

Translate this page into:
New dimensions in triazolo[4,3-a]pyrazine derivatives: The land of opportunity in organic and medicinal chemistry
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
Synthesis of novel and potent hit molecules have endless demand. The triazolo[4,3-a]pyrazine scaffold is used as an essential building block/scaffold in organic synthesis, which works as a key template for the development of various therapeutic agents. It displays a wide spectrum of potential biological activities that have been progressively studied in recent years on account of their versatile structure and the diverse derivatives that can be synthesized from it to ensure functional motifs. This review article gives an inclusive account of the synthetic utility of triazolo[4,3-a] pyrazine derivatives employed in the design and synthesis of different types of compounds containing various active heterocyclic rings with greater emphasis in recent literature.
Keywords
ABT-341
Anti-diabetic activity
Diabetes mellitus
DPP-IV inhibitor
Sitagliptin
Triazolo[4,3-a]pyrazine

1 Introduction
Diabetes mellitus (DM) is an illness that shows the highest morbidity across the world. It is a metabolic disorder of the endocrine system, which is being displayed an inequity between spending and energy consumption. (Zhou et al., 2017) It is an enormously increasing global epidemic that influences millions of people. DM is a group of biological dysfunctions characterized by hyperglycemia causing directly from excessive glucagon secretion, inadequate insulin secretion, or insulin resistance. (Namoto et al., 2014; Zhou et al., 2017) It is triggered by various reasons and can be characterized by divided levels of blood glucose in the quick or post-glucose challenge state. It is increasing at the highest level owing to the aged people in developed countries and the fast appearance (79%) in low- and middle-income countries. (Namoto et al., 2014) As stated by the international diabetes federation (IDF), (http://www.idf.org/news/169:diabetes-atlas-report-463-million-with-diabetes.html) the worldwide number of diabetes mellitus patients are 463 million in adults between the age of 20–79 years in 2019 and is likely to upsurge to 578 and 700 million in 2030, 2045 (http://www.idf.org/news/169:diabetes-atlas-report-463-million-with-diabetes.html) respectively. There are two types of diabetes can be usually known: Type 1 diabetes mellitus (T1DM), in which patients generate none or slight insulin, which cut down glucose, and Type 2 diabetes mellitus (T2DM), wherein patients can produce insulin usually, but this insulin has a poor effect in regulating glucose consumption. (Pan et al., 2015) Among them, T2DM is a long-lasting metabolic illness distinguished by hyperglycemia in the body, leading to blindness, heart failure, amputations, acute vascular complications, obvious morbidity, mortality, and renal disease. (Xie et al., 2018)
The glucagon-like peptide 1, (also known as GLP-1) is an incretin hormone. (Jiang et al., 2015) It is an interesting topic for research associated with type 2 diabetes cure. (Kim et al., 2005) It came from the L cells of the small intestine, (Zhu et al., 2013) which activated glucose-induced insulin excretion, inhibits glucagon secretion, (Toft-Nielsen et al., 2001), and delays the gastric emptying for overall monitoring of the blood glucose. (Pei et al., 2006) It is one of the starting materials of DPP-IV (dipeptidyl peptidase-4). (Costante et al., 2015) (Zhu et al., 2013) The active form of GLP-1(7–37) peptide, is quickly deactivated by dipeptidyl peptidase IV (DPP-IV), a serine protease, through cleavage of a dipeptide from the N-terminus of GLP-1. (Zhu et al., 2013) As a result, inhibition of DPP-IV improves endogenous GLP-1 activity by lessening the degradation rate of GLP-1, which represents a promising approach to the treatment of type II diabetes (Zhu et al., 2013) in the many years since the metformin introduced. From the recent literature survey, there are a number of an active small molecule available for DPP-IV inhibitors, such as vildagliptin, (Villhauer et al., 2002) linagliptin, (Eckhardt et al., 2007) sitagliptin, (Kim et al., 2005) alogliptin, (Feng et al., 2007) and saxagliptin, (Augeri et al., 2005) (Fig. 1) has arisen as a constructive class of agents for the treatment of type 2 diabetes. The existing anti-diabetic agents such as sodium-glucose co-transporter-2 (SGLT-2) inhibitors, (Villhauer et al., 2002) α-glucosidase inhibitors, (Knudsen, 2004) insulin analogs, insulin sensitizers, (Zhu et al., 2013) incretins, dipeptidyl peptidase-4 (DPP-IV) inhibitors. (Kim et al., 2005) Nevertheless, currently used T2DM treatments typically originated various lateral effects, gastrointestinal reactions, urinary tract infections, and hypoglycaemia. (Augeri et al., 2005) Thus, there is an urgent need to discover novel oral anti-diabetic therapeutics with improved safety and efficacy profiles.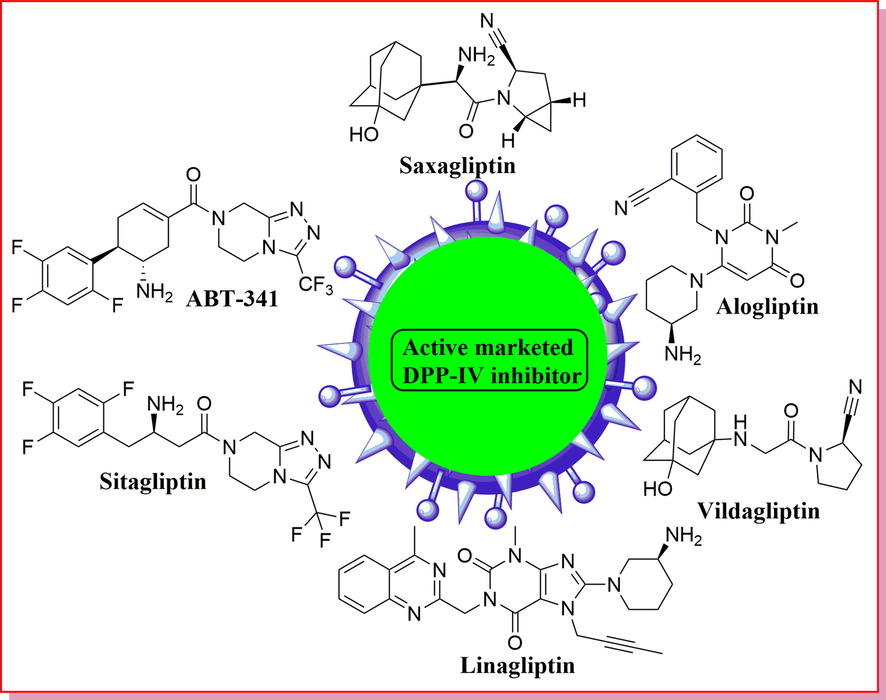
Marketed inhibitors of DPP-IV.
Heterocyclic compounds are a noticeable class of compounds in organic and medicinal chemistry. (Mannam et al., 2019) It has pharmaceutical importance because of their vital role in metabolism, and industrial applications. It can be used as an active structural unit in vitamins, many natural products, and a wide variety of drugs. (Jethava et al., 2019; Mannam et al., 2019) The chemistry of heterocyclic compounds is the most important field for medicinal chemistry research. It plays a remarkable role in the development of numerous potent biological agents in medicinal chemistry. Heterocyclic compounds mainly those with S, N, P, and O atom work as a unique and versatile scaffold for drug discovery of novel compounds. (Jethava et al., 2019) Nitrogen atoms are existing in most natural products and>80% of medicinal drugs permitted by the US-FDA. (Bao, Bayeh, & Tambar, 2013; Henkel, Brunne, Müller, & Reichel, 1999; Hili & Yudin, 2006) The significant properties in potent and hit small molecules due to the presence of nitrogen atoms which leads to improving water solubility under physiological conditions, desirable medicinal properties, hydrogen-bonding interactions with amino acid residues, and including favourable polar surfaces. (Bao et al., 2013) Consequently, many chemical methods have been established for the combination of nitrogen atoms into small molecules for drug discovery. (Bao et al., 2013)
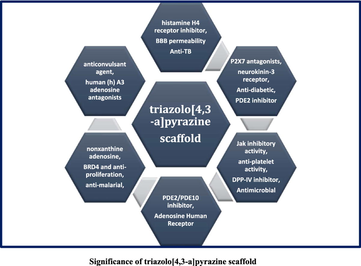
Fused heterocycle in which two or more dissimilar pharmacophore units are joint together in one molecule. The chemistry of fused scaffolds is considered to be a significant area of research which occupied a unique place in the realm of medicinal chemistry. (Bhoi et al., 2019a, 2014, 2019b; Jethava et al., 2019) As a result, they have found to be displayed improved pharmacological activity against various biological strains. Triazole is one of the key heterocyclic compounds which has several biological properties. (Tornøe, Christensen, & Meldal, 2002) Its derivatives have led to widespread use as significant intermediates in organic and medicinal chemistry research. It is an essential constituent of many alkaloids, pesticides, drugs, and dyes. (H. Li, Aneja, & Chaiken, 2013) This moiety has been applied in β-turn mimetic peptides as well as in isostere of the amide bond. (Azzurra et al., 2019; Costante et al., 2015; H. Li et al., 2013; Tornøe et al., 2002; Turner, et al., 2007) The biological profile and metabolic stability of the novel heterocyclic entities have been increased due to the presence of triazole moiety. (Azzurra et al., 2019; H. Li et al., 2013) The triazolo[4,3-a]pyrazine is one of the derivatives of triazole which is used as a precursor compound in the current study is a remarkable fused heterocycle of piperazine and 1,2,4-triazole. (Boucher et al., 2009; Mannam et al., 2019) It is mainly used in very important drug sitagliptin for diabetes. The scaffold, triazolo[4,3-a]pyrazine which has gained remarkable attention in drug discovery that used in the structural modification by using target compounds or drug intermediates with a combination of various functional groups that can be lead to a new drug molecule. From the present literature survey, triazolo[4,3-a]pyrazine analogues have presented numerous biological activities such as anticonvulsant activity, (Unciti-Broceta et al., 2005) human P2X7 antagonists, (Rudolph et al., 2015) human (h) A3 adenosine receptor antagonists, (Falsini et al., 2017) BRD4 inhibitory and anti-proliferation activity, (Xing et al., 2019) histamine H4 receptor (H4R) , (Jethava et al., 2019) adenosine human receptor, (Falsini et al., 2017) blood–brain barrier permeability, (Chrovian et al., 2016) neurokinin-3 receptor (NK-3) , (Jethava et al., 2019) non-xanthine adenosine receptor, (Unciti-Broceta et al., 2005) anti-diabetic, (Wei et al., 2016) brain Penetrant Phosphodiesterase 2 (PDE2) inhibitor, (Rombouts et al., 2015) anti-platelet aggregations, (Jethava et al., 2019; Xie et al., 2018) human renin activity, (Bradbury et al., 1990; Bradbury & Rivett, 1991; Roberts et al., 1990) good jak inhibitor, (Friedman et al., 2015) DPP-IV inhibitors, (Kim et al., 2005) anti-malarial, (Jethava et al., 2019) anti-tuberculosis, (Jethava et al., 2019) anti-bacterial, (Jethava et al., 2019) and anti-fungal, (Jethava et al., 2019) etc. in medicinal chemistry.
By inspiring the aforementioned significant biological activity of triazolo[4,3-a]pyrazine scaffold with active heterocyclic analogous, the present review article is intended to review on recent advance research progress concerning the synthesis of various triazolo[4,3-a]pyrazine derivatives through diverse methodologies along with biological activity which is a greater emphasis in current literature.
2 Synthesis of triazolo[4,3-a]pyrazine scaffold and it’s biological activity
2.1 Scheme 1
Kim and his co-workers have synthesized a novel, potent, β-amino amides and fused heterocycle containing a various derivative of triazolopiperazines as a DPP-IV inhibitor. (Kim et al., 2005) They have prepared triazolopiperazines derivative by typical peptide coupling reaction of β-amino acids with fused heterocycles. (Kim et al., 2005) They have initially focused on the synthesis of desired stereochemistry contained β-amino acids that were synthesized through the Arndt-Eistert homologation of the conforming R-amino acid (ee > 99% by chiral HPLC) which were prepared by Evans’s asymmetric azidation (Evans et al., 1990) or Schollkopf’s bis-lactam method(Schöllkopf et al., 1981) or one spot synthesis using silver benzoate. (Müller et al., 1998) They have prepared various fused heterocycles using pyrazine derivatives via the sequential reaction such as substitution and cyclization reaction. They have evaluated all the synthesized compounds as dipeptidyl peptidase IV (DPP-IV) inhibitors for the treatment of type 2 diabetes (Scheme 1). Among them, the phosphate salt of (2R)-4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl) butan-2-amine (MK-0431) is an effective, orally active DPP-IV inhibitor (IC50 = 18 nM) with excellent selectivity and good pharmacokinetic profile, as compared to others. It elected for the further development against type 2 diabetes mellitus due to their DPP-IV potency, in vivo efficacy, selectivity, and good drug-likeness profile.![Synthesis of (R)-3-amino-1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-phenylbutan-1-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig3.png)
Synthesis of (R)-3-amino-1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-phenylbutan-1-one derivatives.
2.2 Scheme 2
A new, potent, and large-scale synthesis of sitagliptin (MK-0431 compound, 13), have been described by Hansen and his colleagues as a DPP-IV inhibitor. It showed potential for the advanced treatment of type 2 diabetes. (Hansen et al., 2005) They have reported the synthesis of a triazolopyrazine scaffold containing a fragment of sitagliptin with only 26% yield after four successive synthesis steps using the medicinal chemistry synthesis route in their previous study. They have done research and development (R&D) and set key process step in the first step as well as the addition of hydrazine to chloropyrazine for the synthesis of 3-trifluoromethyl[1,2,4]triazolo- [4,3-a]piperazine with good safety operation on a larger scale-up reaction. They have also prepared a β-amino acid fragment of sitagliptin through asymmetric reduction of β-ketoester subsequently a two-step elaboration to an N-benzyloxy β-lactam. Hydrolysis of the lactam followed by direct coupling to the triazolopiperazine afforded the final product of sitagliptin with 52% practical yield over eight consecutive synthesis steps (Scheme 2a-2b). (Hansen et al., 2005)![Synthesis of methyl (R)-3-((benzyloxy)amino)-4-(2,4,5-trifluorophenyl)butanoate and 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine hydrochloride.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig4.png)
Synthesis of methyl (R)-3-((benzyloxy)amino)-4-(2,4,5-trifluorophenyl)butanoate and 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine hydrochloride.
![Synthesis of phosphate salt of (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,3,5-trifluorophenyl)butan-1-one.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig5.png)
Synthesis of phosphate salt of (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,3,5-trifluorophenyl)butan-1-one.
2.3 Scheme 3
A well-organized method for the synthesis of potent, selective, orally active and a novel series of β-amino amides bearing triazolopiperazines have been firstly discovered by Kim et al. and assessed as dipeptidyl peptidase IV (DPP-IV) inhibitors with wide-ranging SAR studies around the triazolopiperazine scaffold (Scheme 3a-3c) (Kim et al., 2008). It was revealed that a β-aminoacids in combination with triazolopiperazines with suitable substitution afford enormously potency of DPP-IV inhibitors which displayed high selectivity over other related enzymes, good pharmacokinetic profiles, and high in vivo ability in an OGTT in lean mice. It was also confirmed that (R)-stereochemistry containing isomer at the 8th position substitution of the triazolopiperazine moiety was mightily preferred over (S) isomer, concerning DPP-IV inhibition contrary to DPP-IV enzyme. They have also made a structural substitution for fine-tuning of relative inhibitory properties against DPP-IV and DPP-VIII by variations of substituents (R1, R2, and R3) around the triazolopiperazine moiety. Among these three different substituents, R3 substituents with (R)-stereochemistry containing isomer at the 8th-position, has of critical significance for the greater strength and more effective as compared to either with R1 at the 5th position or R2 at the 6th position. Among all synthesized compounds, Compound (R)-8-methyl derivative of sitagliptin (27b) (IC50 = 4.3 nM), displayed a four-fold increase in DPP-IV activity over sitagliptin, which showed noticeable in vivo efficacy in the lean mice OGTT. (Kim et al., 2008) While compound (R)-8-(4-fluorophenyl-substituted) sitagliptin (39b), which was notable for its greater potency (IC50 = 0.18 nM) activity against DPP-IV. It has been confirmed that (R)-stereochemistry containing isomer at the 8th position of triazolopiperazines has strongly preferred over (S) for DPP-IV inhibition from an X-ray crystal structure of compounds 27b and 39b in complex with DPP-IV enzyme. (Kim et al., 2008)![Synthesis of (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig6.png)
Synthesis of (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.
![Synthesis of (R)-3-amino-1-(5,5-dimethyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one and (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig7.png)
Synthesis of (R)-3-amino-1-(5,5-dimethyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one and (R)-3-amino-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.
![Synthesis of (3R)-3-amino-1-(8-subsituted-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig8.png)
Synthesis of (3R)-3-amino-1-(8-subsituted-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butan-1-one derivatives.
2.4 Scheme 4
As per the current literature survey, glucose regulating hormones such as GLP-1 and GIP are deactivated by Dipeptidyl peptidase IV (DPP-IV). Because of this reason, DPP-IV inhibition is one of the important biological targets for type 2 diabetes.(Costante et al., 2015) Keeping on this view, Pei and his co-workers(Pei et al., 2006) have done optimization with the help of previously reported hit molecules, by the high-throughput screening methods which lead three potent compounds such as 2-(4-((2-((2R,5R)-2-ethynyl-5-methylpyrrolidin-1-yl)-2-oxoethyl)amino)-4-methylpiperidin-1-yl)isonicotinic acid, (1R,2S)-2′-chloro-1,2,3,6-tetrahydro-[1,1′-biphenyl]-2-amine, and TFA salt of (1′R,2′S)-2-chloro-1′,2′,3′,6′-tetrahydro-[1,1′:4′,1′'-terphenyl]-2′-amine that led to the discovery of ((4R,5S)-5-Amino-4-(2,4,5-trifluorophenyl)cyclohex-1-enyl)-(3-(trifluoro-methyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone (ABT-341, 17). It is an extremely potent (IC50 = 1.3 nM against DPP-IV), a very selective, orally efficacious, and potentially active third-generation DPP-IV inhibitor (Scheme 4a-4b). They have also noted that when ABT-341 dosed orally, it’s worked dose-dependent that increased active GLP-1 levels, decreased glucagon levels, and reduced glucose excursion in ZDF rats. It was very safe in a cordless both in vitro and in vivo safety tests and might signify a new therapeutic agent against the treatment of type 2 diabetes. (Pei et al., 2006)![Synthesis of (1R, 2S)-4-(aminomethyl)-1,2,3,6-tetrahydro-[1,1′-biphenyl]-2-aminium TFA salt derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig9.png)
Synthesis of (1R, 2S)-4-(aminomethyl)-1,2,3,6-tetrahydro-[1,1′-biphenyl]-2-aminium TFA salt derivatives.
![Synthesis of various derivative of (1R, 2S)-4-(aminomethyl)-1,2,3,6-tetrahydro-[1,1′-biphenyl]-2-aminium TFA salt.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig10.png)
Synthesis of various derivative of (1R, 2S)-4-(aminomethyl)-1,2,3,6-tetrahydro-[1,1′-biphenyl]-2-aminium TFA salt.
2.5 Scheme 5
Mannam and his colleagues(Mannam et al., 2019) have established an efficient, simple, convenient, and rapid synthetic method for the synthesis of various urea (13a-13e) and thiourea derivatives (13f-13j) of 3-(trifluoromethyl)-5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-a]pyrazine, from 2-chloro-pyrazine through the successive chemical transformations such as substitution, induction, and ring reduction with overall 75–88% practical yield (Scheme 5). A new series of all the synthesized compounds (Mannam et al., 2019) have been characterized by various spectroscopic methods like IR, 1H NMR, 13C NMR, LCMS, and HRMS analysis. The main merit of the present protocol was a simple work-up, purification of intermediates via recrystallization processes, and good yield. They have screened all synthesized compounds for their in vitro antimicrobial activity against five bacterial and two fungi stains. Several compounds in these series, for example, compounds 7a, 7d, 7 g, 5i, and 7j displayed potential activity against bacterial and fungi strains with the good MIC values in the range of 6.25–25.0 µg/mL. They noted that thiourea derivatives showed better activity than urea derivatives. They have also carried out molecular docking studies (binding energies −8.1 to −9.8 kcal/mol) with no adverse effect in the active site of protein of poly(ADPribose)polymerase 15 (ARTD7, BAL3), macro domain 2 in complex with the adenosine-5-diphosphoribose enzyme (3V2B) and also calculated Lipinski’s rules of five, drug score and drug-likeness properties.![Synthesis of N-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxamide and N-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carbothioamide compound derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig11.png)
Synthesis of N-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carboxamide and N-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine-7(8H)-carbothioamide compound derivatives.
