

Translate this page into:
Recent advances of PROTACs technology in neurodegenerative diseases
⁎Corresponding authors. 459233223@qq.com (Yujing Zhang), xdm_tsinghua@163.com (Dongming Xing)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Neurodegenerative diseases, like Alzheimer's disease, Huntington's disease, Parkinson's disease, progressive supranuclear palsy, and frontotemporal dementia are among the refractory diseases that lack appropriate drugs and treatments. Numerous disease-causing proteins in neurodegenerative diseases are undruggable for traditional drugs. Many clinical studies of drugs for Alzheimer's disease have failed, and none of the substances that slowed the amyloid-β (Aβ) accumulation process have been approved for use in clinical trials. A novel approach to addressing this issue is Proteolysis targeting chimeras (PROTACs) technology. PROTACs are heterogeneous functional molecules joined by a chemical linker and include binding ligands for the target protein and recruitment ligands for the E3 ligand. When a PROTAC binds to a target protein, the E3 ligand enzyme is brought into close contact and the target protein begins to be polyubiquitinated, followed by proteasome-mediated degradation. Numerous neurodegenerative disease-related targets, including α-Synuclein, mHTT, GSK-3, LRRK2, Tau, TRKA, and TRKC have been successfully targeted by PROTACs to date. This article presents a comprehensive overview of the development of PROTACs in neurodegenerative diseases. These PROTACs' chemical structures, preparative routes, in vitro and in vivo activities, and pharmacodynamics are outlined. We also offer our viewpoint on PROTACs' probable challenges and future prospects.
Keywords
Neurodegenerative diseases
Novel drugs
PROTACs
Targeted degradation
Therapy

1 Introduction
More than 10 million people worldwide are affected by neurodegenerative diseases (NDs) each year, including Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), progressive supranuclear palsy (PSP), and frontotemporal dementia (FTD) (Fig. 1). These diseases are the most common and difficult to treat affecting the central nervous system (CNS) because of their crippling effects (Alzheimer's Association, 2010; Deng et al., 2018; GBD 2016 Neurology Collaborators, 2019; Gribkoff et al., 2017; Scheltens et al., 2021). As life expectancy continues to increase and the number of patients grows each year, the need for novel drugs to treat chronic NDs is becoming more urgent. Unfortunately, much remains to be learned about the pathogenesis of these complex brain disorders, and the ability to treat them is often hampered by the difficulties associated with diagnosis, resulting in interventions that come too late. These diseases' heterogeneity adds to their complexity; frequently, it is this accumulation of toxic proteins that causes severe, gradual neuronal death (Breijyeh and Karaman, 2020; Maher, 2019; Inuzuka et al., 2022). Small-molecule inhibitors and immunotherapies that target disease-causing proteins are among the current treatments for NDs, however, they can only alleviate the symptoms of these diseases rather than reverse their progression. Many new NDs therapies are being explored, such as miRNA therapies, autophagy, and stem cell therapies, which also face problems such as low BBB permeability, target bias, and inability to act on intracellular proteins. The fact that the Food and Drug Administration (FDA) is about half as likely to approve CNS drugs as peripheral targets, and the approval process takes almost twice as long as for oncology drugs, further complicates the situation (Banks, 2016; Charvériat et al., 2021). Finally, many pharmaceutical companies have left the neuroscience research and development space as a result of the failure of numerous advanced clinical trials in the field of AD and other brain diseases, which has decreased the resources being invested to solve the issue. A portion of this failure can be related to a lack of knowledge about the illness itself, which makes it challenging to accurately target the main biological pathways that cause it (Farrell and Jarome, 2021; Ma et al., 2021). To date, a vast majority of NDs still lack effective therapeutic options.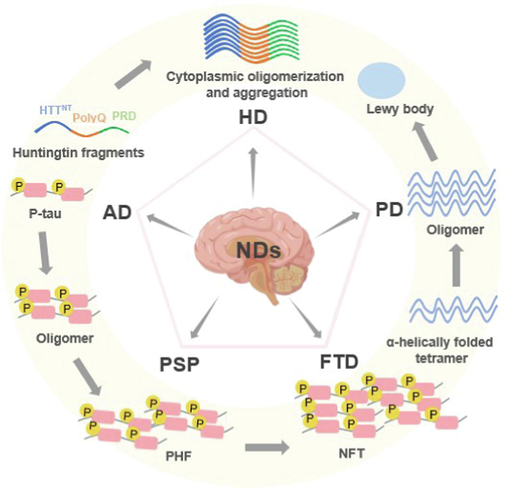
Pathological sites and pathogenesis of pathogenic proteins of common neurodegenerative diseases.
Normal cells acquire the ability to regulate their self-controlled quality processes in order to maintain homeostasis by avoiding long-lasting damage as a result of external factors. Misfolded proteins that aggregate into β-sheet structures are a major cause of neurodegenerative diseases. Typically, the ubiquitin proteasome system (UPS) is the main defense mechanism that cells have against aggregated and misfolded proteins to maintain homeostasis (Pohl and Dikic, 2019). The UPS system, which is primarily made up of the protein ubiquitin (Ub), three catalytic enzymes, proteasomes, and certain substrates, is crucial to many biological processes. A series of enzymatic processes, including the cooperative action of Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), and Ub-ligase enzyme (E3), cause ubiquitination. Once the substrate protein has been polyubiquitinated, the proteasome will detect it and begin to break it down, and UPS can then break the protein down into minute peptides (Deol et al., 2019). E3 appears to have the ability to specifically recognize substrate proteins, and as a result, E3 is crucial in determining the specificity of Ub-mediated protein degradation.
Proteolysis targeting chimeras (PROTACs) are emerging as a novel technology to treat a variety of diseases and are considered the next “blockbuster” therapy for neurodegenerative diseases and other diseases with undruggable protein targets (An and Fu, 2018; Wang et al., 2021; Xiao et al., 2022). PROTACs are heterobifunctional compounds that use cellular degradation systems to degrade a target protein (Fig. 2). With the optimal linker, PROTACs recruit the target protein to the E3 ubiquitin ligase (e.g. CRBN, IAP, MDM2, VHL ligase, etc.). In eukaryotic cells, this complex formation between the protein of interest (POI) and the E3 ligases causes POI ubiquitination, which is then followed by POI degradation by the 26S proteasome. The use and advancement of PROTACs technology have significantly increased in both industry and academia following the verification of the proof of concept (Gadd et al., 2017; Martín-Acosta and Xiao, 2021; Zeng et al., 2021). This is primarily due to the following aspects of PROTACs: 1) ability to eliminate the target proteins that are “undruggable,” 2) ability to decrease both the non-enzymatic and enzymatic functions of proteins, 3) reversible and rapid elimination of protein targets, 3) enhancing selectivity and specificity, and 5) promise in overcoming drug resistance. Drug resistance is a significant issue in treating diseases, therefore PROTACs technology will provide more potent medications to combat it. Because of its new mechanism of action for the reduction of target protein levels while using less dosage of the drug than traditional ones, the use of PROTACs should also be very advantageous (Gu et al., 2018; Sun et al., 2019; Toure and Crews, 2016; Wang et al., 2021). Currently, PROTACs have been successfully used to degrade different types of target proteins associated with various diseases, including neurodegenerative diseases, cancer, viral infections, and immune disorders (Fig. 3). Some of the reported cases include Bristol-Myers Squibb's PROTACs targeting the androgen receptor (AR), Dialectic's PROTACs targeting the B-cell lymphoma extra large (BCL-XL), Kymera's PROTACs targeting the interleukin-1 receptor-associated kinase 4 (IRAK4) and STAT3, Nurix’ PROTACs targeting the BTK, C4 Therapeutic's PROTACs targeting the epidermal growth factor receptor (EGFR), and Cullgen's PROTACs targeting the tropomyosin receptor kinases (TRKs) (Garber, 2022; Mullard, 2021). In short, PROTAC molecules have the potential to treat a variety of diseases, including neurodegenerative diseases (Table 1).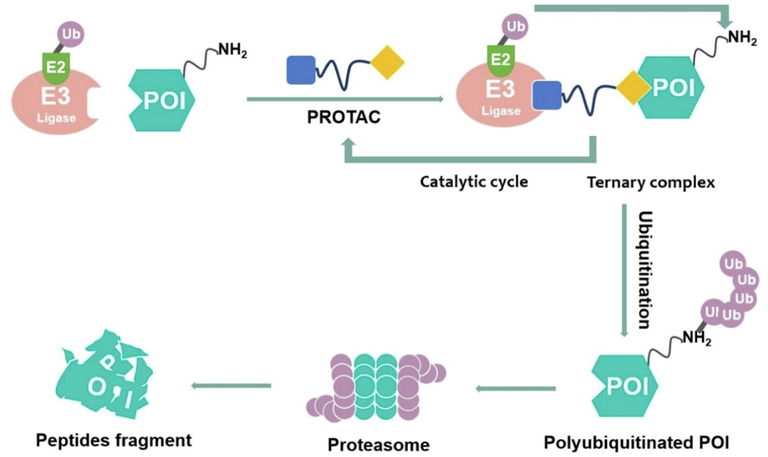
The proposed mechanism of action of PROTACs.
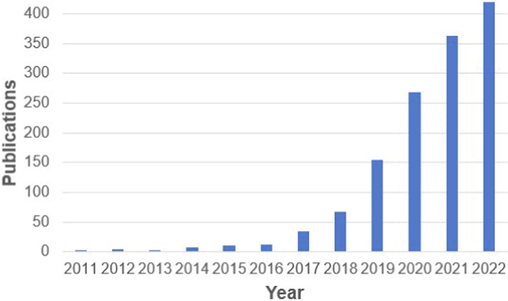
The number of publications on PROTACs in PubMed (accessed on 27/11/2022).
PROTAC
Small-Molecule Inhibitor
Target scaffolding functions
Yes
No
Potential to treat undruggable proteins
Yes
No
Broad tissue penetration
Yes
No
Iterative mechanism of action
Yes
No
Orally bioavailable
Yes
Yes
Ease of manufacturing
Yes
Yes
Preclinical validation, proof-of-concept established
Yes
Yes
Clinical validation
Yes
Yes
The concept of PROTACs was first introduced by Craig M. Crews and Raymond J. Deshaies in 2001. The first generation of PROTACs was a bifunctional molecule consisting of the 10 amino acid phosphopeptide DRHDSGLDSM and the angiogenesis inhibitor ovalicin. This peptide sequence could be recognized by the Fbox protein β-TRCP E3 ubiquitin protein ligase. The mechanism of the first generation PROTACs was the induction of MetAP2 degradation through the recruitment of β-TRCP E3 ligase. The problem with first generation PROTACs was that they have low cell permeability and stability in biological systems. In 2008, to address the aforementioned problem, Craig M. Crews and coworkers developed the first small-molecule PROTACs capable of successfully inducing intracellular degradation of the androgen receptor (AR) in human cervical cancer cells (Benowitz et al., 2021). This PROTAC associated the selective androgen receptor modulator SARM with nutlin, recruiting the E3 ubiquitin ligase human homolog of mouse double molecule 2 (MDM2). Recently, the study of new small-molecule PROTACs with cereblon (CRBN), inhibitors of apoptosis (IAPs), von Hippel-Lindau (VHL), DDB1 and CUL4-associated factors (DCAF15, DCAF16), and ring finger protein (RNF114) ligands had progressed rapidly, offering the potential for the development of new therapeutic drugs. Updated through November 2022, there were approximately a dozen PROTACs in clinical development globally (Table 2). Among them, Arvinas’ ARV-471 has entered clinical phase III and is the most advanced PROTAC drug in clinical development. Some start-ups have gained close attention from large global pharmaceutical companies, such as Gilead, Merck, Pfizer, Roche, Sanofi, etc. Additionally, Chinese companies are also actively involved in the development, including Hesco, Yaxin, Jinto, Novocain Jianhua, Hengrui, etc. It can be seen that the clinical application is currently in a state of explosive growth. Today, PROTAC technology has evolved into a new approach to the development of novel drugs and the treatment of diseases. A crucial time for the development of PROTACs will be in the upcoming years. To date, more than a dozen PROTACs molecules have entered preclinical and clinical studies. Future advances in PROTAC technology are anticipated to help the treatment of human disease and overall well-being (Chirnomas et al., 2023; Cao et al., 2022; Garber, 2022).
