

Translate this page into:
Synthesis and antimicrobial evaluation of some novel pyrido[3,2-f][1,2,3]thiadiazaphosphepinone compounds, bearing a pyridine moiety
⁎Corresponding author. a.algohary@mu.edu.sa (Ayman M. Algohary), mmhassan12@yahoo.com (Mohamed M. Hassan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
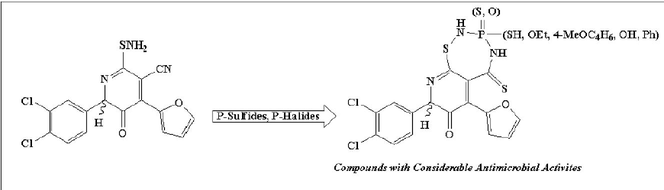
Several pyridothiadiazaphosphepinones (3–9) were synthasized by combining tetraphosphorus decasulfide, Lawesson's reagent, phosphorus tribromide, and P,P-dichlorophenyl phosphine with 2-(aminosulfanyl)-6-(3,4-dichlorophenyl)-4-(furan-2-yl)-5-oxo-5,6-dihydropyridine-3-carbonitrile (2). The reaction mechanisms for these products were discussed. On the basis of elemental analysis and spectrum data, the structures of the newly synthesized compounds were determined. The biological activity of every synthesized compound was examined against various microorganisms using the disc diffusion method. The bulk of the microorganisms tested are effectively inhibited by the newly synthesized chemicals.
Keywords
Pyridine
Tetraphosphorus decasulfide
Lawesson’s reagnt
Thiadiazaphosphepinone
Organophosphorous compounds

Data Availability Statement
All data are available in the manuscript and from the corresponding author upon request.
1 Introduction
Because of the numerous biological and pharmacological uses for phosphorus heterocycles, particularly those phosphorus connected to sulphur atoms, their synthesis has received a lot of attention. Anticancer (EOJ, 2000; Gholivand and Shariatinia, 2012), antibacterial (Karpagam and Guhanathan, 2014), insecticidal (Fagadar et al., 2006), herbicidal (Abdel Rahman, 2002), and antibody properties were found in some of these heterocycles (Stewart et al., 1993). Phosphorus heterocycles with multiple rings are also used as polymer stabilizers, lubricating oil additives, and biocatalysts in organic synthesis (Bartlett et al., 1990). Contrarily, because they are used as intermediates in numerous synthetic, biological, and pharmacological processes, six and seven membered phosphorus heterocyclic rings have attracted a lot of attention (Aweda et al., 2015). Researchers have recently become interested in the synthesis of thiazaphosphinine heterocycles because of their important biological role and several applications in synthetic chemistry (Younes et al., 2013a,b). In the light of the foregoing facts, as well as our ongoing research into the production of new biologically active heterocyclic organophosphorus compounds (Ayman and Mohamed, 2022; Ali et al., 2018; Ali et al., 2019a,b; Hassan et al., 2017; Ali and Hassan 2017), we herein report an effective approach for the synthesis of novel pyrido[3,2-f][1,2,4,3]thiadiazaphosphepinone compounds bearing a pyridine moiety, extending our applications to phosphorus reagents in addition to their antimicrobial potencies.
2 Results and discussion
2.1 Synthetic strategies
2-(aminosulfanyl)-6-(3,4-dichlorophenyl)-4-(furan-2-yl)-5-oxo-5,6-dihydropyridine-3-carbonitrile (2) was synthesized from amination of our reported starting material 6-(3,4-Dichlorophenyl)-4-(furan-2-yl)-5-oxo-2-thioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (1) (Ayman et al., 2021). On an ice bath, pale yellow crystals precipitated when an aqueous solution of hydroxylamine-O-sulfonic acid (HOSA) with an equimolecular amount of potassium hydroxide was dropwisely added to an aqueous solution of 2-thioxocarbonitrile 1 and potassium hydroxide (Scheme 1). Compound 2’s infrared spectrum displayed the NH2 and C≡N functions at 3460, 3320, and 2195 cm−1. 1H NMR spectrum showed a singlet signal at δ 5.42 ppm which is assigned to the chiral H-6 proton. Also, the distinctive NH2 proton was found as a singlet at δ 6.62 ppm. Additionally, the carbon atom CN was seen in its 13C NMR spectra at a level of 115 ppm. Additionally, the molecular ion peak of compound 2 at m/z 377 was observed, supporting the hypothesized structure.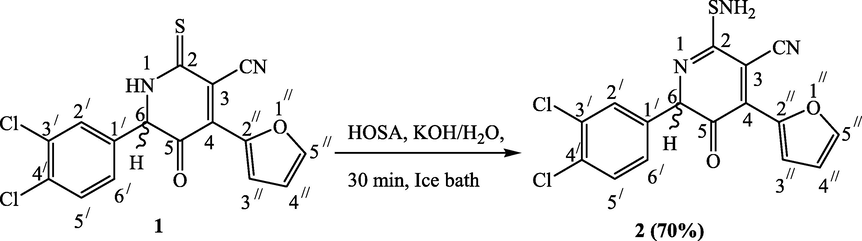
Synthesis of compound 2.
