

Translate this page into:
Investigating chlorothiophene-based chalcones as anticancer agents: Synthesis, biological evaluation, and computational insight via the p53-targeted pathway
*Corresponding author: E-mail address: tutikdw@ugm.ac.id (T.D. Wahyuningsih)
-
Received: ,
Accepted: ,
Abstract
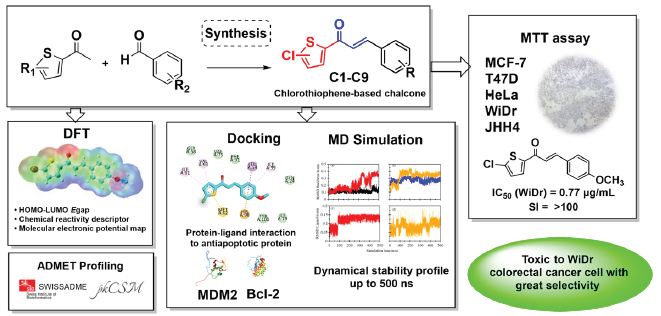
Chalcone, a prominent flavonoid known for its diverse biological effects, has been widely used in anticancer drug development to create compounds with improved selectivity. This study synthesized nine chlorothiophene-based chalcone analogs to explore their anticancer activity and selectivity via structural optimization and bio-isosteric replacement. Our compounds were synthesized using the Claisen–Schmidt condensation reaction and this method was proven to be highly effective, yielding our desired compounds at levels ranging from 68% to 96%. These compounds exhibited toxicity against various cancer cell lines, with compounds C4 and C6 exerting the strongest toxicity on WiDr colorectal cancer cells (IC50 = 0.77 and 0.45 µg/mL) and promising selectivity toward normal cells. Compounds C4 and C6 were selected for computational analysis to understand their inhibitory mechanisms. The analysis revealed binding energy values ranging from −6.2 to −6.6 kcal/mol, indicating moderate interactions with the anti-apoptotic proteins MDM2 and Bcl-2. Molecular dynamics simulation trajectories of C4 revealed stability profiles for Bcl-2 of up to 500 ns. Meanwhile, Density functional theory (DFT) studies provided insights into the designed compounds’ Highest occupied molecular orbital-Lowest unoccupied molecular orbital (HOMO-LUMO) energy gap and Molecular electrostatic potential (MEP) plot. In addition, ADMET (Adsorption, Distribution, Metabolism, Excretion, and Toxicity) analyses indicated the potential for oral drug development across all compounds.
Keywords
Anticancer
Bcl-2
Chalcone
p53-MDM2
Thiophene

1. Introduction
Cancer significantly contributes to global mortality, posing a primary obstacle to advances in life expectancy. The comprehensive 2020 report from the World Health Organization (WHO) reviews the current global impact of cancer, estimating an annual occurrence surpassing 18.1 million cases and foreseeing a doubling by 2040 [1]. The most substantial mortality rates are linked to lung, colorectal, and prostate cancers in men and breast cancer in women [2,3]. Although smaller molecules remain the predominant choice for anticancer therapies, their potential toxicity to normal cells and tissues poses a drawback. This study explored the potential of certain functional groups of chlorothiophene-based chalcones to exhibit selectivity toward cancerous and normal cells. Disruptions of the p53 tumor suppressor protein pathway are frequent occurrences in carcinogenesis, rendering this protein among the most pertinent targets for cancer therapeutics. The activation of p53 in response to various stressors is crucial for the survival of normal cells and the prevention of tumors. Major p53-targeting therapeutic strategies can be divided into two categories: (1) restoring wild-type (wt) p53 functions and (2) eliminating mutp53 [4]. For tumors carrying the wt p53, the primary approach to reactivate p53 involves inhibiting MDM2, a key down-regulator of the tumor suppressor p53 protein. The murine double minute 2 (MDM2) interferes with p53 by binding to its transactivation domain, which hinders p53 from binding to its target DNAs and impairs its function as a transcription factor for gene expression [5]. Conversely, B-cell lymphoma serves as a protective protein that blocks cell death by preventing the disruption of the mitochondrial outer membrane. This protein, when overexpressed, helps cells evade death resulting in uncontrolled, prolonged survival.This occurs because of the capability of Bcl-2 to stop cytochrome c from leaving the mitochondria, contributing to the immortality of cancer cells [6]. Studies have demonstrated that the concurrent inhibition of MDM2 and Bcl-2 enhances the sensitivity of cancer cells to treatment and results in a cooperative induction of apoptosis (e.g., venetoclax/idanasanutlin combination) [7-9].
This study examines the potential of chalcone, a member of the flavonoid family with myriad biological effects, to simultaneously target Bcl-2 and MDM2—highlighting that their concurrent inhibition may effectively initiate p53 activation in cancer cells. Chalcones are known for their diverse biological activities, including antimalarial, antioxidant, antimicrobial, antihistaminic, anti-inflammatory, and antitumor properties, as documented by numerous studies [10-12]. The effectiveness of chalcones against tumors appears to be influenced by various cellular and molecular mechanisms, one of which involves disrupting the p53 pathway. Many chalcones, both natural and synthetic, with different arrangements of substituents—particularly hydroxy, methoxy, and chloro groups—have been shown to affect the p53 pathway (Figure 1) [4,13-15]. A synthetic chalcone with a chloro group has been pinpointed using multidimensional nuclear magnetic resonance (NMR) spectroscopy and enzyme-linked immunosorbent assay for its capability to activate p53. The p53 is activated by releasing it from its binding with MDM2, effectively destabilizing the latter [16]. These findings indicate that chalcone compounds could be the basis for creating innovative inhibitors that target the interaction between p53-MDM2 and Bcl-2.
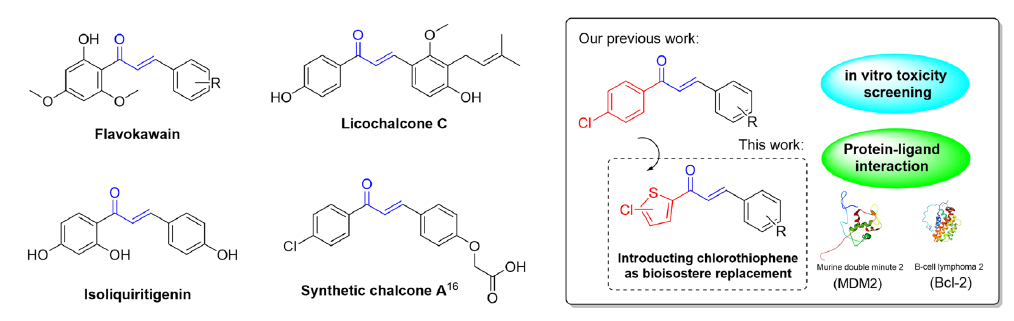
- Natural and synthetic chalcones induce apoptosis via the p53-targeted pathway (left), and our experimental scheme (right).
Our previous investigation primarily examined the effectiveness of chloro-substituted chalcones, revealing their capability to selectively target and exhibit toxicity against breast and colorectal cancer cells with good selectivity toward normal cells [17,18]. Our previous investigation further explored the impact of introducing chlorothiophene as bio-isostere to the chalcone structure to evaluate their toxicity and physiochemical characteristics. These modifications often result in distinct properties compared with their counterparts with an all-carbon ring structure [19,20]. Thiophene and its derivatives represent significant heterocyclic compounds with varied properties and applications, some of which are already used in commercially available drugs [21]. We investigated these compounds through in vitro studies on predominant cancer cell lines, supported by in silico analysis, to understand their binding affinity, molecular interactions, and stability with MDM2 and Bcl-2 proteins.
2. Materials and Methods
2.1. General procedure of the synthesis of chlorothiophene-based chalcones
A mixture of 2-acetyl-5-chlorothiophene or 2-acetyl-4,5-dichlorothiophene (0.01 mol) and the corresponding aromatic aldehyde (0.01 mol) in methanol (20 mL) was stirred for 24 h in the presence of 40% KOH (4 mL). The reaction was monitored by thin layer chromatography (TLC) with the eluent n-hexane/ethyl acetate (7:3). The workup procedure was performed by pouring the mixture into crushed ice and acidifying it with 5% HCl. The resulting solid was washed and purified using a recrystallization process with ethyl acetate and then dried in a desiccator. All chemicals and reagents were obtained from both Sigma-Aldrich Co. LLC and Shanghai Macklin Biochemical Co., Ltd. and utilized without further purification. Electrothermal’s IA9100 melting point apparatus was used to determine melting points (m.p.). Purity level analysis was conducted using a Dionex Ultimate 3000 system with a C18 column and a UV detector set at 254 nm. The mobile phase consisted of 0.1% formic acid in water (v/v) (Phase A) and 0.1% formic acid in acetonitrile (v/v) (Phase B). The gradient program was structured as follows: starting with 32.5% B in A, transitioning to 40% B in A at 4 mins, maintaining 40% B in A until 8 mins, increasing to 45% B in A at 10 mins, reaching 95% B in A at 15 mins, holding at 95% B in A until 18 mins, and then returning to the initial 32.5% B in A by 19 mins, maintaining this condition until the run ended at 35 mins. The flow rate was set at 1 mL/min. The injection volume for the analysis was 10 µL, with samples prepared at a concentration of 1 mg/mL, dissolved in acetonitrile [22]. Molecular mass was obtained using gas chromatography with mass spectrometry (Shimadzu QP-2010S). 1H NMR (500 MHz) and 13C NMR (125 MHz) spectroscopy were performed using the JEOL JNM-ECA 500 NMR spectrometer. The chemical shifts (δ) were expressed in parts per million (ppm) downfield from tetramethyl silane as an internal standard, and the coupling constant (J) was expressed in Hertz (Hz). Infrared spectra were obtained from PerkinElmer® spectrum two attenuated total reflectance-infrared (ATR-IR) spectrometer. TLC on silica gel Al foil was visualized under a UV lamp.
2.1.1. (E)-1-(5-chlorothiophen-2-yl)-3-phenylprop-2-en-1-one (C1)
Compound C1 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and benzaldehyde. The resulting product yielded 79% white–greenish powder, with a purity of 93.46%, retention time (Rt) = 17.80 minutes, and m.p. 98°C. 1H NMR (500 MHz, chloroform-d) δ 7.83 (d, J = 15.6 Hz, 1H), 7.62 (dd, J = 9.2, 3.6 Hz, 3H), 7.46–7.37 (m, 3H), 7.31 (d, J = 15.6 Hz, 1H), 7.00 (d, J = 3.8 Hz, 1H). 13C NMR (125 MHz, chloroform-d) δ 181.16, 144.63, 144.34, 139.90, 134.60, 131.31, 130.91, 129.12, 128.65, 127.82, 120.40. IR (ATR): νmax 1,644, 1,587, 1,418, 998, 757. MS (EI): m/z calcd for C13H9ClOS, 248.
