazole derivatives have better pharmacological activity compared with traditional clinical drugs. However, these compounds have not established complete structure–activity relationship studies. Besides, the mechanism of action of many natural product-isoxazole derivatives is not clear, and in vivo activity evaluation has not been carried out, so it is necessary to further study these derivatives.
In conclusion, with the increase of research and development of natural products-isoxazole derivatives, there will be more and more isoxazoles with good efficacy, low toxicity, and superior pharmacokinetic properties applied in clinical practice, and make outstanding contributions to the prevention and protection of human health.
CRediT authorship contribution statement
Jin Wang: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – original draft, Writing – review & editing. Dong-Bo Wang: Conceptualization, Data curation, Writing – original draft. Li-Li Sui: Formal analysis, Investigation, Methodology. Tian Luan: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Acknowledgements
This study received funding from the Scientific Research Project of the Department of Education of Liaoning Province, China (No. LJKMZ20221801; No. LJKQZ20222274), the PhD Start-up Foundation of Liaoning Province, China (No. 2022-BS-341), the Doctoral Research Foundation of Shenyang Medical College (No. 20205041).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis of furo[3,2-g]chromones under microwave irradiation and their antitumor activity evaluation. J. Heterocycl. Chem.. 2019;57:731-743.
- [Google Scholar]
- Structure-activity relationships of natural quinone vegfrecine analogs with potent activity against VEGFR-1 and -2 tyrosine kinases. J. Antibiot. (tokyo). 2021;74:734-742.
- [Google Scholar]
- Screening of curcumin-derived isoxazole, pyrazoles, and pyrimidines for their anti-inflammatory, antinociceptive, and cyclooxygenase-2 inhibition. Chem. Biol. Drug Des.. 2018;91:338-343.
- [Google Scholar]
- Sulfonamides containing curcumin scaffold: Synthesis, characterization, carbonic anhydrase inhibition and molecular docking studies. Bioorg. Chem.. 2018;76:218-227.
- [Google Scholar]
- Synthesis, structural analysis and antitumor activity of novel 17α-picolyl and 17(E)-picolinylidene A-modified androstane derivatives. Bioorg. Med. Chem.. 2015;23:1557-1568.
- [Google Scholar]
- Imaging the spread of reversible brain inactivations using fluorescent muscimol. J. Neurosci. Methods. 2008;171:30-38.
- [Google Scholar]
- A new pregnenolone analogues as privileged scaffolds in inhibition of CYP17 hydroxylase enzyme. Synthesis and in silico molecular docking study. Steroids. 2015;100:52-59.
- [Google Scholar]
- Anticancer activity profiling of parthenolide analogs generated via P450-mediated chemoenzymatic synthesis. Bioorg. Med. Chem.. 2018;26:1365-1373.
- [Google Scholar]
- Synthesis and evaluation of electron-rich curcumin analogues. Bioorg. Med. Chem.. 2009;17:360-367.
- [Google Scholar]
- Pyrrolo-isoxazole: a key molecule with diverse biological actions. Mini-Rev. Med. Chem.. 2014;14:623-627.
- [Google Scholar]
- Effects of Danazol on gonadotropins and steroid blood levels in normal and anovulatory women. Am. J. Obstet. Gynecol.. 1975;121:817.
- [Google Scholar]
- Exploring mitochondria-mediated intrinsic apoptosis by new phytochemical entities: an explicit observation of cytochrome c dynamics on lung and melanoma cancer cells. J. Med. Chem.. 2019;62:8311-8329.
- [Google Scholar]
- Facile synthesis and docking studies of 7-hydroxyflavanone isoxazoles and acrylates as potential anti-microbial agents. Med. Chem. Res.. 2020;29:217-228.
- [Google Scholar]
- Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivatives. Eur. J. Med. Chem.. 2015;96:425-435.
- [Google Scholar]
- Synthesis and antimicrobial screening of some novel chromones and pyrazoles with incorporated isoxazole moieties. J. Heterocycl. Chem.. 2013;50:999-1004.
- [Google Scholar]
- Synthesis, screening and docking analysis of hispolon pyrazoles and isoxazoles as potential antitubercular agents. Curr. Top. Med. Chem.. 2019;19:662-682.
- [Google Scholar]
- Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J. Nat. Prod.. 2005;68:711-715.
- [Google Scholar]
- 14β-(Isoxazol-3-yl)methylestrane steroids: chemoselective synthesis and transformations with heterocyclic ring opening. Russ. J. Org. Chem.. 2019;55:202-214.
- [Google Scholar]
- Transformations, NMR studies and biological testing of some 17β-isoxazolyl steroids and their heterocyclic ring cleavage derivatives. Steroids. 2021;166:108768
- [Google Scholar]
- Isoxazolo(aza)naphthoquinones: A new class of cytotoxic Hsp90 inhibitors. Eur. J. Med. Chem.. 2012;53:64-75.
- [Google Scholar]
- Medicinal chemistry perspective of fused isoxazole derivatives. Curr. Top. Med. Chem. (sharjah, United Arab Emirates). 2016;16:2863-2883.
- [Google Scholar]
- Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCgamma1- and Ca2+-dependent apoptosis. Blood. 2008;111:2354-2363.
- [Google Scholar]
- Synthesis, evaluation and quantitative structure-activity relationship (QSAR) analysis of Wogonin derivatives as cytotoxic agents. Bioorg. Med. Chem. Lett.. 2017;27:1012-1016.
