

Translate this page into:
Molecular Docking approach on the effect of Site- Selective and Site-Specific Drugs on the Molecular Interactions of Human Serum Albumin (HSA) -Acridinedione dye complex
⁎Corresponding authors. govindasamy420700@gmail.com (Mani Govindasamy), Kumaranwau@rediffmail.com (Kumaran Rajendran)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Molecular Docking (Mol.dock) of resorcinol based acridinedione dyes (ADR1 and ADR2) with a globular protein, Human Serum Albumin (HSA) were carried out. Docking studies reveal that ADR2 dye binding with HSA is energetically more stable and feasible than ADR1 dye. ADR1 dye predominantly resides in site I and III of HSA rather than binding site II wherein, ADR1 dye acts as hydrogen bonding (HB) acceptor through its carbonyl oxygen. On the contrary, ADR2 dye resides in all the binding sites of HSA such that the dye acts as the HB donor through the N—H hydrogen atom and the carbonyl oxygen of the amino acid acts as the HB acceptor. The stability of dye-protein complex in the presence of several non-steroidal anti-inflammatory drugs (NSAIDs) was carried out by employing specific site selective drugs (Sudlow binding site drugs). The energetics and the bimolecular interactions of various drugs with ADR1-HSA and ADR2-HSA were generated to ascertain the influence of drug and its governance on the binding affinity of dye-protein complex. Sudlow site I binding drugs were effective in decreasing the energetics of ADR1 dye-HSA complex whereas site II binding drugs predominantly decreases the affinity of ADR2 dye with HSA. However, the dyes efficiently displaces the site specific drugs from their specific binding sites of HSA which was not observed in the case of drugs on the displacing ability over dyes situated in different domains of protein. Mol.dock studies are employed as an authentic, reliable and most effective tool to ascertain the binding stability of host–guest complex as well as to ascertain the most probable location of several competing ligands in various domains of HSA.
Keywords
Human Serum Albumin
Molecular Docking
Acridinedione dyes
Binding energy: site specific drugs

1 Introduction
Albumins are widely categorized as a large family of globular proteins that are commonly found in blood plasma, egg white, milk, and plants (Kalra et al., 2013; El-Fakharany et al., 2013; Abeyrathne et al., 2013; Zilic et al., 2011). Among the proteins, Serum albumin (SA) is the most abundant protein in the blood plasma of all vertebrates (Peters, 1995; Fanali et al., 2012) and are widely explored for their physiological and probing their denaturation properties. The most widely studied albumins in the context of dye-protein, drug-protein, metal-protein and ligand–protein interactions are Bovine Serum Albumin (BSA) followed by Human Serum Albumin (HSA). HSA is a globular, heart-shaped protein with a repeating series of six helical subdomains (He and Carter, 1992; Sugio et al., 1999). HSA is present in both extravascular and intravascular spaces (Merlot et al., 2014; Evans, 2002) that provides a larger space of binding towards guest molecules. Albumin performs a variety of essential functions such that it regulates the osmotic pressure and pH of the blood (Peters, 1995). It also binds and facilitate the transportation of various bioactive molecules, including proteins, peptides, fatty acids, hormones, amino acids, drugs, nutrients, and metal ions (Fanali et al., 2012; Ghuman et al., 2005).
HSA and BSA share 76 % identity (Huang et al., 2004), while, overall SA obtained from various sources share more than 62 % sequence identity (Majorek et al., 2012). This structural similarity is one of striking feature of HSA which can be substituted with BSA or albumins derived from other sources in cell culture practices (Francis, 2010). Further, there is a remarkable degree of sequence and structural similarity as well as surface charge distribution between HSA from BSA(Huang et al., 2004; Bujacz, 2012; Ketrat et al., 2020). At a sequence level, molecular dynamics analysis of all serum albumins indicates that motion of domains I and III play a key role in defining the properties of the albumins (Ketrat et al., 2020; Mishra and Heath, 2021).
HSA has a single polypeptide chain consisting of 583 amino acids with molecular weight of 66.5 kDa. HSA is an alpha helical globular protein (Carter et al., 1989)and consist of three separate functional domains I, II, and III (He and Carter, 1993) such that the overall structure is predominantly hydrophobic in nature. Additionally, the binding sites are restricted to the subdomains IA, IB, IIA, IIB, IIIA, and IIIB, each of which has six helices that comprise a hydrophobic amino acid residue. (Sudlow et al., 1975; Sudlow et al., 1976)in which various drugs and fluorescent probes bind. The domains vary on the length of the amino acid chain and also on the composition of the amino acid (polar and non-polar). The combination of several bimolecular interactions presumably coexists with variation in extent of stability in several protein molecules which is a key factor on the denaturation process involving solutes (Kumaran and Ramamurthy, 2011; Kumaran and Ramamurthy, 2006). Domain I is made up of 195 amino acids, from ASP to LYS, while domain II is made up of 187 amino acids, from GLN196 to GLU383. Additionally, domain III is made up of the amino acid sequence from PRO384 to LEU583 (Meloun et al., 1975; Wang et al., 2015). The sites IIA, IIB, IIIA, and IIIB, which various guest molecules attach to, are the most preferred locations for HB, hydrophobic contacts, and other weak forces of attraction in the domains with the amino acid sequence 197–583.
Interestingly, in the context of medicinal chemistry and drug absorption, ligands binding with protein does not favour site III and predominantly prefer site I and II. Two important binding sites of HSA are Sudlow’s sites namely Site I and Site II which is of significance in several studies involving photophysical methods that employ fluorescence tools with denaturing agents. Even though these domains have similar structure, each domain exhibit different ligand binding affinities and functions. Sudlow binding site I have a preferential binding affinity for bulky heterocyclic compounds such as azapropazone (AZA), phenylbutazone (PBZ) and warfarin (WAR) which are well established NSAID in pharmaceutical chemistry. Sudlow site II preferentially binds with aromatic compounds such as ibuprofen (IBU), which is an excellent antipyretic agent (Ghuman et al., 2005; Deng et al., 2022).
The important function of HSA is to regulate the colloid osmotic pressure and other physiological functions are binding with wide variety of endo or exogeneous substance, and also facilitate transport of anti-oxidant and anti-inflammatory actions (Evans, 2002). HSA is found in nearly all mammals (Wang et al., 2015; Rabbani et al., 2017)and is the most abundant carrier protein in human plasma (Rabbani et al., 2017). Further, it also supports the enzymatic activities related to HSA (Wang et al., 2015). New molecular insight for determination of stability of complexes has generated a wider interest on the binding of ligands, drugs and probes.
In our present study we have employed two competing ligands namely a fluorophore and drug with HSA as similar to that of BSA. The fluorophore is an intramolecular charge transfer (ICT) based resorcinol derived acridinedione dyes namely ADR1 and ADR2 (Fig. 1), which varies in the 9th position of the basic dye structure.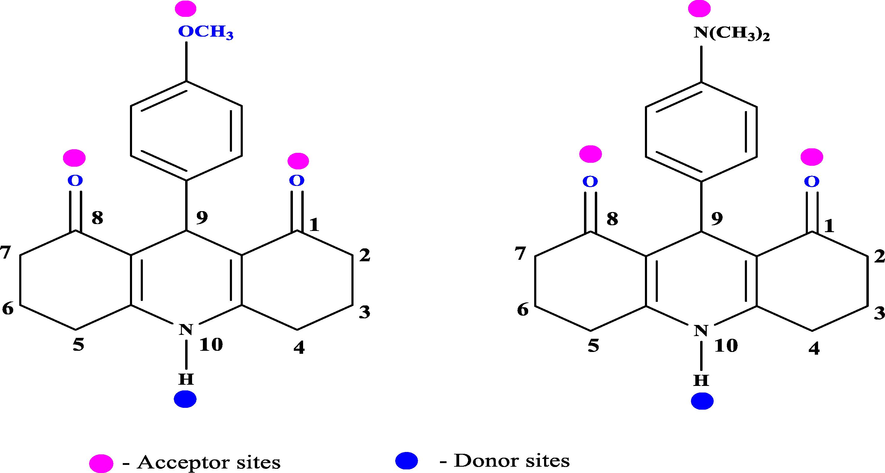
Structure of ADR1 and ADR 2 dyes representing the HB donor and acceptor sites.
Our reports on the photophysical studies of ADR dyes with BSA provides the illustration of the dye molecules in various microheterogenous environment of the protein molecule, the exact location and the preferred binding domains could not be authenticated in the aqueous phase (Kumaran and Ramamurthy, 2010; Kumaran et al., 2015). In order to explore in depth regarding the molecular interactions and the binding energies of dye-protein complex in the absence and presence of drugs, we employed an efficient theoretical approach through Mol.dock techniques. In the case of BSA we authenticated that ADR1 dye stabilizes the protein more efficiently than ADR2 dye and on the addition of site selective drugs to the dye-protein complex ADR1 dye effectively displaces site I drugs from its active site whereas, ADR2 dye displaces site II drugs from its binding sites. Herein, we closely examine the binding efficiency and affinity of similar protein molecule homologs to BSA with a PET and non-PET dye in the presence specific Sudlow binding drugs. (warfarin(WAR), Azapropazone(AZA), Phenylbutazone(PBZ), Oxyphenylbutazone(OPBZ), Indomethacin(IMET), 3-carboxyl-4-methyl-5-propyl-2-furanpropanic acid(CMPF), Ibuprofen(IBU), Diflusinal(DIFLU), Diazepam(DIAZ), Flufenamic acid (FA) and Indoxyl sulphate (INSL)).
The HB donor and acceptor sites of these drugs are provided in the supporting information figure S1 and the structure of protein used for docking purpose is provided in supporting information figure S2.
2 Molecular docking (Mol.dock) techniques
i) ADR1, ADR2 dye and the structures of drugs were drawn and optimised using Chemsketch and saved in mdl mol format and converted to pdb format using open babel molecular converter program. The SMILES format was generated using Chemsketch and their properties were calculated using Molinspiration tool (Lipinski et al., 1997).
ii) Mol.dock studies of ADR dyes with HSA.
The protein databank (PDB) (https://www.rcsb.org/pdb, PDB ID: 6HSC, A Chain), also employed in our earlier experiments with the ICT based dye DDP (Tamaraiselvan et al., 2021). This structure was chosen based on the resolution of the crystal structure. The methodology (Daina et al., 2017; Ruth Huey et al., 2007; Biovia Discovery Studio Visualizer BIOVIA, 2019) was followed as carried out for BSA-dye-drug (Seba Merin Vinod, n.d.) system and is provided in the supporting information section (SC).
