

Translate this page into:
Effect of oxygen-containing functional groups on the wettability of coal through DFT and MD simulation
⁎Corresponding author at: College of Safety Science & Engineering, Liaoning Technical University, Fuxin 123000, China. LXQab161616@163.com (Xiaoqing Liu),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
To investigate the wettability of different oxygen-containing functional group (OFG) surfaces, graphite substrates were used as a model for coal adsorbents. The substrates were modified with —COOH, —OH, —C⚌O, and —OCH3. The adsorption-diffusion behavior of H2O molecules/water droplets on different OFG surfaces was investigated using molecular dynamics (MD) simulations with frontier orbital energy difference as a metric for different surface wettability degrees in quantum chemical analysis. The results indicated that the frontier orbital energy difference of the H2O molecule was 3.480, 3.491, 3.631, and 3.680 eV for Ph—COOH, Ph—OH, Ph—C⚌O, and Ph—OCH3, respectively. In addition, the equilibrium contact angle, interaction energy, and number of hydrogen bonds after the adsorption equilibrium of water droplets for —COOH, —OH, —C⚌O, and —OCH3 surfaces were 22.34°, −5.03 kcal/mol, and 652; –23.72°, −4.19 kcal/mol, and 450; 68.01°, −0.79 kcal/ mol, and 61; 90.51°, −0.50 kcal/mol, and 28, respectively. The smaller the energy difference between the frontier orbitals of the H2O molecule and the OFG, the smaller the equilibrium contact angle between the water droplet and the OFG surface, the more hydrogen bonds were formed, and the larger the absolute value of the interaction energy, the better the wettability of the surface of the OFG. The order of wettability of the different OFG surfaces was —COOH > —OH > —C⚌O > —OCH3, which is consistent with the radial distribution function and the analysis results for the extended area, etc. The results of density functional theory (DFT) calculations and MD simulations exhibited identical patterns, indicating the reasonableness of the simulations. This study may serve as a reference for the suppression of hydrophilicity in low-order coal and the enhancement of the flotation effect.
Keywords
Oxygen-containing functional groups
Electrostatic potential
Frontier orbital
Equilibrium contact angle
Number of hydrogen bonds

1 Introduction
Flotation is an important means of coal quality and ash reduction and mineral separation (Gui et al., 2017; Gui et al., 2017). It occurs when hydrophilic fine coal particles are retained in a slurry and hydrophobic particles adhere to the air bubbles as mineralized gas flocs float up to form high-quality fine coal (Gui et al., 2015; Xia et al., 2017; Prakash et al., 2018). The hydrophilicity and hydrophobicity of the coal surface are influenced by pore structure, the degree of metamorphism, oxygen-containing functional groups, the degree of defects, and other factors (Crawford et al., 1994; Xia et al., 2016; Wang et al., 2020). Low-order coals account for 50 % of the coal resources of the world (Osman et al., 2011). Low-order coals require short coal formation time and have high moisture, low heat content, high volatile matter, more micropores, and well-developed structures; thus, low-order coals burn easily and have a strong adsorption capacity for water molecules (Wang et al., 2009). In addition, low-order coals have high oxygen content, primarily in the form of OFGs, such as —OH, —COOH, and —OCH3, which had a more notable influence on the hydrophilicity and hydrophobicity of coal surfaces (Wang et al., 2013; Xin et al., 2013). Strong polar OFGs easily interact with water molecules to form hydrogen bonds, increasing the water content on the surface of low-order coal and hindering the flotation of the coal. Therefore, treatment with OFGs affects the flotation of low-order coal, and the interaction between water molecules and OFGs in coals should be investigate to enhance flotation and realize the safe and efficient utilization of coal (Tang et al., 2015; Cui and Chen, 2021; Wang et al., 2022).
Several physical testing methods, including X-ray diffraction (Li et al., 2016), X-ray photoelectron spectroscopy (Kozlowski, 2004; Kelemen et al., 2002), infrared spectroscopy (Baysal et al., 2016; Liu et al., 2019), and nuclear magnetic resonance carbon spectroscopy (Erdenetsogt et al., 2010), have been applied to characterize OFGs on coal surfaces. Ma et al. investigated the molecular structure of anthracite from Xifeng County, Shaanxi Province, through NMR carbon spectroscopy, infrared spectroscopy, and X-ray photoelectron spectroscopy. The results revealed that anthracite is composed of aromatic structures, aliphatic carbon, and various functional groups and that OFGs are dominated by phenolic hydroxyl and ether oxygens and also contain some carbonyl groups (Ma et al., 2019). Chen et al. investigated the characteristics of OFGs in coals with different degrees of metamorphism through infrared spectroscopy experiments and concluded that the aromaticity of coal increases and the length of fatty chains and the number of OFGs decrease with increasing coal grade. Condensed aromatic domains are commonly found in coals of higher grades (Chen et al., 2012). All the aforementioned studies performed experiments, and this work is not only wasteful of financial and material resources, but also inefficient. By contrast, molecular simulation techniques and related software are mature technologies and are extensively used for research in the fields of biology, physics, and chemistry (Zhao et al., 2017). In addition to high visualization abilities and cost savings, molecular simulations can be used to obtain static structures of different adsorption systems and to elucidate the dynamic adsorption processes in those systems (Neyts and Bogaerts, 2013). Zhao et al. performed DFT simulations to investigate the interaction of water molecules with different OFGs and concluded that water molecules have the strongest interaction with carboxyl groups and the weakest interaction with ether groups by comparing the bond levels, interaction energies, and differential electron densities of different OFGs bonded to water molecules (Zhao and Liu, 2022). Wang et al. analyzed the adsorption of OFGs on coal surfaces with H2O molecules through a combination of experimental and DFT calculations; and the results indicated that the adsorption of OFGs with H2O molecules is attributable to the combined effects of van der Waals forces and hydrogen bonding (Wang et al., 2021). Gao et al. investigated the interaction of single water molecules and multilayer water with the surface of lignite based on DFT theory; the results indicated that the interaction of single water molecules and multilayer water with lignite is primarily caused by the adsorption of water molecules in OFGs (Gao et al., 2017). Zhang et al. performed contact angle measurement experiments and MD simulations to analyze the wettability of coal surfaces with different degrees of metamorphism and concluded that a higher number of OFGs causes a higher hydrophilicity of low-order coal surfaces (Zhang et al., 2020). Cheng et al. analyzed the microscopic adsorption of water molecules by OFGs using DFT calculations and grand canonical ensemble Monte Carlo (GCMC) simulations; the results indicated that the adsorption strength of water molecules with carboxyl groups was stronger than that of hydroxyl, carbonyl, and ether groups (Cheng et al., 2021). Gao et al. investigated the interaction between lignite and H2O molecules through DFT and CP molecular dynamics, and the ESP charges obtained indicate that the interaction of —COOH with H2O molecules is stronger than that of other OFGs (Gao et al., 2019). Liu and Wu et al. investigated the correlation between dehydration and OFGs in coal molecules through molecular simulations; their analysis revealed that the ability of coal molecules to adhere water decreases significantly with the decrease in the number of OFGs, indicating that OFGs are the main adsorption sites for water molecules (Wu et al., 2017; Liu et al., 2016). However, few studies have used MD simulations to analyze coal surface wettability. Therefore, the effects of different OFGs on the wettability of coal should be analyzed by selecting suitable charge and force fields for MD simulations and efficient flotation.