- [Google Scholar]
- Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother.. 2005;49:619-626.
- [Google Scholar]
- Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov.. 2006;5:769-784.
- [Google Scholar]
- Herbicidal effects of sulfamethoxazole in Lemna gibba: using p-aminobenzoic acid as a biomarker of effect. Environ. Sci. Technol.. 2008;42:8965-8970.
- [Google Scholar]
- Synthesis of novel forskolin isoxazole derivatives with potent anti-cancer activity against breast cancer cell lines. Bioorg. Med. Chem. Lett.. 2017;27:4314-4318.
- [Google Scholar]
- Development of HSP90 inhibitors-SN38 conjugates for cancer treatment. Bioorg. Chem.. 2023;137:106582
- [Google Scholar]
- New bisindole alkaloids of the topsentin and hamacanthin classes from the Mediterranean marine sponge Rhaphisia lacazei. J. Nat. Prod.. 2000;63:447-451.
- [Google Scholar]
- Stable and potent analogues derived from the modification of the dicarbonyl moiety of curcumin. Biochemistry. 2013;52:7449-7460.
- [Google Scholar]
- Isoxazole analogs of curcuminoids with highly potent multidrug-resistant antimycobacterial activity. Eur. J. Med. Chem.. 2010;45:4446-4457.
- [Google Scholar]
- αGal-conjugated anti-rhinovirus agents: chemo-enzymatic syntheses and testing of anti-Gal binding. J. Chem. Soc., Perkin Trans.. 2001;1:1716-1722.
- [Google Scholar]
- Synthesis and biological investigation of coumarin piperazine (piperidine) derivatives as potential multireceptor atypical antipsychotics. J. Med. Chem.. 2013;56:4671-4690.
- [Google Scholar]
- Synthesis and evaluation of new coumarin derivatives as potential atypical antipsychotics. Eur. J. Med. Chem.. 2014;74:427-439.
- [Google Scholar]
- Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur. J. Med. Chem.. 2019;183:111722
- [Google Scholar]
- Design and synthesis of heterocyclic malonyl-CoA decarboxylase inhibitors. Bioorg. Med. Chem. Lett.. 2006;16:695-700.
- [Google Scholar]
- Synthesis and evaluation of 3-ylideneoxindole acetamides as potent anticancer agents. Eur. J. Med. Chem.. 2015;98:1-12.
- [Google Scholar]
- Natural products: a lead for drug discovery and development. Phytother. Res.. 2021;35:4660-4702.
- [Google Scholar]
- Regiospecific synthesis, anti-inflammatory and anticancer evaluation of novel 3,5-disubstituted isoxazoles from the natural maslinic and oleanolic acids. Ind. Crops Prod.. 2016;85:287-299.
- [Google Scholar]
- Structural basis for selective inhibition of Cyclooxygenase-1 (COX-1) by diarylisoxazoles mofezolac and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6) Eur. J. Med. Chem.. 2017;138:661-668.
- [Google Scholar]
- New C25 carbamate rifamycin derivatives are resistant to inactivation by ADP-ribosyl transferases. Bioorg. Med. Chem. Lett.. 2007;17:522-526.
- [Google Scholar]
- Design and synthesis of a new series of 3,5-disubstituted isoxazoles active against Trypanosoma cruzi and Leishmania amazonensis. Eur. J. Med. Chem.. 2017;128:25-35.
- [Google Scholar]
- New azole derivatives of [17(20)E]-21-norpregnene: Synthesis and inhibition of prostate carcinoma cell growth. Steroids. 2019;147:10-18.
- [Google Scholar]
- Anti-mitotic properties of indirubin-3'-monoxime, a CDK/GSK-3 inhibitor: induction of endoreplication following prophase arrest. Oncogene. 2001;20:3786-3797.
- [Google Scholar]
- Synthesis and antimicrobial evaluation of some isoxazole based heterocycles. Heterocycles. 2014;89:1393-1411.
- [Google Scholar]
- Binding of isoxazole and pyrazole derivatives of curcumin with the activator binding domain of novel protein kinase C. Bioorg. Med. Chem.. 2011;19:6196-6202.
- [Google Scholar]
- Recent developments in the discovery of FFA1 receptor agonists as novel oral treatment for type 2 diabetes mellitus. Bioorg. Med. Chem. Lett.. 2014;24:2991-3000.
- [Google Scholar]
- Synthesis and biological evaluation of 2-(4,5,6,7-tetrahydrobenzo[c]Isoxazol-3-yl)-4H-chromen-4-ones. Polycyclic Aromat. Compd.. 2022;42:6337-6351.
- [Google Scholar]
- Actin-curcumin interaction: insights into the mechanism of actin polymerization inhibition. Biochemistry. 2015;54:1132-1143.
- [Google Scholar]
- Synthesis and antitumor activity of 2,5-bis(3'-indolyl)-furans and 3,5-bis(3'-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem.. 2010;18:4524-4529.
- [Google Scholar]
- Synthesis and characterization of some heteroaromatic derivatives of 3-but-2-enoyl-chromen-2-one and their potential as anti-inflammatory agents. J. Heterocycl. Chem.. 2013;50:1431-1436.
- [Google Scholar]
- Synergistic antifungal effects of curcumin derivatives as fungal biofilm inhibitors with fluconazole. Chem. Biol. Drug Des.. 2021;97:1079-1088.