3 Results and Discussion
3.1 Binding of ADR dyes with HSA
The energetics related to the docking of ADR1 and ADR2 dye with HSA resulting in various conformers is provided in tables 1 and 2 respectively. The free energy (ΔG) obtained due to complex formation between dye and protein is categorized based on the thermodynamic parameters of which the free energy is referred as the B.E in docking studies. Apart from B.E, there are several other parameters involved in stabilization of the complex. They are intermolecular, electrostatic and torsional energy that attribute to the formation and stabilization of the complex. Further, the energies attributed to several molecular interactions like van der Waals’s force of interaction, desolvation energy and HB also contribute for the stabilization of the dye-protein complex. The ten different conformers generated for ADR1 with HSA are coined as ADR1HSA1-10 respectively. Based on the –ΔG values, the conformers are arranged in the decreasing order of stability accounting for the inhibitory constant values and parameters involving ligand efficiency. As carried out for ADR1 dye, the conformers for ADR2 with HSA are labeled as ADR2HSA1-10 and are arranged based on their binding energies (ΔG). The larger the negative value of B.E/ΔG indicates the ease of formation and stability of the complex. #Ligand efficiency value is of the range −0.25 ± 0.4 and Tortional energy value is 0.6. #Ligand efficiency value is of the range −0.18 ± 0.6 and Tortional energy value is 0.6.
Conformation
Binding Energy (B.E)
Inhibitory constant, Ki (μm)
Intermolecular energy
vdW + H bond + desolv Energy
Electrostatic energy
Total internal Unbound Energy
ADR1HSA1
−7.2
7.2
−7.61
−7.31
−0.31
−0.95
ADR1HSA2
−6.69
12.53
−7.28
−6.96
−0.33
−1.01
ADR1HSA3
−6.49
17.43
−7.09
−6.97
0.12
−0.96
ADR1HSA4
−6.39
20.55
−6.99
−6.89
−0.1
−1.00
ADR1HSA5
−5.68
68.43
−6.28
−6.42
0.15
−1.02
ADR1HSA6
−5.65
72.24
−6.25
−5.96
−0.28
−1.01
ADR1HSA7
−5.5
92.69
−6.1
−6.03
−0.07
−0.75
ADR1HSA8
−5.47
98.07
−6.07
−5.89
−0.17
−0.98
ADR1HSA9
−5.21
150.71
−5.81
−5.82
0.01
−0.95
ADR1HSA10
−5.21
152.81
−5.8
−5.77
−0.03
−1.01
Conformation
Binding Energy (B.E)
Inhibitory constant, Ki (μm)
Intermolecular energy
vdW + H bond + desolv Energy
Electrostatic energy
Total internal Unbound Energy
ADR2HSA1
−7.87
1.7
−8.47
−8.69
−0.22
−1.06
ADR2HSA2
−6.75
11.32
−7.34
−7.1
−0.25
−1.06
ADR2HSA3
−6.66
13.07
−7.26
−7.13
0.13
−1.03
ADR2HSA4
−6.57
15.28
−7.17
−7.0
−0.17
−1.02
ADR2HSA5
−6.52
16.65
−7.12
−6.96
0.15
−1.06
ADR2HSA6
−6.49
17.48
−7.09
−6.93
−0.15
−1.06
ADR2HSA7
−6.35
22.21
−6.95
−6.91
−0.04
−1.03
ADR2HSA8
−5.33
123.43
−5.93
−5.96
−0.03
−1.04
ADR2HSA9
−4.77
318.72
−5.37
−5.16
0.2
−1.05
ADR2HSA10
−4.4
591.89
−5.0
−4.82
−0.18
−1.04
It is well known that several molecular interactions exist between dye and protein molecule even in the absence of water molecules (Tamaraiselvan et al., 2021; Tamaraiselvan et al., 2022; Vinod et al., 2022). The exclusion of water molecule in our study paves way for the exact location of the dye in various sub domains of protein and the molecular interaction site in which the amino acids that are involved in bonding (HB, hydrophobic and unfavorable interactions). In Mol.dock studies, the larger negative value of ligand efficiency is ascertained to greater stability of the complex formation and a positive value of the conformers correspond to less stable complex formation. A very high inhibitory constant existing between the ligand and the guest decreases the docking efficiency. As a result of these parameters the most and least favoured conformers are accounted in our studies which play a significant role on their binding sites and domains in the protein molecule.
Docking of ADR1 dye with HSA resulted in the existence of nine unique conformers and they were ascertained based on their binding sites and domains in HSA. Further, based on the energetics associated parameters, the ligand efficiency of all the ten conformers were found to be in the range of −0.25 ± 0.4. The inhibitory constant value of ADR1HSA1 conformer was found to be the lowest compared to that of all other conformers computed. The variation in the ΔG value of the most stable conformer (ADR1HSA1) with that of the least stable conformers (ADR1HSA8-10) were greater than 2 kcal/mol. Mol.dock studies clearly visualizes that there exist several conformers that substantially differ on energetics although all conformers coexist with difference in the binding site and molecular interaction. The 2D structures of the unique conformers of ADR1HSA are provided in Fig. 2 and their corresponding 3D structures of these conformers are provided in supporting information figure S4. Further, through docking studies, it is evident that there exists no unfavorable interaction between ADR1 dye and protein in all the conformers generated which signifies that the guest molecule enjoys a favourable position to reside in various domains of the protein molecule.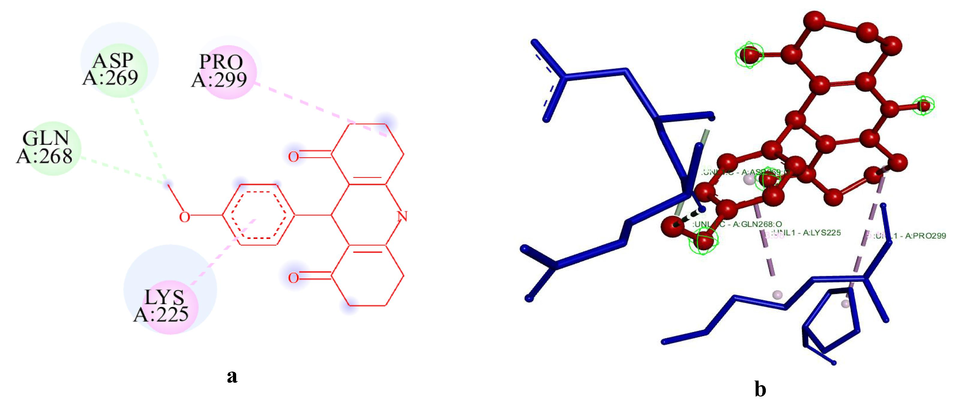
(a-b): The 2D and 3D diagram of the unique conformers of ADR1HSA1 visualized using Biovia Discovery Studio visualizer. ADR1 dye is represented in ball and stick model in red colour. #Key amino acids contributing to interactions are shown in circles; ADR1 dye is represented in ball and stick model in red colour. Green colour dotted line indicates hydrogen bonds with electronegative elements like N and O atoms, light green colour dotted line indicates carbon-hydrogen bonds; light purple colour indicates pi-alkyl interactions, violet colour dotted line indicates pi-sigma interaction. Light green colour amino acids without interactions represent van der Waals interactions. The blue halo surrounding the interacting residues represents the solvent accessible surface that is proportional to its diameter#.
A larger negative value of intermolecular energy, energy resulted due to HB and electrostatic energy favours the binding stability of the conformers. The above-mentioned parameters play a prominent and crucial role in docking studies and have been well established in our earlier studies involving a well-known ICT fluorophore, DDP dye with globular proteins like BSA (Tamaraiselvan et al., 2021), ovalbumin (Tamaraiselvan et al., 2022) and β-lactoglobulin(Vinod et al., 2022).
Based on Mol.dock studies, the docking of ADR1 dye with HSA is energetically least favourable and comparatively less stable than that of ADR2 dye with HSA. The 2D and 3D structures of the unique conformers of ADR2HSA are provided in Fig. 3 and supporting information figure S5 respectively. The formation of most stable conformer ADR2HSA1 is −7.87kJmol-, whereas that of ADR1HSA1 results in lesser B.E than that of ADR2 dye. Interestingly, this behavioral pattern was not observed in the case of BSA even though BSA and HSA are homologous in nature and exhibit almost a similar sequence. The ligand efficiency values for both ADR1 and ADR2 dyes were quite similar, whereas the intermolecular energy and energy associated with various bimolecular interactions of ADR1HSA1 conformer are lesser than that of ADR2HSA1 conformer. A striking difference on the presence of unfavourable interactions in ADR2HSA was evident in ADR2HSA7 conformer which was not observed in ADR1HSA system. Even though both these dyes are structurally similar, a variation in the binding pattern and bimolecular interactions is evident through docking studies, which would have not been possible to be investigated in the solution phase comprising several interactions.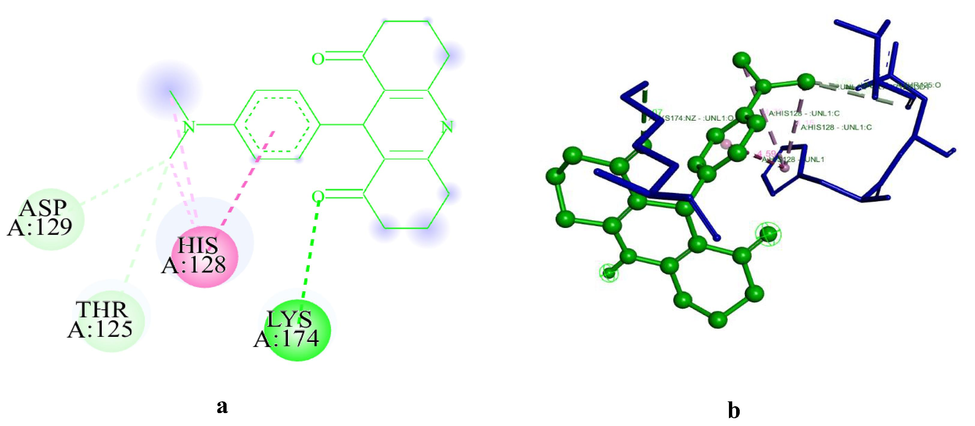
(a-b): The 2D and 3D diagram of ADR2HSA1, visualized using Biovia Discovery Studio visualizer. ADR2 dye is represented in in light green colour.#.