In this study, the wettability degrees of four surfaces with different OFGs, namely —COOH, —OH, —C⚌O, and —OCH3 were investigated using MD. The influence of H2O molecules on the wettability of different OFGs was comprehensively analyzed from the perspective of electrostatic potential and frontier orbitals by evaluating the equilibrium contact angle, radial distribution function, the number of hydrogen bonds, and interaction energy. This study can provide theoretical guidance for the efficient flotation of low-order coal.
2 Calculation methods
2.1 DFT calculations
The 3 × 3 carbon model structure on the graphite sheet layer was selected for the DFT calculations process and constructed as shown in Fig. 1. Later four OFGs, —COOH, —OH, —C⚌O, and —OCH3 were grafted to represent the models of different polar coals (Xu et al., 2016), as shown in Fig. 2(a)-(d). The structure of the H2O molecule is displayed in Fig. 2(e). The properties and interaction energies of different OFG models and H2O molecules were analyzed using the Materials Studio software package Dmol3 module. The task item was set to Geometry Optimization, and the precision was set to Fine. Because the local density approximation (LDA) function overestimates the value of interaction energy, the generalized gradient approximation (GGA) function was selected (Liu et al., 2016). The electron exchange correlation potential was calculated using the Perdew-Burke-Ernzerhof (PBE) generalized function based on the GGA (Song et al., 2019; Chen et al., 2017). The Grimme method was used for DFT dispersion correction to improve calculation accuracy (Perdew et al., 1996). The calculations were performed using DFT Semi-core Pseudopots and selected double numerical polarization groups (DNP) (Chen et al., 2016), without restricting electron spins. In order to consider the influence of the surrounding water environment, the solvation model was used. The SCF convergence accuracy was set to Fine, and the cycle period was set to 500. The convergence criteria for force, displacement, and energy were 0.002 Ha/Å, 0.005 Å, and 1.0 × 10-5 Ha, respectively (Tian et al., 2022).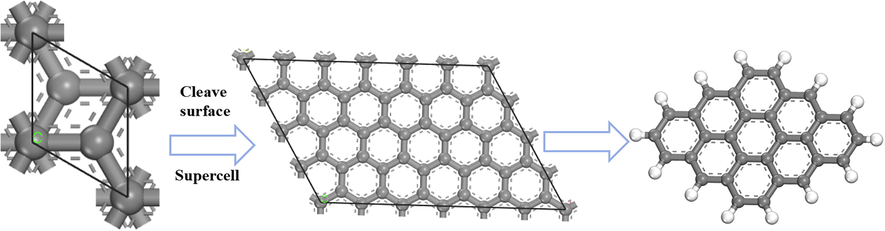
Selected 3 × 3 carbon model structure on graphite.
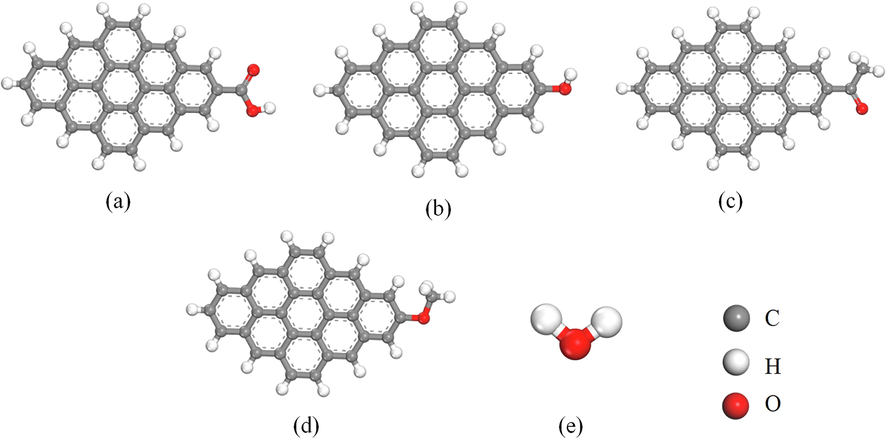
Different OFG models and H2O molecular models. (a) Ph—COOH, (b) Ph—OH, (c) Ph—C⚌O, (d) Ph—OCH3; (e) H2O molecule.
2.2 MD simulation
A graphite lamellar structure was used as the adsorbent substrate owing to the similarity of this structure to the coal skeleton (Zhang et al., 2018). The adsorption distribution process of water droplet on the surface of OFGs was analyzed by grafting different OFGs on the basis of graphite (Xia et al., 2020; Zhao et al., 2019). The specific simulation details are displayed in Fig. 3. First, the graphite was cleaved along the (0 0 1) surface, and four layers were cut and orthogonalized. Then, the adsorbent substrates were modified with four different OFGs, namely —COOH, —OH, —C⚌O, and —OCH3. The final grafted model consisted of a 25 × 15 extended cell, and the grafted substrate consisted of 195 OFGs, each of which had a length of 20 Å. The surface dimensions of the established model were 61.50 Å×63.91 Å×20.20 Å. To simulate the adsorption of water clusters on the surfaces of different OFGs, a spherical cluster of water molecules, which is a spherical water droplet with a radius of 15 Å and composed of 483 water molecules, was created using the Nanocluster function in the software. Water droplets with a diameter of 30 Å have been used in the literature and the simulation results were in good agreement with the experimental values (Zhang et al., 2020; Li et al., 2018). The initial distance between the water clusters and the surfaces of different OFGs was set to the same value. The water cluster was placed in a cell with the same length and width dimensions as the grafted OFG. Using three-dimensional periodic boundary conditions, the OFG model and the water droplet model were combined together using the Build Layer tool. To eliminate the influence of the periodic structure on the substrate surface, a vacuum layer with a height of 50 Å was added by extending the Z-axis, and the dimensions of all four systems were approximately 61.50 Å×63.91 Å×98.56 Å. The construction process of the final model is illustrated in Fig. 4. To save time, the graphite substrates in the four different systems were immobilized, and the grafted OFGs and water droplets were allowed to move.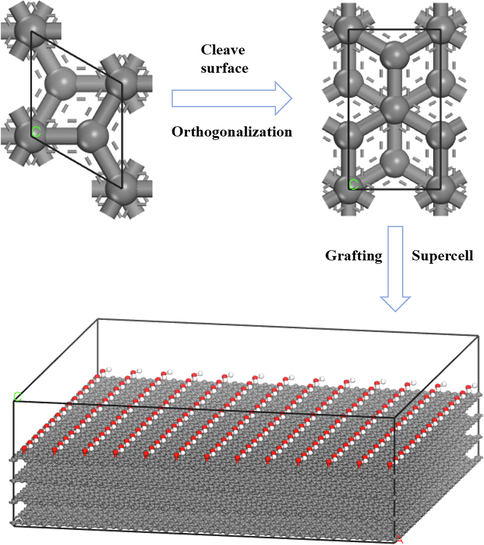
Construction of graphite substrate modified with OFGs.