- [Google Scholar]
- Synthesis, structural analysis and antiproliferative activity of nitrogen-containing hetero spirostan derivatives: oximes, heterocyclic ring-fused and furostanes. ChemistrySelect. 2022;7
- [Google Scholar]
- Synthesis of Benzalacetophenone-based Isoxazoline and Isoxazole Derivatives. Curr. Org. Chem.. 2022;26:679-692.
- [Google Scholar]
- Synthesis of pregnenolone-danazol-ethylenediamine conjugate. Relationship between descriptors log P, π, Rm, and Vm and its antibacterial activity in S. aureus and V. cholerae. Med. Chem. Res.. 2011;20:847-853.
- [Google Scholar]
- Synthesis of new harmine isoxazoles and evaluation of their potential anti-alzheimer, anti-inflammatory, and anticancer activities. Med. Chem.. 2016;12:184-190.
- [Google Scholar]
- Synthesis of triterpene A-condensed azoles. Chem. Heterocycl. Compd.. 2014;50:65-75.
- [Google Scholar]
- The Synthesis of glycyrrhetinic acid derivatives containing a nitrogen heterocycle and their antiproliferative effects in human leukemia cells. Molecules. 2010;15:4439-4449.
- [Google Scholar]
- Development of coumarine derivatives as potent anti-filovirus entry inhibitors targeting viral glycoprotein. Eur. J. Med. Chem.. 2020;204:112595
- [Google Scholar]
- Design, synthesis, and antifungal activities of hymexazol glycosides based on a biomimetic strategy. J. Agric. Food Chem.. 2022;70:9520-9535.
- [Google Scholar]
- Analogue based drug design, synthesis, molecular docking and anticancer evaluation of novel chromene sulfonamide hybrids as aromatase inhibitors and apoptosis enhancers. Eur. J. Med. Chem.. 2016;124:946-958.
- [Google Scholar]
- N-Hydroxy-(4-oxime)-cinnamide: a versatile scaffold for the synthesis of novel histone deacetylase [correction of deacetilase] (HDAC) inhibitors. Bioorg. Med. Chem. Lett.. 2009;19:2346-2349.
- [Google Scholar]
- Carbohydrate triazoles and isoxazoles as inhibitors of galectins-1 and -3. Chem. Commun. (camb.) 2006:2379-2381.
- [Google Scholar]
- Utility of 3-acetyl-6-bromo-2H-chromen-2-one for the synthesis of new heterocycles as potential antiproliferative agents. Molecules. 2015;20:21826-21839.
- [Google Scholar]
- Inhibition of dihydroorotate dehydrogenase by the immunosuppressive agent leflunomide. Biochem. Pharmacol.. 1995;50:861.
- [Google Scholar]
- Preparation of novel ring-A fused azole derivatives of betulin and evaluation of their cytotoxicity. Eur. J. Med. Chem.. 2017;125:629-639.
- [Google Scholar]
- Synthesis and biological evaluation of novel flavanone derivatives as potential antipsychotic agents. Chem. Biol. Drug Des.. 2017;89:353-364.
- [Google Scholar]
- Differential effect of heterocyclic D-ribofuranoside derivatives on human prostate cancer cell viability and cell cycle progression. Biomed. Pharmacother.. 2014;68:847-854.
- [Google Scholar]
- Indole alkaloid marine natural products: an established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci.. 2005;78:442-453.
- [Google Scholar]
- Betulinic acid derivatives: a new class of α-glucosidase inhibitors and LPS-stimulated nitric oxide production inhibition on mouse macrophage RAW 264.7 cells. Nat. Prod. Res.. 2019;33:2618-2622.
- [Google Scholar]
- Synthesis of carbohydrate-substituted isoxazoles and evaluation of their antitubercular activity. Heterocycl. Commun.. 2017;23:225-229.
- [Google Scholar]
- Synthesis, DFT study, and antitumor activity of some new heterocyclic compounds incorporating isoxazole moiety. J. Chin. Chem. Soc. (weinheim, Ger.). 2017;64:1203-1212.
- [Google Scholar]
- Design, synthesis and biological evaluation of stilbene derivatives as novel inhibitors of protein tyrosine phosphatase 1B. Molecules. 2016;21
- [Google Scholar]
- Design, synthesis of betulin derivatives containing 5-phenyl-3-isoxazole and their inhibitory activities against protein tyrosine phosphatase 1B. Chinese J. Org. Chem.. 2016;36:2888-2894.
- [Google Scholar]
- Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol.. 1999;1:60-67.
- [Google Scholar]
- Tanshinone IIA: pharmacology, total synthesis, and progress in structure-modifications. Curr. Med. Chem.. 2022;29:1959-1989.
- [Google Scholar]
- Chemoselective synthesis of enamino-coumarin derivatives identified as potent antitumor agents. J. Heterocycl. Chem.. 2016;53:1318-1323.
- [Google Scholar]
- Ilaria, L., Roberto, G., Daniele, 2021. Isoxazole derivatives as inducers of fetal hemoglobin in erythroid precursor cells from beta-thalassemic patients, US11077116, 2021-08-03.
- Synthesis of novel estrogen receptor antagonists using metal-catalyzed coupling reactions and characterization of their biological activity. J. Med. Chem.. 2013;56:2779-2790.
- [Google Scholar]
- Synthesis and anticancer activity of heteroaromatic linked 4β-amido podophyllotoxins as apoptotic inducing agents. Bioorg. Med. Chem. Lett.. 2013;23:273-280.