2.6 Scheme 6
New series of 4-((3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl) methyl)benzenamine derivatives (6a-6o) have designed and synthesized by Madje and his colleagues through the microwave-assisted protocol as well as conventional heating method (Scheme 6). (Patil et al., 2019) They established a rapid, cheap, safe method for the desired final compound with amide linkage without any kind of a base. They have observed that the reaction rate increased with reduced reaction time (8 h to 15 min.) and improved yield (50% to 98%) in the case of microwave irradiation compared to the classical heating method. They have confirmed final compounds by different spectroscopic methods and subjected for their in vitro anti-tubercular activity against mycobacterium tuberculosis H37Ra using an established XTT reduction menadione assay. Base on the biological results, compounds 6i, 6j, and 6 k showed great activity with an IC50 value (<2.0 μg/mL) and MIC value (>15 μg/mL). (Patil et al., 2019)![Synthesis of 4-((3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methyl)benzenamine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig12.png)
Synthesis of 4-((3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methyl)benzenamine derivatives.
2.7 Scheme 7
The optimization step is a key step for successful drug molecules. By using a similar strategy, Letavic et al. have described optimization efforts that directed them to a novel and hit molecular series of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl) methanone derivatives (4a-4y) which have a potential effect against not only the rat but also human P2X7 antagonists. They have prepared racemic and chirally pure desired compounds (4a-4y) using different chemical transformation such as protection, cyclization, de-protection, acid–amine as well as halo-amine reaction. They have selected some compounds for characterized in more detail and showed drug-likeness properties, which were capable of high P2X7 receptor tenancy in the rat by oral administration. Some compounds 4n (IC50 = 7.7 nM) and 4u (IC50 = 7.7 nM) showed best activity against rat and human P2X7 target. They also noted that some of the compounds were effective candidates for P2X7 PET tracers (Scheme 7a-7c) (Rudolph et al., 2015).![Synthesis of derivative of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig13.png)
Synthesis of derivative of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.
![Synthesis of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig14.png)
Synthesis of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone derivatives.
![Various derivative of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl) methanone.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig15.png)
Various derivative of methyl-substituted 1-(5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl) methanone.
2.8 Scheme 8
By pointing out the similar biological drug target of the P2X7 inhibitor, a new synthetic method for the synthesis of a various derivative of 8-phenyl substituted 5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazine as a potent P2X7 antagonist has been revealed by Chrovian and his colleagues(Chrovian et al., 2016) (Scheme 8a-8b). They have optimized for a potent compound for good blood–brain barrier penetrability and high P2X7 target engagement in the brain of rats. They have confirmed that the substituent at the 3rd position of the final compound has an important for both P2X7 rat potency and efflux tendency. They also found that -CF3 group remarkably improved brain infiltration as compared to other functional groups like alkyl groups. They have also studied structure–activity relationships (SAR), in vitro ADME properties, and in vivo pharmacokinetic data for important compounds. Compound (R)-(2-chloro-3-(trifluoromethyl)phenyl)(8-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone (20) (huP2X7 IC50 = 9 nM; rat P2X7 IC50 = 42 nM) loaded dose orally at 10 mg/kg in rats which attained 80% receptor occupancy for 6 h as measured by ex vivo radio-ligand binding autoradiography.![Synthesis of various derivatives of (2-chloro-3-(trifluoromethyl)phenyl)(8-phenyl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig16.png)
Synthesis of various derivatives of (2-chloro-3-(trifluoromethyl)phenyl)(8-phenyl-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.
![Synthesis of (S)-(2-chloro-3-(trifluoromethyl)phenyl)(8-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig17.png)
Synthesis of (S)-(2-chloro-3-(trifluoromethyl)phenyl)(8-phenyl-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone derivatives.
2.9 Scheme 9
Likewise, the synthesis, preclinical description, and structural activity relationship of a novel and biologically active series of 6,7-dihydro[1,2,4]triazolo[4,3]pyrazin-8(5H)-one derivatives (11a-11 t) have been described by Ameriks et al. as a P2X7 receptor antagonist. They have prepared achiral, racemic, and chiral compounds by the use of different organic synthetic transformation such as protection, cyclization, de-protection, and halo-amine reaction. They have introduced –CH3 group in the 6th position dramatically improved potency in not only in humans but also in the rat. As from the biological result, Compound 11d (hP2X7 IC50 = 0.7 ± 0.3 nM and rP2X7 IC50 = 79 ± 39 nM) has great potential, orally active, very selective, and brain-penetrant P2X7R antagonist in rat to enable broadly in vivo profiling (Scheme 9a-9c) (Ameriks et al., 2016).![Synthesis of 7-(2-chloro-3-(trifluoromethyl)benzyl)-6-substituted-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig18.png)
Synthesis of 7-(2-chloro-3-(trifluoromethyl)benzyl)-6-substituted-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one derivatives.
![Synthesis of (S)-3-amino-7-(2-chloro-3-(trifluoromethyl)benzyl)-6-methyl-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig19.png)
Synthesis of (S)-3-amino-7-(2-chloro-3-(trifluoromethyl)benzyl)-6-methyl-6,7-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-8(5H)-one derivatives.
![Synthesis of (S)-7-substituted-6-methyl-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4] triazolo[4,3-a]pyrazin-8(5H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig20.png)
Synthesis of (S)-7-substituted-6-methyl-3-(pyrazin-2-yl)-6,7-dihydro-[1,2,4] triazolo[4,3-a]pyrazin-8(5H)-one derivatives.
2.10 Scheme 10
The triazolo[4,3-a]pyrazine derivative has a superior interest because of their greater significant activity in contradiction of several diseases. Looking for appropriate effective drug candidates, Jethava et al. have presented the design, synthesis, characterization, biological activities, and computation study of novel triazolo[4,3-a]pyrazine derivatives (Scheme 10a-10c) (Jethava et al., 2019). All the synthesized compounds were carried out in their in vitro biological activities such as anti-malarial, anti-tuberculosis, anti-bacterial, and anti-fungal activities against plasmodium falciparum, H37Rv, various bacterial and fungal strains, correspondingly. They have also carried out a computational study such as molecular docking (using the active site of aspartic proteinase enzyme, PDB: 1PFZ), molecular dynamics (between 1PFZ-12c), ADMET properties, and Lipinski rule of five for the development of new Anti-mycobacterial agents.![Synthesis of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine hydrochloride.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig21.png)
Synthesis of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine hydrochloride.
![Synthesis of various derivatives of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig22.png)
Synthesis of various derivatives of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine.
![Synthesis of various derivatives of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig23.png)
Synthesis of various derivatives of 3-(trifluoromethyl)-5,6,7,8-tetrahydro-[1,2,4]triazolo[4,3-a]pyrazine derivatives.
2.11 Scheme 11
Colotta and his colleague have identified novel compound 1,2,4-triazolo[4,3-a]pyrazin-3-one as a new multipurpose scaffold against adenosine human receptor (hAR) antagonists. They applied the molecular simplification approach of their formerly reported series of 1,2,4-triazolo[4,3-a]quinoxalin-1-one derivatives (Scheme 11a-11b) (Falsini et al., 2017). By inspiring this practical approach, a novel set of 8-amino-2-aryl-1,2,4-triazolopyrazin-3-one derivatives which contained diverse substituents on the 2-phenyl ring and at 6th position, have been designed and synthesized for targeting the hA2A adenosine receptor. Many synthesized compounds were showed greater nano-molar (nM) affinities along with good selectivity against hA1, hA2A, and hA3 AR subtypes. It was noted that few derivatives of 8-amino-2-phenyl-1,2,4-triazolopyrazin-3-one which contained p-OR substituents on the 6-Ph ring, displayed high affinity (Ki = 2.9–10.6 nM) for completely selective in contradiction of hA2A AR subtype.![Synthesis of 8-amino-2-(4-aminophenyl)-6-phenyl-[1,2,4]triazolo[4,3-a]pyrazin-3(2H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig24.png)
Synthesis of 8-amino-2-(4-aminophenyl)-6-phenyl-[1,2,4]triazolo[4,3-a]pyrazin-3(2H)-one derivatives.
![Synthesis of 8-amino-2,6-diphenyl-[1,2,4]triazolo[4,3-a]pyrazin-3(2H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig25.png)
Synthesis of 8-amino-2,6-diphenyl-[1,2,4]triazolo[4,3-a]pyrazin-3(2H)-one derivatives.
2.12 Scheme 12
The various novel derivative of 2-arylpyrido[2,3-e]-1,2,4-triazolo[4,3-a]pyrazin-1-one with 4-oxo (series A) and 4-amino-substituted (series B) have been introduced against a new biological target of human (h) A3 adenosine receptor antagonists. They have designed molecules in such a way that contained hydrogen bond acceptor functional group like -OMe, –OH, -F, -COOEt on the 2-phenyl ring in series A and cycloalkyl and acyl residues on the 4-amino group in series B. Among all synthesized compounds, some compounds revealed high hA3 AR affinities (Ki < 50 nM) and selectivity vs both hA1 and hA2A receptors. The elected compound 4-benzoyl amino-2-(4-methoxyphenyl)pyrido[2,3-e]-1,2,4-triazolo[4,3-a]pyrazin-1-one (31) (Ki value 4.54 ± 0.2 nM) was tested in an in vitro activity against a rat model of cerebral ischemia which displayed to be effective in preventing the failure of synaptic activity induced by oxygen and glucose deprivation in the hippocampus (Scheme 12a-12b) (Colotta et al., 2009).![Synthesis of 2-(4-hydroxyphenyl)pyrido[2,3-e][1,2,4]triazolo[4,3-a]pyrazine-1,4(2H, 5H)-dione and 4-(1,4-dioxo-4,5-dihydropyrido[2,3-e][1,2,4]triazolo[4,3-a]pyrazin-2(1H)-yl)benzoate.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig26.png)
Synthesis of 2-(4-hydroxyphenyl)pyrido[2,3-e][1,2,4]triazolo[4,3-a]pyrazine-1,4(2H, 5H)-dione and 4-(1,4-dioxo-4,5-dihydropyrido[2,3-e][1,2,4]triazolo[4,3-a]pyrazin-2(1H)-yl)benzoate.
![synthesis of N-benzoyl-N-(1-oxo-2-phenyl-1,2-dihydropyrido[2,3-e] [1,2,4]triazolo[4,3-a]pyrazin-4-yl)benzamide compound and 4-amino-2-phenylpyrido[2,3-e] [1,2,4]triazolo[4,3-a]pyrazin-1(2H)-one derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig27.png)
synthesis of N-benzoyl-N-(1-oxo-2-phenyl-1,2-dihydropyrido[2,3-e] [1,2,4]triazolo[4,3-a]pyrazin-4-yl)benzamide compound and 4-amino-2-phenylpyrido[2,3-e] [1,2,4]triazolo[4,3-a]pyrazin-1(2H)-one derivatives.
2.13 Scheme 13
An isomer of formycin which was synthesized by two-step synthesis process of 8-amino-3-β-D-ribofuranosyl-1,2,4-triazolo[4,3-a]pyrazine (3), similar to 3-deazaadenosine, have been recognized and described by Schneller (Schneller et al., 1984) et al. They noted that compound (3) which was a very poor substrate for adenosine deaminase and an unalterable inhibitor of S-adenosyl-homocysteinase in the reaction route direction. It was inhibited the L1210 cell growth culture and not converted up to the nucleotide level in erythrocytes. However, it showed inhibition not only in the cellular uptake of nucleic acid precursors but also in their amalgamation into the nucleic acids of L1210 cells. It was confirmed that compound 3 was a very feeble antiviral agent and coronary vasodilator (Scheme 13).![Synthesis of (2R, 3S, 4R, 5R)-2-(8-amino-[1,2,4]triazolo[4,3-a]pyrazin-3-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol and 3-((2R, 3S, 4R, 5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-[1,2,4]triazolo[4,3-a]pyrazin-8(7H)-one.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig28.png)
Synthesis of (2R, 3S, 4R, 5R)-2-(8-amino-[1,2,4]triazolo[4,3-a]pyrazin-3-yl)-5-(hydroxymethyl)tetrahydrofuran-3,4-diol and 3-((2R, 3S, 4R, 5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-[1,2,4]triazolo[4,3-a]pyrazin-8(7H)-one.
2.14 Scheme 14
To determine the best approach to identify selective Jak1 inhibitors, Friedman and his co-worker had worked on initial optimization of their previously reported(Argiriadi et al., 2012) work about tricyclic pyrrolopyrazines as a kinase core inhibitor. They discovered a novel series of 1-substituted 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine derivatives which showed jak inhibitory activity. In this, they also designated their innovative efforts for the development of orally bioavailable analogs of 1-cyclohexyl-6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine which was very selective of Jak1 over Jak2 inhibitory activity (Scheme 14a-14b) (Friedman et al., 2015).![Synthesis of 1-substituted 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig29.png)
Synthesis of 1-substituted 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine derivatives.
![Synthesis of various derivative of 1-substituted 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig30.png)
Synthesis of various derivative of 1-substituted 6H-pyrrolo[2,3-e][1,2,4]triazolo[4,3-a]pyrazine.
2.15 Scheme 15
Synthesis of the novel, potent, and hit molecules has an everlasting demand. The design, synthesis, and biological evaluation of novel derivatives of pyrido[4,3-e][1,2,4]triazolo[4,3-a] pyrazines have been reported by Rombouts and his co-worker (Scheme 15). (Rombouts et al., 2015). All synthesized compounds showed the good biological result of brain penetrant PDE2 inhibitors after introducing lipophilic moiety on the m-position of the aromatic ring pendant from the triazolo scaffold. They have also described molecular dynamics calculations, free energy perturbation, ADMET properties, and Lipinski rule of 5 to forecast the SAR of original molecules from the phenyl/pyridyl group, its substitution, orientation, and capability to access the Leu770 lipophilic roof pocket. Among the all synthesized compounds, 1-(2-chlorophenyl)-4-methylpyrido[4,3-e][1,2,4]triazolo[4,3-a]pyrazine (11) (ED50 = 21 mg/kg) and 1-(2-chloro-5-((2,2,2-trifluoroethoxy) methyl)phenyl)-4-methyl-pyrido[4,3-e][1,2,4]triazolo[4,3-a]pyrazine (15) (ED50 value 3.6 mg/kg) presented good rat pharmacokinetic and in vivo receptor occupancy data.![Synthesis of pyrido[4,3-e][1,2,4]triazolo[4,3-a]pyrazines derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig31.png)
Synthesis of pyrido[4,3-e][1,2,4]triazolo[4,3-a]pyrazines derivatives.
2.16 Scheme 16
An efficient, one-pot, multi-component synthesis of 9-alkyl-6-chloropyrido[3,2-e][1,2,4] triazolo[4,3-a]pyrazines has been accomplished by Unciti-broceta (Unciti-Broceta et al., 2005) et al. which was easily prepared through the nucleophilic displacement of the 6-chloro group by hydrazides (Scheme 16). This designated approach was appropriate for superior libraries of compounds as tricycle nonxanthine adenosine antagonists. These were vital for the sympathetic of the different adenosinic receptors such as A1, A2a, A2b, and A3 along with their pharmacological activities.![Synthesis of 9-alkyl-6-chloropyrido[3,2-e][1,2,4]triazolo[4,3-a]pyrazines derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig32.png)
Synthesis of 9-alkyl-6-chloropyrido[3,2-e][1,2,4]triazolo[4,3-a]pyrazines derivatives.
2.17 Scheme 17
To find out the best approach to identify selective anticonvulsant agents, Kelley et al. have reported the synthesis of a novel, hit, and potentially active 11-substituted 8-amino-3-benzyl-1,2,4-triazolo[4,3-a]pyrazines derivatives (Scheme 17) (Kelley et al., 1995). They have prepared the desired compounds in four different organic synthetic steps such as substitution, addition, and cyclization reaction by using starting material (phenylacetonitriles 1) to get the final desired compounds. They have screened all the compounds by anticonvulsant activity on rats using maximal electroshock-induced seizures (MES). Amongst them, compounds 3-(2-fluorobenzyl)-N-methyl-[1,2,4]triazolo [4,3-a]pyrazin-8-amine (7) (oral ED50S = 2.6 ± 0.8 mg/kg) and 3-(2,6-difluorobenzyl)-N-methyl-[1,2,4]triazolo[4,3-a]pyrazin-8-amine (15) (oral ED50S = 3 ± 2 mg/kg) appeared as a best anticonvulsant agent.![Synthesis of 11-substituted 8-amino-3-benzyl-1,2,4-triazolo[4,3-a]pyrazines derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig33.png)
Synthesis of 11-substituted 8-amino-3-benzyl-1,2,4-triazolo[4,3-a]pyrazines derivatives.
2.18 Scheme 18–20
Bradbury and his co-worker (Bradbury & Rivett, 1991) have successfully synthesized two series of novel, efficient derivatives of 1,2,4-triazolo[4,3-a]pyrazine which were evaluated its human renin inhibitory activity. It incorporated with the transition-state mimetics of different compound such as (3S, 4S)- and (3R, 4S)-5-cyclohexyl-3,4-diaminopentanoic acid ((S)- and (R)-CDAPA), and (4S)-4-amino-5-cyclohexyl-2,2-difluoro-3-oxopentanoic acid (ACDFOPA). They have synthesized many compounds that showed good to moderate human renin activity. (Scheme 18a-18c).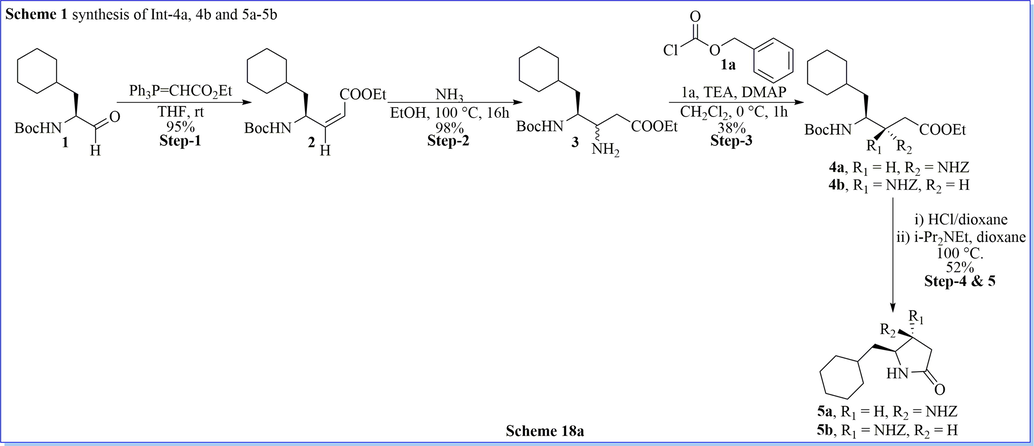
Synthesis of (R)-5-(cyclohexylmethyl)pyrrolidin-2-one derivatives.
![Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig35.png)
Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.
![Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig36.png)
Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.
As keeping on this view, Robert and his associates have also stated an innovative and efficient derivative of 1,2,4-triazolo[4,3-a]pyrazine which displayed good human renin inhibitory activity. It was integrated derivative of (1S, 2S)-2-amino-1,3-dicyclohexyl-1-hydroxypropane, statine (Sta), as well as (3S, 4S)-4-amino-5-cyclohexyl-3-hydroxy-pentanoic acid. They have also worked on the structure–activity relationship (SAR study) based on inhibitory activity (Scheme 19) (Roberts et al., 1990).![Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig37.png)
Synthesis of 1,2,4-triazolo[4,3-a]pyrazine derivatives.
By a similar study, Bradbury et al. has also reported synthesis which was an amalgamation between 2-(8-propyl-6-pyridin-3-yl-1,2,4-triazolo[4,3-a]pyrazin-3-yl)-3-pyridin-3-yl-propionic acid moiety and hydroxy ethylene isostere which contained amidic bond of (2S, 4S, 5S)-5-amino-6-cyclohexyl-4-hydroxy-2-isopropylhexanoic acid (Scheme 20a-20b) (Bradbury et al., 1990). They have been characterized well by various spectroscopic methods and evaluated their human renin inhibitory activity. The number of compounds was the effective candidate of this series that exhibited better inhibitory activity. Several compounds in these series, for instance, compound 6d was one of the best and potent molecules that presented excellent activity (IC50 value 0.2 nM), and also some derivatives (6e and 6 h-6 k) demonstrated IC50S < 1 nM.![Synthesis of [1,2,4]triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig38.png)
Synthesis of [1,2,4]triazolo[4,3-a]pyrazine derivatives.
![Synthesis of [1,2,4]triazolo[4,3-a]pyrazine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig39.png)
Synthesis of [1,2,4]triazolo[4,3-a]pyrazine derivatives.