2.1.2. (E)-1-(5-chlorothiophen-2-yl)-3-(2-methoxyphenyl) prop-2-en-1-one (C2)
Compound C2 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 2-methoxybenzaldehyde. The resulting product yielded 70% pale yellow powder, with a purity of 92.20%, Rt = 17.96 minutes, and m.p. 135°C. 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 15.7 Hz, 1H), 7.63–7.57 (m, 2H), 7.43 (d, J = 15.8 Hz, 1H), 7.38 (ddd, J = 8.3, 7.3, 1.7 Hz, 1H), 7.02–6.96 (m, 2H), 6.94 (dd, J = 8.3, 1.0 Hz, 1H), 3.92 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 181.79, 159.06, 144.64, 140.30, 139.38, 132.10, 131.07, 129.69, 127.70, 123.66, 121.34, 120.86, 111.36, 55.65. IR (ATR): νmax 1,643, 1,583, 1,416, 1,224, 1,015, 809, 750. MS (EI): m/z calcd, for C14H11ClO2S, 278.
2.1.3. (E)-1-(5-chlorothiophen-2-yl)-3-(3-methoxyphenyl) prop-2-en-1-one (C3)
Compound C3 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 3-methoxybenzaldehyde. The resulting product yielded 79% army green powder, with a purity of 94.74%, Rt = 17.73 minutes, and m.p. 99°C. 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 15.6 Hz, 1H), 7.64 (d, J = 4.0 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.30 (d, J = 15.5 Hz, 1H), 7.23 (dt, J = 7.6, 1.2 Hz, 1H), 7.14 (dd, J = 2.5, 1.7 Hz, 1H), 7.01 (d, J = 4.0 Hz, 1H), 6.98 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 3.86 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ 181.13, 160.05, 144.55, 144.30, 139.94, 135.97, 131.34, 130.11, 127.82, 121.24, 120.71, 116.56, 113.70, 55.48. IR (ATR): νmax 1,644, 1,590, 1,578, 1,416, 1,220, 1,021, 778. MS (EI): m/z calcd, for C14H11ClO2, 278.
2.1.4. (E)-1-(5-chlorothiophen-2-yl)-3-(4-methoxyphenyl) prop-2-en-1-one (C4)
Compound C4 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 4-methoxybenzaldehyde. The resulting product yielded 81% pale yellow powder, with a purity of 94.01%, Rt = 17.65 minutes, and m.p. 118°C. 1H NMR (500 MHz, chloroform-d) δ 7.81 (d, J = 15.5 Hz, 1H), 7.62 (d, J = 4.1 Hz, 1H), 7.61–7.57 (m, 2H), 7.20 (d, J = 15.5 Hz, 1H), 7.00 (d, J = 4.0 Hz, 1H), 6.96–6.92 (m, 2H), 3.86 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ 181.19, 161.98, 144.61, 144.45, 139.43, 130.91, 130.48, 129.10, 127.74, 127.33, 118.02, 114.56, 113.77, 55.55. IR (ATR): νmax 1,643, 1,582, 1,574, 1,417, 1,226, 1,216, 1,169, 1,016, 815, 794. MS (EI): m/z calcd, for C14H11ClO2S, 278.
2.1.5. (E)-1-(5-chlorothiophen-2-yl)-3-(3,4-dimethoxyphenyl) prop-2-en-1-one (C5)
Compound C5 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 3,4-dimethoxybenzaldehyde. The resulting product yielded 96% yellow powder, with a purity of 97.40%, Rt = 16.98 minutes, and m.p. 120°C. 1H NMR (500 MHz, chloroform-d) δ 7.79 (d, J = 15.4 Hz, 1H), 7.64 (d, J = 4.1 Hz, 1H), 7.23 (dd, J = 8.3, 2.0 Hz, 1H), 7.18 (d, J = 15.5 Hz, 1H), 7.13 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 4.1 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H), 3.95 (s, 3H), 3.94 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ 181.13, 151.74, 149.34, 144.78, 144.54, 139.50, 130.98, 127.73, 127.57, 123.46, 118.23, 111.20, 110.21, 56.13, 56.08. IR (ATR): νmax 1,639, 1,567, 1,509, 1,415, 1,222, 1,135, 1,019, 971, 792, 712. MS(EI): m/z calcd, for C15H13ClO3S, 308.
2.1.6. (E)-3-(2-chlorophenyl)-1-(5-chlorothiophen-2-yl) prop-2-en-1-one (C6)
Compound C6 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 2-chlorobenzaldehyde. The resulting product yielded 75% black powder (cotton-like texture), with a purity of 96.26%, Rt = 18.33 minutes, and m.p. 146°C. 1H NMR (500 MHz, chloroform-d) δ 8.20 (d, J = 15.7 Hz, 1H), 7.71 (dd, J = 7.4, 2.0 Hz, 1H), 7.63 (d, J = 4.1 Hz, 1H), 7.46–7.42 (m, 1H), 7.36–7.30 (m, 2H), 7.30–7.25 (m, 1H), 7.00 (d, J = 4.1 Hz, 1H). 13C NMR (125 MHz, chloroform-d) δ 181.04, 144.03, 140.43, 140.23, 135.74, 133.00, 131.61, 131.51, 130.50, 127.93, 127.86, 127.20, 126.47, 123.21. IR (ATR): νmax 1,646, 1,596, 1,423, 799, 748. MS (EI): m/z calcd, for C13H8Cl2OS, 282.
2.1.7. (E)-3-(4-chlorophenyl)-1-(5-chlorothiophen-2-yl) prop-2-en-1-one (C7)
Compound C7 was prepared based on the general procedure of the reaction between 2-acetyl-5-chlorothiophene and 4-chlorobenzaldehyde. The resulting product yielded 83% yellow-orange powder, with a purity of 92.39%, Rt = 18.27 minutes, and m.p. 155°C. 1H NMR (500 MHz, chloroform-d) δ 7.78 (d, J = 15.6 Hz, 1H), 7.64 (d, J = 4.1 Hz, 1H), 7.58–7.55 (m, 2H), 7.42–7.37 (m, 2H), 7.29 (d, J = 15.6 Hz, 1H), 7.02 (d, J = 4.1 Hz, 1H). 13C NMR (125 MHz, chloroform-d) δ 180.88, 144.15, 143.13, 140.16, 136.82, 133.08, 131.41, 129.77, 129.41, 129.22, 128.74, 127.86, 120.82. IR (ATR): νmax 1,646, 1,589, 1,556, 1,419, 800. MS (EI): m/z calcd, for C13H8Cl2OS, 282.
2.1.8. (E)-1-(4,5-dichlorothiophen-2-yl)-3-(4-methoxyphenyl) prop-2-en-1-one (C8)
Compound C8 was prepared based on the general procedure of the reaction between 2-acetyl-4,5-dichlorothiophene and 4-methoxybenzaldehyde. The resulting product yielded 68% yellow powder, with a purity of 92.83%, Rt = 18.620 minutes, and m.p. 141°C. 1H NMR (500 MHz, chloroform-d) δ 7.83 (d, J = 15.4 Hz, 1H), 7.65–7.54 (m, 3H), 7.15 (d, J = 15.5 Hz, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ 180.43, 162.24, 145.43, 141.84, 133.81, 130.68, 130.39, 127.07, 125.27, 125.17, 117.08, 114.63, 114.30, 55.57. IR (ATR): νmax 1,642, 1,585, 1,568, 1,409, 1,293, 1,216, 1,177, 978, 817, 797. MS (EI): m/z calcd, for C14H10Cl2O2S, 312.
2.1.9. (E)-1-(4,5-dichlorothiophen-2-yl)-3-(3,4-dimethoxyphenyl) prop-2-en-1-one (C9)
Compound C9 was prepared based on the general procedure of the reaction between 2-acetyl-4,5-dichlorothiophene and 3,4-dimethoxybenzaldehyde. The resulting product yielded 90% yellow powder, with a purity of 91.37%, Rt = 18.21 minutes, and m.p. 135°C. 1H NMR (500 MHz, chloroform-d) δ 7.80 (d, J = 15.4 Hz, 1H), 7.61 (s, 1H), 7.23 (dd, J = 8.3, 2.0 Hz, 1H), 7.12 (d, J = 2.0 Hz, 1H), 7.12 (d, J = 15.5 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 3.96 (s, 3H), 3.94 (s, 3H). 13C NMR (125 MHz, chloroform-d) δ 180.38, 152.02, 149.39, 145.76, 141.80, 133.90, 130.41, 127.30, 123.84, 117.26, 111.20, 110.15, 56.15, 56.11. IR (ATR): νmax 1,641, 1,577, 1,514, 1,422, 1,283, 1,269, 1,208, 1,150, 1,042, 803. MS m/z C15H12Cl2O3S found 342.
2.2. Anticancer activity
Approximately 10,000 cells per 100 µL were seeded into 96-well microplates and incubated in a CO2 incubator for 24 h. The media were removed, and the cells were first treated with a serial dilution solution of the compounds and then incubated for 24 h. Subsequently, 100 µL of an 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) solution was added to each well. The plate was left to incubate for 4 h. To prevent crystal formation, 100 µL of sodium dodecyl sulfate (SDS) stopper solution was added to each well, and the plates were left overnight. Following the complete dissolution of all crystals, the absorbance (A) was measured at 595 nm using a microplate reader. Then, the percentage of dead cells was calculated using Eq. (1):
The obtained percentage of cell death is transformed into probit numbers, creating a curve of log concentration (x) vs. probit number (y) using the equation y = ax + b. When y equals 5, the corresponding x value represents the Log IC50. The IC50 is determined by calculating the antilog of the x value, representing the minimum compound concentration required to induce 50% cell death. The selectivity index was calculated using Eq. (2):
2.3. Computational methods
2.3.1. Molecular docking
Following our previous work [23], ligands were initially sketched using GaussView 5.0 and subsequently optimized by the Gaussian 09W software [24] utilizing the density functional theory/Becke three-parameter Lee–Yang–Parr (DFT/B3LYP) method with a 6-31G basis set. In addition, crystal structures of the proteins were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (PDB) for p53-MDM2 (PDB ID: 4HG7) and Bcl2 (PDB ID: 6QGK). These structures, along with their co-crystallized ligands, were prepared using Chimera [25]. This preparation included adding hydrogen atoms and Kollman charges [26] to the structures to ensure accurate representation in subsequent computational analyses. Docking calculations were conducted utilizing AutoDock Vina [27], with the graphical user interface provided by AutoDock Tools. To validate docking for both MDM2 and Bcl-2 proteins, redocking of the native ligand into the active site of the target protein was performed using a cubic grid box of 15 Å, with a spacing of 1.000 Å centered on the ligand. The coordinate settings were −23.945, 8.091, and −13.667 (x, y, and z, respectively) for the MDM2 protein and 1.861, 0.439, and 19.153 (x, y, and z, respectively) for Bcl-2, with an exhaustiveness value of 16. We followed a similar procedure by substituting the native ligands with our ligand, ensuring that the grid box sizes and positions remained consistent. The protein-ligand interactions were visualized using the Discovery Studio Visualizer (DSV) 2017 [28].