- [Google Scholar]
- Design and synthesis of pyrazole/isoxazole linked arylcinnamides as tubulin polymerization inhibitors and potential antiproliferative agents. Org. Biomol. Chem.. 2015;13:10162-10178.
- [Google Scholar]
- ER alpha selective chromone, isoxazolylchromones, induces ROS-mediated cell death without autophagy. Chem. Biol. Drug Des.. 2019;94:1352-1367.
- [Google Scholar]
- Kim, B., Lee, Y., Kim, B.H., Lee, Y.S., 2003. New pyrimidine nucleoside analogs containing an isoxazoline or isoxazole ring, useful as antiviral agents in the treatment of e.g. vesicular stomatitis virus (VSV) and Cox B3 virus, WO2003018577, 2003-03-06.
- Synthesis and cytotoxicity of 2-cyano-28-hydroxy-lup-1-en-3-ones. Bioorg. Med. Chem. Lett.. 2009;19:2168-2171.
- [Google Scholar]
- Synthesis, characterization and biological evaluation of some novel 17-isoxazoles in the estrone series. Steroids. 2012;77:1075-1085.
- [Google Scholar]
- Synthesis of pyrano isoxazoline/isoxazole annulated coumarins via intramolecular nitrile oxide cycloaddition and their cytotoxicity. Russ. J. Gen. Chem.. 2017;87:1857-1863.
- [Google Scholar]
- New class of glutamate agonist structurally related to ibotenic acid. Nature (lond.). 1980;284:64.
- [Google Scholar]
- Synthesis of new arylisoxazole-oxindole conjugates as potent antiproliferative agents. Chem. Biol. Drug Des.. 2017;89:634-638.
- [Google Scholar]
- New histone deacetylase inhibitors and anticancer agents from Curcuma longa. Med. Chem. Res.. 2019;28:1773-1782.
- [Google Scholar]
- Design, synthesis, and evaluation of 3C protease inhibitors as anti-enterovirus 71 agents. Bioorg. Med. Chem.. 2008;16:7388-7398.
- [Google Scholar]
- Biological activity, design, synthesis and structure activity relationship of some novel derivatives of curcumin containing sulfonamides. Eur. J. Med. Chem.. 2013;64:579-588.
- [Google Scholar]
- The use of the monoamine oxidase inhibitor isocarboxazide in treatment-resistant depression. Ugeskr. Laeger. 2015;177:V06150499.
- [Google Scholar]
- Synthesis and biological evaluation of naphthoquinone analogs as a novel class of proteasome inhibitors. Bioorg. Med. Chem.. 2010;18:5576-5592.
- [Google Scholar]
- Synthesis, antiproliferative activities and telomerase inhibition evaluation of novel asymmetrical 1,2-disubstituted amidoanthraquinone derivatives. Eur. J. Med. Chem.. 2012;47:323-336.
- [Google Scholar]
- Discovery of novel simplified isoxazole derivatives of sampangine as potent anti-cryptococcal agents. Bioorg. Med. Chem.. 2019;27:832-840.
- [Google Scholar]
- Novel 12N-substituted matrinanes as potential anti-coxsackievirus agents. Bioorg. Med. Chem. Lett.. 2017;27:829-833.
- [Google Scholar]
- β-lactam antibiotics: An overview from a medicinal chemistry perspective. Eur. J. Med. Chem.. 2020;208:112829
- [Google Scholar]
- Linker Optimization and therapeutic evaluation of phosphatidylserine-targeting zinc dipicolylamine-based drug conjugates. J. Med. Chem.. 2019;62:6047-6062.
- [Google Scholar]
- C-3 benzoic acid derivatives of C-3 deoxybetulinic acid and deoxybetulin as HIV-1 maturation inhibitors. Bioorg. Med. Chem.. 2016;24:1757-1770.
- [Google Scholar]
- New therapeutic aspects of flavones: the anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treat. Rev.. 2009;35:57-68.
- [Google Scholar]
- β-D-Glucosyl conjugates of highly potent inhibitors of blood coagulation factor Xa bearing 2-chorothiophene as a P1 Motif. ChemMedChem. 2012;7:1669-1677.
- [Google Scholar]
- A novel synthesized sulfonamido-based gallate-JEZTC blocks cartilage degradation on rabbit model of osteoarthritis: an in vitro and in vivo study. Cell. Physiol. Biochem.. 2018;49:2304-2319.
- [Google Scholar]
- Electrosynthesis of stable betulin-derived nitrile oxides and their application in synthesis of cytostatic lupane-type triterpenoid-isoxazole conjugates. Eur. J. Org. Chem.. 2021;2021:2557-2577.
- [Google Scholar]
- Vinblastine 20' amides: synthetic analogues that maintain or improve potency and simultaneously overcome pgp-derived efflux and resistance. J. Med. Chem.. 2017;60:7591-7604.
- [Google Scholar]
- Synthesis and biological evaluation of heterocyclic ring-fused dammarane-type ginsenoside derivatives as potential anti-tumor agents. Bioorg. Chem.. 2021;116:105365
- [Google Scholar]
- Design, molecular docking study of synthesised N-heteroaryl substituted gallamide derivatives and their antibacterial assessment. Nat. Prod. Res.. 2022;36:5575-5583.