PROTAC
Target protein
Usage
Company
Condition
ARV-110
AR
Prostate cancer
Arvinas
Phase II
ARV-766
AR
Prostate cancer
Arvinas
Phase I
CC-94676
AR
Prostate cancer
Bristol Myers Squibb
Phase I
ARV-471
ER
Breast cancer
Arvinas
Phase II
DT2216
BCL-XL
Liquid and solid tumors
Dialectic
Phase I
FHD-609
BRD9
Synovial sarcoma
Foghorn
Phase I
NX-2127
BTK, Ikaros, Aiolos
B-cell malignancies
Nurix
Phase I
BGB-16673
BTK
B-cell malignancies
BeiGene
Phase I
NX-5948
BTK
B-cell malignancies
Nurix
Phase I
HK29116
BTK
B-cell malignancies
Haisco
Phase I
KT-474
IRAK4
Atopic Dermatitis
Kymera
Phase I
KT-413
IRAK4
B-cell NHL
Kymera
Phase I
LNK-01002
Ras GTPase
Myelofibrosis or Myeloid Leukemia
Lynk
Phase I
Target proteins associated with neurodegenerative diseases have been successfully degraded by PROTACs. We discovered a comprehensive list of these targets after searching the literature over the previous few years, mainly including α-Synuclein, mutant huntingtin (mHTT), glycogen synthase kinase 3β (GSK-3β), leucine-rich repeat kinase 2 (LRRK2), Tau, TRKA, and TRKC. Thus, PROTACs are today thought to be one of the most promising alternatives to chemical interventions in pharmacology and as potential therapeutic tools. The major turning points and events in the evolution of PROTACs associated with neurodegenerative diseases are shown in Fig. 4. According to drug target class criteria and alphabetical target order, we present PROTACs for drug targets for neurodegenerative diseases in this review. In addition, we focus on E3 ligase ligands utilized in successful PROTACs in NDs. We also discuss potential problems specific to this field. We hope that this article will serve as a complement to previous papers on protein degradation.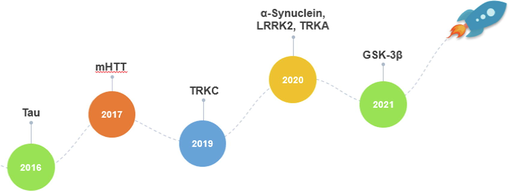
Timeline and major milestones for the development of neurodegenerative diseases.
2 E3 ligase ligands
2.1 CRBN
CRBN is a 442-amino acid protein that associates with the adaptor molecule damaged DNA binding protein 1 (DDB1) to generate the Cullin-4-RING E3 ubiquitin ligase (CRL4) complex (Chamberlain et al., 2014). CRBN functions as a substrate-specificity receptor inside the CRL4 complex. Thalidomide and other immunomodulatory imide drugs (IMiDs) are known CRBN ligands. The E3 ubiquitin ligase activity of CRBN is remodulated when IMiDs attach to it (Zhu et al., 2011). The transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) are consequently recruited more frequently, which results in their subsequent ubiquitination and proteasomal degradation. To date, PROTAC has been successful in using CRBN as the E3 ligase to target NDs-associated proteins. The CRBN targeting NDs PROTACs employ pomalidomide, 4-hydroxythalidomide, and 5-aminothalidomide (Fig. 5).
Chemical structures of CRBN ligase ligands used in NDs PROTACs.
The synthetic routes to obtain CRBN E3 ligands are presented in Scheme 1a, Scheme 1b, Scheme 1c. Typically, 3-nitrophthalic anhydride was used as the starting compound in the synthesis of pomalidomide. 4-nitrothalidomide was created by condensing 3-nitrophthalic anhydride with the glutarimide ring. In comparison to employing iron-ammonium chloride or Pd/C and ammonium formate as reducing agents (Steinebach et al., 2018), the subsequent reduction to pomalidomide was significantly more efficient when using Pd/C-catalyzed hydrogenation with reports of nearly quantitative yield (Chen et al., 2018). A different approach was described by Huang et al. in which the 3-nitrophthalimide was first reacted with glutamine via amination to produce a carboxylic acid intermediate. Next, it was reduced to an amine intermediate and subsequently cyclized under CDI-mediated conditions (Huang et al., 2016). Low reduction conversion led to a low pomalidomide yield, which was the main cause of this.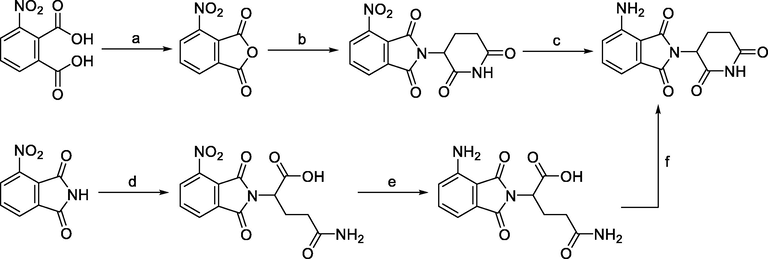
Synthesis of pomalidomide. Reagents and conditions: a) Ac2O, reflux, 2 h; b) tert-butyl (2,6-dioxopiperidin-3-yl)carbamate, NaOAc, AcOH, reflux, 6 h; b) 3-aminopiperidine-2,6-dione hydrochloride, NaOAc, AcOH, 130 °C, 48 h; c) Pd/C, H2, DMF, r.t., 24 h; d) glutamine, DMF, 80–87 °C, 8 h; e) Pd/C, H2, 50 psi, MeOH, 2.5 h; f) CDI, MeCN, reflux, 4.5 h.
Synthesis of 4-hydroxythalidomide. Reagents and conditions: a) 3-aminopiperidine-2,6-dione hydrochloride, KOAc, AcOH, reflux, 24 h; b) Linker-Br, KI, NaHCO3, DMF, 60 °C, 12 h.
Synthesis of 5-aminothalidomide. Reagents and conditions: a) 3-aminopiperidine-2,6-dione trifluoroacetate, AcOH, reflux, 2 h; b) Pd/C, H2, r.t., 20 h.
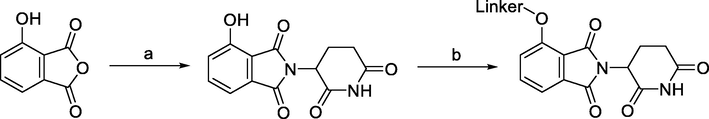
3-hydroxyphthalic anhydride was typically used as the starting compound in the preparation of 4-hydroxythalidomide. With different reaction conditions, the condensation with the glutarimide ring was achievable using 3-hydroxyphthalic anhydride as the starting material. While yields for other reported procedures ranged between 83 and 90% (Burslem et al., 2018; Papatzimas et al., 2019), which were comparable to the reactions used to synthesize 4-fluoro- and 4-nitrothalidomide, the highest yield for the desired product 4-hydroxythalidomide was described to be 96% (KOAc, AcOH, reflux) (Robb et al., 2017). By adding a bromo group as a linker terminus, it was feasible to derivatize 4-hydroxythalidomide and create a derivative that had an ether bond. Through a Mitsunobu reaction, a linker with a terminal hydroxyl group could be joined to 4-hydroxythalidomide (Chessum et al., 2018).
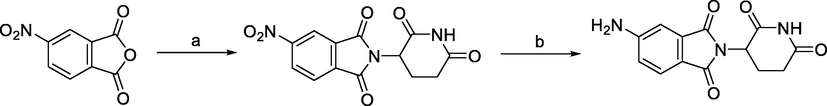
Derivatives of 5-aminothalidomide were less frequently used in PROTACs, although this substitution pattern remained a valid option for targeting CRBN. 5-aminothalidomide was synthesized by condensation of 4-nitrophthalic anhydride with 3-aminopiperidine-2,6-dione trifluoroacetate to form 5-nitrothalidomide, which was then reduced to the desired product 5-aminothalidomide (Capitosti et al. 2003).
2.2 IAP
IAP are a group of apoptosis-inhibiting regulators that work by preventing caspase from activating. There are eight members of the IAP family; the three most well researched members are c-IAP1, c-IAP2, and XIAP. IAP are members of the E3 ubiquitin ligase family and are important targets for cancer therapy. The term “ specific and nongenetic IAP-dependent protein erasers (SNIPERs)” is also used to describe IAP-based PROTACs. IAP inhibitors and their derivatives are frequently employed as SNIPERs E3 ligase ligands. The cIAP1-binding ligands for the reported NDs PROTACs recruiting this E3 ligase were bestatin and MV1 (Fig. 6) (Ma et al., 2021). In comparison to early bestatin-based compounds, the effectiveness of SNIPERs was increased by the subsequent development of high-affinity IAP ligands and their incorporation into bifunctional molecules. The representative synthetic routes to obtain IAP E3 ligands are presented in Scheme 2a, Scheme 2b.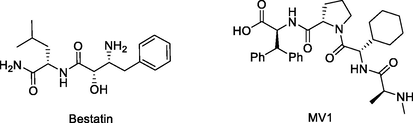
Chemical structures of IAP ligase ligands used in NDs PROTACs.
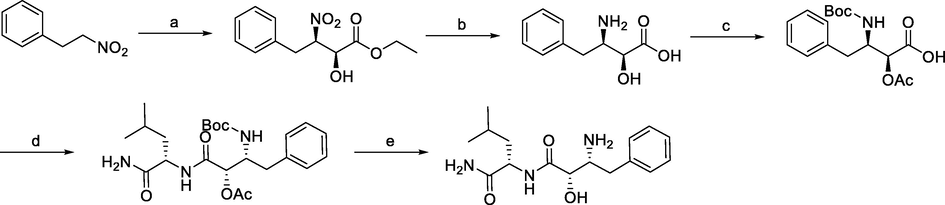
Synthesis of bestatin. Reagents and conditions: a) Ethyl glyoxalate, La-(R)-BINOL, THF, −50 °C; b) i) acetylation; ii) Pd/C, NaBH4, H2, MeOH, r.t.; c) (Boc)2O, NaHCO3, H2O, EtOAc, r.t.; d) i) L-leucine benzyl ester, N-ethylmorpholine, isobutyl chloroformate, THF, −10 °C; ii) Pd/C, H2, MeOH, r.t.; e) i) K2CO3, MeOH, r.t.; ii) TFA, MeOH, r.t..
Synthesis of MV1. Reagents and conditions: a) i) Fmoc-L-3,3-diphenylalanine, DIPEA, DCM, r.t., 2 days; ii) 20% piperidine in DMF, r.t., 10 min; b) i) Fmoc-L-Pro-OH, HCTU, HOBt, DIPEA, DMF, r.t., 1.5 h; ii) 20% piperidine in DMF, r.t., 10 min; c) i) Fmoc-L-cyclohexylglycin, HCTU, HOBt, DIPEA, DMF, r.t., 1.5 h; ii) 20% piperidine in DMF, r.t., 10 min; d) i) Fmoc-L-Ala-OH, HCTU, HOBt, DIPEA, DMF, r.t., 1.5 h; ii) 20% piperidine in DMF, r.t., 10 min; e) 1% TFA in DCM, r.t., 5 min.
The representative synthetic route to bastein involved the treatment of (2-nitroethyl)benzene and ethyl glyoxalate in Shibasaki's asymmetric Henry reaction catalyzed by optically active lanthanum-(R)-binaphthol complex. Then, ethyl (2S,3R)-2-hydroxy-3-nitro-4-phenylbutanoate was O-acetylated prior to the reduction of the nitro group to give (2S,3R)-3-amino-2-hydroxy-4-phenylbutyric acid. After N-Boc protection, the phenylbutanoic acid derivative was coupled to L-leucine benzyl ester followed by immediate deprotection of the terminal carboxylic acid group. Both protecting groups of the precursor of bestatin were removed to give bestatin in an overall yield of 26% (Gogoi et al., 2005).