3 Optimization for the synthesis of various functionalized triazolo[4,3-a]pyrazine derivatives using different reagents/catalyst
3.1 Scheme 21
Sitagliptin is a potent and very selective DPP-IV inhibitor for the cure of type 2 diabetes mellitus. A novel, green, one-pot, very well-organized synthesis of sitagliptin have been established by Karl and his co-workers (Hansen et al., 2009). The most useful intermediate dehydrositagliptin (8) was synthesized by using one pot, three steps reaction with good practical isolated yield (82%) along with good compound purity (>99.6%) as well (Scheme 21a-21b) (Hansen et al., 2009). They have used a very low amount of Rh(COD)2OTf/t-Bu-JOSIPHOS (15 mol%) for extremely enantioselective hydrogenation of dehydrositagliptin (8), to give sitagliptin as phosphate salt with good chemically (99.9%) and chirally/optically (>99.9% ee) purity. The advantages of this method include atom-efficient, high-yield, simple high efficiency, mild reaction conditions, convenient operation, and environmentally benign conditions which lessens the total waste generated per kilogram of sitagliptin produced as compared to the first-generation synthetic route. After completion of the reaction, the recovery of valuable rhodium metal made this method highly cost-effective which has been applied to manufacturing scale-up reaction. They got the presidential green chemistry award and the ICHEME Astra-Zeneca award for this efficient process in terms of essential reductions in waste.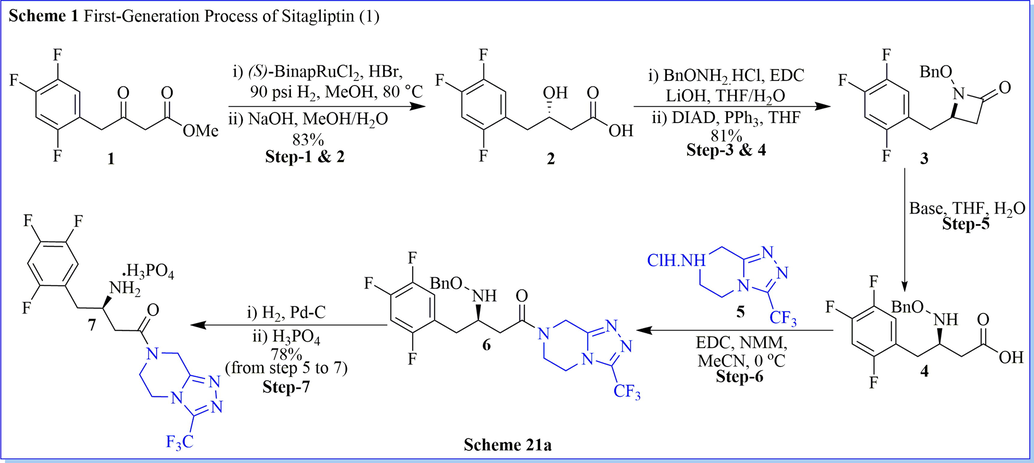
Synthesis of sitaglipttin.
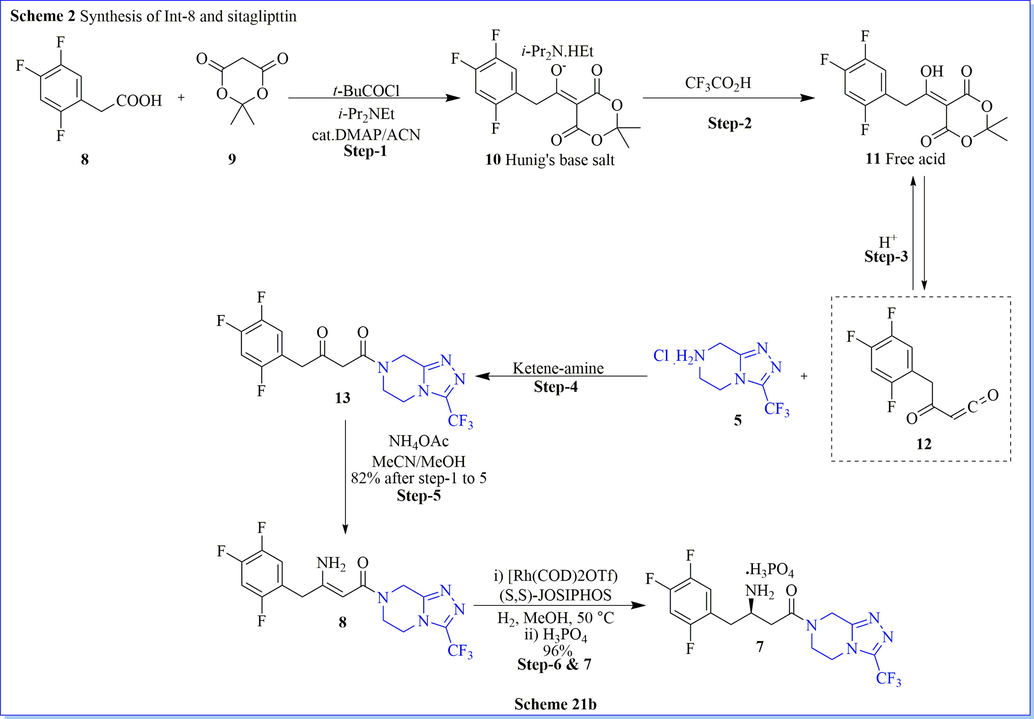
Synthesis of Int-8 and sitaglipttin.
3.2 Scheme 22
The asymmetric organo-catalysis have had significant importance in asymmetric synthesis. It has been established for organic synthetic reactions related to the pharmaceutical industry due to its high efficiency, selectivity, simple operational, and economic benign. Keeping in this view, Song (Bae et al., 2016) et al. have established extremely enantioselective mannich reactions using cinchona-based-squaramide as organo-catalyst, resulting in varied chiral mannich products with good practical yield. They have developed dibenzyl dithiomalonate (DTMs) as organo-catalyst which was a very effective mannich donor with good stereo-selectivity as well as chemical reactivity. They have reduced catalyst loading (up to 0.1 mol%) along with outstanding (≤99% ee) enantioselectivity on account of the superior reactivity of dithiomalonate (DTMs) comparison with the conventional malonates. Additionally, they have successfully synthesized very potent antidiabetic drug (R)-(-)-sitagliptin by the use of a DTMs (Scheme 22a). Likewise, Davies (Davies et al., 2012) et al. have reported the chiral synthesis of (R)-(-)-sitagliptin after seven successful steps using the highly diastereoselective protecting group strategies using not only lithium (R)-N-benzyl-N-(α-methylbenzyl)amide (yield = 43%) but also lithium (R)-N-benzyl-N-(α-methyl-p-methoxybenzyl)amide (yield = 42%) with good enantioselectivity (Scheme 22b-22c).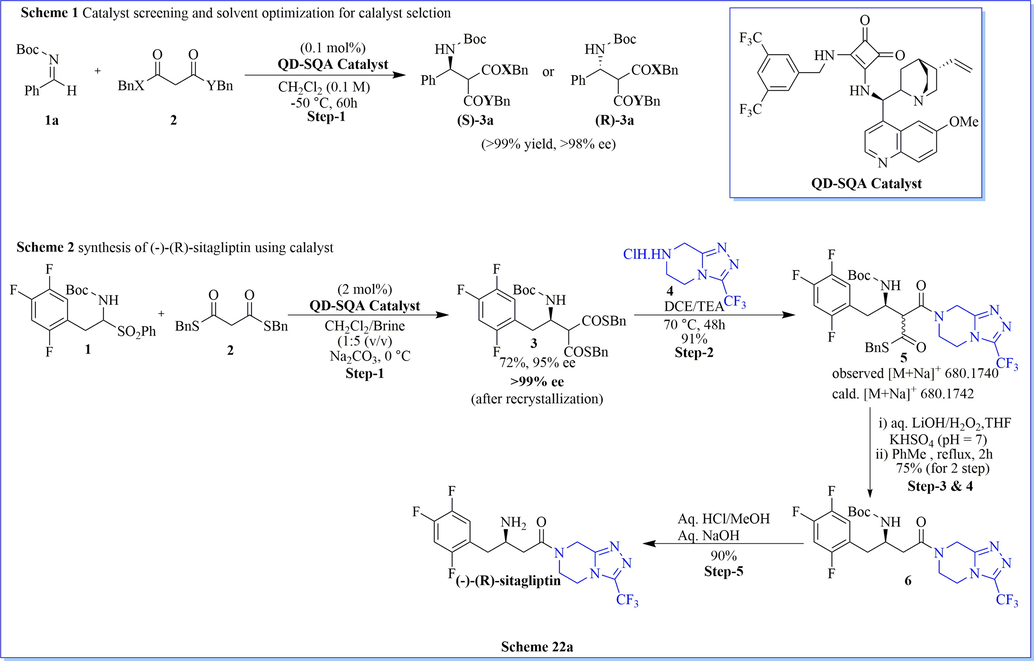
Synthesis of (-)-(R)-sitagliptin.
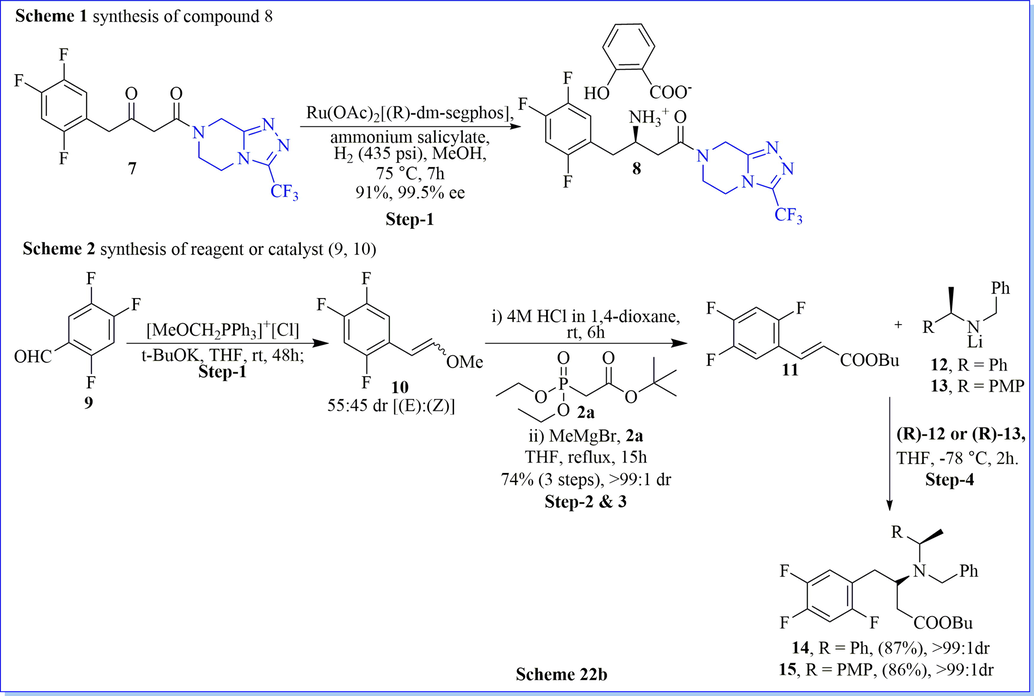
Synthesis of reagent or catalyst 8–10.
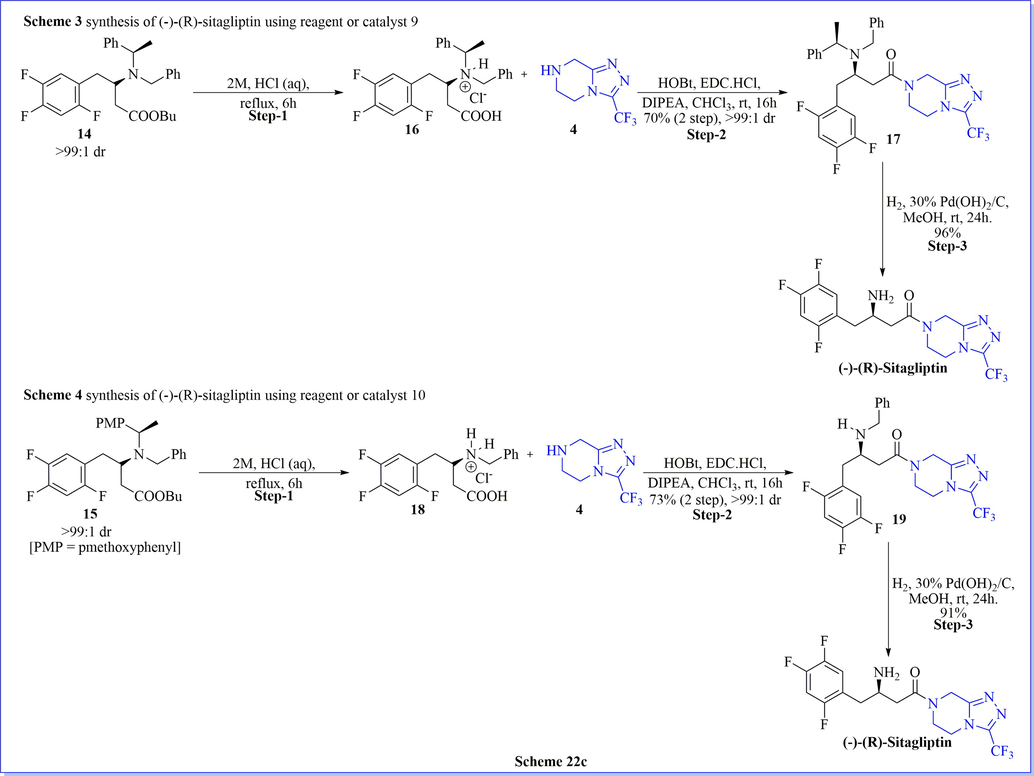
Synthesis of (-)-(R)-sitagliptin using catalyst 9 and 10.
3.3 Scheme 23
The chemical resolution of chiral compounds is one of a remarkable method for the separation of stereoisomer from racemic compounds/mixture. Base on the similar concept, an efficient, maiden, environmentally benign, and practicality possible advanced chemical resolution method for resolution by the reaction between N, C-unprotected β-amino acids (β-AA) and NiCl2 to form in situ enantioselective nickel(II) complexes of β-AA Schiff bases with disassembly, as a result providing the target enantiomerically pure β-AAs compounds in good isolated yield and excellent enantioselectivity (up to > 99:1). The main merit of this method was a simple operation, a two-step process, high efficiency, mild reaction conditions without specific requirements such as inert atmosphere, controlled low temperature, no particularly reagents and no dry solvents require, etc.. This method showed broad synthetic generality as β-heteroaryl-, various β-aryl-, and β-alkyl-containing β–amino acids could be efficiently resolved to enantiomerically pure form. The designed chiral ligands (S)- and (R)-N-(2-benzoylphenyl)-1-benzylpyrrolidine-2-carboxamide derivatives were very cheap, quantitatively recycled, and reusable. This method was used for the synthesis of (R)-sitagliptin with a good enantiomeric pure form which can be dependably reproduced with a larger scale, with even better performance (Scheme 23) (Zhou et al., 2014).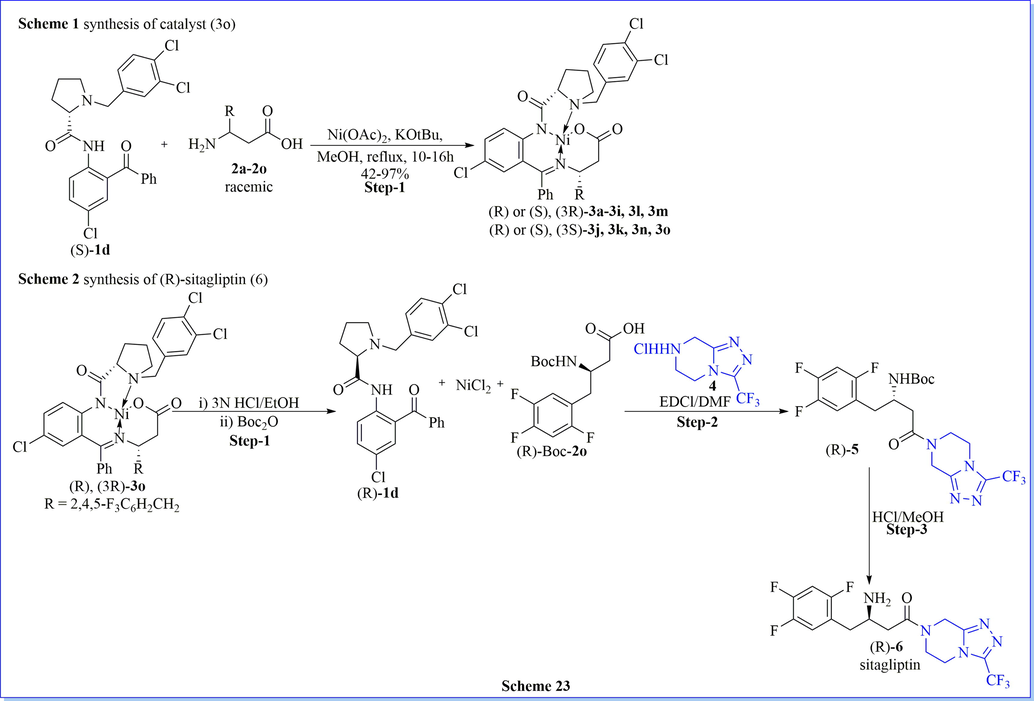
Synthesis of sitagliptin using Ni-complex catalyst.
3.4 Scheme 24
Asymmetric synthesis of sitagliptin is everlasting demand as of now. A scalable and efficient process of sitagliptin has been established by Kozlowski and his co-workers(Gutierrez et al., 2015) to access advanced sitagliptin intermediate by the use of homogeneous reagent-based reduction (NaBH4/MsOH) and followed by hydrogenation with H2 (105 PSI), 10% Pd/C (50% wet) in i-PrOH/water as a solvent with excellent diastereoselectivity. They have confirmed the mechanism of the asymmetric orientation of the desired product by density functional theory (DFT) study. DFT calculations disclosed that two noncovalent interactions (intramolecular hydrogen bond and novel C-H···F interaction) were liable for the higher diastereoselectivity (Scheme 24) (Gutierrez et al., 2015). In a similar study, Savile et al. (Savile et al., 2010) have synthesized sitagliptin from diketo-derivative with good stereo-selectively using transaminase/PLP, isopropylamine, and solvent at room temperature.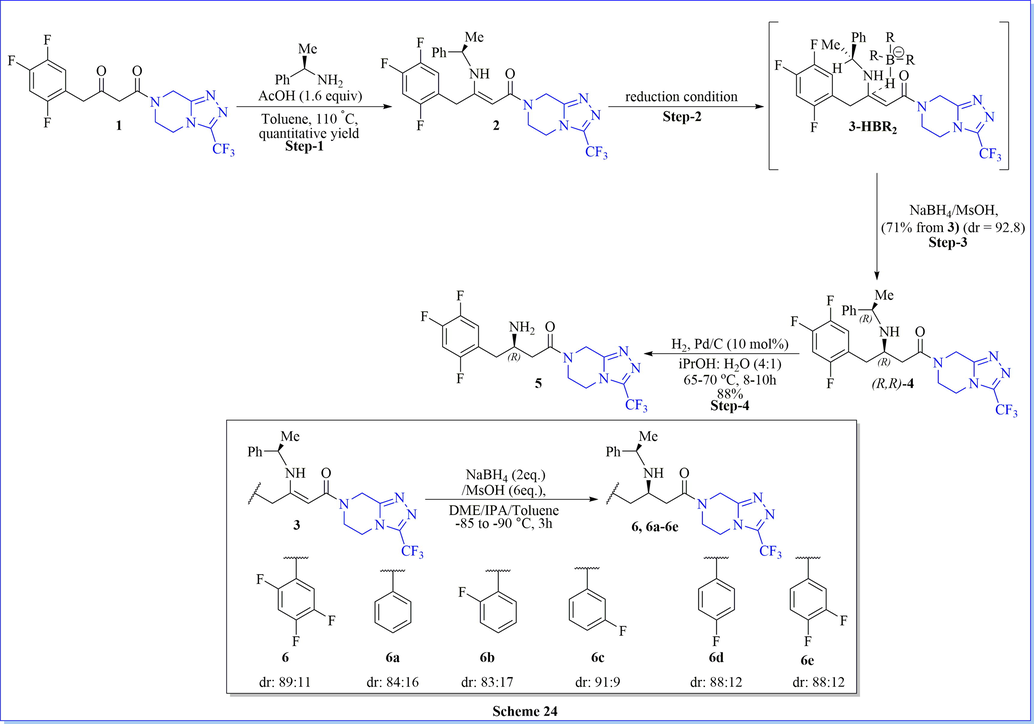
Synthesis of (R)-sitagliptin using NaBH4/MsOH.
