

Translate this page into:
Optimization of pyrazolo[1,5-a]pyrimidine based compounds with pyridine scaffold: Synthesis, biological evaluation and molecular modeling study
⁎Corrsponding author. mhmdgwd@ju.edu.sa (Mohamed A. Abdelgawad),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Background
Pyrazolopyrimidine heterocycle and its isosteres represent the main scaffold for many pharmacologically active drugs including anti-inflammatory agents. The COX-2 inhibitors are the principal gate for the design of new safe and potent anti-inflammatory agents.
Methods
Novel derivatives of pyrazolo[1,5-a] pyrimidines were synthesized and screened in vivo and in vitro for their anti-inflammatory potential.
Results
Within the constructed compounds, compound 11 was the most active compound on IL-6 and TNF-α (percentage inhibition = 80 and 89%, respectively). In addition, compound 12 displayed the most inhibitory effect towards COX-2 (IC50 = 1.11 µM), whereas compound 11 recorded the highest COX-2 selectivity (S.I = 8.97). The target derivatives 11–14 displayed good edema inhibitory potential (46–68%) and compound 11 was the most potent candidate (ED50 = 35 mg/kg). Additionally, the most potent sPLA2-V inhibitors were compounds 11 and 13 (IC50 = 1 and 1.7 µM respectively). Regarding activity towards 15-LOX, derivative 12 was the most active compound with IC50 = 5.6 µM revealing higher inhibitory activity than nor-dihydroguaiaretic acid (IC50 = 8.5 µM). To confirm the anti-inflammatory potential of the target derivatives, molecular modeling was performed inside COX-2 and 15-LOX active sites.
Conclusion
Display discoveries increment the plausibility that these pyrazolo[1,5-a]pyrimidines might act as a beginning point for the improvement of anti-inflammatory agents.
Keywords
Pyrazolo[1,5-a]pyrimidines
Molecular modeling
Cyclooxygenase
Pro-inflammatory cytokines
TNF-α

1 Introduction
Arachidonic acid and its metabolites have sparked the interest of many researchers because of their role in inflammation (Wang et al., 2019). The main targets for novel anti-inflammatory agents design involve inhibiting the essential enzymes included in the metabolism of arachidonic acid as cyclooxygenase (COX) (Tratrat et al., 2021), phospholipase A2 (sPLA2) (Meyer et al., 2005), lipoxygenase (LOX) (Meng et al., 2015) and pro-inflammatory cytokines (Arshad et al., 2017).
Pro-inflammatory cytokines are produced by activated macrophages which included within pathogenesis and favoring inflammation and autoimmune disorders (Tedgui and Mallat, 2006; Chaudhry et al., 2013). Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) are essential pro-inflammatory cytokines implicated in many disorders as osteoarthritis, endotoxemia, psoriasis and multiple sclerosis. (Shen et al., 2005; Chen, 2010) Apart from these cytokines pro-inflammatory functions, they also have many physiological properties, for example, TNF-α is responsible for apoptosis induction and stimulate many cytokines release as IL-6, IL-1 and IL-10 (Horowitz and Lorenzo, 2002; Tsatsanis et al., 2005). Dysregulation of TNF-α has been recorded in many pathological disorders, including congestive heart failure, cancer, Crohn,s disease, Alzeheimer,s and rheumatoid arthritis (Kumar et al., 2004; Zidi et al., 2010; Medeiros and LaFerla, 2013). So, targeting TNF-α can be used for the control of the above mentioned diseases. Until now, no orally available drug has been approved as TNF-α inhibitor, only some proteins (Etanercept, Anakinra, and Infliximab) had been used in clinics.
IL-6 is a pro-inflammatory cytokine responsible for initiation of inflammatory process (Sultani et al., 2012). It is generated by many cells at the region of inflammation and submits signal to the body (Tanaka et al., 2014). IL-6 not only controls inflammation and auto-immunity problems, but it also included in bone loss, neuronal loss and neuronal differentiation (Ataie-Kachoie et al., 2013; Noack and Miossec, 2017). Dysregulation of IL-6 was recorded in inflammation and auto-immunity problems (Wong et al., 2008). So designing bioactive agents as IL-6 suppressors will be effective in curing immune-mediate diseases.
On the other hand, prostaglandin endoperoxide synthase (cyclooxygenase, COX) is an essential enzyme for prostanoid biosynthesis (prostaglandin, prostacyclin and thromboxane) from arachidonic acid (Smyth et al., 2009). One isoform of cyclooxygenase enzyme is COX-1 which is the constitutive form and required for biosynthesis of thromboxane A2 and prostaglandin (RA Abdellatif et al., 2017), while the other isoform is COX-2 that is activated as a result of pro-inflammatory stimulus (Abdelgawad et al., 2017b). Traditional non-steroidal anti-inflammatory drugs (NSAID) suppress both COX isoforms leading to kidney damage and gastrointestinal ulceration (Ghoneim et al., 2018). In endeavor to avoid these drawbacks of nonselective NSAID, medicinal chemists designed selective COX-2 inhibitors as valdecoxib, rofecoxib and celecoxib that devoid of gastrointestinal side effects (Bäck et al., 2012; Piper and Garelnabi, 2020). Unfortunately, some selective COX-2 inhibitors as valdecoxib and rofecoxib recorded adverse cardiovascular effects that led to their withdrawal from the market (Brueggemann et al., 2009).
Phospholipase A2 (PLA2) is an enzyme that stimulates glycerophospholipids hydrolysis to yield arachidonic acid which is a precursor of eicosanoids like prostaglandins, leukotrienes and thromboxane. The same reaction yields also lysophospholipid, which could be converted into pro-inflammatory mediator called platelet activating factor (Rink and Khanna, 2011; Szefel et al., 2011). So, suppression of PLA2 is regarded as an attractive target for the discovery of novel anti-inflammatory drugs.
LOX enzymes metabolize arachidonic acid to leukotrienes and lipoxins. Leukotrienes enhance the in-inflammatory process while, lipoxins have primarily anti-inflammatory effect (Wisastra and Dekker, 2014).
Pyrazolo[1,5-a]pyrimidine fused heterocycle was documented in literature to exhibit many biological potential as anticancer (Liu et al., 2016; Hassan et al., 2017), antioxidant (Metwally et al., 2012; Cherukupalli et al., 2017), serotonin antagonist (Ivachtchenko et al., 2011), antimicrobial (Abdelhamid et al., 2010) and anti-inflammatory (Kaping et al., 2016; Naik et al., 2020). Almansa et al. (Almansa et al., 2001) designed and prepared a new group of pyrazolo[1,5-a]pyrimidines and screened these novel derivatives for their activity towards COX isoforms. The tested compounds demonstrated good activity especially compound 1 (Fig. 1) which recorded promising COX-2 selectivity (IC50 = 0.012 µM). In addition, compound 2 (Fig. 1) was prepared and exhibited anti-inflammatory potential (edema inhibition percentage = 26.5% after 4 h.) comparable to that recorded by indomethacin (edema inhibition percentage = 28.7% after 4 h.) (Shaaban et al., 2008). On the other hand, pyridine heterocycle attracted medicinal chemists interest due to its biological importance as anticancer (Abadi et al., 2009), antimicrobial (Elkanzi et al., 2019), antihypertensive (Laneri et al., 2011), antimalarial (UEYAMA et al., 1994), LOX inhibitors (Elzahhar et al., 2018) and anti-inflammatory (Abdelgawad et al., 2018). New pyridine derivatives 3a-d (Fig. 1) were constructed and screened for their anti-inflammatory potential using formalin stimulated paw edema procedure (Abdellatif et al., 2015). The record of this study displayed that these candidates 3a-d exhibited anti-inflammatory activity with ED50 = 339–809 µM/Kg po. Furthermore, compound 4 (Fig. 1) revealed similar COX-2 selectivity index to that recorded by celecoxib (S.I = 7.46) (Renard et al., 2014). It was thought that the hybridization of pyrazolo[1,5-a]pyrimidine and pyridine moieties would yield molecular hybrids, which might be developed as lead anti-inflammatory agents (Fig. 1). Energized by pyrazolo[1,5-a]pyrimidine and pyridine anti-inflammatory potential and in continuation of our past work for designing and preparing more selective anti-inflammatory agents (Bakr et al., 2016; Abdelgawad et al., 2017a; Abdellatif et al., 2017; Abdelgawad et al., 2018; Oraby et al., 2018; Abdelgawad et al., 2019; Al-Sanea et al., 2019; Bakr et al., 2019; Elkanzi and Bakr, 2020; Abdelgawad et al., 2021; Abdelgawad et al., 2022), we decided to synthesize and evaluate novel pyrazolo[1,5-a]pyrimidine-pyridine hybrids for their inhibitory potential towards cyclooxygenase, lipoxygenase, IL-6, TNF-α and phospholipase A2-V with the hope of discovering novel safe anti-inflammatory agents. For suggesting the action mode of these novel hybrids as anti-inflammatory agents, molecular docking simulation study was carried out.![Some anti-inflammatory agents having pyrazolo[1,5-a]pyrimidines (1, 2) and pyridine derivatives (3a-d, 4) and target compounds 9 and 11–14.](/content/184/2022/15/8/img/10.1016_j.arabjc.2022.104015-fig1.png)
Some anti-inflammatory agents having pyrazolo[1,5-a]pyrimidines (1, 2) and pyridine derivatives (3a-d, 4) and target compounds 9 and 11–14.
