

Translate this page into:
Discovery of novel indene-based hybrids as breast cancer inhibitors targeting Hsp90: Synthesis, bio-evaluation and molecular docking study
⁎Corresponding author. thoraya-f@hotmail.com (Thoraya A. Farghaly) thoraya-f@cu.edu.eg (Thoraya A. Farghaly)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
New indene derivatives targeting Hsp90 were synthesized, and biologically evaluated. Five new indene derivatives displayed significant antitumor effect especially on MCF-7 cell line compared to doxorubicin. The most active compound 8a showed reduced cytotoxic effect against WI‐38 normal cells, suggesting its high safety profile. The Hsp90 enzyme assay of 8a displayed IC50 = 18.79 ± 0.68 nM relative to Alvespimycin as a reference drug. The molecular modeling of the most active compounds in the Hsp90 binding site was done presenting agreement with the in vitro anti-Hsp90 activity.
Abstract
Inhibition of Heat-shock protein 90 (Hsp90) is considered an attractive route in fighting against cancer proliferation. Herein, new indene derivatives targeting Hsp90 were synthesized, and biologically evaluated. The new series of indeno-pyrimidine and indeno-pyridine were synthesized from the reaction of indene-enaminone with various heterocyclic amines and active methylene derivatives. Two breast cancer cell lines were used to examine the new compounds in vitro for their anticancer activity, namely, MCF-7 and MDA-MB231 cancer cells. The new indene derivatives 8a-c, 17a, and 25 displayed significant antitumor effect especially on MCF-7 cell line compared to doxorubicin. Derivative 8a was further subjected to Hsp90 enzyme assay aiming to ensure the inhibitory potential of such compound on Hsp90, it displayed IC50 = 18.79 ± 0.68 nM relative to Alvespimycin as a reference drug. Finally, molecular modeling of the most active compounds in the Hsp90 binding site was done presenting agreement with the in vitro anti-Hsp90 activity.
Keywords
Enaminones
Hetero-amines
Aminopyrazoles
Aminotriazoles
Breast cancer inhibitors

1 Introduction
Heat shock protein 90 (Hsp90) is a molecular chaperone that is a member of the heat shock protein family and plays an important role in cell’s vital processes including cell growth, differentiation, and survival (Whitesell and Lindquist, 2005; Taipale et al., 2010; Biebl and Buchner, 2019). Consequently, inhibiting Hsp90 might cause cancer cells to collapse or become much weaker (Neckers and Mollapour, 2015; Zhang, et al., 2021). Breast cancer is still the most commonly diagnosed malignancy in women and the second biggest cause of cancer-related fatalities. Despite continued efforts to find novel therapies, breast cancer mortality rates have just lately begun to fall, and this is most likely due to early detection of the disease (Berry et al., 2005). However, it was reported that inhibiting Hsp90 causes client proteins to degrade, resulting in inhibition of both cancer cell growth and proliferation, especially those proteins related to breast cancer which account for about 20 % of breast cancers and have violent clinical aspects, leading to poor diagnosis and clinical consequences, therefore high expression of Hsp90 is correlated with high expression of these client protiens, such correlation makes targeting Hsp90 a considerable approach to overcome breast cancer (Nguyen et al., 2021). Hsp90′s therapeutic potential is also increased by the fact that cancer cells express it 2–10 times more than normal, healthy cells do. Targeting Hsp90 can also get around the well-known cancer resistance problem due to its potential for affecting numerous oncogenic protein pathways (El-Shafey et al., 2020; Lianos et al., 2015). Therefore, Hsp90 has consequently become a desirable and focused target for the development of new medications (Gimenez Ortiz and Montalar Salcedo, 2010; Liu et al., 2022). Hsp90 inhibitors include a number of natural products and their derivatives as well as fully synthetic compounds (Li et al., 2020). A new selective Hsp90 inhibitor called Pimitespib recently demonstrated therapeutic activity in patients with advanced gastrointestinal stromal tumors resistant to conventional therapies (Sheridan, M. H., 2022; Akira et al., 2022).
Different indene derivatives were reported as analogues of Hsp90 inhibitors exerting marked inhibitory effect on Hsp90 with cytotoxic effect on different cell lines (Chaudhury et al., 2021). Fusion between indene scaffold and nitrogen bearing moiety was reported and such hybrid proved marked antiproliferative effect against different cancer cell lines, where some new derivatives displayed marked effect especially against MCF-7 breast cancer cell line (Fayed et al., 2020). Because of their chemical and biological relevance, indeno-pyrimidines have been investigated. They have been discovered to have numerous pharmacological characteristics, including antitumor effect. A large number of structurally new indeno-pyrimidines were shown to exhibit significant anticancer effect both in vitro and in vivo (Ghorab and Alsaid, 2015). Besides, Indeno-pyridines have been reported for their anticancer activity especially against breast cancer cell line (MCF7) (Ghorab and Alsaid, 2012). Concerning pyrimidine scaffold, the anticancer action of pyrimidine derivatives has been described through a number of mechanisms, among which is the inhibition of Hsp90 enzyme (Davies et al., 2012). Interestingly, fused pyrimidines such as pyrazolo-pyrimidine, triazolo-pyrimidine and benzimidazole-pyrimidine showed significant anti-MCF-7 activity (Aastha et al., 2021). From our experience over 20 years to design and synthesis of biologically active heterocyclic derivatives (Alsaedi et al., 2022; Abbas et al., 2022; Alamri et al., 2023; Almehmadi et al., 2021; Farghaly et al., 2015, 2021; Althagafi et al., 2019; Bayazeed et al., 2022), we decided to utilize the indeno-pyrimidine scaffold in the synthesis of new derivatives, and examine their anticancer activities especially against breast cancer cell lines.
2 Results and discussion
Herein, our project was directed to synthesize novel series of indeno-pyrimidines or indeno-pyridine moieties via the reaction of 5-bromo-2-dimethylaminomethylene-indan-1-one with heterocyclic amines and active methylene derivatives. Such project started with the synthesis of enaminone 3 through the condensation of indanone derivative 1 with DMF-DMA in dry xylene as illustrated in Scheme 1. The structural identity of the novel enaminone derivative 3 was confirmed by various spectroscopic analyses. The 1H NMR spectrum of enaminone derivative 3 (Fig. 1) revealed three characteristic singlet signals for the two CH3 of N(CH3)2, the CH2 and = CH groups at δH: 3.19, 3.85 and 7.54 ppm in addition to the remarkable signals for three protons of aromatic ring. Also, the 13C NMR showed characteristic carbon signals for the two chemically equivalent two methyl groups of N(CH3)2 and C⚌O groups at δC: 31.1 and 191.0 ppm, respectively.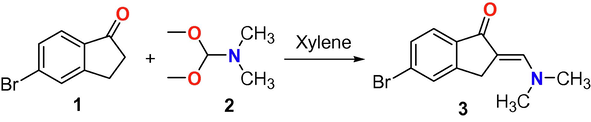
The synthesis of enaminone derivative 3.