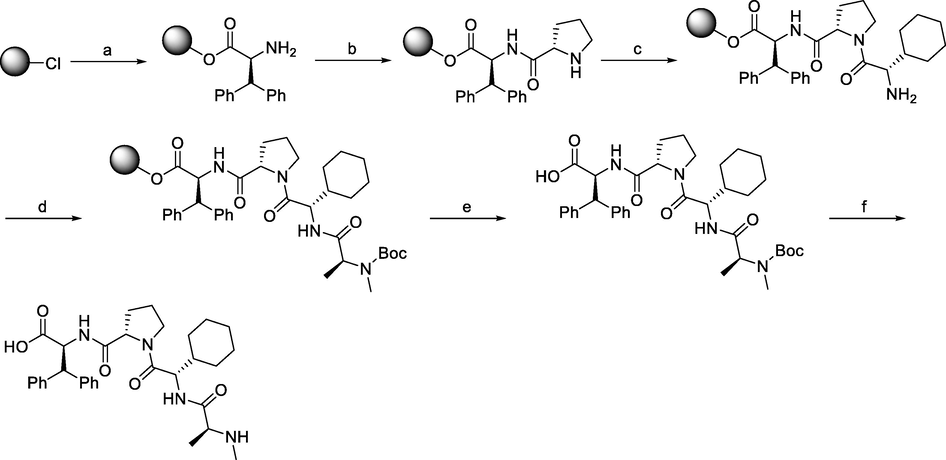
Scheme 2a, Scheme 2b outlines the solid-phase peptide synthesis of IAP ligand B derivative on a 2-chlorotrichloromethane resin. It was performed stepwise, using HCTU, HOBt, and DIPEA for coupling, followed by the addition of 20% piperidine in DMF and removal of the Fmoc moiety after each step. Finally, the Boc-protected IAP ligand was treated with 1% TFA in DCM to remove the resin to obtain MV1 (Ohoka et al., 2017).
2.3 VHL
The VHL protein is part of a multiprotein complex with elongin B and C, cullin 2, and Rbx-1, which have E3 ubiquitin ligase activity. In the complex, VHL folds into two structural domains, one of which is responsible for binding specific substrates (Van Molle et al., 2012), most notably hypoxia-inducible factor (HIF)-1α, leading to its ubiquitination and proteasomal degradation. VHL, similar to CRBN, is a broad target for NPs PROTACs (Fig. 7). The synthetic routes to obtain VHL E3 ligands (VHL ligands 1, 2, and 3) are shown in Scheme 3a, Scheme 3b, Scheme 3c.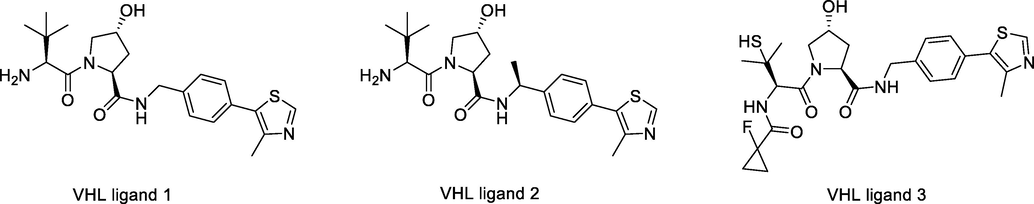
Chemical structures of VHL ligase ligands used in NDs PROTACs.
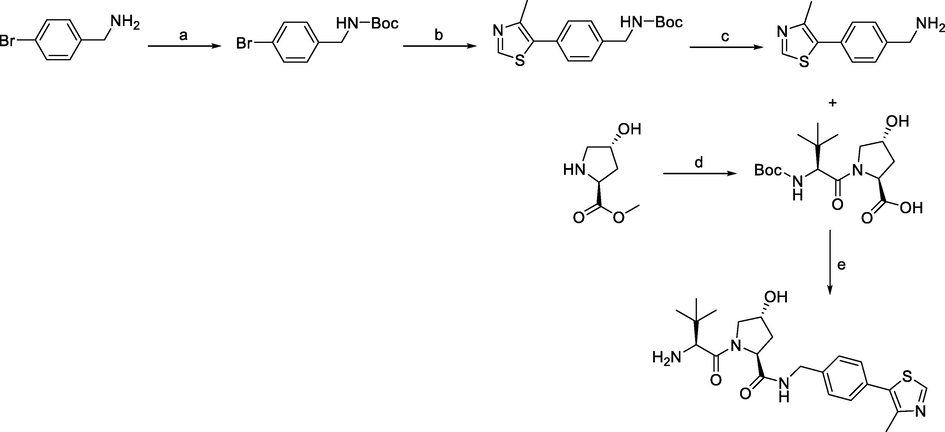
Synthesis of VHL ligand 1. Reagents and conditions: a) (Boc)2O, NaHCO3, EtOAc/H2O, r.t., 1 h; b) 4-methylthiazole, Pd(OAc)2, KOAc, DMF, 90 °C, 2 h; c) TFA, DCM, r.t., 30 min; d) i) Boc-Tle-OH, HATU, DIPEA, DMF, r.t., overnight; ii) LiOH, THF/H2O, r.t., 30 min; e) i) HATU, DIPEA, DMF, r.t., overnight; ii) TFA, DCM, r.t., 30 min.
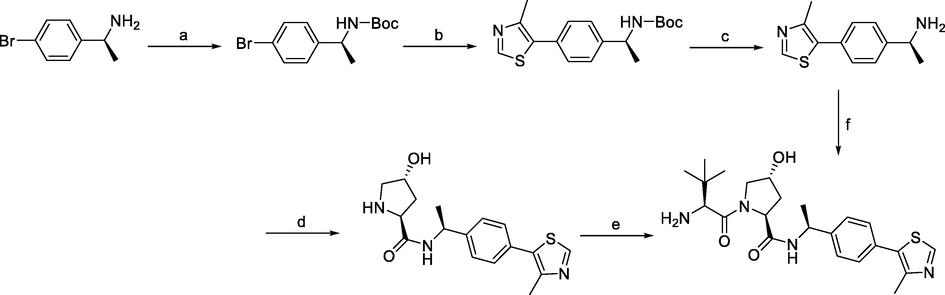
Synthesis of VHL ligand 2. Reagents and conditions: a) (Boc)2O, NaHCO3, H2O/EtOAc, r.t., 2 h; b) 4-methylthiazole, Pd(OAc)2, KOAc, DMA, 90 °C, 18 h; c) 4 M HCl in MeOH, r.t., 3 h; c) 4 M HCl in dioxane, MeOH, r.t., 12 h; d) i) Boc-Hyp-OH, HATU, DIPEA, DMF, 0 °C-r.t., 12 h; ii) 4 M HCl in dioxane, MeOH, r.t., 12 h; e) i) Boc-Tle-OH, HATU, DIPEA, DMF, 0 °C-r.t., 12 h; ii) 4 M HCl in dioxane, MeOH, r.t., 12 h; f) i) dipeptide, HATU, DIPEA, THF, r.t., 2 h; ii) 4 M HCl in MeOH, r.t., 3 h.
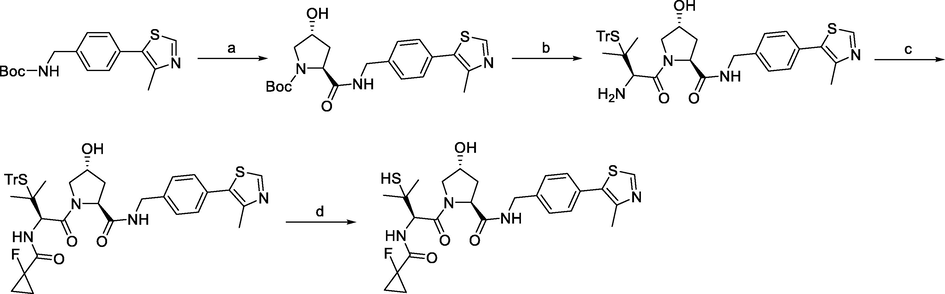
Synthesis of VHL ligand 3. Reagents and conditions: a) 2 M HCl in DCM/MeOH, r.t., overnight; (b) (2S,4S)-1-(tert-butoxycarbonyl)-4-hydroxypyrrolidine-2-carboxylic acid, HATU, Et3N, DCM, r.t., overnight; (c) Fmoc-S-trityl-l-penicillamine, HATU, TEA, DMF r.t., overnight; (d) 1-fluorocyclopropane-1-carboxylic acid, HATU, Et3N, DCM, r.t., overnight; (e) TFA, triisopropylsilane, DCM, 0 °C, 30 min.
The most efficient procedure for the synthesis of VHL ligand 1 started with 4-bromobenzylamine, which was first protected by Boc to tert-butyl (4-bromobenzyl)carbamate, and then underwent the Heck reaction and Boc deprotection to give the key intermediate (4-(4-methylthiazol-5-yl)phenyl)methanamine. The final VHL ligand 1 was prepared by coupling (4-(4-methylthiazol-5-yl)phenyl)methanamine with the dipeptide, which was prepared from N-Boc-L-tert-leucine and hydroxyproline methyl ester. Incorporating an element of convergent synthesis helped to increase the overall yield (Han et al., 2019).
In the design and optimization of VHL ligands, the introduction of an (S)-methyl group on the benzyl carbon atom improved the binding affinity for VHL. That was, the potency of the methyl-substituted ligand was three times better than that of the non-substituted VHL ligand 1 (Han et al., 2019). The synthesis of the important intermediate (S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethan-1-amine was similar to the synthetic route described in Scheme 3a, using (S)-(-)-4-bromo-α-methylbenzylamine as the starting material. The left-hand side dipeptide moiety could be assembled by linear synthesis (Hu et al., 2019) or convergent synthesis (Raina et al., 2016) to obtain VHL ligand 2.
The synthesis of VHL ligand 3 started with tert-butyl (4-(4-methylthiazol-5-yl)benzyl)carbamate (Liu et al., 2022). The important intermediate, tert-butyl (2S,4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidine-1-carboxylate was generated from tert-butyl (4-(4-methylthiazol-5-yl)benzyl)carbamate and Boc-L-hydroxyproline, which was then Boc-deprotected and coupled with tributyl-protected penicillamine to give the sulfur-based intermediate which was derivatized into the precursor of VHL ligand 3. VHL ligand 3 was obtained from removing the trityl group.
3 PROTACs for neurodegenerative diseases
3.1 α-Synuclein
α-Synuclein is a protein that is found in abundance in neurons and is particularly enriched in presynaptic nerve terminals (Shahnawaz et al., 2020). α-Synuclein has been linked to PD and other neurodegenerative diseases through neuropathology and genetic research (Schwab et al., 2020). Numerous neurodegenerative diseases known as synucleinopathies are characterized by the accumulation of misfolded oligomers and larger aggregates of α-synuclein.
In 2020, a recent patent filing from Andrew P Crew and coworkers disclosed PROTAC molecules that are claimed to degrade α-Synuclein. Exemplified compounds incorporated CRBN and VHL-binding ligands as well as chemically diverse binders to α-Synuclein, and they were shown to significantly reduce α-Synuclein in HEK293 TREX u-syn A53T cells which had been treated with pre-formed α-Synuclein fibrils to induce insoluble α-Synuclein aggregates (Kargbo, 2020). The authors found that representative compounds PROTAC1 (Fig. 8), PROTAC2 (Fig. 8), and PROTAC3 (Fig. 8) could induce to degrade α-Synuclein, and the degradation activities were 30–65% at 1 μM, demonstrating that neuronal diseases associated with α-Synuclein aggregation and accumulation (dementia, PD, AD, etc.) could be treated by targeted degradation of α-Synuclein. PROTAC1, PROTAC2, and PROTAC3 were synthesized via synthetic pathways shown in Scheme 4a, Scheme 4b, Scheme 4c.
Representative PROTACs targeting α-synuclein.
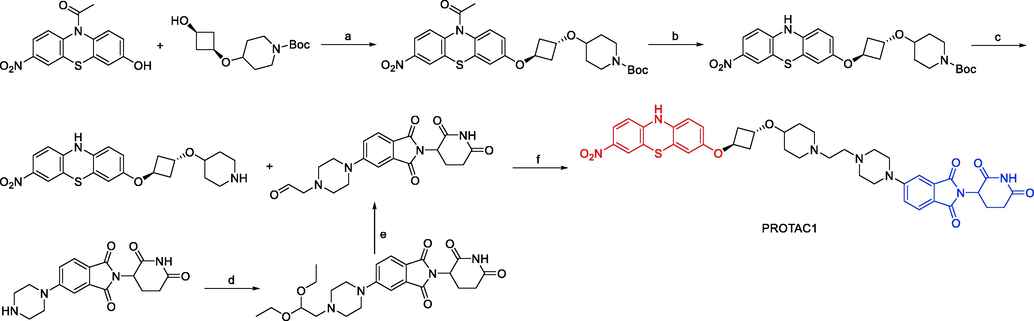
Synthesis of PROTAC1. Reagents and conditions: a) ADDP, Bu3P, Toluene, 15–110 °C, 16 h; b) K2CO3, MeOH, 15 °C, 10 h; c) 4 M HCl, Dioxane, 20 °C, 1 h; d) 2-bromo-1,1-diethoxy-ethane, DIEA, DMF, 100 °C, 2 h, m.w.; e) 2 M H2SO4, THF, 60 °C, 14 h; f) NaBH(OAc)3, Et3N, DCM, MeOH, 0–20 °C, 6 h.
