

Translate this page into:
Comparative spectroscopic and electrochemical study of N-1 or N-2-alkylated 4-nitro and 7-nitroindazoles
⁎Corresponding author. domenico.spinelli@unibo.it (Domenico Spinelli)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Our research groups are by long time involved in the study of the reactivity and the pharmacological activity of nitrogen-containing heterocyclic compounds: in this line we have now examined the behaviour of some substituted 4- and 7-nitroindazoles. Considering the fact that nitroreduction processes are often essential steps for the biological activity of nitro compounds and remembering that some nitroindazoles show interesting biological activities, we have collected nuclear magnetic resonance, electron spin resonance, and cyclic voltammetry data and carried out density functional theory computations on the above compounds thus obtaining an accurate picture of electronic distribution and reduction processes of the examined substrates as a function of their chemical structure. Looking also to our previous results obtained examining the behaviour of 5- and 6-nitroindazoles, we have confirmed the different general behaviour of 1- and 2-alkyl substituted nitroindazoles strictly related to the known different electronic distribution in these two classes of compounds. Interestingly, cyclic voltammetry data have confirmed the ability of N-1—H nitroindazoles to give rise to the formation of dimers, already observed by us studying 5- and 6-nitroindazoles.
Keywords
Nitroindazoles
Electron spin resonance measurements
NMR spectroscopy
Cyclic voltammetry
Density functional theory computations
Radical anions

- NMR
-
nuclear magnetic resonance
- ESR
-
electron spin resonance
- CV
-
cyclic voltammetry
- DFT
-
density functional theory
- QM
-
quantum mechanics
Abbreviations
1 Introduction
The indazole (1), a bicyclic heteroaromatic system containing a pyrazole ring condensed with a benzene one (aromaticity index 144, according with Bird’s classification) (Bird, 1992), has a planar structure, but its derivatives show different bond lengths distribution depending on their structures. As shown in Fig. 1, non-substituted and N-1 substituted indazoles (2) are characterized by an aromatic character of the benzene ring; in contrast, N-2 substituted indazoles (3) are characterized by an aromatic character of the pyrazole ring.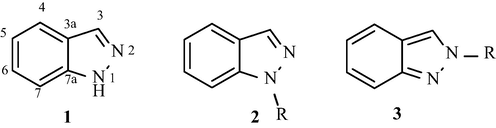
Indazole (1) and its N1 (2) and N2 (3) substituted derivatives.
Recently, we have examined some physicochemical properties of 5- and 6-nitroindazoles as well as of several 1- and 2-alkyl derivatives collecting data concerning their nuclear magnetic resonance (NMR), electron spin resonance (ESR) and cyclic voltammetry (CV) behaviour (Kouakou et al., 2015). The experimental data collected have been able to furnish a deep picture of the electronic distribution and reduction processes as a function of their structures. The interpretation of the obtained data has received strong support from density functional theory (DFT) computations.
Considering the interest for several nitroindazoles because of their variegate chemical reactivity and their potential pharmacological activities, we have now enlarged our study to some 4- and 7-nitroindazoles and their 1- or 2-alkyl derivatives. Good evidence exists that some electrochemical properties of nitro compounds, for example, their reduction potential, can be related to the pharmacological effects of these molecules (Olea-Azar et al., 2006; Pérez-Cruz et al., 2009).
We like to remember that 7-nitroindazole, by itself, is a selective inhibitor for neuronal nitric oxide synthase which is able to affect the oxidative stress leading to a possible decrease of some kinds of nerve damage (Babbedge et al., 1993; Castagnoli et al., 1997; Di Monte et al., 1997; Hantraye et al., 1996; Schulz et al., 1995).
Moreover, several indazole derivatives have been described as inhibitors of protein kinases: the interaction within the ATP-binding pocket often involves directly the indazole moiety (Gavara et al., 2010, 2011a, 2011b, 2013; Suchaud et al., 2013).
Interestingly, several indazole derivatives, whose biological activity has been found by rational approaches or by screenings, are characterized by nice selectivity profiles.
A very recent review (Giraud et al., 2014) has given a complete picture of synthetic methodologies and kinase inhibitory potency of indazole derivatives.
Looking at the chemical structure of 4- and 7-nitroindazoles, the peri-like position of the nitro group with respect to substituents at C-3 (hydrogen or chlorine) or to substituents at N-1 (hydrogen or alkyls), respectively, can affect their properties. As a matter of fact the nitro group at C-7 can interact with the proton at N-1 giving hydrogen bond interactions, or with alkyls at N-1 giving steric repulsive interactions, capable to move the nitro group out of the plane of the indazole ring. Similar steric interactions can occur between the nitro group at C-4 and the chlorine at C-3.
In the framework of our interests for the study of physicochemical and biological properties of heteroaromatic compounds (thiophenes, furans, azoles, and so on) (Bianchi et al., 2007; Carosati et al., 2015; Frenna et al., 2015; Ruccia et al., 1981; Spinelli et al., 1991), we have planned to examine the behaviour of 4- and 7-nitroindazoles. Data were collected on their physicochemical properties (NMR, EPR, CV) as a function of their structures for: (a) unsubstituted at N-1; (b) alkyl substituted at N-1; (c) alkyl substituted at N-2; (d) substituted at C-3 with a chlorine, as depicted in Fig. 2. All the obtained experimental results have received support by DFT calculations.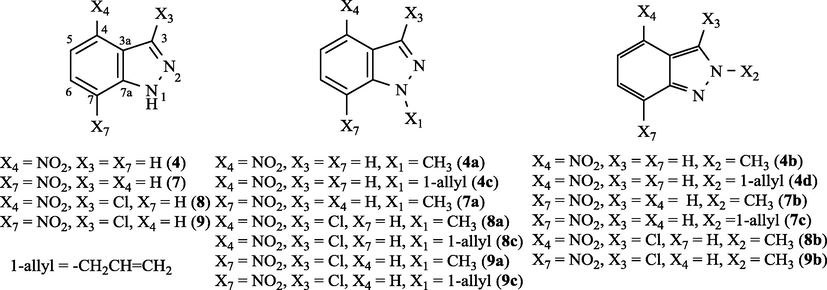
4-Nitroindazole, 7-nitroindazole, and their 3-chloro- and N-1 and N-2 alkyl derivatives.
2 Materials and methods
1H and 13C NMR spectra were recorded at 600 and 150.82 MHz, respectively, in CDCl3. Chemical shifts are reported in δ (ppm) with respect to the solvent (7.26 and 77.0 ppm for 1H and 13C NMR spectra, respectively). Coupling constants are reported in Hertz (Hz). Multiplicities of 13C NMR signals were assigned with distortionless enhancement by polarization transfer (DEPT) and, in some cases, by NOESY-1D and gradient heteronuclear single quantum coherence (g-HSQC) or heteronuclear multiple bond correlation (HMBC) experiments.
ESR spectra were recorded at room temperature using an ELEXYS E500 spectrometer equipped with an NMR Gaussmeter for the calibration of the magnetic field and a frequency counter for the determination of g-factors that were corrected against that of the perylene radical cation in concentrated sulphuric acid (g = 2.002583). The electrochemical cell was home-made and consisted of an ESR flat cell (Wilmad WG-810) equipped with a 25 × 5 × 0.2 mm platinum gauze (cathode), and a platinum wire (anode). The current was supplied and controlled by an AMEL 2051 general-purpose potentiostat (Alberti et al., 2000; Boga et al., 2012). In a typical experiment, the cell was filled with an acetonitrile (ACN) solution of the appropriate substrate (ca. 1 mM) containing tetrabutylammonium perchlorate (ca. 0.1 M) as supporting electrolyte. After thoroughly purging the solution with N2, spectra were recorded at different potential settings in the range from 0 to −3.0 V. An iterative least squares fitting procedure, based on the systematic application of the Monte Carlo method, was performed in order to obtain the experimental spectral parameters of the radical species (Franchi et al., 2014).