Because of the versatility of α-aminocarbonitrile substrate 2 in terms of electrophilic and nucleophilic attack, we decided to investigate its chemical reactivity towards phosphorus reagents. To synthesize our desired thiadiazaphosphepinone heterocycles, Compound 2 was allowed to react with tetraphosphorus decasulfide and Lawesson's reagent (LR) under various circumstances. As a result, reaction of compound 2 and the molar equivalent of tetraphosphorus decasulfide in dry toluene produced the compound 8-(3,4-dichlorophenyl)-6-(furan-2-yl)-3-sulfanyl-5-thioxo-2,3,4,5-tetrahydropyrido[3,2-f][1,2,4,3]. Thiadiazaphosphepin-7(8H)-one-3-sulfiden (3) in a yield of 73% (Scheme 2). Compound 3’s IR spectra showed the presence of a novel C=S group at 1120 cm−1 and the absence of a prominent typical nitrile band. The sulfanyl proton and two NH protons were detected in the 1H NMR spectra of compound 3 at δ 3.84, 9.03 and 10.20 ppm, respectively. The carbon atom of C=S was located at about 168.43 ppm in the 13C NMR spectra, which provided strong support for the hypothesized structure. Compound 3’s 31P NMR spectra showed a singlet at about 17.32 ppm. Additionally, its mass spectrum showed the anticipated molecular ion peak at m/z 505 (M+, 20%). A rational justification for the synthesis of compound 3 is presented in (Scheme 2). The reaction suggested to proceed via two steps. First, in the nonisolable intermediate B, P4S10 was added to the nitrile group, converting it to a thioamide group. Then, the new pyridothiadiazaphosphepinonesulfide system 3 is produced by the heterocyclization of the nearby amino groups by a reaction with the PS2 cation via C intermediate.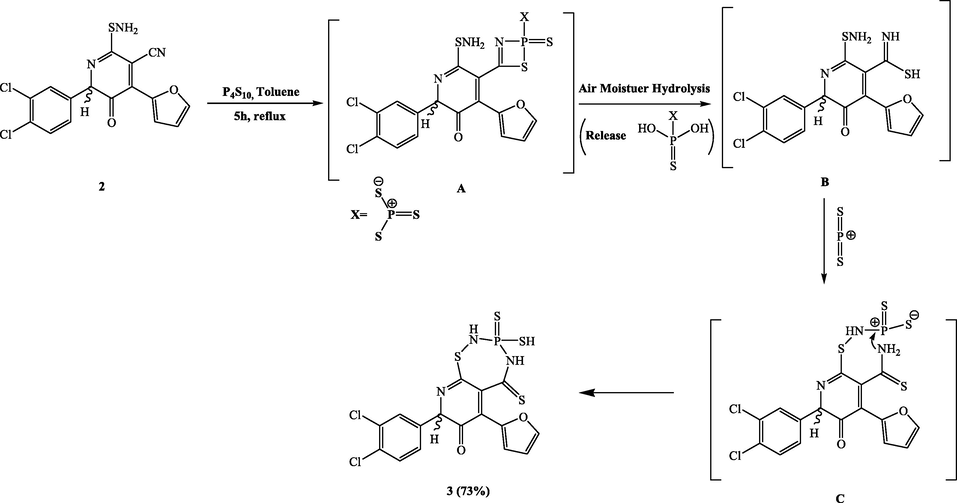
Synthesis of compound 3.
The equivalent thiadiazaphosphepinone product 4 was produced when compound 2 reacted with O,O-diethyldithiophosphoric acid (which was created in situ from the reaction of tetraphosphorus decasulfide with absolute ethanol) (Scheme 3). Compound 4’s mass spectrum revealed a molecular ion peak at m/z 517, and data from its 1H- and 13C NMR spectra confirmed the presence of the unique ethoxy group. The thiol group's nucleophilic addition to the nitrile group, which creates the nonisolable intermediate D, can be used to explain how the thiadiazaphosphepinone ring is created. Next, the amino functional group attacks the phosphorus atom. The nonisolable intermediate E was produced by this intramolecular cyclization, and it was then modified by a Dimroth rearrangement to produce the desired fused heterocyclic system (Scheme 3).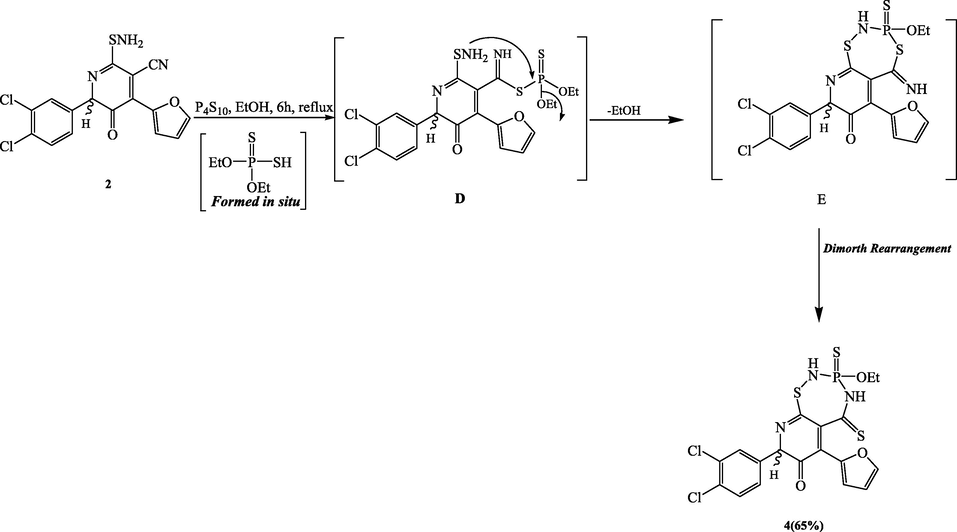
Synthesis of compound 4.