2 Results and discussion
2.1 Chemistry
The newly prepared derivatives (9, 11–14) were constructed as demonstrated in Scheme 1. Reacting malononitrile derivative (5) with 4-methoxypyridin-ylamine (6) in presence of three drops of triethylamine using absolute ethanol as a solvent yielded 2-[(4-methoxypyridin-2-ylamino)-methylsulfanyl-methylene]malononitrile (7) in 45% yield. IR displayed the presence of two peaks for NH and CN groups at 3376 and 2217 cm−1. Moreover, 1HNMR revealed the existence of SCH3 and OCH3 protons as two singlet signals at 2.54 and 4.12 ppm respectively. In addition, the presence of the three pyridine protons as a multiplet peak at 6.12–8.02 ppm in 1HNMR proved the structure. Cyclizing compound 7 with hydrazine hydrate constructed pyrazole-4-carbonitrile derivative 8 in a good yield (60%). 1HNMR of compound 8 revealed the disappearance of SCH3 protons at 2.54 ppm and existence of NH2 and NH protons as singlet peaks exchanged with D2O at 6.78 and 12.73 ppm. Mass spectrum of compound 8 demonstrated molecular ion peak at 230 (54.22%). Treating the key intermediate 8 with 2-(bis-methylsulfanyl-methylene) malononitrile (5) using acetone and few drops of triethylamine produced compound 9 in 66% yield. IR spectrum of the derivative 9 displayed two CN groups at 2220 and 2215 ppm. Moreover, 1HNMR of derivative 9 recorded the existence of SCH3 protons at 2.61 ppm. On the other hand, refluxing compound 8 with ethyl acrylate derivative 10 in ethanol yielded compound 11 in good yield (63%). Compound 11 structure was proved by spectral outcomes and elemental analyses. 1HNMR chart recorded the existence of two singlet peaks at 2.62 and 11.76 ppm attributed to SCH3 and OH, sequentially. Besides, molecular ion peak at m/z 353 (14.11%) was recorded in mass chart of compound 11. In addition, the candidate 12 was constructed by coupling compound 8 with acetyl acetone. 1HNMR spectrum of derivative 12 recorded the appearance of two methyl groups at 2.35 ppm. Finally, 7-ethoxypyridopyrazolopyrimidine-3-carbonitrile 13 and 5-ethoxy-7-aminopyridopyrazolo[1,5-a]pyrimidine-3-carbonitrile 14 were constructed from coupling the key intermediate 8 with ethyl acetoacetate and ethyl cyanoacetate, respectively. Compounds 13 and 14 were elucidated by spectral and elemental analysis. Mass spectra of 13 and 14 recorded molecular ion peaks at 324 (27.54%) and 325 (34.83%), respectively.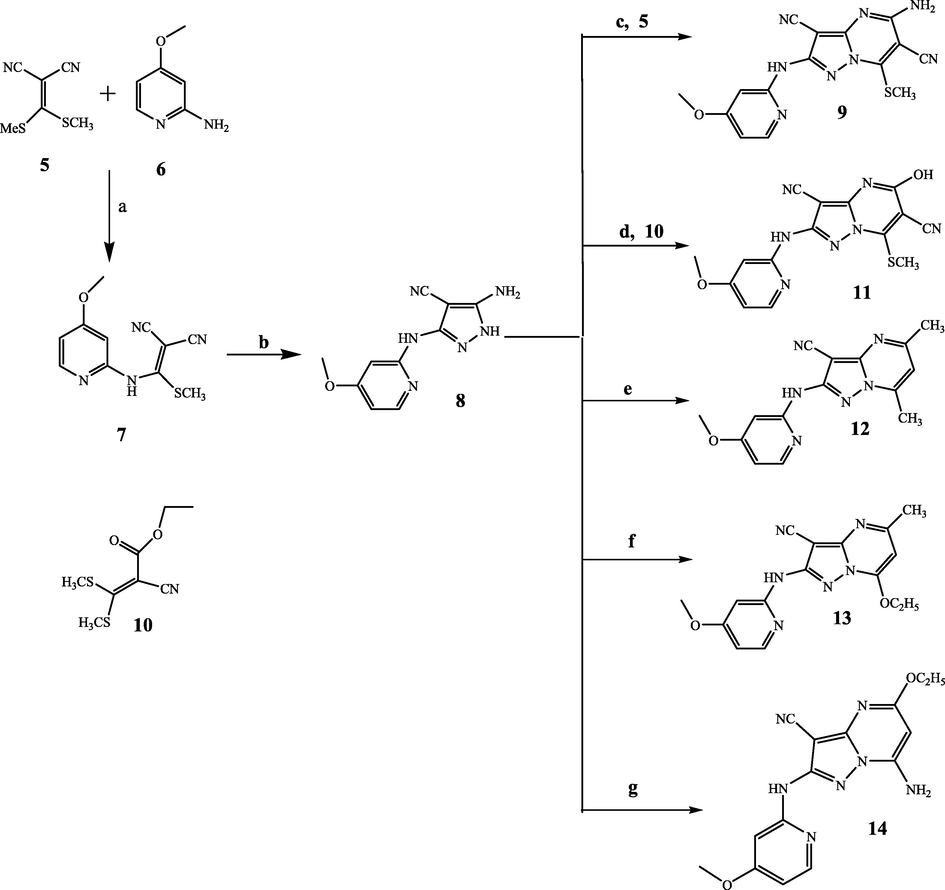
Reagents and conditions: a) ethanol, (C2H5)3N, reflux 3 days at 180 °C, b) NH2NH2·H2O, 8 h, c) acetone, (C2H5)3N, reflux 7 h, d) ethanol, (C2H5)3N, reflux 9 h, e) acetyl acetone, ethanol, acetic acid, reflux 5 h, f) ethyl acetoacetate, ethanol, acetic acid, reflux 7 h, g) ethyl cyanoacetate, DMF, acetic acid, reflux 6 h.
2.2 Biological studies
2.2.1 TNF-α and IL-6 suppression assay
The outcomes results of this assay were recorded in Fig. 2 and Fig. 3 which revealed that all the tested derivatives suppressed both TNF-α and IL-6 with various percentages. They suppressed TNF-α release with percentage inhibition range = 37–89% and IL-6 release with percentage inhibition range = 40–80% to LPS control. The most suppressor effect was achieved by compound 11, it was able to inhibit both TNF-α (percentage inhibition = 89%) and IL-6 (percentage inhibition = 80%) release in RAW264.7 mouse macrophages. The least inhibitory result was assigned by the pyrazole derivative 8 towards IL-6 and TNF-α secretion with inhibitory percentage 37% and 40%, sequentially. So, fusing the pyrazole ring with pyrimidine scaffold markedly increased the TNF-α and IL-6 inhibitory potential.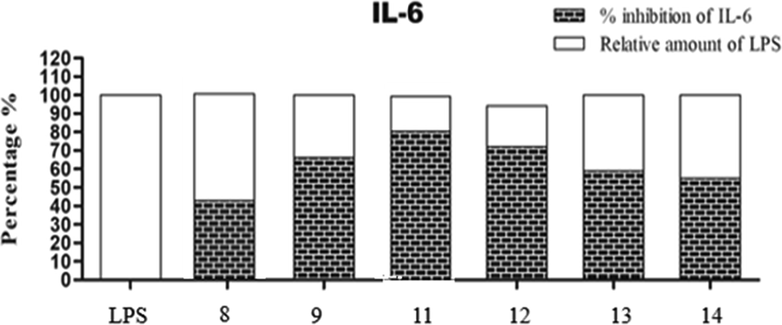
IL-6 suppression results using compounds 8, 9 and 11–14 in RAW264.7 mouse macrophages. Note: The results are expressed as LPS control percent of LPS control. Each column represents mean ± SE of three independent experiments. Acronyms: LPS: lipopolysaccharides; IL-6: interleukin-6; SE: standard error.
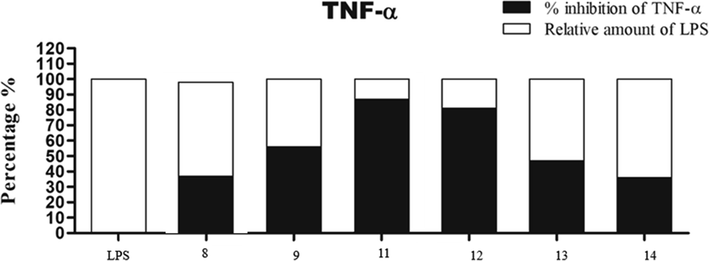
TNF-α suppression results using compounds 8, 9 and 11–14 in RAW264.7 mouse macrophages. Note: The results are expressed as LPS control percent of LPS control. Each column represents mean ± SE of three independent experiments. Acronyms: LPS: lipopolysaccharides; TNF-α: tumor necrosis factor-alpha; SE: standard error.
2.2.2 Suppression of sPLA2-V
One of strategies used for treating inflammation is inhibiting sPLA2-V (Yedgar et al., 2006). The obtained data from this screening is reported in Table 1, which showed that all the screened compounds displayed good sPLA2-V inhibitory potential with IC50 range = 1–3.2 µM compared with dexamethasone (IC50 = 0.59 µM). Moreover, 5-hydroxy-2-(4-methoxypyridin-2-ylamino)-7-methylsulfanylpyrazolopyrimidinedicarbonitrile (11) was the most active sPLA2-V suppressor (IC50 = 1 µM), followed by 7-ethoxy-2-(4-methoxypyridin-2-ylamino)pyrazolopyrimidine carbonitrile (13) which exerted sPLA2-V inhibition with IC50 = 1.7 µM. Replacing the hydroxyl group in compound 11 (IC50 = 1 µM) with amino group in compound 9 (IC50 = 2.32 µM) decreased the inhibitory potential against sPLA2-V. It is apparent that the existence of ethoxy group in compound 13 (IC50 = 1.7 µM) was advantageous than methyl substitution in compound 12 (IC50 = 3.2 µM) for achieving better sPLA2-V inhibitory potential. Values are the mean ± SD; n = 3.