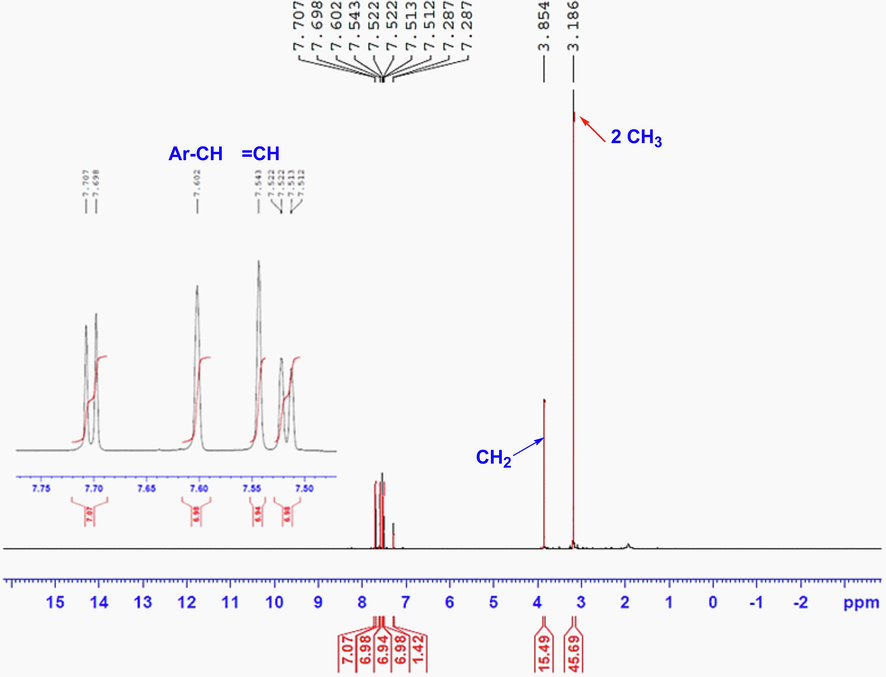
The 1H NMR spectrum of enaminone derivative 3 in CDCl3.
The second step in the synthesis of a series of indanopyrimidine is the reaction of enaminone derivative 3 with 3,5-diamino-4-arylazopyrazoles 4a-d in acetic acid as depicted in Scheme 2. This reaction proceeded through the formation of three intermediates 5–7 to form four derivatives of indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine 8a-d. The other products 12a-d with their intermediates 9–11 were discarded based on the previous fact that proofed using X-ray single crystal analyses (Farghaly et al., 2010), that the reaction between exocyclic-enaminone and heteroamine derivatives proceeded via at first the addition of exocyclic amino group to the double bond of = CH-N(Me)2 followed with elimination of NH(Me)2 then cyclization through nucleophilic attack of the cyclic-NH to the carbon of C⚌O of the enaminone moiety with elimination of water molecule. The structure of indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine derivatives 8a-d was confirmed from interpretation of their IR, Mass, and NMR spectral data with the suggested structure. For example, the mass spectrum of derivative 8a (Fig. 2) showed the exact molecular weight as well as several fragments corresponding to the fragmentation species of that derivative. In addition, the 1H NMR of the same derivative 8a showed the correct signals corresponding to 13 protons for CH2 (3.60), NH2 (7.25), Ar-H (7.35–8.01) and pyrimidine- H (8.54) ppm.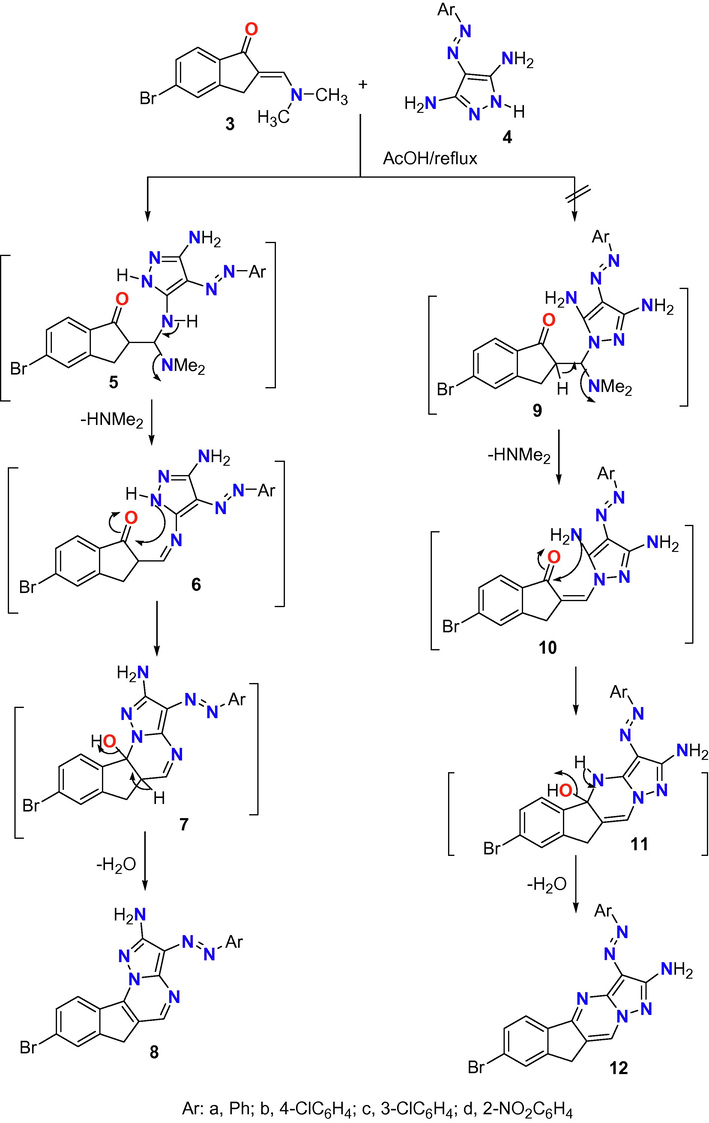
Reaction of enaminone 3 with heterocyclic amines 4.
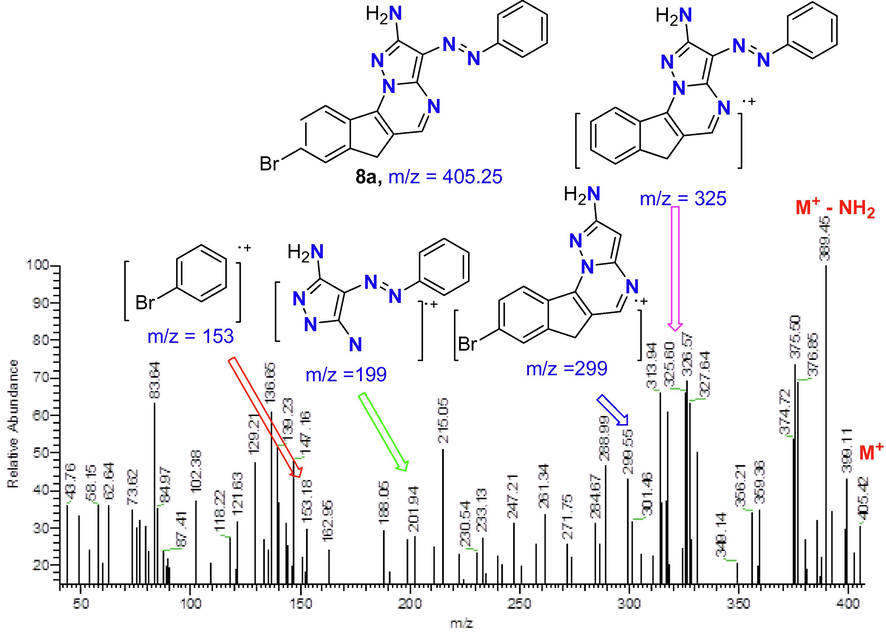
The mass spectrum with fragmentation of derivative 8a.