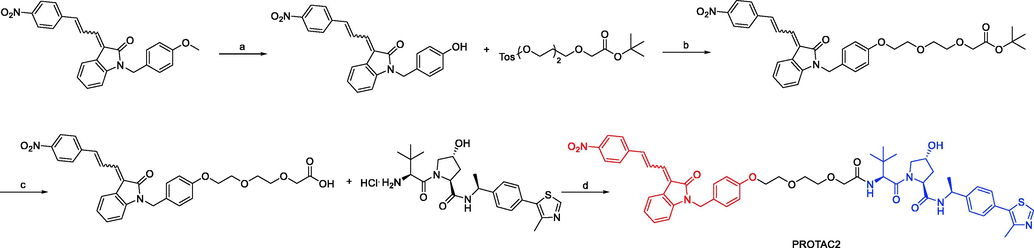
Synthesis of PROTAC2. Reagents and conditions: a) BBr3, DCM, −60–10 °C, 15 min; b) Cs2CO3, DMF, 85 °C, 1 h; c) TFA, DCM, 12 °C, 12 h; d) EDCI, HOBT, DMF, 12–30 °C, 12 h.
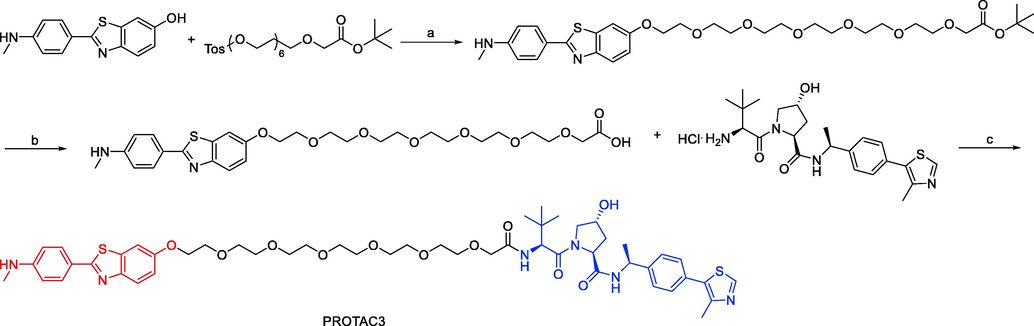
Synthesis of PROTAC3. Reagents and conditions: a) Cs2CO3, DMF, 85 °C, 1 h; b) TFA, DCM, 12 °C, 12 h; c) EDCI, HOBT, DMF, 12–30 °C, 12 h.
3.2 mHtt
The neurodegenerative disease HD is inherited autosomal dominant and characterized by a triad of psychiatric, cognitive, and motor features. All cases of HD are brought on by an abnormally expanded CAG repeat close to the huntingtin gene's N terminus, which results in the synthesis of mHTT on translation (Reiner et al., 2011). Since the genetic mutation was discovered in 1993, 30 years have passed, and extensive studies have revealed numerous cellular pathogenic pathways driving disease onset. The mHTT protein, which is very universally expressed and is thought to cause disease through a predominate harmful gain-of-function mechanism, is mostly responsible for all of them (Sánchez et al., 2003). Reducing mHTT enhances disease phenotypes and reverses neuropathology in HD animal models, demonstrating the significance of mHTT lowering as a treatment strategy (Bauer et al., 2010).
In 2017, Shusuke Tomoshige and coworkers developed two mHtt PROTACs (PROTAC4 and PROTAC5, Fig. 9), each of which was made up of the cIAP1 ligand BE04 coupled to either BTA or PDB (Tomoshige et al., 2017). They examined how PROTAC4 and PROTAC5 affected the levels of mHtt in fibroblasts taken from two HD patients (HD fb). It appeared that PROTAC4 and PROTAC5 function efficiently as mHtt down-regulators because they decreased the levels of mHtt in both sets of HD fb at a concentration of 10 μM. The mHtt level was decreased by PROTAC4 in a dose-dependent manner. There is mounting evidence that neurodegenerative diseases are affected by UPS. The design idea of posttranslational knockdown utilizing particular ligands to target proteins is different from the design concept of targeting the aggregated state of proteins. They thought that PROTACs technology could be used to target other proteins that are prone to aggregation and are responsible for neurodegenerative illnesses like PD, AD, and other polyQ diseases.
Representative PROTACs targeting mHtt.
To test the impact of changing the E3 ligand, Shusuke Tomoshige and colleagues developed a new Htt degrader called PROTAC6 in 2018 (Fig. 9). This version used PDB coupled to MV1 rather than BE04, which was used in their previously reported Htt degrader PROTAC5 (Tomoshige et al., 2018). PROTAC6 successfully decreased both wtHtt level and mHtt level. The efficacy of PROTAC6 was lower than that of PROTAC4 or PROTAC5, despite the fact that MV1 was thought to have a higher affinity than BE04 for IAPs, indicating that the linker length of PROTAC6 might not be optimal for protein knockdown of Htt. They confirmed that PROTAC6 induced an interaction between GST-BIR3 and insoluble model aggregates of 62Q peptides, supporting the idea that PROTAC6 worked by forming a non-physiological complex consisting of Htt aggregate, IAPs, and PROTAC6. Additionally, PROTAC6 induced an interaction between GST-BIR3 and soluble GST-62Q, which was the observation that it decreased the levels of both mHtt and wtHtt. Overall, their findings show that substituting the E3 ligase ligand for protein knockdown of Htt in hybrid molecules does not change the mode of action but may affect the efficacy. In these hybrids, it might be crucial to optimize the linker length for each unique E3 ligand. PROTAC4, PROTAC5, and PROTAC6 were synthesized via synthetic pathways shown in Scheme 5a, Scheme 5b.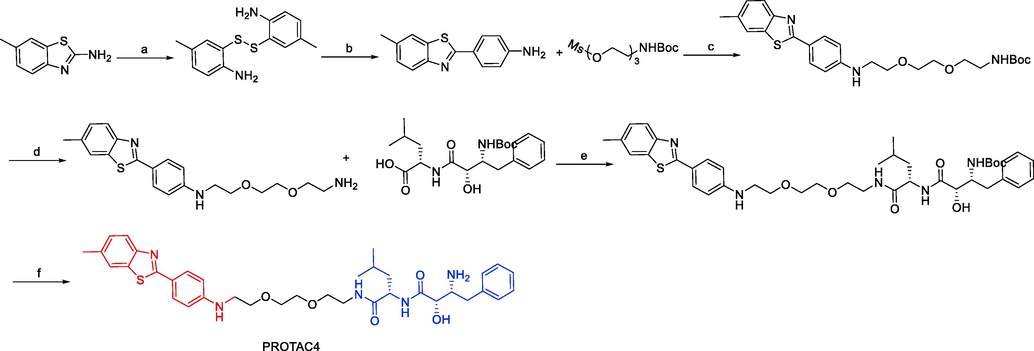
Synthesis of PROTAC4. Reagents and conditions: a) 10 M NaOH, reflux, 18.5 h; b) PPh3, 4-aminobenzoic acid, polyphosphoric acid, 220 °C, 4 h; c) K2CO3, KI, DMF, 90 °C, 3.5 h; d) 4 M HCl, 1,4-dioxane, DCM, r.t., 3 h; e) EDCI, HOBt, DIEA, DMF 0 °C-r.t., 24 h; f) 4 M HCl, 1,4-dioxane, DCM, r.t., 3 h.
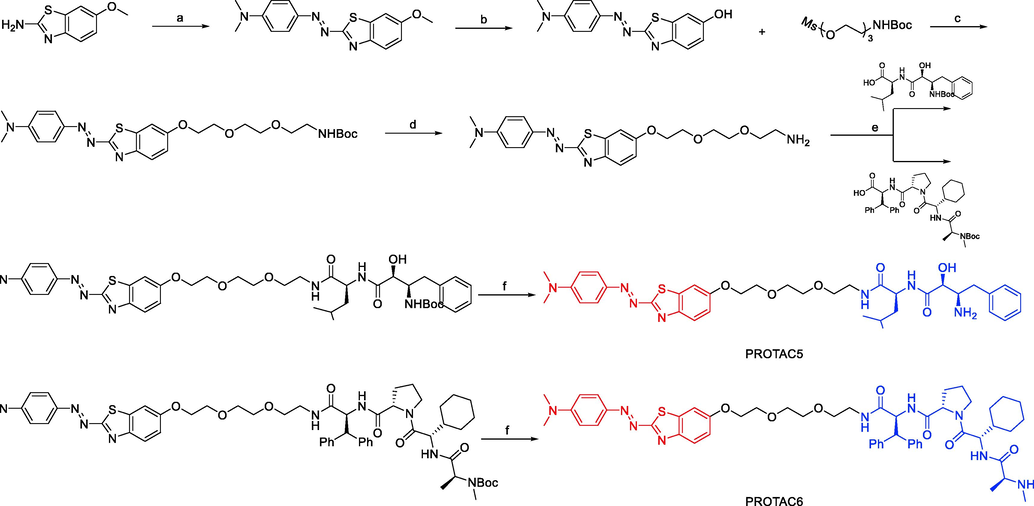
Synthesis of PROTAC5 and PROTAC6. Reagents and conditions: a) i) NaNO2, H2SO4, AcOH, 0 °C, 10 min; ii) N, N-dimethylaniline, 1.4 M HCl, 0 °C, 15 min; b) BBr3, DCM, r.t., overnight; c) K2CO3, KI, DMF, 100 °C, 7 h; d) 4 M HCl, 1,4-dioxane, DCM, r.t., overnight; e) EDCI, HOBt, DIEA, DMF 0 °C-r.t., 24 h; f) 4 M HCl, 1,4-dioxane, DCM, r.t., 6 h.
3.3 GSK-3β
A highly conserved serine/threonine kinase is GSK-3β. Tau is made hyperphosphorylated by GSK-3β, which is a crucial step in the development of neurofibrillary tangles (NFTs). GSK-3β is the most abundant isoform in the CNS, and the expression level of GSK-3β increases with age (Embi et al., 1980). According to recent research, AD has higher levels of total and active GSK-3β than healthy controls. GSK-3β is active in pre-tangle neurons, preferentially co-localizes with NFTs, and aids in the development of paired helical filaments (PHF) in the AD brain. The efficacy of several GSK-3β inhibitors in the treatment of AD has been established. Clinical trial findings on the ATP noncompetitive GSK-3β inhibitor tideglusib showed that it could improve cognition and lower phospho-Tau levels in cerebrospinal fluid in AD patients (Eldar-Finkelman, 2002). Therefore, GSK-3β is considered a potential target for the treatment of AD.
Through the coupling of GSK-3 inhibitor G1 and CRBN ligand thalidomide, Xueyang Jiang and coworkers reported a family of GSK-3β PROTACs in 2021 (Jiang et al., 2021). They produced a potent small molecule GSK-3β degrader called PROTAC7 by extensively optimizing the length and composition of the linker. According to Western blot results, PROTAC7 (Fig. 10) could effectively and dose-dependently degrade GSK-3β, leading to 44% protein degradation at a concentration of 2.8 μM. Their mechanistic analysis revealed that to cause GSK-3β degradation, PROTAC7 must rely on the intracellular UPS. PROTAC7 displayed neuroprotective effects, according to preliminary in vitro cytotoxicity tests. This is the first instance of PROTAC targeting the GSK-3β protein. PROTAC7 is anticipated to be used as a cutting-edge tool to examine the function of GSK-3β in various pathological states because of its good kinase selectivity.
Representative PROTACs targeting GSK-3β.