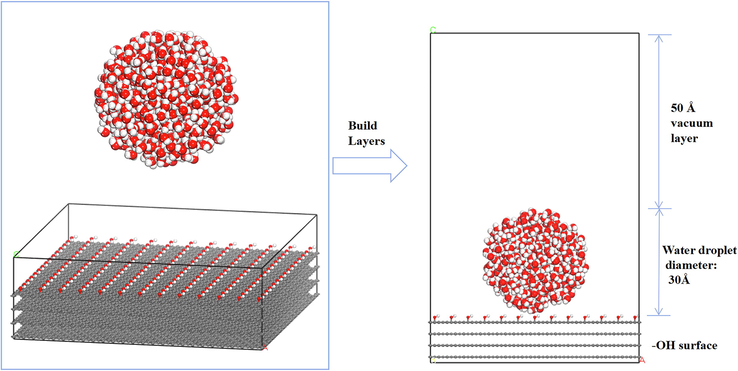
Initial configurations of water droplets adsorbed on the surface of different OFGs.
Before the MD simulations, the different OFGs/water droplet systems were assigned charges by COMPASS II force field using Materials Studio software, and a 5000-step geometry-optimized energy minimization simulation was performed to reach a stable structure, followed by a 1 ns MD simulation using COMPASS II force field on the Forcite module. A large body of literature has been published using the COMPASS II force field to study coal systems (Meng et al., 2021; Xia et al., 2019), and the results showed that the data obtained using the COMPASS II force field were in good agreement with the experimental results. PCFF can also be used to study coal systems (Zhang et al., 2015), but the COMPASS II force field is more widely applicable. After 500 ps, the interaction energy, contact angle, temperature and energy of different OFG/water droplet systems reached equilibrium, and the contact angle, hydrogen bonding, and interaction energy were analyzed in the next 500 ps. The parameter settings for the MD simulations are displayed in Table 1.
Parameter settings
Quality
Medium
Ensemble
Canonical ensemble NVT(Zhao and Liu, 2022)
Temperature
298 K
Temperature control method
Nose(Song et al., 2017)
Electrostatic
Ewald
Accuracy of electrostatic force
1.0 × 10-3 kcal/mol
van der Waals
Atom based
Truncation radius
12.5 Å(Li et al., 2019)
Time step
1.0 fs
3 Results and discussion
3.1 DFT calculation results
3.1.1 Electrostatic potential (ESP) analysis
By analyzing the surface ESP distribution of different OFGs and H2O molecules, the location of hydrogen bond formation and the strength of the interactions between the OFGs and H2O molecules can be predicted. Fig. 5 displays the ESP distribution of different OFGs and H2O molecules, with positive values lying in the red region and negative values in the blue region. The darker the red or blue color in the figure, the greater the absolute value of the ESP and the greater the chance of causing electrostatic attractive interactions (Nie et al., 2022). To facilitate observation, the value of the ESP isosurface were uniformly selected as 0.017 a.u.. As illustrated in Fig. 5, the ESP maximum point of each OFG were all near the hydrogen atom, and the order of magnitude was Ph-OH (71.25 kcal/mol) > Ph-COOH (66.65 kcal/mol) > Ph-C⚌O (30.79 kcal/mol) > Ph-OCH3 (29.40 kcal/mol). The ESP minima were all near the oxygen atom, and the order of magnitude was Ph-OCH3 (-24.61 kcal/mol) > Ph-OH (-26.41 kcal/mol) > Ph-COOH (-36.13 kcal/mol) > Ph-C⚌O (-41.57 kcal/mol). Thus, the structure of H2O molecule adsorbed on the H atoms of Ph-COOH and Ph-OH was the most stable, while the structure of H2O molecules adsorbed on O atom of Ph-C⚌O and Ph-OCH3 was the most stable. Furthermore, the aforementioned structures easily form hydrogen bonds. The positive ESP in Ph-COOH and Ph-OH was greater than that of H2O molecules, and the absolute value of the negative ESP was smaller than that of H2O molecules, indicating that the strongly polar OFGs easily interacted with H2O molecules and were easily wetted by water. The positive ESP values for Ph-C⚌O was smaller than that of H2O molecules, and the absolute value of the negative ESP was similar to that of the H2O molecule, indicating that the interaction of Ph-C⚌O with the H2O molecule was weaker than that of the strongly polar OFGs. The absolute values of both positive and negative ESP in Ph-OCH3 were smaller than those of H2O molecules, indicating that Ph-OCH3 had the least wettability and the weakest interaction with H2O molecules.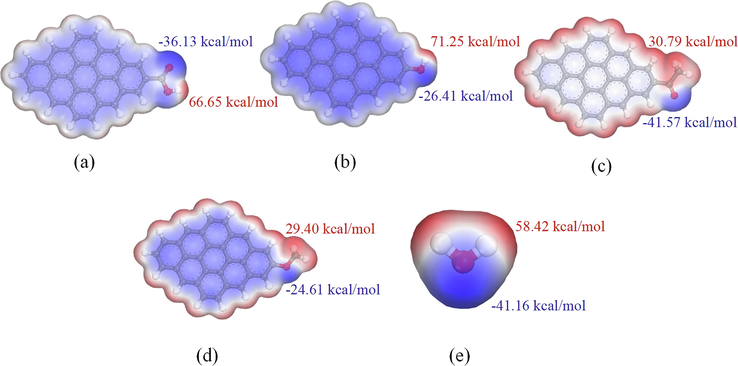
ESP distribution of different OFGs and H2O molecules. (a) Ph—COOH; (b) Ph—OH; (c) Ph—C⚌O; (d) Ph—OCH3; (e) H2O molecule.