Further, on evaluation and analysis of the various conformers generated for both these dyes with HSA, we affirm that ADR1 dye does not dock in Sudlow site II and the dye is confined to other binding sites of HSA, whereas ADR2 dye is confined to all six major binding domains of HSA. Interestingly, unlike specific site selective drugs and ligands interaction with HSA, ADR1 and ADR2 dye predominantly reside in Sudlow site I and site III (site III is not Sudlow binding site).
Literature reports reveal that most of these drugs either prefer Sudlow binding site I or site II and does not favour site III. However, in our studies. Site III is found to be the most favourable site for ADR dyes compared to Sudlow sites (Ghuman et al., 2005). Among the ten conformers of ADR1HSA generated, an equal contribution of the conformers preferring site I and site III is observed which was found to be entirely different from that of ADR2HSA complex. In the case of ADR2HSA complex, the dye preferentially resides in site III (non-Sudlow binding site) rather than the usual binding domains of HSA. This was ascertained based on the first three energetically stable conformers. The illustration and compilation of the various conformers of both these dyes with HSA corresponding to the binding domains are provided in Table 3 and Table 4 respectively. The overall schematic representation of the conformers generated for ADR1 and ADR2 dyes located in the various subdomains of HSA are provided in Figs. 4 and 5 respectively.
CONFORMERS
SUB-DOMAINS
SITES
ADR1HSA1
ADR1HSA7
ADR1HSA10IIA
I
ADR1HSA2
IB
III
ADR1HSA3
IIA & IIB
I
ADR1HSA4
ADR1HSA8
ADR1HSA9IA
III
ADR1HSA5
IIB
In between site I and site II
CONFORMERS
SUB-DOMAINS
SITES
ADR2HSA1
IB
III
ADR2HSA2
ADR2HSA3
ADR2HSA4
ADR2HSA7IA
III
ADR2HSA5
IIIA
II
ADR2HSA6
ADR2HSA8
ADR2HSA9IIA & IA
I &III
ADR2BSA10
IB, IIA, IIB & IIIA
I, II,III
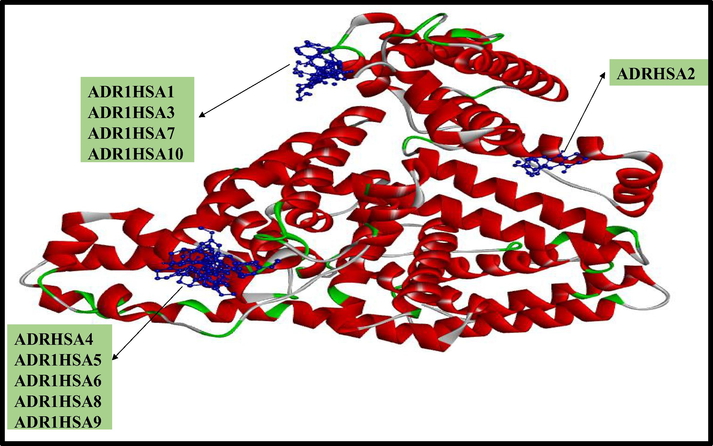
Ten conformations of ADR1 dye (depicted in blue colour ball and stick model) with three dimensional structures of HSA (Coloured based on solid ribbon model; Red colour represents helix; green and white colour represents turns and coils).
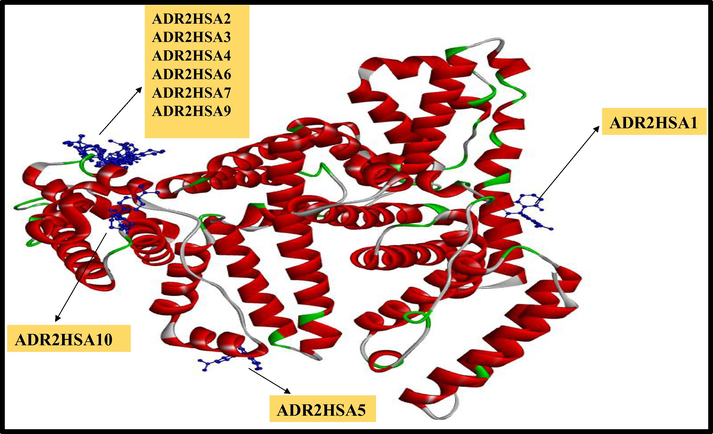
Ten conformations of ADR2 dye (depicted in blue colour ball and stick model) with three dimensional structures of HSA (Coloured based on solid ribbon model; Red colour represents helix; green and white colour represents turns and coils).
4 Molecular interactions of dye-protein complex
4.1 ADR1-HSA conformers
HB interaction accompanied with several hydrophobic interactions (Pi-alkyl, Alkyl, Pi-sigma, Pi-Pi) exists in all these conformers generated. However, the enhanced stability of ADR1HSA complex is attributed to several HB and hydrophobic interactions. The number of HB (non-conventional) and hydrophobic interactions for ADR1HSA1 are two and two respectively. However, the most stable conformer is not influenced by conventional HB interaction. In the case of ADR1 dye with HSA, several modes of interaction were obtained resulting stability of the conformer. The molecular interaction table representing various interactions existing between ADR1 dye and HSA is provided in detail in Table 5. In ADR1HSA1 conformer, two amino acids that are involved in HB interactions with the dye are GLN and ASP located at 268 and 269th position of the protein sequence (Site I of domain II). Both these amino acids are polar in nature. Further, the amino acids that are involved in hydrophobic interactions of ADR1HSA1 conformer are LYS and PRO wherein LYS is polar and PRO is non-polar amino acid. Interestingly, the second and third most stable conformers namely ADR1HSA2 and ADR1HSA3 are stabilized through conventional HB interaction assisted by hydrophobic interactions. The amino acids LYS, PRO, LEU, ALA and ARG are involved in molecular interactions are from site III of sub domain IA.. Interestingly these two conformers are predominantly involved in bonding with both polar and non-polar amino acids through more than one binding site. A detailed investigation on the molecular interaction of other conformers clearly reveals the existence of HB interactions with both polar and non-polar amino acids. The variation on the docking of ADR1 dye with BSA was predominantly through HB whereas in the case of HSA, hydrophobic interactions predominate over HB interaction. Docking studies affirmates that the binding stability is governed by several molecular interactions resulting in location of the dye in all the domains of protein.
Conformation
Binding Energy
Hydrogen-Bonding Interaction Donor –Acceptor Amino acid-Dye
Bond Distance
Hydrophobic Interactions
Bond Distance
ADR1HSA1
−7.02
ASP269 (C…..O)GLN268
(C….O)3.30
3.25
Pi-Alkyl
LYS225
Alkyl
PRO2293.98
5.14
ADR1HSA2
−6.69
LYS162(NZ….O)
LYS159(CA….O)3.05
Pi-Alkyl
LYS159
Alkyl
ALA158
LEU135
LEU139
LYS1365.08
5.09
4.42
5.29
3.88
ADR1HSA3
−6.49
ARG336(NH1….O)ARG336
(NH2….O)3.31
3.23
Pi-Alkyl
PRO299
Alkyl
PRO299
LYS2255.24
4.26
4.03
ADR1HSA4
−6.39
ASN99(ND2….O)
3.17
Pi-Alkyl
PRO96
HIS67
Alkyl
PRO96
PRO96
4.33
5.29
4.35
4.68
ADR1HSA5
−5.68
LYS225
2.87
Alkyl
LYS2253.74
ADR1HSA6
−5.65
HIS67(ND1…..O)
2.83
Pi-alkyl
PRO96
HIS67
Pi-Anion
GLU252
Pi-Pi Stacked
HIS674.70
4.06
4.98
4.33
ADR1HSA7
−5.5
GLU227(OE…..C)
SER2703.23
4.05
Alkyl
LYS225
Pi-Anion
GLU2274.16
3.97
ADR1HSA8
−5.47
HIS67 (ND1…..O)
ASN99(ND2…..O)2.51
3.06
Pi-Alkyl
PRO96
Alkyl
PRO96
Pi-Pi stacked
HIS674.65
4.72
4.25
ADR1HSA9
−5.21
–
–
Pi-Alkyl
PRO96
Alkyl
PRO96
Amide-Pi stacked
GLU954.03
4.09
4.71
ADR1HSA10
−6.34
ASP269 (C…..O)GLN268
(C….O)3.23
3.11
Pi-Alkyl
LYS224.01
Further a closer analysis on the amino acids that are involved in molecular interaction are located in all the binding sites of protein, such that ADR1 dye resides in all the domains (except site II) with variation in their binding energies and dock predominantly at the non-Sudlow binding sites of HSA. This clearly supports our earlier elucidation on the existence of 9 unique conformers (Fig. 4).
4.2 ADR2-HSA conformer
As observed in the case of ADR1HSA complex, HB interaction accompanied with several hydrophobic interactions also coexist in all the conformers of ADR2HSA. The number of HB interactions is fewer in all the conformers of ADR2HSA compared to that of ADR1HSA complex. The most stable conformer, ADR2HSA1 possesses only three HB (both conventional and non-conventional) sites for interaction. The amino acids that are responsible for HB interaction are LYS, THR and ASP amino acid. However, with HIS128 it forms hydrophobic interaction resulting in enhanced stability of the complex. On the contrary, all the amino acids that are involved in the stability of this conformer are polar amino acids only which was not resulted in the case of ADR1 dye. The molecular interaction table of ADR2HSA conformer is provided in Table 6 which comprises the HB donor and acceptor moieties. The conformers ADR2HSA2 and ADR2HSA3 are stabilized by hydrophobic interactions and are docked at the same binding site of HSA, as observed in the case of ADR1dye. Interestingly, all the stable conformers predominantly are located in site III of HSA, however the binding domains differ for the individual conformers. The amino acids that are involved in HB interaction with dye is ASN located at 99th position of HSA and the amino acids HIS and PRO located sat 67th and 96th positions are involved in hydrophobic interactions with HSA. All these amino acids are polar in nature which was not in the case of BSA. Both ADR2 dye and the amino acids form HB through the donor and acceptor sites. The nature of this interaction was not visualized in the case of ADR1 dye even though both these dyes have similar structural features except on the nature of the functional group in the para position of the phenyl ring attached at the 9th position (Fig. 1). A comparative investigation on the molecular interactions of all the conformers of ADR1 and ADR2 dyes with HSA authenticates the stability of the conformers are attributed to hydrophobic interaction only.