3.5 Scheme 25
An efficient, simple, and applied route for the asymmetric synthesis of sitagliptin phosphate monohydrate (Scheme 25) (Gao et al., 2018) was disclosed by using chiral hemiacetal as the key intermediate starting from (E)-4-(2,4,5-trifluorophenyl)but-2-enal with 54% overall yield. The chiral hemiacetal part was built via tandem aza-michael/hemiacetal reaction catalyzed with 20 mol% of p-nitrobenzoic acid (PNBA) and (S)-2-(diphenyl((trimethylsilyl)oxy) methyl)pyrrolidine as organo-catalyst with good practical yield.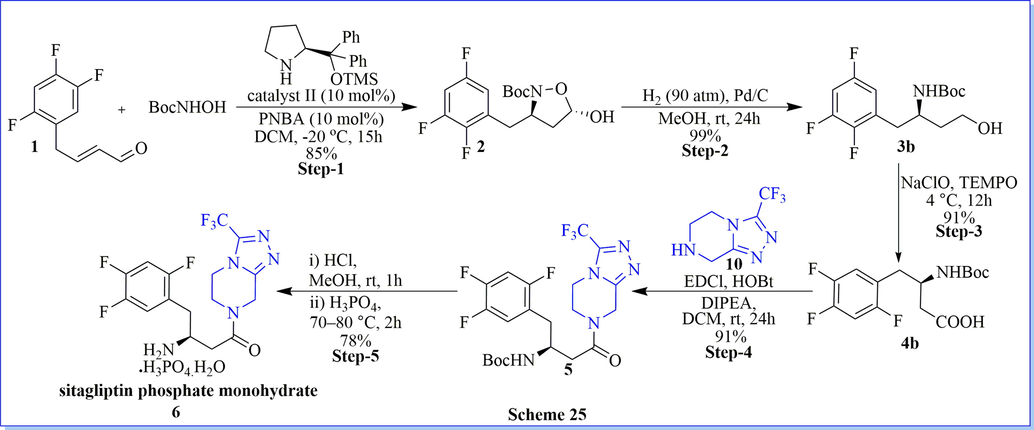
Synthesis of sitagliptin using (S)-2-(diphenyl((trimethylsilyl)oxy)methyl) pyrrolidine.
3.6 Scheme 26
β-amino acid derivatives are valuable chiral building blocks in the synthesis of pharmaceutical intermediates. An atom efficient reaction step, cost-effective approach, and the practical yield has been a comprehensive substrate scope nowadays. Keeping this view, a novel, well-organized, highly enantioselective reductive amination of β-keto amides to β-amino amides for the synthesis of sitagliptin (91% yield) by asymmetric induction. The advantage of this method was attributed to the very important properties of the Ru catalyst (Ru(OAc)2((R)-dm-Segphos) with extraordinary chemoselectivity as well as enantioselectivity (Scheme 26) (Steinhuebel et al., 2009).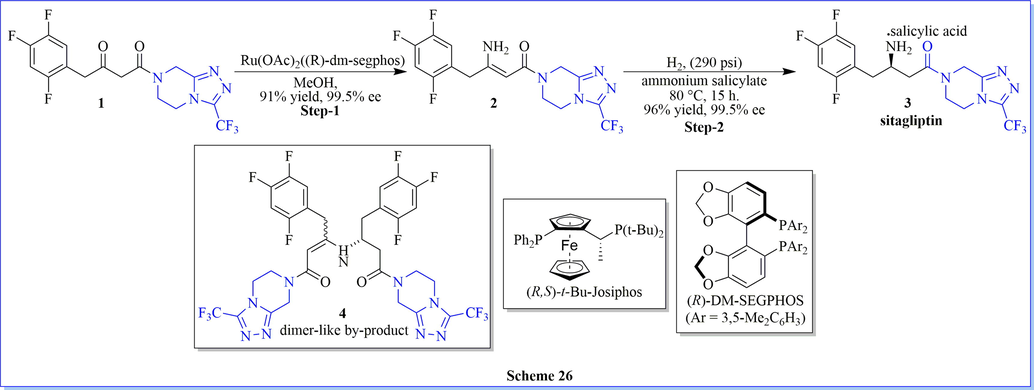
Synthesis of sitagliptin using Ru(OAc)2(R)-dm-segphos).
3.7 Scheme 27-31
An environment-friendly, synthesis of sitagliptin phosphate monohydrate as a dipeptidyl peptidase 4 (DPP-IV) inhibitor, have been completed after four successive steps from 1-{3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl-4-(2,4,5-trifluorophenyl) butane-1,3-dione with an overall 42.4% yield (Scheme 27) (Lin et al., 2013). The key step (stereoselective reduction) of this method was performed by using NaBH4/HCOOH with avoiding costly as well as venomous catalysts or ligands. Likewise study, many researched have worked on the synthesis of sitagliptin phosphate monohydrate by using different chiral/achiral reagent or catalysts such as chiral synthon (1,4-bis[(R)-1-phenylethyl]piperazine-2,5-dione) (scheme 28) (Subbaiah & Haq, 2014), chiral aziridine derivative, (scheme 29) (Pan et al., 2013), NaBH4/ZnCl2 (scheme 30) (Pan et al., 2015) and ammonium chloride (Scheme 31) (Clausen et al., 2006).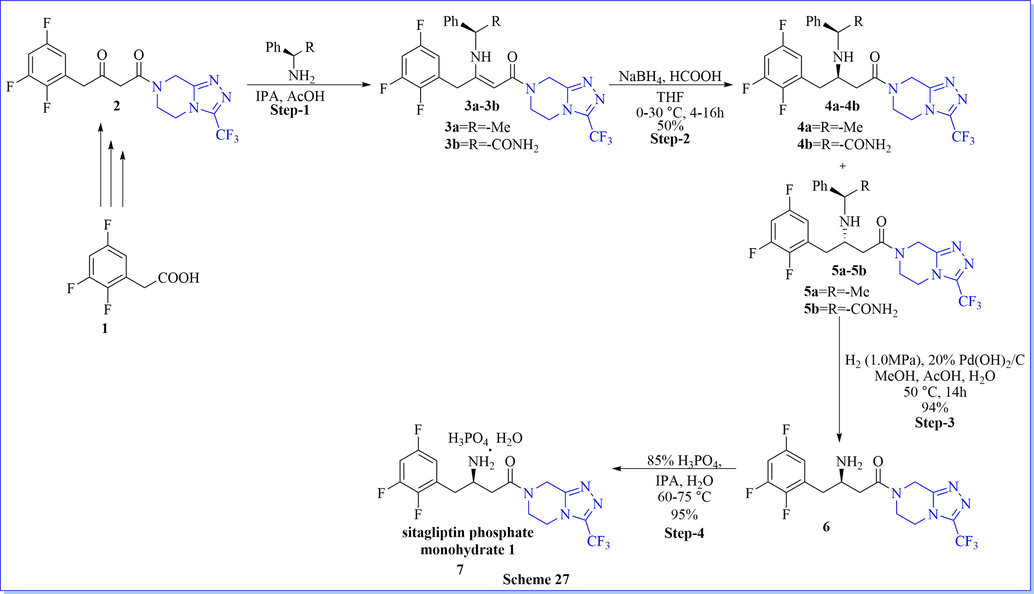
Synthesis of sitagliptin using NaBH4/HCOOH.
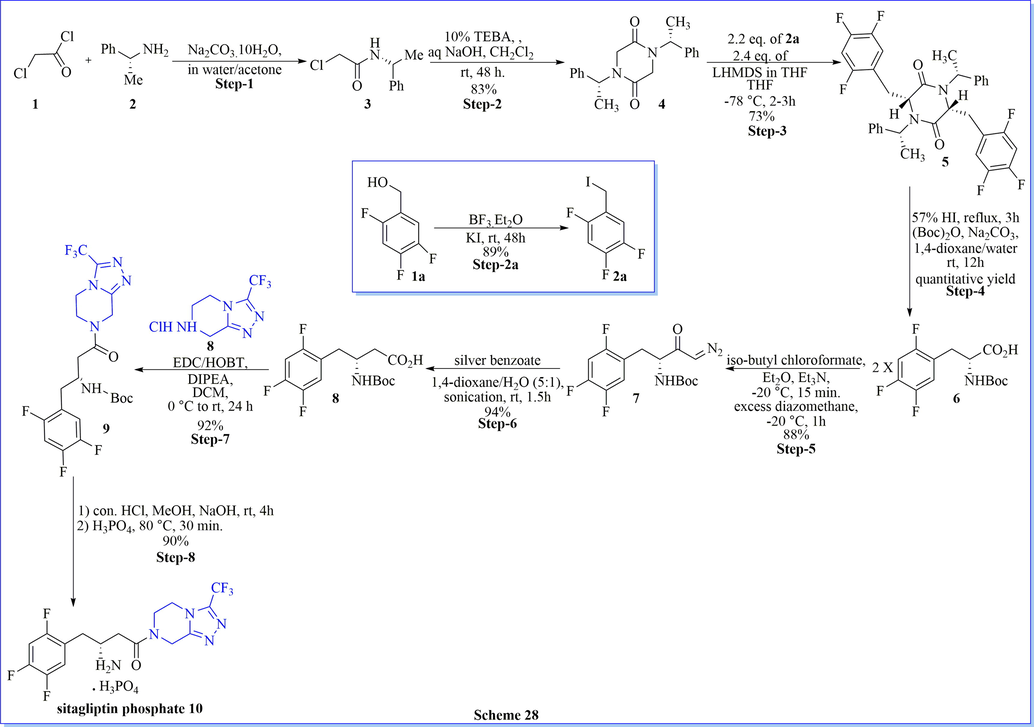
Synthesis of sitagliptin using different chiral/achiral reagent or catalysts.
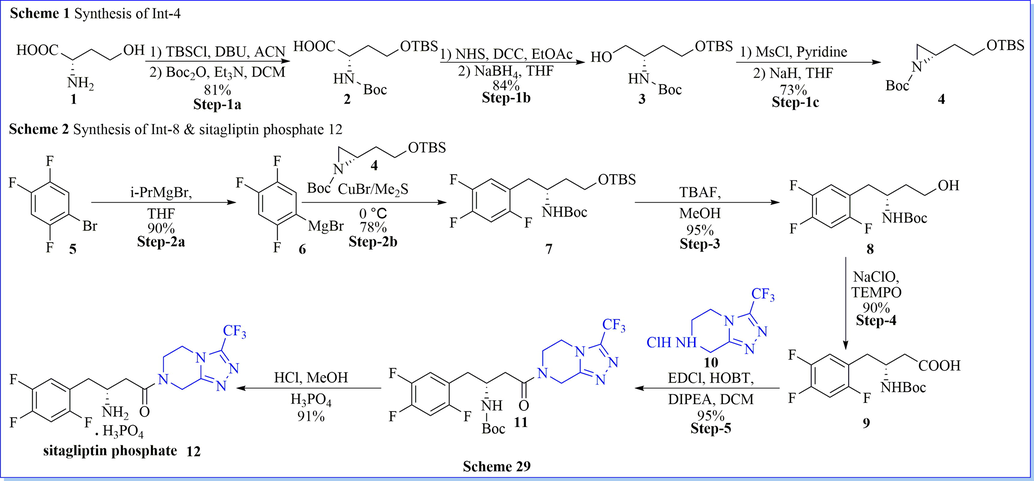
Synthesis of sitagliptin phosphate.
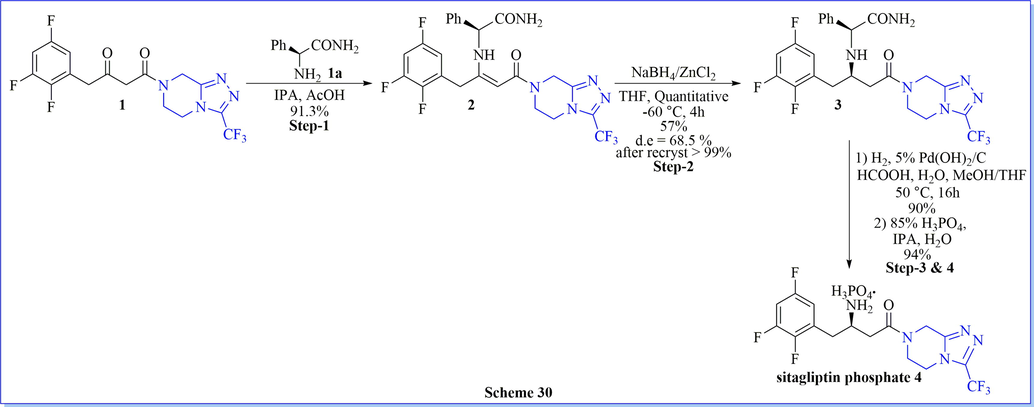
Synthesis of sitagliptin phosphate using NaBH4/ZnCl2.
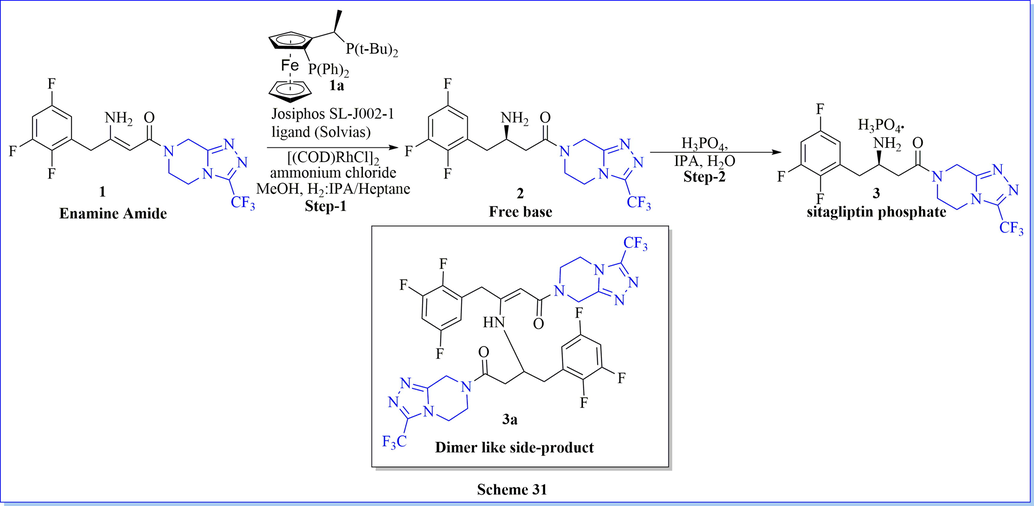
Synthesis of sitagliptin phosphate using ammonium chloride.
3.8 Scheme 32
Kang (2017) et al. have accomplished an extremely stereoselective and brief synthesis of chirally pure sitagliptin (>99.9% ee) with a 40.9% overall isolated yield after six reaction steps from the readily obtainable 2,4,5-trifluorobenzaldehyde as starting material. The main feature of this synthesis method which included embellishment of the chiral β-amino acid moiety of sitagliptin by the use of a highly stereoselective enolate addition of allyl ethyl malonate to the chiral sulfinyl imine in>99:1dr ratio. The merit of this method was cheap, highly efficient, and ecologically friendly (Scheme 32) (Kang et al., 2017).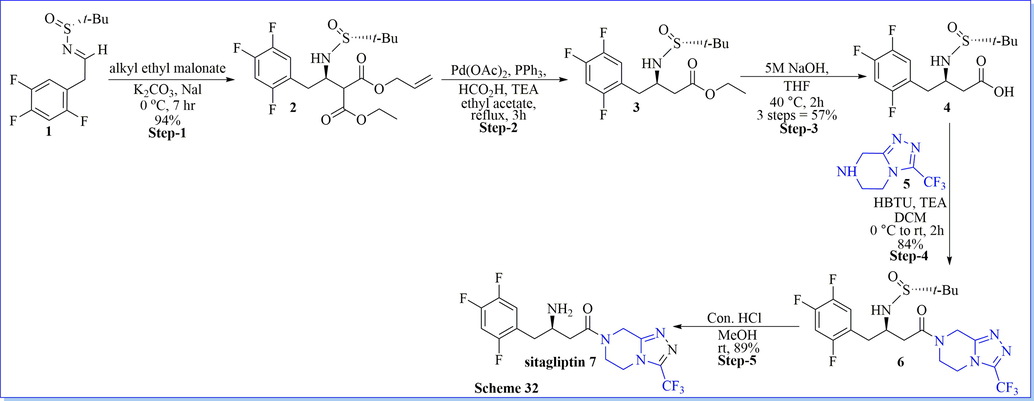
Synthesis of sitagliptin.
3.9 Scheme 33
Nitrogen atoms are significantly existing in nature as well as in>80% of pharmacological drugs accepted by the U.S. FDA. Most of the heterocyclic derivatives which contain nitrogen atom is work as a unique and versatile scaffold for the development of enantiomerically enriched amines. (Bhoi et al., 2014, 2019b) A similar concept was used for the enantioselective synthesis of ent-sitagliptin reported by using enantioselective allylic amination of a non-activated terminal olefin as a frugally effective and ecologically benign substitute for the manufacture of pharmaceutical drugs. This method was very useful for the synthesis of other nitrogen-containing pharmaceutical agents from low-cost and plentiful non-activated olefins (Scheme 33) (Bao et al., 2013).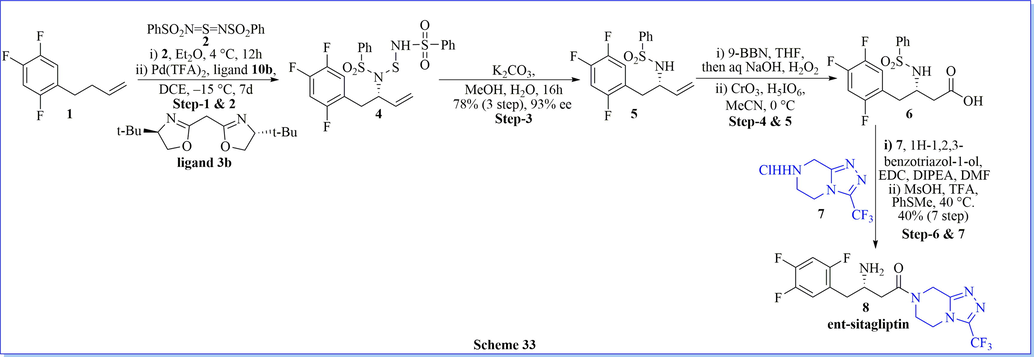
Synthesis of ent-sitagliptin.
4 Potential medicinal use of triazolo[4,3-a]pyrazine derivatives along with various biological activities
4.1 Scheme 34
Type 2 diabetes (included hyperglycemia) is considered by abnormal glucose production from the liver and utilized in the peripheral tissues. The production and consumption of glucose contribute to glycaemic control in diabetic patients. Base on this target, design, synthesis, and biologically evaluation of novel series of 4,7,12,12a-tetrahydro-5H-thieno[3′,2′:3,4]pyrido[1,2-b]isoquinolines (berberine derivatives) have been first presented as AMPK indirect activators for the treatment of type 2 diabetes. They have also carried out a SAR study base on the biological result. From the biological result, three compounds 4aa, 4bq, and 4bv reduced gluconeogenesis (up to 85%), improved pharmacokinetic profiles (bioavailability 45%), and enthused glucose consumption (1.8 to 2.3 time), compared to berberine as a standard drug (Scheme 34) (Zhou et al., 2017).![synthesis of 4,7,12,12a-tetrahydro-5H-thieno[3′,2′:3,4]pyrido[1,2-b] isoquinolines derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig56.png)
synthesis of 4,7,12,12a-tetrahydro-5H-thieno[3′,2′:3,4]pyrido[1,2-b] isoquinolines derivatives.
4.2 Scheme 35
Xing (Xing et al., 2019) et al have worked on bromodomain-containing protein 4 (BRD4) which emerged as a promising therapeutic target for many diseases including heart failure, cancer, and inflammation, etc.. They have previously worked on nitroxoline, which is a US-FDA approved antibiotic, possessed potential BRD4 inhibitory activity and anti-proliferation activity against leukemia cell lines. By rational approach of the previous study, a novel class of 5-((1H-imidazol-1-yl)methyl)quinolin-8-ol derivatives were designed and synthesized to find favorable drug-likeness properties of BRD4 inhibitors (Scheme 35) (Xing et al., 2019). They have also carried out SAR activity of synthesized compounds and also optimized it by using computational approaches such as docking (PDB: 6AFR), ADME/T profiles, etc.. At last, three auspicious drug molecule of BRD4 inhibitors such as compound 16, 17, and 19 were developed for potential new therapies for the treatment of selectivity multiple myeloma, acute monocytic-leukemia, and basal type triple-negative breast cancer.
Synthesis of 5-((1H-imidazol-1-yl)methyl) quinolin-8-ol derivatives.