3.1.2 Frontier orbital analysis
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), which are called frontier orbitals and determine the reactivity between the two molecules (Yekeler and Yekeler, 2006). The energies of the frontier orbitals of different OFGs and H2O molecules were calculated; the results are summarized in Table 2. The smaller the difference in frontier orbital energy (ΔE) between the HOMO of the H2O molecule and the LUMO of the OFG, the more favorable the adsorption of the H2O molecule on the OFG and the more the wettability (Bao et al., 2022). As listed in Table 2; ΔE of the strongly polar OFGs Ph-COOH and Ph-OH with H2O molecules was smaller than that of weakly polar OFGs, indicating that H2O molecules are easily adsorbed on the strongly polar OFGs; this is consistent with the results of ESP analysis. ΔE = |EH2O HOMO − EOFGs LUMO|, where EH2O HOMO denotes the HOMO energy of H2O molecules and EOFGs LUMO denotes the LUMO energy of different OFGs.
Models
Frontier orbital energy/eV
Frontier orbital energy difference/eV
HOMO
LUMO
ΔE
Ph-COOH
−4.602
−3.264
3.480
Ph-OH
−4.443
−3.253
3.491
Ph-C⚌O
−4.589
−3.113
3.631
Ph-OCH3
−4.392
−3.064
3.680
H2O
−6.744
1.053
3.1.3 Interaction energy calculations
Interaction energy represents the strength of molecular interactions and can be used to characterize the surface wettability of different OFGs. The interaction energy can be defined as (Dong et al., 2019; Xiang and Lei, 2021):
The optimal configurations of H2O molecules adsorbed by different OFGs are illustrated in Fig. 6, and the corresponding adsorption parameters are listed in Table 3. As depicted in Fig. 6 and Table 3, the interaction energies of the strong polar OFGs Ph-COOH and Ph-OH with H2O molecules were −43.631 and −39.632 kJ/mol, respectively, and the absolute values of the interaction energies were much larger than those of the weak polar OFGs Ph-C⚌O and Ph-OCH3. This was similar to the results of Cheng et al. who noted that the interaction energies of Ph-COOH with H2O molecules was −41.818 kJ/mol (Cheng et al., 2021). This can be attributed to the strong polarity of Ph-COOH and Ph-OH and the weak polarity of Ph-C⚌O and Ph-OCH3. The H2O molecule is polar molecule and easily interacts with strongly polar OFGs to form hydrogen bonds with lengths of 1.692/1.861 Å and 1.841 Å, respectively. Furthermore, the interaction of Ph-COOH with H2O molecules was stronger than that of Ph-OH because of the presence of double hydrogen bonds between Ph-COOH and H2O molecules. Comparison between the interaction energy and hydrogen bond length revealed that the order of wettability was Ph-COOH > Ph-OH > Ph-C⚌O > Ph-OCH3, which was the order of polarity of OFGs and was consistent with the order of adsorption strength reported by Xia and Lun et al (Xia et al., 2016; Lun et al., 2022). There was a correlation between the interaction energy of H2O molecules on different OFGs and ESP with an R2 of 0.878, and the fitted results are shown in Fig. 7. DFT calculations showed that the adsorption of H2O molecules in strongly polar OFGs was favorable and easily wettable.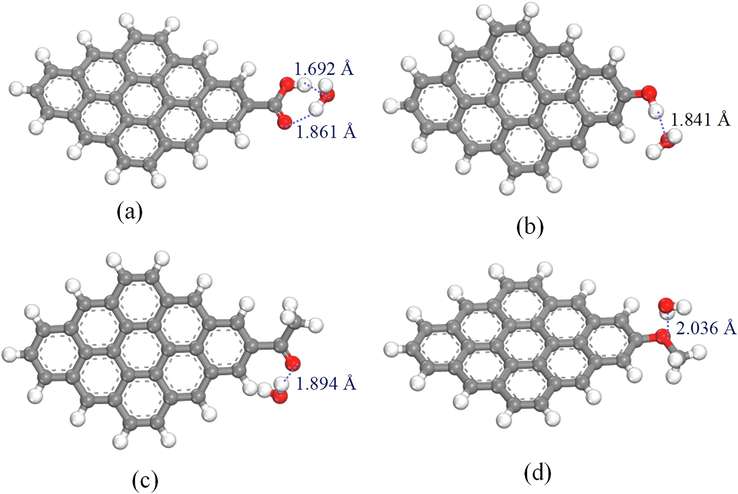
Optimal conformation of H2O molecules adsorbed by different OFGs. (a) Ph—COOH/H2O; (b) Ph—OH/H2O; (c) Ph—C⚌O/H2O; (d) Ph—OCH3/H2O.
Small molecule
OFGs
Hydrogen bond length/Å
Interaction energy/(kJ·mol−1)
H2O
Ph-COOH
1.692/1.861
−43.631
Ph-OH
1.841
−39.632
Ph-C⚌O
1.894
−26.596
Ph-OCH3
2.036
−19.169
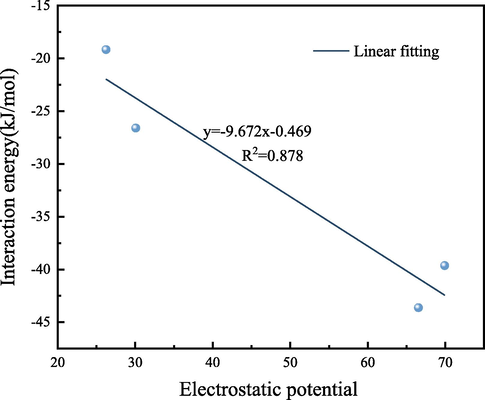
Linear fit between ESP and H2O interaction energy.