CV measurements were performed under nitrogen atmosphere at room temperature in ACN (from Chromanorm Prolabo, HPLC grade, freshly distilled over CaH2) solution containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as supporting electrolyte, with the studied compound at a concentration of about 2 mM. The CVs were recorded using a CHI 660c (CHInstruments, Austin, Texas) potentiostat controlled by a personal computer via CHInstruments original software, enabling the solution resistance compensation option. A single compartment, three electrode cell was employed. The working electrode was a Pt disc electrode (1.6 mm diameter, from BASi, West Lafayette, Indiana), and the counter electrode was a Pt wire. As reference was used a Ag+/Ag electrode, with a silver wire immersed in a solution of 0.1 M AgNO3 and 0.1 M TBAPF6 in distilled ACN, and an external junction containing 0.1 M TBAPF6 in distilled ACN hindering Ag+ release in the working solution. All potentials throughout the paper are reported versus this reference electrode. The stability of the potential of the reference electrode was checked daily by performing a CV in a solution of 2 mM ferrocene in the same electrolyte and solvent employed for all measurements. Prior to experiments, the surface of the Pt working electrode was polished first on a wet fine sandpaper, then with alumina slurry on a wet polishing cloth, thoroughly rinsing the electrode surface after each step. A further polishing step was carried out by electrochemical route, performing 250 cycles in 0.1 M H2SO4 solution between −0.35 e +1.35 V versus SCE (Amel Instruments, Milan, Italy), at 1.0 V s−1. Before performing each measurement in organic medium, the surface of the Pt electrode was rinsed with acetonitrile in order to remove any residual trace of water. For all the tested compounds the values of the cathodic peak potentials (Ep,c), recorded at 0.02 V s−1 have been reported. When a redox couple displayed a practically reversible behaviour the value of the formal potential (E°′) was also calculated from the half-sum of the cathodic and anodic peak potentials.
Indazole (1) and nitroindazoles (4 and 7) were commercially available, and general procedures for the synthesis of other nitroderivatives are described below.
2.1 General procedure for the synthesis of 3-chloro-nitroindazoles 8 and 9
A solution of nitroindazole 4 or 7 (6 mmol) and N-chlorosuccinimide (12 mmol) in ACN was heated at reflux for 1–4 h. The solvent was removed in vacuo and the residue was washed with water (2 × 100 mL), then filtered and dried. The residue was purified by flash chromatography on silica gel eluting with ethyl acetate/n-hexane (1/9). Compounds 8 (Benchidmi et al., 1979) and 9 (Bouissane et al., 2005) were recovered in 82% and 74% yields, respectively, and the related physicochemical data agree with those reported in the literature.
2.2 General procedure for the synthesis of N-alkyl-nitroindazoles 4a–d, 7a–c, 8a–c, and 9a–c
Potassium hydroxide (18 mmol) was added to a solution of nitroindazole 4 (or 7, 8, 9) (12 mmol) in acetone (40 mL). After 15 min of stirring, alkyl halide (CH3I and allyl bromide 12 mmol) was added dropwise and the reaction mixture was stirred at room temperature for 6–8 h. Upon disappearance of the starting material, as indicated by thin layer chromatography (TLC), the resulting mixture was evaporated. The crude material was dissolved with ethyl acetate (100 mL), washed with water and brine, and dried over MgSO4 and the solvent was evaporated in vacuo. The resulting residue was purified by column chromatography (ethyl acetate/n-hexane 1/4). Yields of compounds are reported below in parenthesis and the related physicochemical data agree with those reported in the literature. 1-Methyl-4-nitroindazole (4a) (Abbassi et al., 2012) and 2-methyl-4-nitroindazole (4b) (Abbassi et al., 2012) (64% and 30%); 1-methyl-7-nitroindazole (7a) (Benchidmi et al., 1979; Bouissane et al., 2005) and 2-methyl-7-nitroindazole (7b) (Benchidmi et al., 1979) (54% and 38%); 1-allyl-4-nitroindazole (4c) (Abbassi et al., 2012) and 2-allyl-4-nitroindazole (4d) (Abbassi et al., 2012) (58% and 37%); 2-allyl-7-nitroindazole (7c) (Kouakou et al., 2013) (65%); 3-chloro-1-methyl-4-nitroindazole (8a) (Benchidmi et al., 1979) (68%); 3-chloro-2-methyl-4-nitroindazole (8b) (Benchidmi et al., 1979) (25%) yield; 3-chloro-1-methyl-7-nitroindazole (9a) (Benchidmi et al., 1979; Bouissane et al., 2005) and 3-chloro-2-methyl-7-nitroindazole (9b) (Benchidmi et al., 1979) (57% and 34%); 1-allyl-3-chloro-4-nitroindazole (8c) (62%); and 1-allyl-3-chloro-7-nitroindazole (9c) (66%) are new compounds; their 13C and 1H NMR data are reported in Tables 2 and 4, respectively; other data are as follows: 8c: m.p.: 139–140 °C, EI-HRMS m/z: Calc for C10H8ClN3O2: 237.0305; found: 237.0303; 9c: m.p.: 39–40 °C, EI-HRMS m/z: Calc for C10H8ClN3O2: 237.0305; found: 237.0307.
2.3 Computational details
All density functional theory (DFT) computations here reported were performed with the Gaussian 09 series of programs (Frisch et al., 2009) using the M06-2X functional (Zhao and Truhlar, 2008) and the 6-311++G∗∗ basis set (Frisch et al., 1984). The geometries of the various critical points on the potential surface were fully optimized with the gradient method available in Gaussian 09, and harmonic vibrational frequencies were computed to evaluate the nature of all critical points. Since NMR and ESR experiments were carried out in deuterated chloroform and in ACN, respectively, the solvent effects were taken into account during optimization using the polarizable continuum model (PCM) adopting the integral equation formalism variant (IEFPCM) (Tomasi et al., 2005). A value of 4.7113 was employed for the dielectric constant ε of chloroform and of 35.688 for ACN. NMR shielding tensors were computed with the gauge-independent atomic orbital (GIAO) method (Wolinski et al., 1990). Isosurfaces spin densities were plotted using an isovalue of 0.004 ea0−3.
3 Results and discussion
NMR, ESR, CV, and DFT calculations have been carried out on 4-nitroindazole (4) and 7-nitroindazole (7) and their derivatives as shown in Fig. 2. The data obtained with these techniques are shown and discussed in separate sub-headings. The results and discussions may be presented individually or combined in a single section with short and informative headings.
3.1 Nuclear magnetic resonance (NMR) data
1H and 13C NMR spectra of 4- and 7-nitroindazoles (4 and 7) and of their derivatives have been recorded in CDCl3 at 25 °C. Experimental data have been collected in Tables 1, 2 and 4 and related DFT data are shown in Fig. 3 and Table 3.