Under stirring conditions, compound 2 interacted with Lawesson's reagent in acetonitrile to yield just single product, 8-(3,4-dichlorophenyl)-6-(furan-2-yl)-2,3,4,5-tetrahydro-3-(4-methoxyphenyl)-3-sulfanylene-5-thioxopyrido[3,2-f][1,2,4,3]thiadiazaphosphepin-7(8H)-one (5). The reaction pathway was assumed to proceed via nucleophilic attack of the amino group on LR, followed by the addition of SH to the cyano group. The amino group was phosphorylated to create the nonisolable intermediate G, which was subsequently cyclized by combining the SH group with the nitrile group to create the intermediate H. The latter intermediate was quickly reorganized into the final product 5 (Scheme 4). The IR spectrum of compound 5 revealed the absence of the NH2 and CN groups of starting material 2, as well as the presence of the NH at 3428 cm−1 as a broad band. Two NH protons were found in the 1H NMR spectra at 9.06 and 10.18 ppm, while a singlet of OCH3 protons was found at 3.81 ppm. The carbon atom bound to the phosphorus atom, which resonant as a doublet at 134.71 ppm with a coupling constant of 129 Hz, was of particular significance in its 13C NMR spectra. It was also established that the methoxy group, at 55.62 ppm, was present. It also has a singlet at 19.14 ppm in its 31P NMR spectra. We found that the isolated product was compound 5 in a greater yield when the preceding reaction was conducted in 100% ethanol rather than acetonitrile. The LR reagent may react with absolute ethanol to create the nonisolable 4-MeOC6H4P(S)(SH)(OEt), which subsequently reacted with the starting material 2 to produce the nonisolable intermediate F as shown in the hypothesis for the production of compound 5 by this method (Scheme 4).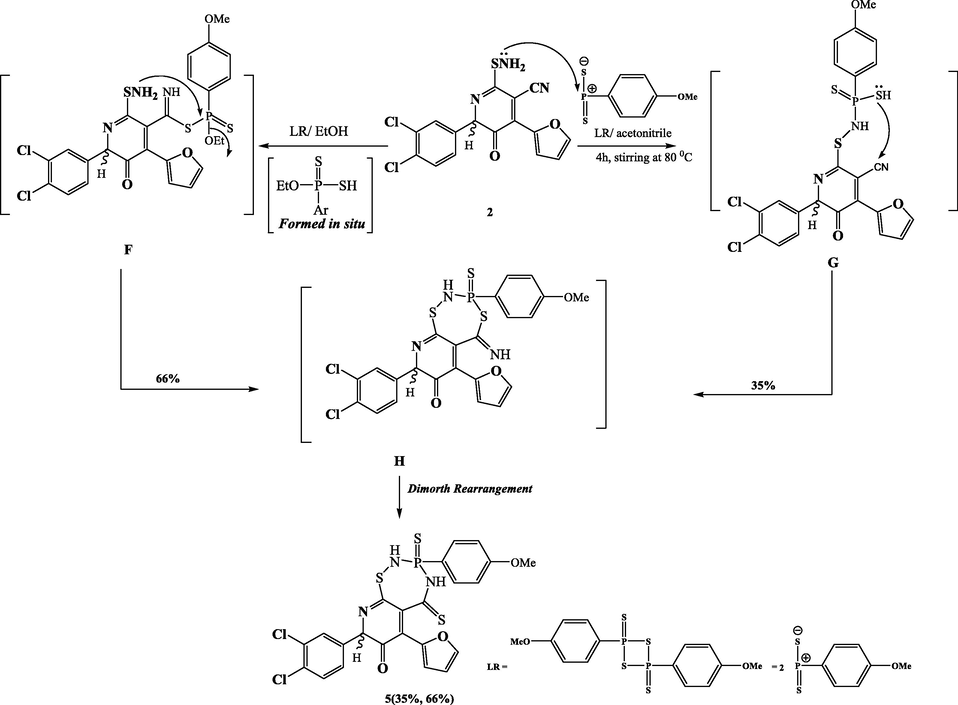
Synthesis of compound 5.
Compound 2 was treated with P-(4-methoxyphenyl)-N,N-diphenylphosphonothioic diamide 6, which was made by reacting LR with distilled aniline as described in the procedure (Clausen et al., 1981). The isolated product 7 (50 %) was obtained by refluxing the later compound 6 with α-aminocarbonitrile substrate 2 in dry toluene for 6 h. A nucleophilic attack by the amino group on the phosphorus atom of reagent 6 can be used to explain how compound 7 is formed. This attack results in the formation of the nonisolable intermediate I and the simultaneous elimination of an aniline moiety. The ring is then closed by adding NH to the nitrile group. It was evident that both amino and nitrile groups were involved when the matching signals from the 1H- and 13C NMR spectra disappeared. The 1H NMR spectra of product 7 also showed NH protons at 9.51 and 11.20 ppm, multiplets at 6.30–7.86 ppm that were likely caused by the aromatic protons introduced by the aniline used and LR, as well as a singlet at 3.75 ppm relevant to the methoxy group. The 13C NMR results provide unambiguous evidence for the produced product 7. The LR at 56.70 ppm added a new singlet methoxy group. The proposed structure is supported by the phosphorus atom signal at 20.10 ppm in the 31P NMR spectra of this molecule. The chemical formula and the mass spectra were closely matched (Scheme 5).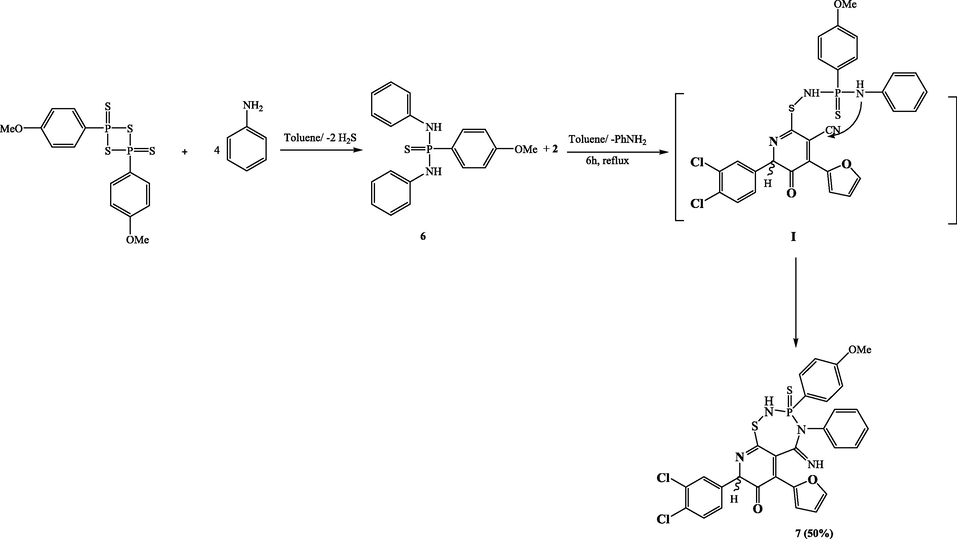
Synthesis of compound 7.