Compound
sPLA2-V
IC50 (µM)15-LOX
IC50 (µM)COX-1
IC50 (µM)COX-2
IC50 (µM)COX-2 S.I
8
3.1 ± 0.8
14.2 ± 2.8
9.1 ± 0.5
4.7 ± 1.4
1.93
9
2.6 ± 0.3
27.3 ± 4.5
10.2 ± 1.6
4.4 ± 0.9
2.32
11
1.0 ± 0.8
18.4 ± 0.7
12.56 ± 2.6
1.4 ± 0.5
8.97
12
3.2 ± 1.7
5.6 ± 1.2
8.5 ± 0.2
1.11 ± 0.7
7.66
13
1.7 ± 0.5
8.4 ± 1.4
7.9 ± 0.9
2.7 ± 1.3
3.59
14
3.2 ± 0.4
8.2 ± 1.2
8.5 ± 0.7
2.2 ± 1.3
3.86
Dexamethasone
0.59 ± 0.04
–
–
–
–
Celecoxib
–
–
7.34 ± 0.2
1.11 ± 0.6
6.61
Indomethacin*
–
–
0.29 ± 0.05
3.65 ± 0.4
0.08
NDGA**
–
8.5 ± 0.5
–
–
–
2.2.3 15-Lipoxygenase (15-LOX) inhibitory assay
All the estimated derivatives suppressed 15-LOX enzyme with IC50 range equal to 5.6–27.3 µM. Compound 12 was the most potent 15-LOX suppressor with IC50 = 5.6 µM revealing higher inhibitory activity than NDGA (IC50 = 8.5 µM). In addition, the pyrazolo[1,5-a]pyrimidines 13 (IC50 = 8.4 µM) and 14 (IC50 = 8.2 µM) demonstrated comparable activity to that exerted by NDGA (IC50 = 8.5 µM) (Table 1). The obtained results pointed out that the disubstituted pyrimidine derivatives 12 (IC50 = 5.6 µM), 13 (IC50 = 8.4 µM) and 14 (IC50 = 8.2 µM) possessed favorable 15-LOX suppressing potential than trisubstituted analogues 9 (IC50 = 27.3 µM) and 11 (IC50 = 18.4 µM).
2.2.4 In vitro cyclooxygenase suppression estimation
The newly designed pyrazole derivative 8 and pyrazolo[1,5-a]pyrimidines 9 and 11–14 were screened in vitro against cyclooxygenase inhibitory potential. The suppression potential of these candidates was estimated by calculating the target concentration producing 50% cyclooxygenase inhibition (IC50). Selectivity index of COX-2 (S.I) for each tested compound was also measured and compared to indomethacin as a standard. The obtained results are reported in Table 1. All the target compounds were weak to moderate potent COX-1 suppressor (IC50 = 7.9–12.56 µM) and moderate to high potent COX-2 suppressor (IC50 = 1.11–4.7 µM). All tested compounds 8, 9 and 11–14 exhibited more activity in suppressing COX-2 than COX-1 isozyme. Furthermore, the target derivatives 11 (IC50 = 1.4 µM), 12 (IC50 = 1.11 µM), 13 (IC50 = 2.7 µM) and 14 (IC50 = 2.2 µM) recorded higher COX-2 inhibitory effect than exhibited by indomethacin (IC50 = 3.65 µM). The dimethylpyrazole 12 showed the most potency towards COX-2 inhibition (IC50 = 1.1 µM) which is similar as displayed by celecoxib (IC50 = 1.1 µM), whereas 5-hydroxy-7-methylsulfanyl-pyrazolopyrimidinedicarbonitrile (11) recorded the highest selectivity to COX-2 (S.I = 8.97) versus celecoxib (S.I. = 6.61) and indomethacin (IC50 = 3.65 µM, S.I = 0.08). The pyrazole derivative 8 exhibited the least COX-2 suppressor potential with IC50 = 4.7 µM, and S.I = 1.93. Moreover, replacing the methyl group in compound 12 (IC50 = 1.1 µM) with ethoxy group in compound 13 (IC50 = 2.7 µM) decreased the activity. In addition, the appendage of hydroxyl group instead of amino group markedly reduced the suppressive activity and selectivity to COX-2. This is evident upon comparing compound 11 (IC50 = 1.4 µM, S.I. = 8.97) with 9 (IC50 = 4.4 µM, S.I. = 2.32). Finally, hybridizing pyrazole ring 8 (IC50 = 4.7 µM, and S.I = 1.93) with pyrimidine scaffold enhanced the inhibitory activity towards COX-2 as revealed by 11 (IC50 = 1.4 µM, S.I. = 8.97), 12 (IC50 = 1.11 µM, S.I. = 7.66), 13 (IC50 = 2.7 µM, S.I. = 3.59).
2.2.5 In-vivo anti-inflammatory potential
The anti-inflammatory potential of derivatives 8, 9 and 11–14 were estimated via the use of procedure for inducing rat paw edema with carrageenan. Before the induction of inflammation by subcutaneous administration of carrageenan, each compound was taken at dose 50 mg/kg orally. The anti-inflammatory potential was measured according to paw volume changes after 3 h from carrageenan administration and the data obtained was listed in Table 2. Looking at the data, all the tested derivatives displayed AI activity with percentage inhibition range = 22–68%. The pyrazolopyrimidine derivatives 11–14 recorded higher AI potential (AI = 46–68%) than exhibited by the pyrazole derivative 8 and 5-aminopyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile (9) (AI = 22–34%) in contrast to celecoxib (AI = 72%). Furthermore, the ED50 values for the most active AI candidates 11–14 were measured using celecoxib as a standard. Compound 11 was the most potent candidate showing the same potency as that recorded by celecoxib (ED50 = 35 mg/kg). Values represent mean ± SEM (n = 3), Significance levels.
Compound no.
(% of anti-inflammatory potential AI ± SEM)a
ED50 (mg/kg)b
8
0.28 ± 0.08*** (22%)
NDc
9
0.24 ± 0.06 * (34%)
NDc
11
0.12 ± 0.03 *** (68%)
35
12
0.18 ± 0.05 *** (50%)
38
13
0.10 ± 1.40 ** (48%)
52
14
0.09 ± 1.45 *** (46%)
54
Celecoxib
0.09 ± 0.75 *** (72%)
35
2.2.6 Ulcerogenic liability
The most active candidates 11–14 were tested for their ulcerogenic liability using the low ulcerogenic standard celecoxib as present in Table 3. Both candidates 11 and 12 displayed the least ulcerogenic potential similar to the ulcerogenic effect of celecoxib (ulcer index = 0.33). The least ulcerogenic effect of compounds 11 and 12 might be due to their low effect to COX-1 (IC50 = 12.56, 8.5 µM, respectively) beside their high selectivity indices to COX-2 (S.I. = 8.97 and 7.66, respectively).
Compound no.
Ulcer index
Relative ulcerogenicity to celecoxib
11
0.33
1
12
0.33
1
13
1.67
5.06
14
1
3.03
Celecoxib
0.33
1
2.3 Docking study
To better understand the foundation of the diverse inhibitory activities of compounds 8–14 against COXs and 15-LOX enzymes, a docking study was conducted using AutoDock Vina 1.2.0 (Trott and Olson, 2010; Eberhardt et al., 2021). Meanwhile, this study delineates the structural features, key interactions, binding modalities adopted by a representative set of highly potent and less potent compounds inside the active pockets of both COX-2 and 15-LOX enzymes. The version of AutoDock Vina which is completely free software possesses a highly developed scoring function, expanded forcefield and can reproduce the improved docking performance compared with the original Autodock4 engine.
To perform this study, the highest resolution (1.73 Ǻ) 3D crystal structure of COX-2 (PDB ID: 3NT1, complexed with naproxen) and 15-LOX 3D crystal structure (PDB code: 1LOX, resolution = 2.40 Ǻ, complexed with 3-(2-oct-1-yn-1-ylphenyl)acrylic acid, RS7) were downloaded from the Protein Data Bank website (https://www.rcsb.org). Initially, the reliability of the docking protocol was validated by removing the co-crystallized naproxen ligand from COX-2 3D complex and and re-docking it into the active pocket of the same enzyme. Upon visual inspection, it showed good alignment and similar conformations with low RMSD value of 0.4 Ǻ, Fig. 4(A). The overlay of the top docking poses of the most active, 12 (IC50 = 1.11 µM) and the least active compound, 8 (IC50 = 4.7 µM) along with naproxen into the active pocket of COX-2 revealed an occupation of the same part of the active site with remarkable shape complementarity and alignment, Fig. 4(B).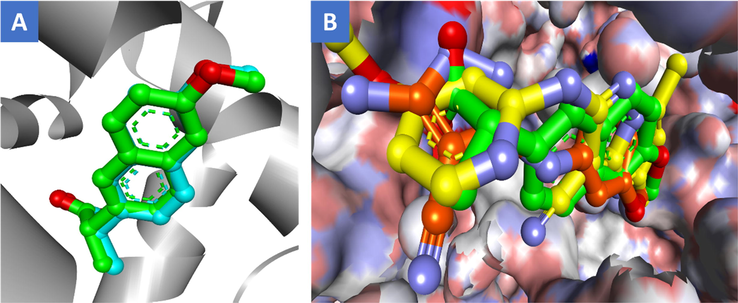
(A) Overlay of co-crystallized ligand, naproxen (green) and its redocked pose (cyan) inside the active site of COX-2 enzyme (PDB code: 3NT1); (B) Superimposition of the top docked poses of compounds 12 (yellow), 8 (orange), and naproxen (green) into the binding pocket of COX-2 where the 3D protein is shown as solvent-style surface.