Entry
Compound
C-3
(Δδ)C-4
(Δδ)C-5
(Δδ)C-6
(Δδ)C-7
(Δδ)C-7a
(Δδ)C-3a
(Δδ)
1
Indazole (1)
134.8
120.8
120.9
126.8
109.7
140.1c
123.2c
2
4-Nitroindazole (4)b
134.7
(−0.1)140.8
(+20)118.7
(−2.2)126.1
(−0.7)116.9
(+7.2)141.7
(+1.6)116.5c
(−6.7)
3
5-Nitroindazole (5)b
137.5
(+2.8)118.7
(−2.1)142.9
(+22)122.1
(−4.7)110.0
(+0.3)141.8
(+1.7)122.6c
(−0.6)
4
6-Nitroindazole (6)b
135.3
(+0.5)121.7
(+0.9)116.1
(−4.8)147.0
(+20.2)106.5
(−3.2)138.7
(−1.4)126.4c
(+3.2)
5
7-Nitroindazole (7)b
136.2
(+1.4)129.4
(+8.6)120.7
(−0.2)123.8
(−3.0)132.5
(+22.8)132.9
(−7.2)127.0c
(+3.8)
6
3-Chloro-4-nitroindazole (8)d
133.9
(−0.9)142.5
(+1.7)119.0
(+0.3)127.1
(+1.0)115.9
(+1.0)143.0
(+1.3)112.1
(−4.4)
7
3-Chloro-7-nitro indazole (9)d
137.4
(+1.2)128.1
(−1.3)121.2
(+0.5)125.0
(+1.2)132.5
(0.0)134.0
(+1.1)124.3
(−2.7)
Entry
Compound
C-3
(Δδ)C-4
(Δδ)C-5
(Δδ)C-6
(Δδ)C-7
(Δδ)C-7a
(Δδ)C-3a
(Δδ)Others
(Δδ)
1
4-NO2-1-Me-indazoleb (4a)
132.4
(−2.3)140.6
(−0.2)118.0
(−0.7)125.3
(−0.8)116.0
(−0.9)141.4
(−0.3)116.9
(+0.4)36.0
(NCH3)
2
4-NO2-2-Me-indazoleb (4b)
124.3
(−10.4)140.5
(−0.3)120.5
(−1.8)125.8
(−0.3)125.2
(+8.3)150.0
(+8.3)115.1
(−1.4)40.9
(NCH3)
3
4-NO2-1-Me-3-Cl-indazole (8a)c
130.7
(−1.7)142.2
(+1.6)118.5
(+0.5)126.3
(+1.0)115.2
(−0.8)142.7
(+1.3)112.1
(−4.8)36.3 (NCH3)
(+0.3)
4
4-NO2-2-Me-3-Cl-indazole (8b)c
120.6
(−3.7)141.4
(+0.9)121.4
(+0.9)124.5
(−1.3)125.4
(+0.2)148.9
(−1.1)110.3
(−4.8)38.9 (NCH3)
(−2.0)
5
4-NO2-1-allylindazole (4c)b
132.9
(−1.8)140.7
(−0.1)118.2
(−0.5)125.4
(−0.7)116.4
(−0.5)141.0
(−0.7)117.3
(+0.8)52.3 (NCH2)
118.5 (⚌CH2)
132.0 (⚌CH)
6
4-NO2-2-allylindazole (4d)b
124.1
(−10.6)140.4
(−0.4)120.3
(+1.6)124.0
(−2.1)125.8
(+8.9)149.6
(+7.9)114.7
(−1.8)56.4 (NCH2)
120.2 (⚌CH2)
131.3 (⚌CH)
7
4-NO2-1-allyl-3-Cl-indazolec (8c)
131.0
(−1.9)142.1
(+1.4)118.6
(+0.4)126.3
(+0.9)115.5
(−0.9)142.2
(+1.2)112.3
(−5.0)52.4NCH2) (+0.1)
119.0 (⚌CH2) (+0.5)
131.2 (⚌CH) (−0.1)
8
7-NO2-1-Me-indazole (7a)b
134.0
(−2.2)127.9
(−1.5)119.7
(−1.0)124.6
(+0.8)135.1
(+2.6)131.1
(−1.8)128.9
(+1.9)40.9 (NCH3)
9
7-NO2-2-Me-indazole (7b)b
126.2
(−10.0)128.8
(−0.6)120.0
(−0.7)125.0
(+1.2)137.1
(+4.6)140.6
(+7.7)125.6
(−1.4)41.0 (NCH3)
10
7-NO2-1-Me-3-Cl-indazole (9a)c
132.4
(−1.6)126.7
(−1.2)120.3
(+0.6)125.8
(+1.2)135.2
(+0.1)134.4
(+3.3)125.7
(−3.2)41.3 (NCH3)
(+0.4)
11
7-NO2-2-Me-3-Cl-indazole (9b)c
123.3
(−2.9)127.2
(−1.6)120.5
(+0.5)126.1
(+1.2)137.3
(+0.2)139.7
(−0.9)122.4
(−3.2)38.2 (NCH3)
(−2.8)
12
7-NO2-1-allyl-3-Cl-indazole (9c)c
131.7
126.7
120.6
125.8
135.6
135.0
126.0
55.8 (NCH2), 118.4 (⚌CH2)
132.4 (⚌CH)
13
7-NO2-2-allylindazole (7c)b
124.9
(−11.3)128.8
(−0.6)120.0
(−0.7)125.1
(+1.3)137.4
(+4.9)140.5
(+7.6)125.6
(−1.4)56.7 (NCH2), 120.7 (⚌CH2)
131.3 (⚌CH)
Bond
4
7
4a
7a
4b
7b
N2—C3
1.315
1.309
1.317
1.345
1.308
1.336
C3—C3a
1.427
1.431
1.424
1.394
1.424
1.398
C3a—C4
1.406
1.400
1.406
1.414
1.400
1.409
C4—C5
1.375
1.384
1.375
1.368
1.380
1.373
C5—C6
1.409
1.403
1.409
1.420
1.403
1.415
C6—C7
1.378
1.380
1.378
1.370
1.373
1.371
C7—C7a
1.403
1.404
1.403
1.415
1.411
1.424
C7a—C3a
1.401
1.412
1.412
1.428
1.417
1.433
C7a—N1
1.360
1.351
1.359
1.342
1.360
1.338
N1—N2
1.341
1.351
1.341
1.329
1.354
1.339
N-1—H (kcal mol−1)
N-2—H (kcal mol−1)
N-1—H (Boltzmann population ratio) (%)
N-2—H (Boltzmann population ratio) (%)
Indazole (1)
0
+4.7
99.96
0.04
4-Nitroindazole (4)
0
+3.1
99.47
0.53
7-Nitroindazole (7)
0
+8.2
100
0
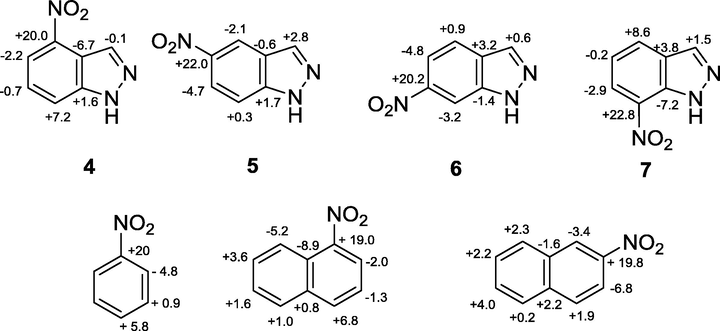
Top: Chemical shift differences between 13C NMR resonances herein found for 4–7 with respect to 1 Down: Chemical shift differences of nitrobenzene and 1-nitro and 2-nitronaphthalene with respect to benzene (Wehrli and Wirthlin, 1978) and naphthalene, respectively (Ernst, 1976).
3.1.1 13C NMR data of indazole (1) and N-1/N-2 unsubstituted 4- and 7-nitroindazoles 4–9
Data shown in Table 1 provide an overview on the trend of chemical shifts on changing the position of the nitro group along the series of substituted nitroindazoles 4–7 and on the effect of the substitution in position 3 by a chlorine atom.
In general, the assignment of the signals was made with the aid of DEPT experiments and, in some cases, of g-HSQC and/or HMBC experiments. When appropriate, they were also compared with literature data collected in DMSO-d6 (Bouchet et al., 1977). In all cases, the chemical shift of the signal belonging to the carbon atom bound to the nitro group is very close to that due to the C-7a carbon atom and, in principle, their attribution might be reversed: on the basis of the well-known signal broadening caused by the quadrupole nitrogen effect, we ascribed the C—NO2 resonance to the broader signal (Ernst, 1976; Ganapathy et al., 1981). Regarding 7-nitro-substituted derivatives, their C-7a signal is considerably more up-field shifted with respect to that of the other isomers. The comparison of the data related to nitroindazoles 4–9 with those of the indazole (1) was further made explicit by the values calculated as difference of the chemical shift (in ppm) shown in parentheses in Table 1 (entries 1–5) and evidenced, for compounds 4–7, in Fig. 3. Also chemical shift differences (in ppm) from the corresponding signal of the 3-unsubstituted parent compound for 3-chloro derivatives are shown in parentheses in Table 1 (entries 6–7).
The highest effect produced by the nitro substitution is on the chemical shift of the carbon atom directly bound to the nitro group that moves downfield the signal of about 20–23 ppm. This effect is in line with that observed for nitrobenzene (Δδ = +20.0 ppm from benzene) (Wehrli and Wirthlin, 1978), 1-nitro- and 2-nitronaphthalene (Δδ = +19.01 and +19.78 ppm, respectively, from naphthalene) (Ernst, 1976), as well as for the same series of compounds 4–7 in DMSO-d6 (Bouchet et al., 1977; Cohen-Fernandes et al., 1982).