In the same way, the enaminone derivative 3 was reacted with another two derivatives of aminopyrazolone 13a,b (Scheme 3) to afford the indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2(6H)-one derivatives 17a,b. Such reaction was achieved through Michael type addition of NH2 group to the double bond of enaminone moiety (=CH-N(Me)2) with abstraction of NH(Me)2 to afford the intermediate 15 followed by cyclization through nucleophilic attack of the cyclic NH to the carbonyl carbon with removal of H2O molecule from intermediate 16.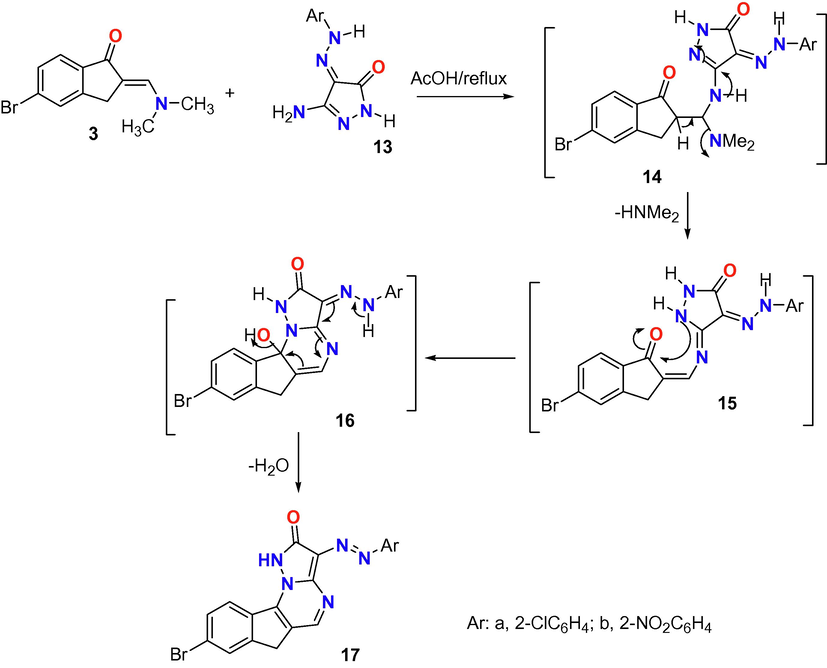
Reaction of enaminone 3 with heterocyclic amines 13a,b.
In Scheme 4, another two heteroamines 18 and 19 were reacted with the enaminone derivative 3 in refluxed acetic acid to give two indinopyrimidine derivatives fused with triazole ring 20 or benzimidazole 21. The structure of the two products was proved from their spectral data as illustrated in experimental section.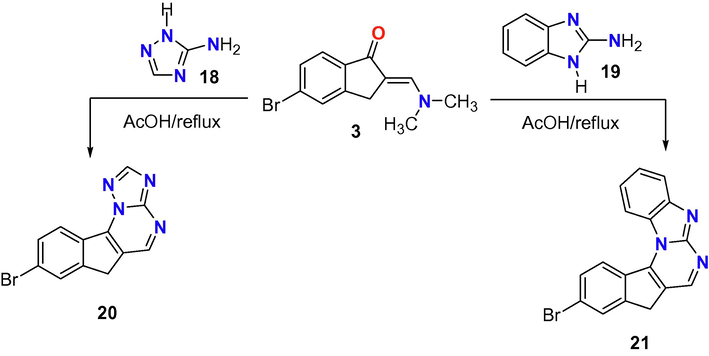
Reaction of enaminone 3 with heterocyclic amines 18 and 19.
Recently published research has drawn our attention to the biological significance of the indeno[1,2-b]pyridine derivatives as anticancer (Kadayat et al., 2015, 2016), topoisomerase inhibitory (Shrestha et al., 2018; Kadayat et al., 2015), anti-aggression (Bell and Brown, 1979) and antimicrobial activities (Brahmbhatt, et al., 2015). From these reports we interested herein to synthesize three new indeno[1,2-b]pyridine derivatives 25, 26 and 27 from the reaction of enaminone derivative 3 with active methylene 22–24 in acetic acid and ammonium acetate under reflux (Scheme 5).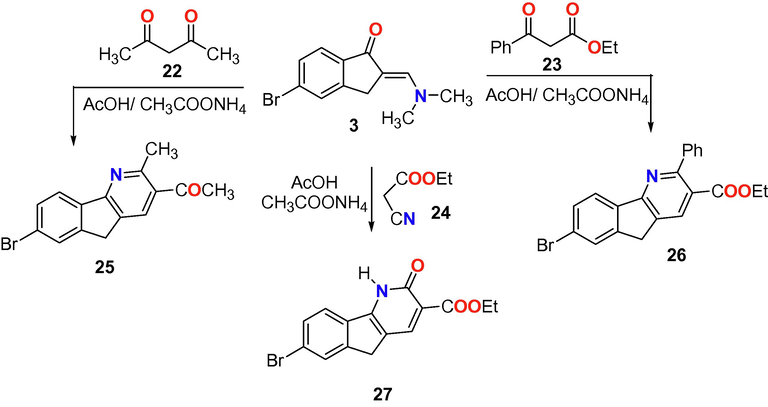
Reaction of enaminone 3 with active methylene derivatives 22–24.
2.1 Biological activity
2.1.1 In-vitro cytotoxicity
In this study the cytotoxic impact of 12 novel compounds was investigated in vitro against two breast cancer cell lines (MCF7 and MDA-MB231) using the MTT assay. The IC50 values of such compounds were calculated as shown in Table 1. A well-known chemotherapeutic drug (doxorubicin) and Alvespimycin (17-DMAG) were used as reference drugs. 17-DMAG displayed potent activity against both tested cell lines with IC50 = 7.43 μM and 6.57 μM against both cell lines respectively. Compounds 8a-c, 17a, and 25 in general, showed considerable cytotoxic activity against MCF7 revealing IC50 ranging from 26.44 to 39.62 μM. In particular, compound 8a was the most active, comparing its cytotoxic effect against both cell lines relative to doxorubicin as a reference drug. From the obtained results, it is worth mentioning that all the tested compounds displayed lower activities against MDA-MB231 cancer cell line comparing to reference drug except compound 8a among them showed IC50 = 69.95 μM exceeding that of doxorubicin having IC50 = 74.32 μM. Additionally, fusion of indene ring with nitrogen bearing heterocyclic rings markedly improved the activity especially against MCF7 cancer cell line as found in compound 3, it exerted very low activity comparing with the reference drug, and yet marked improvement in cytotoxicity is recorded upon fusion. Data are expressed as the mean ± SD of three independent experiments.
Compound no.
IC50 (µM)
MCF7
MDA-MB231
3
> 100
> 100
8a
26.44 ± 4.47
69.95 ± 2.45
8b
37.01 ± 7.54
> 100
8c
39.62 ± 10.21
> 100
8d
> 100
> 100
17a
38.41 ± 4.30
> 100
17b
> 100
> 100
20
61.62 ± 2.34
93.58 ± 1.39
21
76.31 ± 2.61
88.85 ± 1.55
25
32.85 ± 3.97
> 100
26
> 100
> 100
27
> 100
78.52 ± 2.24
Doxorubicin
31.16 ± 3.43
74.32 ± 1.10
Alvespimycin (17-DMAG)
7.43 ± 1.02
6.57 ± 2.01
It is observed that compounds 8a-c bearing 2-amino and 3- phenyl diazenyl substituents on the pyrazolo[1,5-a]pyrimidine scaffold displayed significant inhibitory activities, where the un-substituted phenyl ring in 8a exerted the highest activity against MCF7 with IC50 = 26.44 μM comparing with doxorubicin having IC50 = 31.16 μM. Introduction of electron withdrawing group in both para (p-Cl) and meta (m-Cl) positions of the phenyl ring in 8b and 8c respectively exerted lower inhibitory effects with IC50 = 37.01 and 39.62 μM respectively on MCF7 cancer cell line than the plain phenyl ring. Interestingly the presence of 2-C⚌O instead of 2-amino, and o-Cl phenyl diazenyl substituents retained a moderate activity as in 17a revealing IC50 = 38.41 μM. Furthermore, marked drop-in activity was noticed upon introducing o-NO2 group as in compounds 8d, 17b. On the other hand, both 20 and 21 displayed moderate activities against both tested cell lines. Regarding indeno-pyridine derivatives, the presence of a 2-CH3 group on the pyridine ring as in compound 25 was significantly better in terms of activity compared to its counterparts 26, 27 which held 2-phenyl and 2-C⚌O groups, respectively. The IC50 of compound 25 was 32.85 M, which was nearly equivalent to the reference drug.