- [Google Scholar]
- Synthesis of novel ring-A fused hybrids of oleanolic acid with capabilities to arrest cell cycle and induce apoptosis in breast cancer cells. Eur. J. Med. Chem.. 2014;74:398-404.
- [Google Scholar]
- N-acylated derivatives of sulfamethoxazole and sulfafurazole inhibit intracellular growth of Chlamydia trachomatis. Antimicrob. Agents Chemother.. 2014;58:2968.
- [Google Scholar]
- Curcumin is an inhibitor of calcium/calmodulin dependent protein kinase II. Bioorg. Med. Chem.. 2012;20:6040-6047.
- [Google Scholar]
- Recent advances in the treatment of rhinovirus infections. Curr. Opin. Pharmacol.. 2001;1:477-481.
- [Google Scholar]
- Survey on the pharmacodynamics of the new antipsychotic risperidone. Psychopharmacology (Berlin). 1994;114:9-23.
- [Google Scholar]
- Discovery and optimization of 4,5-diarylisoxazoles as potent dual inhibitors of pyruvate dehydrogenase kinase and heat shock protein 90. J. Med. Chem.. 2014;57:9832-9843.
- [Google Scholar]
- Clinical pharmacology and therapeutic drug monitoring of zonisamide. Ther. Drug Monit.. 1998;20:593-597.
- [Google Scholar]
- Triterpenoid Hydroxamates as HIF Prolyl Hydrolase Inhibitors. J. Nat. Prod.. 2018;81:2235-2243.
- [Google Scholar]
- Synthesis and exploration of novel curcumin analogues as anti-malarial agents. Bioorg. Med. Chem.. 2008;16:2894-2902.
- [Google Scholar]
- Computational studies and sever apoptotic bioactivity of new heterocyclic cyanoacrylamide based p-fluorophenyl and p-phenolic compounds against liver carcinoma (Hepg2) Bioorg. Chem.. 2021;114:105147
- [Google Scholar]
- The Knoevenagel reactions of pregnenolone with cyanomethylene reagents: Synthesis of thiophene, thieno[2,3-b]pyridine, thieno[3,2-d]isoxazole derivatives of pregnenolone and their in vitro cytotoxicity towards tumor and normal cell lines. Steroids. 2013;78:1209-1219.
- [Google Scholar]
- Synthesis and cytotoxicity of novel 14α-O-(andrographolide-3-subsitutedisoxazole-5-carboxylate) derivatives. Nat. Prod. Res.. 2021;35:3738-3744.
- [Google Scholar]
- Synthesis and structure-activity relationships of novel indirubin derivatives as potent anti-proliferative agents with CDK2 inhibitory activities. Bioorg. Med. Chem.. 2006;14:237-246.
- [Google Scholar]
- Curcumin-derived pyrazoles and isoxazoles: swiss army knives or blunt tools for Alzheimer's disease? ChemMedChem. 2008;3:165-172.
- [Google Scholar]
- Novel hydrazide-hydrazone and amide substituted coumarin derivatives: synthesis, cytotoxicity screening, microarray, radiolabeling and in vivo pharmacokinetic studies. Eur. J. Med. Chem.. 2018;151:723-739.
- [Google Scholar]
- Discovery and anti-diabetic effects of novel isoxazole based flavonoid derivatives. Fitoterapia. 2020;142:104499
- [Google Scholar]
- Synthesis and cytotoxic activity of dihydroquercetin aryl derivatives. Pharm. Chem. J.. 2015;49:78-81.
- [Google Scholar]
- Synthesis and bioactivity of novel isoxazole chalcone derivatives on tyrosinase and melanin synthesis in murine B16 cells for the treatment of vitiligo. Bioorg. Med. Chem.. 2016;24:5440-5448.
- [Google Scholar]
- Ring distortion of vincamine leads to the identification of re-engineered antiplasmodial agents. ACS Omega. 2021;6:20455-20470.
- [Google Scholar]
- Complications of the COX-2 inhibitors parecoxib and valdecoxib after cardiac surgery. N. Engl. J. Med.. 2005;352:1081-1091.
- [Google Scholar]
- Small-molecule quadruplex-targeted drug discovery. Bioorg. Med. Chem. Lett.. 2014;24:2602-2612.
- [Google Scholar]
- Design, synthesis of new phenyl acetylene and isoxazole analogues of arjunolic acid as potent tyrosinase and alpha glucosidase inhibitors. Nat. Prod. Res.. 2023;37:1092-1097.
- [Google Scholar]
- Design, synthesis, cytotoxic effect evaluation and molecular docking of (R)-camphor-based thiazolidinone isoxazole and thiazolidinone-1,2,3-triazole hybrids“. ChemistrySelect. 2023;8:e202203349.
- [Google Scholar]
- Inhibition of 4-hydroxyphenylpyruvate dioxygenase: the mode of action of the herbicide RPA 201772 (isoxaflutole) Pestic. Sci.. 1997;50:83-84.
- [Google Scholar]
- Structure-based drug design and identification of H(2)O-soluble and low toxic hexacyclic camptothecin derivatives with improved efficacy in cancer and lethal inflammation models in vivo. J. Med. Chem.. 2018;61:8613-8624.
- [Google Scholar]
- Oleanolic acid derivatives induce apoptosis in human leukemia K562 cell involved in inhibition of both Akt1 translocation and pAkt1 expression. Cytotechnology. 2015;67:821-829.