Furthermore, the resonances of C-7a and C-3a gradually shift downfield going away from the nitro group. The data related to the 4-nitroindazole (4) reveal that the C-7 chemical shift, being related to a para-like and quinonoid carbon (Well’s nomenclature) (Wells et al., 1974) suffers downfield shift (+7.2 ppm) with respect to the resonance of the same carbon atom in the parent compound 1, as expected by considering that some canonical resonance structures of 4 have a positive charge on the C-7 carbon atom. Analogous considerations can be made for 7. In this case, the para-like carbon (C-4) suffers a downfield shift of +8.6 ppm. The shifts induced by the nitro-substituent at the para-like position for both, 4 and 7, are similar to the para-effect observed for nitrobenzene (Wehrli and Wirthlin, 1978) and 1-nitronaphthalene in acetone-d6 (Ernst, 1976).
A comparison with the effect on the para-like position, C-7a and C-3a for 5 and 6, respectively, shows that in these cases, this is smaller with respect to that found in 4 and 7, thus indicating that the localization of the positive charge at the junction carbons in 6 and 7 is not very important, since in this case the π-electron sextet of the heterocyclic fused ring is not retained in the corresponding canonical structures. This consideration agrees with the parallel behaviour observed for the C-10 resonance, in a para-like position, of the 2-nitronaphthalene with respect to naphthalene (see Fig. 3).
About the ortho-effects, the observed trends agree with the localization of the positive charge in the related resonance structures. Actually, a comparison of the values of the two ortho effects in each compound, indicates that the charge is withdrawn more from C-5 than C-3a in 4 and more from C-6 than C-7a in 7, a trend similar to that found for 1-nitronaphthalene, whereas the ortho-effects shown by 5 and 6 resemble those reported for 2-nitronaphthalene. Also for the compounds here investigated, as well as for nitrobenzene and 1-nitro- and 2-nitronaphthalene, a downfield shift for the resonance of the ortho-carbon atom was expected on the only basis of the mesomeric effect, but in all cases the resulting chemical shift differences are negative, and this indicates that other factors may be responsible for the upfield shifts. Tentative explanations in this regard, invoking different effects, such as the length of the C—C bond (Bouchet et al., 1977), twisting of nitro group out of the plane of the aromatic system, or electric field effect of the nitro group have been hypothesized by several authors (Günther, 1973; Stothers, 1972). In line with what reported for nitrobenzene and nitronaphthalenes, the meta-like position of compounds 4–7 undergoes the smallest influence from the nitro substituent, replicating the situation observed in 1- and 2-nitronaphthalenes.
The quantum mechanics (QM) calculations are able to reproduce quantitatively the chemical shift variations and the ortho, meta, and para effects observed by NMR experiments for compounds 4 and 7 (Fig. 4).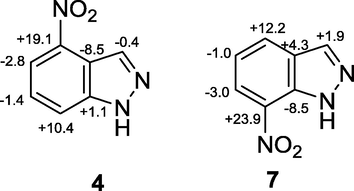
Calculated chemical shift differences between 13C NMR resonances of compounds 4 and 7 with respect to 1.
Any simple explanation based on single parameters such as (i) variation of the C—C bonds length (the bond length are very similar for the two compounds), (ii) net charge variation (ESP charges do not strictly correlate with the observed chemical shift variations), (iii) out of the plane rotation of the nitro group (the nitro group remains planar for both compounds) or (iv) electric field effect due the nitro group (the effect of the nitro substitution on the MEP potential of the carbon atoms of 4 and 7 is always descreening if compared with the indazole) can reproduce efficiently the shielding/deshielding effects caused by the introduction of the nitro group in 4 and 7, showing that the variations in chemical shifts are due to a complex interplay of the different inductive and mesomeric effects. Concerning the effect of the introduction of a chlorine atom in position 3 of nitroindazoles 4 and 7, as already observed for 5 and 6 (Kouakou et al., 2015), the main difference is in the resonance of C-3a that undergoes an upfield shift of 4.4 ppm (entry 6) and 2.7 ppm (entry 7) for 4 and 7, respectively, due to the presence of the halogen atom.
3.1.2 13C NMR data of 1-alkyl and 2-alkyl substituted 4-and 7-nitroindazoles and their 3-chloro-derivatives
In Table 2 13C NMR data of N-1 and N-2 alkyl substituted 4-nitroindazoles 4a–d and 7a–c and of 3-chloro derivatives 8a–c and 9a–c are collected. For N-1 and N-2 alkyl substituted compounds, the difference of chemical shift (in ppm) from the corresponding signal of the unsubstituted parent compound is shown in parenthesis whereas in the case of 3-chloro derivatives, the difference of chemical shift from the corresponding signal of the 3-H N1 or N2 alkyl substituted parent compound is reported in parenthesis.
From an overview of the data reported in Table 2 it emerges that 13C NMR spectra of 1-alkylindazoles are almost superimposable with those of the corresponding N—H parents 4 and 7, while the spectra of 2-alkylindazoles are different from those of 1-alkylsubstituted. An up-field shift in the range of 8–10 ppm of the C-3 resonance and a downfield shift of the C-7a resonance (∼8.0 ppm for 4- and 7-nitro derivatives, and ∼5 ppm for 3-chloro substituted) were observed for all compounds on going from N1 to N2 alkylated derivatives (compare entries 1–2, 3–4, 5–6, 8–9, and 10–11). The C-3 upfield shift is more pronounced on going from 1-alkyl to 2-alkyl substituted derivatives, probably due to a greater C⚌C double bond character between C-3 and C-3a in N-2 substituted compounds with respect to the greater C⚌N character of the C-3 carbon in N-1 substituted compounds. This situation is well in line with the different nature of the C-3/C-3a bond in the two classes of compounds (see Table 3): it goes from a single bond character in unsubstituted and 1-substituted compounds (1.427 Å in 4, 1.431 Å in 7, 1.424 Å in 4a and 7a) to a double bond character in 2-substituted compounds (1.394 Å in 4b and 1.398 Å in 7b). Looking at C-3/N2 bond an analogous, but opposite situation in 1-substituted and 2-substituted compounds has been observed: a greater double bond character in unsubstituted and 1-substituted compounds (1.315 Å in 4, 1,309 Å in 7, 1.317 Å in 4a and 1,308 Å in 7a) with respect to 4b and 7b (1.345 Å in 4b and 1.336 Å in 7b).
Moreover, a down-field chemical shift of ∼10 ppm was observed for the C-7 resonance of 2-alkyl substituted 4-nitroindazoles (compare entries 1–2, 3–4, and 5–6), while the substitution of the methyl with the allyl group did not cause sensible variation of chemical shifts along the series of N-1 (compare entries 1, 3, 5, 7, 8, 10, 12) and N-2 substituted derivatives (entries 2, 4, 6, 9, 11, 13), with the exception of the resonances of C-3 and C-3a in 3-chloro substituted compounds. As already observed for 5- and 6-nitroderivatives (Kouakou et al., 2015), the methyl signals of methyl substituted 4-nitroindazoles shift downfield changing the position of the methyl substituent from the N-1 to N-2 (Table 2, compare entry 1 with 2, entry 3 with 4), while for 7-nitro derivatives the resonances of the methyl group are very close (entries 8, 9, and 10), likely due to the electron-withdrawing effect of the nitro-substituent on C-7.
In the whole, the 13C NMR chemical shift trend observed in CDCl3 for the compounds here studied is comparable with that previously reported in other solvents and agrees with a large, if not unique, prevalence of a N-1—H structure, in which the homocyclic ring has an aromatic character.
DFT calculations confirmed the NMR results, in fact if we compare the energy differences between N-1—H and N-2—H tautomers (see Table 4) of the unsubstituted indazole and of the 4- and 7-nitro derivatives (compounds 4 and 7) the N-1—H structure is practically the only populated (from 99.47% for the 4-nitroindazole to the 100% of the 7-nitroindazole). In the 7-nitroindazole the population of the N-1—H is 100% because an additional factor contributes to the stability of N-1—H tautomer, and the formation of an intra-molecular hydrogen bond occurs between the nitro group in position 7 and the hydrogen atom in position 1, that is not present in the N-2—H tautomer.