3.2 MD simulation results
3.2.1 Conformation of water droplets on the surface of OFGs
To elucidate the effects of the type of OFGs on the adsorption-diffusion of water droplets, the adsorption-diffusion behavior of water droplets on functional groups was simulated by grafting four OFGs of different polarities on graphite. The simulated snapshots of the contact between different OFGs and water droplets are displayed in Fig. 8, which illustrates the adsorption configurations of water droplets on different OFGs. The OFGs of the initial configuration of the systems did not interact with water droplets, and the contact lines of the water droplets on —COOH, —OH, —C⚌O, and —OCH3 surfaces increased after interactions, indicating that the water droplets were adsorbed on the surfaces of all four OFGs and diffusion occurred during the simulation. The water droplets were completely spread on the surfaces with strongly polar OFGs namely —COOH and —OH, and the contact lines extend to the cell width of 63.91 Å, forming a dense hydration film. By contrast, the water droplets were hemispherical droplets on the surfaces with weakly polar OFGs, namely —C⚌O and —OCH3, and the contact lines with the surfaces at the end of the simulation were 53.10 Å and 38.60 Å, respectively.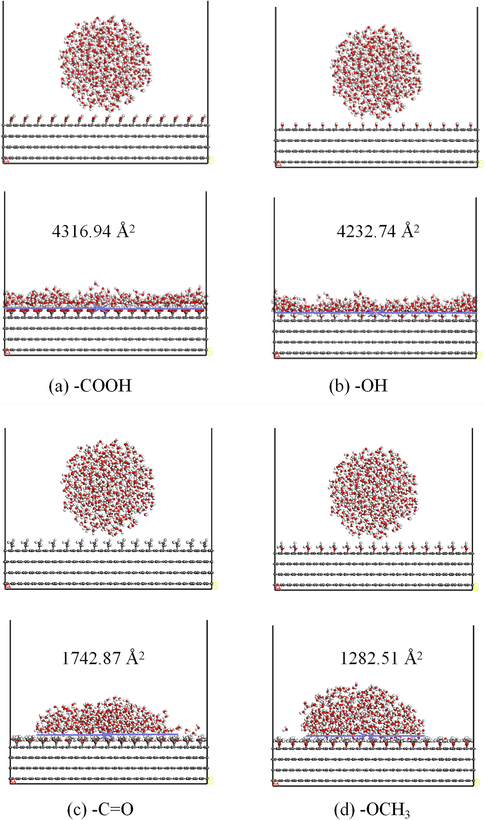
Initial (top) and stable (bottom) configurations of water droplets on different OFGs surfaces. (a) —COOH; (b) —OH; (c) —C⚌O (d) —OCH3.
The equilibrium contact angle can be used to characterize the wettability of different OFGs on the surface and quantify the adsorption-diffusion behavior of water droplets on the surface. The contact angles of water droplet adsorbed on different OFGs surfaces after MD simulation equilibrium are displayed in Fig. 9. The simulation results yielded contact angles of 22.34°, 23.72°, 68.01°, and 90.51° for water droplet adsorption on —COOH, —OH, —C⚌O, and —OCH3 surfaces, respectively, with contact angles less than or equal to 90°. These results indicated that the surfaces were wetted by water droplets in all four systems, but the degree of wetting varied. This was consistent with the results of Li et al. who reported that the equilibrium contact angle of water droplet on the —OH surface was 23.70° (Li et al., 2018). The equilibrium contact angle of water droplets on strongly polar OFGs surfaces was much smaller than the equilibrium contact angle on weakly polar OFGs surfaces, and the order of contact angle was as follows: —OCH3 > —C⚌O > —OH > —COOH, which is the order of hydrophobicity of OFGs. The surfaces with —COOH and —OH were hydrophilic, and water droplets could easily spread on the surfaces with small equilibrium contact angles.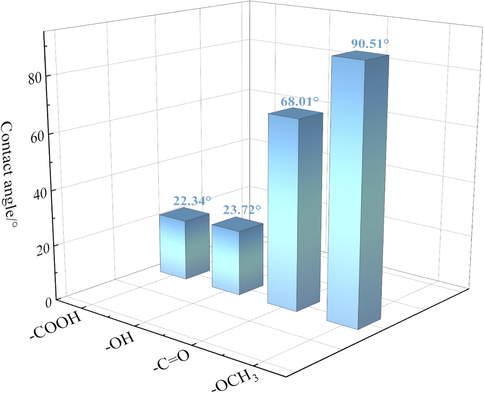
Equilibrium contact angle of water droplets on different OFGs surfaces.
Fig. 10 displays the top view of water droplets adsorbed in stable adsorption configurations on the surfaces of different OFGs. The surface of the strongly polar OFG, the water droplets diffused to the whole crystal surface, and the H2O molecules in the water droplet were dispersed from each other with large gaps between the H2O molecules. On the —C⚌O surface, the top view of the water droplet changes from a round shape to an irregular shape. The H2O molecules in the water droplet were more aggregated compared with that the strongly polar OFG system. On the —OCH3 surface, the water droplets in the crystal surface were only slightly diffused outward from the center, and the model was still a liquid droplet. The highest degree of aggregation of water molecules was observed in the water droplets.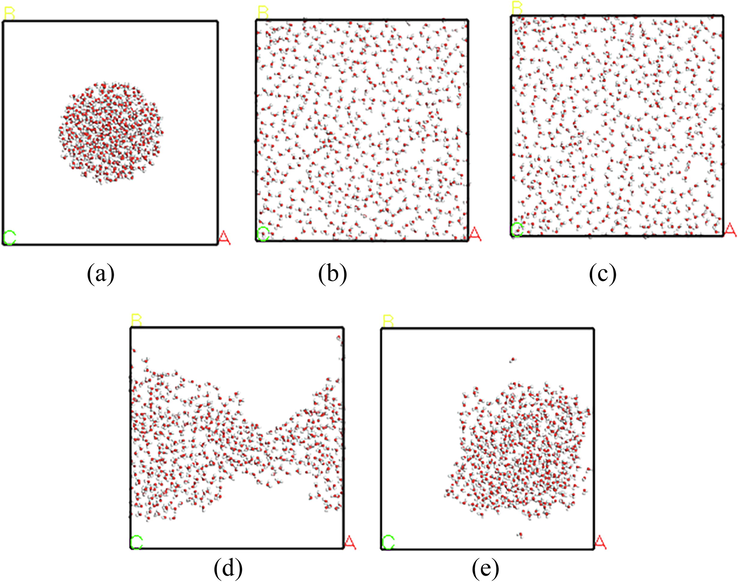
Top view of water droplets on OFGs surfaces. (a) Initial state; (b) Equilibrium state of - COOH; (c) Equilibrium state of —OH; (d) Equilibrium state of —C⚌O; (e) Equilibrium state of —OCH3.
The adsorption-diffusion behavior of water droplets on different OFG surfaces was analyzed using contact lines, equilibrium contact angles, and top views of water droplets. The water droplets had strong interactions with strongly polar OFG surfaces and easily diffused to wet the surfaces. By contrast, the interactions of the weakly polar OFG surface with water molecules were weaker, the diffusion was difficult, and the degree of wetting was weaker.