4.3 Scheme 36–37
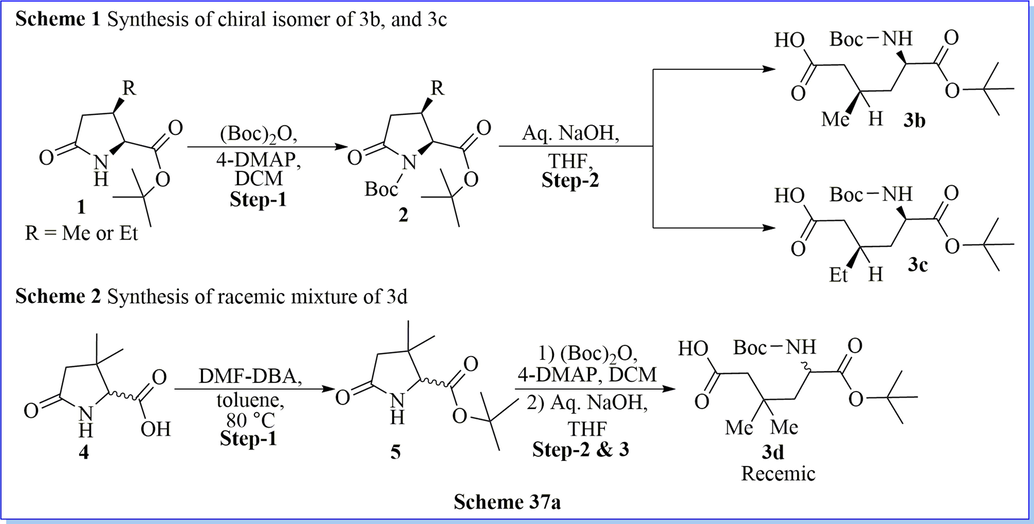
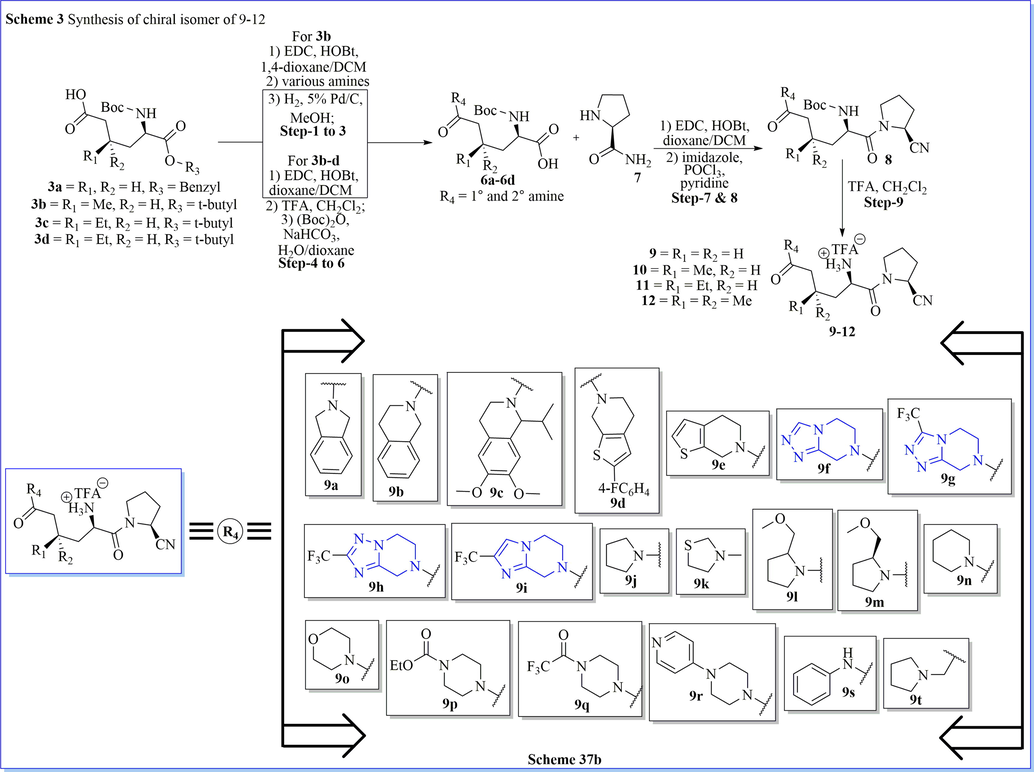
A superficial method for the synthesis of chiral and racemic 3,3-dimethylglutamic acid derivative (5/5a/5b) has been prepared after five successful steps of synthesis with easy protocol and good practical yield. By using 3,3-dimethylglutamic acid derivative (5b) as a building block, the author efficiently revealed a novel class of DPP-IV inhibitors, glu-pro-nitrile dipeptide mimics of (2S)-1-(2-amino-3,3-dimethyl-5-oxopentanoyl)pyrrolidine-2-carbonitrile derivatives, with high potency (IC50 value < 40 nM). They have also evaluated aqueous stability study of these DPP-IV inhibitors in different buffer solutions. From biological profile result, compound (2S)-1-(2-amino-5-((R)-2-(methoxymethyl)pyrrolidin-1-yl)-3,3-dimethyl-5-oxopentanoyl)pyrrolidine-2-carbonitrile (13c) is the most stable compound in this class of DPP-IV inhibitors (Scheme 36a-36b) (Hsu et al., 2011). Likewise study, Jiaang and his colleagues have worked on the preparation of (2S)-cyanopyrrolidines with glutamic acid derivatives and evaluated as inhibitors of dipeptidyl peptidase IV (DPP-IV). Compound 9 m which contained 3,3-dimethyl substituents showed IC50 = 14 nM DPP-IV inhibitors with an excellent selectivity profile over FAP (IC50 > 20 µM), DPP-II (IC50 > 20 µM) and DPP-VIII (IC50 = 14 µM) with admirable aqueous stability. It demonstrated the aptitude towards both significantly decrease the glucose excursion and inhibit plasma DPP-IV activity. Compound 9 m is used as a selective, potent, and orally obtainable inhibitor of DPP-IV (Scheme 37a-37b)(Tsai et al., 2009).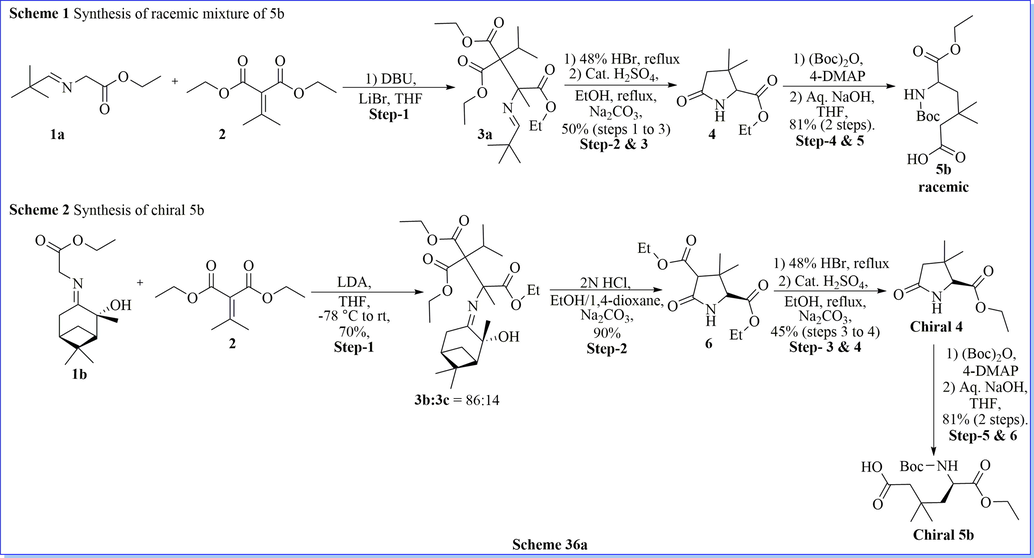
Synthesis of chiral and racemic compound of 5-((tert-butoxycarbonyl)amino)-6-ethoxy-3,3-dimethyl-6-oxohexanoic acid.
![Synthesis of chiral and racemic compound of (2R)-1-(2-amino-4,4-dimethyl-6-oxo-6-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)hexanoyl)pyrrolidine-2-carbonitrile.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig59.png)
Synthesis of chiral and racemic compound of (2R)-1-(2-amino-4,4-dimethyl-6-oxo-6-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)hexanoyl)pyrrolidine-2-carbonitrile.
Synthesis of chiral and racemic compound of 6-(tert-butoxy)-5-((tert-butoxycarbonyl)amino)-3-ethyl-6-oxohexanoic acid.
Synthesis of (2S)-cyanopyrrolidines derivatives.
4.4 Scheme 38
The improvement and progression of diabetic complications because of its many roles in the vascular system have been solved by nitric oxide (NO) dysfunction. Keeping on this view, xie (Xie et al., 2018) et al. described a design, synthesis of a novel series of NO-donating sitagliptin derivatives, and assessed as potential multifunctional anti-diabetic agents. All of the synthetic compounds have shown notable biological activity against in vitro dipeptidyl peptidase IV (DPP-IV) and worked as excellent hypoglycemic activities in diabetic mice, close to the standard drug sitagliptin. Synthesized compounds (4–7) revealed different degrees of NO-releasing aptitudes and powerful in vivo antioxidant abilities (Scheme 38) (Xie et al., 2018). Based on the screening data of DPP-IV, compound 7 was recognized as a potent DPP-IV inhibitor with the IC50 = 0.060 μM. Additionally, all the compounds showed different levels of NO-releasing capabilities and admirable anti-platelet aggregation in vitro activity. The docking study (PDB: 1X70) also gave support to the biological activity of compound 7 which has a favorable binding mode with the protein target.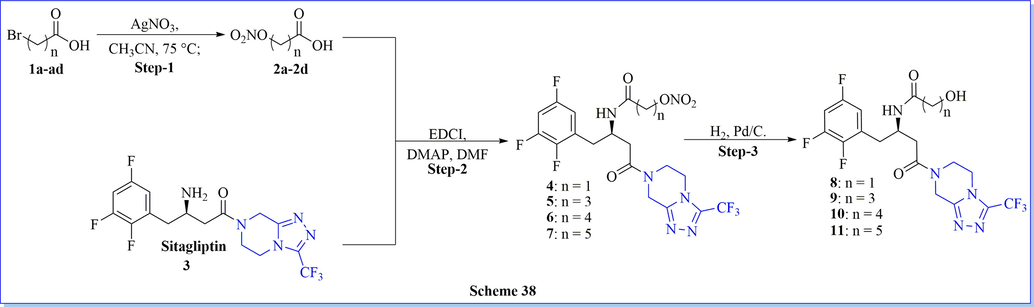
Synthesis of NO-donating sitagliptin derivatives.
4.5 Scheme 39
Synthesis of imidazo[1,2-a]pyridine derivatives has been reported by various reaction transformations such as bromination, cyclization, formylation, and acid–amine coupling reaction, etc. to get desired final compounds as DPP-IV inhibitors. Based on biological activity data and docking study (PDB: 4JH0), compound (2-(2,4-dichlorophenyl)imidazo[1,2-a]pyridin-3-yl)methanamine (6d) contained 2,4-dichlorophenyl group at the 2-position which worked as a novel hit (IC50 = 0.13 μM), very selective with the ratio of DPP-VIII/DPP-IV = 215, DPP-IX/DPP-IV = 192, and in vivo effective DPP-IV inhibitor (Scheme 39) (Q. Li et al., 2015).![Synthesis of imidazo[1,2-a]pyridine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig63.png)
Synthesis of imidazo[1,2-a]pyridine derivatives.
4.6 Scheme 40
With the help of the molecular modelling rational approach, the design and synthesis of a new, effective, and orally bioavailable cyclohexylamine derivative as a DPP-IV inhibitor was designated by using molecular modelling study of the x-ray crystal structure of sitagliptin. Compounds 13a and 13b were estimated in vitro activity for inhibition of DPP-IV, DPP-VIII, DPP-IX, QPP, and FAP activity. It exposed related strength to sitagliptin, excellent pharmacokinetic properties as well as an excellent profile in an OGTT assay (Scheme 40a-40b) (Biftu et al., 2007b). Among them, Compound (1S, 2R, 5S)-5-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4] triazolo[4,3-a]pyrazin-7(8H)-yl)-2-(2,4,5-trifluoro-phenyl)cyclohexan-1-amine (13a) was originated to be a potent DPP-IV inhibitor with IC50 value 21 nM by admirable in vivo activity as well as pharmacokinetic profile.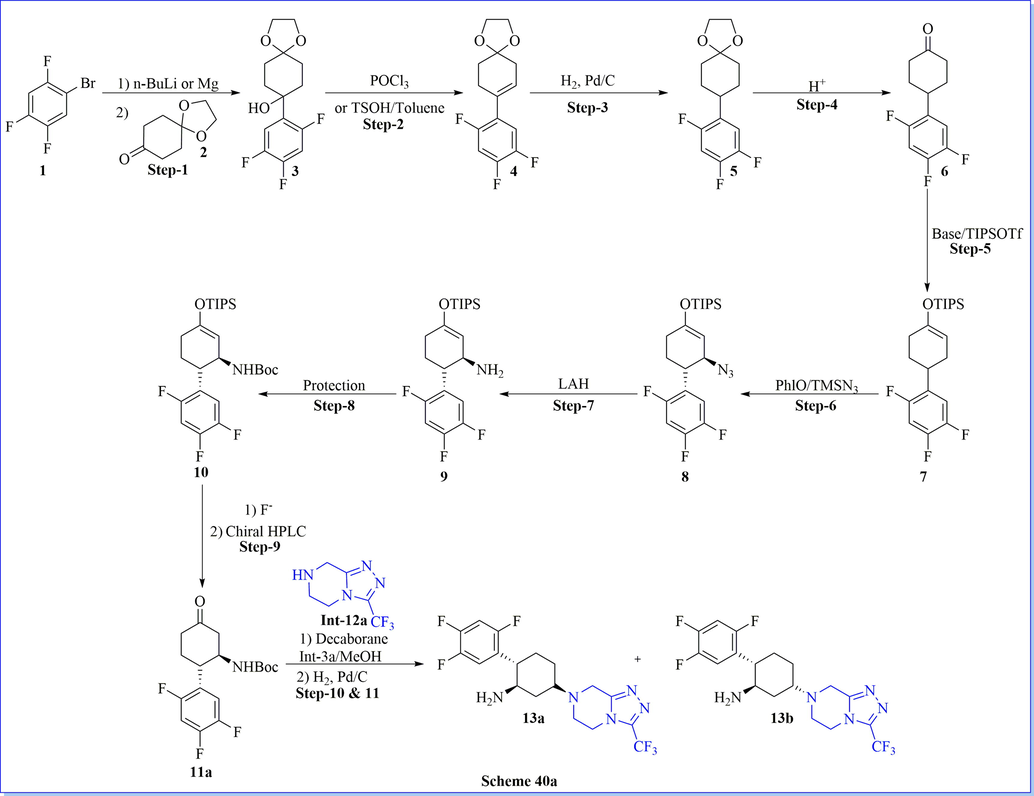
Synthesis of cyclohexylamine derivative.
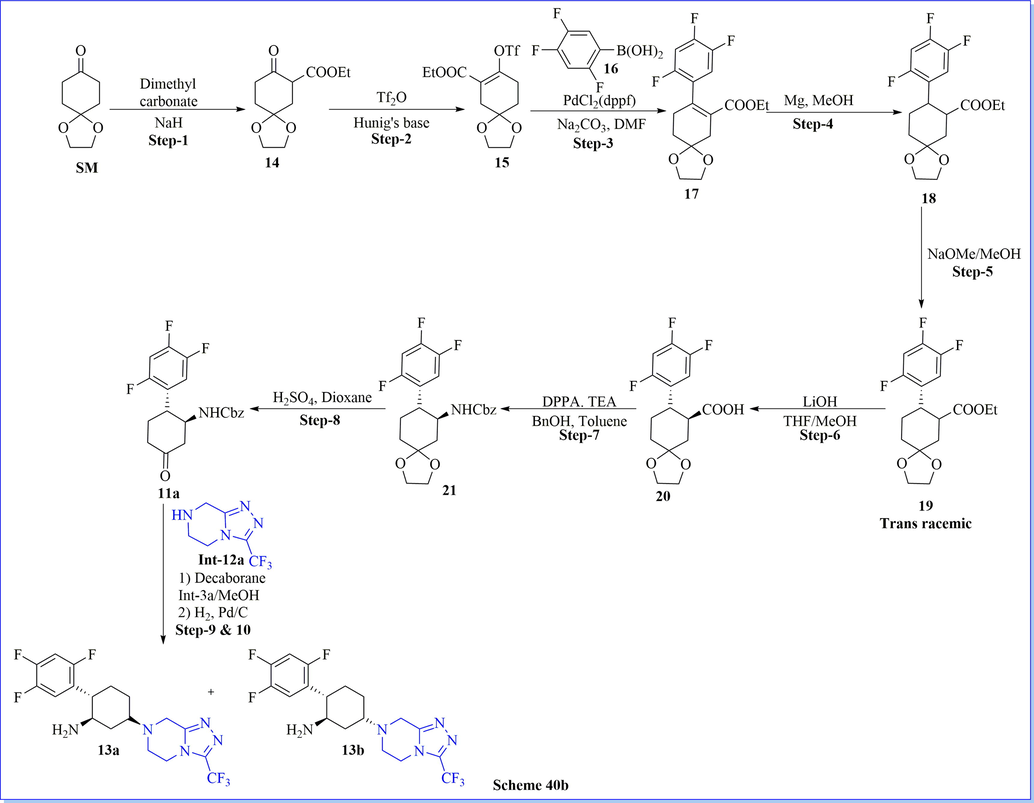
Synthesis of cyclohexylamine derivative.
4.7 Scheme 41
Dipeptidyl peptidase-4 (DPP-IV) inhibitors are accepted as a favorable class of agents for the treatment of type 2 diabetes. Discovery of a novel C-(1-aryl-cyclohexyl)-methylamine derivatives have been prepared for DPP-IV inhibition via six synthetic steps with good practical yield. They have carried out an antidiabetic activity and performed structure–activity relationship (SAR) for optimization to synthesized compounds which led to successful improvement of target potency in this novel chemo-type of compounds. As per the SAR concerned, compound ((1S,4S)-4-(cinnamyloxy)-1-phenylcyclohexyl)methanamine (syn-7aa) found to be potent, selective and orally bioavailable DPP-IV inhibitors (IC50 = 11.9 nM). The best hit derivative, 2-((1S, 4S)-4-(aminomethyl)-4-(3-chlorophenyl)-cyclohexyl)-6-(pyridin-3-yl)pyridazin-3(2H)-one revealed the expressively improved duration of DPP-IV inhibition at the low-nM level and possessed an excellent oral pharmacokinetic outline in the rat compared with the standard drug sitagliptin (Scheme 41a-41b) (Namoto et al., 2014).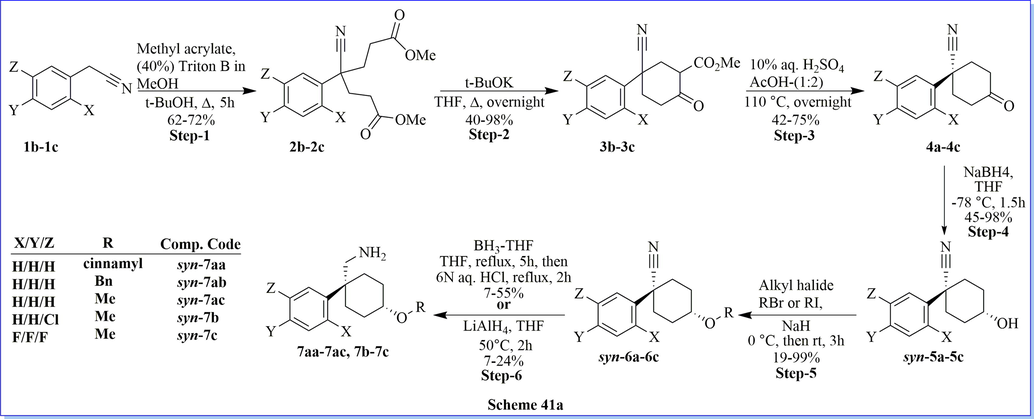
Synthesis of C-(1-aryl-cyclohexyl)-methylamine derivatives.