The chemical reactivity of compound 2 towards some phosphorus halides was then investigated. However, when compound 2 was reacted with phosphorus tribromide in dry dioxane with a catalytic quantity of triethylamine, we obtained the novel 7-(3,4-dichlorophenyl)-5-(furan-2-yl)-3-hydroxy-4-imino-3,4-dihydro-2H-pyrido[3,2-e][1,2,3]thiazaphosphinin-6(7H)-one-3-oxide (8) (Scheme 6). Compound 8’s IR spectra revealed specific absorption bands of 3380 (OH), 3200–3100 (2 NH), and 1620 (C=N). Two NH protons were found at 9.15 and 10.02 ppm in the 1H NMR spectra, as well as a typical singlet of P–OH proton at 5.80 ppm. The C=NH at the expense of nitrile carbon resulted in a singlet at 160.82 ppm in the 13C NMR spectrum. The molecular ion peak at m/z 441 (M+, 100 %) was visible in the ESI mass spectrum data for 8. Furthermore, the proposed structures' 31P NMR spectra matched the proposed structures' chemical shift at δ 16.30 ppm. It was once assumed that compound 8 was created through a straightforward condensation reaction that eliminated hydrogen bromide and created the nonisolable intermediate J. The latter intermediate's nitrile group underwent a nucleophilic addition of the phosphorus atom to produce the nonisolable intermediate K, which was then hydrolyzed by distilled water to produce the intermediate L, which was then rearranged to produce the product 8.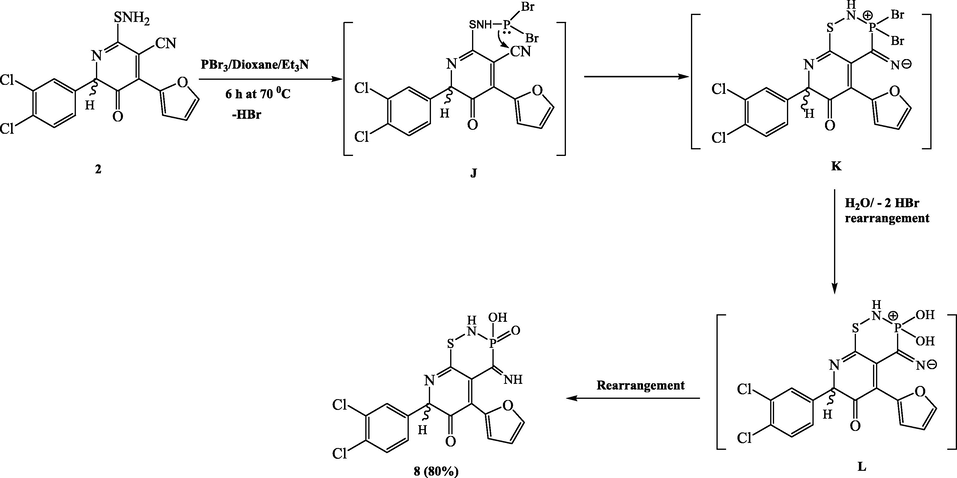
Synthesis of compound 8.
compound 2 was allowed to react with equimolar amounts of P,P-dichlorophenyl phosphine in dry pyridine at 70–80 °C for 8–10 h to give 7-(3,4-dichlorophenyl)-5-(furan-2-yl)-4-imino-3-phenyl-3,4-dihydro-2H-pyrido[3,2-e][1,2,3]thiazaphosphinin-6(7H)-one 3-oxide (9) (Scheme 7). Compound 9’s IR spectrum showed that it exhibits absorption bands at 1250, 1625, and 3250–3100 cm−1, which correspond to P=O, C=N, and (2 NH), respectively. Two singlet’s in its 1H NMR spectrum, corresponding to two NH protons, were seen at 9.51 and 10.50 ppm. Compound 9’s 31P NMR chemical shift showed a singlet at approximately 19.88 ppm, which agreed with the thiazaphosphinine chemical shift values. The 13C NMR spectrum showed the characteristic signals of all carbons, particularly those associated with the thiazaphosphinine ring. Special attention was paid to the carbon atom that was bound to the phosphorus atom and resonated as a doublet at a frequency of 128 Hz and 130.13 ppm. Additionally, compound 9’s mass spectrum confirmed its structure by displaying the proper molecular ion peak at m/e 501. By nucleophilically attacking the phosphorus atom of P,P-dichlorophenyl phosphine and removing one molecule of HCl, the intermediate M was produced, from which the target product 9 was created. The latter intermediate's nitrile group underwent a nucleophilic addition of phosphorous atoms to produce the nonisolable intermediate N, which was hydrolyzed by distilled water to release the intermediate O, which was then rearranged to produce the product 9.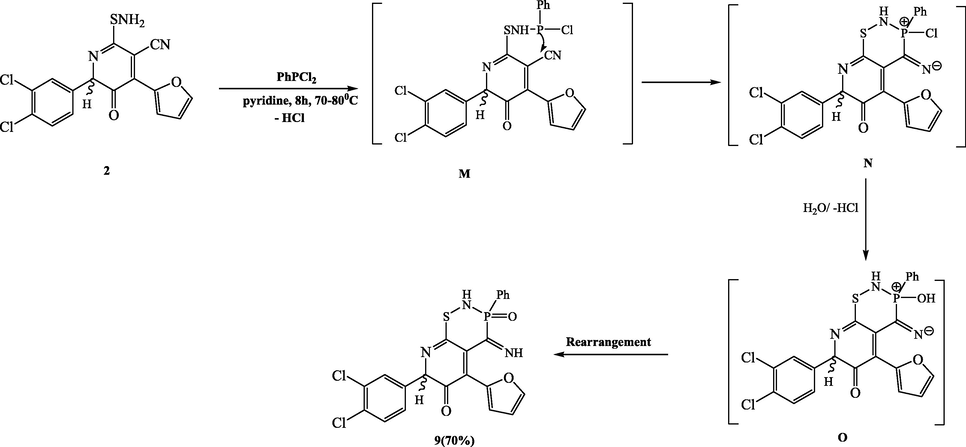
Synthesis of compound 9.
2.2 Biological screening (Microbiology)
The activity of the synthesised compounds against the vulnerable organisms Staphylococcus aureus and Bacillus subtilis as Gram-positive bacteria, Salmonella typhimurium and Escherichia coli as Gram-negative bacteria, and Candida albicans as a fungus strain was assessed using the standardised disc agar diffusion method (Gross and DeVay, 1977). To achieve a concentration of 100 g mL−1, the chemicals were dissolved in DMSO, which has no inhibitory activity. The medium potato dextrose agars (PDA), which comprise an infusion of 250 g potatoes, 5 g dextrose, and 20 g agar, were used for the test discs of filter paper of the same size (Bauer, et al., 1966), three per component, were carefully placed on the inoculated agar surface and impregnated with an equivalent volume (15 L) of the particular concentration of the dissolved tested compounds. The amount of inhibition of the organisms was measured and used to determine the mean of inhibition zones after incubation for 48 h at 25 °C in the case of bacteria and for 72 h at 22 °C in the case of fungi.