3.2.2 Radial distribution function (RDF)
The RDF can be used to evaluate the distribution of other particles in space, given the coordinates of a certain particle (Zhao and Liu, 2022; Xiang et al., 2014). The RDF can also be used to determine the order of water droplets on the surface of different OFGs and is calculated as follows.
where
denotes the number density of class y particles; dN is the average number of particles of class x to class y within a certain distance from r + dr. To further elucidate the distribution of H2O molecules in water droplets, the RDF of oxygen atoms in H2O molecules in different systems was calculated; the results are displayed in Fig. 11. For different OFG surfaces, the positions of the RDF peaks for oxygen atoms in H2O molecules were observed at 2.7 Å, and only the heights of the peaks were different. The high and sharp peaks indicated a strong interatomic order, a high probability of finding another atom within a certain distance from a given atom, and close relationships between atoms. The RDF peaks for between oxygen atoms in H2O molecules on the surface of weakly polar OFGs were higher than that for the surface of strongly polar OFGs. This indicates that on the surface of weakly polar OFGs, the probability of finding oxygen atoms within a certain distance of oxygen atoms in H2O molecules was higher and H2O molecules were more aggregated. The H2O molecules interacted weakly with the surface of weakly polar OFGs, making them ordered and not easily diffused to the surface. The lowest RDF peak for oxygen atoms adsorbed on the surface of —COOH indicated that the distance between OH2O and OH2O was the largest, the interaction was the weakest, and the H2O molecules were not easily aggregated with each other. Therefore, the H2O molecule was completely wetted on the —COOH surface; this was consistent with the simulated stable conformation displayed in Fig. 8. By calculating the height of the peak, the order of the strength of the surface wettability of different OFGs was —COOH > –OH > —C⚌O > —OCH3, which is consistent with the visualization results for adsorption conformation.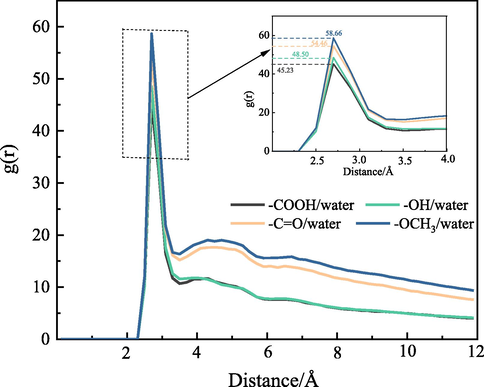
RDF between oxygen atoms in H2O molecules adsorbed on different OFGs surfaces.
3.2.3 Extended area between water droplets and surfaces with different OFGs
In addition to parameters such as RDF, the parameter extended area was introduced to quantify the adsorption-diffusion of water droplets on different OFGs. The extended area (EA) of water droplets on different OFG surfaces was calculated by using a probe with a radius of 1.4 Å, which can be calculated using Equation (3).
3.2.4 Interaction energy between water droplets and different OFGs surfaces
The interaction energy (Eint) between the surfaces of different OFGs and water droplets were calculated using Eq. (1); the same calculations were performed for the energy components van der Waals force (Evdw) and electrostatic force (Eelec). The calculation results of the interaction energy and energy components are displayed in Table 4. The interaction energies of the —COOH and —OH surfaces with each H2O molecule in the water droplets were −5.03 and −4.19 kcal/mol; these interactions were considerably larger than those of —C⚌O and —OCH3 surfaces, indicating that the water droplets were not favorable for diffusion on weakly polar OFGs surfaces. There is a linear correlation between the interaction energy of different OFG surfaces with each H2O molecule in the water droplet and the contact angle, with an R2 of 0.901, and the fitting results are shown in Fig. 12. From Fig. 12, it can be concluded that the larger the absolute value of the interaction energy, the smaller the contact angle and the easier it is to diffuse on the surface of the OFG. In terms of energy fraction, for —COOH and —OH surfaces, the adsorption of water droplets on —COOH and —OH surfaces was controlled by the electrostatic force due to the high percentage of Eelec in Eint, 99.08 % and 91.88 %, respectively. For —C⚌O, —OCH3 surfaces, the percentage of Evdw exceeds that of Eelec, and the sharp decrease in the percentage of Eelec was responsible for the relatively weak interaction of water droplets with -C⚌O, —OCH3 surfaces, while the higher percentage of Evdw was a weak interaction. In addition, the percentage of electrostatic interactions varied for different OFGs/water droplet systems because of the differences in the charges of the grafted OFGs. The charge distribution of different OFGs and H2O molecules is displayed in Fig. 13. The charges of the two O atoms in the strongly polar OFG —COOH were −0.455 e and −0.450 e, respectively, and the charges of the O atom in —OH was −0.570 e, whereas the charges of the O atoms in the weakly polar OFGs -C⚌O and —OCH3 were −0.419 e and −0.320 e, respectively. The negative charges of the O atoms in strongly polar OFGs are higher than those in the weakly polar OFGs. Comparing the positive charges of H and C atoms in different OFGs also revealed that the positive charges of both H and C atoms in strongly polar OFGs are greater than those in weakly polar OFGs. The charge of the H2O molecule is the same for the different OFGs investigated in this study; the higher charge of —COOH and —OH caused higher electrostatic force in the system. Electrostatic interactions are crucial to the interaction of OFGs with water droplets. The —CH3 in the weak OFG causes a spatial site barrier effect in the system, which was also the reason for the weak interaction of water droplets on the —C⚌O and —OCH3 surfaces.
Systems
Eint(kcal/mol)
Eelec(kcal/mol)
Evdw(kcal/mol)
–COOH/water droplet
−5.03
−4.98 (99.08 %)
−0.05 (0.92 %)
–OH/water droplet
−4.19
−3.85 (91.88 %)
−0.34 (8.12 %)
-C⚌O/water droplet
−0.79
−0.37 (46.33 %)
−0.42 (53.67 %)
–OCH3/water droplet
−0.50
−0.21 (42.75 %)
−0.29 (57.25 %)
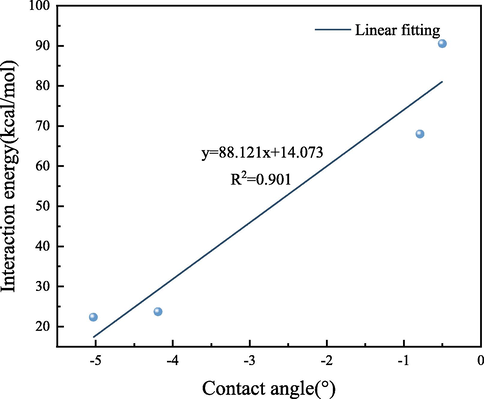
Linear fit of the interaction energy of H2O to the contact angle.
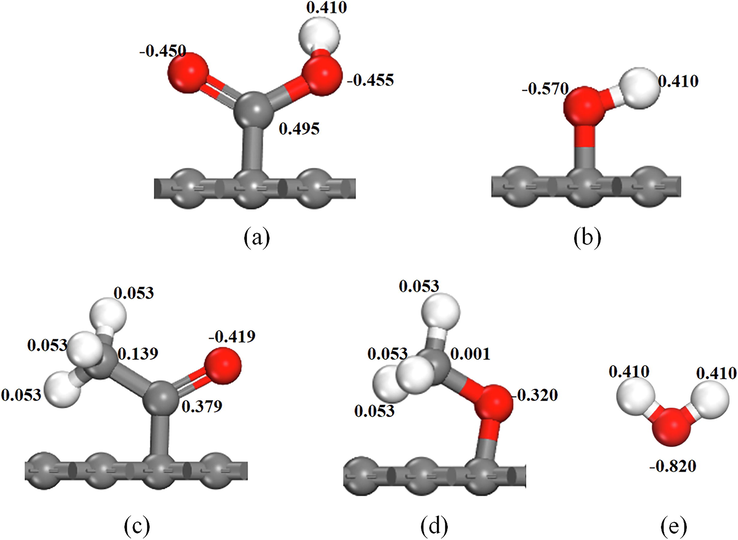
Charge of grafted OFGs and H2O molecules. (a) —COOH; (b) —OH; (c) —C⚌O; (d) —OCH3; (e) H2O.