Through the optimization of GSK-3β ligands, linkers, and E3 ligase ligands based on a click chemistry platform, several GSK-3β PROTACs were developed (Qu et al., 2021). The work of Lailiang Qu and coworkers produced numerous efficient GSK-3β PROTACs, with PROTAC8 serving as an example (Fig. 10). In SH-SY5Y cells, PROTAC8 was able to degrade GSK3α and GSK3β with DC50 values of 28.3 and 34.2 nM, respectively. The SPR experiment demonstrated that PROTAC8 has a strong affinity for GSK-3β (KD = 12.41 nM), which underlies the strength of its GSK-3β degradation ability. And the proteomic investigation revealed that PROTAC8 was able to specifically promote the degradation of GSK-3β. Moreover, they confirmed that CRBN and the proteasome were responsible for the degradation of GSK-3β. In SH-SY5Y and HEK GSK3 293 T cells, PROTAC8 could efficiently reduce GSK-3β and Aβ (amyloid β) regulated tau hyperphosphorylation in a dose-dependent manner and shield SH-SY5Y cells from Aβ-induced cell damage. Compared to the conventional GSK-3β inhibitor AZD2858, PROTAC8 was more effective for a longer period. Additionally, they verified that PROTAC8 might improve learning and memory deficits in AD model rats and decrease okadaic acid (OA)-induced Tau hyperphosphorylation. Overall, PROTAC8 is a powerful GSK-3β degrader and may hold promise as a therapy for AD. PROTAC7 and PROTAC8 were synthesized via synthetic pathways shown in Scheme 6a, Scheme 6b.
Synthesis of PROTAC7. Reagents and conditions: a) 8-decamethylene glycol, PPh3 DIAD, THF, r.t., 24 h; b) i) 1.5 M LiOH, THF/MeOH, 0 °C-r.t., 6 h; ii) methylamine, DIPEA, HATU, DMF, r.t., 24 h; c) p-tosyl chloride, Et3N, DCM, r.t., 24 h; d) i) NaN3, DMF, 80 °C, 10 h; ii) sodium ascorbate, Et3N, CuSO4, n-butanol, r.t., 8 h.
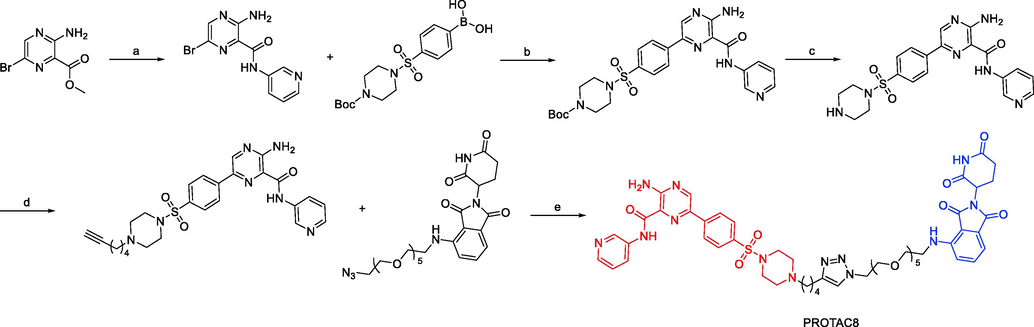
Synthesis of PROTAC8. Reagents and conditions: a) 3-aminopyridine, DBU, 70 °C, 4 h; b) Pd(dppf)Cl2, DCM, Na2CO3, toluene/EtOH, 80 °C, overnight; c) TFA, DCM, r.t., 2 h; d) haloalkynyl, DIPEA, DMF, 80 °C, 2 h; e) CuSO4, NaVic, t-BuOH/DCM/DMF/H2O, r.t., 8 h.
3.4 LRRK2
LRRK2 is a large protein that includes multiple structural domains and consists of 2527 amino acids. Interestingly, it has a GTPase domain and a kinase domain (Nichols et al., 2005). The majority of the mutations, which increase kinase activity and autophosphorylation, are found in the kinase and GTPase domains. LRRK2 mutations have been connected to a considerable variety of neurodegenerative diseases; five missense variants (G2019S, I2020T, R1441C, R1441G, and Y1699C) are associated with the development of PD. Importantly, it was recently discovered that postmortem brain tissue from patients with idiopathic PD had increased LRRK2 activity (Thaler et al., 2009). Targeting LRRK2 may therefore be advantageous for PD in general as it is thought to be a key factor in the etiology of PD.
The LRKK2 inhibitors (GNE-7915 and PF-06447475) and a CRBN ligand were the basis for certain PROTACs targeting LRKK2 that were described by Markella Konstantinidou and coworkers in 2020 (Konstantinidou et al., 2021). They discovered that none of the PROTACs could degrade LRKK2. The efficacy of different PROTACs to degrade LRRK2 in LRRK2 parental RAW 264.7 cells was then assessed. The RAW 264.7 cells were exposed to PROTAC9 at two concentrations (1 and 10 μM) for 24, 48, and 72 h during the initial screening (Fig. 11). Western blotting results showed no discernible differences in the amounts of LRRK2 protein between cells treated with PROTAC9 and cells treated with the original kinase inhibitors, proving that PROTACs were unable to degrade LRRK2. Although PROTACs have so far been successful in challenging targets, the development of degraders is not an easy task. To further explore the possibility of degrading LRRK2, which continues to be a difficult drug target, and to comprehend the reasons for the observed differences when highly effective LRRK2 inhibitors were modified into potential degraders, a better understanding of its structure and conformational changes, which have just recently been reported, is required. Overall, they wanted to highlight the difficulties of degrading a target whose complete structure is unknown in this work. They think that further research will make it possible to rationally create an effective LRRK2-targeting PROTAC after gaining a better understanding of the structure and dynamics of LRRK2.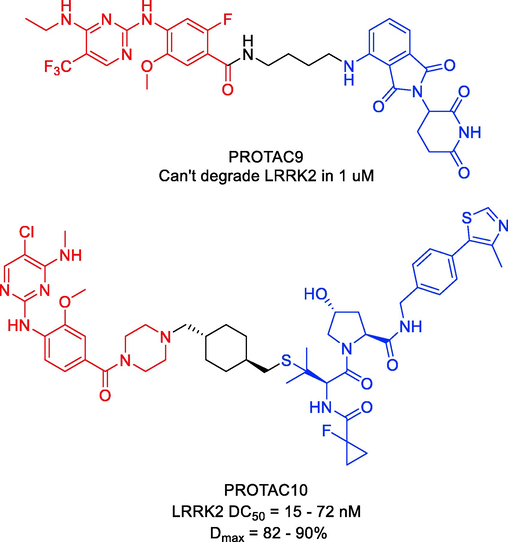
Representative PROTACs targeting LRKK2.
As an alternate LRRK2-targeting strategy, Xingui Liu and colleagues reported the development of LRRK2 PROTACs in 2022, which culminated in the identification of degrader PROTAC10 (Fig. 11) (Liu et al., 2022). The most effective degrader contained thioether-conjugated VHL ligand VH101, as determined by initial designs and tests of PROTACs based on ligands for E3 ligases VHL, CRBN, and cIAP. PROTAC10 was identified as a potent and powerful LRRK2 degrader in several cell lines during a second round of medicinal chemistry exploration, with DC50 values between 15 and 72 nM, Dmax values between 82 and 90%, and degradation half-lives spanning from 0.6 to 2.4 h. PROTAC10 exhibited high cell permeability and formed a positively cooperative ternary complex with VHL and LRRK2 (α = 5.7), which compensated for a substantial loss of binary binding affinities to VHL and LRRK2, underscoring its strong degradation performance in cells. Importantly, PROTAC10 could penetrate the blood–brain barrier after being administered to mice either oral or parenteral dosing (F = 15%). When taken as a whole, these studies demonstrate the suitability of PROTAC10 as a degrader probe to investigate the noncatalytic and scaffolding roles of LRRK2 in vitro and in vivo, and they provide an alluring starting point for potential future therapeutic development. PROTAC9 and PROTAC10 were synthesized via synthetic pathways shown in Scheme 7a, Scheme 7b.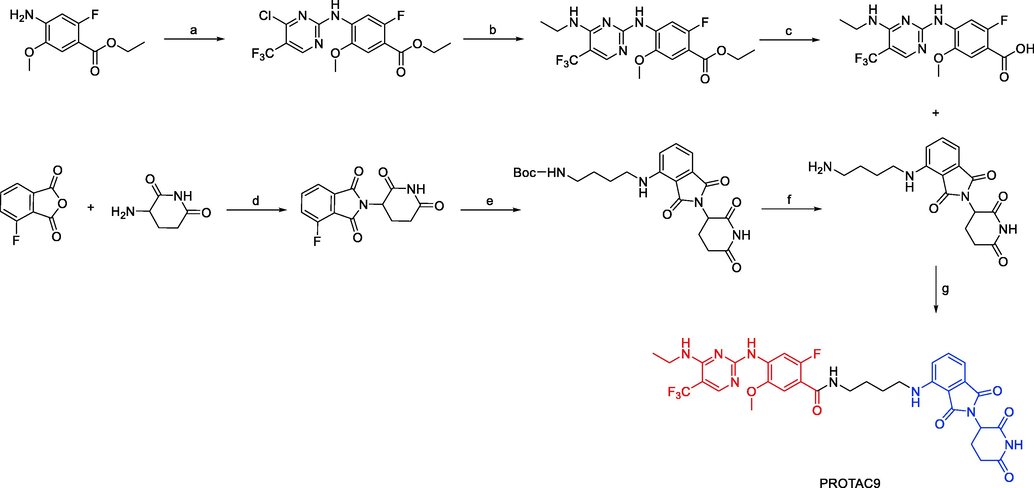
Synthesis of PROTAC9. Reagents and conditions: a) 2,4-dichloro-5-(trifluoromethyl)pyrimidine, zinc chloride, diethyl ether, DCM, tert-butanol, triethylamine, 0 °C-r.t., 48 h; b) ethanamine, triethylamine, THF, 0 °C - r.t.,1h; c) LiOH, THF/H2O, 1 h; d) TFA, DCM, r.t., 2 h; d) sodium acetate, acetic acid, reflux, overnight; e) N-Boc-butane-1,4-diamine, DIPEA, DMF, reflux, overnight; f) 4 M HCl, 1,4-dioxane, DCM, r.t., 6 h; g) EDCI, HOBt, DIEA, DMF 0 °C-r.t., 24 h.
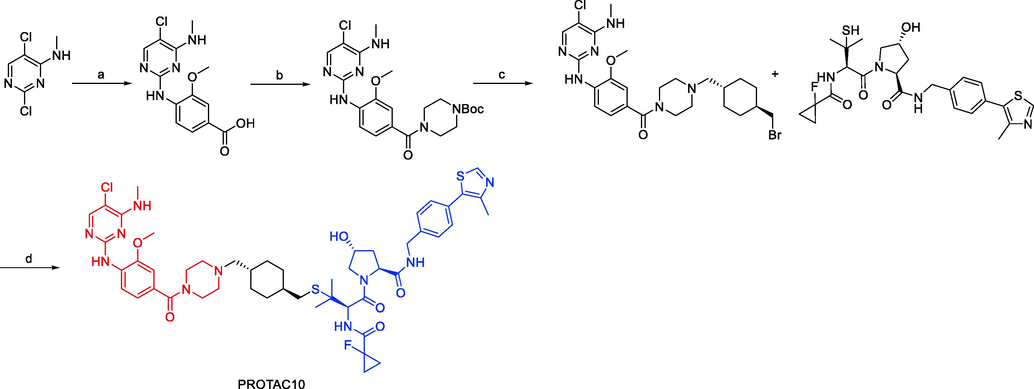
Synthesis of PROTAC10. Reagents and conditions: a) 4 M HCl, 1,4-dioxane, H2O, 4-amino-3-methoxybenzoic acid, 100 °C, overnight; b) EDCI, HOBt, DIPEA, 1-Boc-piperizine, DMF, r.t., 16 h; c) i) 2 M HCl, 1,4-dioxane, DCM, r.t., overnight; ii) trans-1,4-bis(bromomethyl)cyclohexane, K2CO3, acetone, 50 °C, overnight; d) DBU, THF r.t., overnight.
3.5 Tau
Tau is a protein that is associated with microtubules and is typically assumed to control microtubule stability and participate in axonal transport (Jeremic et al., 2021). As an essential pathogenic protein in AD, Tau has the ability to mediate the toxicity of Aβ. It has been suggested that Tau is a particularly alluring target for future treatments of neurodegenerative disorders (Congdon and Sigurdsson, 2018). As a result, lowering Tau with chemical compounds might present a new approach to treating AD.