Furthermore, the chemical shift resonances of N-unsubstituted indazoles are similar to those of 1-alkyl substituted derivatives, again indicating for the latter compounds a structure with the homocyclic ring aromatic in character. The significant differences observed between 2-substituted and 1-substituted nitroindazoles can be explained by considering that in 2-alkyl substituted derivatives the six-membered carbocyclic ring has a strong dienic character.
3.1.3 1H NMR data of indazole (1), 4-nitroindazole (4), 7-nitroindazole (7) and their derivatives
Table 5 reports 1H NMR data in CDCl3 of indazole (1) and its nitro derivatives herein studied, and includes, for comparison, data of 5-nitroindazole (5) and 6-nitroindazole (6).
Entry
Comp.
H-3
H-4
H-5
H-6
H-7
Others
1
1
8.14 d J = 0.9
7.79 ddd J = 8.2, 0.9, 0.9
7.19 ddd J = 8.3, 8.2, 0.9
7.40 ddd J = 8.3, 8.2, 0.9
7.52 dddd J = 8.3, 1.8, 0.9, 0.9
10.92 (NH)b
2
4
8.74
NO2
8.19 d J = 7.6z
7.55 d J = 8.1, 8.1
7.89 d J = 8.3
10.62 (NH)b
3
4a
8.60
NO2
8.10 d J = 7.6
7.49 tc J = 8.3
7.76 d J = 8.3
4.16 (CH3)
4
4b
8.54
NO2
8.17 d J = 7.6
7.39 dd J = 8.4, 7.6
8.06 d J = 8.4
4.31 (CH3)
5
4c
8.62
NO2
8.14 d J = 8.2
7.49 tc J = 8.2
7.77 dd J = 8.2
6.10–6.00 (m, CH)
5.28 (d, 1H, J = 10.4)
5.16 (d, 1H, J = 17.0)
5.11 (dt, 2H, J = 5.7, 1.5)
6
4d
8.49
NO2
8.06d J = 7.7
7.30 tc J = 8.7
7.99 d J = 7.8
6.14–6.05 (m, CH)
5.34 (d, 1H, J = 10.4)
5.33 (d, 1H, J = 17.0)
5.05 (dt, 2H, J = 6.4, 1.2)
7
5
8.30
8.78
NO2
8.32 dd J = 9.1, 1.7
7.59 d J = 9.1
10.39 (NH)b
8
6
8.24
7.90 d J = 8.7
8.07 d J = 8.7
NO2
8.49
10.60 (NH)b
9
7
8.30
8.17 d J = 7.9
7.34 dd J = 7.9, 7.5
8.37 d J = 7.5
NO2
11.53 (NH)b
10
7a
8.13
8.01 dd J = 7.7, 0.8
7.21 tc J = 7.8
8.10 dd J = 7.8, 0.8
NO2
4.24 (CH3)
11
7b
8.18
8.01 d J = 8.0
7.15 ddc J = 7.6, 8.0
8.28 d J = 7.6
NO2
4.32 (CH3)
12
7c
8.21
8.02 dd J = 8.2, 0.7
7.16 dd J = 7.5, 0.7
8.30c dd J = 7.5, 8.2
NO2
6.18–6.10 (m, CH)
5.41–5.39 (m, 1H)
5.37 (dq, J = 10.2, 1.3)
5.16 (dt, 2H, J = 6.5, 1.3)
13
8
Cl
NO2
7.91 d J = 7.3
7.55 tc J = 7.8
7.78 d J = 8.2
14
8a
Cl
NO2
7.83 d J = 7.4
7.49 tc J = 8.1
7.67 d J = 8.6
4.10 (CH3)
15
8b
Cl
NO2
8.00 dd J = 7.7, 0.5
7.96 dd J = 8.5, 0.5
7.35 dd J = 8.5, 7.7
4.24 (CH3)
16
8c
Cl
NO2
7.81 d J = 7.6
7.47 d J = 8.5
7.68 dd J = 8.5, 7.6
6.03–5.94 m (CH)
5.27 (d, 1H, J = 10.3)
5.18 (d, 1H, J = 17.1)
5.02 dt (d, 2H, J = 5.9, 1.5)
17
9
Cl
8.10 d J = 7.8
7.40 tc J = 7.8
8.41 d J = 7.9
NO2
11.32 (NH)
18
9a
Cl
7.95 d J = 7.7
7.27 tc J = 7.7
8.16 d J = 7.7
NO2
4.19 (CH3)
19
9b
Cl
7.93 dd J = 8.2, 0.9
7.22 dd J = 8.2, 7.6
8.35 dd J = 8.2, 7.6
NO2
4.28 (CH3)
20
9c
Cl
8.01 dd J = 7.9, 0.9
7.31 tc J = 7.8
8.16 dd J = 7.8, 0.9
NO2
5.91–5.82 m (CH)
5.23 (dt, 2H, J = 5.6, 1.5)
5.16 (dq, 1H, J = 10.4, 0.9)
4.98 (dq, 1H, J = 17.2, 0.9)
A comparison of the H-3 chemical shift of compounds 4–9 evidences, as expected, that the presence of the electron-withdrawing substituent on the homocyclic ring shifts downfield the H-3 resonance of an extent dependent on its distance (through the sigma bonds) from the nitro group, as can be seen, for example, for 4 or 7 with respect to 5 and 6 (see entries 2, 7, 8, and 9).
Moreover, the protons belonging to the benzene ring in meta position with respect to the nitro substituent fall at higher fields with respect to the others, as expected by considering the lower influence of the electronic effects of the nitro group in this position. This trend appears quite general.
Furthermore, the presence of the methyl substituent in position 2 causes, see 13C NMR data, a small downfield shift of the methyl signal with respect to that of 1-methyl substituted indazoles. As expected, the substitution of the methyl substituent on N-1 or N-2 with an allyl group does not cause relevant variations of chemical shifts. Similar considerations can be made for 3-chloroderivatives with respect to 3-H derivatives.
A particular behaviour is observed in the instance of 1-allyl-3-chloro-7-nitroindazole (9c). Actually, in all other N-1 and N-2 allyl derivatives (4c, 4d, 7c, 8c, see entries 5, 6, 12, and 16, respectively) the protons belonging to the methylene group bound to the N-1 or N-2 nitrogen atom fall at the highest field between the allyl signals; on the contrary, in 9c, the signals of the terminal methylene protons shift up-field with respect to those of the N—CH2 signal.
3.2 Electrochemical Electron Spin Resonance (E-ESR) data
ESR spectra of radical anions were recorded at room temperature in ACN (0.1 M in tetrabutylammonium perchlorate, as supporting electrolyte). Single electron reduction of nitroindazole derivatives was obtained by applying the corresponding reduction potential (E01) to the ACN solution inside a quartz flat cell positioned inside the ESR cavity. In all cases investigated the electron uptake leads to the formation of strong ESR signals.
3.2.1 ESR data of 4-nitroindazoles derivatives 4, 4a, 4c; 8, 8a, 8c
The ESR spectrum of the unsubstituted derivative 4 consisted of seven different hyperfine splitting coupling constants (hfsc, see Table 6), three of them being attributed to the coupling between the unpaired electron with three magnetically non-equivalent nitrogen atoms (1:1:1 triplet structure for each of them). Based on previous work (Kouakou et al., 2015), the larger nitrogen coupling (aN4 = 10.05 G) was attributed to the nitrogen at the nitro group while the much smaller nitrogen couplings to the N-1 and N-2 atoms. The remaining a-values are related to coupling with four hydrogen atoms (doublet structure for each of them). The larger couplings are with the H-atoms at the C-5 and C-7 positions (aH = 4.80 and 4.56 G). This behaviour indicates a greater electron delocalization at the ortho and para positions (see Fig. 5b) with respect to the nitro group than in the other positions, as expected on the basis of the related resonance structures. The two smaller H-couplings were attributed to H-3 and H-6 atoms, while coupling with H-1 atom was not detected in the ESR spectrum indicating a very low spin density on the 1-position.