The number of hydrogen bonds formed between different OFGs and water droplets was calculated by scripting simulations to react to differences in wettability. The geometrical criteria for hydrogen bond formation were as follows: intermolecular H-acceptor distance less than 3 Å and donor-H-acceptor angle greater than 135°. The total number of hydrogen bonds in different OFG systems is displayed in Fig. 14. From Fig. 14, the —COOH, —OH, —C⚌O and —OCH3 surfaces formed 652, 450, 61, and 28 hydrogen bonds with water droplet respectively. Under the same conditions, the formation of hydrogen bonds was the most favorable on the —COOH surface, with the highest number of hydrogen bonds due to the existence of double hydrogen bonds between a single —COOH and the H2O molecule (see section 3.1.3 for details), whereas the formation of hydrogen bonds of water droplets with the —OCH3 surface was relatively difficult, with the lowest number of hydrogen bonds. The order of wettability of the different surfaces was as follows: —COOH > —OH > —C⚌O > OCH3. This is consistent with the adsorption configuration, the RDF and extended area, and the interaction energy, as well as with the DFT results.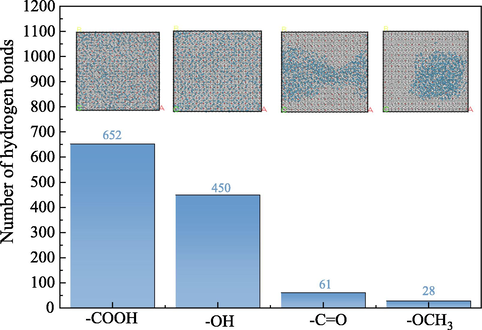
Hydrogen bonding number diagram.
4 Conclusions
In this study, the surface wettability of four different OFGs were predicted through ESP analysis and frontier orbital simulation analysis in DFT. MD simulations were also performed, while the rationality of the simulations was finally determined at the molecular microscopic level by combining the radial distribution function and interaction energy analysis.
H2O molecules complete the adsorption-diffusion on the surface of different OFGs through electrostatic attraction and hydrogen bonding, thus realizing the wetting of the surface of different OFGs.
The surface wettability of different OFGs can be reacted by the ESP and the frontier orbital energy difference. The larger the absolute values of the positive and negative ESP of different OFGs, the higher the wettability of the surfaces. The smaller the energy difference, the higher the wettability of the OFG surface.
The analysis of contact angle, RDF, number of hydrogen bonds, extended area, and interaction energy revealed that the wettability of the strongly polar OFG surface was stronger than that of the weakly polar OFG surface. The order of their wettability was —COOH > —OH > —C⚌O > OCH3, which is the same as that obtained for the ESP and frontier orbital energy difference in DFT.
ESP and frontier orbitals were used to determine the wettability of OFG surfaces and to suppress the hydrophilicity of low-order coals to achieve better flotation.
Acknowledgments
This work was supported by the Liaoning Provincial Department of Education Project(LJ2019JL025). The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Investigation on mechanism of the oleic acid/methyl oleate/diesel ternary compound collector in low-rank coal flotation. Fuel. 2022;320:123894
- [Google Scholar]
- Structure of some western Anatolia coals investigated by FTIR, Raman, 13C solid state NMR spectroscopy and X-ray diffraction. Int. J. Coal Geol.. 2016;163:166-176.
- [Google Scholar]
- Characterization of chemical functional groups in macerals across different coal ranks via micro-FTIR spectroscopy. Int. J. Coal Geol.. 2012;104:22-33.
- [Google Scholar]
- DFT calculations of different amine /ammonium cations adsorption on kaolinite (001) surface. J. China Coal Soc.. 2016;41(12):3115-3121.
- [Google Scholar]
- Experimental investigation and DFT calculation of different amine/ammonium salts adsorption on kaolinite. Appl. Surf. Sci.. 2017;419:241-251.
- [Google Scholar]
- Simulation of the adsorption behavior of CO2/N2/O2 and H2O molecules in lignite. J. China Coal Soc.. 2021;46(S2):960-969.
- [Google Scholar]
- The influence of coal rank and mineral matter content on contact angle hysteresis. Fuel. 1994;73(5):742-746.
- [Google Scholar]
- Insight into mineral flotation fundamentals through the DFT method. Int. J. Min. Sci. Techno.. 2021;31:983-994.
- [Google Scholar]
- Molecular simulation of the preferential adsorption of CH4 and CO2 in middle-rank coal. Mol. Simulat.. 2019;45(1):15-25.
- [Google Scholar]
- Solid-state C-13 CP/MAS NMR study of Baganuur coal, Mongolia: oxygen-loss during coalification from lignite to subbituminous rank. Int. J. Coal Geol.. 2010;82(1):37-44.
- [Google Scholar]
- DFT study of water adsorption on lignite molecule surface. J. mol. Model.. 2017;23:27.
- [Google Scholar]
- Car-Parrinello molecular dynamics study on the interaction between lignite and water molecules. Fuel. 2019;258:116189
- [Google Scholar]
- Coal preparation technology: Status and development in China. Energ. Environ-UK. 2015;26(6–7):997-1014.
- [Google Scholar]
- Fine coal flotation process intensification: part 1-A general overview of the state-of-the-art of the related research work conducted both within and abroad. Coal Prep. Technol.. 2017;1:93-107.
- [Google Scholar]
- Coal flotation process intensification: part II-Study on mechnasim of difficulty in flotation of low-rank and oxidized coal. Coal Prep. Technol.. 2017;2:79-83.
- [Google Scholar]
- Characterization of organically bound oxygen forms in lignites, peats, and pyrolyzed peats by X-ray photoelectron spectroscopy (XPS) and solid-state 13C NMR methods. Energ. Fuel.. 2002;16(6):1450-1462.
- [Google Scholar]
- XPS study of reductively and non-reductively modified coals. Fuel. 2004;83(3):259-265.
- [Google Scholar]
- Molecular simulation of adsorption of gas in coal slit model under the action of liquid nitrogen. Fuel. 2019;255:115775
- [Google Scholar]
- Effect of oxidation on the wetting of coal surfaces by water: experimental and molecular dynamics simulation studies. Physicochem. Probl. Ml.. 2018;54(4):1039-1051.