2.3.2. Molecular dynamics simulation
The molecular dynamics (MD) simulation of the protein-ligand complex was performed for apoprotein and complexes using the GROMACS 2022.4 software [29], which employs the AMBER99SB [30] force field. This force field is renowned for providing a reliable parameter set for protein simulations within an efficient, additive molecular mechanics model compared with other AMBER force field versions [30,31]. The topology and ligand parameters were generated using the ACPYPE external tool [32]. The complex was enclosed within a cube under periodic boundary conditions and immersed in the transferable intermolecular potential 3P (TIP3P) water model [33]. The combination of the AMBER99SB force field and the TIP3P water model is widely regarded as the optimal choice for computing ∆Gbind, yielding values that are consistent with the experimental results [34] and the widely accepted recommendations from the GROMACS 2022.4 software. Na+ ions were added to neutralize the system to balance the negative charges, and Cl− ions were added to balance the positive charges, ensuring overall system neutrality. The long-range electrostatic interactions were corrected using the particle mesh Ewald (PME) method [35] with a 1.2 nm cutoff. The simulation commenced with energy minimization employing 1,000 steepest descent steps. Then, the system was equilibrated in the canonical (NVT) ensemble at 298.15 K using velocity rescaling [36] to stabilize the temperature for 500 ps. Subsequently, equilibration of the isothermal-isobaric (NPT) ensemble at 1 bar was conducted using the Parrinello–Rahman barostats [37] for 4 ns. The MD production phase was simulated for 500 ns. Afterward, trajectory files were analyzed based on multiple parameters, including root–mean–square deviation (RMSD), root–mean–square fluctuation (RMSF), radius of gyration (RoG), solvent-accessible surface area (SASA) [38,39], number of the hydrogen bonds, and free energy landscape (FEL), utilizing the GROMACS 2022.4 software. Graphical presentations of the MD simulation trajectories were generated using Xmgrace [40] and DSV 2017. Two-dimensional (2D) FEL contour plots were created using Wolfram Mathematica [41]. The validation process for the three-dimensional (3D) protein model involved the generation of a Ramachandran (RAM) plot, which provides insights into the quality of the protein structure via the PDBsum site [42] and the Dictionary of Secondary Structure Protein (DSSP) [43], utilizing the gmx do_dssp tool.
2.3.3. Free binding energy calculation using MM-PBSA
The binding energy was determined using the MM-PBSA protocols facilitated by the gmx_mmpbsa package [44] within the GROMACS 2022.4 software. At its core, the free binding energies () using MM-PBSA are determined by subtracting the free binding energies of the unbound receptor and ligand from the free binding energy of the bound complex [45], as illustrated in Eq. (3):
where each component of ∆Gsolvated is calculated using the parameters shown in Eq. (4):
where ∆Gelec and ∆Gvdw are the contributions from electrostatic and van der Waals interactions, respectively; ∆Gnp and ∆GPB are the nonpolar and polar solvation energies, respectively; and T∆S is the entropic contribution [34]. Only 5,000 frames collected from the last 50 ns MD simulation output are utilized in this computation.
2.3.4. Density functional theory (DFT) calculation
Quantum chemical calculations were conducted using the Gaussian 09W software employing the DFT/B3LYP method with a 6-31G basis set. The optimized structures and electronic properties, such as the highest occupied molecular orbital (EHOMO) and lowest unoccupied molecular orbital (ELUMO) energies, were determined for each compound. Furthermore, some chemical reactivity descriptors were calculated using Eqs. (5) to (9) based on Koopmans’ theorem [46,47].
Thus, other chemical reactivity descriptors were calculated based on I and A. Electronegativity (χ) quantifies the capability of an atom or a group of atoms to attract electrons toward itself and can be calculated using Eq. (7):
Chemical hardness (η) is a vital descriptor indicating the resilience of an atom against charge transfer and can be calculated using Eq. (8):
The electrophilicity index ( measures the capability of a system to attract an electron and can be calculated using Eq. (9):
Finally, to pinpoint the reactive regions within the compounds, where they might be vulnerable to electrophilic and nucleophilic attacks, we generated MEP maps from their corresponding checkpoint files. Multiwfn [48] and Visual Molecular Dynamics (VMD) [49] were utilized to generate the graphical representation of MEP.
2.3.5. Pharmacokinetic analysis
To evaluate drug-likeness, various molecular properties, such as molecular weight (MW), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), number of heavy atoms (Nat), rotatable bonds, topological polar surface area (TPSA), molar refractivity, and Log P were analyzed using the SwissADME predictors [50]. In addition, for a comprehensive assessment of Adsorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties, including gastrointestinal absorption (GIA), blood-brain barrier (BBB) permeability, central nervous system (CNS) permeability, total clearance, and cytochrome enzyme inhibition, the pkCSM prediction [51] was utilized. Furthermore, the adherence of the compounds to Lipinski’s rule of five [52] and Veber’s criteria [53] was categorized to assess their suitability for oral drug development.
3. Results and Discussion
3.1. Structural design
Recent advancements in anticancer drug design have placed considerable emphasis on modifying the structure of molecules containing heteroaryl substitutions. Bio-isosteric replacement has emerged as a key strategy to retain comparable properties within heteroaromatic systems while enhancing biological activity [54]. Our research has been consistently dedicated to analyzing and developing heteroaryl chalcones and their derivatives, specifically those containing five-member rings. We observed a trend indicating that thiophene significantly enhances the toxicity of a compound to cancer cells compared to other five-member rings heterocycles such as pyrrole and furan, especially against HeLa cervical cancer cells [55-57]. Thiophene is commonly employed as a substitute for benzene in drug discovery because of its recognized similar aromaticity, rendering it an effective mimic of the phenyl moiety.
Moreover, our previous research considered the impact of two substituents, i.e., chloro and methoxy groups, as crucial functional groups influencing the toxicity and selectivity of the compound [58]. Several studies have investigated the introduction of substituents in thiophene and have observed alterations in activity [59-61]. Incorporating a chlorine atom has been noted to significantly improve the stability, potency, and specificity of the binding site. This enhancement arises from the capability of the chlorine atom to fit into tight and deep hydrophobic pockets within biological targets, thereby facilitating stronger and more selective interactions [62,63]. This attribute is particularly advantageous because our binding target consists of hydrophobic pockets. However, achieving selectivity is a significantly challenging task. Hence, we explore other substituents to optimize selectivity, aiming for compounds that remain inactive in normal cells. Although a comprehensive study of anticancer activity on this scaffold has not yet been conducted, we consider it the most promising starting point. Therefore, we focus further optimization efforts on this framework.
3.2. Chemistry
The chalcone scaffold is highly regarded as a versatile pharmacophore in medicinal chemistry for designing diverse therapeutic agents. In this study, we expanded upon our previous work [18] with the chalcone scaffold by successfully synthesizing a series of chlorothiophene-based chalcone derivatives. The synthetic route employed is straightforward, as illustrated in Figure 2.
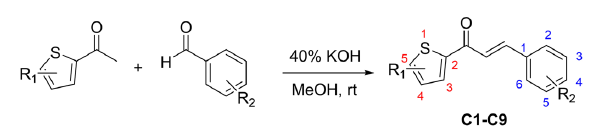
- Reaction scheme.
The required starting material was the chloro-substituted acetyl thiophene (2-acetyl-5-chlorothiophene and 2-acetyl-4,5-dichlorothiophene) and appropriate aldehydes with methoxy and chloro substituents. Then, chalcone compounds C1–C9 were synthesized using base-catalyzed aldol with aqueous KOH (40%) in the presence of methanol as a solvent at room temperature by stirring. This condition is sufficient to produce high-yield products of chalcones, as shown in Table 1. The chemical features of the product analogs were determined using spectrophotometric techniques.
| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| C1 | -5-Cl | -H | 79 |
| C2 | -5-Cl | -2-OCH3 | 70 |
| C3 | -5-Cl | -3-OCH3 | 79 |
| C4 | -5-Cl | -4-OCH3 | 81 |
| C5 | -5-Cl | -3,4-diOCH3 | 96 |
| C6 | -5-Cl | -2-Cl | 75 |
| C7 | -5-Cl | -4-Cl | 83 |
| C8 | -4,5-diCl | -4-OCH3 | 68 |
| C9 | -4,5-diCl | -3,4-diOCH3 | 90 |
Both 1H NMR and 13C NMR spectroscopy were used to analyze the synthesized chalcone compounds. These compounds have a conjugated enone group at the center of the molecule. The alkene C=C double bond positions are designated as the α and β positions, with the carbonyl C=O group referred to as the α position and the carbonyl C=O group referred to as the β position. In all compounds, the protons corresponding to these positions appeared as doublets because of their coupling with each other, with the coupling constant (JHα–Hβ) ranging from 15.4 to 15.7 ppm, indicating a trans configuration of alkene proton [64,18]. The carbon in the C=O group typically appeared in the most downfield region of all-carbon signals, with chemical shifts ranging from 180.38 to 181.88 ppm. The IR spectra also support the presence of this carbonyl system by showing strong absorbance at approximately 1,660 cm−1, indicating a stretching vibration of the C=O carbonyl bond.
By analyzing the yield trend, we observed that benzaldehyde substitution at position 4 (para-position) resulted in higher yields compared with benzaldehyde substitution at position 2 (C4 vs. C7). Moreover, aldehydes with electron-withdrawing chloro substituents exhibited greater yield than those with electron-donating methoxy groups, attributed to an electron-withdrawing effect that enhances benzaldehyde reactivity. Conversely, the introduction of an additional chloro group to the thiophene system led to decreased yield (C4 vs. C8). The dichloro group in the thiophene system exerted a significant electron-withdrawing influence via an inductive effect, thereby reducing the reactivity of acetyl during the reaction.