Conformation
Binding Energy
Hydrogen-Bonding Interaction Donor –Acceptor Amino acid-Dye
Bond Distance
Hydrophobic Interactions
Bond Distance
Other Interactions
ADR2HSA1
−7.87
LYS174 (NZ…..O)THR125
(C….O)ASP129
(C….OD)3.07
3.61
3.06
Pi-Alkyl
HIS128
HIS128
Pi-Pi Stacked
HIS1284.79
4.15
4.59–
ADR2HSA2
−6.75
ASN99(ND….O)
3.13
Pi-Alkyl
PRO96
HIS67
Alkyl
PRO96
PRO964.22
5.01
4.85
4.27–
ADR2HSA3
−6.66
ASN99(ND….O)
3.13
Pi-Alkyl
PRO96
HIS67
Alkyl
PRO96
PRO964.22
5.01
4.85
4.27–
ADR2HSA4
−6.57
GLU95(C….O)
3.41
Pi-Alkyl
PRO96
HIS67
Alkyl
PRO96
PRO964.42
4.19
4.99
4.66–
ADR2HSA5
−6.52
ARG445(NH1….O)
3.34
Pi-Alkyl
LYS389
Alkyl
ARG445
ILE388
5.26
4.89
4.16
ADR2HSA6
−6.49
GLU252(OE1…..H)
GLU95(C….O)
GLU95(C….O)2.09
3.61
3.76
Pi-alkyl
PRO96
Alkyl
PRO96
Pi-Sigma
HIS674.91
4.09
3.46–
ADR2HSA7
−6.35
–
Alkyl
TYR844.37
Donor-donor (GLN33)
ADR2HSA8
−5.33
GLU252(OE1…..H)
GLU95(C….O)2.73
3.48
Pi-Alkyl
PRO96
HIS67
Alkyl
PRO96
PRO962.56
4.23
4.63
5.36–
ADR2HSA9
−4.77
GLU252(OE1…..H)
GLU95(C….O)
GLU95(C….O)2.09
3.61
3.76
Pi-alkyl
PRO96
Alkyl
PRO96
Pi-Sigma
HIS674.91
4.09
3.46–
ADR2HSA10
−4.4
–
Pi-Alkyl
LEU481
TRF214
VAL344
Alkyl
LEU457
LEU457
ARG484
ARG484
ARG485
LEU481
LEU481
LEU481
LYS199
LYS1954.33
4.26
5.19
5.10
4.15
4.05
3.98
4.25
5.07
5.25
4.33
4.49
4.32–
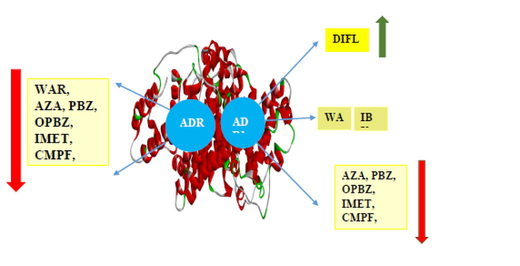
Further in order to ascertain the extent of binding stability of these dyes in various binding domains of HSA, we incorporated several sites selective and site specific Sudlow binding site drugs (Kumaran and Ramamurthy, 2006). These drugs possess enormous binding affinity towards specific binding domains of the protein molecule such that they have the capability in displacing competing ligands from the binding sites. The drugs WAR, AZA, PBZ, OPBZ, IMET and CMPF which has the greater binding affinity towards site I of HSA has an enhanced property on displacing a competing ligand from binding site I. This pattern has been evident from several fluorophore interaction with globular proteins in the presence of drugs. Similarly, the drugs IBU, DIFLU, DIAZ, FA and INSL has a largest affinity towards binding site II and displaces the guest molecule to various other domains. The ligand in the present study is ADR dyes. The molecular interaction table representing various interactions existing between ADR2 dye and HSA is provided in detail in Table 6.
4.3 Binding stability of ADR1-HSA in the presence of site I and site II drugs
WAR, AZA, PBZ, OPBZ, IMET and CMPF (Site I binding drug) and five site II binding drugs IBU, DIFLU, DIAZ, FA and INSL were docked to dye-protein complex. These specific site selective drugs were employed in our study particularly to evaluate the extent of displacement of the dye from the binding domains of protein and also to ascertain the binding stability and the energetics of ADR1 with HSA in the presence of drug. These drugs are generally referred as Sudlow binding site drugs and have have been well established in the literature confined to drug-protein binding characteristics (Wang et al., 2015). The structure of site I and Site II selective drugs are provided in the supporting information figure S2 and S3 respectively. The drugs chosen in the present study satisfies the Lipinski rule of five PSA (Kumaran et al., 2015). The well-known site I and site II binding drugs possesses both HB donor and acceptor sites along with rotatable single bond characteristics that enable the drug molecule to dock in the various position of the protein molecule (helix/Sheet/Coils/Turns).
The presence of two competing ligands which are generally classified as guest molecules (fluorophore and drug) with protein is explored in depth through variation in the pattern of binding stability (increase or decrease) and the various energetics involved in the ease of formation of ADR1HSA complex versus ADR2HSA site I and site II binding drugs by employing MOL.DOCK studies. The energetics and molecular interactions of drug-dye-protein complex are tabulated in Tables 7 and 8. Further, we also probe the role of drug binding with dye-protein complex and vice versa based on its most probable docking domains and sites. The energetically most preferred active binding sites of various subdomains of the protein for which the site I and site II drugs reside along with dye in the various subdomains of HSA is discussed in depth pertaining to the amino acids involved in bimolecular interactions. The role and influence on the extent of displacement of the dye by drug and vice-versa is authenticated and elucidated by theoretical studies. The exact location of ADR1 dye and drug in the domains of HSA is established from docking studies, along with the possible molecular interactions existing in it resulting in stability of the complex. The extent of stabilisation or destabilisation of the dye-protein complex in the presence of drugs are explored based on the B.E along with intermolecular energy and other energies attributed to molecular interactions. A detailed outcome on the simultaneous docking of drugs with ADR1HSA is provided below.
-
Site I drug efficiently destabilizes the binding affinity of ADR1HSA complex than Site II drug. This pattern was exactly the opposite of dye docking with BSA.
-
Compared to all other drugs considered in our study, CMPF (site I binding drug) destabilizes the binding affinity of dye-protein complex by 35 % and whereas FA (site II binding drug) results in decrease in the stability of dye-protein complex by 25 % only.
-
The drug DIFLU alone enhances the binding stability of dye-protein complex. All other drugs doesn’t govern the binding nature of dye with HSA in comparison to that of CMPF, FA and DIFLU. A pictorial representation of the extent of destabilization of dye-protein complex is provided in Fig. 6.
-
Except CMPF, all other site I binding drugs almost exhibits a similar proportion of destabilization of the dye-protein complex.
-
Apart from CMPF, FA (Site II) has the better efficiency in decreasing the binding affinity of dye- protein interaction as observed in the case of HSA.
-
There is no direct dye-drug interaction when docked simultaneously with protein and the existence of dye-protein and drug-protein is only resulted. The 2D and the ribbon structure representation of ADR1 dye in the presence of drugs is provided in Fig. 7 and Fig. 8 respectively. The 3D images are given in supporting information S5. Table 9 provides the evidence for the combined forces of interaction of a ternary system comprising of drug-dye-protein.
-
Site I and site II-specific drugs does not displace the dye from the Sudlow binding sites, rather the dye governs the most probable location of the drugs predominantly to non-Sudlow binding sites of HSA.