2.1.2 Effect on normal cells
The promising new compound 8a was further assessed by examining its cytotoxicity against noncancerous normal cells (WI‐38 cells), as presented in Table 2. From cytotoxicity results it was obvious that MCF-7 cell line was more sensitive than MDA-MB231. The obtained results showed that 8a recorded IC50 value = 78.34 ± 2.33 μM higher than that recorded against MCF-7 cell line and nearly equal to the corresponding IC50 of MDA-MB231 cell line, Therefore selective toxicity should be improved before further development of such compound. Our promising new compound was further subjected to an examination of the possible anticancer mechanism of action. Data are expressed as the mean ± SD of three independent experiments.
Compound
IC50 (µM) WI-38
8a
78.34 ± 2.33
Doxorubicin
66.15 ± 1.78
2.1.3 Effect of target compound 8a on Hsp90 enzyme
The inhibitory effect of compound 8a on Hsp90 enzyme was assessed via using the Hsp90 inhibitor Alvespimycin (17-DMAG) as a reference drug. Consistent with the cytotoxicity results, compound 8a revealed marked inhibitory effect on Hsp90 enzyme with IC50 = 18.79 ± 0.68 nM exceeding that of the reference drug as presented in Table 3. Data are expressed as the mean ± SD of three independent experiments.
Compound
Hsp90 IC50 (nM)
8a
18.79 ± 0.68
Alvespimycin (17-DMAG)
62.00 ± 1.78
2.2 Molecular modeling inside the active binding site of Hsp90
In order to perform molecular docking of the most active compounds (8a-c, 17a, and 25) revealing the highest activity against MCF7 cancer cells, especially compound 8a with in vitro inhibitory effect on Hsp90 enzyme, we utilized MOE 2014.0901 [33] aiming to find a possible fitting inside the Hsp90 reported active site. Molecular modeling inside the active binding site of Hsp90 was performed on the crystal structure of Hsp90 downloaded from the protein data bank (PDB: 2xjx) (Woodhead, et al., 2010). The most important amino acids in the active site which are considered the conserved regions and may be accountable for Hsp90 inhibitory activity are (Leu48, Asn51, Ser52, Asp54, Ala55, Lys58, Ile91, Asp93, Gly97, Met98, Asp102, Asn106, Lys112, Gly135, Gly137, Phe138, Val150, Ile151, Thr184, Lys185 and Val186) (Saxena et al., 2010).
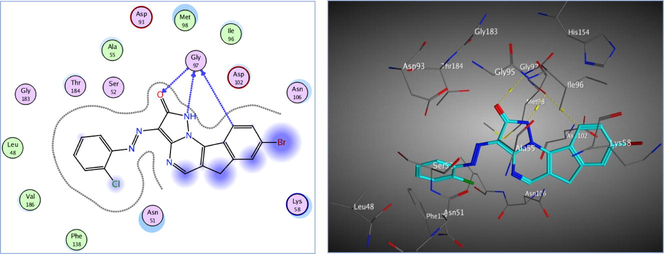
Redocking of the co-crystalized ligand was first done for validation. It was found that the carbonyl group was involved in the binding to the key residues; Thr4184 and Gly97 via two hydrogen bonds in the active binding site of the Hsp90. Additionally; Asp93 was able to form one H bond with OH group and another C—H bond with the phenyl ring. It displayed docking score = -7.103 kcal/mol and the (RMSD) = 1.05 Å (Fig. 3). The obtained docking results of the most active hits showed their ability to fit in the binding site of the enzyme by a significant number of interactions explaining their potency displayed by both scores and binding poses Table 4, Figs. 4-8. Compounds 8a-c, 17a, and 25 were docked perfectly inside the Hsp90 binding site with binding scores = -8.556, −8.213, −7.982, −8.651, and −7.028 kcal/mol, respectively. Interestingly compounds 8a and 8b exhibited similar binding behavior within the active site, where in both compounds Asp93 residue formed hydrogen bond with the phenyl ring of indene moiety, in addition to another hydrogen bond between the NH2 group on the pyrazolo-pyrimidine scaffold and Gly97.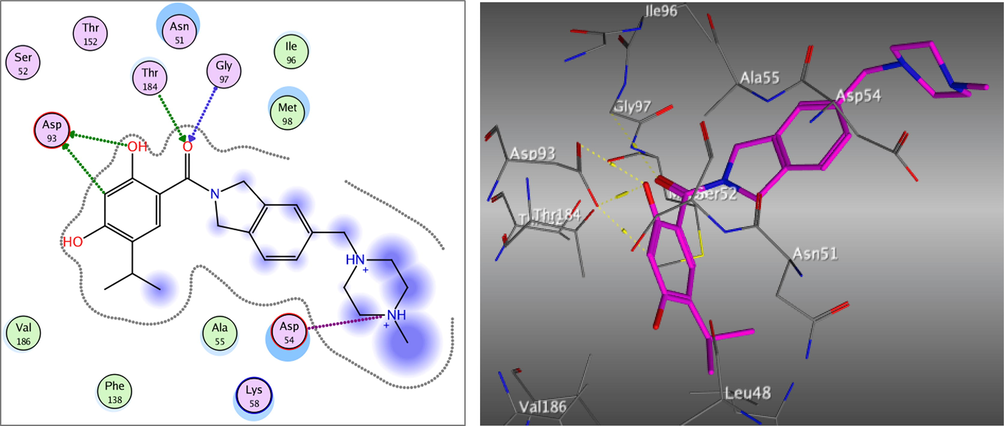
The 2D and 3D proposed binding modes of the co-crystalized ligand inside Hsp90 receptor active site.
Compound
Docking score (Kcal/mol)
Interacting amino acids (Type of interaction)
8a
8b
8c
17a
25
Ligand−8.556
−8.213
−7.982
−8.651
−7.028
−7.103Asp93, Gly97 (2H-bonds)
Asp93, Gly97 (2H-bonds)
Gly97 (1H-bond), His154 (pi-H)
Gly97 (3H-bonds)
Asp93, Lys112 (2H-bonds), Asn51 (pi-H)
Asp54,Asp93, Gly97, Thr184 (5H-bonds)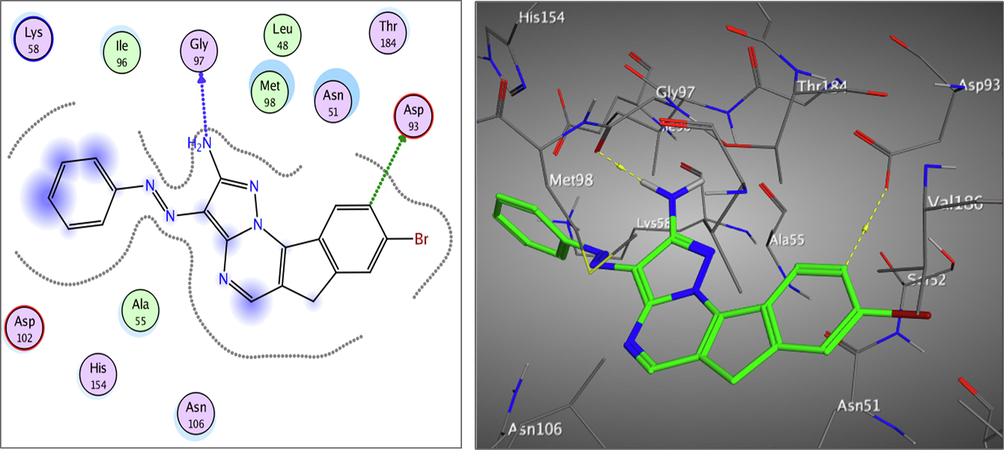
The 2D and 3D proposed binding modes of 8a inside Hsp90 receptor active site.
The 2D and 3D proposed binding modes of 8b inside Hsp90 receptor active site.
The 2D and 3D proposed binding modes of 8c inside Hsp90 receptor active sit.