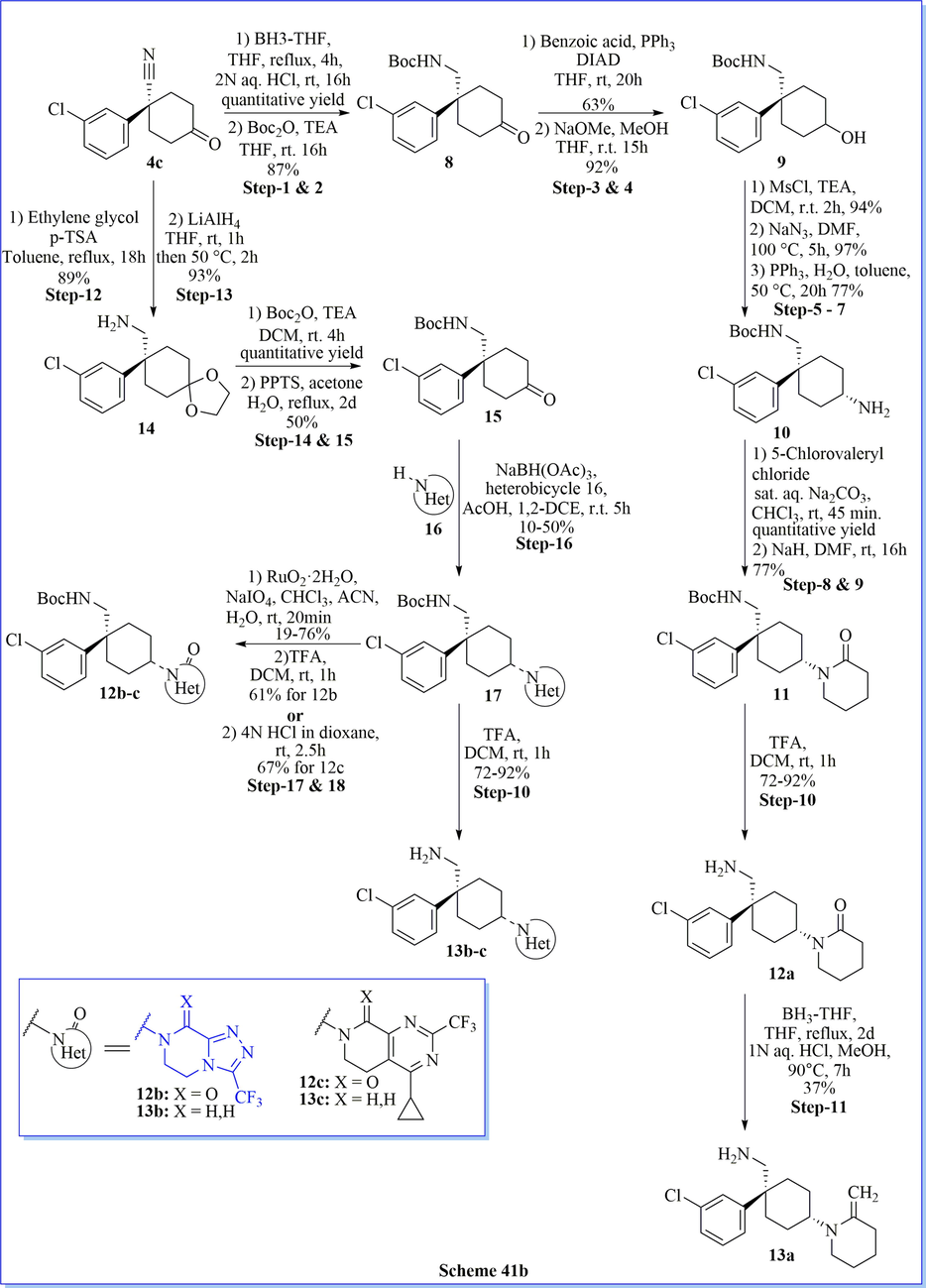
Synthesis of C-(1-aryl-cyclohexyl)-methylamine derivatives.
4.8 Scheme 42
Likewise similar biological profile, a novel combinational series of 4-amino cyclohexanes and 4-substituted piperidines derivatives have been prepared and screened for their biological activity of DPP-IV inhibition. These classes of compounds meaningfully improved selectivity in contradiction of proline based particular enzymes such as QPP, DPP-IIIV, and DPP-IX, etc.. Among these analogs, compound N-methyl-(2,5-dimethylisoxazol-yl)sulphonamide presented the greatest favorable potency (IC50 value 55 nM) and selectivity (>46 µM) as a DPP-IV inhibitor with a lack of good pharmacokinetic properties. While derivative of 4-(1,2,3-thiadiazolyl) benzenesulfonamide demonstrated extensive half-life and good oral bioavailability in the rat. The best effective derivative, 3-(5-aminocarbonylpyridyl)piperidine, displayed outstanding DPP-IV activity (IC50 = 18 nM) with respectable selectivity (IC50 = 18–27 µM) contrasted with other proline enzymes (Scheme 42) (Chen et al., 2011).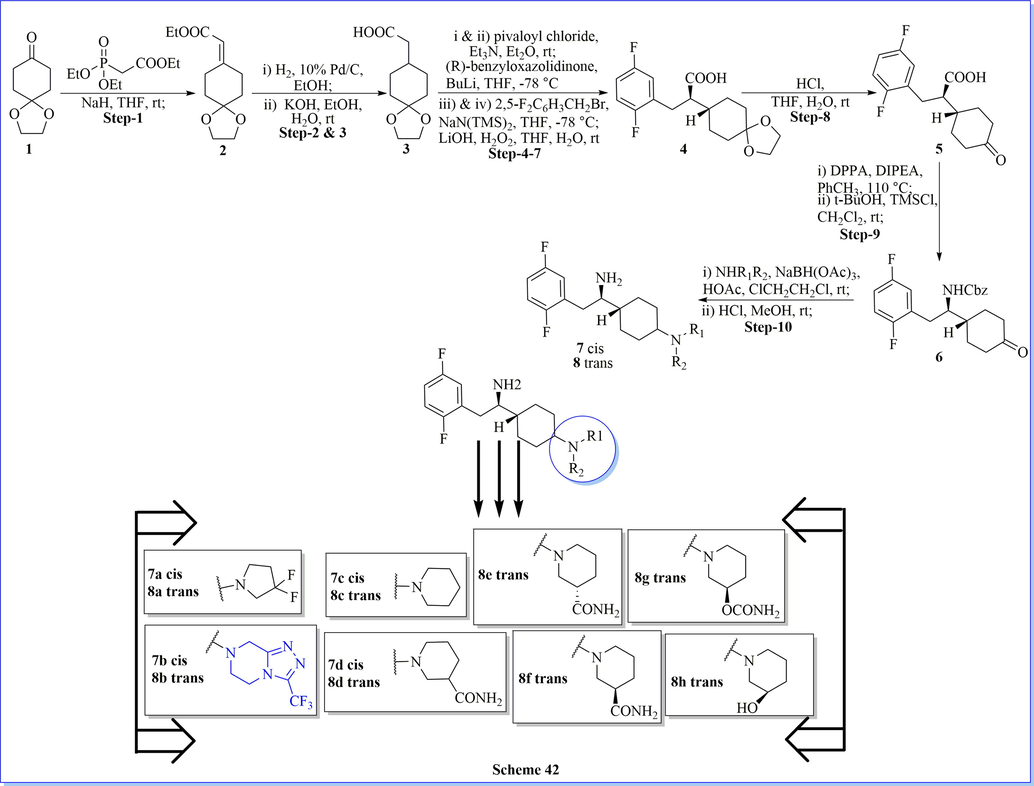
Synthesis of (R)-1-cyclohexyl-2-(2,5-difluorophenyl)ethan-1-amine derivatives.
4.9 Scheme 43
Molecular modification in the currently marketed drug is necessary with the aim of getting desired biological activity on protein targets. With the similar objective, a new series of effective diazepanone ring containing derivatives as DPP-IV inhibitors have been discovered by Biftu (Biftu et al., 2007a) et al. They have done molecular modification approach by using the triazolopiperazine ring of standard sitagliptin drug (IC50 = 18 nM as DPP-IV) with 3-(2,2,2-trifluoroethyl)-1,4-diazepan-2-one provided good IC50 value = 2.6 nM against dipeptidyl peptidase IV inhibitor. Among them, (3R)-4-[(3R)-3-amino-4-(2,4,5-trifluorophenyl)butanoyl]-3-(2,2,2-trifluoroethyl)-1,4-diazepan-2-one (18) was selective, hit molecule, and efficient an oral glucose tolerance test in mice with good preclinical development as a potential back-up candidate similar to sitagliptin against type 2 diabetes mellitus (Scheme 43a-43b) (Biftu et al., 2007a).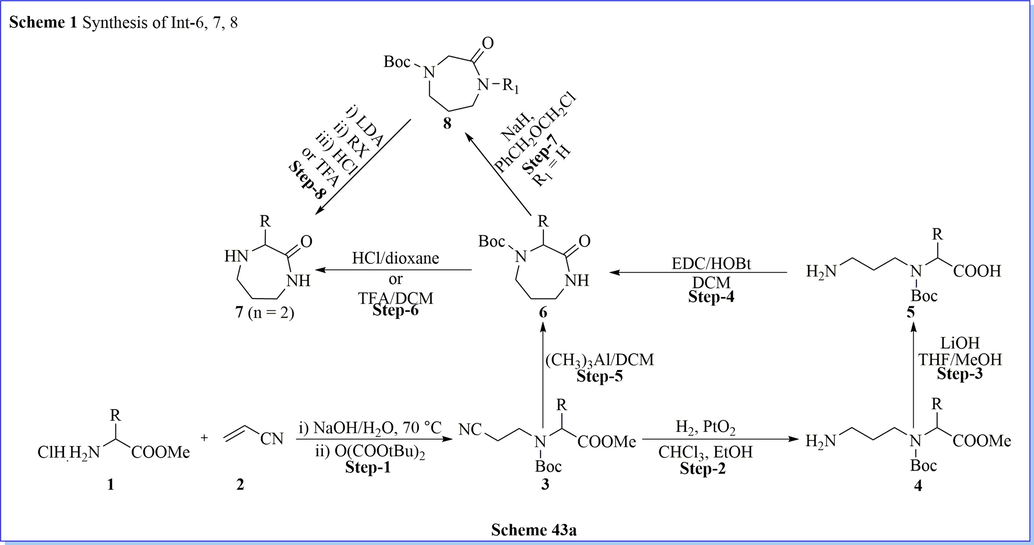
Synthesis of diazepanone derivatives.
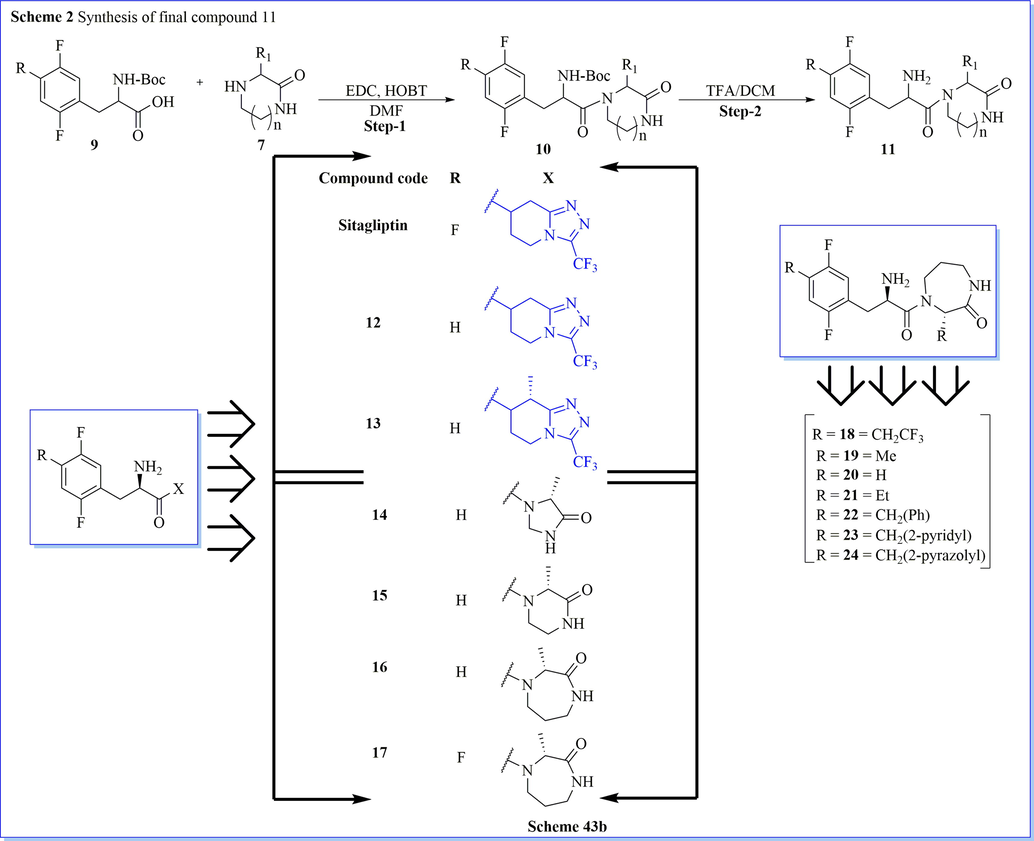
Synthesis of various derivative of diazepanone.
4.10 Scheme 44
Compound 2-azabicyclo[2.2.1]hept-5-en-3-one underwent a hydroarylation reaction and followed by nucleophilic ring-opening of N-BOC lactams was discovered as a simple and an efficient method to get chiral tri-substituted cyclopentane derivatives with practically good yields (Scheme 44)(Polivkova & Piotrowski, 2013). This method became very useful by using a variety of nucleophiles such as amines, hydride, hydroxide, grignard reagents, and alkoxide. By using this cyclopentane derivative, they have also reported a facile synthesis of a constrained analog of sitagliptin as a DPP-IV inhibitor.![Synthesis of ((1S,3S,4R)-3-amino-4-(2,4,5-trifluorophenyl)cyclopentyl)(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig71.png)
Synthesis of ((1S,3S,4R)-3-amino-4-(2,4,5-trifluorophenyl)cyclopentyl)(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)methanone.
4.11 Scheme 45
Hepatitis C virus (HCV) is presently available at a high risk of emerging liver cirrhosis and hepatocellular carcinoma. There is an urgent need for the development of a novel inhibitor against it. By aiming similar target, design, and discovery of a new-fangled class of thieno[2,3-b]pyridine analogues have been firstly reported by Wang et al. as HCV inhibitors. They have prepared the desired compound with good practical yield by using various synthetic steps such as enamine/addition reaction, ring formation, cyclization, hydrolysis, and acid–amine reaction. Earlier cell-based screening of their advantaged small molecule library led to the discovery of compound 3-amino-6-phenyl-4-(trifluoromethyl)benzo[b]thiophene-2-carboxamide characterized by a thienopyridine core (Scheme 45a-45b)(Wang et al., 2014). They also carried out a structure–activity relationship study (SAR) for the potent compound. From SAR study, compound 12c (EC50 = 3.3 µM), 12b (EC50 = 3.5 µM), 10 l (EC50 = 3.9 µM), 12o (EC50 = 4.5 µM), showed good in vitro antiviral potency and high selectivity index (SI) SI > 30.3, SI > 28.6, SI > 25.6, and SI > 22.2, correspondingly.![Synthesis of thieno[2,3-b]pyridine analogues.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig72.png)
Synthesis of thieno[2,3-b]pyridine analogues.
![Synthesis of thieno[2,3-b]pyridine derivatives.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig73.png)
Synthesis of thieno[2,3-b]pyridine derivatives.
4.12 Scheme 46
Biftu (Biftu et al., 2013) et al. have designed and synthesized new series of tri-2,3,5-substituted tetrahydropyran derivatives via cyclization and acid–amine reaction with a good yield which were effective and very selective DPP-IV inhibitors for type 2 diabetes. They have carried out the optimization of the present series providing inhibitors with good DPP-IV strength and selectivity over other peptidase targets such as DPP-8, FAP, and QPP. Compound (2R,3S,5S)-2-(2,5-difluorophenyl)-5-((S)-1,4,5,6-tetrahydrocyclopenta[c]pyrazol-5-yl)tetrahydro-2H-pyran-3-amine was a hit, selective, effective against PD model in diabetes with outstanding pharmacokinetic outline (Scheme 46a-46c) (Biftu et al., 2013).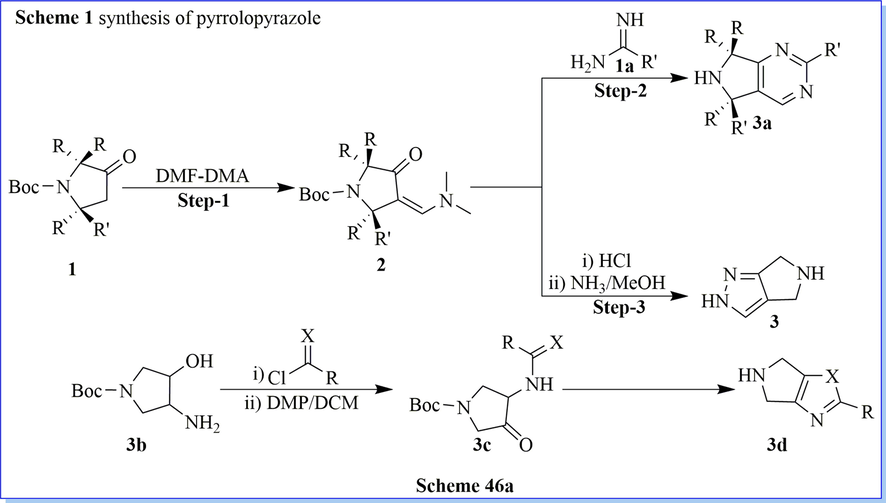
Synthesis of pyrrolopyrazole derivatives.
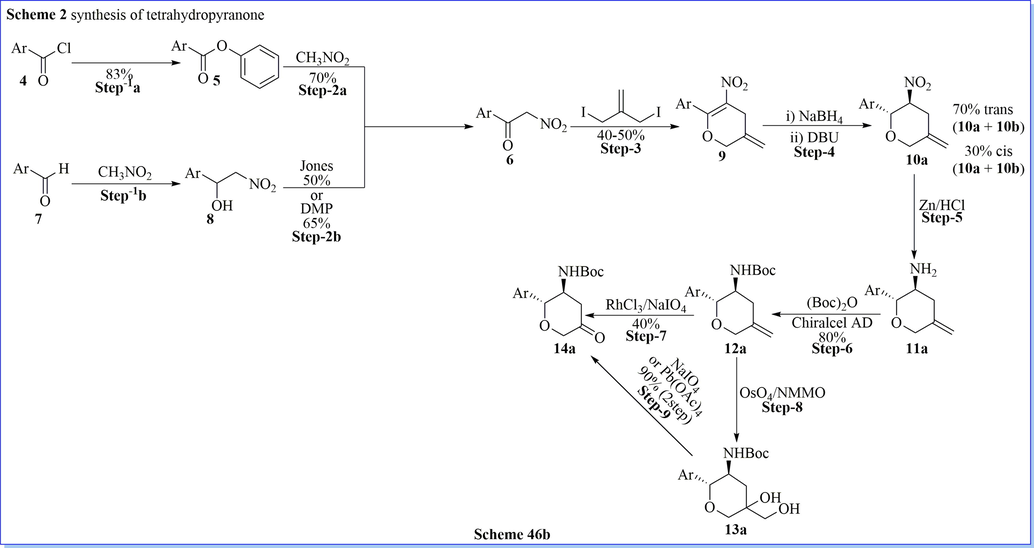
Synthesis of tetrahydropyranone derivatives.
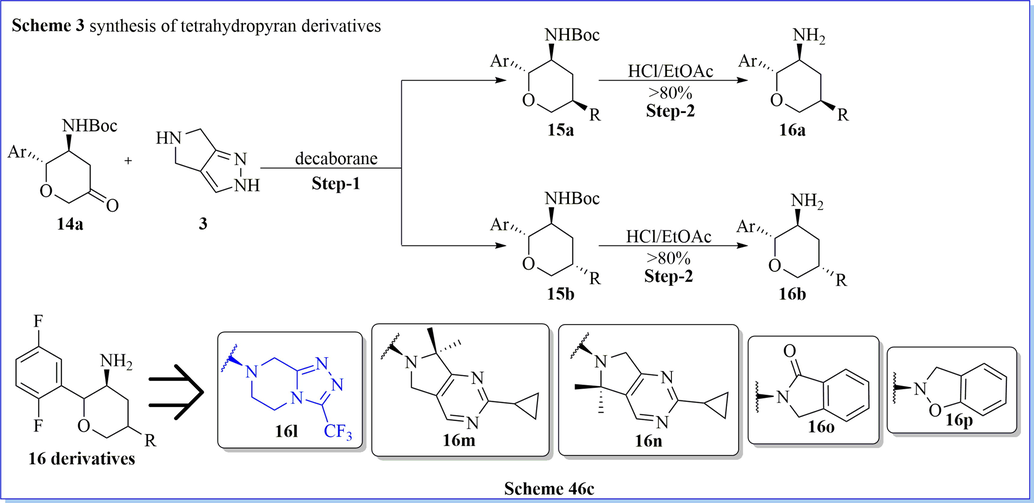
Synthesis of tetrahydropyranone derivatives.