2.3 Antimicrobial activities
The data shown in Table 1 indicate a range of actions against all types of microorganisms, which may indicate that structural changes have an impact on how well they grow. The findings showed that certain chemicals under investigation had low to strong antibacterial action versus Gram-positive bacteria, Gram-negative bacteria, and a fungal strain (Table 1). Baciillus subtilis and candida albicans were both very active against both compouns 5 and 7 in both concentrations. Candida albicans, Escherichia coli, and Bacillus subtilis were all highly active against by compound 4. Additionally, compound 3 had strong action against candida albicans and Bacillus subtilis. Surprisingly, compound 8 showed considerable action at both doses against Staphylococcus aureus, salmonella typhimurium and Aspergillus fumigatus. At both concentrations, compound 2 and compound 9 had negligible effects on some of the tested microorganisms. The considerable variances in these compounds' antibacterial qualities further support the study's objectives. Designing more effective antimicrobial drugs for therapeutic application may benefit from further research into the links between structure and activity, toxicity, and biological consequences. * = Calculate from 3 values. ** = identified on the basis of routine cultural, morphological and microscopical characteristics. – = No effect. Low activity (Inhibition zone 7–10 mm) = Mean of zone diameter ≤ 1/3 of mean zone diameter of control. Intermediate activity (Inhibition zone 11–20 mm) = Mean of zone diameter ≤ 2/3 of mean zone diameter of control. High activity (Inhibition zone 21–28 mm) = Mean of zone diameter > 2/3 of mean zone diameter of control. #: Chloramphencol in the case of Gram-positive bacteria, Cephalothinin the case of Gram-negative bacteria and cycloheximide in the case of fungi.
Mean* of zone diameter, nearest whole mm.
Organism
Gram-positive bacteria
Gram-negative bacteria
Yeasts and Fungi**
Staphylococcus aureus
Bacillus subtilis
Salmonella typhimurium
Escherichia coli
Candida albicans
Aspergillus fumigatus
Comp.no.
1
mg/ml0.5 mg/ml
1 mg/ml
0.5 mg/ml
1 mg/ml
0.5 mg/ml
1 mg/ml
0.5 mg/ml
1 mg/ml
0.5 mg/ml
1
mg/ml0.5
mg/ml
2
9
8
–
–
11
8
–
–
10
7
–
–
3
11
8
19
16
–
–
14
10
20
14
–
–
4
12
10
21
18
–
–
20
19
21
18
14
12
5
–
–
23
21
5
11
19
16
25
21
–
–
7
–
–
26
22
–
–
8
10
24
22
13
12
8
16
12
–
–
16
14
10
8
–
–
18
14
9
10
8
–
–
–
–
10
6
–
–
11
10
Control #
34
25
32
25
35
24
35
26
33
26
35
25
3 Experimental
3.1 Instruments and reagents
An automated melting point technique called Optimelt was used to calculate the uncorrected melting points. The IR spectra were captured using a Perkin-Elmer 1800 Series FT-IR spectrometer. The samples were assessed using thin films on KBr plates. 1H- and 13C-NMR spectra were captured at ambient temperature in base-filtered DMSO‑d6 and/or CDCl3 as solvents and TMS (δ) as an internal standard on a Bruker Avance III-400 MHz instrument running at 400 MHz for protons, 100 MHz for carbon, and 240 MHz for phosphorus nuclei. Electrospray ionization (ESI) mass spectra were captured on a Micromass LC-ZMD single quadrupole liquid chromatograph-mass spectrometer. Analytical thin-layer chromatography (TLC) was carried out using aluminum-backed, 0.2-mm-thick silica gel 60 F254 plates. Using a UV lamp with a wavelength of 254 nm and/or by giving the plates a suitable dip and then heating them, eluted plates may be observed. The retardation factor (Rf) values have been rounded to the nearest whole number. With silica gel 60 (40–63 lm) as the stationary phase, column chromatographic separations were carried out using AR- or HPLC-grade solvents. Starting materials, reagents, drying agents, and other inorganic salts could all be purchased commercially and used according to instructions. 6-(3,4-Dichlorophenyl)-4-(furan-2-yl)-5-oxo-2-thioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile (1) has been prepared according to the reported method (Ayman et al., 2021).
3.2 Synthesis
3.2.1 Synthesis of 2-(aminosulfanyl)-6-(3,4-dichlorophenyl)-4-(furan-2-yl)-5-oxo-5,6-dihydropyridine-3-carbonitrile (2)
In water (20 ml) that also contained potassium hydroxide (0.168 g, 3 mmol), HOSA (0.339 g, 3 mmol) was dissolved. To a solution of compound 1 (3.61 g, 10 mmol) in water (20 ml) in the presence of potassium hydroxide (0.112 g, 2 mmol) on an ice bath under nitrogen atmosphere, this solution was added dropwise. After 30 min, the pale yellow precipitate was filtered and dried at low pressure. Column chromatography was used to separate compound 2 from the residue.
Recryst. Solvent: hexane; pale yellow crystals, Rf = 0.45 (EtOAc/hexane, 1:2); Yield: 70%; mp: 210–212 °C; Anal. Calc. (%) for C16H9CL2N3O2S (376.98): C, 50.81; H, 2.40; N, 11.11; found: C, 50.87; H, 2.38; N, 11.09; IR (KBr), ν (cm−1): 3460, 3322 (NH2), 3070 (C–Harom), 2930 (C–Haliph), 2215 (C≡N), 1610(C=Ocarbonyl), 1590 (C=N), 1525 (C=C), 1165 (C-O-C); 1H NMR (400 MHz, DMSO- d6): 5.42 (s, 1H, H-6), 6.62 (s, 2H, NH2), 7.21–7.34 (d, 2H, Ar–H),7.69–7.77(m, 2H, Ar–H),8.26–8.28 (d, 1H, Ar–H), 8.50(s, 1H, Ar–H);13C NMR (100 MHz, DMSO- d6): 24.14, 114.89(CN), 116.38, 118.03, 132.26, 133.42, 133.44, 135.45, 138.48, 144.21, 147.32, 150.01, 154.50, 160.13, 161.09, 166.30; MS(ESI) m/z 377[(M)+, 7%], 400[(M+Na)+, 15%].
3.2.2 Synthesis of 8-(3,4-dichlorophenyl)-6-(furan-2-yl)-3-sulfanyl-5-thioxo-2,3,4,5-tetrahydropyrido[3,2-f][1,2,4,3]thiadiazaphosphepin-7(8H)-one 3-sulfide (3)
Tetraphosphorus decasulfide (1.11 g, 2.5 mmol) and compound 2 (0.94 g, 2.5 mmol) were combined and heated under reflux for 5 h in dry toluene (20 ml). The resulting orange solid was filtered out, washed with water, and then column chromatographed was used to produce product 3.