Finding small molecule compounds that will selectively cause Tau degradation is difficult. The first study on the precise knockdown of endogenous Tau protein via peptide-directed ubiquitin–proteasome degradation was published in 2016 by Tingting Chu and coworkers (Chu et al., 2016). They designed and produced several multifunctional compounds that contained moieties for interacting with E3 ligases, identifying Tau protein, and penetrating cell membranes. PROTAC11 (Fig. 12) was the synthetic compound that promoted intracellular Tau degradation the most. It managed to get through the cell membrane and enhance Tau's polyubiquitination in cells. Furthermore, the cytotoxicity brought on by Aβ was lessened by Tau degradation mediated by PROTAC11. Additionally, it controlled the Tau level in the mouse model of AD. In a nutshell, their research has described a cutting-edge AD treatment approach. They believe that the use of their synthetic molecules will make it easier to create AD treatments that work.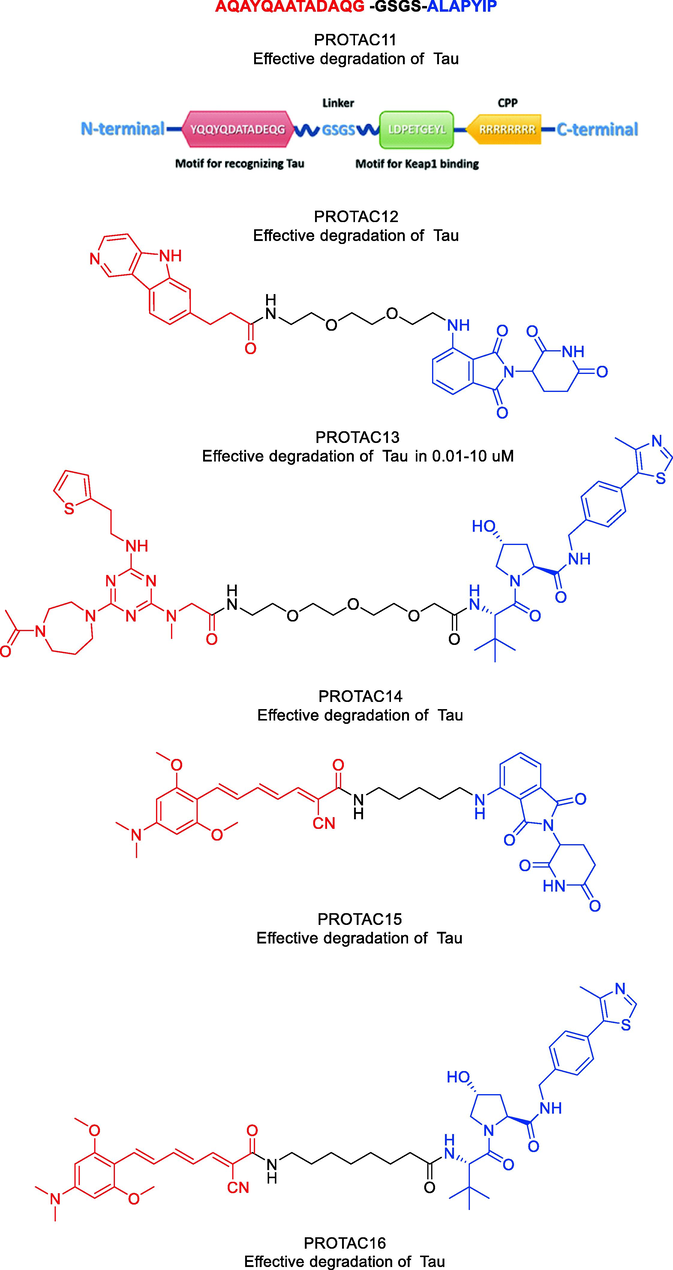
Representative PROTACs targeting Tau.
Another peptide PROTAC was created and used to degrade intracellular Tau by recruiting Keap1-Cul3 ubiquitin E3 ligase (Lu et al., 2018). Keap1 and Tau exhibited robust in vitro interaction with PROTAC12 (Fig. 12). PROTAC12 was discovered to colocalize with cellular Keap1 with appropriate cell permeability, which led to the coimmunoprecipitation of Tau and Keap1. It was determined through the use of flow cytometry and western blotting assays that PROTAC12 has the ability to reduce intracellular Tau levels in a manner that is both concentration- and time-dependent. PROTAC12′s ability to encourage Tau's poly-ubiquitination and proteasome-dependent degradation was validated by the use of Keap1 siRNA silencing and the proteasome inhibitor MG132. The findings suggested that employing PROTACs to enlist Keap1 and trigger Tau breakdown may have therapeutic potential for treating neurodegenerative diseases. Additionally, their research suggested that Keap1 could be a useful E3 ligase adaptor for the creation of new PROTACs. To test against therapeutically relevant protein phenotypes and species, their approach made use of already-existing PET probe binders in degrader design and well-characterized patient-derived neural models. PROTAC13 (Fig. 12) (Silva et al., 2019), a useful new research tool for examining Tau-mediated processes in human tauopathies, was the result of these efforts. Between 0.01 µM and 10 µM, PROTAC13 could effectively degrade both wild-type and mutant Tau in neurons. Moreover, when compared to healthy cells, PROTAC13 could preferentially degrade Tau in FTD neurons. In a mouse pharmacokinetic study to test PROTAC13′s blood–brain barrier penetration, it was shown that a dose of 30 mg/kg produced compound brain levels that were equal to those used in in vitro experiments where Tau degradation was seen. According to their research, a promising method for improving our comprehension of human neurodegenerative disease and developing tailored treatments is small molecule-mediated protein degradation.
In 2021, Weijin Wang and coworkers designed a small molecule PROTAC, named PROTAC14 (Fig. 12) (Wang et al., 2021). Three components make up PROTAC14: a VHL ligand, a linker, and a Tau binder. They showed that PROTAC14 could effectively stimulate Tau protein clearance in both healthy and pathological conditions through in vitro and in vivo testing. Remarkably, even single-dose or seldom (once per 6 days) subcutaneous treatment of PROTAC14 achieved a sustained Tau decrease in the brain of 3xTg-AD and hTau transgenic mouse models. Low Tau levels can reduce the neurotoxicity caused by Aβ, and Tau knockdown in mice does not cause obvious abnormalities. PROTAC14 is a promising therapeutic candidate for AD and the associated tauopathies as a result.
A small number of exemplified Tau-degrading PROTACs were disclosed by Mingliang Ma and colleagues in 2022 as part of a wider set of claims for CRBN-binders (Ma et al., 2022). In N2a/APP cells, the depicted Tau PROTAC (PROTAC15, Fig. 12) was found to be a moderately effective degrader. Similarly, a small number of exemplified Tau-degrading PROTACs in the context of claims describing VHL E3 ligase binders were also disclosed. The VHL-recruiting PROTAC (PROTAC16, Fig. 12) was likewise reported to be moderate to weak Tau-degrader in N2a/APP cells, with Dmax values of 50% at 10 μM. PROTAC13, PROTAC14, PROTAC15, and PROTAC16 were synthesized via synthetic pathways shown in Scheme 8a, Scheme 8b, Scheme 8c, Scheme 8d.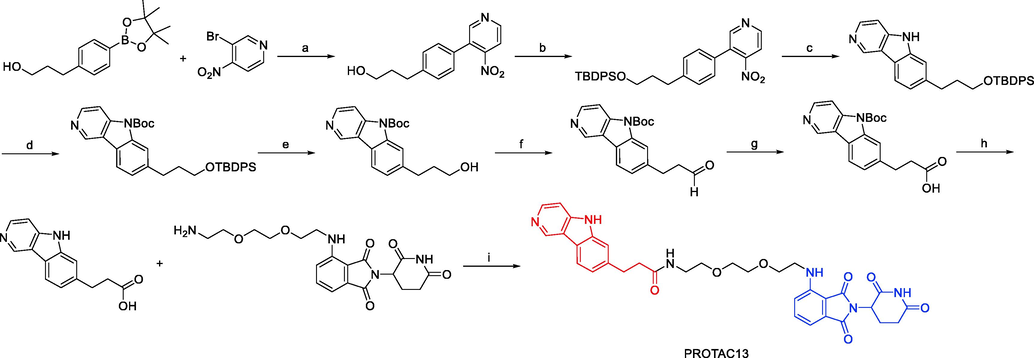
Synthesis of PROTAC13. Reagents and conditions: a) Pd(PPh3)4, Na2CO3, 1,4-dioxane, H2O, 110 °C, 16 h; b) TBDPSCl, imidazole, DCM, r.t., 3 h; c) P(OEt)3, 110 °C, 3 h; d) (Boc)2O, DMAP, Et3N, DCM, r.t., 2 h; e) TBAF, THF, 0 °C, 2 h; f) (COCl)2, DMSO, Et3N, DCM, −78 °C-0 °C, 1.5 h; g) NaClO2, NaH2PO4, 2-methyl butene, THF/t-BuOH/H2O, r.t., 2 h; h) TFA, DCM, r.t., 12 h; i) EDCI, DMAP, DIPEA, DCM, r.t., 4 h.
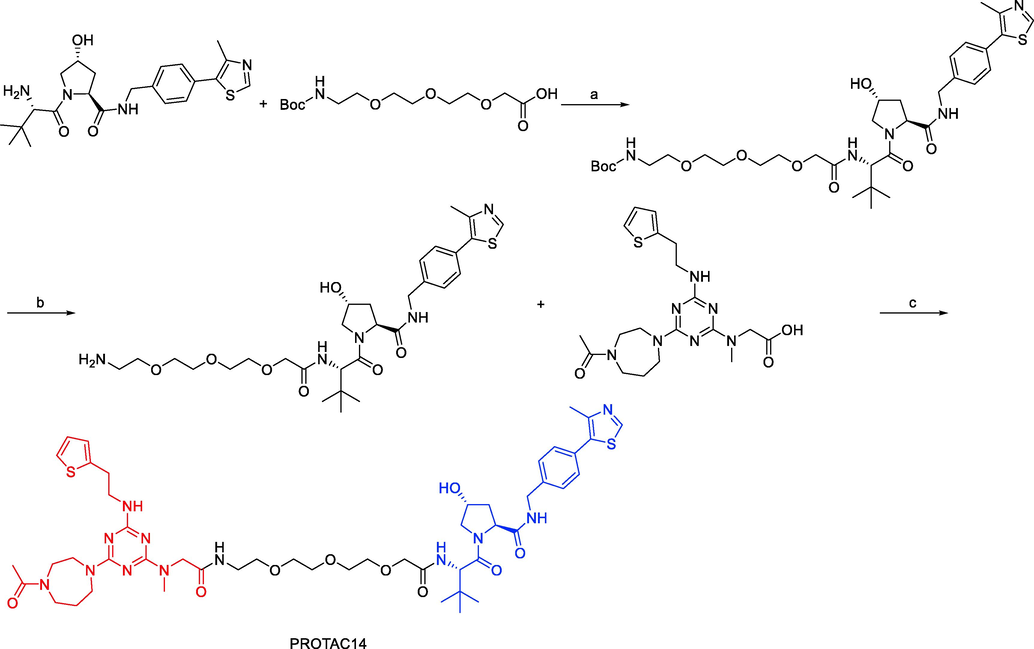
Synthesis of PROTAC14. Reagents and conditions: a) DIPEA, HATU, DMF, r.t., overnight; b) 2 M HCl/EA, DCM, r.t., 1 h; c) DIPEA, HATU, DMF, r.t., overnight.

Synthesis of PROTAC15. Reagents and conditions: a) Dimethyl sulfate, K2CO3, DMF, −4°C, 4–6 h; b) POCl3, DMF, 0 °C, 12–14 h; c) ((1,3-Dioxolan-2-yl)methyl) triphenylphosphonium bromide, CH3ONa/MeOH (25%), THF, 78 °C, 12–16 h; d) ((1,3-Dioxolan-2-yl)methyl) triphenylphosphonium bromide,CH3ONa/MeOH (25%), THF, 78 °C, 12–16 h; e) cyanoacetic acid, K2CO3, MeOH, 70 °C, 12 h; f) DIPEA, DMAP, HATU, r.t., 2 h.