Precursor
X1
X3
X4
X7
a(N)/G
a(H)/G
4
H
H
NO2
H
10.05, 0.71, 0.25
4.96, 4.60, 1.35, 0.41
8
H
Cl
NO2
H
9.54
5.12, 4.74a
4a
Methyl
H
NO2
H
8.02, 1.19
5.05, 4.65, 1.28, 0.47
8a
Methyl
Cl
NO2
H
7.34, 1.16, 0.26
5.22, 4.70, 1.21
4c
Allyl
H
NO2
H
8.39, 1.14
5.00, 4.70, 1.28, 0.44
8c
Allyl
Cl
NO2
H
7.33, 1.16, 0.29
5.17, 4.66, 1.18
7
H
H
H
NO2
8.98, 0.38
5.41, 5.26, 1.43, 0.73
9
H
Cl
H
NO2
8.37, 0.46
5.57, 5.55, 1.48
7a
Methyl
H
H
NO2
9.42
5.00, 4.95a
9a
Methyl
Cl
H
NO2
8.79, 0.30
5.29, 5.28, 1.41
9c
Allyl
Cl
H
NO2
8.75, 0.29
5.30, 5.33, 1.46
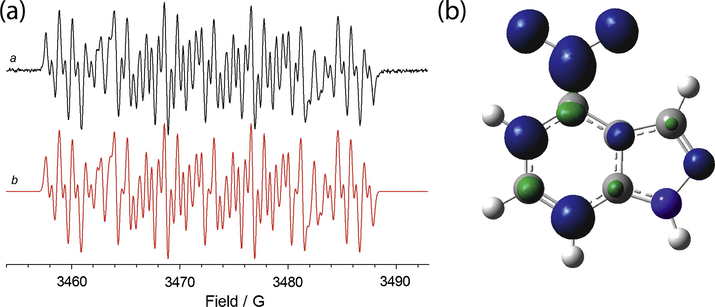
(a) ESR spectrum of the radical species electrogenerated from 4a (1 mM) in 0.1 M Bu4NClO4—CH3CN. Eappl = E01 (a). The corresponding theoretical simulation is reported in red (b); (b) isosurfaces spin densities for the radical anion of 4.
The introduction of a chlorine atom in position 3 (compound 8) leads to a small decrease of the aN4 value in the ESR spectrum of the corresponding radical anion (aN4 = 9.54 G). On the contrary, the larger hydrogen coupling does not change significantly after the addition of a chlorine atom in position 3; this observation leads to the conclusion that the presence of a chlorine atom has a little effect on spin density distribution.
The introduction of an alkyl on N-1 results in a significant decrease of coupling constant between the unpaired electron and N atom of the nitro group compared to the analogous unsubstituted derivatives (compounds 4a and 4c). Negligible differences in the hydrogen coupling constants were instead observed with respect to the unsubstituted compound 4 (as an example see Fig. 5a).
Finally, the simultaneous substitution of the C-3 and of the N-1 atoms with a chlorine atom and an alkyl group (compounds 8a and 8c, respectively), further emphasizes the decrease of the N-5 coupling above described.
3.2.2 ESR data of 4-nitroindazoles derivatives 4b–4d; 8b
The substitution at N-2 with an alkyl causes a small but detectable redistribution of the spin density in the corresponding radical anions. The higher effect is observed for the coupling between the unpaired electron and N-5 which decreases more than 1 G both with the methyl and the allyl substituent compared to the unsubstituted derivative 4 (4b and 4d, see Table 7).
Precursor
X2
X3
X4
X7
a(N)
a(H)
4b
Methyl
H
NO2
H
8.44, 1.53, 0.83
5.32, 4.83, 1.41, 0.83 (3H)
8b
Methyl
Cl
NO2
H
7.79, 1.10, 0.27
5.18, 4.76, 1.27
4d
Allyl
H
NO2
H
8.92, 1.29, 0.79
5.82, 4.84, 1.00, 0.71 (2H)
7b
Methyl
H
H
NO2
7.20
6.42, 6.60a
9b
Methyl
Cl
H
NO2
7.09, 1.35, 0.60
6.31, 5.57, 0.79, 0.93 (3H)
7c
Allyl
H
H
NO2
7.10
6.42, 6.55a
Once again, the introduction of a chlorine atom in position 3 (compound 8b) leads to a decrease of the nitrogen and hydrogen couplings (as an example see Fig. 6a).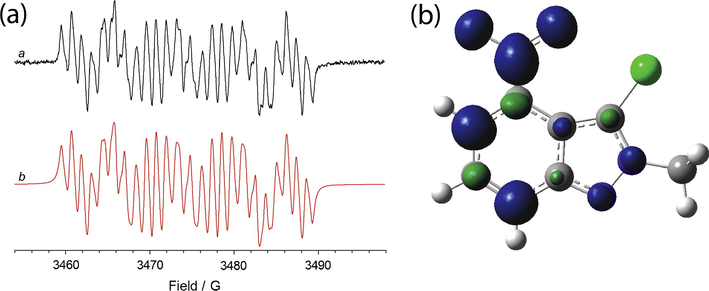
(a) ESR spectrum of the radical species electrogenerated from 8b (1 mM) in 0.1 M Bu4NClO4—CH3CN. Eappl = E01 (a). The corresponding theoretical simulation is reported in red (b); (b) isosurfaces spin densities for the radical anion of 8b.
3.2.3 ESR data of 7-nitroindazoles derivatives (7, 7a, 9, 9a, 9c)
7-Nitroindazole (7) radical anion is characterized by an ESR spectrum showing six different hyperfine coupling constants, two of them being attributed to the coupling between the unpaired electron with the three magnetically non-equivalent nitrogen atoms (see Fig. 7a). Once again, the larger nitrogen coupling (aN7 = 12.24 G) was attributed to the nitrogen at the nitro group, while the smaller coupling to the N-1 atom. The coupling with N-2 is smaller than the line width and cannot be detected. The remaining a-values are related to couplings with four hydrogen atoms. Also in this case, the larger couplings are those with the H-atom at the C-4 and C-6 (aH = 5.41 and 5.26 G) positions, i.e. at the ortho and para position with respect to the nitro group (see Fig. 7b). Comparison with the spectra obtained for the derivative substituted at the 3-position with the chlorine atom (9) suggests that 0.73 G coupling constant is due to the coupling of the unpaired electron with H-3.
(a) ESR spectrum of the radical species electrogenerated from 7 (1 mM) in 0.1 M Bu4NClO4—CH3CN. Eappl = E01 (a). The corresponding theoretical simulation is reported in red (b); (b) isosurfaces spin densities for the radical anion of 7.
In the presence of a methyl on N-1 (compound 7a) an increase of the coupling constant between the unpaired electron and N-7 is observed. This behaviour is the contrary of what observed in the 4-nitro derivatives and in the previous investigated 5- and 6-nitro derivatives (Kouakou et al., 2015) where a decrease of the coupling with the nitrogen at the nitro group was instead observed after the substitution at N-1 atom. A similar trend was observed also in the presence of a chlorine atom at the C-3 position (compounds 9a and 9c). A possible explanation of this different behaviour can be related to the loss of an intra-molecular hydrogen bonding which occurs between the nitro group in position 7 and the hydrogen atom in position 1 in the unsubstituted 7-nitro indazole (Fig. 8). Unfortunately, both N-1 and N-2 couplings were instead too small to be detected.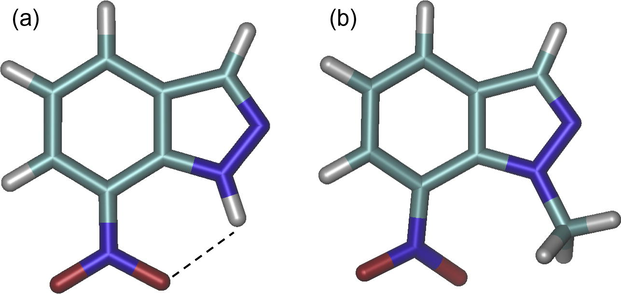
Optimized structure of the compounds 7 (a) and 7a (b).