The docking results of compound 12 against COX-2 active site revealed that the 5,7-dimethyl pyrazolopyrimidine moiety occupies the well-defined central binding pocket formed by Leu-352, Leu-384, Tyr-385, Trp-387, Phe-381, Phe-518, Met-522, Gly-526, and Ser-530 residues establishing a huge network of hydrophobic contacts between the two methyl groups and Phe-381, Leu-384, Tyr-385, Trp-387, Phe-518 and Met-522 amino acids. In addition, the pyrazolopyrimidine nucleus formed more hydrophobic interactions with Leu-352, Val-523, Gly-526 and Ala-527 residues. Also, a remarkable π-π stacking interaction with Phe-518 was noted. The 4-methoxypyridin-2-ylamino moiety lies primarily in the proximal binding pocket surrounded by Arg-120, Val-349, Ser-353, Tyr-355, Val-523, and Ala-527 forming some hydrophobic contacts with Val-116 and one π-π stacking interaction with Tyr-355 residue. Moreover, the amino group formed a crucial hydrogen bonding interaction with the OH of Tyr-355. It was found also that the methoxy group projects downward toward the mouth of the active site where it engaged in an important polar interaction (H-bond) with Arg-120 residue. Intriguingly, the majority of interactions exerted by 12 were hydrophobic in nature with reduced polar contacts, which is in a complete agreement with the well-documented binding patterns of most of known COX-2 selective inhibitors. The 2D/3D binding pattern and key interactions of 12 within the active site of COX-2 enzyme were depicted in Fig. 5 (A&B).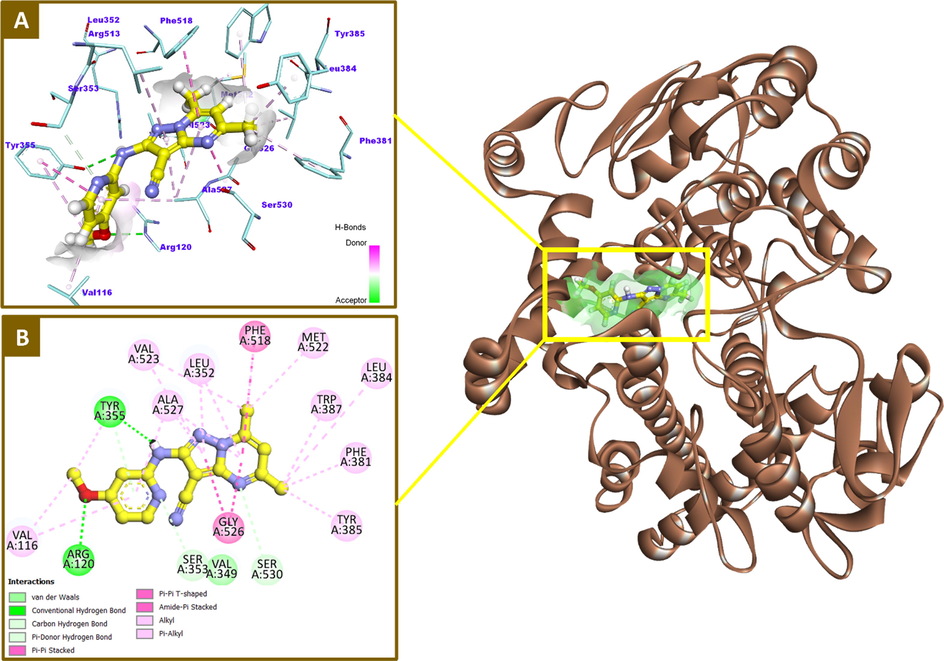
2D and 3D binding modalities of compound 12 (Ball & stick with carbons colored in yellow) within the catalytic active site of COX-2 enzyme (PDB code: 3NT1); A) 3D binding pattern of 12 into the active pocket of COX-2; B) 2D binding pattern of 12 into the active pocket of COX-2; H-bonds were represented as dashed green lines. π-Stacking and hydrophobic interactions were showed in dashed dark pink and light violet lines, respectively. H-bond surfaces around ligands were created. The 3D protein is displayed as secondary structure in ribbon solid style colored in tan.
On the other hand, the less potent compound, 8 was found to bind into COX-2 active site with an inverted conformation where the 4-methoxypyridine located in the central binding pocket rather the proximal one as in compound 12. This 180° rotation could be explained by the less steric hinderance existed in compound 8 which forced the 4-methoxypyridine to be deeply extended into the central pocket away from the constriction. After cyclization of 8 to furnish compound 12 bearing the bulkier dimethylpyrazolopyrimidine moiety, the constriction could not accommodate it and the whole structure adopted different conformation. The methoxy group of 8 extended upward toward the apex of active site forming some hydrophobic contacts with Phe-381, Tyr-385 and Trp-387 residues. However, a part of pyridine ring lies in the proximal pocket and interacted with Leu-352, Val-553 and Glu-356. Notably, pyrazole-4-carbonitrile occupies the proximal binding pocket establishing two important interactions (H-bond and π-π stacking) with OH and phenyl ring of Tyr-355, respectively. However, an unfavorable clash with Arg-120 was observed instead of forming the important H-bond which might be the reason behind the reduction in activity beside the inverted binding pattern of compound 8 comparing with 12, Fig. 6 (A-C).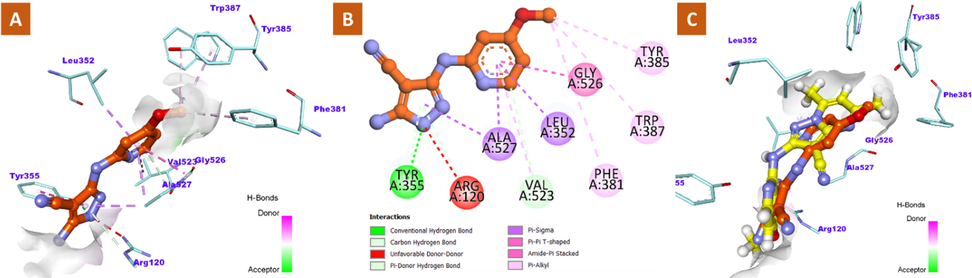
2D and 3D binding modalities of compound 8 (Ball & stick with carbons colored in orange) within the catalytic active site of COX-2 enzyme (PDB code: 3NT1); A) 3D binding pattern of 8 into the active pocket of COX-2; B) 2D binding pattern of 12 into the active pocket of COX-2; C) Overlay of 8 (orange) and 12 (yellow) inside the active site of COX-2 enzyme. Unfavorable clashes were showed in dashed red lines. H-bonds were displayed as dashed green lines.
Despite the COX-2 active site comprises four pockets; central, proximal, distal and the special side pocket lined by His-90, Gln-192, Leu-352, Ser-353, Tyr-355, Arg-513 and Val-523 (Malkowski et al., 2000), our active compounds occupy and interact with only two binding pockets. Indeed, it is not surprising with such a wide variety of chemical structures of the reported and well-known COXs inhibitors which exhibit several different binding patterns depending on their size and orientation and establish contacts with various numbers of binding pockets (Rouzer and Marnett, 2020). Also confirmed that the stabilization of interactions between ligand and COX isoenzymes is of greater importance than access to a greater number of binding pockets. Therefore, the preferential COX-2 inhibitory activity of our compounds could be now properly justified, and their adopted binding mode resembles that of some reported COXs inhibitors. Furthermore, it can be conceptualized obviously that it is not necessary for the COX inhibitor to interact with the characteristic COX-2 side pocket to achieve the selectivity for COX-2 over COX-1 isoenzyme.
Meanwhile, the most potent compound 12 (IC50 = 5.60 µM) against 15-LOX was docked into the catalytic domain of its active site using the mammalian 3D crystal structure (PDB: 1LOX) after removing the co-crystallized ligand, RS7. To compare the results and to have a robust explanation for the divergence of activity, the least potent compound, 9 was selected to be docked as well.
Upon examination of the docking results, it indicates that compound 12 adopted a typical orientation and occupies the hydrophobic part of the active site in a similar manner to that of RS7. The 4-methoxypyridin-2-ylamino functionality was laid near the catalytic iron ion with formation of a critical π-charge interaction which might play a significant role in the inhibitory activity of the compound. The phenyl ring also was involved in π-π stacking interactions with His-366 residue, in addition to some other hydrophobic contacts with Ilu-400, Ala-404 and Leu-408 amino acids. Due to the high hydrophobicity nature of the 15-LOX active pocket except its opening, it is not surprising to found the 5,7-dimethylpyrazolopyrimidine moiety of 12 located and extended deeply in this region establishing a network of hydrophobic interactions with the surrounding amino acids including Leu-408, Ile-418, Ile-593, Val-594 and Leu-597. Moreover, it provides two π-anion interactions with Glu-357 residue and π-π stacking with Phe-353. It interacts also with Met-419 through a π-sulfur bond. The stabilization of the complex of 12 and 15-LOX could be attributed to these hydrophobic contacts and π-charge interaction with catalytic iron. Fig. 7(A&B) illustrates the 2D and 3D interactions of compound 12 with the catalytic pocket of 15-LOX.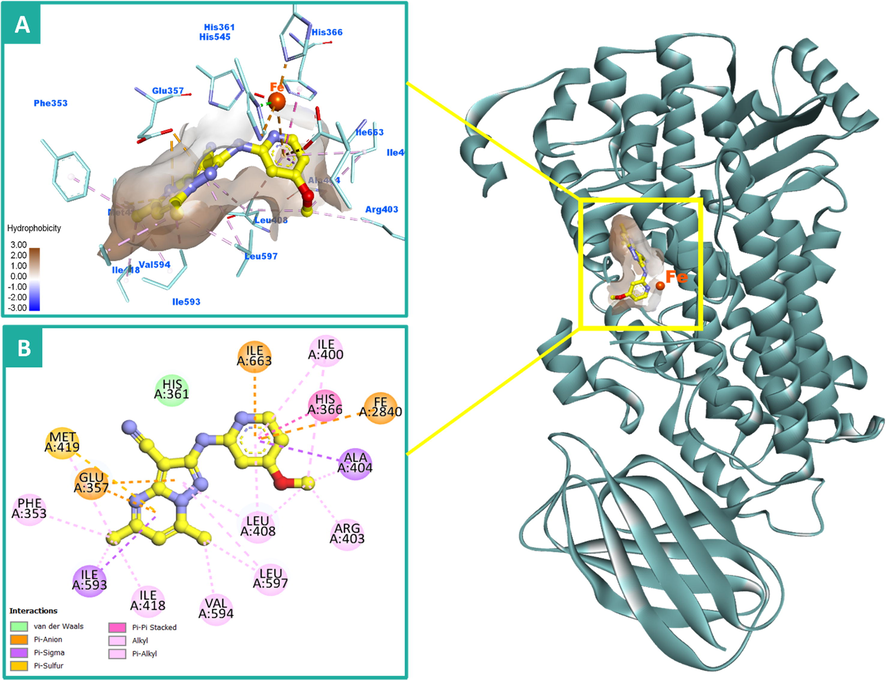
2D and 3D binding modalities of compound 12 (Ball & stick with carbons colored in yellow) within the catalytic active site of 15-LOX enzyme (PDB code: 1LOX); A) 3D binding pattern of 12 into the active pocket of 15-LOX; B) 2D binding pattern of 12 into the active pocket of 15-LOX. π-Stacking, π-charge and hydrophobic interactions were showed in dashed dark pink, orange, and light violet lines, respectively. Hydrophobic surface around ligand was created. The 3D protein is displayed as secondary structure in ribbon solid style colored in dark cyan. The iron (Fe) atom was displayed as ball colored in dark orange. Hydrogens were deleted for clarity purpose.