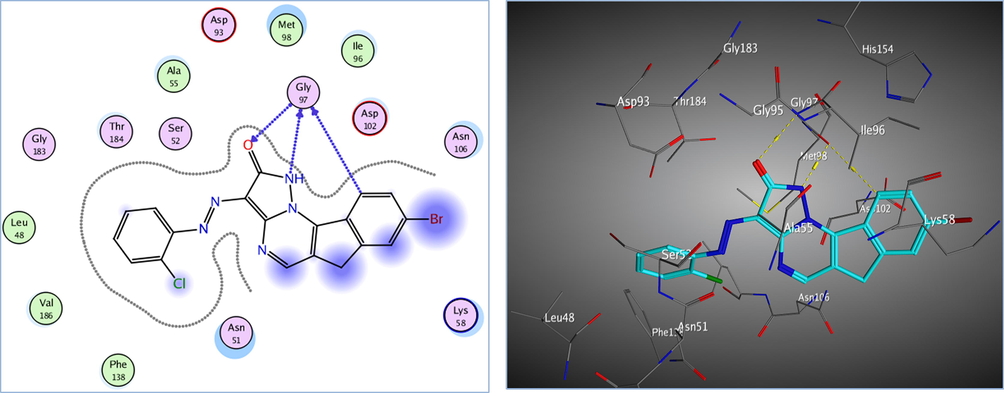
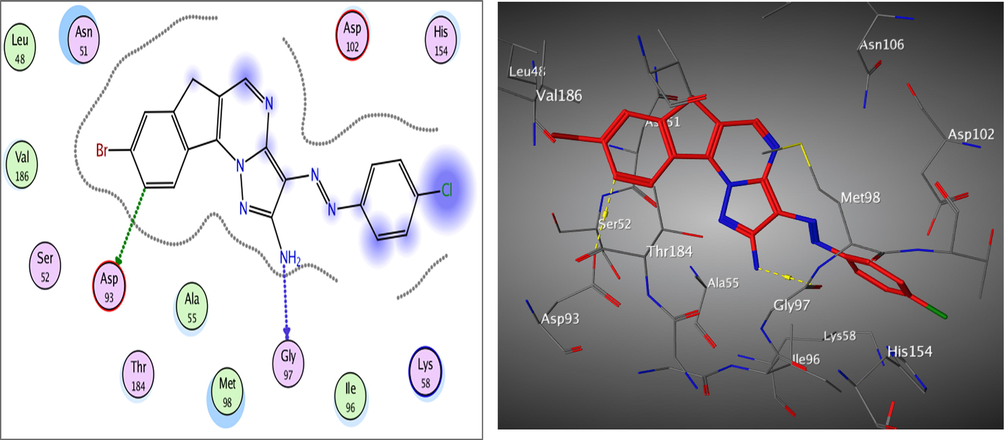
The 2D and 3D proposed binding modes of 17a inside Hsp90 receptor active site.
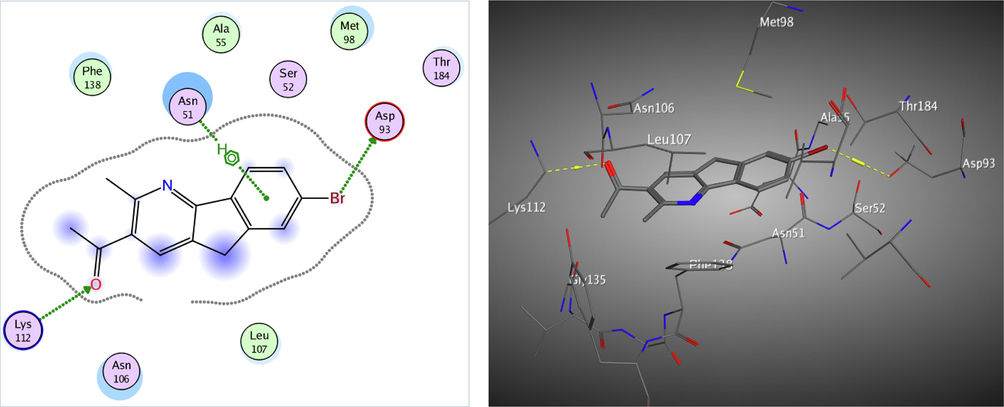
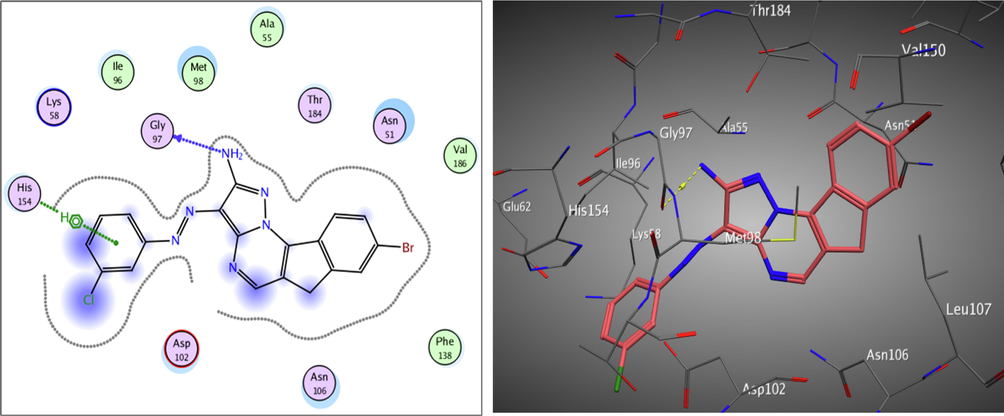
The 2D and 3D proposed binding modes of 25 inside Hsp90 receptor active site.
3 Conclusion
New indene derivatives were developed as Hsp90 inhibitors and their cytotoxicity was assessed on two breast cancer cell lines. Besides, the inhibitory effect of the most active compound was examined on Hsp90 enzyme. Results revealed that 8a exhibited the most potent cytotoxic effect on MCF7 cell line having IC50 = 26.44 ± 4.47 μM with significant inhibitory activity on Hsp90 enzyme with IC50 = 18.79 ± 0.68 nM comparing to reference drug. Furthermore, molecular modeling revealed that 8a is fitted inside the active site of the enzyme with high score = -8.556 Kcal/mol. On the basis of the obtained results compound 8a could be considered as Hsp90 inhibitor, and may be useful for further investigation as a hopeful compound for the development of anti-Hsp90 hits.
4 Experimental
4.1 Instrumentations
Recording melting points
Gallenkamp apparatus
IR spectra
KBr / Pye-Unicam SP300 spectrophotometer
1H and 13C NMR spectra
DMSO‑d6 / Varian Gemini 300 NMR spectrometer (300 MHz for 1H NMR and 75 MHz for 13C NMR)
Mass spectra
GCMS-Q1000-EX Shimadzu and GCMS 5988-A HP spectrometers
4.2 Synthesis of 5-bromo-2-((dimethylamino)methylene)-2,3-dihydro-1H-inden-1-one (3)
In 50 mL round flask, we added 0.005 mol of 5-bromo-indan-1-one in 15 mL dry xylene and 0.005 mol of DMF-DMA and the whole mixture was refluxed for 5 h. The progress of the condensation reaction was followed with TLC technique. The colored solid enaminone was collected by filtration and crystalized from ethanol to afford rosy solid, (89 % yield), mp 219–220 °C; IR (KBr) νmax 3055 (SP2 CH), 2917 (SP3 CH), 1672 (C⚌O), 1583 (C⚌C), 1438, 1378, 1201, 1128, 1092, 1057 cm−1;1H NMR ((CDCl3) 3.19 (s, 6H, 2CH3), 3.85 (s, 2H, CH2), 7.51(dd, J = 8.5, 0.85 Hz, 1H, Ar-H), 7.54 (s, 1H, =CH), 7.60 (s, H, Ar-H), 7.69 (d, J = 8.5 Hz, 1H, Ar-H). 13C NMR (CDCl3) 31.1 (CH3), 103.6, 124.3, 126.1, 128.4, 130.3, 139.1, 147.5, 149.2, 191.0 (C⚌O) one carbon overlapped; MS m/z (%): 266 (M+, 26), 256 (83), 264 (23), 235 (55), 208(45), 183 (24), 109 (27), 98 (84), 78 (70), 57 (1 0 0). Anal. Calcd. for C12H12BrNO (266.13): C, 54.16; H, 4.54; N, 5.26. Found: C, 54.03; H, 4.46; N, 5.17 %.