| Conformer | Binding Energy | Ligand efficiency | Inhibitory Constant | vDW enegy + H-bond energy + desolvation energy | Inter molecular energy | HSA Sub domain | Binding site | Difference in B.E |
|---|---|---|---|---|---|---|---|---|
| ADR1HSA1 | −7.2 | −0.29 | 7.2 | −7.31 | −7.61 | IIB | – | 0 |
| ADR1HSA1WAR | −7.18 | −0.31 | 5.46 | −8.35 | −8.67 | IA | III | 0.02 |
| ADR1HSA1AZA | −6.39 | −0.29 | 20.78 | −7.28 | −7.28 | IIIA, IB | II | 0.9 |
| ADR1HSA1PBZ | −6.61 | −0.29 | 14.2 | −8.16 | −8.11 | IIIA, IIA, IIB | I &II | 0.59 |
| ADR1HSA1OPBZ | −6.78 | −0.28 | 10.78 | −8.33 | −8.57 | IIB | – | 0.42 |
| ADR1HSA1IMET | −4.48 | −0.28 | 314.54 | −4.94 | −7.16 | IB | III | 2.72 |
| ADR1HSA1CMPF | −4.48 | −0.28 | 314.54 | −4.94 | −7.16 | IB | III | 2.72 |
| ADR1HSA1IBU | −7.15 | −0.48 | 5.69 | −7.33 | −8.65 | IB | III | 0.05 |
| ADR1HSA1DIAZ | −6.09 | −0.3 | 34.36 | −7.09 | −7.58 | IIIA | II | 1.11 |
| ADR1HSA1DIFLU | −7.91 | −0.44 | 1.6 | −7.79 | −9.1 | IIIA,IB | II,III | −0.71 |
| ADR1HSA1FA | −5.07 | −0.3 | 35.51 | −7.02 | −7.56 | IA | III | 1.13 |
| ADR1HSAINSL | −6.41 | −0.39 | 108.15 | −5.75 | −6.31 | IA | III | 1.79 |
| Conformation | Binding Energy | Hydrogen-Bonding Interaction Donor –Acceptor Amino acid-Dye | Bond Distance | Hydrophobic interactions | Bond Distance | Other Interactions |
|---|---|---|---|---|---|---|
| ADR1HSA1 | −7.2 | ASP269 (C…..O)GLN268 (C….O) |
3.30 3.25 |
Pi-Alkyl
LYS225 Alkyl PRO229 |
3.98 5.14 |
– |
| ADR1HSA WAR | −7.18 | SER65(OG….O) | 2.86 |
Pi-Alkyl
PRO96 PRO96 PRO96 Pi-Pi stacked HIS67 |
5.12 4.87 4.49 4.22 |
– |
| ADR1HSA AZA | −6.39 | TYR452 | 4.12 |
Pi-Alkyl
ALA191 Alkyl ALA191 VAL455 VAL456 LYS436 Pi-Pi T Shaped TYR452 TYR452 |
4.59 3.47 4.79 4.42 4.04 4.83 4.66 |
– |
| ADR1HSA PBZ | −6.61 | SER480(OG2….O) | 2.85 |
Pi-Alkyl
ALA210 ALA213 TRP214 LEU347 |
5.07 3.44 4.75 5.15 |
– |
| ADR1HSA OPBZ | −6.78 |
Pi-Alkyl
PRO299 ALA300 |
4.89 4.82 |
|||
| ADR1HSA IMET | −6.07 | GLU184(OE2…..C) TYR452 |
3.53 3.89 |
Pi-alkyl
VAL455 ALA191 Pi-Anion ASP187 GLU188 Pi-Cation LYS432 Pi-Pi T Shaped TYR452 TYR452 |
4.83 5.07 4.97 4.21 4.96 5.07 5.19 |
– |
| ADR1HSA CMPF | −4.48 | LEU115(O…..H)ARG114 (NH1…..O)LYS190 (NZ…..O) |
2.23 3.11 2.88 |
Alkyl
ARG186 Pi-Cation ARG114 |
4.91 4.24 |
Donor-donor (ARG117-2.49) |
| ADR1HSA8 IBU | −7.15 | ARG117 (NH1…..O) | 3.05 |
Pi-Alkyl
TYR161 TYR138 Alkyl ILE142 LEU182 ARG117 MET123 PRO118 Pi-Pi stacked TYR138 TYR161 |
3.97 4.04 4.46 5.02 4.14 4.50 5.34 3.97 3.92 |
– |
| ADR1HSA DIFLU | −7.91 |
Pi-Alkyl
ALA194 LYS190 VAL456 Pi-sigma VAL455 Halogen ASN429(CG…..F) |
4.47 4.31 5.23 3.52 3.43 |
– | ||
|
ADR1HSA DIAZ ADR1HSA FA ADR1HSA INSL |
−6.09 −5.07 −6.41 |
SER489 (OG…..O)TYR411 (OH….H)ILE388 (CG….F) TYR411 HALOGENLEU387 (O….F)SER65 (OG…..F)SER65 (N…..F)THR68 (OG…..F)GLU95 (OE…..H) HALOGENASP63 (O…..F)GLU92 (OE2…..F) GLU95 (OE2…..H)GLU95 (O…..H) |
2.54 2.13 3.21 3.71 3.66 3.25 3.35 2.99 2.25 3.52 3.65 2.24 3.00 |
Pi-Alkyl
ARG485 LEU453 Alkyl CYS392 Pi-Pi T shaped TYR411 Pi-Alkyl PRO96 Pi-Anion GLU95 GLU6 Pi-Alkyl PRO96 PRO96 Pi-Pi Stacked HIS67 |
5.21 5.46 5.06 5.81 4.94 4.82 4.99 4.45 4.09 4.64 |
Donor-donor (ARG410- 2.66) |
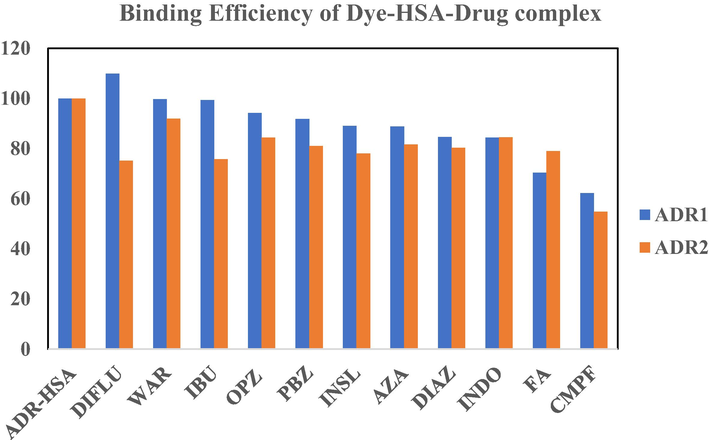
- A pictorial represtantation of decrease in binding affinity of dye-protein complex after the addition of site I and site II binding drugs. The X axis represents the Dye-protein complex in the presence of drugs and the Y axis represents the binding efficiency of the dye-protein complex in the presence of drugs. Blue colour represents ADR1 dye and orange color represents the ADR 2 dye.
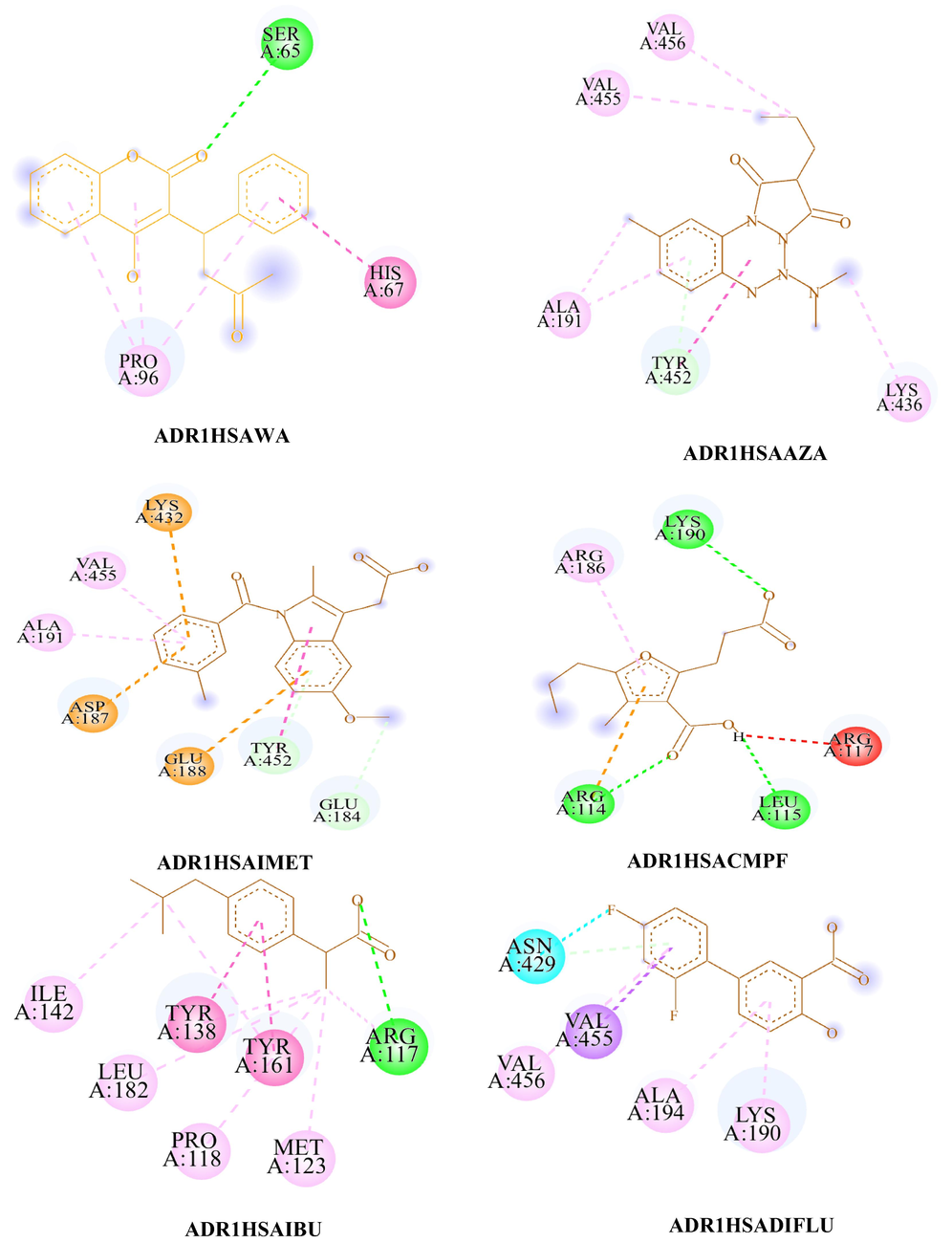
- The 2D diagram of ADR1HSAWAR, ADR1HSAAZA, ADR1HSAPBZ, ADR1HSAOPBZ, ADR1HSAINDO, ADR1HSACMPF, ADR1HSAIBU, ADR1HSADIFLU, ADR1HSADIAZ, ADR1HSAFA and ADR1HSAINSL visualized using Biovia Discovery Studio visualizer. #.