Conversely, the replacement of methyl groups on pyrazolopyrimidine moiety of compound 12 with polar amino and cyano group in compound 9 resulted in a sharp drop in potency (IC50 = 27.30 µM). The possible explanation for that is upon the increased polarity of 9, it was forced to protrude outside the pocket toward the polar opening and exhibited a different conformation to avoid the highly hydrophobic character of the deep active site. However, the 4-methoxypyridine functionality was positioned near the catalytic iron, it failed to be engaged in any kind of interactions like the corresponding moiety in compound 12 due to a significant increase in the distance with and Fe cation. This could also participate in explanation of the remarkable decrease in the potency of 9 comparing to 12, Fig. 8 (A&B).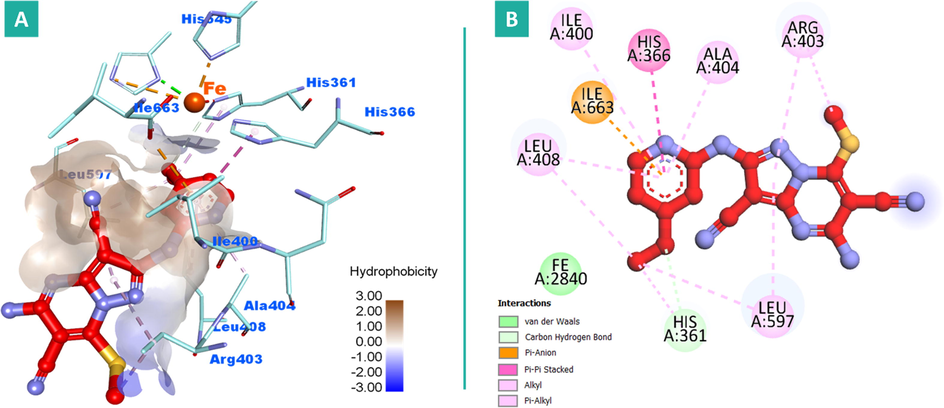
2D and 3D binding modalities of compound 9 (Ball & stick with carbons colored in red) within the catalytic active site of 15-LOX enzyme (PDB code: 1LOX); A) 3D binding pattern of 9 into the active pocket of 15-LOX; B) 2D binding pattern of 9 into the active pocket of 15-LOX. Hydrophobic surface around ligand was created. The iron (Fe) atom was colored in dark orange. Hydrogens were deleted for clarity purpose.
The dominant conformations of 9, 12, and RS7 inside the active cavity of 15-LOX were superimposed to show the different dispositions and orientations of these compounds, Fig. 9. Interestingly, the docking scores were in line with the visual investigations where compound 12 exerted higher score than that of the co-redocked co-crystallized ligand, RS7 with recorded ΔG values of −8.39 and −7.68 kcal/mol, respectively. However, compound 9 recorded ΔG value of −6.71 kcal/mol.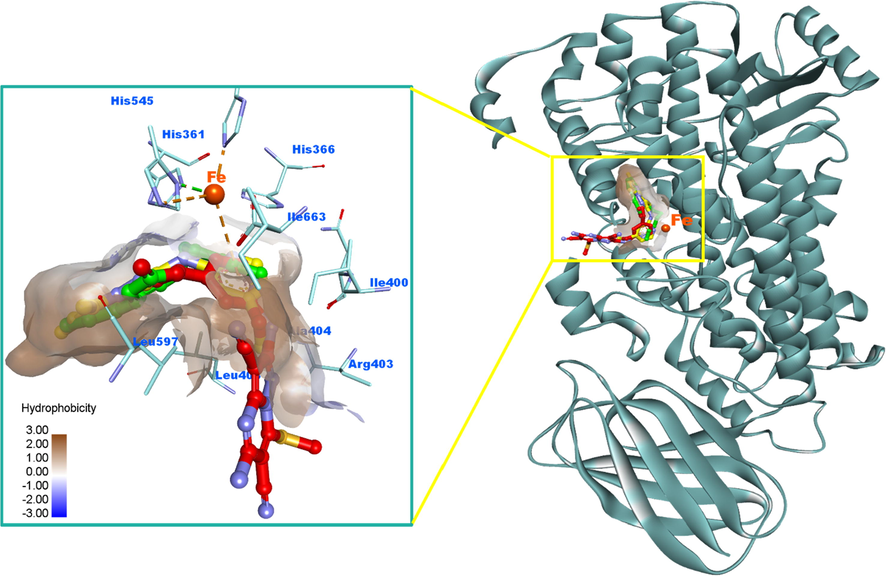
Overlay of 9 (red), 12 (yellow) and RS7 (green) to show the shape complementarities, difference in orientation and torsions exhibited by the docked poses within the active site of 15-LOX. The iron (Fe) atom was displayed as ball colored in dark orange. Pi-sulfur and hydrophobic interactions were showed in dashed orange and light violet lines, respectively. Hydrogens were deleted for clarity purpose. Hydrophobic surfaces around ligands were created. The 3D protein is displayed as secondary structure in ribbon solid style colored in dark cyan.
Collectively, these results strongly endorsed the in vitro inhibitory potential of our newly synthesized derivatives against both COX-2 and 15-LOX enzymes. We herein pointed out the possible explanation for the potency divergence, the crucial structural features and key-interactions that might greatly contribute to further design and develop potential leads as promising anti-inflammatory agents with dual inhibitory activity.
3 Conclusion
In conclusion, new pyrazole derivative 8 and pyrazolo[1,5-a]pyrimidines 9 and 11–14 were constructed and assayed for their anti-inflammatory potential in vivo and in vitro towards certain pro-inflammatory mediators (IL-6 and TNF-α) and by estimating COX isoforms, 15-LOX, sPLA2-V, inhibitory potential. Excellent dual suppressor potential was recorded by the target compound 11 towards IL-6 and TNF-α release (percentage inhibition = 89, 80%, respectively). In addition the target compounds 11, 12, 13 and 14 exhibited higher COX-2 inhibitory activity (IC50 = 1.4, 1.11, 2.7 and 2.2 µM, respectively) than displayed by indomethacin (IC50 = 3.65 µM). Moreover, 5-hydroxy-2-(4-methoxypyridin-2-ylamino)-pyrazolo[1,5-a]pyrimidinedicarbonitrile (11) was the most active sPLA2-V suppressor (IC50 = 1 µM). Regarding 15-LOX inhibitory potential, derivative 12 was the most potent 15-LOX suppressor (IC50 = 5.6 µM) showing higher inhibitory activity than NDGA (IC50 = 8.5 µM) and compounds 13 (IC50 = 8.4 µM) and 14 (IC50 = 8.2 µM) demonstrated comparable 15-LOX inhibitory activity to that exerted by NDGA. Furthermore, the pyrazole compound 8 was the least potential target candidate against all the assayed enzymes and pro-inflammatory cytokines revealing that fusing pyrazole ring with pyrimidine scaffold increased the anti-inflammatory potential. Finally, molecular modeling studies displayed that compounds 12 and 11 were the most fitted derivatives within the active site of COX-2 forming 4 to 5 hydrogen bondings with Arg513, His90, Tyr385, Ser530, Arg120, Tyr355 and Gln192 amino acids.
4 Materials and methods
4.1 Chemistry
The utilized chemicals had been purchased from Aldrich Company for chemicals. To check progress of the reactions and products purity, we used silica gel percolated with F254Merck plates (Darmstadt, Germany). Griffin apparatus had been utilized for recording melting points and was uncorrected. Bruker-Vector 22 spectrophotometer was used to get IR spectra. 1H and 13C NMR had been estimated using rianMercuryVXR-400 at 400 and 125 MHz by the use of DMSO as a selected solvent. Hewlett Packard MS-EI5988 spectrometer had been used to estimate mass spectra and electron impact at 70 eV, at Cairo University. Perkin-Elmer 2400 at Cairo University had been utilized to estimate the microanalysis. The ketene S,S acetals 5 and 10 were constructed as documented (Chandrashekar et al., 2011).