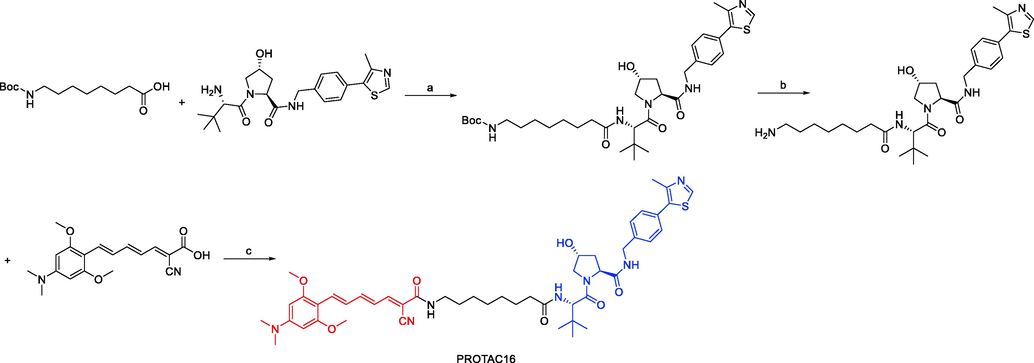
Synthesis of PROTAC16. Reagents and conditions: a) DIPEA, HATU, DMAP, THF, 25 °C, 2–3 h; b) 2 M HCl/EtOH, CH3OH, 25 °C, 6–8 h; c) DIPEA, DMAP, HATU, r.t., 2 h.
3.6 TRKs
The NTRK1, NTRK2, and NTRK3 genes, respectively, encode the three members of the TRKs receptor family: TRKA, TRKB, and TRKC. TRKs are receptor tyrosine kinases that are primarily involved in the development and function of neuronal tissue (Martin-Zanca et al., 1986). Nerve growth factor (NGF) for TRKA, brain-derived neurotrophic factor (BDNF) for TRKB, and neurotrophins for TRKC are the major ligands of TRKs. Induced dimerization and activation of the intracellular kinase domains by the binding of ligands to the extracellular domains of TRKs provides signals to downstream pathways, including RAF/MEK/ERK, PI3K/AKT, and phospholipase C gamma (PLCγ) (Rydén et al., 1996). There have been reports of chromosomal translocation, point mutations, amplification, and alternative splicing among other genetic aberrations of NTRK genes. These modifications cause the TRK pathway to be constitutively activated, which encourages cellular growth, survival, and malignant transformation.
The first TRKC PROTACs were described by Bosheng Zhao and coworkers in 2019 and were based on the TRKC binder IY-IY (Kd = 112 nM) and various E3 ligase ligands (Zhao et al., 2019). With an estimated DC50 of 0.1–1.0 μM, the pomalidomide-based degrader PROTAC17 (Fig. 13) demonstrated improved TRKC degradation activity. TRKC degraders may present prospects for alternate therapeutic approaches for TRKC-related diseases.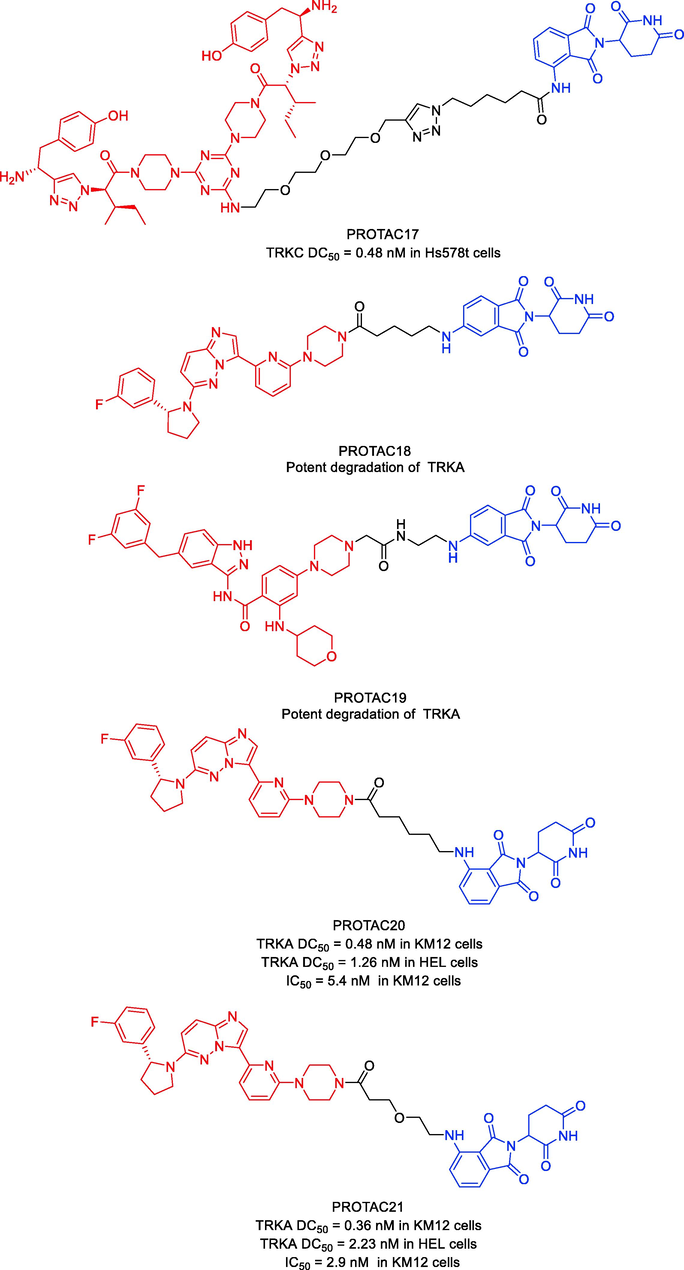
Representative PROTACs targeting TRKs.
Jing Liu and coworkers published a patent filing disclosing TRKA degraders, comprising more than 200 exemplars of TRK PROTACs (Liu et al., 2020). Exemplified compounds in this filing all employ binders to CRBN and VHL as the E3 ligases recruiting moiety. Representative compounds PROTAC18 and PROTAC19 (Fig. 13) could effectively and selectively inhibit the proliferation of KM12 cells (PROTAC18 IC50 = 0.9 nM, PROTAC19 IC50 = 0.9 nM). PROTAC18 and PROTAC19 could degrade TRKA protein in a dose-dependent manner by the ubiquitin–proteasome pathway. This filling also contains voluminous biological data to support the potential utility of these TRK degraders to treat NDs and oncology.
In 2020, Liqun Chen and coworkers developed a variety of putative protein PROTACs molecules with various types of linkers based on the X-ray cocrystal structure of TRKA in complex with an analog of GNF-8625 (Chen et al., 2020). The most promising degraders that target the intracellular kinase domain of TRKs are PROTAC20 and PROTAC21 (Fig. 13). In KM12 colorectal cancer cells, PROTAC20 (DC50 = 0.48 nM) and PROTAC21 (DC50 = 0.36 nM) decreased tropomyosin 3 (TPM3)-TRKA fusion protein levels and blocked downstream PLC1 signaling at sub-nanomolar concentrations. Both degraders also degraded human wild-type TRKA with DC50 values of 1.26 nM and 2.23 nM in HEL cells. It is interesting to note that in KM12 cells, both degraders, particularly PROTAC21, showed selectivity for the degradation of endogenous TPM3-TRKA over ectopically produced ATP/GTP binding protein-like 4 (AGBL4)-TRKB or ETS variant transcription factor 6 (ETV6)-TRKC fusion proteins. Assays using a global proteome profiler showed that PROTAC20 is very selective for the intended target. It was further established that CRBN and the ubiquitin–proteasome system are responsible for the TPM3-TRKA protein degradation caused by PROTAC20 and PROTAC21. Both degraders showed more potency for preventing the proliferation of KM12 cells as compared to the parental TRKs kinase inhibitor. Additionally, both PROTAC20 and PROTAC21 showed good plasma exposure levels in mice. Therefore, PROTAC20 and PROTAC21 are potential chemical tool compounds to study the in vivo function of TRKs fusions during tumorigenesis. This study also points the way for the pharmacological degradation of TRKs. PROTAC17, PROTAC18, PROTAC19, PROTAC20, and PROTAC21 were synthesized via synthetic pathways shown in Scheme 9a, Scheme 9b, Scheme 9c, Scheme 9d.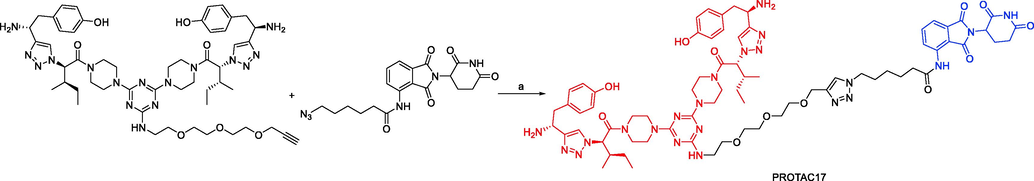
Synthesis of PROTAC17. Reagents and conditions: a) CuSO4, DMSO/H2O = 2/1, r.t., 24 h.
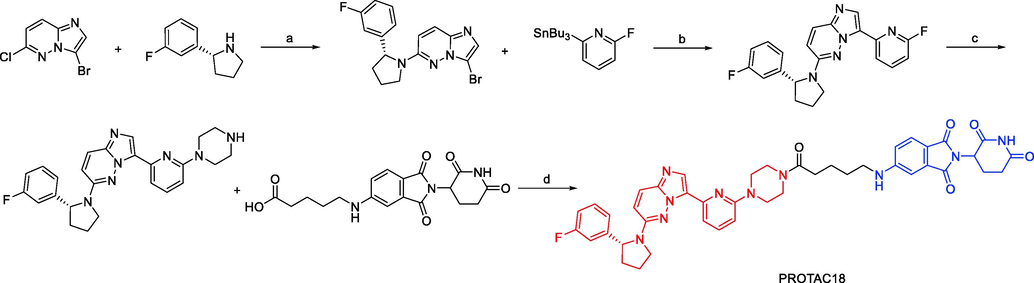
Synthesis of PROTAC18. Reagents and conditions: a) KF, DMSO, 100 °C, 12 h; b) Pd(PPh3)4, toluene,110 °C, 12 h; c) piperazine, KF, DMSO, 130 °C, 12 h; d) DIPEA, DMAP, HATU, r.t., 2 h.
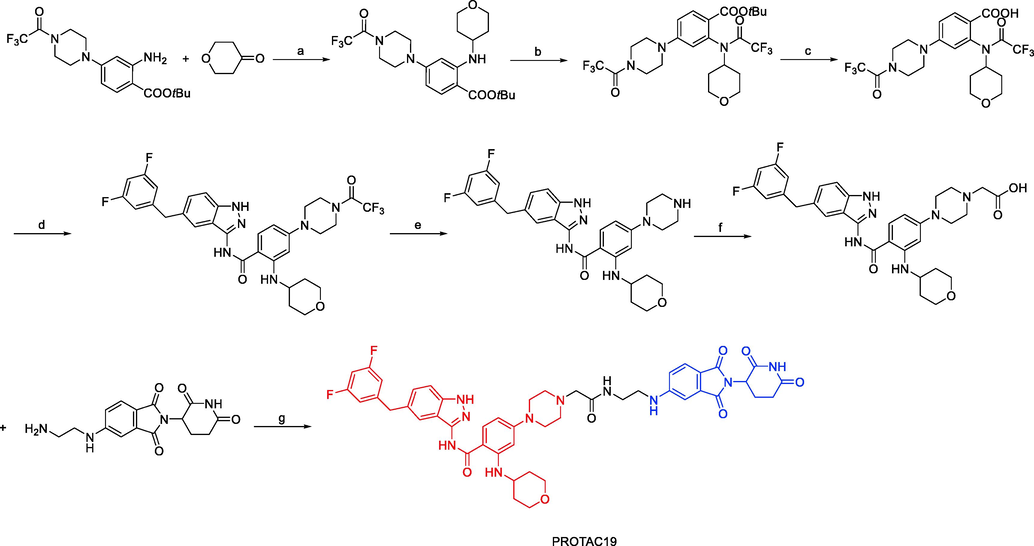
Synthesis of PROTAC19. Reagents and conditions: a) TFA, Me4N+BH(OAc)3, DCM, r.t., 16 h; b) TFAA, Et3N, DCM, r.t., 1 h; c) TFA, DCM, r.t., 16 h; d) i) (COCl)2, DCM, 0 °C, 30 min; ii) 5-(3,5-difluorobenzyl)-1H-indazol-3-amine, Et3N, DCM, r.t., 16 h; e) K2CO3, MeOH, r.t., 2 h; f) i) tert-butyl-2-bromoacetate, K2CO3, DMF, r.t., 3 h; ii) TFA, DCM, 3 h; g) DIPEA, DMAP, HATU, r.t., 2 h.