3.2.4 ESR data of 7-nitroindazoles derivatives (7b–7c; 9b)
The substitution at N-2 with an alkyl causes effects similar to those observed with 4-nitro substituted derivatives in the spin density distribution: the N-7 coupling is smaller compared to the parent compound 7, while a significant increase of the aH4 value was observed. Among the N-2 substituted compounds, it was not possible to determine the smaller hydrogen coupling constant values with derivative 7c. Also in this case the introduction of a chlorine atom in position 3 (compound 9b, see Fig. 9) gives rise to little effects on spin density distribution.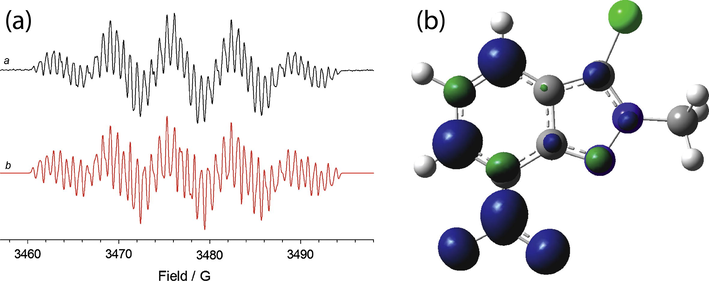
(a) ESR spectrum of the radical species electrogenerated from 9b (1 mM) in 0.1 M Bu4NClO4—CH3CN. Eappl = E01 (a). The corresponding theoretical simulation is reported in red (b); (b) isosurfaces spin densities for the radical anion of 9b.
3.3 Electrochemical Voltammetric (CV) data
None of the tested compounds showed an electrochemical behaviour which can be named as truly reversible if the criteria of the cathodic to anodic peak current ratio and the peak potential difference are taken into account to evaluate reversibility.
The majority of the compounds displayed an electrochemical behaviour consisting of one forward cathodic peak and one backward anodic one, with peak currents of almost equal intensities and peak potential differences ranging between 0.070 and 0.200 V. We decided to tag that behaviour as ‘practically reversible’. Such compounds were those bearing alkyl substituents on N-1 and N-2. The voltammetric peaks can be ascribed to the reduction and subsequent re-oxidation of the nitro group.
As an example, Fig. 10 reports the CV recorded for 2-methyl-7-nitroindazole (compound 7b).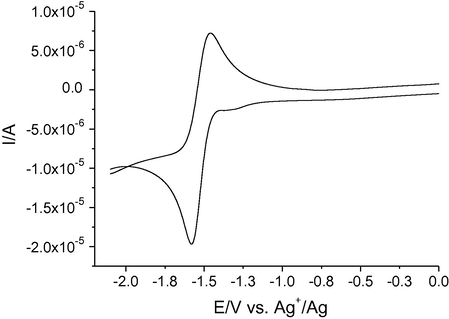
CV recorded at bare Pt electrode in ACN solution containing 0.1 M TBAPF6 and 2 mM compound 7b, at 0.050 V s−1. The potential window has been shrunk to 0 V, since the voltammograms showed no significant features at potentials more anodic than 0 V.
We found that the characterization of this kind of compounds showed a diffusion-limited electrochemical behaviour, since the intensities of the cathodic and anodic peak currents relevant to CVs recorded at different scan rates (0.020, 0.050, 0.100, 0.200, 0.500 V s−1) were linearly dependent on the square root of the scan rate. Fig. 11 shows, as an example, this trend observed for 2-methyl-4-nitroindazole (compound 4b).
Dependence of the cathodic (triangles) and anodic (circles) peak currents on the square root of the scan rate, relevant to CV characterizations of compound 4b carried out at five different scan rates.
Differently, the compounds containing a hydrogen atom at N-1 (‘unsubstituted’ nitroindazoles) showed two systems of peaks. The former, less cathodic, had an irreversible behaviour, while the latter, more cathodic, displayed a partially reversible behaviour.
As a typical example relevant to an ‘unsubstituted’-type indazole, Fig. 12 reports the CV recorded for 4-nitroindazole (4).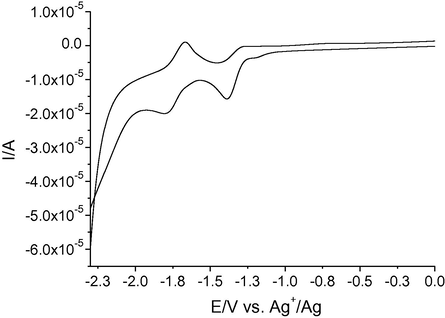
CV recorded at bare Pt electrode in ACN solution containing 0.1 M TBAPF6 and 2 mM compound 4. The potential window has been shrunk to 0 V, since the voltammograms showed no significant features at potentials more anodic than 0 V.
This particular kind of voltammogram had already been observed by us in the voltammetric characterization of 5- and 6-nitroindazoles. We had attributed that feature to the possible establishment of a rapid equilibrium between species, resulting either from the shift of a proton from N-1 to N-2 of the ring (tautomeric equilibrium) or more likely from an inter-molecular chemical interaction between the indazoles themselves (dimerization equilibrium). In the first case, it would be assumed that the hydrogen at N-1 atom is mobile, while in the second case that the hydrogen atom would be blocked by a hydrogen bond established with either the nitro group or with a nitrogen atom of another nitroindazole molecule. The fact that the presence of a hydrogen atom bound to N-1 of the ring affects the electrochemical response so much is confirmed by the voltammetric characterization of compound 7, which is the only ‘unsubstituted’ nitroindazole, displaying a single peak system, like the ‘substituted’ ones. For this molecule, the proximity of the nitro group in position 7 to the hydrogen atom on N-1 is likely to permit the formation of an intra-molecular hydrogen bonding (see Fig. 8) which makes the hydrogen unavailable for other possible interactions. The stability of this hydrogen bond was also previously discussed (see Table 4 and Fig. 8). The formation of this intramolecular hydrogen bond would produce, for the nitro group in compound 7, an electrochemical ‘activation’ facilitating its reduction. As a consequence the reduction of the nitro group of compound 7 occurs at a potential less cathodic than those relevant to the nitro groups of compounds 7a, 9a and 9c (all of them substituted with an alkyl on N-1).
The characteristics of the voltammetric characterizations of all compounds are summarized in Table 8, which reports, for each cathodic peak, the potential value (Ep,c) recorded at a scan rate of 0.020 V s−1. For every peak system displaying partially reversible behaviour, the E°′ value (calculated as the half-sum of the cathodic and anodic peak potentials) was also added.
Compound
Electrochemical behaviour
4-Nitroindazole (4)
I peak system: irreversible, Ep,c = −1.372 V
II peak system: practically reversible, Ep,c = −1.769 V, E°′ = −1.723 V
4-Nitro-2-methylindazole (4b)
Practically reversible, Ep,c = −1.459 V, E°′ = −1.408 V
4-Nitro-1-methylindazole (4a)
Practically reversible, Ep,c = −1.418 V, E°′ = −1.384 V
4-Nitro-2-allylindazole (4d)
Practically reversible, Ep,c = −1.438 V, E°′ = −1.391 V
4-Nitro-1-allylindazole (4c)
Practically reversible, Ep,c = −1.438 V, E°′ = −1.389 V
4-Nitro-3-chloroindazole (8)
I peak system: irreversible, Ep,c = −1.385 V
II peak system: practically reversible, Ep,c = −1.791 V, E°′ = −1.759 V
4-Nitro-2-methyl-3-chloroindazole (8b)
Practically reversible, Ep,c = −1.489 V, E°′ = −1.421 V
4-Nitro-1-methyl-3-chloroindazole (8a)
Practically reversible, Ep,c = −1.449 V, E°′ = −1.406 V
4-Nitro-1-allyl-3-chloroindazole (8c)
Practically reversible, Ep,c = −1.441 V, E°′ = −1.392 V
7-Nitroindazole (7)
Practically reversible, Ep,c = −1.423 V, E°′ = −1.378 V
7-Nitro-2-methylindazole (7b)
Practically reversible, Ep,c = −1.587 V, E°′ = −1.516 V
7-Nitro-1-methylindazole (7a)
Practically reversible, Ep,c = −1.507 V, E°′ = −1.461 V
7-Nitro-2-allylindazole (7c)
Practically reversible, Ep,c = −1.541 V, E°′ = −1.484 V
7-Nitro-3-chloroindazole (9)
I peak system: irreversible, Ep,c = −1.326 V
II peak system: practically reversible, Ep,c = −2.129 V, E°′ = −2.028 V
7-Nitro-2-methyl-3-chloroindazole (9b)
Practically reversible, Ep,c = −1.571 V, E°′ = −1.436 V
7-Nitro-1-methyl-3-chloroindazole (9a)
Practically reversible, Ep,c = −1.515 V, E°′ = −1.401 V
7-Nitro-1-allyl-3-chloroindazole (9c)
Practically reversible, Ep,c = −1.449 V, E°′ = −1.402 V
Looking at the cathodic potentials relevant to the reduction of the nitro group in the ‘substituted’-type indazoles, some observations can be made. The compounds for which the reduction appears more difficult are 7b and 9b, which are both substituted with a methyl group on N-2. This behaviour can be included into a more general trend for which the compounds bearing a methyl group and those having a substituent at N-2 display more negative reduction potentials with respect to those bearing an allyl group and those substituted at N-1. Exceptions to the first trend (methyl versus allyl) are compound 4a with respect to 4d and 4c; exceptions to the second trend (position 2 versus 1) are compounds 4d and 4c which display equal reduction potential values.