4.1.1 Synthesis of 2-[(4-methoxypyridin-2-ylamino)-methylsulfanyl-methylene]-malononitrile (7)
A blend of 2-(bis-methylsulfanyl-methylene)malononitrile (5) (1.70 g, 0.01 mol), 4-methoxypyridin-2-ylamine (6) (1.27 g, 10 mmol), triethylamine (3 drops) and ethyl alcohol (25 mL) was refluxed for 3 days at 180 °C. Ethanol had been evaporated to half its volume followed by collecting the residue formed, then dried and purified by crystallization to afford derivative 7 using methanol.
Yield 45%; m.p.: 112–114 °C. IR (KBr): 3376 (NH), 2256, 2217 (2CN) cm−1; 1HNMR: δ 2.55 (s, SCH3, 3H), 3.73 (s, OCH3, 3H), 6.12–8.02 (m, 3H, Ar-H), 12.20 (s, NH, 1H, exchangeable by D2O); 13C NMR (DMSO–d6): δ 15.7, 55.3, 95.3, 98.2, 149.6, 159.8, 160.7, 43.8, 192.1, 116.2; EIMS (m/z, %) 246 (M+, 85%). Anal. Calcd for C11H10N4OS (246.29): C, 53.64; H, 4.09; N, 22.75. Found: C, 53.61; H, 4.03; N, 22.50.
4.1.2 Preparation of 5-amino-3-(4-methoxypyridin-2-ylamino)-1H-pyrazole-4-carbonitrile (8)
A blend of 2-[(4-methoxypyridin-2-ylamino)-methylsulfanyl-methylene]malononitrile 7 (2.46 g, 10 mmol) and 99.99% hydrazine hydrate (0.64 g, 20 mmol) was heated under reflux using water bath for 8 h and then cooling the reaction mixture. The formed precipitate was filtered, washed using ethyl alcohol, dried then purified by crystallization using dimethylformamide/ water to give pyrazole derivative 8.
Yield 60%; m.p.: 125–127 °C. IR (KBr): 3367–3231 (2NH, NH2), 2216 (CN) cm−1; 1HNMR: δ 3.73 (s, OCH3, 3H), 6.78 (s, NH2, 2H), 6.32–8.14 (m, 3H, Ar-H), 12.35 (s, NH, 1H), 12.73 (s, NH, 1H); 13C NMR: δ = 56.1, 61.2, 95.3, 98.2, 115.1, 149.6, 152.3, 156.5, 160.7; EIMS (m/z, %) 230 (M+, 54.22, %). Anal. Calcd for C10H10N6O (230.23): C, 52.17; H, 4.38; N, 36.50. Found: C, 51.99; H, 4.29; N, 36.46.
4.1.3 Synthesis of 5-amino-2-(4-methoxypyridin-2-ylamino)-7-methylsulfanyl-pyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile (9)
To a solution of key intermediate (8) (2.30 g, 10 mmol) in dry acetone (30 mL), 2-(bis-methylsulfanyl-methylene)malononitrile 5 (1.70 g, 10 mmol) and triethylamine (3 drops) were added, followed by refluxing the blend for 7 h. The product was obtained through filtration then dried and purified by crystallization from dimethylformamide to afford immaculate product 9.
Yield 66%; m.p.: 181–183 °C. IR (KBr): 3400–3100 (NH,NH2) and 2215, 2220 (2CN) cm−1; 1H NMR: δ = 2.61 (s, SCH3, 3H), 3.72 (s, OCH3, 3H), 7.01–8.21 (m, 3H, Ar-H), 11.33 (s, 2H, NH2), 12.52 (s, NH, 1H); 13C NMR: δ = 14.2, 56.1, 95.3, 98.2, 149.6, 156.5, 160.7, 76.9, 147.6, 91.2, 172.7, 152.9, 164.8, 116.5. EIMS (m/z, %) 352 (M+, 12.19%). Anal. Calcd for C15H12N8OS (352.37):C, 51.13; H, 3.43; N, 31.80. Found: C, 51.10; H, 3.33; N, 31.73.
4.1.4 Synthesis of 5-hydroxy-2-(4-methoxypyridin-2-ylamino)-7-methylsulfanyl-pyrazolo[1,5-a]pyrimidine-3,6-dicarbonitrile (11)
A blend of derivative 8 (2.30 g, 10 mmol), ethyl 2-cyano-3,3-bis(methylthio)acrylate (10) (2.17 g, 0.01 mol), trimethyl amine (4 drops) and ethyl alcohol (20 mL) was heated under reflux for 9 h. The formed pyrazolopyrimidine 11 was collected then dried and purified through crystallization by ethanol and dimethylformamide blend (1:1).
Yield 63%; m.p.: 189–190 °C. IR (KBr): 3400–3100 (NH, OH), 2216, 2224 cm−1 (2CN) and; 1H NMR: δ = 2.62 (s, 3H, SCH3), 3.73 (s, 3H, OCH3), 6.41–8.14 (m, 3H, Ar-H), 11.76 (s, 1H, OH), 12.02 (s, 1H, N-H); 13C NMR (DMSO–d6): δ = 14.2, 56.1, 95.3, 98.2, 149.6, 156.5, 160.7, 76.9, 147.6, 102.5, 172.9, 153.1, 176.5, 117.3. EIMS (m/z, %) 353 (M+, 14.11%). Anal. Calcd for C15H11N7O2S (353.36): C, 50.99; H, 3.14; N, 27.75. Found: C, 50.94; H, 3.09; N, 27.66.
4.1.5 Synthesis of 2-(4-methoxypyridin-2-ylamino)-5,7-dimethylpyrazolo[1,5-a]pyrimidine-3-carbonitrile (12)
A blend of pyrazole-4-carbonitrile derivative (8) (2.30 g, 0.01 mol) and acetylacetone (1 g, 0.01 mol) in ethanol (25 mL) and acetic acid glacial (3 drops) was heated for 7 h under reflux. The reaction blend was cooled then poured into 30 mL cold water then the isolated product was collected, dried well followed by crystallization from DMF to afford the target 12.
Yield 89%; m.p. 219–220 °C. IR (KBr): 3400–3100 (NH), 2215 (CN) cm−1; 1H NMR: δ 2.35 (s, 6H, 2CH3), 3.72 (s, 3H, OCH3), 7.06–8.32 (m, 4H, Ar-H), 12.03 (s, NH, 1H); 13C NMR: δ 17.7, 28.0, 56.1, 95.3, 98.2, 116.4, 149.6, 156.5, 160.7, 76.9, 147.6, 119.8, 146.7, 153.1, 161.30, EIMS (m/z, %) 294 (M+, 4.11%). Anal. Calcd for C15H14N6O (294.31): C, 61.22; H, 4.79; N, 28.55. Found: C, 61.00; H, 4.50; N, 28.38.
4.1.6 7-Ethoxy-2-(4-methoxypyridin-2-ylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (13)
A blend of pyrazole 8 (2.30 g, 10 mmol) and ethyl acetoacetate (1.30 g, 10 mmol) in ethanol (20 mL) and glacial acetic acid (3 drops) was heated for 6 h under reflux. After cooling, the reaction mixture was poured onto ice and the isolated compound was collected, then dried and purified by crystallization using EtOH/ DMF to yield compound 13.
Yield: 66%; m.p.: 197–199 °C. IR (KBr): 3375 (NH), 2215 (CN) cm−1; 1H NMR: δ 1.29 (t, CH3, 3H), 2.75 (s, CH3, 3H), 3.73 (s, OCH3, 3H), 4.43 (q, CH2, 2H), 6.42–8.02 (m, 4H, Ar-H), 12.03 (s, NH, 1H); 13C NMR: δ = 15.2, 28.03, 49.6, 56.10, 65.02, 95.3, 98.2, 156.5, 160.7, 76.9, 112.2, 116.45, 147.6, 153.1, 163.4, 172. EIMS (m/z, %) 324 (M+, 27.54%). Anal. Calcd for C16H16N6O2 (324.34): C, 59.25; H, 4.97; N, 25.91. Found: C, 58.79; H, 5.00; N, 26.00.
4.1.7 Synthesis of 7-amino-5-ethoxy-2-(4-methoxypyridin-2-ylamino)pyrazolo[1,5-a]pyrimidine-3-carbonitrile (14)
A blend of key intermediate 8 (2.30 g, 10 mmol) and ethyl cyanoacetate (1.13 g, 10 mmol) in dimethylformamide (15 mL) and glacial acetic acid (3 drops) was refluxed for 8 h. After cooling, the blend was poured over crushed ice, and the isolated compound was collected then dried well and crystallized using acetic acid to afford derivative 14.
Yield 67%; m.p.: 222–224 °C. IR (KBr): 3415–3266 (NH2 & NH), 2216 (CN) cm−1; 1H NMR: δ = 1.35 (t, CH3, 3H), 3.73 (s, OCH3, 3H), 4.45 (q, CH2, 2H), 7.42–8.02 (m, 4H, Ar-H), 12.02 (s, 1H, N-H); 13C NMR: δ 15.3, 56.1, 65.02, 95.3, 98.2, 114.03, 116.12, 149.6, 156.5, 160.7, 176.9, 147.6, 119.3, 143.2, 153.1, 170.2. EIMS (m/z, %) 325 (M+, 34.83%). Anal. Calcd for C15H15N7O2 (325.33): C, 55.38; H, 4.65; N, 30.14. Found: C, 55.50; H, 5.00; N, 30.00.