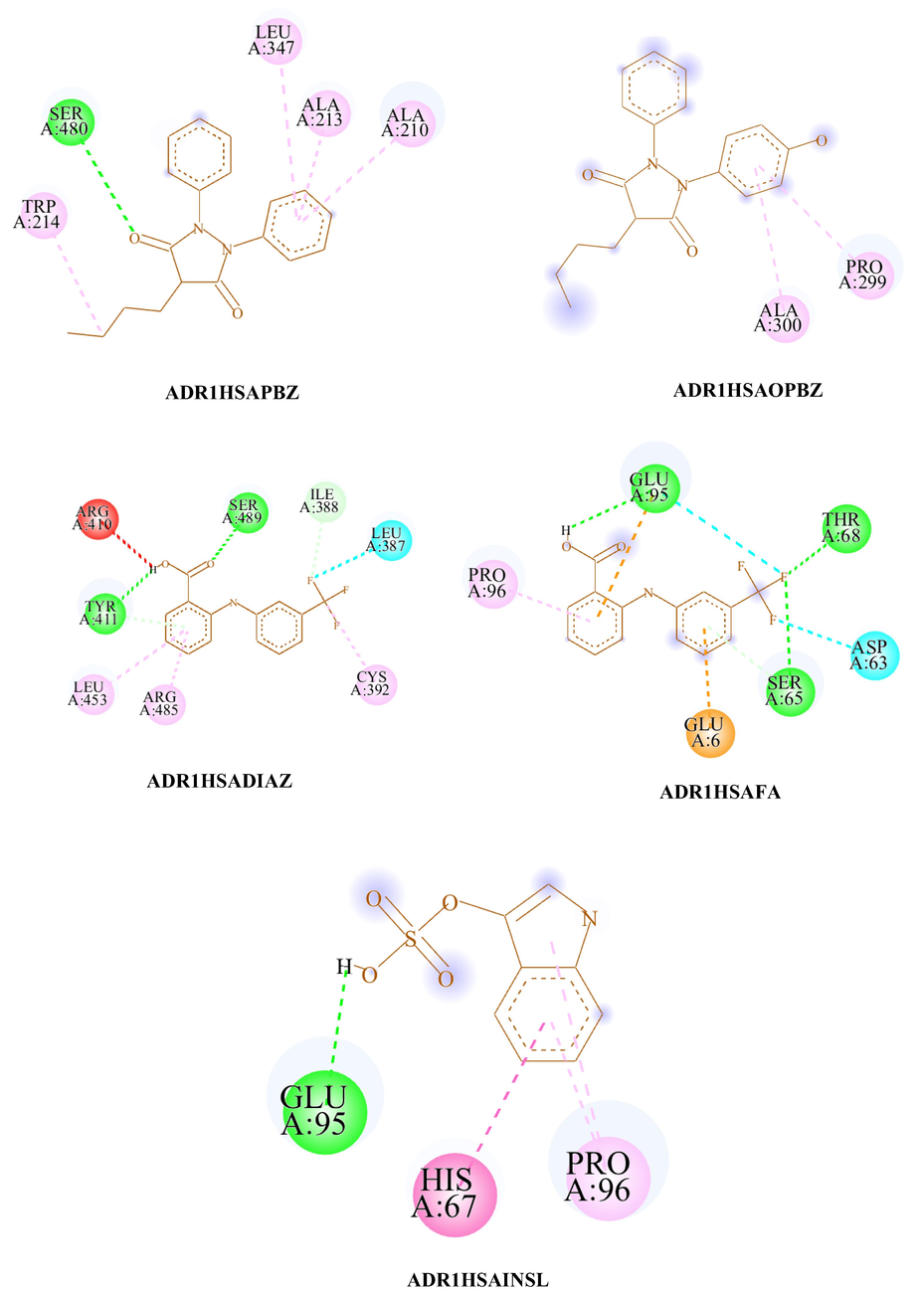
- The 2D diagram of ADR1HSAWAR, ADR1HSAAZA, ADR1HSAPBZ, ADR1HSAOPBZ, ADR1HSAINDO, ADR1HSACMPF, ADR1HSAIBU, ADR1HSADIFLU, ADR1HSADIAZ, ADR1HSAFA and ADR1HSAINSL visualized using Biovia Discovery Studio visualizer. #.
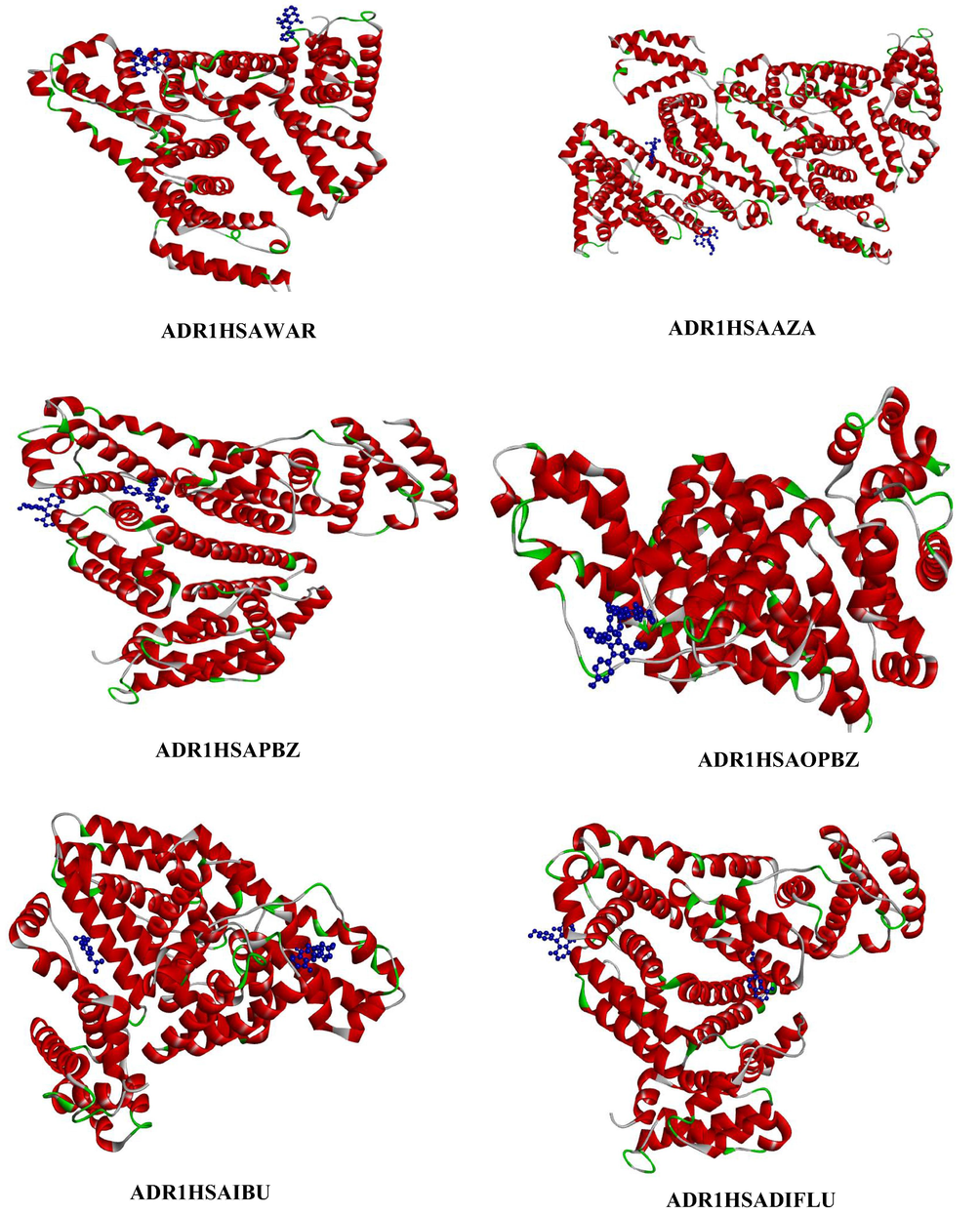
- Represents the three-dimensional structure of site I and site II drugs (depicted in blue colour ball and stick model) with ADR1HSA complex, where the dye is depicted in blue colour ball and stick model and the HSA is coloured based on solid ribbon model; Red colour represents helix; green and white colour represents turns and coils.
- Represents the three-dimensional structure of site I and site II drugs (depicted in blue colour ball and stick model) with ADR1HSA complex, where the dye is depicted in blue colour ball and stick model and the HSA is coloured based on solid ribbon model; Red colour represents helix; green and white colour represents turns and coils.
| CONFORMER | Conventional HB | Non-Conventional Hydrogen Bonding | Pi-Alkyl | Alkyl | Pi-Sigma | Pi-Pi | Pi- Cation | Pi-Anion | Unfavorable (acceptor–acceptor) | Halogen |
|---|---|---|---|---|---|---|---|---|---|---|
| ADR1HSA1 | – | 2 | 1 | 1 | – | – | – | – | – | – |
| ADR1HSA1WAR | 1 | – | 3 | – | – | 1 | – | – | – | – |
| ADR1HSA1AZA | – | 1 | 1 | 4 | – | 2 | – | – | – | – |
| ADR1HSA1PBZ | 1 | – | 4 | – | – | – | – | – | – | – |
| ADR1HSA1OPBZ | – | – | 2 | – | – | – | – | – | – | – |
| ADR1HSA1MET | – | 2 | 2 | – | – | 2 | 1 | 2 | – | – |
| ADR1HSA1CMPF | 3 | – | – | 1 | – | – | 1 | – | 1 | – |
| ADR1HSA1IBU | 1 | – | 2 | 5 | – | 2 | – | – | – | – |
| ADR1HSA1DIAZ | – | – | 3 | – | 1 | – | – | – | – | – |
| ADR1HSA1DIFLU | 2 | 2 | 2 | 1 | – | 1 | – | – | 1 | 1 |
| ADR1HSA1FA | 4 | – | 1 | – | – | – | – | – | 2 | – |
| ADR1HSAINSL | 2 | – | 2 | – | – | 1 | – | – | – | – |
4.4 Binding stability of ADR2-HSA in the presence of site I and site II drugs
A comparison on the simultaneous docking of the drug with ADR2 dye was also carried out and the outcome resulted in an entirely different approach of the action of drugs on the displacement of dye from the binding domains of HSA. Site II drugs effectively destabilizes the binding stability of dye-protein complex than Site I drugs. The energetics and molecular interactions of drug-dye-protein complex is tabulated in Table 10 and 11.
-
The drugs, CMPF (Site I) and DIFLU (Site II) exhibits better destabilization efficiency than other site-specific drugs.
-
The addition of either site I or site II drugs to ADR2 dye results in the removal of unfavorable interaction existing in ADR2 protein complex.