- [Google Scholar]
- XRD characterization of structural evolution in low-middle rank coals. J. Fuel Chem. Technol.. 2016;44(7):777-783.
- [Google Scholar]
- Molecular simulation of CH4, CO2, H2O and N2 molecules adsorption on heterogeneous surface models of coal. Appl. Surf. Sci.. 2016;389:894-905.
- [Google Scholar]
- Study on FTIR features of middle and high rank coal structure in north part of Qinshui Basin. Coal Sci. Technol.. 2019;531(2):186-192.
- [Google Scholar]
- Removal of oxygen functional groups in lignite by hydrothermal dewatering: an experimental and DFT study. Fuel. 2016;178:85-92.
- [Google Scholar]
- Studying the micromechanism of water injection to suppress coal and gas protrusion. AIP Adv.. 2022;12:125014
- [Google Scholar]
- Model construction and optimization of molecule structure of high-rank coal in Feng County, Shaanxi Province. J. China Coal Soc.. 2019;44(6):1827-1835.
- [Google Scholar]
- Study of the wetting mechanism of SDBS solution on Zhaozhuang coal surface. J. China Univ. Min. Technol.. 2021;50(2):381-388.
- [Google Scholar]
- Combining molecular dynamics with Monte Carlo simulations: implementations and applications. Theor. Chem. Acc.. 2013;132(2):1-12.
- [Google Scholar]
- Study on the effect of different surfactants on the wettability of coal dust based on Dmol3 module. J. China Coal Soc. 2022
- [Google Scholar]
- Drying of Low-Rank Coal (LRC)-A Review of Recent Patents and Innovations. Dry. Technol.. 2011;29:1763-1783.
- [Google Scholar]
- Generalized Gradient Approximation Made Simple. Phys. Rev. Lett.. 1996;77:3865-3868.
- [Google Scholar]
- Flotation technique: Its mechanisms and design parameters. Chem. Eng. Process.. 2018;127:249-270.
- [Google Scholar]
- Molecular simulation of CH4/CO2/H2O competitive adsorption on low rank coal vitrinite. Phys. Chem. Chem. Phys.. 2017;19:17773-17788.
- [Google Scholar]
- Competitive adsorption of CO2/N2/CH4 onto coal vitrinite macromolecular: effects of electrostatic interactions and oxygen functionalities. Fuel. 2019;235:23-38.
- [Google Scholar]
- Theoretical study on the interactions between the lignite monomer and water molecules. Russ. J. Phys. Chem. A+. 2015;89:1605-1613.
- [Google Scholar]
- Density functional simulation study of surface wettability of coal molecules with different degrees of defects. ACS omega. 2022;7:47031-47039.
- [Google Scholar]
- Quantum chemistry study on thermochemical properties of coal. J. China Coal Soc.. 2009;34(9):1239-1243.
- [Google Scholar]
- Investigation of collector mixtures on the flotation dynamics of low-rank coal. Fuel. 2022;327:125171
- [Google Scholar]
- Effect of vacancy defects on electronic properties and wettability of coal surface. Appl. Surf. Sci.. 2020;2020(511):145546
- [Google Scholar]
- Investigation of interactions between oxygen-containing groups and water molecules on coal surfaces using density functional theory. Fuel. 2021;287:119556
- [Google Scholar]
- Study on oxygen containing functional groups in coal by solid state nuclear magnetic resonance. J. Fuel Chem. Technol.. 2013;41(12):1422-1426.
- [Google Scholar]
- Moisture removal mechanism of low-rank coal by hydrothermal dewatering: physicochemical property analysis and DFT calculation. Fuel. 2017;187:242-249.
- [Google Scholar]
- Mechanism of water molecule adsorption by oxygen-containing functional groups on the surface of lignite. Coal Conversion. 2016;39(4):1-5.
- [Google Scholar]
- Clean low-rank-coal purification technique combining cyclonicstatic microbubble flotation column with collector emulsification. J. Clean. Prod.. 2017;153:657-672.
- [Google Scholar]
- Improving the adsorption of oily collector on the surface of low-rank coal during flotation using a cationic surfactant: an experimental and molecular dynamics simulation study. Fuel. 2019;235:687-695.
- [Google Scholar]
- Studying interactions between undecane and graphite surfaces by chemical force microscopy and molecular dynamics simulations. Fuel. 2020;269:117367
- [Google Scholar]
- Study on influence of coal surface functional groups on methane and carbon dioxide adsorption properties. Coal Sci. Technol.. 2021;49(6):145-151.
- [Google Scholar]
- Molecular simulation of the CH4/CO2/H2O adsorption onto the molecular structure of coal. Sci. China Earth Sci.. 2014;44(7):1418-1428.
- [Google Scholar]
- Distribution and quantum chemistry analysis of surface functional groups of lignite. Chinese J. Eng.. 2013;35(2):135-139.
- [Google Scholar]
- CO2 adsorption assisted CH4 desorption on carbon models of coal surface: a DFT study. Appl. Surf. Sci.. 2016;375:196-206.
- [Google Scholar]
- A density functional study on the efficiencies of 2-mercaptobenzoxazole and its derivatives as chelating agents in flotation processes. Colloid. Surface. A.. 2006;286(1–3):121-125.
- [Google Scholar]
- Molecular dynamics simulations of interaction between sub-bituminous coal and water. Mol. Simulat.. 2018;44(9):769-773.
- [Google Scholar]
- Adsorption of collectors on model surface of Wiser bituminous coal: a molecular dynamics simulation study. Miner. Eng.. 2015;79:31-39.
- [Google Scholar]
- New insight into surface wetting of coal with varying coalification degree: an experimental and molecular dynamics simulation study. Appl. Surf. Sci.. 2020;511:145610
- [Google Scholar]
- Effect of ionic surfactants on flotation of low-rank coal: a DFT calculation and MD simulation study. Mol. Phys.. 2022;120(24):e2140719.
- [Google Scholar]
- Monte Carlo and molecular dynamics simulations of CH4 molecules adsorption behavior in bituminous coal. Int. J. Low-Carbon Tec.. 2022;17:879-887.
- [Google Scholar]
- Density functional calculation of H2O/CO2/CH4 for oxygen-containing functional groups in coal molecules. ACS Omega. 2022;7:17330-17338.
- [Google Scholar]
- Molecular dynamics simulation on adsorption performances of phenol by oxygenic functional groups on adsorbent surface. J. China Coal Soc.. 2019;44(S1):296-304.
- [Google Scholar]
- Overview of polymer nanocomposites: computer simulation understanding of physical properties. Polymer. 2017;133:272-287.
- [Google Scholar]