Recryst. Solvent: ethanol; yellow crystals, Rf = 0.4 (acetone/hexane, 1:3); Yield: 73%; mp: 230–232 °C; Anal. Calc. (%) for C16H10Cl2N3O2PS4 (504.88): C, 37.95; H, 1.99; N, 8.30; found: C, 37.90; H, 1.95; N, 8.27; IR (KBr), ν (cm−1): 3350, 3300 (br, 2 NH), 3020 (C–Harom), 2900 (C–Haliph), 2600 (br, SH), 1612(C=Ocarbonyl), 1580 (C=N), 1570 (C=C), 1130 (C=S), 1050 (C-O–C), 690 (P=S); 1H NMR (400 MHz, CDCl3): 3.84 (s, 1H, SH); 5.45 (s, 1H, H-6), 7.42–7.44 (d, 2H, Ar–H),7.99–8.01(m, 2H, Ar–H),8.15–8.17 (d, 1H, Ar–H), 8.20(s, 1H, Ar–H), 9.03(brs, 1H, NH); 10.20 (brs, 1H, NH); 13C NMR (100 MHz, CDCl3): 24.55, 118.93, 119.74, 128.65, 129.56, 130.63, 133.40, 135.34, 141.50, 142.31, 145.67, 152.18, 157.36, 160.17, 161.03, 168.43; 31P NMR (240 MHz, DMSO‑d6): 17.3 ppm; MS(ESI) m/z 505[(M)+, 49%].
3.2.3 Synthesis of 8-(3,4-dichlorophenyl)-3-ethoxy-6-(furan-2-yl)-5-thioxo-2,3,4,5-tetrahydropyrido[3,2-f][1,2,4,3]thiadiazaphosphepin-7(8H)-one-3-sulfide (4)
O,O-diethyldithiophosphoric acid was produced in situ by heating under reflux a solution of tetraphosphorus decasulfide (1.11 g, 2.5 mmol) in absolute ethanol (20 ml) for one hour. The preceding ethanolic solution was supplemented with Compound 2 (0.94 g, 2.5 mmol). The mixture was heated for 6 h while in reflux. The reaction mixture was reduced to half its original volume and then allowed to cool. Column chromatography was used on the residue to produce product 4.
Recryst. Solvent: ethanol; Orange solid; Rf = 0.50 (EtOAc/hexane, 1:2); Yield: 65%; mp: 205–207 °C; Anal. Calc. (%) for C18H14Cl2N3O3PS3 (516.93): C, 41.70; H, 2.72; N, 8.11; found: C, 41.75; H, 2.70; N, 8.13; IR (KBr), ν(cm−1): 3420 (br, 2NH), 3030 (C–Harom), 2920 (C–Haliph), 1615(C=Ocarbonyl), 1590 (C=N), 1540(C=C), 1125 (C=S), 1020, 1010 (O–C), 730 (P=S); 1H NMR (400 MHz, DMSO‑d6): 1.45 (t, 3H, J = 7.2 Hz, CH3); 3.40 (q, 2H, J = 7.2 Hz, OCH2), 5.40 (s, 1H, H-6), 7.31–7.43 (d, 2H, Ar–H),7.69–7.77(m, 2H, Ar–H),8.26–8.28 (d, 1H, Ar–H), 8.60(s, 1H, Ar–H),8.83 (brs, 1H, NH); 9.27 (brs, 1H, NH); 13C NMR (100 MHz, DMSO‑d6): 15.24, 24.11, 60.11, 116.38, 118.03, 125.20, 128.45, 132.24, 133.50, 133.83, 135.63, 138.50, 142.00, 145.50, 150.11, 154.50, 160.17, 161.03; 31P NMR (240 MHz, DMSO‑d6): 16.94 ppm; MS(ESI) m/z 517[(M)+, 4%], 518[(M+H)+, 28%], 540[(M+Na)+, 29%].
3.2.4 Synthesis of 8-(3,4-dichlorophenyl)-6-(furan-2-yl)-2,3,4,5-tetrahydro-3-(4-methoxyphenyl)-3-sulfanylene-5-thioxopyrido[3,2-f][1,2,4,3]thiadiazaphosphepin-7(8H)-one (5)
Method A: A magnetically agitated solution of compound 2 (0.94 g, 2.5 mmol) in dry acetonitrile (20 ml) kept at 80 °C was given Lawesson's reagent (1 g, 2.5 mmol). The reaction mixture was agitated for 4 h before being decreased in pressure and concentrated. The residue was extracted with ethyl acetate after being diluted with water (20 ml). The mixed organic phases were dried (Na2SO4), filtered, and concentrated under reduced pressure after being washed with water (2 × 50 ml) and brine (2 × 100 ml). Column chromatography was used on the residue to produce orange solid with a yield of 35%.
Method B: Lawesson's reagent (1.0 g, 2.5 mmol) was dissolved in absolute ethanol (20 ml), and the mixture was heated over reflux for 4 h. The preceding ethanolic solution was mixed with Compound 2 (0.94 g, 2.5 mmol). The mixture was heated for 6 h while in reflux. The reaction mixture was reduced to half its original volume and then allowed to cool. With order to produce orange solid in a 66 % yield, the produced solid was filtered out and recrystallized from diluted ethanol.
Recryst. Solvent: ethanol; orange solid; Rf = 0.30 (EtOAc/hexane, 1:1); mp: 218–220 °C. Anal. Calc. (%) for C23H16Cl2N3O3PS3 (578.95): C, 47.59; H, 2.78; N, 7.24; found: C, 47.75; H, 2.20; N, 7.28; IR (KBr), ν (cm−1): 3428 (br, 2 NH), 3010 (C–Harom), 2900 (C–Haliph), 1611(C=Ocarbonyl), 1585 (C=N), 1520(C=C), 1120 (C=S), 1020, 1030 (O–C), 705 (P=S); 1H NMR (400 MHz, CDCl3): 3.81 (s, 3H, OCH3), 5.46 (s, 1H, H-6), 5.66–5.68 (dd, 2H, J = 8 and 4 Hz, Ar–H), 7.39–7.40 (d, 2H, Ar–H), 7.41–7.48 (m, 2H, Ar–H), 7.95–8.19 (d, 1H, Ar–H), 8.21 (s, 1H, Ar–H), 8.51–8.53 (dd, 2H, J = 8 and 4 Hz, Ar–H), 9.06 (brs, 1H, NH), 10.18 (brs, 1H, NH); 13C NMR (100 MHz, CDCl3): 21.59, 55.62, 115.71 (C-3′, 5′aryl),116.29, 119.67, 122.85,125.83, 126.45 (C-2′,6′aryl), 126.86, 128.93, 129.76,133.90, 134.71 (d, Jpc = 129 Hz, C–1phenyl), 138.37, 139.91, 142.56, 144.37, 159.71,165.55, 167.44, 169.47; 31P NMR (240 MHz, DMSO‑d6): 19.14 ppm; MS(ESI) m/z 579[(M)+, 16%], 602[(M+Na)+, 18%].