- [Google Scholar]
- Synthesis and in vitro assay of new triazole linked decursinol derivatives showing inhibitory activity against cholinesterase for Alzheimer's disease therapeutics. J. Korean Chem. Soc.. 2016;60:125-130.
- [Google Scholar]
- Translational impact of novel widely pharmacological characterized mofezolac-derived COX-1 inhibitors combined with bortezomib on human multiple myeloma cell lines viability. Eur. J. Med. Chem.. 2019;164:59-76.
- [Google Scholar]
- Synthesis of novel caffeic acid derivatives and their protective effect against hydrogen peroxide induced oxidative stress via Nrf2 pathway. Life Sci.. 2020;247:117439
- [Google Scholar]
- Synthesis of multivalent neoglycoconjugates by 1,3 dipolar cycloaddition of nitrile oxides and alkynes and evaluation of their lectin-binding affinities. Tetrahedron. 2005;61:9338-9348.
- [Google Scholar]
- Effect of mofezolac-galactose distance in conjugates targeting cyclooxygenase (COX)-1 and CNS GLUT-1 carrier. Eur. J. Med. Chem.. 2017;141:404-416.
- [Google Scholar]
- Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis.. 2011;2:e182.
- [Google Scholar]
- The antitumor activities of curcumin and of its isoxazole analogue are not affected by multiple gene expression changes in an MDR model of the MCF-7 breast cancer cell line: analysis of the possible molecular basis. Int. J. Mol. Med.. 2007;20:329-335.
- [Google Scholar]
- Synthesis of usnic acid derivatives and evaluation of their antiproliferative activity against cancer cells. J. Nat. Prod.. 2019;82:1768-1778.
- [Google Scholar]
- Lipid accumulation inhibitory activities of novel isoxazole-based chenodeoxycholic acids: Design, synthesis and preliminary mechanism study. Bioorg. Med. Chem. Lett.. 2018;28:2879-2884.
- [Google Scholar]
- Structure-guided design and synthesis of isoflavone analogs of GW4064 with potent lipid accumulation inhibitory activities. Bioorg. Med. Chem. Lett.. 2018;28:3726-3730.
- [Google Scholar]
- A facile synthesis, anti-inflammatory and analgesic activity of isoxazolyl-2,3-dihydrospiro[benzo[f]isoindole-1,3'-indoline]-2',4,9-triones. Bioorg. Med. Chem. Lett.. 2013;23:3954-3958.
- [Google Scholar]
- Synthesis and biological evaluation of novel flavone/triazole/benzimidazole hybrids and flavone/isoxazole-annulated heterocycles as antiproliferative and antimycobacterial agents. Mol. Diversity. 2018;22:803-814.
- [Google Scholar]
- Design and synthesis of novel β-carboline linked amide derivatives as anticancer agents. Russ. J. Gen. Chem.. 2019;89:511-516.
- [Google Scholar]
- Synthesis of some novel methyl β-orsellinate based 3, 5-disubstituted isoxazoles and their anti-proliferative activity: Identification of potent leads active against MCF-7 breast cancer cell. Bioorg. Chem.. 2020;105:104374
- [Google Scholar]
- The inhibitory potency of isoxazole-curcumin analogue for the management of breast cancer: A comparative in vitro and molecular modeling investigation. Chem. Pap.. 2021;75:5995-6008.
- [Google Scholar]
- Treatment of potentially life-threatening enterovirus infections with pleconaril. Clin. Infect. Dis.. 2001;32:228-235.
- [Google Scholar]
- Synthesis and structure-activity relationship of new chalcone linked 5-phenyl-3-isoxazolecarboxylic acid methyl esters potentially active against drug resistant Mycobacterium tuberculosis. Eur. J. Med. Chem.. 2021;222:113580
- [Google Scholar]
- Synthesis of new halogenated flavonoid-based isoxazoles: in vitro and in silico evaluation of α-amylase inhibitory potential, a SAR analysis and DFT studies. J. Mol. Struct.. 2022;1247:131379
- [Google Scholar]
- Tivozanib in renal cell carcinoma: a new approach to previously treated disease. Ther. Adv. Med. Oncol.. 2020;12 1758835920923818
- [Google Scholar]
- Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho[2,3-d]isoxazole-4,9-diones. Bioorg. Med. Chem. Lett.. 2010;20:193-195.
- [Google Scholar]
- Design, synthesis, biological evaluation and molecular docking of curcumin analogues as antioxidant, cyclooxygenase inhibitory and anti-inflammatory agents. Bioorg. Med. Chem. Lett.. 2005;15:1793-1797.
- [Google Scholar]
- Antimicrobial, antioxidant, and anticancer activities of some novel isoxazole ring containing chalcone and dihydropyrazole derivatives. Molecules. 2020;25
- [Google Scholar]
- Synthesis and biological evaluation of novel isoxazoles and triazoles linked 6-hydroxycoumarin as potent cytotoxic agents. Bioorg. Med. Chem. Lett.. 2014;24:4243-4246.
- [Google Scholar]
- Mechanochemical synthesis and antioxidant activity of curcumin-templated azoles. Arch. Pharm. (weinheim). 2015;348:908-914.
- [Google Scholar]
- Design, synthesis and cytotoxic activities of scopoletin-isoxazole and scopoletin-pyrazole hybrids. Bioorg. Med. Chem. Lett.. 2017;27:147-151.