3.3. Anticancer activity
Cytotoxicity tests were conducted using assays on various predominant types of cancer cells to assess the anticancer effects of chalcone compounds. Doxorubicin, cisplatin, and 5-fluorouracil (5-FU) were utilized as reference drugs. Table 2 shows that most compounds exhibit low IC50 values against WiDr colorectal cancer cells, surpassing the efficacy of the positive control (i.e., 5-FU). Compounds C4 and C6 exhibited the most promising IC50 value for this cell line at 0.77 and 0.45 µg/mL, respectively. However, C6 was observed to have moderate toxicity to normal cells, whereas C4 was non-toxic.
| Compound | IC50 (µg/mL) | Selectivity index WiDr to Vero | |||||
|---|---|---|---|---|---|---|---|
| Cancer cell | Normal cell | ||||||
| Breast cancer | Cervix | Colorectal | Liver | ||||
| T47D | MCF-7 | HeLa | WiDr | JHH4 | Vero | ||
| C1 | 1.78 | 7.06 | 5.61 | 0.81 | 4.80 | 78.16 | 96.36 |
| C2 | 1.43 | 26.27 | 77.26 | 1.60 | 78.98 | 407.47 | >100 |
| C3 | 7.34 | 2.26 | 7.72 | 5.97 | >100 | 246.40 | 41.24 |
| C4 | 76.88 | >100 | 86.86 | 0.77 | 58.12 | 227.81 | >100 |
| C5 | >100 | 44.48 | 65.38 | 1.57 | 42.83 | 14.19 | 9.06 |
| C6 | 12.66 | 1.11 | 66.02 | 0.45 | 47.18 | 21.73 | 47.87 |
| C7 | >100 | 23.28 | 60.65 | 6.48 | >100 | 256.71 | >100 |
| C8 | 66.23 | 81.63 | 84.07 | 6.19 | >100 | 34.61 | 5.59 |
| C9 | 4.64 | 15.94 | 79.97 | 9.64 | >100 | 6.19 | 0.64 |
| Doxorubicin | 50.84 | 14.63 | – | – | 9.30 | – | – |
| Cisplatin | – | – | 15.87 | – | – | – | – |
| 5-fluorouracil | – | – | – | 14.44 | – | – | – |
C1 exhibited low IC50 values across all cancer cell lines, with moderate toxicity toward normal cells. The observed trend was the addition of one methoxy substituent (C2–C4) led to reduced toxicity toward normal cells, indicating a better selectivity index. However, this trend was not observed with the addition of the dimethoxy group (C5). Compounds with multiple chloro substituents did not yield favorable results, e.g., C6. Similarly, C8 and C9 exhibited more toxicity to normal cells. Thus, the optimal conditions lead to good activity toward WiDr colorectal cancer cells, as shown in Figure 3.
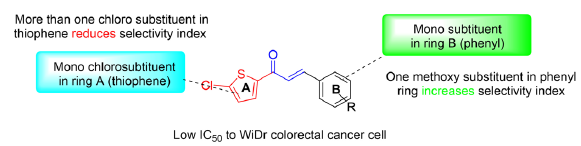
- Optimized structures of compounds that potentially show good selectivity.
3.4. Molecular docking
The docking protocol commenced with the validation process using the native ligand. The redocking procedure was conducted with a predefined grid box size, yielding RMSD values of 0.385 Å for MDM2 and 0.583 Å for Bcl-2 (Table S1). To validate the adequacy of the grid box size used in the docking procedure, our approach includes employing an RMSD threshold of <2 Å. Figure S1 illustrates the stacking condition of the native ligand before and after the validation process. For further analysis, we selected two samples, i.e., C4 and C6, known for their cytotoxicity against WiDr colorectal cancer cells.
In the current investigation, Table 3 shows the binding affinity of the compounds, and the protein-ligand interactions compared with each native ligand. The binding affinity of both compounds to both MDM2 and Bcl-2 proteins did not exhibit higher values than their respective native ligands. In complex to MDM2, the binding affinity values of −6.2 kcal/mol for C4 and −6.6 kcal/mol for C6 were observed. In comparison, the native ligand nutlin-3a has a value of −8.8 kcal/mol, which indicates that our compound has weaker binding than nutlin-3a. This trend is similarly reflected in the Bcl-2 protein, with C4 and C6 exhibiting values of −6.4 and −6.6 kcal/mol, respectively, whereas the native ligand has a value of −8.5 kcal/mol. C6 exhibits a slightly higher value than C4, indicating potentially stronger binding strength, as evidenced by its smaller IC50 value.
| Complex | Binding affinity (kcal/mol) | Interaction |
|---|---|---|
| MDM2 | ||
| C4-MDM2 | −6.2 |
Pi-cation: His96 (3.974 A) Hydrophobic: Val93, Leu54 Ile99, Ile61 Pi-sulfur: Met62 |
| C6-MDM2 | −6.6 |
Pi-cation: His96 (4.188 A) Hydrophobic: Val93, Leu, 54, Ile99, Ile61 Pi-sulfur: Met62 |
| Nutlin-3a-MDM2 | −8.8 |
Pi-cation: His96 (3.685 A) Hydrophobic: Val93, Leu54, Tyr100, Ile99, Leu57, Phe91, Val75 |
| Bcl-2 | ||
| C4-Bcl-2 | −6.4 |
Hydrophobic: Phe112, Met115, Phe153, Val133, Ala149, Arg146, Phe104 Pi-sulfur: Phe104 |
| C6-Bcl-2 | −6.6 |
Hydrophobic: Phe112, Met115, Arg146, Ala149, Phe104 Pi-sulfur: Phe104 |
| THIQ-PP-Bcl-2 | −8.5 | Hydrophobic: Phe112, Tyr108, Met115, Phe104, Ala149, Leu137, Arg146, Val113 |
Note: MDM2: murine double minute 2, Bcl-2: B-cell lymphoma 2, THIQ-PP: tetrahydroisoquinoline phenyl pyrazole, Ala: alanine, Arg: arginine, His: histidine, Leu: leucine, Ile: isoleucine, Met: methionine, Phe: phenylalanine, Tyr: tyrosine, Val: valine.
In addition to assessing binding energies, we also evaluated the protein-ligand interactions that are essential for inhibiting the selected proteins. This comprehensive approach enabled us to consider the specific molecular interactions that contribute to the inhibition of target proteins, providing valuable insights into the efficacy of potential inhibitors. Before delving into further interactions, we need to comprehend the binding area of the p53 peptide to the MDM2 protein. The MDM2 domain consists of structural repetitions, comprising two portions, each spanning 45 and 38 amino acids, with similar structures (residues 26–70 and 71–108) which are linked by an approximate dyad axis of symmetry. The crystal structure of MDM2 bound to a p53 peptide elucidated the significance of Phe19, Trp23, and Leu26 as key residues, establishing a locally confined, hydrophobic interaction site within p53. Moreover, the MDM2 cleft comprises 14 hydrophobic and 11 aromatic amino acids, facilitating numerous van der Waals contacts with p53, including those with His96, Ile99, and Tyr100 [65]. The compact nature of the p53-MDM2 interface has facilitated the design of not only peptides but also small organic compounds capable of blocking this interaction. A compound such as nutlin-3a, along with other established cancer drugs, exert their inhibitory effects by targeting the hydrophobic cleft formed by specific amino acids, such as Leu54, Gly58, Ile61, Val93, His96, and Ile99 [66,67]. This targeted binding disrupts the interaction between MDM2 and p53, thereby restoring the tumor-suppressive functions of p53 and offering promising therapeutic strategies for cancer treatment.
Figure 4(a) and 4(b) present the 3D visualization of docked compounds within the binding pocket of MDM2. This docking protocol shows that both compounds lack hydrogen bonding interactions, like nutlin-3a. Instead, both compounds interact electrostatically (Pi-cation) with the amino acid His96, with distances of 3.974 and 4.188 Å for C4 and C6, respectively. Moreover, the thiophene moiety contributes to a Pi-sulfur interaction with Met62. Hydrophobic contacts are observed with Leu54, Ile61, Val93, and Ile99. These hydrophobic interactions play a crucial role in anchoring compounds to the binding site and contribute to the overall stability of the protein-ligand complex.
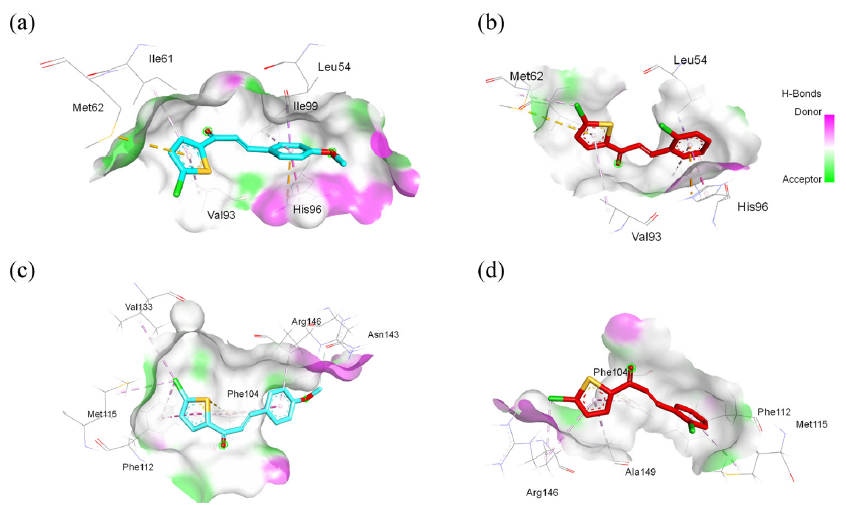
- 3D interaction visualization of murine double minute 2 (MDM2) complexed with (a) C4 and (b) C6. 3D interaction visualization of B-cell lymphoma 2 (Bcl-2) complexed with (c) C4 and (d) C6. Note: Ala: alanine, Arg: arginine, His: histidine, Leu: leucine, Ile: isoleucine, Met: methionine, Phe: phenylalanine, Tyr: tyrosine, Val: valine.