3.2.5 Synthesis of 8-(3,4-dichlorophenyl)-6-(furan-2-yl)-2,3,4,5-tetrahydro-5-imino-3-(4-methoxyphenyl)-4-phenyl-3-sulfanylenepyrido[3,2-f][1,2,4,3]thiadiazaphosphepin-7(8H)-one (7)
A 2 h reflux-heated mixture of dry toluene, LR (0.5 g, 1.25 mmol), and distillated aniline (0.5 ml, 5 mmol). The previous solution was heated under reflux for an additional 6 h after compound 2 (0.94 g, 2.5 mmol) was added. The produced solid was filtered and dried after cooling. By using column chromatography, the obtained raw material was purified.
Recryst. Solvent: methanol; pale brown solid; Rf = 0.4 (acetone/hexane, 3:2); Yield: 50%; mp: 180–182 °C. Anal. Calc. (%) for C29H21Cl2N4O3PS2 (638.02): C, 54.47; H, 3.31; N, 8.76;; found: C, 54.44; H, 3.30; N, 8.77; IR (KBr), ν (cm−1): 3410 (br, 2NH), 3025 (C–Harom), 2950, 2810 (C–Haliph), 1615(C=Ocarbonyl), 1600 (C=N), 1565 (C=C), 1020 (O–C), 650 (P=S); 1H NMR (400 MHz, CDCl3): 3.75 (s, 3H, OCH3), 5.25 (s, 1H, H-6), 6.30 (d, 2H, J = 7.2 Hz, H-3,5), 7.10 (d, 2H, J = 8.0 Hz, H-2,6), 7.28–7.29 (m, 5H, Ar–H), 7.31(s, 1H, Ar–H), 7.41(d, 2H, Ar–H), 7.51(t, 1H, Ar–H), 7.86(d, 2H, Ar–H), 9.51 (brs, 1H, NH), 11.20 (brs, 1H, NH); 13C NMR (100 MHz, CDCl3): 21.74, 56.70, 111.15(C-3′, 5′aryl), 116.12, 117.26, 123.98, 126.38(C-2′,6′aryl), 128.03, 128.27, 128.46, 128.57, 130.00, 130.42, 135.38 (d, Jpc = 129 Hz, C–1phenyl), 135.58, 136.07,138.95, 144.62, 145.52, 148.06, 150.04, 151.90,158.32, 160.74, 162.68, 165.28,168.95; 31P NMR (240 MHz, DMSO‑d6): 20.10 ppm; MS(ESI) m/z, 638[(M)+, 100%], 639[(M+H)+, 38%], 661[(M+Na)+, 4%].
3.2.6 Synthesis of 7-(3,4-dichlorophenyl)-5-(furan-2-yl)-3-hydroxy-4-imino-3,4-dihydro-2H-pyrido[3,2-e][1,2,3]thiazaphosphinin-6(7H)-one-3-oxide (8)
In a magnetically agitated solution of compound 2 (1.88 g, 5 mmol) in dry dioxane (30 ml) containing a catalytic quantity of triethylamine (1.4 ml, 10 mmol) for 30 min at 5–10 °C, phosphorus tribromide (0.47 ml, 5 mmol) was added dropwise. For 6 h, the reaction mixture was held at 70 °C. The resulting orange solid was filtered off, washed with water, and then submitted to column chromatography to produce the desired product.
Recryst. Solvent: DMF/EtOH; yellow solid; Rf = 0.4 (EtOAc/hexane, 1:2); Yield: 80%; mp: 110–112 °C. Anal. Calc. (%) for C16H10Cl2N3O4PS (440.95): C, 43.46; H, 2.28; N, 9.50; found: C, 43.44; H, 2.260; N, 9.57; IR (KBr), ν (cm−1) 3380 (br, OH), 3200–3100 (br, 2NH), 3010 (C– Harom), 1610(C=Ocarbonyl), 1620 (C=N), 1600, 1550 (C=C), 1220 (P=O); 1H NMR (400 MHz, CDCl3): 4.90 (s, 1H, H-6), 5.80 (brs, 1H, P-OH); 7.45–7.52 (m, 2H, Ar–H),8.01–8.03(dd, 2H, Ar–H),8.10–8.12 (dd, 1H, Ar–H), 8.74(s, 1H, Ar–H), 9.15 (s, 1H, NH), 10.02 (s, 1H, NH); 13C NMR (100 MHz, CDCl3): 24.10,128.02, 129.60, 130.71, 132.91, 134.09, 142.33, 144.00, 146.33, 147.37, 154.48, 155.55, 157.59, 160.82, 165.47, 166.24; 31P NMR (240 MHz, DMSO‑d6): 16.30 ppm; MS(ESI) m/z, 441[(M)+, 100%], 442[(M+H)+, 14%], 464[(M+Na)+, 12%].
3.2.7 Synthesis of 7-(3,4-dichlorophenyl)-5-(furan-2-yl)-4-imino-3-phenyl-3,4-dihydro-2H-pyrido[3,2-e][1,2,3]thiazaphosphinin-6(7H)-one 3-oxide (9)
P,P-dichloro-(phenyl)phosphine was added to compound 2 (1.88 g, 5 mmol) in dry pyridine (30 ml) under stirring for 15 min at 10 °C, followed by heating under reflux for 8 to 10 h at 70 to 80 °C. Drops of strong HCl were used to neutralise the solution after it had been cooled and placed into cold water. Column chromatography was used on the produced solid to produce product 9.
Recryst. Solvent: ethanol; yellow solid; Yellow solid; Rf = 0.4 (acetone/hexane, 1:4); Yield: 70%; mp: 210–212 °C. Anal. Calc. (%) for C22H14Cl2N3O3PS (500.99): C, 52.60; H, 2.81; N, 8.37; found: C, 52.64; H, 2.84; N, 8.39; IR (KBr), ν (cm−1) 3250–3100 (br, 2 NH), 3020 (C–Harom), 1610(C=Ocarbonyl), 1625 (C=N), 1520 (C=C), 1250 (P=O); 1H NMR (400 MHz, CDCl3): 5.56 (s, 1H, H-6); 7.12–7.14 (d, 2H, Ar–H), 7.25–7.30 (d, 1H, Ar–H), 7.38–7.51(m, 4H, Ar–H), 7.70–7.75(d, 2H, Ar–H), 7.86–7.88(d, 2H, Ar–H), 9.51 (s, 1H, NH), 10.50 (s, 1H, NH); 13C NMR (100 MHz, CDCl3): 21.22, 121.66, 122.92, 123.96, 124.69,126.29, 127.83(C-3,5phenyl), 128.48 (C-2,6phenyl), 130.13 (d, J = 128 Hz, C–1phenyl), 135.17, 135.63, 136.17, 137.35, 141.64, 143.49, 145.72, 149.09, 161.68, 165.71, 166.81; 31P NMR (240 MHz, DMSO‑d6): 19.88 ppm; MS(ESI) m/z, 501[(M)+, 38%], 524[(M+Na)+, 10%].