- [Google Scholar]
- New Bis(Indole) alkaloids of the topsentin class from the sponge spongosorites genitrix. J. Nat. Prod.. 1999;62:647-649.
- [Google Scholar]
- Optimization of the in vitro cardiac safety of hydroxamate-based Histone deacetylase inhibitors. J. Med. Chem.. 2011;54:4752-4772.
- [Google Scholar]
- The design, synthesis and structure-activity relationships of novel isoindoline-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett.. 2011;21:4909-4912.
- [Google Scholar]
- Antitumor effects of curcumin and structurally beta-diketone modified analogs on multidrug resistant cancer cells. Bioorg. Med. Chem. Lett.. 2008;18:845-849.
- [Google Scholar]
- Structure-activity relationship studies leading to the identification of (2E)-3-[l-[(2,4-dichlorophenyl)methyl]-5-fluoro-3-methyl-lH-indol-7-yl]-N-[(4,5-dichloro-2-thienyl)sulfonyl]-2-propenamide (DG-041), a potent and selective prostanoid EP3 receptor antagonist, as a novel antiplatelet agent that does not prolong bleeding. J. Med. Chem.. 2010;53:18-36.
- [Google Scholar]
- Evaluation of cholinesterase inhibitory activity and cytotoxicity of synthetic derivatives of di- and triterpene metabolites from Pinus silvestris and Dipterocarpus alatus resins. Med. Chem. Res.. 2020;29:1478-1485.
- [Google Scholar]
- Reexamining hydroxamate inhibitors of botulinum neurotoxin serotype A: extending towards the β-exosite. Bioorg. Med. Chem. Lett.. 2012;22:3754-3757.
- [Google Scholar]
- Fungal hallucinogens psilocin, ibotenic acid, and muscimol: analytical methods and biologic activities. Ther. Drug Monit.. 2013;35:420-442.
- [Google Scholar]
- Design, synthesis and evaluation of novel polypharmacological antichlamydial agents. Eur. J. Med. Chem.. 2015;101:595-603.
- [Google Scholar]
- Synthesis and antibacterial evaluation of benzofuran based 3,5-disubstituted isoxazoles. Russ. J. Gen. Chem.. 2018;88:2669-2674.
- [Google Scholar]
- Synthesis and antimicrobial evaluation of bis-3,5-disubstituted isoxazoles based chalcones. Russ. J. Gen. Chem.. 2018;88:1904-1911.
- [Google Scholar]
- Synthesis and in vitro evaluation of dihydro-6H-chromeno[4,3-b]isoxazolo[4,5-e]pyridine derivatives as potent antidiabetic agents. Res. Chem. Intermed.. 2017;43:5433-5451.
- [Google Scholar]
- Design, synthesis and biological evaluation of diaziridinyl quinone isoxazole hybrids. Eur. J. Med. Chem.. 2016;117:85-98.
- [Google Scholar]
- Sulfisoxazole, an endothelin receptor antagonist, protects retinal neurones from insults of ischemia/reperfusion or lipopolysaccharide. Neurochem. Int.. 2006;48:708-717.
- [Google Scholar]
- Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem.. 2017;137:292-309.
- [Google Scholar]
- Curcuminoid derivatives enhance telomerase activity in an in vitro TRAP assay. Bioorg. Med. Chem. Lett.. 2014;24:5242-5246.
- [Google Scholar]
- 4-[5-Methyl-3-phenylisoxazol-4-yl]- benzenesulfonamide, Valdecoxib: A Potent and Selective Inhibitor of COX-2. J. Med. Chem.. 2000;43:775-777.
- [Google Scholar]
- Oxazole and isoxazole: From one-pot synthesis to medical applications. Tetrahedron. 2022;119:132813
- [Google Scholar]
- Synthesis and antiproliferative activity of 3α-hydroxyl-3β-methoxymethyl-5α-pregnan-20-one with a C-21 hydrophilic substituent. Heteroat. Chem.. 2017;28
- [Google Scholar]
- Chemoenzymatic synthesis and antileukemic activity of novel C9- and C14-functionalized parthenolide analogs. Bioorg. Med. Chem.. 2016;24:3876-3886.
- [Google Scholar]
- Rescue of pulmonary hypertension with an oral sulfonamide antibiotic sulfisoxazole by endothelin receptor antagonistic actions. Hypertens. Res.. 2008;31:1781-1790.
- [Google Scholar]
- Lack of correlation between the central anti-nociceptive and peripheral anti-inflammatory effects of selective COX-2 inhibitor parecoxib. Brain Res. Bull.. 2009;80:56-61.
- [Google Scholar]
- Design, synthesis and biological evaluation of novel naphthoquinone-4-aminobenzensulfonamide/carboxamide derivatives as proteasome inhibitors. Eur. J. Med. Chem.. 2021;209:112890
- [Google Scholar]
- The mode of action of isoxaflutole II. Characterization of the inhibition of carrot 4-hydroxyphenylpyruvate dioxygenase by the diketonitrile derivative of isoxaflutole. Pestic. Biochem. Physiol.. 1998;62:125-134.
- [Google Scholar]
- Synthesis and evaluation of novel isoxazolyl chalcones as potential anticancer agents. Bioorg. Chem.. 2014;54:38-43.
- [Google Scholar]
- Synthesis, structure-activity relationships and biological evaluation of caudatin derivatives as novel anti-hepatitis B virus agents. Bioorg. Med. Chem.. 2012;20:2877-2888.