4.3 Reaction of enaminone 3 with heterocyclic amines 4a-d, 13a,b, 18 and 19
Reaction of 0.0025 mol of rosy enaminone 3 with the same number of moles of each of heterocyclic amines 4a-d or 13a,b or 18 or 19 in 20 mL acetic acid was refluxed for 5 h. The solid colored product was collected by usual way and washed with ethanol, crystalized with ethanol/dioxane mixture to give a series of indenoazolopyrimidines 8a-d, 17a,b, 20 and 21.
4.3.1 8-Bromo-3-(phenyldiazenyl)-6H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine (8a)
Dark blue solid, (84 % yield), mp above 300 °C; IR (KBr) νmax 3420, 3312 (NH2), 3057 (SP2 CH), 2952 (SP3 CH), 1592 (C⚌N), 1437, 1327, 1212, 1035 cm−1; 1H NMR (DMSO‑d6) 3.60 (s, 2H, CH2), 7.25 (s, 2H, NH2), 7.35–7.54 (m, 5H, Ar-H), 7.75 (d, J = 8.5 Hz, 1H, Ar-H), 7.82 (s, 1H, Ar-H), 8.01 (d, J = 8.5 Hz, 1H, Ar-H), 8.54 (s, 1H, pyrimdine- H). MS m/z (%): 405 (M+, 31), 402 (24), 389 (1 0 0), 375 (74), 326 (66), 299 (43), 274 (22), 190 (18), 84 (63), 77 (32). Anal. Calcd. for C19H13BrN6 (405.26): C, 56.31; H, 3.23; N, 20.74. Found: C, 56.28; H, 3.05; N, 20.53 %.
4.3.2 8-Bromo-3-((4-chlorophenyl)diazenyl)-6H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine (8b)
Brown solid, (85 % yield), mp above 300 °C; IR (KBr) νmax 3405 (NH2), 1600 (C⚌N), 1545, 1475, 1373, 1225, 1075 cm−1;1H NMR (CDCl3) 3.85 (s, 2H, CH2), 6.54 (s, 2H, NH2), 7.21 (d, J = 8.5 Hz, 2H, Ar-H), 7.51 (d, J = 8.5 Hz, 1H, Ar-H), 7.74 (d, J = 8.5 Hz, 2H, Ar-H),7.83 (s, 1H, Ar-H), 7.92 (d, J = 8.5 Hz, 1H, Ar-H), 8.10 (s,1H, pyrimidine-H); MS m/z (%): 440 (M+, 12), 400 (60), 361 (1), 301 (2), 282 (6), 237 (10), 192 (2), 179 (7), 154 (13), 139 (4), 129 (22), 105 (1 0 0), 73 (22). Anal. Calcd. for C19H12BrClN6 (439.70): C, 51.90; H, 2.75; N, 19.11. Found: C, 51.85; H, 2.67; N, 19.03 %.
4.3.3 8-Bromo-3-((3-chlorophenyl)diazenyl)-6H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine (8c)
Brown solid, (79 % yield), mp above 300 °C; IR (KBr) νmax 3266 (NH2), 1599 (C⚌N), 1533, 1411, 1351, 1240, 1103, 1051 cm−1;1H NMR DMSO‑d6) 3.82 (s, 2H, CH2), 7.48–8.22 (m, 7H, Ar-H), 8.37 (s, 2H, NH2), 8.75 (s, 1H, pyrimdine- H).MS m/z (%): 439 (M+, 28), 404 (7), 357 (4), 329 (13), 299 (4), 236 (8), 220 (22), 190 (7), 154 (16), 141 (7), 58 (1 0 0), 40 (1 0 0).Anal. Calcd. for C19H12BrClN6 (439.70): C, 51.90; H, 2.75; N, 19.11. Found: 51.83; H, 2.61; N, 19.04 %.
4.3.4 8-Bromo-3-((2-nitrophenyl)diazenyl)-6H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2-amine (8d)
Brown solid, (82 % yield), mp above 300 °C; IR (KBr) νmax 3441, 3210 (NH2), 2936 (SP3 CH), 1585(C⚌N), 1434, 1305, 1203, 1039 cm−1; MS m/z (%): 451 (M+, 23), 418 (20), 243 (31), 132 (72), 89 (79), 62 (1 0 0), 41 (54). Anal. Calcd. for C19H12BrN7O2 (450.26): C, 50.68; H, 2.69; N, 21.78. Found: C, 50.51; H, 2.48; N, 21.67 %.
4.3.5 8-Bromo-3-((2-chlorophenyl)diazenyl)-1H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2(6H)-one (17a)
Dark blue solid, (86 % yield), mp above 300 °C; IR (KBr) νmax 3200 (NH), 1691 (C⚌O), 1533, 1379, 1208, 1096 cm−1;1H NMR (DMSO‑d6) 3.74 (s, 2H, CH2), 6.95–7.63 (m, 8H, Ar-H and pyridine-H), 8.82 (s, 1H, NH); MS m/z (%): 444 (M+ + 4, 40), 439 (M+, 28), 436 (88), 422 (82), 361 (41), 353 (80), 330 (35), 302 (41), 284 (78), 242 (29), 228 (28), 219 (21), 193 (36), 151 (49), 128 (1 0 0), 108 (36). Anal. Calcd. for C19H11BrClN5O (440.69): C, 51.79; H, 2.52; N, 15.89. Found: C, 51.65; H, 2.42; N, 15.78 %.
4.3.6 8-Bromo-3-((2-nitrophenyl)diazenyl)-1H-indeno[2,1-e]pyrazolo[1,5-a]pyrimidin-2(6H)-one (17b)
Brown solid, (90 % yield), mp above 300 °C; IR (KBr) νmax 3205 (NH), 1736 (C⚌O), 1502, 1358, 1210, 1150 cm−1; 1H NMR (DMSO‑d6) 3.74 (s, 2H, CH2),7.37 (t, J = 8.5 Hz, 1H, Ar-H), 7.62 (s, 1H, Ar-H), 7.87 (s,1H, pyrimidine-H), 7.91 (t, J = 8.5 Hz, 2H, Ar-H), 8.23 (d, J = 8.5 Hz, 1H, Ar-H), 8.28 (d, J = 8.5 Hz, 1H, Ar-H), 8.33 (d, J = 8.5 Hz, 1H, Ar-H),11.61 (s, 1H, NH); MS m/z (%): 450 (M+, 10), 430 (82), 405 (25), 385 (83), 371 (24), 344 (1 0 0), 324 (66), 303 (46), 277 (82), 228 (54), 192 (7). Anal. Calcd. for C19H11BrN6O3 (451.24): C, 50.57; H, 2.46; N, 18.62. Found: C, 50.64; H, 2.39; N, 18.58 %.
4.3.7 8-Bromo-6H-indeno[2,1-e][1,2,4]triazolo[1,5-a]pyrimidine (20)
Yellow solid, (90 % yield), mp 290–291 °C; IR (KBr) νmax 3057 (SP2 CH), 2970 (SP3 CH), 1600 (C⚌N), 1553, 1437, 1331, 1205, 1035 cm−1; 1H NMR (DMSO‑d6) 3.69 (s, 2H, CH2), 7.57–7.64 (m,2H), 7.85 (s, 1H, pyrimdine- H), 8.11(s, 1H, Ar-H), 8.41 (s, 1H, triazole-H). 13C NMR (DMSO‑d6) 29.79 (CH2), 124.63, 127.10, 129.77, 130.74, 135.74, 139.35, 144.04, 150.46, 152.49, 160.46, 172.49; MS m/z (%): 287 (M+, 47), 268 (83), 261 (41), 253 (85), 244 (50), 153 (93), 127 (56), 119 (23), 104 (1 0 0), 63 (33). Anal. Calcd. for C12H7BrN4 (287.12): C, 50.20; H, 2.46; N, 19.51. Found: C, 50.14; H, 2.39; N, 19.28 %.