The Bcl-2 proteins fold into a distinctive multiple α-helices bundle [68]. The hydrophobic groove within the anti-apoptotic protein Bcl-2, characterized by the presence of conserved Bcl-2 homology domains BH1 and BH2, serves as a critical binding site for regulating apoptotic pathways. This specialized region is pivotal in mediating protein-protein interactions and modulating cellular responses to apoptotic stimuli. This groove works like a hiding spot, trapping the BH3 domain of pro-apoptotic Bcl-2 proteins. This BH3 domain plays a critical role in correctly folding a hydrophobic pocket within the Bcl-2 protein. To disrupt the function of Bcl-2, an approach involving small molecules that can bind to this groove area is utilized. These small molecules target one or more P1–P4 sub-pockets within the BH3 groove of anti-apoptotic proteins [69]. By binding to these sub-pockets, these molecules induce the release of pro-apoptotic BH3-only proteins, which subsequently activate the apoptotic pathway, leading to cell death. Polar residues Tyr108 and Arg146 act as important anchor points, helping to stabilize the attached ligands [70,71].
Figure 4(c) and 4(d) show the 3D visualization of the docked compound inside the binding pocket of Bcl-2. Interestingly, neither the docked compound nor the native ligand forms hydrogen bonds in the complexes. However, the crucial amino acid Arg146 is observed to interact hydrophobically with the docked compound, supported by interactions with Phe112, Met115, and Val133. In addition, the thiophene moiety contributes to a Pi-sulfur interaction with Phe104. These interactions indicated that our compound had moderately bound to the Bcl-2 protein. Further exploration of this protein-ligand interaction will be discussed more comprehensively through MD simulation studies.
3.5. Molecular dynamics simulation
After the docking investigation, MD simulation was utilized to evaluate the dynamic stability of the compound in a solvent system. C4 was chosen as the most promising candidate for MD simulation because of its high toxicity and better selectivity. The simulation was conducted over 500 ns, with its trajectory result compared with their apoproteins. To validate the MD simulation and free binding energy calculation method, we attempted to replicate the system used by Kumar et al. [72] which employed AMBERFF. The summarized results are presented in Table S2.
Figure 5(a) depicts the stability of the C4-MDM2 complex, exhibiting a consistent RMSD profile within the range of 0.10–0.20 nm for the initial 300 ns, followed by an increase to 0.40 nm toward the end of the simulation. This deviation is noticeably larger than that of the stable RMSD of the apoprotein throughout the simulation. Interestingly, previous studies focusing on the development of potential ligands for MDM2 via MD simulation have shown a similar trend up to a maximum of 200 ns, which is in the same range of RMSD values [73,74]. However, the results obtained up to 500 ns provide new insights for analysis, offering information about events occurring after 300 ns of simulation. Figure 5(b) shows that the C4-Bcl2 complex exhibits a more consistent RMSD trend, like its apoprotein counterpart, and achieves equilibrium within the first 100 ns, with an RMSD increment from 0.10 nm to 0.40 nm. Compared with numerous studies of Bcl-2 complex MD simulations, typically spanning 100–200 ns, the range of RMSD values of our study was considered acceptable [75,76]. The stability persisted until the end of the simulation. This behavior was also noted in the apoprotein simulation, indicating that the introduction of the ligand did not significantly alter the protein backbone.
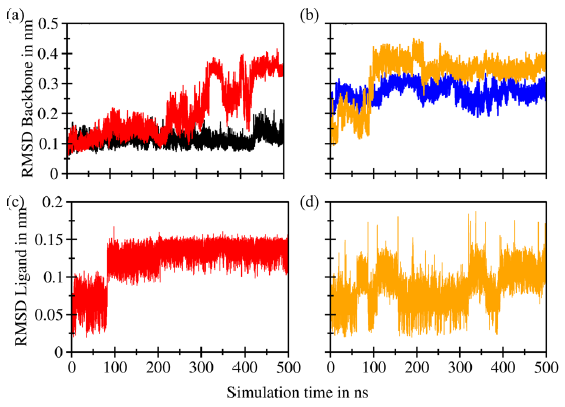
- Root mean square deviation (RMSD) superimposition of the (a) backbones of murine double minute 2 (MDM2) (black) and C4-MDM2 (red), (b) the backbones of B-cell lymphoma 2 (Bcl-2) (blue) and C4-Bcl-2 (orange), (c) ligand C4 complexed with MDM2 (red), and (d) ligand C4 complexed with Bcl-2 (orange).
The RMSD value of the ligand C4 complexed with MDM2 ranges from 0.05 to 0.15 nm (Figure 5c), with a significant change occurring at 100 ns. This finding is attributed to the formation of a hydrogen bond between the carbonyl group of the chalcone moiety and the amino acid residue Tyr67. Subsequently, the RMSD remains stable until the end of the simulation. Similarly, the RMSD value of ligand C4 complexed with Bcl-2 exhibits fluctuations within the same range (Figure 5d), i.e., it fluctuated along the simulation, with the highest deviation occurring at approximately 150 and 350 ns, reaching 0.18 nm.
The RMSF pattern of the protein backbone of the C4-MDM2 complex, depicted in Figure 6, exhibits fluctuations within the range of 0.10–0.20 nm. A significant deviation up to 0.23 nm is observed, which is attributed to the formation of a hydrogen bond between the carbonyl group of the chalcone and Tyr97, consistent with the RMSD profile findings. The RMSF pattern of the protein backbone of the C4-Bcl2 complex closely resembles the apoprotein. Minor discrepancies are noticed around amino acid residues 130–140 ns, particularly in the binding pocket area, which will be further explained by the DSSP result. In summary, the RMSF profile of the protein backbone during the simulation indicates that the introduction of the ligand has minimal impact on the protein structure.
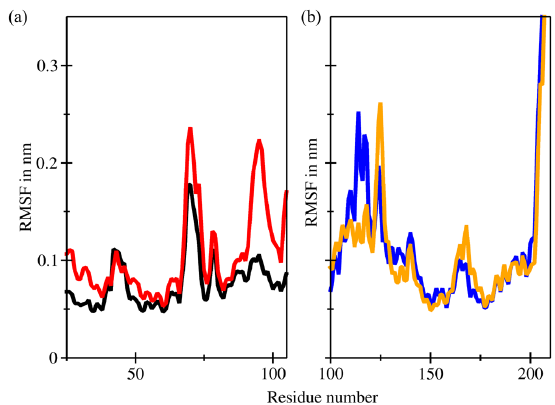
- Root mean square fluctuations (RMSF) superimposition of the (a) backbones of murine double minute 2 (MDM2) (black) and C4-MDM2 (red) and (b) backbones of Bcl-2 (blue) and C4-Bcl-2 (orange).
The RoG serves as a vital metric for assessing the structural stability of proteins and reflecting their compactness level. Large RoG values indicate a protein molecule with a loosely packed structure, whereas small RoG values indicate a protein molecule with a tightly packed structure. In the case of the MDM2 protein, the RoG values ranged from 1.20 nm to 1.35 nm (Figure 7a). An increase in RoG values was observed after a simulation time of 300 ns, indicating significant structural changes consistent with the RMSD profile findings. This result indicates that the simulation of the C4-MDM2 complex may not be stable beyond 300 ns. The RoG values of the protein backbone of the C4-Bcl2 complex ranged from 1.40 to 1.55 nm and remained stable throughout the simulation (Figure 7b). This finding supports the notion that the C4-Bcl2 complex has greater stability and compactness than the C4-MDM2 complex.
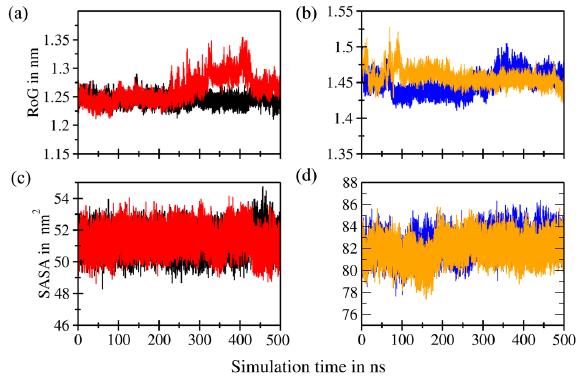
- Radius of gyration (RoG) superimposition of the (a) backbones of murine double minute 2 (MDM2) (black) and C4-MDM2 (red) and (b) backbones of B-cell lymphoma 2 (Bcl-2) (blue) and C4-Bcl-2 (orange). Solvent accessible surface area (SASA) superimposition of the (c) backbones of MDM2 (black) and C4-MDM2 (red) and (d) backbones of Bcl-2 (blue) and C4-Bcl-2 (orange).
Determining the SASA parameters is pivotal for understanding the structural alterations occurring in proteins during the binding process. The SASA values, quantified in nm2, provide insights into the flexibility of protein structures. An increase in SASA signifies the expansion of receptor macromolecules, whereas a decrease in SASA indicates greater compactness. In the case of the C4-MDM2 complex, the SASA values ranged from 45 to 54 nm2 (Figure 7c). By contrast, in the case of the C4-Bcl2 complex, the SASA values ranged from 78 to 86 nm2 (Figure 7d). Both were consistent during the simulation and reflected stability comparable to the apoprotein simulation.
Hydrogen bonds are pivotal in facilitating the formation of protein-ligand complexes, thereby influencing the stability and specificity of interactions. Figure 8(a) and 8(b) illustrate the bonding patterns observed in both complexes. The C4-Bcl-2 complex had a larger number of potential hydrogen bonds, i.e., a total of 4, than the C4-MDM2 complex, which exhibited only two potential hydrogen bonds. Specifically, in the C4-MDM2 complex, hydrogen bonds involving interactions with amino acids Tyr67 and Lys94 were captured at 167 ns (Figure 9a). The crucial amino acid His96 was weakly involved, primarily via van der Waals interactions. Conversely, in the C4-Bcl-2 complex, three potential hydrogen bonds were captured at 194 ns (Figure 9b), involving amino acids Leu137, Asn143, and Arg146, which are considered crucial for binding. The interactions involved the carbonyl moiety of the ligand and hydrogen atoms in the amino acid residues. Overall, the hydrogen bond analysis confirms that C4 exhibits a stronger binding affinity for Bcl-2 than MDM2 protein, highlighting its potential as a promising candidate for further investigation in anticancer drug development.
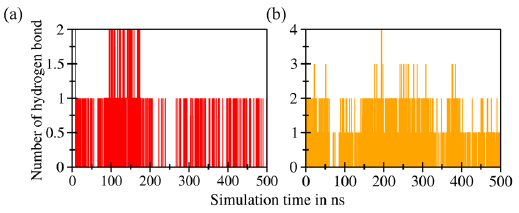
- Number of hydrogens detected in the complexes of (a) C4-MDM2 and (b) C4-Bcl-2.