Differently from what expected, taking into account the electron-withdrawing inductive effect typical of halogen substituents, no significant differences could be found among the Ep,c values depending on the presence or not of a chlorine substituent on C-3.
4 Conclusions
Because of the interest for their chemical behaviour and for some promising pharmacological properties we have investigated the NMR, ESR, and CV behaviour of several derivatives of 4- and 7-nitroindazole.
The 13C NMR data have been confirmed for indazoles unsubstituted at nitrogen (4 and 7) a N-1—H structure, with the homocyclic ring having an aromatic character. DFT calculations have confirmed a significant energy difference between N-1—H and N-2—H tautomers for these compounds. Moreover, the CV data for indazoles unsubstituted at nitrogen (4 and 7) have shown the existence of a fast chemical equilibrium (two systems of peaks, time and concentration dependent, are observed) according to a dimer formation, which makes the two nitro groups no more electrochemically equivalent. In the whole the experimental data have given us a nice picture of the chemical behaviour and electronic distribution of all the studied nitroindazoles: the experimental data appear strongly supported by the results of DFT computations.
Acknowledgements
Work was supported by Alma Mater Studiorum – University of Bologna (RFO funds) and by University of Sultan Moulay Slimane, Beni-Mellal, Morocco for financial support.
References
- Synthesis, antiproliferative and apoptotic activities of N-(6(4)-indazolyl)-benzenesulfonamide derivatives as potential anticancer agents. Eur. J. Med. Chem.. 2012;57:240-249.
- [Google Scholar]
- J. Chem. Soc., Perkin Trans.. 2000;2:1908-1913.
- Inhibition of rat cerebellar nitric oxide synthase by 7-nitro indazole and related substituted indazoles. Br. J. Pharmacol.. 1993;110:225-228.
- [Google Scholar]
- Synthèse et réactivité de nitro indazoles. J. Heterocycl. Chem.. 1979;16:1599-1603.
- [Google Scholar]
- Oxidative nucleophilic substitution of hydrogen versus ring-opening in the reaction of 4-R-2-nitrothiophenes with amines. The crucial effect of 4-alkyl groups. J. Org. Chem.. 2007;72:5771-5777.
- [Google Scholar]
- Electron reduction processes of nitrothiophenes. A systematic approach by DFT computations, cyclic voltammetry and E-ESR spectroscopy. Org. Biomol. Chem.. 2012;10:7986-7995.
- [Google Scholar]
- N.m.r. studies in the heterocyclic series: XVII—carbon-13 spectra of indazole derivatives. Org. Magn. Res.. 1977;9:716-718.
- [Google Scholar]
- New and efficient synthesis of bi- and trisubstituted indazoles. Tetrahedron. 2005;61:8218-8225.
- [Google Scholar]
- Synthesis and L-type calcium channel blocking activity of new chiral oxadiazolothiazinones. Eur. J. Med. Chem.. 2015;92:481-489.
- [Google Scholar]
- The neuronal nitric oxide synthase inhibitor 7-nitroindazole also inhibits the monoamine oxidase-B-catalyzed oxidation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Chem. Res. Toxicol.. 1997;10:364-368.
- [Google Scholar]
- Carbon-13 NMR study of some isomeric n-nitroindazole derivatives. Org. Magn. Res.. 1982;19:225-227.
- [Google Scholar]
- Inhibition of monoamine oxidase contributes to the protective effect of 7-nitroindazole against MPTP neurotoxicity. J. Neurochem.. 1997;69:1771-1773.
- [Google Scholar]
- 13C NMR spectroscopy of polycyclic aromatics VII. Naphthalenes carrying electron-withdrawing substituents. Correlations between substituent-induced shifts and INDO MO charge densities. J. Magn. Res.. 1976;22:279-287.
- [Google Scholar]
- SOMO–HOMO conversion in distonic radical anions: an experimental test in solution by EPR radical equilibration technique. J. Am. Chem. Soc.. 2014;136:1250-1252.
- [Google Scholar]
- On the rearrangement of some Z-arylhydrazones of 3-benzoyl-5-phenylisoxazoles into 2-aryl-4-phenacyl-2H-1,2,3-triazoles: a kinetic study of the substituent effects in Boulton–Katritzky reactions. Tetrahedron. 2015;71:7315-7322.
- [Google Scholar]
- Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys.. 1984;80:3265-3269.
- [Google Scholar]
- Gaussian 09, Revision A.02. Wallingford, CT: Gaussian, Inc.; 2009.
- Paramagnetic doping as an aid in obtaining high-resolution carbon-13 NMR spectra of biomolecules in the solid state. J. Am. Chem. Soc.. 1981;103:6011-6015.
- [Google Scholar]
- Synthesis of 1,6-dihydropyrrolo[2,3-g]indazoles using Larock indole annulation. Tetrahedron. 2011;67:7330-7335.
- [Google Scholar]
- Synthesis and biological activities of pyrazolo[3,4-g]quinoxaline derivatives. Eur. J. Med. Chem.. 2010;45:5520-5526.
- [Google Scholar]
- Regioselective synthesis of novel substituted indazole-5,6-diamine derivatives. Tetrahedron. 2011;67:1633-1639.
- [Google Scholar]
- Identification of pyrrolo[2,3-g]indazoles as new Pim kinase inhibitors. Bioorg. Med. Chem. Lett.. 2013;23:2298-2301.
- [Google Scholar]
- Advances in the synthesis and Kinase inhibitory potencies of non-fused indazole derivatives. In: Attanasi O.A., Noto R., Spinelli D., eds. Targets in Heterocyclic Systems – Chemistry and Properties. Vol vol. 18. Roma: Società Chimica Italiana; 2014. p. :1-28.
- [Google Scholar]
- NMR Spektroscopie. Stuttgart: Thieme-Verlag; 1973.
- Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat. Med.. 1996;2:1017-1021.
- [Google Scholar]
- Spectroscopic and electrochemical properties of 1- or 2-alkyl substituted 5- and 6-nitroindazoles. Curr. Org. Chem.. 2015;19:1526-1537.
- [Google Scholar]
- ESR and electrochemical study of 5-nitroindazole derivatives with antiprotozoal activity. Spectrochim. Acta. 2006;A63:36-42.
- [Google Scholar]
- Molecular encapsulation of 5-nitroindazole derivatives in 2,6-dimethyl-β-cyclodextrin: electrochemical and spectroscopic studies. Bioorg. Med. Chem.. 2009;17:4604-4611.
- [Google Scholar]
- Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J. Neurochem.. 1995;64:936-939.
- [Google Scholar]
- Gronowitz S., ed. The Chemistry of Heterocyclic Compounds, Thiophene and its Derivatives. Vol vol. 44. New York: John Wiley & Sons; 1991. p. :295-396.
- Carbon-13 NMR Spectroscopy. New York: Academic Press; 1972.
- Identification of 1,6-dihydropyrazolo[4,3-c]carbazoles and 3,6-dihydropyrazolo[3,4-c]carbazoles as new Pim kinase inhibitors. Bioorg. Med. Chem.. 2013;21:4102-4111.
- [Google Scholar]
- Interpretation of Carbon-13 NMR Spectra. London: Heyden; 1978.
- Carbon-13 chemical shifts in substituted naphthalenes. J. Chem. Soc. Perkin. 1974;2:1745-1749.
- [Google Scholar]
- Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc.. 1990;112:8251-8260.
- [Google Scholar]
- The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc.. 2008;120:215-241.
- [Google Scholar]