4.13 Scheme 47
Cancer is one of the chief foremost reasons for decease across the world. Because of this important target, various novel derivative of 1-cyano-2-amino-benzimidazole derivatives has been produced through intramolecular cyclization with moderate to good practical yield (Scheme 47) (Hu et al., 2014). All the compounds have shown potent in vitro cytotoxic activities in contradiction of three human cancer cells lines such as A549, K562, and PC-3. From the SAR study and cell cycle analysis, compound 2-(4-Methylpiperazin-1-yl)-1H-benzo[d]imidazole-1-carbonitrile (4d) have demonstrated notable arrest of the cell cycle at the G2/M phase.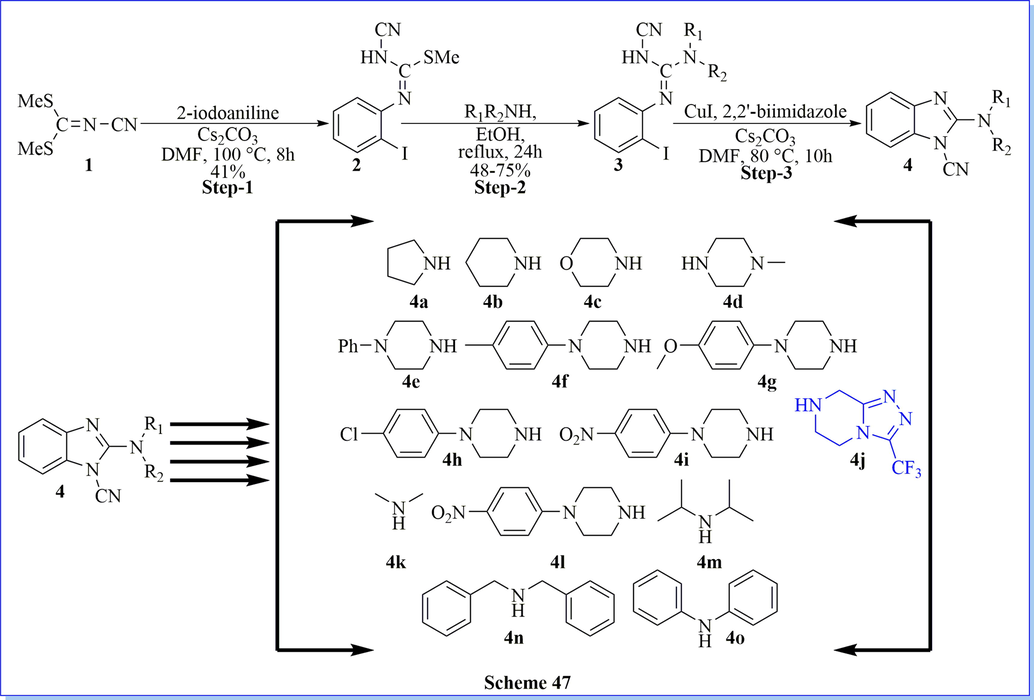
Synthesis of 1-cyano-2-amino-benzimidazole derivatives.
4.14 Scheme 48
With the help of computational studies to discover new DPP-IV inhibitors, Shen and his colleagues (Zhu et al., 2013) have designed and developed a novel series of triazolo containing 4-(2,4,5-trifluorophenyl)butane-1,3-diamine derivatives as DPP-IV inhibitors. They have evaluated enzyme activity assays and crystallographic confirmation of the binding interaction with PDB (proteins) and found that triazole derivatives of desired compounds inhibited DPP-IV with good IC50 values (0.031 ± 0.002). Based on the SAR and pharmacokinetics profiles result, compound (R)-N-(3-amino-4-(2,4,5-trifluorophenyl)butyl)-3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a] pyrazine-7(8H)-carboxamide (17r) found to be most potent, selective, and orally bioavailable DPP-IV inhibitors with good attributes such as shorter t1/2, longer mean residence time, good tmax value (fourfold slower), good metabolic stability, similar efficacy (from oral glucose tolerance test (OGTT)) as compared to the standard marketed drug (Scheme 48a-48b) (Zhu et al., 2013).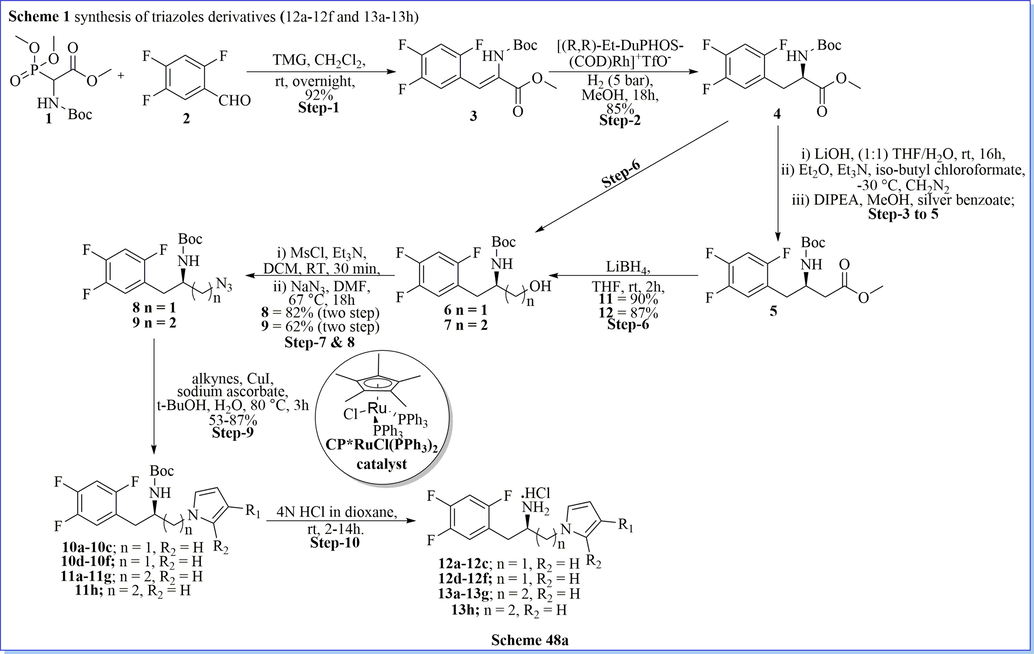
Synthesis of 4-(2,4,5-trifluorophenyl)butane-1,3-diamine derivatives.
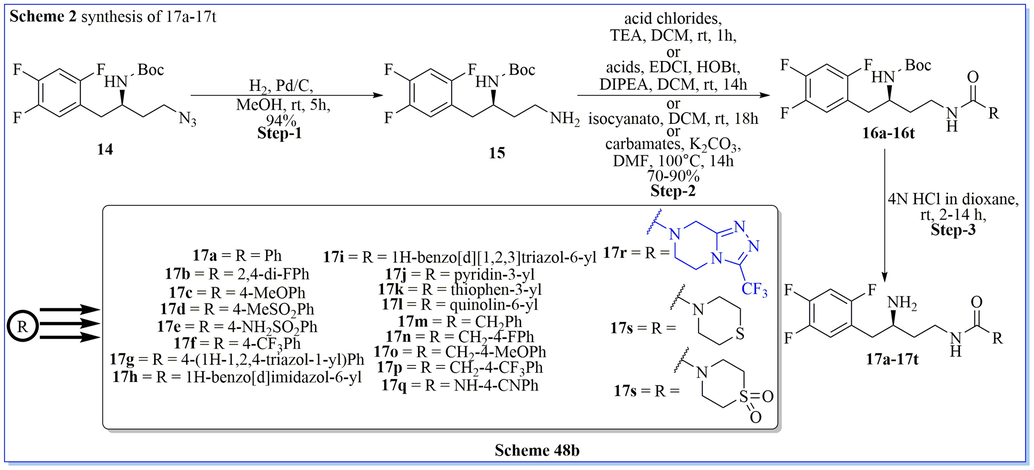
Synthesis of (R)-(3-amino-4-(2,4,5-trifluorophenyl)butyl)carbamate.
4.15 Scheme 49
Based on the computational analysis of a huge volume of crystal structure data obtainable in the protein data bank (PDB), a new and efficient fused β-homophenylalanine derivatives have been designed, synthesized, and assessed as potent, effective, and selective DPP-IV inhibitors. Compounds 9aa, 18a, and 18 m presented an admirable DPP-IV inhibitory activity, and good in vivo effectiveness in an OGTT in ICR mice. Additionally, compound 4-((S)-3-amino-3-((S)-5,6-difluoro-2,3-dihydro-1H-inden-1-yl)propanoyl)-1-methyl-1,3,4,5-tetrahydro-2H-benzo[e][1,4] diazepin-2-one (18 m) have meaningfully superior strength comparison with sitagliptin (Scheme 49) (Jiang et al., 2015).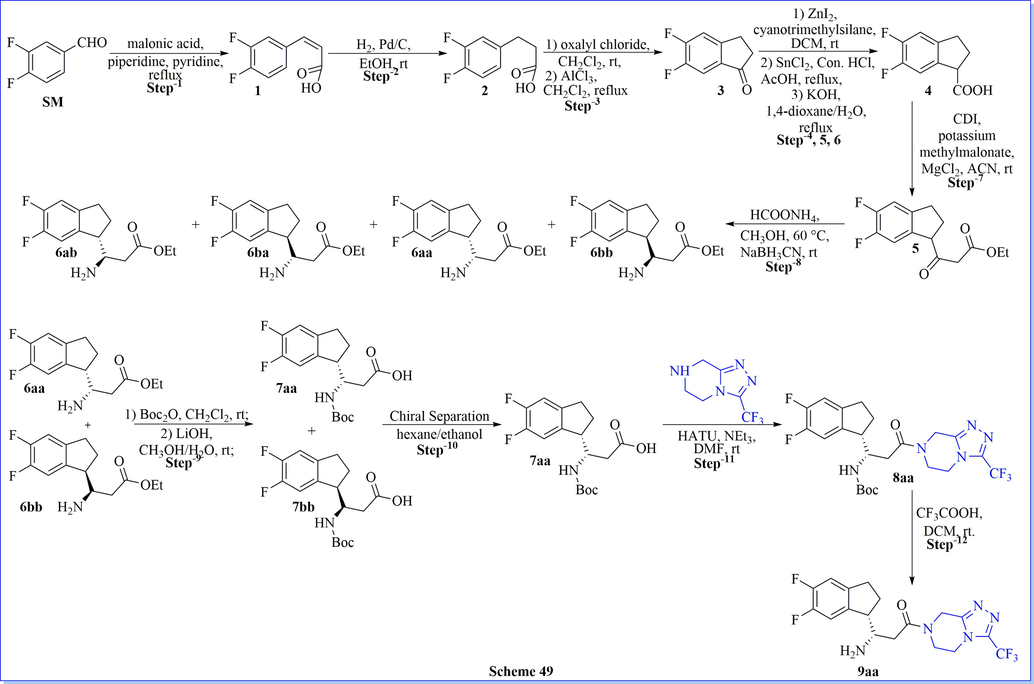
Synthesis of β-homophenylalanine derivatives.
5 One-pot synthesis of triazolo[4,3-a]pyrazine derivatives
5.1 Scheme 50-51
One-pot synthesis or multi-component reactions is a powerful tool that attracts much more focus on synthetic organic reactions because of the complex molecules with a varied range of complexity where the starting materials are easily available. (Bhoi et al., 2019b) With the inspiration from the current methodology, simple operation, efficient, and enantioselective total synthesis of ABT-341 have been described by Ishikawa and his co-workers(Ishikawa et al., 2011) via a one-pot asymmetric reaction in the presence of organo-catalysts, as well as diphenylprolinol silyl ether. The present synthetic protocol has several advantages such as excellent yield, one-pot method, very cheaper way of ABT-341, covering different name reaction like michael reaction, horner–wadsworth-emmons reaction, retro-aldol reaction, etc.. base-catalysed isomerization, easily formed amide bond by reaction between acid–amine as starting material, appropriate reaction conditions, less side product and mostly desired product formation (Scheme 50) (Ishikawa et al., 2011). Similar to the synthesis of ABT-341 described above, Lu and his co-workers (Weng et al., 2012) have described an efficient, short method for the preparation of ABT-341 and its analogs as DPP-IV inhibitor with good yield (85%, after 3 steps,) by using well-organized asymmetric domino nitro-michael/HWE reaction by using nitro phosphonates and α, β-unsaturated aldehydes as starting material. This method was a simple, mild, and scalable reaction with great diastereo- and enantioselectivity (dr up to > 20:1, 83–92% ee) (Scheme 51) (Weng et al., 2012). While Christmann (Vaxelaire et al., 2011) et al. have given the valuable discussion on one spot synthesis and conventional stepwise synthesis of Tamiflu and ABT-341 (1). They have described their discussion based on mass intensity, reaction pot, the yield of the reaction, and solvent used during the reaction.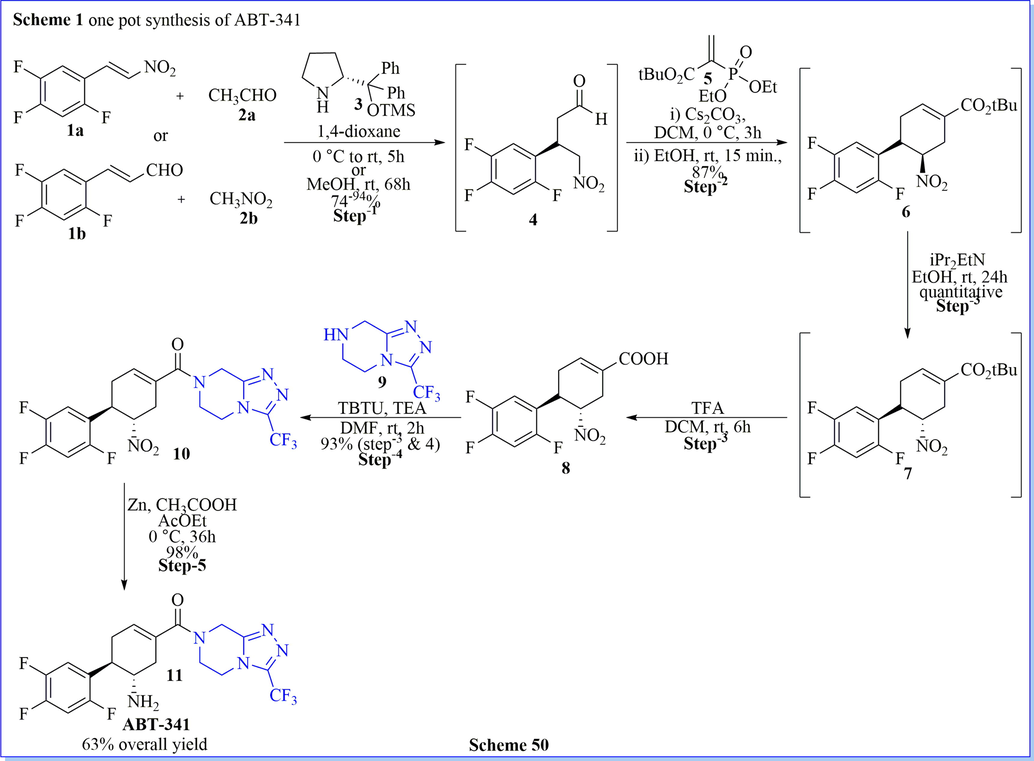
Synthesis of ABT-341.
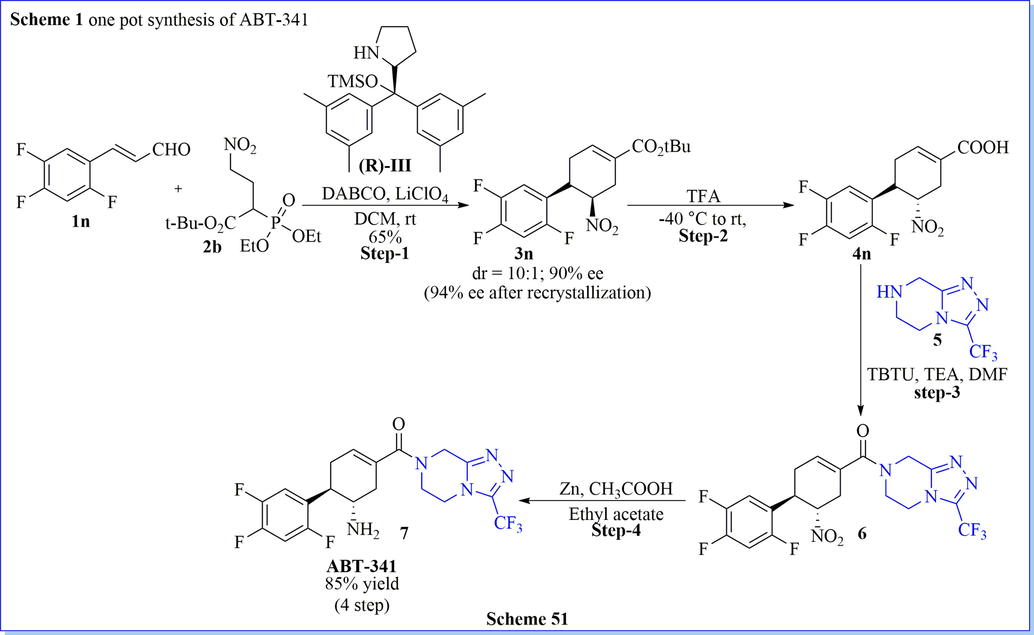
Synthesis of ABT-341.
6 Miscellaneous
6.1 Scheme 52
By using pyridinium radical cation, directly late-stage aryl C-H amination reaction for the synthesis of complex primary arylamines C-N bond conversion was first reported in the presence of the photo-redox catalyst Ru(bpy)3(PF6)2 with moderate to good yields (Scheme 52) (Ham et al., 2019).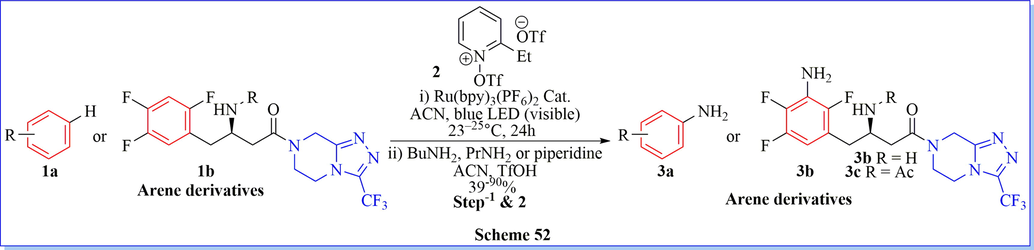
Synthesis of amine derivative using pyridinium radical cation.
6.2 Scheme 53
Resolution is an important method to get chirally pure isomer from racemic mixtures. An effective, enantioselective, bio-catalytic chiral synthetic method for a chiral intermediate of sitagliptin was developed by Sun and his co-workers (Sun et al., 2016). The author has worked on isolation of a fungal strain from a soil sample and recognized it as rhizopus microsporus var. rhizopodiformis ZJPH1308. It was used in bio-catalytic chiral synthesis of keto-amide 4-oxo-4-[3-(trifluoro-methyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-1-(2,4,5-trifluorophenyl)butan-2-one (OTPP) to (S)-3-hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluoro-phenyl)butan-1-one (S-HTPP) showing excellent enantioselectivity (>99.9 ee). They applied the optimum conditions (15 nM of OTPP, 45 °C, 24 h) for good practically possible bio-catalytic reduction with good yield (93.2%). The rhizopodiformis ZJPH1308 was used as an auspicious biocatalyst for the industrial production of some valuable chiral intermediates (Scheme 53) (Sun et al., 2016).![Bio-catalytic chiral synthetic method for a chiral intermediate of (S)-3-Hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluoro-phenyl)butan-1-one (S-HTPP) using rhizopodiformis ZJPH1308.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig84.png)
Bio-catalytic chiral synthetic method for a chiral intermediate of (S)-3-Hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluoro-phenyl)butan-1-one (S-HTPP) using rhizopodiformis ZJPH1308.
6.3 Scheme 54
Likewise, Wei and his colleagues(Wei et al., 2016) have created a method for microbial bio-reduction of (S)-3-hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-4-(2,4,5-trifluorophenyl)butan-1-one(S)-1 from 4-oxo-4-[3-(trifluoromethyl)-5,6-dihydro[1,2,4]-triazolo[4,3-a]pyrazin-7(8H)-yl)-1-(2,4,5-trifluorophenyl)butan-2-one by using pseudomonas pseudoalcaligenes XW-40 with the best practical yield (90%) and good enantioselectivity (ee 99%) (Scheme 54) (Wei et al., 2016). It was isolated from soil using acetophenone as the sole carbon source. Christopher (Savile et al., 2010) et al. also described a well-organized bio-catalytic process for the preparation of sitagliptin with the help of enzymes (transaminase), which gave direct amination of prositagliptin ketone to sitagliptin (99.95% ee).![Microbial bio-reduction of (S)-3-hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-4-(2,4,5-trifluorophenyl)butan-1-one(S)-1 using pseudomonas pseudoalcaligenes XW-40.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig85.png)
Microbial bio-reduction of (S)-3-hydroxy-1-(3-(trifluoromethyl)-5,6-dihydro[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl]-4-(2,4,5-trifluorophenyl)butan-1-one(S)-1 using pseudomonas pseudoalcaligenes XW-40.