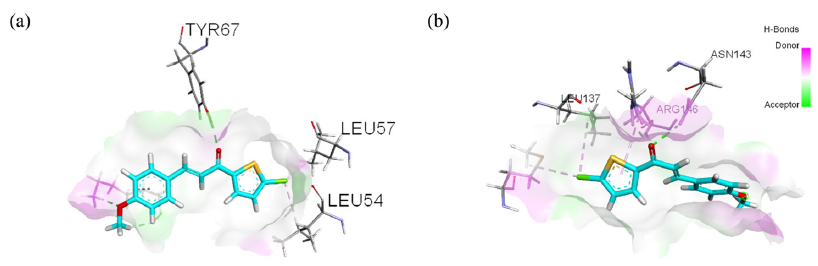
- Visualization of hydrogen bonds in (a) C4-MDM2 and (b) C4-Bcl-2 (green lines). Note: Leu: leucine, Tyr: tyrosine.
The stability of the protein structures was evaluated using the RAM plot, which visually represents the statistical distribution of amino acids. For the MDM2 protein (Figure 10a), 88.9% (72 amino acids) of the 81 residues were in the favored region (red area), indicating a favorable conformational arrangement. Moreover, 11.1% (9 amino acids) was detected in the additional allowed region (yellow area), indicating minor deviations from the ideal conformation. Similarly, for the Bcl-2 protein post-MD simulation, the RAM plot revealed that 85.8% (115 amino acids) of the 134 residues were situated in the favored region (Figure 10b). Furthermore, 12.7% (17 amino acids) was observed in the additional allowed region, signifying acceptable structural deviations. Only 1.5% (2 amino acids) were in the generously allowed region, indicating minimal structural strain. None of the amino acid residues in each protein were in the disallowed area (white area) after the MD simulation, indicating the overall structural stability of the proteins under investigation.
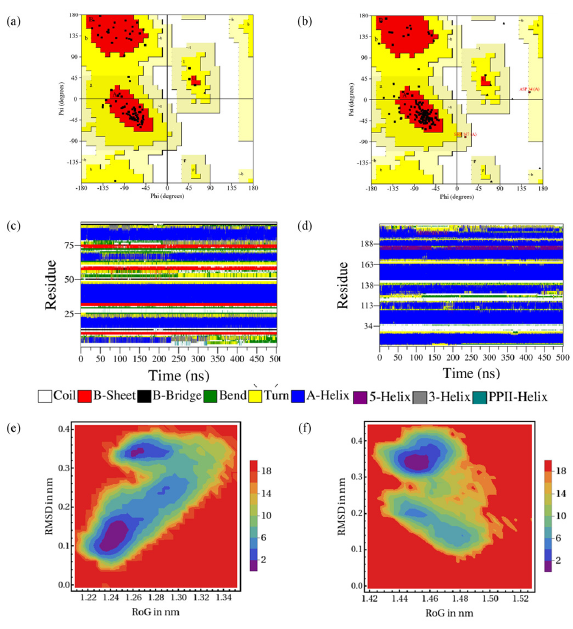
- Ramachandran plot (RAM) plots of (a) MDM2 and (b) Bcl-2, and DSSP plots of (c) MDM2 and (d) Bcl-2, and FEL plots of RMSD vs RoG after simulation of (e) MDM2 and (f) Bcl-2 complexed with C4 after MD simulation.
Supporting the RMSD and RMSF profile findings, Figure 10(c) and 10(d) illustrate the DSSP plots of both MDM2 and Bcl-2 proteins after MD simulation using C4. The increase in RMSD value after 300 ns is confirmed to be influenced by the loss of helical structure around amino acid residues 18–25, detected by a change in color from blue to yellow in the DSSP spectrum. As shown in Figure S2a, the helical structure loosens into a coil structure. The crucial amino acid residue His96 also shows slight changes, as is observed at 343 ns, when the helical structure begins to loosen. These two changes are predicted to contribute to the fluctuation of the RMSF profile. In the DSSP profile of the C4-Bcl-2 complex, the essential binding area is dominated by α-helical loops. In the binding pocket of Arg146, the structure remains consistent throughout the simulation, indicating no structural change around this area. However, slight changes were observed in the amino acid Tyr108 area, and helical loop opening was observed in amino acid residues above 200 (Figure S2b).
Using the RoG and RMSD values extracted from the trajectories, we constructed a contour map of the FEL, enabling us to pinpoint a dominant cluster encompassing all complexes. In Figure 10(e) and 10(f), the blue area denotes the minimum free binding energy state, whereas the cyan, green, and yellow areas denote the metastable conformational states. From each complex, a single frame was selected from the cluster with the lowest energy level, ensuring acceptable structural deviations. The C4-MDM2 complex has the lowest free binding energy structures (∆G = 0) with confirmation located around RoG = 1.24519 nm and RMSD = 0.12813 nm. This confirmation was captured from the frames at 124.64 ns, where the RMSD is at a local minimum energy, indicating high consistency with its starting point. However, after 300 ns, the increasing RMSD indicates structural changes, including the opening of helices in the crucial amino acid residue His96 area and other residues in the tail side from 15 to 18. This finding indicates that, after 300 ns, the introduction of ligand C4 compromises its stability.
Meanwhile, the C4-Bcl2 complex exhibits the lowest free binding energy structure (∆G = 0) with conformation at RoG = 1.45387 nm and RMSD = 0.32822 nm. The local minima with RMSD = 0.3822 nm were captured at 240.75 ns, indicating structural changes around amino acid residues 200–211, which are not within the crucial binding pocket area. This change starts occurring at 90–100 ns, consistent with the RMSD profile. Meanwhile, the rest of the protein structure remains stable, including the binding pocket area, indicating that the introduction of the ligand did not significantly alter the protein structure. This stability is observed until the end of simulation. All trajectories, including secondary structure analyses and FEL interpretations, confirmed the stability of the C4-Bcl2 complex compared with the C4-MDM2 complex.
3.6. MM-PBSA calculation
The MM-PBSA method, known for its advanced capabilities in calculating free binding energy, was employed to validate the results obtained from molecular docking. Using this methodology, we computed the free binding energy (∆Gbind) of the anticancer C4-MDM2 and C4-Bcl2 complexes. Then, the total energies of these complexes were computed, and the results are presented in Table 4. The calculation was taken from the last 50 ns of the entire simulation. The binding energy of the C4-MDM2 complex was determined to be −21.26 kcal/mol. This finding falls within the range of free-binding energies reported by Li et al. [77] for the p53-MDM2 complex (−28.93 kcal/mol) and another peptide inhibitor (−28.22 kcal/mol), as calculated using a similar MM-PBSA method. In comparison to that of the C4-MDM2 complex, the free binding energy of the C4-Bcl-2 complex was calculated to be −15.70 kcal/mol. This value is close to the reported free binding energy of a small molecule inhibitor, i.e., α-muurolene, investigated for its inhibitory action on Bcl-2, which was reported to be −10.33 kcal/mol [78]. The difference indicates that the binding site may favor the formation of complexes with the ligands.
| Complex | ∆ Eelea | ∆ EvdWa | ∆ Gnpa | ∆ GPBa | ∆ Gbinda |
|---|---|---|---|---|---|
| C4-MDM2 | −5.22 | −29.23 | −2.96 | 16.15 | −21.26 |
| C4-Bcl-2 | −6.12 | −27.45 | −2.99 | 20.87 | −15.70 |
3.7. DFT and MEP analyses
The DFT calculations were performed in the gas phase using the B3LYP method with a 6-31G basis set. These calculations provided crucial parameters for analyzing the chemical reactivity of the synthesized compounds, such as energy gap, electronegativity (χ), chemical hardness (η), and electrophilicity index (ω). Frontier molecular orbitals encompass the EHOMO and ELUMO, which are pivotal quantum chemical parameters governing the reactivity of molecules and are utilized to compute various chemical reactivity descriptors. A high difference (∆Egap) between the HOMO and LUMO energy values indicates low reactivity of the compound, resulting in higher stability. By computing the energy gap, C6 was determined to have the most substantial gap with a value of ∆Egap = 4.0129 eV, indicating its superior stability relative to all other compounds examined. Following that, C1, C7, and C4 were ranked in decreasing order of stability. Interestingly, the trend indicates that the incorporation of a methoxy substituent in the phenyl group decreases the energy gap. The addition of another chloro group to thiophene, as observed in C8 and C9, further decreases the energy gap, resulting in the lowest energy gap in these two compounds. This finding indicates that the additional chloro group in thiophene destabilizes the system and renders the compound more reactive. The electronegativity (χ) tendencies indicate that C6 and C7 exhibit the highest electron-attracting capabilities, whereas C5 exhibits the lowest electronegativity. The additional chemical reactivity descriptors are summarized in Table S3.
To validate the evidence regarding the inhibitory properties of the compound, the MEP/ESP contour plot provides insights into the size, shape, and identification of reactive sites based on the electrostatic potential of the molecule. Figure S3 depicts the molecular electronic potential of all proposed compounds. Across all examined compounds, the red regions encircling the oxygen of the α, β unsaturated system, signify areas with the most substantial negative charge, indicative of high electron density available for electrophilic attack. Interestingly, a subtle increase in electron density is noted at sites where chlorine is substituted in the thiophene ring, indicating an enhanced capacity for interaction with electrophiles because of the addition of chlorine. However, oxygen from the carbonyl group remains the central focus of the interaction. The addition of one methoxy group results in a negative electron density around the oxygen. However, no significant increase in these red areas using dimethoxy is detected, as observed in C5 and C9. This finding confirms that the best compound design involves one substituent in the phenyl moiety (refer to Figure 3).
3.8. ADMET estimation
Drug-likeness is a critical attribute for compounds to become effective therapeutic candidates. Two approaches were employed to assess the drug-likeness of the compounds, namely, Lipinski’s rule of five and Veber’s criteria. Lipinski’s rule of five is based on five fundamental principles, such as (i) MW < 500 g/mol, (ii) lipophilicity or partition coefficient (Log P) < 5, (ii) TPSA < 140 Å2, (iv) number of HBD < 5, and (v) number of HBA < 10. Molecules meeting at least three rules were anticipated to exhibit favorable absorption properties. According to Veber’s criteria, compounds possessing ≤10 rotatable bonds and a polar surface area of ≤140 Å2 are predicted to exhibit good oral bioavailability. This study used data from SwissADME predictions to generate information on several drug-likeness parameters.