-
The site II drugs FA and DIFLU exhibits interactions predominantly through halogen atom. The 2D,3D and the ribbon structure representation of ADR2 dye in the presence of drugs are provided in supporting information S6, S7 and S8 respectively. Table 12 provides the evidence for the combined forces of interaction of a ternary system comprising of drug-dye-protein.(See Tables 13 and 14)
| Conformer | Binding Energy | Ligand efficiency | Inhibitory Constant | vDW enegy + H-bond energy + desolvation energy | Inter molecular energy | HSA Sub domain | Binding site | Difference in B.E |
|---|---|---|---|---|---|---|---|---|
| ADR2HSA1 | −7.87 | −0.31 | 1.7 | −8.69 | −8.47 | IB | III | 0 |
| ADR2HSA1WAR | −7.23 | −0.31 | 5.0 | −8.72 | −8.73 | IIA,IIB, IIIA | I & II | 0.64 |
| ADR2HSA1AZA | −6.42 | −0.29 | 19.57 | −7.24 | −7.32 | IIIA | II | 1.45 |
| ADR2HSA1PBZ | −6.38 | −0.28 | 21.2 | −7.89 | −7.89 | IA | III | 1.49 |
| ADR2HSA1OPBZ | −6.64 | −0.28 | 13.54 | −8.32 | −8.43 | IIIA | II | 1.23 |
| ADR2HSA1MET | −6.65 | −0.27 | 13.27 | −7.88 | −8.15 | IB | III | 1.22 |
| ADR2HSA1CMPF | −4.31 | −0.17 | 687.57 | −5.5 | −5.81 | IIIA | II | 3.56 |
| ADR2HSA1IBU | −5.96 | −0.4 | 42.85 | −7.38 | −7.45 | IIIA, IIA, IB | I &II | 1.91 |
| ADR2HSA1DIAZ | −6.32 | −0.32 | 23.2 | −7.26 | −7.81 | IIIA | II | 1.55 |
| ADR2HSA1DIFLU | −5.92 | −0.33 | 15.63 | −7.83 | −8.05 | IA | III | 1.95 |
| ADR2HSA1FA | −6.22 | −0.31 | 27.36 | −6.68 | −5.52 | IIA | I | 1.65 |
| ADR2HSAINSL | −6.14 | −0.44 | 31.83 | −5.55 | −7.03 | IIA | I | 1.73 |
| Conformation | Binding Energy | Hydrogen-Bonding Interaction Donor –Acceptor Amino acid-Dye | Bond Distance | Hydrophobic interactions | Bond Distance | Other Interactions |
|---|---|---|---|---|---|---|
| ADR2HSA1 | −7.87 | LYS174 (NZ…..O)THR125 (C….O)ASP129 (C….OD) |
3.07 3.61 3.06 |
Pi-Alkyl
HIS128 HIS128 Pi-Pi Stacked HIS128 |
4.79 4.15 4.59 |
– |
| ADR2HSA WAR | −7.23 | TRP214(NE1….O) GLU450(OE2….H) |
2.51 2.70 |
Pi-Alkyl
LEU481 LEU481 ARG484 VAL344 Pi-Sigma LEU481 |
4.04 4.62 5.06 5.12 3.66 |
– |
| ADR2HSA AZA | −6.42 | ARG445(NH1….O) ARG445(CD….O) |
2.93 2.70 |
Pi-Alkyl
LYS389 Alkyl LYS389 ARG445 ILE388 Pi- Anion GLU442 GLU442 Pi- Cation LYS38 |
4.85 3.47 4.39 4.39 3.87 3.33 3.83 |
– |
| ADR2HSA PBZ | −6.38 |
Alkyl
PRO96 |
4.19 | – | ||
| ADR2HSA OPBZ | −6.64 | GLU188(OE1….H) TYR452 |
2.36 4.07 |
Pi-Alkyl
LYS436 Alkyl VAL453 VAL456 Pi-Pi T Shaped TYR452 |
5.11 4.37 5.16 5.10 |
|
| ADR2HSA IMET | −6.65 | ARG145(CD…..O) | 3.07 |
Pi-alkyl
ARG114 LEU115 HIS146 ARG145 Alkyl ARG117 ARG114 ARG145 |
4.46 5.35 4.70 4.06 3.99 4.42 4.08 |
– |
| ADR2HSA CMPF | −4.31 | LYS389(NZ….O) | 2.59 |
Pi-Alkyl
PHE395 Alkyl CYS392 LYS349 CYS438 Pi-Sigma GLU396 |
4.91 3.78 4.41 3.49 3.44 |
|
| ADR2HSA8 IBU | −5.96 | LYS199(O…..H) | 2.32 |
Pi-Alkyl
LEU198 LEU198 LEU481 Alkyl LEU481 LEU198 LYS199 |
3.87 5.45 4.13 4.61 3.87 4.33 |
– |
| ADR2HSA DIFLU | −5.92 | GLN94 | 4.00 | – | ||
|
ADR2HSA DIAZ ADR2HSAFA ADR2HSA INSL |
−6.32 −6.22 −6.14 |
SER419 (OG…..H)THR422 (OG1….H)THR422 (OG1….H)SER419 (N….F)GLU227 (OE2…..H)ARG257 (H…..O)ARG257 (H…..O)HIS242 (CE1…..O) |
2.38 2.15 2.24 3.09 2.05 2.35 3.34 3.73 |
Pi-Alkyl
VAL469 Alkyl VAL469 Halogen GLN417 THR467 PRO468 Alkyl LYS225 Pi-Anion GLU227 Halogens SER270 LYS225 LYS225 ASP269 ASP269 Pi-Alkyl LEU238 LEU238 VAL241 LEU290 Pi-Sigma LEU260 LEU260 |
5.31 3.72 3.07 3.09 3.68 2.36 3.61 3.42 3.19 2.82 2.86 3.45 5.27 5.37 4.66 5.26 3.82 3.99 |
| CONFORMER | Conventional HB | Non-Conventional Hydrogen Bonding | Pi-Alkyl | Alkyl | Pi-Sigma | Pi-Pi | Pi- Cation | Pi-Anion | Unfavorable (acceptor–acceptor) | Halogen |
|---|---|---|---|---|---|---|---|---|---|---|
| ADR2HSA1 | 1 | 2 | 2 | – | – | 1 | – | – | – | – |
| ADR2HSA1WAR | 2 | – | 4 | – | 1 | – | – | – | – | – |
| ADR2HSA1AZA | 2 | – | 1 | 3 | – | – | 1 | 2 | – | – |
| ADR2HSA1PBZ | – | – | – | 1 | – | – | – | – | – | – |
| ADR2HSA1OPBZ | 2 | – | 1 | 2 | – | 1 | – | – | – | – |
| ADR2HSA1INDO | – | 2 | 4 | 3 | – | – | – | – | – | – |
| ADR2HSA1CMPF | 1 | – | 1 | 3 | 1 | – | – | – | – | – |
| ADR2HSA1IBU | 1 | – | 3 | 3 | – | – | – | – | – | – |
| ADR2HSA1DIAZ | 1 | – | – | – | – | – | – | – | – | – |
| ADR2HSA1DIFLU | 4 | – | 1 | ‘1 | – | – | – | – | – | 3 |
| ADR2HSA1FA | 1 | – | – | 1 | – | – | – | 1 | – | 5 |
| ADR2HSAINSL | 3 | – | 4 | – | 2 | – | – | – | – | – |
| DRUGS | HSA-Drugs Binding Energy | HSA-Drugs + ADR1 Binding Energy | Percentage increase in Binding energy | HSA-Drugs + ADR2 Binding Energy | Percentage increase in Binding energy |
|---|---|---|---|---|---|
| WAR | −6.98 | −7.16 | 2.5 % | −7.51 | 7 % |
| AZA | −5.95 | −7.64 | 22 % | −7.28 | 18 % |
| PBZ | −6.88 | −8.52 | 18 % | −8.94 | 23 % |
| OPBZ | −5.8 | −7.4 | 21.6 % | −7.45 | 22 % |
| IMET | −6.62 | −7.14 | 7.2 % | −8.06 | 17 % |
| CMPF | −4.96 | −8.36 | 41 % | −7.45 | 33 % |
| IBU | −5.87 | −8.93 | 34 % | −8.38 | 30 % |
| FA | −8.56 | −9.83 | 12 % | −10.33 | 21 % |
| DIAZ | −8.39 | −9.35 | 10 % | −9.38 | 11 % |
| DIFLU | −8.72 | −8.85 | 1.4 % | −10.7 | 19 % |
| INSL | −6.92 | −9.73 | 29 % | −10.8 | 36 % |
| DRUGS | HSA-Drugs Binding Site | HSA-Drugs + ADR1 Binding site | HSA-Drugs + ADR2 Binding Site |
|---|---|---|---|
| WAR | I & II | I | I |
| AZA | III | III | III |
| PBZ | III | II | III |
| OPBZ | II | I | I |
| IMET | III | III | III |
| CMPF | I & II | I | I |
| IBU | II | I & III | III |
| FA | II | III | III |
| DIAZ | III | III | III |
| DIFLU | II | I & III | III |
| INSL | III | I & III | I & II |
Based on the extent of destabilization of dye-protein complex by various drugs, we establish that CMPF results in a larger extent of decreasing the B.E of the dye-protein complex. The order of decreasing binding affinities of ADR1HSA complex by drugs is of the order CMPF > INDO > AZA > PBZ > OPBZ > WAR in the case of site I drugs, whereas in the case of site II drugs FA has maximum efficiency in decreasing the binding affinity of dye-protein complex. Apart from FA, all other site II drugs exhibit a similar proportion of destabilization of the dye-protein complex.
Likewise, in the case of ADR2HSA the order of destabilization by site I drugs is as follows CMPF > PBZ > AZA > OPBZ > PBZ > INDO > WAR. Interestingly the order of destabilization of ADR1HSA by site I drug is entirely different from ADR2HSA system. Similarly, in the case of site II drugs the order of destabilization is DIFLU > IBU > INSL > FA > DIAZ.
In order to establish a variation in the binding of amino acids of HSA with ADR1 and ADR2 dyes in the presence of these drugs a detailed account on the molecular interaction table was formulated and analyzed.
Non-conventional HB interactions accompanied with several Pi-Alkyl, Alkyl, Pi-Pi interactions results in several conformers of dye-protein interaction in the presence of drug. Among these drugs, CMPF destabilizes dye protein interaction efficiently compared to all other drugs. A significant change in the molecular interaction is visualized in the presence of CMPF. ADR1HSACMPF conformer possesses several conventional HB interactions along with unfavourable interaction which was not observed in any of the drug systems. Similar to CMPF, FA (site II) drug results in four conventional HB interactions. Interestingly, docking of DIFLU resulted in larger stabilization of ADR1 dye-protein complex which was not observed in the case of BSA. Through docking studies, it is evident that the drug CMPF which destabilizes ADR1HSA complex proceeds through HB interactions whereas the drug that promotes ADR1HSA binding occurs through hydrophobic interactions. From Table 12 it is evident that site II drugs promote several HB interactions except DIFLU accompanied with several Pi-Alkyl interactions. In the case of ADR2HSA complex, there is no unfavorable interactions. Except PBZ and indomethacin all the other drugs induces conventional HB interactions supported by hydrophobic interactions.
Even though studies regarding interaction of drugs with HSA provide the exact nature of interaction and the binding domains, we have established the role of dye governance on the drug binding with protein molecule. The literature reports on HSA docking with site 1 and site II drugs in the absence of ADR1 dye illustrates the amino acids that are involved in HB or hydrophobic interactions are entirely different. The amino acids that are predominantly involved in bonding for site I drugs are ARG257, ARG222, ARG218, TRP214, LYS195, LYS199, HIS242 and TYR150. Whereas with that for site II drugs are ARG348, GLU383, GLU450, ARG485, ARG410, SER489, LEU387, LYS414, LEU453, TYR411, LEU430, VAL433 and ASN391. The above studies authenticate that ADR dye which are neither site specific nor site selective ligand governs the position of drug binding with HSA.