6.4 Scheme 55
One pot, three-component, an efficient, high-yielding process for the synthesis of β-keto amides has been developed by Xu (Xu et al., 2004) et al. using a sequential reaction with the carboxylic acid, Meldrum’s acid, and amine. They have carried out kinetic studies via online ir observing and following major constituent analysis with a depth understanding of the mechanism of the multistep process. It confirmed that the development of β-keto amides from acyl meldrum’s adducts was produced through the R-oxoketene intermediate-2 (Scheme 55) (Xu et al., 2004). Similar to the biological discussion on Sitagliptin, Diniz et al. have done depth biological study and discussion of it against type 2 diabetes (Pinheiro et al., 2017).![Synthesis of 1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butane-1,3-dione.](/content/184/2020/13/12/img/10.1016_j.arabjc.2020.09.038-fig86.png)
Synthesis of 1-(3-(trifluoromethyl)-5,6-dihydro-[1,2,4]triazolo[4,3-a]pyrazin-7(8H)-yl)-4-(2,4,5-trifluorophenyl)butane-1,3-dione.
7 Conclusion
In this review, we have highlighted the recent advance research development of the triazolo[4,3-a]pyrazine derivatives as DPP-IV and other useful inhibitors. We have also noted that the common considerations in constructing triazolo[4,3-a]pyrazine scaffold/ring by using various starting material such as halo-pyrazine derivatives, piperazine derivatives, tert-butyl (2-aminoethyl)carbamate derivatives, tert-butyl (2-hydroxyethyl)carbamate derivatives, 2-(chloromethyl)-5-substituted-1,3,4-oxadiazole derivatives, 2,4-dihydro-3H-1,2,4-triazol-3-one derivatives, etc.. as per current literature survey. Among them, the most used starting material is various derivatives of halo-pyrazine, piperazine derivatives, and 1,3,4-oxadiazole derivatives for the synthesis of triazolo[4,3-a]pyrazine ring. There are several merits of the many popular synthetic routes for the synthesis of triazolo[4,3-a]pyrazine are starting material obtainability, simple operation, easy purification of intermediates, good yield, high efficiency, mild reaction conditions without specific requirements, cheaper, and ecologically friendly. Synthesis of triazolo[4,3-a]pyrazine derivatives suggest the possibility of SAR study of various substitutions around this adaptable pharmacophore and used in both drug discovery as well as in medicinal chemistry. The triazolo[4,3-a]pyrazine has been demonstrated various biological activities such as DPP-IV inhibitor, anticonvulsant activity, anti-malarial, neurokinin-3 receptor, Anti-tuberculosis, anti-bacterial, and anti-fungal, jak inhibitor, BRD4 inhibitor, etc. and their application especially against diabetes. The triazolo[4,3-a]pyrazine scaffold containing the drugs is already in the market such as sitagliptin, ABT-341, etc.. Therefore, the voyage on triazolo[4,3-a]pyrazine scaffold will be sustained to identify many more biologically active inhibitors on different targets in near future. We believe that the present account absolutely signifies a new episode in the chemistry of triazolo[4,3-a]pyrazine. As far as future perspectives applications of triazolo[4,3-a]pyrazine derivatives concerned, we trust that this scaffold of the organic chemistry family will be more discover in the important areas such as may use in catalysis, drug discovery, medicinal chemistry, etc.. This review article offers wide-ranging and target concerning the information of the core to the researcher for the development of potent and novel triazolo[4,3-a]pyrazine derivatives as a novel inhibitor in the future.
Acknowledgement
The authors are thankful to the department of chemistry, Gujarat University, Ahmedabad, for providing the necessary facilities and also thankful to UGC-Info net & INFLIBNET, Gujarat university for providing e-source facilities.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
The authors have declared that there is not any conflict of interest.
References
- Preclinical characterization of substituted 6, 7-dihydro-[1, 2, 4] triazolo [4, 3-a] pyrazin-8 (5H)-one P2X7 receptor antagonists. Bioorg. Med. Chem. Lett.. 2016;26:257-261.
- [Google Scholar]
- Enabling structure-based drug design of Tyk2 through co-crystallization with a stabilizing aminoindazole inhibitor. BMC struct. Biol.. 2012;12:22.
- [Google Scholar]
- Discovery and preclinical profile of Saxagliptin (BMS-477118): a highly potent, long-acting, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem.. 2005;48:5025-5037.
- [Google Scholar]
- On resin click-chemistry-mediated synthesis of novel enkephalin analogues with potent anti-nociceptive activity. Rep: Sci; 2019. p. :9.
- Direct Catalytic Asymmetric Mannich Reaction with Dithiomalonates as Excellent Mannich Donors: Organocatalytic Synthesis of (R)-Sitagliptin. Chem. Int. Ed.. 2016;55:10825-10829.
- [Google Scholar]
- Catalytic Enantioselective Allylic Amination of Olefins for the Synthesis of ent-Sitagliptin. Synlett.. 2013;24:2459-2463.
- [Google Scholar]
- Synthesis, biological evaluation and computational study of novel isoniazid containing 4H-Pyrimido [2,1-b] benzothiazoles derivatives. Eur. J. Med. Chem.. 2019;177:12-31.
- [Google Scholar]
- Synthetic strategies for fused benzothiazoles: past, present, and future. Synth. Commun.. 2014;44:2427-2457.
- [Google Scholar]
- Novel benzothiazole containing 4H-pyrimido [2, 1-b] benzothiazoles derivatives: One pot, solvent-free microwave assisted synthesis and their biological evaluation. Arab. J. Chem.. 2019;12:3799-3813.
- [Google Scholar]
- (3R)-4-[(3R)-3-Amino-4-(2, 4, 5-trifluorophenyl) butanoyl]-3-(2, 2, 2-trifluoroethyl)-1, 4-diazepan-2-one, a selective dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett.. 2007;17:49-52.
- [Google Scholar]
- Novel tetrahydropyran analogs as dipeptidyl peptidase IV inhibitors: profile of clinical candidate (2R, 3S, 5R)-2-(2, 5-difluorophenyl)-5-[2-(methylsulfonyl)-2, 6-dihydropyrrolo [3, 4-c] pyrazol-5 (4H)-yl] tetrahydro-2H-pyran-3-amine (23) Bioorg. Med. Chem. Lett.. 2013;23:5361-5366.
- [Google Scholar]
- Rational design of a novel, potent, and orally bioavailable cyclohexylamine DPP-4 inhibitor by application of molecular modeling and X-ray crystallography of sitagliptin. Bioorg. Med. Chem. Lett.. 2007;17:3384-3387.
- [Google Scholar]
- Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis.. 2009;48:1-12.
- [Google Scholar]
- 1, 2, 4-Triazolo [4, 3-a] pyrazine derivatives with human renin inhibitory activity. 2. Synthesis, biological properties and molecular modeling of hydroxyethylene isostere derivatives. J. Med. Chem.. 1990;33:2335-2342.
- [Google Scholar]
- 1, 2, 4-Triazolo [4, 3-a] pyrazine derivatives with human renin inhibitory activity. 3. Synthesis and biological properties of aminodeoxystatine and difluorostatone derivatives. J. Med. Chem.. 1991;34:151-157.
- [Google Scholar]
- Synthesis and evaluation of [(1R)-1-amino-2-(2, 5-difluorophenyl) ethyl] cyclohexanes and 4-[(1R)-1-amino-2-(2, 5-difluorophenyl) ethyl] piperidines as DPP-4 inhibitors. Bioorg. Med. Chem. Lett.. 2011;21:1880-1886.
- [Google Scholar]
- Novel phenyl-substituted 5,6-dihydro-[1,2,4] triazolo [4, 3-a] pyrazine P2X7 antagonists with robust target engagement in rat brain. ACS Chem. Neurosci.. 2016;7:490-497.
- [Google Scholar]
- Identification of ammonium chloride as an effective promoter of the asymmetric hydrogenation of a β-enamine amide. Org. Process Res. Dev.. 2006;10:723-726.
- [Google Scholar]
- Pyrido [2, 3-e]-1, 2, 4-triazolo [4, 3-a] pyrazin-1-one as a new scaffold to develop potent and selective human A3 adenosine receptor antagonists. Synthesis, pharmacological evaluation, and ligand− receptor modeling studies. J. Med. Chem.. 2009;52:2407-2419.
- [Google Scholar]
- DPP-4 inhibitors: a patent review (2012–2014) Expert Opin Ther Pat.. 2015;25:209-236.
- [Google Scholar]
- 8-(3-(R)-aminopiperidin-1-yl)-7-but-2-ynyl-3-methyl-1-(4-methyl-quinazolin-2-ylmethyl)-3, 7-dihydropurine-2, 6-dione (BI 1356), a highly potent, selective, long-acting, and orally bioavailable DPP-4 inhibitor for the treatment of type 2 diabetes. J. Med. Chem.. 2007;50:6450-6453.
- [Google Scholar]
- The asymmetric synthesis of. alpha.-amino acids. Electrophilic azidation of chiral imide enolates, a practical approach to the synthesis of (R)-and (S)-. alpha.-azido carboxylic acids. J. Am. Chem. Soc.. 1990;112:4011-4030.
- [Google Scholar]
- The 1, 2, 4-Triazolo[4, 3-a]pyrazin-3-one as a versatile scaffold for the design of potent adenosine human receptor antagonists. Structural investigations to target the A2A receptor subtype. J. Med. Chem.. 2017;60:5772-5790.
- [Google Scholar]
- Discovery of alogliptin: a potent, selective, bioavailable, and efficacious inhibitor of dipeptidyl peptidase IV. J. Med. Chem.. 2007;50:2297-2300.
- [Google Scholar]
- Structure activity optimization of 6H-pyrrolo [2, 3-e][1, 2, 4] triazolo [4, 3-a] pyrazines as JAK1 kinase inhibitors. Bioorg. Med. Chem. Lett.. 2015;25:4399-4404.
- [Google Scholar]
- Practical Asymmetric Synthesis of Sitagliptin Phosphate Monohydrate. Molecules.. 2018;23:1440.
- [Google Scholar]
- Practical, Asymmetric Route to Sitagliptin and Derivatives: Development and Origin of Diastereoselectivity. Org. lett.. 2015;17:1742-1745.
- [Google Scholar]
- Divergent Late-Stage (Hetero) aryl C− H Amination by the Pyridinium Radical Cation. Chem. Int. Ed.. 2019;58:532-536.
- [Google Scholar]
- First generation process for the preparation of the DPP-IV inhibitor sitagliptin. Org. Process Res. Dev.. 2005;9:634-639.
- [Google Scholar]
- Highly efficient asymmetric synthesis of sitagliptin. J. Am. Chem. Soc.. 2009;131:8798-8804.
- [Google Scholar]
- Statistical investigation into the structural complementarity of natural products and synthetic compounds. Chem. Int. Ed.. 1999;38:643-647.
- [Google Scholar]
- Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol.. 2006;2:284-287.
- [Google Scholar]
- Synthesis of 3, 3-Dimethylglutamic Acid Derivatives as DPP-IV Inhibitors and Evaluation of Their Chemical Stability. J. Chin. Chem. Soc.. 2011;58:108-117.
- [Google Scholar]
- http://www.idf.org/news/169:diabetes-atlas-report-463-million-with-diabetes.html
- Synthesis and biological evaluation of 1-cyano-2-amino-benzimidazole derivatives as a novel class of antitumor agents. Med. Chem. Res.. 2014;23:3029-3038.
- [Google Scholar]
- One-pot high-yielding synthesis of the DPP4-selective inhibitor ABT-341 by a four-component coupling mediated by a diphenylprolinol silyl ether. Chem. Int. Ed.. 2011;50:2824-2827.
- [Google Scholar]
- Design, synthesis, biological evaluation and computational study of novel triazolo [4, 3-a]pyrazin analogues. J. Mol. Struct.. 2019;1184:168-192.
- [Google Scholar]
- Design, synthesis, and pharmacological evaluation of fused β-homophenylalanine derivatives as potent DPP-4 inhibitors. ACS med. chem. lett.. 2015;6:602-606.
- [Google Scholar]
- A highly stereoselective and efficient synthesis of enantiomerically pure sitagliptin. Tetrahedron: Asymmetry.. 2017;28:34-40.
- [Google Scholar]
- 8-Amino-3-benzyl-1, 2, 4-triazolo [4, 3-a] pyrazines. Synthesis and anticonvulsant activity. J. Med. Chem.. 1995;38:3676-3679.
- [Google Scholar]
- Discovery of potent and selective dipeptidyl peptidase IV inhibitors derived from β-aminoamides bearing subsituted triazolopiperazines. J. Med. Chem.. 2008;51:589-602.
- [Google Scholar]
- (2 R)-4-Oxo-4-[3-(trifluoromethyl)-5, 6-dihydro [1, 2, 4] triazolo [4, 3-a] pyrazin-7 (8 H)-yl]-1-(2, 4, 5-trifluorophenyl) butan-2-amine: a potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J. Med. Chem.. 2005;48:141-151.
- [Google Scholar]
- Glucagon-like peptide-1: the basis of a new class of treatment for type 2 diabetes. J. Med. Chem.. 2004;47:4128-4134.
- [Google Scholar]
- Design, Synthesis and Biological Evaluation of Imidazo [1,2-a] pyridine Derivatives as Novel DPP-4 Inhibitors. Chem. boil. Drug des.. 2015;86:849-856.
- [Google Scholar]
- Practical and economical approach to synthesize sitagliptin. Synth. Commun.. 2013;43:3281-3286.
- [Google Scholar]
- Urea and thiourea derivatives of 3-(trifluoromethyl)-5, 6, 7, 8-tetrahydro-[1, 2, 4] triazolo [4, 3-a] pyrazine: Synthesis, characterization, antimicrobial activity and docking studies. Phosphorus. Sulfur Silicon Relat. Elem.. 2019;194:922-932.
- [Google Scholar]
- Synthesis of Fmoc-β-homoamino acids by ultrasound-promoted Wolff rearrangement. Synthesis.. 1998;1998:837-841.
- [Google Scholar]
- Discovery of C-(1-aryl-cyclohexyl)-methylamines as selective, orally available inhibitors of dipeptidyl peptidase IV. Bioorg. Med. Chem. Lett.. 2014;24:731-736.
- [Google Scholar]
- Efficient synthesis of sitagliptin phosphate, a novel DPP-IV inhibitor, via a chiral aziridine intermediate. Tetrahedron Lett.. 2013;54:6807-6809.
- [Google Scholar]
- Synthesis of Sitagliptin Phosphate by a NaBH4/ZnCl2-catalyzed Diastereoselective Reduction. Chem. Lett.. 2015;44:1170-1172.
- [Google Scholar]
- Microwave-Assisted Synthesis and Antituberculosis Screening of Some 4-((3-(Trifluoromethyl)-5, 6-dihydro-[1, 2, 4] triazolo [4, 3-a] pyrazin-7 (8H)-l) methyl) benzenamine Hybrids. J. Heterocycl. Chem.. 2019;56:434-442.
- [Google Scholar]
- Discovery of ((4 R, 5 S)-5-Amino-4-(2, 4, 5-trifluorophenyl) cyclohex-1-enyl)-(3-(trifluoromethyl)-5, 6-dihydro-[1, 2, 4] triazolo [4, 3-a] pyrazin-7 (8 H)-yl) methanone (ABT-341), a Highly Potent, Selective, Orally Efficacious, and Safe Dipeptidyl Peptidase IV Inhibitor for the Treatment of Type 2 Diabetes. J. Med. Chem.. 2006;49:6439-6442.
- [Google Scholar]
- Sitagliptin inhibit human lymphocytes proliferation and Th1/Th17 differentiation in vitro. Eur. J. Pharma. Sci.. 2017;100:17-24.
- [Google Scholar]
- Stereodefined Cyclopentanes by Hydroarylation-Ring Opening. Synth. Commun.. 2013;43:1007-1015.
- [Google Scholar]
- 1, 2, 4-Triazolo [4, 3-a] pyrazine derivatives with human renin inhibitory activity. 1. Synthesis and biological properties of alkyl alcohol and statine derivatives. J. Med. Chem.. 1990;33:2326-2334.
- [Google Scholar]
- Pyrido [4, 3-e][1, 2, 4] triazolo [4, 3-a] pyrazines as selective, brain penetrant phosphodiesterase 2 (PDE2) inhibitors. ACS med. chem. lett.. 2015;6:282-286.
- [Google Scholar]
- Novel methyl substituted 1-(5, 6-dihydro-[1, 2, 4] triazolo [4, 3-a] pyrazin-7 (8H)-yl) methanones are P2X7 antagonists. Bioorg. Med. Chem. Lett.. 2015;25:3157-3163.
- [Google Scholar]
- Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science.. 2010;329:305-309.
- [Google Scholar]
- Biological activity and a modified synthesis of 8-amino-3-. beta.-D-ribofuranosyl-1, 2, 4-triazolo [4, 3-. alpha.] pyrazine, an isomer of formycin. J. Med. Chem.. 1984;27:924-928.
- [Google Scholar]
- Enantioselective syntheses of (R)-amino acids using L-valine as chiral agent. Angew. Chem. Int. Ed.. 1981;20:798-799.
- [Google Scholar]
- Efficient stereocontrolled synthesis of sitagliptin phosphate. Tetrahedron: Asymmetry.. 2014;25:1026-1030.
- [Google Scholar]
- Efficient enantioselective biocatalytic production of a chiral intermediate of sitagliptin by a newly filamentous fungus isolate. Appl. Biochem. Biotechnol.. 2016;180:695-706.
- [Google Scholar]
- Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J. Clin. Endocrinol. Metab.. 2001;86:3717-3723.
- [Google Scholar]
- Peptidotriazoles on solid phase:[1, 2, 3]-triazoles by regiospecific copper (I)-catalyzed 1, 3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem.. 2002;67:3057-3064.
- [Google Scholar]
- Rational design and synthesis of potent and long-lasting glutamic acid-based dipeptidyl peptidase IV inhibitors. Bioorg. Med. Chem. Lett.. 2009;19:1908-1912.
- [Google Scholar]
- Click chemistry as a macrocyclization tool in the solid-phase synthesis of small cyclic peptides. Org. lett.. 2007;9:5011-5014.
- [Google Scholar]
- Regioselective One-Pot Synthesis of 9-Alkyl-6-chloropyrido [3, 2-e][1, 2, 4] triazolo [4, 3-a] pyrazines. Reactivity of Aliphatic and Aromatic Hydrazides. J. Org. Chem.. 2005;70:2878-2880.
- [Google Scholar]
- One-Pot Reactions Accelerate the Synthesis of Active Pharmaceutical Ingredients. Chem. Int. Ed.. 2011;50:3605-3607.
- [Google Scholar]
- 1-[2-[(5-Cyanopyridin-2-yl) amino] ethylamino] acetyl-2-(S)-pyrrolidinecarbonitrile: a potent, selective, and orally bioavailable dipeptidyl peptidase IV inhibitor with antihyperglycemic properties. J. Med. Chem.. 2002;45:2362-2365.
- [Google Scholar]
- Discovery and structure–activity relationships study of novel thieno [2,3-b] pyridine analogues as hepatitis C virus inhibitors. Bioorg. Med. Chem. Lett.. 2014;24:1581-1588.
- [Google Scholar]
- Highly enantioselective production of a chiral intermediate of sitagliptin by a novel isolate of Pseudomonas pseudoalcaligenes. Biotechnol. Lett.. 2016;38:841-846.
- [Google Scholar]
- Asymmetric Domino Nitro-Michael/Horner–Wadsworth–Emmons Reaction for Disubstituted Cyclohexenecarboxylate Annulation: Efficient Synthesis of Dipeptidyl Peptidase IV Inhibitor ABT-341 and Influenza Neuraminidase Inhibitor. Adv. Synth. Catal.. 2012;354:1961-1970.
- [Google Scholar]
- Synthesis, nitric oxide release, and dipeptidyl peptidase-4 inhibition of sitagliptin derivatives as new multifunctional antidiabetic agents. Bioorg. Med. Chem. Lett.. 2018;28:3731-3735.
- [Google Scholar]
- Rational design of 5-((1H-imidazol-1-yl) methyl) quinolin-8-ol derivatives as novel bromodomain-containing protein 4 inhibitors. Eur. J. Med. Chem.. 2019;163:281-294.
- [Google Scholar]
- Mechanistic evidence for an α-oxoketene pathway in the formation of β-ketoamides/esters via Meldrum's acid adducts. J. Am. Chem. Soc.. 2004;126:13002-13009.
- [Google Scholar]
- Design, synthesis and biological evaluation of 4, 7, 12, 12a-tetrahydro-5H-thieno [3′, 2’: 3, 4] pyrido [1, 2-b] isoquinolines as novel adenosine 5′-monophosphate-activated protein kinase (AMPK) indirect activators for the treatment of type 2 diabetes. Eur. J. Med. Chem.. 2017;140:448-464.
- [Google Scholar]
- Chemical kinetic resolution of unprotected β-substituted β-amino acids using recyclable chiral ligands. Chem. Int. Ed.. 2014;53:7883-7886.
- [Google Scholar]
- Design and synthesis of 4-(2, 4, 5-trifluorophenyl) butane-1, 3-diamines as dipeptidyl peptidase IV inhibitors. Chem. Med. Chem.. 2013;8:1104-1116.
- [Google Scholar]