4 Conclusions
Starting with 6-(3,4-Dichlorophenyl)-4-(furan-2-yl)-5-oxo-2-thioxo-1,2,5,6-tetrahydropyridine-3-carbonitrile and certain phosphorus reagents in various solvents, simple procedures were developed to synthesis new pyridothiazaphosphininones in one step. We expect that others who are looking for innovative synthetic fragments with unique features for medicinal chemistry will find our technique useful. The findings showed that several of the compounds under investigation have considerable to moderate antibacterial activity against two strains of Gram-negative and two strains of Gram-positive bacteria. Therefore, our discovery has increased the potential for creating more linked heterocycles.
Author Contributions
Participating authors developed the chemistry and performed the chemical analysis described herein. Also they led the writing of this manuscript assisted with the chemistry as well as the write up the manuscript. All authors have read and agreed to the published version of the manuscript.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data are available in the manuscript and from the corresponding author upon request.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to the Deanship of scientific research Majmaah University, Saudi Arabia, for funding this research work through the Project No: R- 2022-328.
References
- Chemoselective heterocyclization of pharmacological activities of new heterocycles a review: synthesis of new phospha heterobicyclic systems containing 1,2,4- triazine moiety, Part IX: straightforward synthesis of new fluorine bearing 5-phospha-1,2,4- triazine/1,2,4-triazepine-3-thiones. Trends Heterocycl. Chem.. 2002;8:187.
- [Google Scholar]
- Reaction of 2-cyano-3-(4-oxo-4H-chromen-3-yl)prop-2-enamide with some phosphorus reagents: synthesis of some novel diethyl phosphonates, 1,2,3-diazaphosphinanes, 1,2,3-thiazaphosphinine and 1,2-azaphospholes bearing a chromone ring. Res. Chem. Intermed.. 2017;44(1):173.
- [Google Scholar]
- Reaction of 2-imino-2H-chromene-3-carboxamide with phosphorus sulfides: Synthesis of novel 2-sulfido-2,3-dihydro-4H-chromeno[2,3-d][1,3,2]diaza-phosphinines. Phosphorus, Sulfur, Silicon Related Elem.. 2018;193(10):651.
- [Google Scholar]
- Reaction of 2-imino-2H-chromene-3-carboxamide with some phosphorus esters: Synthesis of some novel chromenes containing phosphorus heterocycles and phosphonate groups and their antioxidant and cytotoxicity properties. Synth. Commun.. 2019;49(21):2983.
- [Google Scholar]
- Reaction of 2-imino-2H-chromene-3-carboxamide with phosphorus halides: Synthesis of some novel chromeno- [2,3-d][1,3,2]diazaphosphinines and chromeno[4,3-c][1,2]- azaphosphole and their antioxidant and cytotoxicity properties. Heterocycles. 2019;98(5):681.
- [Google Scholar]
- Investigating the pharmacokinetics and biological distribution of silver-loaded polyphosphoester-based nanoparticles using 111Ag as a radiotracer. J. Label Compd. Radiopharm.. 2015;58:234.
- [Google Scholar]
- Ayman, A., Hassan, M., Maher, H., Sameh, R., Manahil, E., Amani, A., 2021. Novel colorimetric chemosensors containing pyridine moiety for detection of some cations in water and crops samples: Design, synthesis, and evaluation. 25(12), 101386.
- Novel phospha-oxazepinoquinazolinyl derivatives of ibuprofen as nitric oxide synthase inhibitors: Synthesis and biological evaluation. Arabian J. Chem.. 2022;3(15):103642
- [Google Scholar]
- Potent inhibition of pepsin and penicillopepsin by phosphorus-containing peptide analogs. J. Org. Chem.. 1990;55:6268.
- [Google Scholar]
- Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol.. 1966;45:493-496.
- [Google Scholar]
- EOJ, B.N., 2000. Isoquino[2,1-c][1,3,2]benzodiazaphosphorine derivatives: new potential agents for cancer chemotherapy. Phosphorus Sulfur Silicon Relat. Elem. 162, 231.
- Synthesis, characterization and correlative biological effects in wheat of a benzoxaza- and a diazaphosphorus (V) heterocycles. J. Serb. Chem. Soc.. 2006;71:1031.
- [Google Scholar]
- Antitumor activities of some new 1,3,2-oxaza- and 1,3,2-diazaphosphorinanes against K562, MDA-MB-231, and HepG2 cells. Med. Chem. Res.. 2012;21:2185.
- [Google Scholar]
- Production and purification of syringomycin, a phytotoxin produced by Pseudomonas syringae. Physiol. Plant Pathol.. 1977;11:13-28.
- [Google Scholar]
- Synthesis and antioxidant properties of some novel 1,3,4,2-oxadiazaphosphepino[6,7-c]quinolinones and pyrazolo[3,4:4′,3′]quinolino[5,1-c][1,4,2]oxazaphosphinine. Phosphorus, Sulfur, Silicon Related Elem.. 2017;192(7):866.
- [Google Scholar]
- Phosphorus based indole and imidazole functionalized hyperbranched polyester as antimicrobial surface coating materials. Prog. Org. Coat.. 2014;77:1901.
- [Google Scholar]
- Reaction mechanisms displayed by catalytic antibodies. Acc. Chem. Res.. 1993;26:396.
- [Google Scholar]
- Studies on Organophosphorus Compounds. Part 1: Synthesis andIn VitroAntimicrobial Activity of Some New Pyrimido[5′,4′:5,6]pyrano[2,3-d][1,3,2]thiazaphosphinine Compounds. Archiv. Der Pharm.. 2013;346(10):727.
- [Google Scholar]
- Studies on Organo-phosphorus Compounds Part II: Synthesis and biological activities of some new benzochromeno[2,3-d][1,3,2]thiazaphosphinine derivatives. Int. J. Pharm. Sci. Rev. Res.. 2013;23(2):15-81.
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104385.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