4.2 Biological activity
4.2.1 TNF-α and IL-6 suppression assay
Dulbecco’s modified Eagles medium having FBS (10%), penicillin (100 U/ml) and streptomycin (100 µg/ml) with carbon dioxide (5%) at 37 °C was used for incubating mouse Raw264.7 Macrophages, The macrophages had been treated with vehicle or test compounds (10 µM) for 2 h then treated with lipopolysaccharide (LPS) (0.5 µM/mL) for 22 h. Cells and culture media were centrifuged for 10 min at 1,000 rpm. Levels of TNF-α and IL-6 levels were estimated via the use of IL-6 and TNF-α ELISA kits following manufacturing guidelines. The supernatant was isolated after centrifugation the deposited till use at −80 °c. Cells had been washed using PBS then extracted later with cell lysis buffer (0.1 SDS, 20 mM Tris-HCl, 20 mM NaF, 2 mM EDTA, 150 mM NaCl and NP40 1%). The blend was agitated forcelly in lysis buffer for 15 min at 0 °C. After centrifuge at 4 Oc for 30 min, protein was collected and concentrations were estimated utilizing Bio-Rad protein assay reagents (Stoll et al., 2006).
4.2.2 Suppression of sPLA2-V
The source of enzyme used in this study was human recombinant sPLA2-V. The target compounds were estimated for their sPLA2-V suppression potential utilizing reported procedure (Jantan et al., 2014). Hydrolysis of 1,2-bis(heptanoylthio)- glycerophosphocholine at sn-2 bond by PLA2 led to free thiols exposure which converts DTNB into 2-nitro-5-thiobenzoic acid that measured at 405 nm photometrically. The assay had been performed using aqueous buffer at pH = 7.5 having NaCl (7 mM), Tris (26 mM), CaCl2 (9 mM) and Triton-X (280 mM). PLA2 and the substrate were re-suspended in buffer. DTNB was dissolved by aq. Tris HCl (pH = 7.5). DTNB and enzyme produced final concentrations of 87 µM and 100 µg/mL, sequentially. The assays were carried out in 96-well microliter plates comprising substrate solution at room temperature, DTNB and the tested compounds. Potential of sPLA2-V was measured 100% by only adding substrate and enzyme. The negative control used in this assay is DMSO (1.7% v/v).
4.2.3 15-Lipoxygenase (LOX) suppression assay
The potential of the prepared candidates 8, 9 and 11–14 towards soybean 15-LOX was estimated by colorimetric assay (Bharate et al., 2008). To carry out the assay Tris-HCl (0.1 M) had been utilized and 15-lipoxygenase had been suspended within the used buffer before this assay. The substrate was dissolved in KOH and vortexed in equal amounts and followed by dilution to get 1 mM concentration. This work had been done in in 96-well microliter plates comprising enzyme, substrate, tested compounds at room temperature. The maximum potential of 15-LOX was determined by adding enzyme and substrate only. To estimate the potential, tested compound (10 µL) and LOX (95 µL) were added. The reaction was started by adding substrate solution to all wells for 5 min. To halt enzyme catalysis, chromogen (100 mL) for 5 min was added. Then, the absorbance was determined at 490 nm by the use of Tecan infinite pro 200 microplate reader (Tecan Group Ltd., Mannedorf, Switzerland).
4.2.4 In vitro cyclooxygenase suppression assay
Suppressing Ovine cyclooxygenase-1 and cyclooxygenase-2 was measured in vitro as reported using enzyme immunoassay (EIA) kit (Abdelgawad et al., 2017b).
4.2.5 In vivo anti-inflammatory studies
We used in this study adult male Wister albino rats weighing 150–180 g. Before any experimental study, rats are given 14 days to acclimate. The rats were kept in a controlled environment with access to water and food. All assays had been done adapting rules for care of animals in lab. Carrageenan initiated paw edema in rat had been used in order to determine the anti-inflammatory potential for the constructed candidates 8, 9 and 11–14 (Bakr et al., 2019). We used in this assay groups male Wister albino rats (body weight = 100–150 g) each of four rats under controlled conditions (humidity 60 ± 10% and temperature 27 ± 2 °C). 1% carrageenan in saline (0.05 mL/rat) was injected subcutaneously into the left hind paw of each rat one hour after administrating the examined compounds (50 mg/kg). After three hours of injecting carrageenan, the paw thickness for each rat was calculated and the thickness change and percent of paw edema inhibition had been measured. The ED50 was calculated for the most active candidates.
4.2.6 Ulcerogenic liability
Ulcerogenic risk for derivatives 11–14 and celecoxib was determined as reported procedure (Cho and Ogle, 1979).
4.3 Docking study
The newly released Autodock Vina program version 1.2.0 was employed to conduct this study as reported (Trott and Olson, 2010). 3D Crystal structures were selected and downloaded from Protein Data Bank. The co-crystallized ligands were removed from the initial crystal structures followed by deleting all water molecules. The docked compounds were energy minimized and rotatable bonds were adjusted and then, they were saved as PDBQT files. The Swiss-PdbViewer software version 4.1.0 was used to prepare both proteins by adding the missing atoms and energy minimization. Then, the polar hydrogens and Gastieger charges were added, and the corresponding charge files were generated, and the structures were saved in PDBQT file format using the Autodock Tools (ADT) version 1.5.6 according to the previous reports (Morris et al., 2009). The exhaustiveness was adjusted to 32. The settings of the two grid boxes were selected to encompass the co-crystallized inhibitors inside the active sites of both targets. The center co-ordinate was obtained from the central atom of the co-crystallized ligand. The COX-2 box was centered toward the coordinates of (X = -40.69, Y = -51.50, Z = –22.39 Å) with size of (X = 40, Y = 20, Z = 20 Å), while 15-LOX box was centered toward the coordinates of (X = -27.89, Y = 151.75, Z = 56.94 Å) with size of (X = 30, Y = 30, Z = 30 Å). The predicted and putative interactions and binding patterns were visualized using Discovery Studio Visualizer (DSV), V21.1 (BioVia, San Diego, CA, USA) after the successful completion of the docking simulation steps (Biovia, 2017).
4.4 Statistical analysis
All tests had been carried out thrice and obtained results are shown as mean ± S.E. IC50 Values were determined from three determinations via use of Graph Pad Prism 5 software. All the data had been analyzed utilizing one way ANOVA. P < 0.05 was considered as statistically significant.
Acknowledgements
The authors of this research would like to acknowledge the financial support offered by Taif University Researchers Supporting Project number (TURSP-2020/50), Taif University, Taif, Saudi Arabia. This publication was supported by AlMaarefa University researchers supporting program (grant number: MA-006), AlMaarefa University, Riyadh, Saudi Arabia. Also, The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: (22UQU4290565DSR25)
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Design, synthesis and biological evaluation of novel pyridine derivatives as anticancer agents and phosphodiesterase 3 inhibitors. Bioorg. Med. Chem.. 2009;17:5974-5982.
- [Google Scholar]
- Docking Study, Synthesis, and Anti-Inflammatory Potential of Some New Pyridopyrimidine-Derived Compounds. J. Inflammat. Res.. 2022;15:451-463.
- [Google Scholar]
- New pyrimidine-benzoxazole/benzimidazole hybrids: Synthesis, antioxidant, cytotoxic activity, in vitro cyclooxygenase and phospholipase A2-V inhibition. Bioorg. Chem.. 2019;92:103218
- [Google Scholar]
- Novel pyrimidine-pyridine hybrids: synthesis, cyclooxygenase inhibition, anti-inflammatory activity and ulcerogenic liability. Bioorg. Chem.. 2018;77:339-348.
- [Google Scholar]
- Discovery of a COX-2 selective inhibitor hit with anti-inflammatory activity and gastric ulcer protective effect. Future Med. Chem.. 2017;9:1899-1912.
- [Google Scholar]
- Design, synthesis and biological evaluation of some novel benzothiazole/benzoxazole and/or benzimidazole derivatives incorporating a pyrazole scaffold as antiproliferative agents. Bioorg. Chem.. 2017;74:82-90.
- [Google Scholar]
- Novel Phenolic Compounds as Potential Dual EGFR and COX-2 Inhibitors: Design, Semisynthesis, in vitro Biological Evaluation and in silico Insights. Drug Design, Development and Therapy. 2021;15:2325.
- [Google Scholar]
- Synthesis and antimicrobial activity of some new 5-arylazothiazole, pyrazolo [1, 5-a] pyrimidine,[1, 2, 4] triazolo [4, 3-a] pyrimidine, and pyrimido [1, 2-a] benzimidazole derivatives containing the thiazole moiety. Phosphorus, Sulfur, Silicon. 2010;185:709-718.
- [Google Scholar]
- Synthesis, cyclooxygenase inhibition, and anti-inflammatory evaluation of novel diarylheterocycles with a central pyrazole, pyrazoline, or pyridine ring. Med. Chem. Res.. 2015;24:2632-2644.
- [Google Scholar]
- Synthesis and biological evaluation of new diarylpyrazole and triarylimidazoline derivatives as selective COX-2 inhibitors. Arch. Pharm.. 2017;350:1600386.
- [Google Scholar]
- Synthesis and biological evaluation of novel 3-(quinolin-4-ylamino) benzenesulfonamides as carbonic anhydrase isoforms I and II inhibitors. J. Enzyme Inhib. Med. Chem.. 2019;34:1457-1464.
- [Google Scholar]
- Synthesis and SAR of a new series of COX-2-selective inhibitors: pyrazolo [1, 5-a] pyrimidines. J. Med. Chem.. 2001;44:350-361.
- [Google Scholar]
- An overview of structure–activity relationship studies of curcumin analogs as antioxidant and anti-inflammatory agents. Future Med. Chem.. 2017;9:605-626.
- [Google Scholar]
- Inhibition of the IL-6 signaling pathway: a strategy to combat chronic inflammatory diseases and cancer. Cytokine Growth Factor Rev.. 2013;24:163-173.