4.3.8 10-Bromo-8H-benzo[4,5]imidazo[1,2-a]indeno[2,1-e]pyrimidine (21)
Dark green solid, (87 % yield), mp 139–140 °C; IR (KBr) νmax 1550 (C⚌N), 1416, 1207, 1049 cm−1; 1H NMR (DMSO‑d6) 3.60 (s, 2H, CH2), 7.08–7.83 (m, 7H, Ar-H), 8.22 (s, 1H, pyrimdine- H). 13C NMR (DMSO‑d6) 31.01 (CH2), 103.17,113.98, 122.0, 124.22, 124.69, 124.79, 125.40, 128.98, 129.93, 130.34, 130.82, 138.94, 139.76, 147.81, 150.24, 150.55. MS m/z (%): 336 (M+, 32), 301 (90), 284 (68), 230 (66), 209 (95), 104 (14), 89 (1 0 0). Anal. Calcd. for C17H10BrN3 (336.19); C, 60.74; H, 3.00; N, 12.50. Found: C, 60.62; H, 2.93; N, 12.42 %.
4.4 Reaction of enaminone derivative 3 with active methylene derivatives 22–24
By usual way for the reaction of enaminone with the active methylene, 0.0025 mol of rosy enaminone 3 reacted with active methylene derivatives 22–24 in 20 mL acetic acid in the presence of 0.5 g CH3COONH4 with reflux for 5 h. The solid colored product was collected by usual way and washed with ethanol, crystalized with ethanol/dioxane mixture to give a series of indenopyridine derivatives 25–27.
4.4.1 1-(7-Bromo-2-methyl-4a,9b-dihydro-5H-indeno[1,2-b]pyridin-3-yl)ethan-1-one (25)
Yellow solid, (88 % yield), mp above 300 °C; IR (KBr) νmax 1676 (C⚌O) 1540, 1251, 1098, 1036 cm−1;1H NMR (CDCl3) 2.19 (s, 3H, CH3), 2.48 (s, 3H, CH3), 3.71 (s, 2H, CH2), 7.53–7.82 (m, 4H, Ar-H and pyridine-H). 13C NMR (CDCl3) 18.35 (CH3), 24.76 (CH3), 30.80 (CH2), 103.46, 116.0, 124.5, 124.8, 126.5, 128.91, 130.81, 131.05, 138.56, 149.23, 154.78, 199.40 (C⚌O). MS m/z (%): 302 (M+, 35), 272 (80), 233 (64), 223 (48), 156 (84), 113 (1 0 0), 94 (63), 71 (25).Anal. Calcd. for C15H12BrNO (302.17): C, 59.62; H, 4.00; N, 4.64. Found: C, 59.50; H, 3.91; N, 4.49 %.
4.4.2 7-Bromo-2-phenyl-5,9b-dihydro-4aH-indeno[1,2-b]pyridine-3-carboxylic acid ethyl ester (26)
Yellow solid, (85 % yield), mp above 300 °C; IR (KBr) νmax 3054 (SP2 CH), 2893 (SP3 CH), 1679 (C⚌O), 1579, 1459, 1415, 1375, 1322, 1202, 1108, 1044 cm−1;1H NMR (DMSO‑d6) 1.12 (t, J = 8.5 Hz, 3H, CH3), 3.51 (s, 2H, CH2), 4.25 (q, J = 8.5 Hz, 2H, CH2), 6.95–8.13 (m, 9H, Ar-H and pyridine-H). Anal. Calcd. for C21H16BrNO2 (394.26): C, 63.97; H, 4.09; N, 3.55. Found: C, 63.83; H, 4.02; N, 3.43 %.
4.4.3 7-Bromo-2-oxo-2,4a,5,9b-tetrahydro-1H-indeno[1,2-b]pyridine-3-carboxylic acid ethyl ester (27)
Yellow solid, (84 % yield), mp above 300 °C; IR (KBr) νmax 3308 (NH), 1745(C⚌O), 1691(C⚌O), 1580, 1416, 1207, 1113, 1046 cm−1;1H NMR (DMSO‑d6) 1.30 (t, J = 8.5 Hz, 3H, CH3), 3.45 (s, 2H, CH2), 4.35 (q, J = 8.5 Hz, 2H, CH2), 7.23–8.01 (m, 4H, Ar-H and pyridine-H), 9.94 (s, 1H, NH); Anal. Calcd. for C15H12BrNO3 (334.16): C, 53.91; H, 3.62; N, 4.19. Found: C, 53.78; H, 3.52; N, 4.04 %.
4.5 In vitro cytotoxicity
MTT assay was utilized to explore the cytotoxic behavior of the tested compounds via using a Sigma in vitro MTT-based assay kit. MCF7 and MDA-MB231 breast cancer cell lines were obtained from the American Type Culture Collection, cultured in DMEM medium with penicillin, FBS, and streptomycin at 5 % CO2 and 37 °C. After treatment with several doses of research compounds, cells were cultured at 37 °C for 48 h, then they were incubated in dark for 4 h with MTT reagent. The absorbance was measured at 560 nm with a plate reader, and cell viability was determined. Finally the IC50 values were then calculated.
4.6 Hsp90 inhibitory activity
The inhibitory effect of compound 8a on the activity of the human heat shock protein 90 ELISA Kit (Cat.No.E3061Hu) was measured. To the standard well, add 50 μl of standard. Then 40 μl of sample should be added to the sample wells, followed by 10 μl of anti-HSP90 antibody and 50 μl of streptavidin-HRP in both the sample and standard wells. Mix well. Apply a sealant to the plate. 60 min at 37 °C of incubation. Remove the sealant, then use a wash buffer to wash the plate five times. For every wash, soak wells in a minimum of 0.35 mL of wash buffer for 30 to 1 min. Aspirate or decant each well for automatic washing, then use wash buffer five times. Place paper towels or another absorbent material nearby to blot the plate. Each well should first receive 50 μl of substrate solution A before receiving 50 μl of substrate solution B. Plate should be incubated for 10 min at 37°Celsius in the dark. Each well's colour will turn to yellow when 50 μl of stop solution is added. Within 10 min of adding the stop solution, measure the optical density of each well using a microplate reader set to 450 nm.
4.7 Molecular docking
In this study, docking was achieved using MOE 2014.09 (MOE, 2014). In order to build structures of the docked compounds, the builder button was used. The tested compounds were docked inside the Hsp90 crystal structure (PDB: 2xjx). To start docking, we used MOE's “Docking” module. Water molecules were deleted throughout the docking steps. The missing hydrogen atoms were retained to correct ionization states in order to be assigned to the protein structure. The “Ligand Interactions” MOE tool was then used for the analysis of docking results by visualization of the ligand–protein interactions in the active binding site.
Acknowledgement
This research was funded by Scientific Research Deanship at University of Ha’il - Saudi Arabia through project number RG-21141.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Pyrimidine: a review on anticancer activity with key emphasis on SAR. Future J. Pharm. Sci.. 2021;7
- [CrossRef] [Google Scholar]
- Design, synthesis, cytotoxicity, and molecular docking studies of novel thiazolyl–hydrazone derivatives as histone lysine acetyl-transferase inhibitors and apoptosis inducers. Arch. Pharm. 2022:e2200076.
- [CrossRef] [Google Scholar]
- PS4-3 A phase III trial of pimitespib (TAS-116) in patients with advanced gastrointestinal stromal tumor: CHAPTER-GIST-301. Ann. Oncol.. 2022;33:S467.