Spectroscopic approach on the ground and excited state properties of both ADR1 and ADR2 dyes with BSA has established the presence of dye in several subdomains which are heterogeneous in nature. HSA and BSA are the two most extensively studied serum albumins regarding their structural aspects on denaturation, binding sites of drugs and fluorophores, yet they differ in the context of sequence of amino acids (Kumaran and Ramamurthy, 2010; Kumaran et al., 2015; Tamaraiselvan et al., 2021; Han et al., 2012). Both albumins share around 76 % sequence homology yet the intra distance between similar amino acids pair of the domain IIA and IIB respectively differ considerably and this accounts for the structural change of HSA with that of BSA (Huang et al., 2004). The fluorescence approach comprising time resolved fluorescence studies and circular dichroism methods establish the presence of dye in more than one microenvironment and predominantly influencing the primary structure of protein by hydrogen bonding interaction. Although these studies could not clearly establish the exact location of the ligand molecule in the subdomains of the protein molecule and the forces operating on the stability of the complex a substantial information on the bimolecular interaction were able to arrive. However, in the absence of large number of water molecules the contribution towards the molecular interactions were largely restricted such that a theoretical approach on 1:1 dye:protein and 1:1:1 dye:protein:drug complex could definitely provide a sufficient information resulting in the forces of attraction, thereby leading to stability of complex. As a consequence, Mol.dock methods has provided the better approach on the docking nature of dye with protein in the absence and presence of drug.
4.5 Binding stability of HSA-drug complex in the presence of ADR1 and ADR2
The comparison of binding stability of drug-protein in the presence of ADR dyes versus the binding affinity of ADR dyes-protein in the presence of drugs provides an interesting and significant information on the protein binding studies as provided in Tables 13 and 14 respectively. On this basis, a comparative docking of HSA-site I and site II drugs in the presence of ADR1 and ADR2 dyes were carried out to study the competitive binding of dye in the presence of drug-protein complex. The energetics and molecular interaction of HSA with site I and site II drugs are provided in supporting information table ST1 and ST2 and the 2D images of the conformers are given in supporting figure S9. It is evident that the addition of dye enhances the B.E of the drug-protein complex which was not observed on the addition of drug to dye-protein complex, rather it decreases the binding affinity of dye-protein complex. Interestingly, both the dye displaces the drug from its specific binding sites, which was found to be more effective in the case of site II binding drugs displaced to other non-specific binding domains. The energetics and molecular interaction of HSA-site I and HSA-site II drugs with ADR1 and ADR2 dyes are provided in supporting information table ST3, ST4, ST5 and ST6. The 2D images of the conformers of HSAsiteI-ADR1 and HSAsiteII-ADR2 are given in supporting figure S10 and S11.
In aqueous solution involving dye-protein-drug interactions, there are several factors influencing the binding interactions of host–guest complex. There are drug related factors pertaining to physiochemical characteristics, concentration and affinity of drug towards a particular binding component. However, the present study clearly focusses on the most probable binding nature of drug with protein in the presence of a competing ligand such that the view on binding sites and domains are established for better understanding regarding number of binding sites, domains and allosteric changes that occur in the protein molecule. Competitive binding of two drugs for a single binding site in albumins have been a larger role of research for several years and the role of acidic drugs and basic drugs vying for similar sites provided an excellent platform for binding sites. In the present report a competing ligand, a water-soluble dye in the presence of drugs provides an easier approach towards the fascinating field of protein chemistry.
5 Conclusion
Compared to ADR1 dye, ADR2 dye dock favourably with the protein as resulted from the energetics. Apart from Sudlow binding sites, the dye largely prefers to reside in site III apart from all other binding sites. ADR dye predominantly docks within the protein molecule in turns and coils rather than in helices of HSA. Both HB and hydrophobic interactions operate together in stabilizing the dye-protein complex. The influence of drug is not significant in the case of simultaneous docking of the dye. Mol.dock studies reveal that dye-protein complex is relatively stable even in the presence of site specific and selective drugs.
Acknowledgement
This work was funded by the Researchers Supporting Project Number (RSP2023R259) King Saud University, Riyadh, Saudi Arabia. This research was also funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2023R19), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The R.K thank Mrs. Shoba Gunasekaran for aiding us in MD techniques. R.K also thank Principal, Dr Dr.S. Santhosh Baboo and Shri. Ashok Kumar Mundra, Secretary, D.G. Vaishnav College (Autonomous), Chennai, for permitting us to avail the instrumentation facilities.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Egg white proteins and their potential use in food processing or as nutraceutical and pharmaceutical agents—a review. Poult. Sci.. 2013;92:3292.
- [Google Scholar]
- Biovia Discovery Studio Visualizer BIOVIA, Dassault Systèmes, Biovia Discovery Studio Visualizer, [3.0], San Diego: Dassault Systèmes, 2019
- Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D Biol. Crystallogr.. 2012;68:1278.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [Google Scholar]
- Probing the serum albumin binding site of fenamates and photochemical protein labeling with a fluorescent dye. Org. Biomol. Chem.. 2022;20(25):5085.
- [Google Scholar]
- The use of human, bovine, and camel milk albumins in anticancer complexes with oleic acid. Protein J.. 2013;37:203.
- [Google Scholar]
- Albumin as a drug—biological effects of albumin unrelated to oncotic pressure. Aliment. Pharmacol. Ther.. 2002;16:6.
- [Google Scholar]
- Albumin as a drug—biological effects of albumin unrelated to oncotic pressure. Aliment. Pharmacol. Ther.. 2002;16(5):6.
- [Google Scholar]
- Albumin and mammalian cell culture: Implications for biotechnology applications. Cytotechnology. 2010;62:1.
- [Google Scholar]
- Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol.. 2005;353(1):38-52.
- [Google Scholar]
- Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol.. 2005;353:38.
- [Google Scholar]
- Spectroscopic, structural and thermodynamic properties of chlorpyrifos bound to serum albumin: a comparative study between BSA and HSA. J. Photochem. Photobiol. B. 2012;109:1.
- [Google Scholar]
- Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom.. 2004;15:1237.
- [Google Scholar]
- Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics. 2013;13:3354.
- [Google Scholar]
- Exploring how structural and dynamic properties of bovine and canine serum albumins differ from human serum albumin. J. Mol. Graph. Model.. 2020;98:107601
- [Google Scholar]
- PET suppression of acridinedione dyes by urea derivatives in water and methanol. J. Phys. Chem. B. 2006;110(47):23783.
- [Google Scholar]
- Photophysical studies of PET based acridinedione dyes with globular protein: Bovine Serum Albumin (BSA) J. Lumin.. 2010;30:1203-1210.
- [Google Scholar]
- Photophysical studies on the interaction of formamide and alkyl substituted amides with photoinduced electron transfer (PET) based acridinedione dyes in water. J. Fluoresc.. 2011;21(6):165.
- [Google Scholar]
- Role of hydrogen-bonding and photoinduced electron transfer (PET) on the interaction of resorcinol based acridinedione dyes with Bovine Serum Albumin (BSA) in water. J. Lumin.. 2015;164:146-153.
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23:3.
- [Google Scholar]
- Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol. Immunol.. 2012;52:174.
- [Google Scholar]
- Unraveling the mysteries of serum albumin-more than just a serum protein. Front. Physiol.. 2014;5:299.
- [Google Scholar]
- Structural and Biochemical features of human serum albumin essential for eukaryotic cell culture. Int. J. Mol. Sci.. 2021;22:8411.
- [Google Scholar]
- (1995) All About Albumin: Biochemistry, Genetics and Medical Application; Academic Press Limited: Cambridge. USA: MA; 1995.
- Biophysical study on the interaction between eperisone hydrochloride and human serum albumin using spectroscopic, calorimetric, and molecular docking analyses. Mol. Pharm.. 2017;14(5):1656.
- [Google Scholar]
- A semiempirical free energy force field with charge-based desolvation. J Comput Chem.. 2007;28:1145.
- [Google Scholar]
- Seba Merin Vinod., Sangeetha, M.S., Anju, K., Keerthiga, R., Pradeep, K., Bharath, K., Kumaran, R. Complexity on the role of various site specific and selective Sudlow binding site drugs on the energetics and stability of Acridinedione dye-Bovine Serum Albumin complex: A Molecular Docking approach, ACS Omega, 10.1021/acsomega.2c07111.
- The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol.. 1975;11:824.
- [Google Scholar]
- Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol.. 1976;12:1052.
- [Google Scholar]
- Crystal structure of human serum albumin at 2.5 A resolution. Protein Eng.. 1999;12:439.
- [Google Scholar]
- Tamaraiselvan, R., Shoba, G., Sangeetha, M.S., Seba Merin Vinod, kumaran, R., Tamizhdurai, P., Amer M., A. and Ganesh, P. S. (2021). Comparative studies on biophysical interactions between 4-dicyanomethylene-2,6-dimethyl-4H-pyran (DDP) with bovine serum albumin (BSA) and human serum albumin (HSA) via photophysical approaches and molecular docking techniques. J. Saudi Chem. Soc. 25(12), 101364.
- Tamaraiselvan, R., Seba Merin Vinod., Sangeetha, M.S., Shoba, G., Tamizhdurai, P., Chinnadurai, R. and kumaran, R. (2022). Molecular docking studies on the binding interaction and stability of ovalbumin with an intramolecular charge transfer dye 4-dicyanomethylene-2,6-dimethyl-4H-pyran in the presence of an antibiotic: Tetracycline. J. Indian Chem. Soc. 99(10), 100681.
- Molecular docking approach on the molecular interactions involving beta-lactoglobulin (βLG)-4-Dicyanomethylene 2,6-Dimethyl-4-Hpyran (DDP) dye in the presence of an antibiotic, norfloxacin. J. Indian Chem. Soc.. 2022;99(6):100477
- [Google Scholar]
- Structure and enzymatic activities of human serum albumin. Curr. Pharm. Des.. 2015;21:1831.
- [Google Scholar]
- Characterization of proteins from grain of different bread and durum wheat genotypes. Int. J. Mol. Sci.. 2011;12:5878.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104701.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