- [Google Scholar]
- Cyclooxygenase-2 inhibitors and cardiovascular risk in a nation-wide cohort study after the withdrawal of rofecoxib. Eur. Heart J.. 2012;33:1928-1933.
- [Google Scholar]
- Synthesis, cyclooxygenase inhibition, anti-inflammatory evaluation and ulcerogenic liability of new 1-phenylpyrazolo [3, 4-d] pyrimidine derivatives. J. Enzyme Inhib. Med. Chem.. 2016;31:6-12.
- [Google Scholar]
- Selective cyclooxygenase inhibition and ulcerogenic liability of some newly prepared anti-inflammatory agents having thiazolo [4, 5-d] pyrimidine scaffold. Bioorg. Chem.. 2019;88:102964
- [Google Scholar]
- Synthesis and evaluation of pyrazolo [3, 4-b] pyridines and its structural analogues as TNF-α and IL-6 inhibitors. Bioorg. Med. Chem.. 2008;16:7167-7176.
- [Google Scholar]
- Biovia, D.S., 2017. Discovery studio visualizer. San Diego, CA, USA 936.
- Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol. Pharmacol.. 2009;76:1053-1061.
- [Google Scholar]
- Cytotoxicity activity of Tragia involucrate. Linn Extracts Am Eurasian J. Toxicol. Sci.. 2011;3:67-69.
- [Google Scholar]
- Immunotherapy of rheumatoid arthritis targeting inflammatory cytokines and autoreactive T cells. Archivum immunologiae et therapiae experimentalis. 2010;58:27-36.
- [Google Scholar]
- An insight on synthetic and medicinal aspects of pyrazolo [1, 5-a] pyrimidine scaffold. Eur. J. Med. Chem.. 2017;126:298-352.
- [Google Scholar]
- Cholinergic-mediated gastric mast cell degranulation with subsequent histamine H1-and H2-receptor activation in stress ulceration in rats. Eur. J. Pharmacol.. 1979;55:23-33.
- [Google Scholar]
- AutoDock Vina 1.2. 0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model.. 2021;61:3891-3898.
- [Google Scholar]
- Microwave Assisted, Antimicrobial Activity and Molecular Modeling of Some Synthesized Newly Pyrimidine Derivatives Using 1, 4-diazabicyclo [2.2. 2] octane as a Catalyst. Lett. Drug Des. Discovery. 2020;17:1538-1551.
- [Google Scholar]
- Design, Synthesis, Molecular Modeling Study, and Antimicrobial Activity of Some Novel Pyrano [2, 3-b] pyridine and Pyrrolo [2, 3-b] pyrano [2.3-d] pyridine Derivatives. J. Heterocycl. Chem.. 2019;56:406-416.
- [Google Scholar]
- Elzahhar, P., Belal, A., Nassra, R., Abu-Serie, M., El-Hawash, S., 2018. Design, synthesis and biological evaluation of new pyridine/bipyridine carbonitriles and some related compounds Interfering with arachidonic acid pathway as potential anti-inflammatory agents.
- Synthesis and studies molecular docking of some new thioxobenzo [g] pteridine derivatives and 1, 4-dihydroquinoxaline derivatives with glycosidic moiety. J. Taibah Univ. Sci.. 2018;12:774-782.
- [Google Scholar]
- Synthesis and antitumor activity of some new pyrazolo [1, 5-a] pyrimidines. Chin. Chem. Lett.. 2017;28:388-393.
- [Google Scholar]
- Horowitz, M.C., Lorenzo, J.A. 2002. Local regulators of bone: IL-1, TNF, lymphotoxin, interferon-γ, IL-8, IL-10, IL-4, the LIF/IL-6 family, and additional cytokines. In: Principles of Bone Biology, Elsevier. pp. 961–977.
- Synthesis and Structure-Activity Relationship (SAR) of (5, 7-Disubstituted 3-phenylsulfonyl-pyrazolo [1, 5-a] pyrimidin-2-yl)-methylamines as Potent Serotonin 5-HT6 Receptor (5-HT6R) Antagonists. J. Med. Chem.. 2011;54:8161-8173.
- [Google Scholar]
- Studies of synthetic chalcone derivatives as potential inhibitors of secretory phospholipase A2, cyclooxygenases, lipoxygenase and pro-inflammatory cytokines. Drug Des., Develop. Therapy. 2014;8:1405.
- [Google Scholar]
- A facile, regioselective synthesis of pyrazolo [1, 5-a] pyrimidine analogs in the presence of KHSO 4 in aqueous media assisted by ultrasound and their anti-inflammatory and anti-cancer activities. Monatshefte für Chemie-Chemical Monthly. 2016;147:1257-1276.
- [Google Scholar]
- Synthesis and antihypertensive action of new imidazo [1, 2-a] pyridine derivatives, non peptidic angiotensin II receptor antagonists. Cardiovas. Haematol. Disorders-Drug Targets (Formerly Current Drug Targets-Cardiovascular & Hematological Disorders). 2011;11:87-96.
- [Google Scholar]
- Discovery of pyrazolo [1, 5-a] pyrimidine TTK inhibitors: CFI-402257 is a potent, selective, bioavailable anticancer agent. ACS Med. Chem. Lett.. 2016;7:671-675.
- [Google Scholar]
- The productive conformation of arachidonic acid bound to prostaglandin synthase. Science. 2000;289:1933-1937.
- [Google Scholar]
- Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol.. 2013;239:133-138.
- [Google Scholar]
- Diverse ways of perturbing the human arachidonic acid metabolic network to control inflammation. Acc. Chem. Res.. 2015;48:2242-2250.
- [Google Scholar]
- 3-Iminobutanenitrile as building block for the synthesis of substituted pyrazolo [1, 5-a] pyrimidines with antitumor and antioxidant activities. Int. J. Modern Organic Chem.. 2012;1:96-114.
- [Google Scholar]
- Phospholipase A2 inhibitors as potential anti-inflammatory agents. Curr. Pharm. Des.. 2005;11:1301-1312.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [Google Scholar]
- Synthesis of novel aryl and coumarin substituted pyrazolo [1, 5-a] pyrimidine derivatives as potent anti-inflammatory and anticancer agents. Chem. Data Collect.. 2020;30:100550
- [Google Scholar]
- Selected cytokine pathways in rheumatoid arthritis. In: Seminars in immunopathology. Springer; 2017. p. :365-383.
- [Google Scholar]
- 2, 4-Disubstituted Phenylhydrazonopyrazolone and Isoxazolone Derivatives as Antibacterial Agents: Synthesis, Preliminary Biological Evaluation and Docking Studies. ChemistrySelect. 2018;3:3295-3301.
- [Google Scholar]
- Eicosanoids: Atherosclerosis and cardiometabolic health. J. Clin. Translat. Endocrinol.. 2020;19:100216
- [Google Scholar]
- RA Abdellatif, K., KA Abdelall, E., B Bakr, R., 2017. Nitric oxide-NASIDS donor prodrugs as hybrid safe anti-inflammatory agents. Current Top. Med. Chem. 17. 941–955.
- N-(3-Arylaminopyridin-4-yl) alkanesulfonamides as pyridine analogs of nimesulide: Cyclooxygenases inhibition, anti-inflammatory studies and insight on metabolism. Eur. J. Med. Chem.. 2014;74:12-22.
- [Google Scholar]
- Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid. Redox Signal.. 2011;14:1889-1903.
- [Google Scholar]
- Structural and chemical biology of the interaction of cyclooxygenase with substrates and non-steroidal anti-inflammatory drugs. Chem. Rev.. 2020;120:7592-7641.
- [Google Scholar]
- Synthesis and analgesic/anti-inflammatory evaluation of fused heterocyclic ring systems incorporating phenylsulfonyl moiety. Bioorg. Med. Chem.. 2008;16:6344-6352.
- [Google Scholar]
- Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17-and TNF-α-induced genes in bone cells. J. Leukoc. Biol.. 2005;77:388-399.
- [Google Scholar]
- Endotoxin, TLR4 signaling and vascular inflammation: potential therapeutic targets in cardiovascular disease. Curr. Pharm. Des.. 2006;12:4229-4245.
- [Google Scholar]
- Anti-inflammatory cytokines: important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemotherapy Res. Practice. 2012;2012
- [Google Scholar]
- Szefel, J., Piotrowska, M., J Kruszewski, W., Jankun, J., Lysiak-Szydlowska, W., Skrzypczak-Jankun, E., 2011. Eicosanoids in prevention and management of diseases. Current Mol. Med. 11, 13–25.
- IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspect. Biol.. 2014;6:a016295
- [Google Scholar]
- Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol. Rev.. 2006;86:515-581.
- [Google Scholar]
- New Substituted 5-Benzylideno-2-Adamantylthiazol [3, 2-b][1, 2, 4] Triazol-6 (5H) ones as Possible Anti-Inflammatory Agents. Molecules. 2021;26:659.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- Adiponectin induces TNF-α and IL-6 in macrophages and promotes tolerance to itself and other pro-inflammatory stimuli. Biochem. Biophys. Res. Commun.. 2005;335:1254-1263.
- [Google Scholar]
- Nonpeptide angiotensin II receptor antagonists. I. Synthesis and biological activity of pyridine derivatives. Chem. Pharm. Bull.. 1994;42:1841-1849.
- [Google Scholar]
- Arachidonic acid metabolism and kidney inflammation. Int. J. Mol. Sci.. 2019;20:3683.
- [Google Scholar]
- Inflammation, cancer and oxidative lipoxygenase activity are intimately linked. Cancers. 2014;6:1500-1521.
- [Google Scholar]
- Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol.. 2008;127:385-393.
- [Google Scholar]
- Yedgar, S., Cohen, Y., Shoseyov, D., 2006. Control of phospholipase A2 activities for the treatment of inflammatory conditions. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1761, 1373–1382.