- [CrossRef] [Google Scholar]
- Synthesis of novel series of heterocyclic compounds having two azoles against Methicillin-sensitive Staphylococcus aureus. J. Mol. Struct.. 2023;1277:134863
- [CrossRef] [Google Scholar]
- Synthesis of a new series of pyrazolo[1,5-a]pyrimidines as CDK2 inhibitors and anti-leukemia. Bioorg. Chem.. 2021;117:105431
- [CrossRef] [Google Scholar]
- Fluorinated azole anticancer drugs: synthesis, elaborated structure elucidation and docking studies. Arab. J. Chem.. 2022;15:103782
- [CrossRef] [Google Scholar]
- Althagafi, I. I., Abouzied, A. S., Farghaly, T. A., Al-Qurashi, N. T., Alfaifi, M. Y., Shaaban, M. R., Abdel Aziz M. R., 2019. Novel Nano-sized bis-indoline Derivatives as Antitumor Agents J. Heterocyclic Chem., 56, 391-399. DOI 10.1002/jhet.3410
- Bayazeed, A., Alenazi, N. A., Alsaedi, A. M.R., Ibrahimd, M. H., Al-Qurashi, N. T., Farghaly, T. A., 2022. Formazan analogous: Synthesis, antimicrobial activity, dihydrofolate reductase inhibitors and docking study Journal of Molecular Structure 1258, 132653. https://doi.org/10.1016/j.molstruc.2022.132653.
- The effects of two “anti-aggressive” compounds, an indenopyridine and benzothiazepin, on shock-induced defensive fighting in rats. Prog. Neuropsychopharmacol.. 1979;3:399-402.
- [CrossRef] [Google Scholar]
- Effect of screening and adjuvant therapy on mortality from breast cancer. N. Engl. J. Med.. 2005;353:1784-1792.
- [CrossRef] [Google Scholar]
- Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harb. Perspect. Biol.. 2019;11:1-32.
- [CrossRef] [Google Scholar]
- An efficient synthesis and antimicrobial screening of new hybrid molecules containing coumarin and indenopyridine moiety. Med. Chem. Res.. 2015;24:1596-1604.
- [CrossRef] [Google Scholar]
- Structure-based design, synthesis, and biological evaluation of Hsp90β-Selective Inhibitors. Chem. Eur. J.. 2021;27:14747-14764.
- [CrossRef] [Google Scholar]
- Targeting conserved water molecules: design of 4-aryl-5-cyanopyrrolo[2,3-d]pyrimidine Hsp90 inhibitors using fragment-based screening and structure-based optimization. Bioorg. Med. Chem.. 2012;20:6770-6789.
- [CrossRef] [Google Scholar]
- Synthetic approaches, anticancer potential, HSP90 inhibition, multitarget evaluation, molecular modeling and apoptosis mechanistic study of thioquinazolinone skeleton: promising antibreast cancer agent. Bioorg. Chem.. 2020;101:103987
- [CrossRef] [Google Scholar]
- Synthesis, anti-HCV, antioxidant, and peroxynitrite inhibitory activity of fused benzosuberone derivatives. Eur. J. Med. Chem.. 2010;45:492-500.
- [CrossRef] [Google Scholar]
- New and efficient approach for synthesis of novel bioactive [1,3,4] thiadiazoles incorporated with 1,3-thiazole moiety. Eur. J. Med. Chem.. 2015;97:320e333.
- [CrossRef] [Google Scholar]
- Synthesis under microwaves irradiation, structure elucidation, docking study for inhibiting COVID-19 and DFT calculations of novel azoles incorporated indole moiety. J. Mol. Struct.. 2021;1244:131263
- [CrossRef] [Google Scholar]
- In vitro cytotoxic activity of thiazole-indenoquinoxaline hybrids as apoptotic agents, design, synthesis, physicochemical and pharmacokinetic studies. Bioorg. Chem.. 2020;100:103951
- [CrossRef] [Google Scholar]
- Anticancer activity of novel indenopyridine derivatives. Arch. Pharm. Res.. 2012;35:987-994.
- [CrossRef] [Google Scholar]
- Synthesis of some tricyclic indeno [1,2-d] pyrimidine derivatives as a new class of anti-breast cancer agents. Biomed. Res.. 2015;26:422-425.
- [Google Scholar]
- Heat shock proteins as targets in oncology. Clin. Transl. Oncol.. 2010;12:166-173.
- [CrossRef] [Google Scholar]
- Modified 2,4-diaryl-5H-indeno[1,2-b]pyridines with hydroxyl and chlorine moiety: Synthesis, anticancer activity, and structure–activity relationship study. Bioorg. Chem.. 2015;62:30-40.
- [CrossRef] [Google Scholar]
- Hydroxylated 2,4-diphenyl indenopyridine derivatives as a selective non-intercalative topoisomerase IIα catalytic inhibitor. Eur. J. Med. Chem.. 2015;90:302-314.
- [Google Scholar]
- Effect of chlorine substituent on cytotoxic activities: design and synthesis of systematically modified 2,4-diphenyl-5H-indeno[1,2-b]pyridines. Bioorg. Med. Chem. Lett.. 2016;26:1726-1731.
- [CrossRef] [Google Scholar]
- Heat shock protein 90 inhibitors: an update on achievements, challenges, and future directions. J. Med. Chem.. 2020;63:1798-1822.
- [CrossRef] [Google Scholar]
- The role of heat shock proteins in cancer. Can. Lett.. 2015;360:114-118.
- [CrossRef] [Google Scholar]
- Liu, Q., Tu, G., Hu, Y., Jiang, Q., Liu,J., Lin, S., Yu, Z., Li, G., Wu, X., Y., Tang, Huang,X., Xu, J., Liu, Y., Wu, L.. 2022. Discovery of BP3 as an efficacious proteolysis targeting chimera (PROTAC) degrader of HSP90 for treating breast cancer, Eur. J. Med. Chem. 228, 114013 https://doi.org/10.1016/j.ejmech.2021.114013
- Molecular Operating Environment (MOE) 2014, Chemical Computing Group Inc., Montreal, Canada http://www.chemcomp.com.
- Neckers, L., Mollapour, M., 2015. Heat shock protein 90 and the proteasome: Housekeeping proteins that are also molecular targets for cancer therapy, V. Molecular Basis of Cancer Therapy (Fourth Edition), 779-788.e3. http://dx.doi.org/10.1016/B978-1-4557-4066-6.00056-1
- Discovery of a simplified deguelin analog as an HSP90 C-terminal inhibitor, for HER2-positive breast cancer. Bioorg. Med. Chem. Lett.. 2021;45:128134
- [CrossRef] [Google Scholar]
- Molecular modelling and docking studies on heat shock protein 90 (Hsp90) inhibitors. SAR QSAR Environ. Res.. 2010;21:1-20.
- [CrossRef] [Google Scholar]
- A new phenolic series of indenopyridinone as topoisomerase inhibitors: design, synthesis, and structure-activity relationships. Bioorg. Med. Chem.. 2018;26:5212-5223.
- [CrossRef] [Google Scholar]
- Hsp90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol.. 2010;11:515-528.
- [CrossRef] [Google Scholar]
- Discovery of (2,4-Dihydroxy-5-isopropylphenyl)-[5-(4-methylpiperazin-1-ylmethyl)-1,3-dihydroisoindol-2-yl]methanone (AT13387), a Novel Inhibitor of the Molecular Chaperone Hsp90 by Fragment Based Drug Design. J. Med. Chem.. 2010;53:5956-5969.
- [CrossRef] [Google Scholar]
- Zhang, Y., Zhang, T., Li, X., Liang, J., Tu, S., Xu, H., Xue, W., Qian, X., Zhang, Z., Zhang, X., Meng, F., 2021. 2-((1-phenyl-1H-1,2,3-triazol-4-yl)methyl)-2-azabicyclo[1 3.2.1]octan-3-one derivatives: simplification and modification of aconitine scaffold for the discovery of novel anticancer agents. Eur. J. Med. Chem., 210, 1122988. https://doi.org/10.1016/j.ejmech.2020.112988.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.104569.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