Table S4 presents calculated molecular descriptors of all nine compounds. Remarkably, no violations were detected for either Lipinski’s rule of five or Veber’s criteria in any of the compounds. MW ranged from 248.73 to 343.22. However, the two comparison ligands, i.e., nutlin-3a and THIQ-PP, exhibited the maximal violation of Lipinski’s rule of five (>500). The number of HBD, number of HBA, TPSA, and Log P values all met the criteria. The Log P values ranged from 2.69 to 3.62. The observed changes in values indicate that the addition of a methoxy group reduces lipophilicity, making the compound more polar than the unsubstituted version (C1). The introduction of a chloro group (C6 and C7) increases lipophilicity. Based on this investigation, we can predict that the compounds may have better bioavailability than nutlin-3a and tetraisohydroquinoline phenyl pyrazole (THIQ-PP), which exhibit some violations and indicate poor bioavailability.
The evaluation of the ADMET parameters is a crucial step in drug development, ensuring the suitability of inhibitors for administration in biological systems. These assessments provide insights into how drugs are absorbed, distributed throughout the body, metabolized, and excreted and their potential for toxicity. To perform these evaluations, we utilized pkCSM, a comprehensive online tool known for its reliability in predicting ADMET properties. The results of these evaluations are summarized in Table S5, shedding light on various aspects of the pharmacokinetics and potential safety profiles of the compounds.
Initially, we evaluated GIA to assess the suitability of the compound for oral administration. The evaluation of the GIA thoroughly explains how a drug dissolves, traverses the gut/lumen interface, diffuses across membranes, and eventually gets eliminated from the bloodstream. Orally administered drugs primarily traverse gastrointestinal membranes via passive diffusion, a process influenced by the physicochemical properties of the molecules. Our compound exhibited a high GIA value (>90%), indicating that it will be absorbed into the bloodstream at a rate exceeding 90% from the intestinal tract.
The inadvertent penetration of the brain by drugs, especially those with CNS activity, can pose significant risks. Hence, the potential permeability of chemical compounds across the BBB needs to be evaluated. Our ligand exhibits high values for BBB and CNS permeability, ranging from 0.26 to 0.49, which are different from the two comparison ligands, with values of −1.59 and −0.92 for nutlin-3a and THIQ-PP, respectively. The addition of a methoxy group decreases these permeability values.
In drug metabolism, our compound is predicted to be a potential inhibitor of certain CYP isoenzymes, namely, CYP1A2, CYP2C19, and CYP3A4. Interestingly, some compounds with methoxy groups still exhibit inhibition activity toward CYP2D6, although none show such activity specifically for CYP2D6. The excretion parameters (total clearance) of our compound ranged from 0.05 to 0.42. Compounds C6 and C7 exhibit significantly lower total clearance than the other compounds, including the comparison ligands. As a result, compounds C6 and C7 are expected to necessitate shorter dosing intervals. In addition, we assessed the potential hepatotoxic effects to evaluate liver damage upon administration. Encouragingly, none of our compounds exhibited potency for this effect, indicating their overall safety profile.
4. Conclusions
Our study assessed nine chalcone compounds, synthesized from 2-acetyl-5-chlorothiophene and 2-acetyl-4,5-dichlorothiophene, for their anticancer potential. These compounds exhibited significant toxicity against cancer cells, particularly the colorectal WiDr colorectal cancer cell line. Compounds C4 and C6 had the lowest IC50 values of 0.77 and 0.45 µg/mL, respectively. Remarkably, compound C4 exhibited better selectivity toward normal cells. Our mechanistic predictions indicated that both C4 and C6 bind moderately to both MDM2 and Bcl-2 proteins with binding energies ranging from −6.2 kcal/mol to −6.6 kcal/mol and interact similarly with their comparable native ligands. MD simulations of C4 with both MDM2 and Bcl-2 proteins indicated a more stable complex formation with Bcl-2 within 500 ns simulation time and calculated free binding energy of −15.70 kcal/mol via MM-PBSA. DFT studies identified compound C6 as the most promising candidate, exhibiting the highest ∆Egap and shedding light on the influence of functional group design through the MEP map. None of the derivatives breached Lipinski’s rule of five and Veber’s criteria, affirming their suitability as active oral drugs. This research emphasizes the potential of these compounds as prospective anticancer agents targeting the p53-targeted pathway, advocating for further comprehensive evaluations of the recommended experiment.
Acknowledgment
We express our special appreciation to Universitas Gadjah Mada for granting access to the Wolfram Mathematica license and to the Austria–Indonesia Centre (AIC) for Computational Chemistry for providing Gaussian 09 licenses. The Indonesian Ministry of Education, Culture, Research, and Technology provided financial support for this work through the Pendidikan Magister Menuju Doktor Untuk Sarjana Unggul scholarship (contract number 2201/UN1/DITLIT/Dit-Lit/PT.01.03/2023) and the Enhancing International Publication Scholarship/PKPI (contract number 106.8/E4.4/KU/2023) awarded to author. We also extend our heartfelt thanks to the School of Chemistry, University of New South Wales for their invaluable collaboration and support for this work.
CRediT authorship contribution statement
Fia Fathiana Wulan: Writing - original draft, methodology, investigation, formal analysis. Niko Prasetyo: Writing - review and editing, supervision. Endang Astuti: Writing - review and editing, supervision. Naresh Kumar: Writing - review and editing, supervision. Tutik Dwi Wahyuningsih: Writing - review and editing, conceptualization, supervision, funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Appendix A. Supplementary material
Supplementary material to this article can be found online at https://dx.doi.org/10.25259/AJC_31_2024
References
- WHO report on cancer: Setting priorities, investing wisely and providing care for all.
- Cancer statistics for the year 2020: An overview. International Journal of Cancer. 2021;149:778-789. https://doi.org/10.1002/ijc.33588
- [CrossRef] [Google Scholar]
- Cancer statistics, 2023. CA. Cancer Journal of Clinical. 2023;73:17-48. https://doi.org/10.3322/caac.21763
- [Google Scholar]
- Chalcones as promising antitumor agents by targeting the p53 pathway: An overview and new insights in drug-likeness. Molecules. 2021;26 https://doi.org/10.3390/molecules26123737
- [CrossRef] [Google Scholar]
- Targeting MDM2 for the development of a new cancer therapy: progress and challenges. Medicinal Chemistry Research. 2023;32:1334-1344. https://doi.org/10.1007/s00044-023-03102-1
- [CrossRef] [Google Scholar]
- Therapeutic development and current uses of BCL-2 inhibition. Hematology (United States). 2020;20:1-9. https://doi.org/10.1182/HEMATOLOGY.2020000154
- [CrossRef] [Google Scholar]
- Dual targeting of MDM2 and BCL2 as a therapeutic strategy in neuroblastoma. Oncotarget. 2017;8:57047-57057. https://doi.org/10.18632/oncotarget.18982
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MDM2 and BCL-2: to p53 or not to p53? Blood. 2023;141:1237-1238. https://doi.org/10.1182/blood.2022018739
- [CrossRef] [PubMed] [Google Scholar]
- Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduction and Targeted Therapy. 2023;8:1-35. https://doi.org/10.1038/s41392-023-01347-1
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Anti-cancer chalcones: Structural and molecular target perspectives. European Journal of Medicinal Chemistry. 2015;98:69-114. https://doi.org/10.1016/j.ejmech.2015.05.004
- [CrossRef] [PubMed] [Google Scholar]
- Exploring pharmacological significance of chalcone scaffold: A review. Current Medicinal Chemistry. 2012;19:209-225. https://doi.org/10.2174/092986712803414132
- [CrossRef] [PubMed] [Google Scholar]
- Recent developments in biological activities of chalcones: A mini review. European Journal of Medicinal Chemistry. 2014;85:758-777. https://doi.org/10.1016/j.ejmech.2014.08.033
- [CrossRef] [PubMed] [Google Scholar]
- New insights into binding of natural chalcones to Bcl-2, Bcl-xL and Mcl-1 anti-apoptotic proteins. Journal of Molecular Structure 2021:1241. https://doi.org/10.1016/j.molstruc.2021.130700
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arabian Journal of Chemistry. 2019;12:4150-4161. https://doi.org/10.1016/j.arabjc.2016.04.015
- [Google Scholar]
- Licochalcone C induces apoptosis via B-cell lymphoma 2 family proteins in T24 cells. Molecular Medicine Reports. 2015;12:7623-7628. https://doi.org/10.3892/mmr.2015.4346
- [CrossRef] [PubMed] [Google Scholar]
- Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry. 2001;40:336-344. https://doi.org/10.1021/bi000930v
- [CrossRef] [PubMed] [Google Scholar]
- A potent EGFR inhibitor, N-Phenyl pyrazoline derivative suppresses aggressiveness and cancer stem cell-like phenotype of cervical cancer cells. Drug Design, Development and Therapy. 2022;16:2325-2339. https://doi.org/10.2147/DDDT.S350913
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Efficient synthesis of chloro chalcones under ultrasound irradiation, their anticancer activities and molecular docking studies. Rasayan Journal of Chemistry. 2019a;12:502-510. https://doi.org/10.31788/RJC.2019.1225020
- [Google Scholar]
- Heteroaryl chalcones: Mini review about their therapeutic voyage. Biomedicine & Preventive Nutrition. 2014;4:451-458. https://doi.org/10.1016/j.bionut.2014.04.003
- [CrossRef] [PubMed] [Google Scholar]
- Synopsis of some recent tactical application of bioisosteres in drug design. Journal of Medicinal Chemistry. 2011;54:2529-2591. https://doi.org/10.1021/jm1013693
- [CrossRef] [PubMed] [Google Scholar]
- Recent strategies in the synthesis of thiophene derivatives: highlights from the 2012–2020 literature. Molecular Diversity. 2021;25:2571-2604. https://doi.org/10.1007/s11030-020-10128-9
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Synthesis, fungal biotransformation, and evaluation of the antimicrobial potential of chalcones with a chlorine atom. Science Reports. 2024;14:1-22. https://doi.org/10.1038/s41598-024-65054-9
- [Google Scholar]
- Towards targeting EGFR and COX-2 inhibitors: comprehensive computational studies on the role of chlorine group in novel thienyl-pyrazoline derivative. Journal of Biomolecular Structure and Dynamics. 2024;42:9857-9872. https://doi.org/10.1080/07391102.2023.2252915
- [CrossRef] [PubMed] [Google Scholar]
- Gaussian 09. Gaussian, Inc; 2016.
- UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computer Chemistry. 2004;25:1605-1612. https://doi.org/10.1002/jcc.20084
- [Google Scholar]
- An approach to computing electrostatic charges for molecules. Journal of Computer Chemistry. 1984;5:129-145. https://doi.org/10.1002/jcc.540050204
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computer Chemistry. 2009;31:NA. https://doi.org/10.1002/jcc.21334
- [Google Scholar]
- BIOVIA, D.S., 2020. Discovery Studio 2017. Dassault Systemes.
- Gromacs: A parallel computer for molecular dynamics simulations – ScienceOpen. In: DeGroot R.A., Nadrchal J., eds. Physics Computing ‘92. Vol 1993. Singapore: World Scientific Publishing; 1993. p. :252-256.
- [Google Scholar]
- Comparison of multiple amber force fields and development of improved protein backbone parameters viktor. Proteins: Structure, Function, and Bioinformatics. 2006;65:712-725. https://doi.org/10.1002/prot.21123
- [Google Scholar]
- Assessment of available AMBER force fields to model DNA-Ligand Interactions. Biointerface Research in Applied Chemistry. 2023;13 https://doi.org/10.33263/BRIAC132.156
- [Google Scholar]
- ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes. 2012;5:1-8. https://doi.org/10.1186/1756-0500-5-367
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79:926-935. https://doi.org/10.1063/1.445869
- [CrossRef] [Google Scholar]
- Effects of water models on binding affinity: Evidence from all-atom simulation of binding of tamiflu to A/H5N1 neuraminidase. The Scientific World Journal 2014:2014. https://doi.org/10.1155/2014/536084
- [CrossRef] [Google Scholar]
- Particle mesh Ewald: An N×log(N) method for Ewald sums in large systems. Journal of Chemical Physics. 1993;98:10089-10092. https://doi.org/10.1063/1.464397
- [Google Scholar]
- Canonical sampling through velocity rescaling. Journal of Chemical Physics 2007:126. https://doi.org/10.1063/1.2408420
- [CrossRef] [Google Scholar]
- Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics. 1981;52:7182-7190. https://doi.org/10.1063/1.328693
- [CrossRef] [Google Scholar]
- van der Waals volumes and Radii. Journal of Physical and Chemical. 1964;68:441-451. https://doi.org/10.1021/j100785a001
- [CrossRef] [Google Scholar]
- The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. Journal of Computer Chemistry. 1995;16:273-284. https://doi.org/10.1002/jcc.540160303
- [CrossRef] [Google Scholar]
- XMGRACE. Center for coastal and land-margin research. Oregon Graduate Institute of Science and Technology; 2005.
- Mathematica. Wolfram Research Inc; 2023.
- PDBsum: Structural summaries of PDB entries. Protein Science. 2018;27:129-134. https://doi.org/10.1002/pro.3289
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577-2637. https://doi.org/10.1002/bip.360221211
- [CrossRef] [PubMed] [Google Scholar]
- Gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. Journal of Chemical Theory and Computation. 2021;17:6281-6291. https://doi.org/10.1021/acs.jctc.1c00645
- [CrossRef] [PubMed] [Google Scholar]
- MMPBSA.py: An efficient program for end-state free energy calculations. Journal of Chemical Theory and Computation. 2012;8:3314-3321. https://doi.org/10.1021/ct300418h
- [CrossRef] [PubMed] [Google Scholar]
- Quantum chemical and monte carlo simulation studies on inhibition performance of caffeine and its derivatives against corrosion of copper. Coatings. 2020;10:1-17. https://doi.org/10.3390/coatings10111086
- [CrossRef] [Google Scholar]
- Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica. 1934;1:104-113. https://doi.org/10.1016/S0031-8914(34)90011-2
- [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. Journal of Computer Chemistry. 2012;33:580-592. https://doi.org/10.1002/jcc.22885
- [CrossRef] [Google Scholar]
- VMD: Visual molecular dynamics. Journal of Molecular Graphics. 1996;14:33-38. https://doi.org/10.1016/0263-7855(96)00018-5
- [CrossRef] [PubMed] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Science Reports. 2017;7:42717. https://doi.org/10.1038/srep42717
- [CrossRef] [Google Scholar]
- pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of Medicinal Chemistry. 2015;58:4066-4072. https://doi.org/10.1021/acs.jmedchem.5b00104
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technology. 2004;1:337-341. https://doi.org/10.1016/j.ddtec.2004.11.007
- [CrossRef] [Google Scholar]
- Molecular properties that influence the oral bioavailability of drug candidates. Journal of Medicinal Chemistry. 2002;45:2615-2623. https://doi.org/10.1021/jm020017n
- [CrossRef] [PubMed] [Google Scholar]
- Bioisosterism: A useful strategy for molecular modification and drug design. Current Medicinal Chemistry. 2012;12:23-49. https://dx.doi.org/10.2174/0929867053363540
- [Google Scholar]
- Synthesis thiophene-based of chalcone and n-phenylpyrazoline derivatives and cytotoxic activity against cancer cell lines. Universitas Gadjah Mada; 2022.
- Design, facile synthesis and characterization of dichloro substituted chalcones and dihydropyrazole derivatives for their antifungal, antitubercular and antiproliferative activities. Molecules. 2020;25:3188-3203. https://doi.org/10.3390/molecules25143188
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Synthesis and biologic activities of some novel heterocyclic chalcone derivatives. Medicinal Chemistry Research. 2013;22:3969-3983. https://doi.org/10.1007/s00044-012-0401-7
- [CrossRef] [Google Scholar]
- Synthesis, cytotoxicity evaluation and molecular docking study of n-phenylpyrazoline derivatives. Indonesian Journal of Chemistry. 2019b;19:1081-1090. https://doi.org/10.22146/ijc.45777
- [Google Scholar]
- Thiophene-based N-phenyl pyrazolines: Synthesis, anticancer activity, molecular docking and ADME study. ,. 2024;14:63-71. https://doi.org/10.7324/JAPS.2024.146832
- [CrossRef] [Google Scholar]
- Chalcone derivatives targeting mitosis: Synthesis, evaluation of antitumor activity and lipophilicity. European Journal of Medicinal Chemistry. 2019;184:111752. https://doi.org/10.1016/j.ejmech.2019.111752
- [CrossRef] [PubMed] [Google Scholar]
- Novel pyrazolines: Synthesis and evaluation of their derivatives with anticancer and anti-inflammatory activities. International Journal of Pharmacy and Pharmaceutical Sciences. 2016;8:247-254. https://www.innovareacademics.in/journals/index.php/ijpps/article/view/7707
- [Google Scholar]
- Effect of “magic chlorine” in drug discovery: an in silico approach. RSC Advances. 2023;13:34922-34934. https://doi.org/10.1039/d3ra06638j
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Integration of fragment screening and library design. Drug Discovery Today. 2007;12:1032-1039. https://doi.org/10.1016/j.drudis.2007.08.005
- [CrossRef] [PubMed] [Google Scholar]
- Isolation and structural characterization of dihydrobenzofuran congeners of licochalcone A. Fitoterapia. 2017;121:6-15. https://doi.org/10.1016/j.fitote.2017.06.017
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation. Science. 1996;274:948-953. https://doi.org/10.1126/science.274.5289.948
- [CrossRef] [PubMed] [Google Scholar]
- When the guardian sleeps: Reactivation of the p53 pathway in cancer. Mutation Research - Reviews in Mutation Research. 2017;773:1-13. https://doi.org/10.1016/j.mrrev.2017.02.003
- [CrossRef] [PubMed] [Google Scholar]
- Identification of FDA-approved drugs that computationally bind to MDM2. Chemical Biology & Drug Design. 2012;80:631-637. https://doi.org/10.1111/j.1747-0285.2012.01428.x
- [PubMed] [Google Scholar]
- Structural biology of the Bcl-2 family of proteins. Biochimica et Biophysica Acta - Molecular Cell Research. 2004;1644:83-94. https://doi.org/10.1016/j.bbamcr.2003.08.012
- [CrossRef] [Google Scholar]
- Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nature Communication. 2019;10 https://doi.org/10.1038/s41467-019-10363-1
- [CrossRef] [Google Scholar]
- Establishing drug discovery and identification of hit series for the anti-apoptotic proteins, Bcl-2 and Mcl-1. ACS Omega. 2019;4:8892-8906. https://doi.org/10.1021/acsomega.9b00611
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Design, synthesis, anticancer activity, and solid lipid nanoparticle formulation of indole-and benzimidazole-based compounds as pro-apoptotic agents targeting bcl-2 protein. Pharmaceuticals. 2021;14:1-37. https://doi.org/10.3390/ph14020113
- [Google Scholar]
- Virtual screening and free energy estimation for identifying Mycobacterium tuberculosis flavoenzyme DprE1 inhibitors. Journal of Molecular Graphics and Modelling. 2021;102 https://doi.org/10.1016/j.jmgm.2020.107770
- [CrossRef] [Google Scholar]
- Benzothiazole derivatives as p53-MDM2 inhibitors: in-silico design, ADMET predictions, molecular docking, MM-GBSA Assay, MD simulations studies. Journal of Biomolecular Structure and Dynamics. 2023;0:1-12. https://doi.org/10.1080/07391102.2023.2294836
- [Google Scholar]
- Molecular docking and molecular dynamics studies reveal the anticancer potential of medicinal-plant-derived lignans as MDM2-P53 interaction inhibitors. Molecules. 2023;28 https://doi.org/10.3390/molecules28186665
- [CrossRef] [Google Scholar]
- In Silico mining of natural products atlas (NPAtlas) database for identifying effective Bcl-2 inhibitors: Molecular docking, molecular dynamics, and pharmacokinetics characteristics. Molecules. 2023;28 https://doi.org/10.3390/molecules28020783
- [CrossRef] [Google Scholar]
- Discovery of a new Bcl-2 inhibitor through synthesis, anticancer activity, docking and MD simulations. Journal of Biomolecular Structure and Dynamics. 2023;0:1-10. https://doi.org/10.1080/07391102.2023.2218934
- [Google Scholar]
- Insight into the binding mechanism of p53/pDIQ-MDMX/MDM2 with the interaction entropy method. Front. Chem.. 2019;7:1-13. https://doi.org/10.3389/fchem.2019.00033
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular docking and dynamics simulation study of bioactive compounds from Ficus carica L. With important anticancer drug targets. PLoS One. 2021;16:1-16. https://doi.org/10.1371/journal.pone.0254035
- [CrossRef] [Google Scholar]