- [Google Scholar]
- Isoxazole/isoxazoline skeleton in the structural modification of natural products: a review. Pharmaceuticals. 2023;16:228.
- [Google Scholar]
- Sulfonamides containing coumarin moieties selectively and potently inhibit carbonic anhydrases II and IX: design, synthesis, inhibitory activity and 3D-QSAR analysis. Eur. J. Med. Chem.. 2013;66:1-11.
- [Google Scholar]
- Discovery of farnesoid X receptor antagonists based on a library of oleanolic acid 3-O-esters through diverse substituent design and molecular docking methods. Molecules. 2017;22 690/691-690/613
- [Google Scholar]
- Clinical pharmacokinetics and pharmacodynamics of micafungin. Clin. Pharmacokinet.. 2018;57:267-286.
- [Google Scholar]
- Novel heterocyclic ring-fused oleanolic acid derivatives as osteoclast inhibitors for osteoporosis. MedChemComm. 2016;7:371-377.
- [Google Scholar]
- The discovery of carboline analogs as potent MAPKAP-K2 inhibitors. Bioorg. Med. Chem. Lett.. 2007;17:4664-4669.
- [Google Scholar]
- Synthesis and biological evaluation of panaxatriol derivatives against myocardial ischemia/reperfusion injury in the rat. Eur. J. Med. Chem.. 2020;185:111729
- [Google Scholar]
- Synthesis and biological evaluation of a series of bile acid derivatives as FXR agonists for treatment of NASH. ACS Med Chem Lett. 2017;8:1246-1251.
- [Google Scholar]
- Synthesis and biological evaluation of heterocyclic ring-fused betulinic acid derivatives as novel inhibitors of osteoclast differentiation and bone resorption. J. Med. Chem.. 2012;55:3122-3134.
- [Google Scholar]
- Construction of cholesterol oxime ether derivatives containing isoxazoline/isoxazole fragments and their agricultural bioactive properties/control efficiency. J. Agric. Food Chem.. 2021;69:8098-8109.
- [Google Scholar]
- Deletion of sigB causes increased sensitivity to para-aminosalicylic acid and sulfamethoxazole in mycobacterium tuberculosis. Antimicrob. Agents Chemother.. 2017;61
- [Google Scholar]
- Non-food bioactive products for pesticides candidates (III): Agricultural properties of isoxazole esters from the plant product podophyllotoxin as botanical pesticides. Ind. Crop. Prod.. 2021;174
- [Google Scholar]
- Novel rhein analogues as potential anticancer agents. ChemMedChem. 2011;6:2294-2301.
- [Google Scholar]
- The synthesis of furoquinolinedione and isoxazoloquinolinedione derivatives as selective Tyrosyl-DNA phosphodiesterase 2 (TDP2) inhibitors. Bioorg. Chem.. 2021;111:104881
- [Google Scholar]
- The structural modification of natural products for novel drug discovery. Expert Opin. Drug Discovery. 2017;12:121-140.
- [Google Scholar]
- Synthesis and biological evaluation of 3-benzisoxazolyl-4-indolylmaleimides as potent, selective inhibitors of glycogen synthase kinase-3β. Molecules. 2013;18:5498-5516.
- [Google Scholar]
- An isoxazole chalcone derivative enhances melanogenesis in B16 melanoma cells via the Akt/GSK3β/β-catenin signaling pathways. Molecules. 2017;22
- [Google Scholar]
- Synthesis and cytotoxic activity of A-ring modified betulinic acid derivatives. Bioorg. Med. Chem. Lett.. 2003;13:3137-3140.
- [Google Scholar]
- Synthesis and structure-activity relationship of furoquinolinediones as inhibitors of Tyrosyl-DNA phosphodiesterase 2 (TDP2) Eur. J. Med. Chem.. 2018;151:777-796.
- [Google Scholar]
- Synthesis and biological evaluation of oxygen-containing heterocyclic ring-fused 23-hydroxybetulinic acid derivatives as antitumor agents. Chem. Biol. Drug Des.. 2015;86:424-431.
- [Google Scholar]
- The synthesis and antistaphylococcal activity of N-sulfonaminoethyloxime derivatives of dehydroabietic acid. Bioorg. Med. Chem. Lett.. 2018;28:1943-1948.
- [Google Scholar]
- Aromatic heterocyclic esters of podophyllotoxin exert anti-MDR activity in human leukemia K562/ADR cells via ROS/MAPK signaling pathways. Eur. J. Med. Chem.. 2016;123:226-235.
- [Google Scholar]
- Recent advances in syntheses of oxazole compounds. Chin. J. Org. Chem.. 2011;31:1963-1976.
- [Google Scholar]
- Discovery of 4H-chromen-4-one derivatives as a new class of selective rho kinase (ROCK) inhibitors, which showed potent activity in ex vivo diabetic retinopathy models. J. Med. Chem.. 2019;62:10691-10710.
- [Google Scholar]
- The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem.. 2018;26:3065-3075.
- [Google Scholar]
- Design, synthesis, and biological evaluation of HSP90 inhibitor-SN38 conjugates for targeted drug accumulation. J. Med. Chem.. 2020;63:5421-5441.
- [Google Scholar]
- New synthetic isoxazole derivatives acting as potent inducers of fetal hemoglobin in erythroid precursor cells isolated from β-thalassemic patients. Molecules. 2024;29:8.
- [Google Scholar]