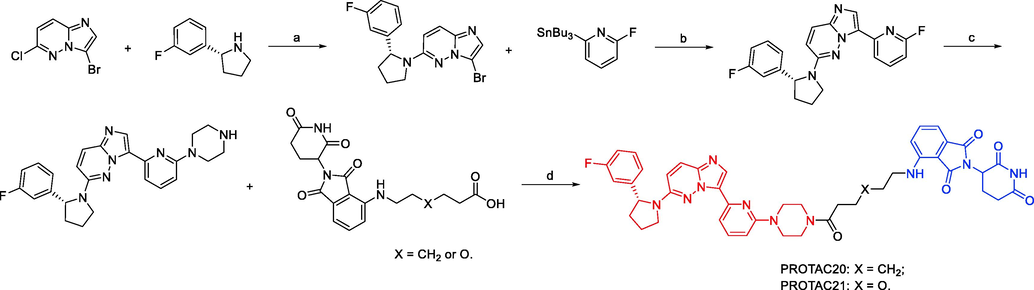
Synthesis of PROTAC20 and PROTAC21. Reagents and conditions: a) KF, DMSO, 100 °C, 12 h; b) Pd(PPh3)4, toluene,110 °C, 12 h; c) piperazine, KF, DMSO, 130 °C, 12 h; d) EDCI, HOAt, NMM, DMSO, r.t., overnight.
4 Conclusion and perspectives
NDs are heterogeneous, currently incurable diseases that cause nerve cells to gradually deteriorate. There is presently no truly effective treatment for NDs, despite the fact that the World Health Organization predicts they will overtake heart disease as the second most common cause of mortality in the next 20 years. Treatments for NDs, such as reliance on small molecule drug therapy and immunotherapy targeting disease-causing proteins, can only alleviate the symptoms of the disease and cannot achieve reversal of disease progression. Even while innovative NDs treatment modalities like autophagy therapy, miRNA therapy, and stem cell therapy have been established, they still have limitations like poor BBB penetration, off-target effects, and the inability to affect intracellular proteins. PROTACs are a potent and alluring technique that has been researched and developed in both academia and industry. A critical property of PROTACs is the ability to degrade proteins that cannot be typically targeted by small-molecule inhibitors due to the lack of active sites. Due to this characteristic, the PROTACs method may be used to treat a number of neurodegenerative illnesses caused by the toxic accumulation of proteins including α-synuclein, GSK-3β, LRRK2, mHTT, Tau, TRKA, and TRAC. There have been more in-depth studies done on the treatments for NDs, and the advancement of PROTACs technology has demonstrated significant promise in addressing the issue of undruggable protein targeted degradation. As shown by the aforementioned instances, PROTAC technology has been flourishing in recent years, displaying successful outcomes in the induction of the degradation of a variety of NDs proteins and overcoming some of the issues related to traditional small-molecule inhibitors. Unlike traditional small molecule inhibitors, PROTACs function through a unique event-driven model in which they induce the formation of ternary complexes and cycle through many rounds of target protein recruitment. In addition to being able to degrade undruggable targets, PROTACs have a number of distinct benefits over traditional small-molecule inhibitors due to their degradation mechanism. First off, PROTACs' catalytic properties make it possible to remove proteins even at sub-stoichiometric concentrations. Because of their catalytic character, PROTACs can be used without the need for high medication doses, which are frequently linked to toxicity and side effects. Moreover, PROTACs offer a higher level of target selectivity than POI ligands: for instance, the degree to which E3 and POI cooperate can affect the PROTACs’ target selectivity. As a result, by carefully choosing linkers and E3 ligands, it is possible to design selective PROTACs for specific proteins over related homologues. Furthermore, PROTAC technology can overcome the resistance mechanisms common to small-molecule inhibitors. Ibrutinib (Timofeeva, et al., 2021), for instance, is frequently ineffective in treating AML when it causes the BTK C481S mutation; yet, PROTACs based on ibrutinib can still effectively degrade BTKC481S (Wang, et al., 2021). Additionally, protein overexpression is a common form of resistance to treatment with small-molecule inhibitors; however, degradation induced by PROTACs can offer an effective and prolonged effect due to their event-driven model, as shown by the fact that the KRASG12C-targeting PROTAC demonstrated longer KRASG12C downregulation than the parent inhibitor (MRTX849) did (Farrell, et al., 2021). Finally, in contrast to inhibitors, PROTACs can block both the scaffolding and enzymatic functions of POIs. PROTACs may significantly improve the limited clinical efficacy of traditional small-molecule inhibitors, offering novel therapeutic possibilities for the treatment of neurodegenerative diseases.
Despite the fact that neurodegenerative diseases PROTACs present a promising strategy for a variety of areas, such as drug discovery and solutions to biological issues, difficulties still lie ahead for these PROTACs in the future. Only a select few E3 ligases have thus far been exploited in the development of useful PROTACs, despite the fact that different E3 ligases have demonstrated varying activity and selectivity for protein degradation. To achieve selectivity in the subcellular or cellular degradation of particular proteins, it is possible to take advantage of the different localization and expression variations among the E3 ligases. The varied protein–protein interactions in various situations may further enlist distinctive selectivity of protein degradation at particular contexts, a trait not conceivable with genetic ablation of proteins, although this has not yet been confirmed. The linker's chemical makeup and physical attributes appear to have a significant impact on PROTAC activity. Linker design essentials like composition and length have not yet been fully understood. Work must be completed fast in order to obtain the greatest linkers. Traditional guidelines like Lipinski's rule might not be appropriate to make a go/no go judgment in a drug discovery project due to their unique mechanisms of action. Even though we noted above that many endogenous targets were efficiently degraded, other proteins have been found to be resistant to PROTAC-mediated degradation. In addition, more investigation is needed to evaluate the absorption, distribution, metabolism, excretion, and toxicity of PROTACs for neurodegenerative diseases. This is because the mechanism underlying PROTACs is not well understood. PROTACs for neurodegenerative diseases have the potential to be developed as therapeutic agents for many deadly diseases, but they confront significant challenges that must be solved.
Author contributions
Concept design: CW, YZ, and DX; CW, YZ, and SY wrote the manuscript; CW and DX directed the study.
Funding
This work was supported by grants from the Natural Science Foundation of Shandong (ZR2021QH156), the Medical and Health Science and Technology Development Plan Project of Shandong (202113051140), and the Key Project of Qingdao University Medical Group Scientific Research Project (YLJT20211014).
Acknowledgments
We thank the laboratory of Professor Dongming Xing for generously supplying reagents, technical assistance, stimulating discussions, and funding.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alzheimer's Association, (2010). 2010 Alzheimer's Disease Facts and Figures. Alzheimer's & dementia: The Journal of the Alzheimer's Association 6 (2), 158-194. doi:10.1016/j.jalz.2010.01.009
- Small-molecule PROTACs: an emerging and promising approach for the development of targeted therapy drugs. EBioMedicine. 2018;36:553-562.
- [CrossRef] [Google Scholar]
- From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat. Rev. Drug Discov.. 2016;15(4):275-292.
- [CrossRef] [Google Scholar]
- Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol.. 2010;28(3):256-263.
- [CrossRef] [Google Scholar]
- The therapeutic potential of PROTACs. Expert Opin. Ther. Pat.. 2021;31(1):1-24.
- [CrossRef] [Google Scholar]
- Comprehensive review on Alzheimer's disease: causes and treatment. Molecules (Basel, Switzerland). 2020;25(24):5789.
- [CrossRef] [Google Scholar]
- Efficient synthesis of immunomodulatory drug analogues enables exploration of structure-degradation relationships. ChemMedChem. 2018;13:1508-1512.
- [CrossRef] [Google Scholar]
- Chemistries of bifunctional PROTAC degraders. Chem. Soc. Rev.. 2022;51(16):7066-7114.
- [CrossRef] [Google Scholar]
- Facile synthesis of an azido-labeled thalidomide analogue. Org. Lett.. 2003;5:2865-2867.
- [CrossRef] [Google Scholar]
- Structure of the human Cereblon-DDB1-Lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat. Struct. Mol. Biol.. 2014;21(9):803-809.
- [CrossRef] [Google Scholar]
- Innovative approaches in CNS drug discovery. Therapie. 2021;76(2):101-109.
- [CrossRef] [Google Scholar]
- Chemically induced degradation of CK2 by proteolysis targeting chimeras based on a ubiquitin proteasome pathway. Bioorg. Chem.. 2018;81:536-544.
- [CrossRef] [Google Scholar]
- Discovery of first-in-class potent and selective tropomyosin receptor kinase degraders. J. Med. Chem.. 2020;63(23):14562-14575.
- [CrossRef] [Google Scholar]
- Demonstrating in-cell target engagement using a pirin protein degradation probe (CCT367766) J. Med. Chem.. 2018;61:918-933.
- [CrossRef] [Google Scholar]
- Protein degraders enter the clinic-a new approach to cancer therapy. Nat. Rev. Clin. Oncol.. 2023;20(4):265-278.
- [CrossRef] [Google Scholar]
- Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem. Biol.. 2016;23(4):453-461.
- [CrossRef] [Google Scholar]
- Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol.. 2018;14(7):399-415.
- [CrossRef] [Google Scholar]
- Advances in automation technologies for lower extremity neurorehabilitation: a review and future challenges. IEEE Rev. Biomed. Eng.. 2018;11:289-305.
- [CrossRef] [Google Scholar]
- Enzymatic logic of ubiquitin chain assembly. Front. Physiol.. 2019;10:835.
- [CrossRef] [Google Scholar]
- Glycogen synthase kinase 3: an emerging therapeutic target. Trends Mol. Med.. 2002;8(3):126-132.
- [CrossRef] [Google Scholar]
- Glycogen synthase kinase-3 from rabbit skeletal muscle. separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem.. 1980;107(2):519-527.
- [Google Scholar]
- Is PROTAC technology really a game changer for central nervous system drug discovery? Expert Opin. Drug Discov.. 2021;16(8):833-840.
- [CrossRef] [Google Scholar]
- Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat. Chem. Biol.. 2017;13(5):514-521.
- [CrossRef] [Google Scholar]
- GBD 2016 Neurology Collaborators, 2019. Global, Regional, and National Burden of Neurological Disorders, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurology 18 (5), 459-480. doi:10.1016/S1474-4422(18)30499-X
- A total synthesis of (-)-bestatin using Shibasaki’s asymmetric henry reaction. Tetrahedron Lett.. 2005;46:7581-7582.
- [CrossRef] [Google Scholar]
- The need for new approaches in CNS drug discovery: why drugs have failed, and what can be done to improve outcomes. Neuropharmacology. 2017;120:11-19.
- [CrossRef] [Google Scholar]
- PROTACs: an emerging targeting technique for protein degradation in drug discovery. Bioessays. 2018;40(4)
- [CrossRef] [Google Scholar]
- Discovery of ARD-69 as a highly potent proteolysis targeting Chimera (PROTAC) degrader of Androgen Receptor (AR) for the treatment of prostate cancer. J. Med. Chem.. 2019;62:941-964.
- [CrossRef] [Google Scholar]
- Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of Estrogen Receptor (ER) J. Med. Chem.. 2019;62:1420-1442.
- [CrossRef] [Google Scholar]
- New synthesis route for the preparation of Pomalidomide. Synth. Commun.. 2016;46:1343-1348.
- [CrossRef] [Google Scholar]
- PROTACs technology for treatment of Alzheimer's disease: advances and perspectives. Acta Mater. Med.. 2022;1(1):24-41.
- [CrossRef] [Google Scholar]
- Past, present and future of therapeutic strategies against amyloid-Β peptides in Alzheimer's disease: a systematic review. Ageing Res. Rev.. 2021;72
- [CrossRef] [Google Scholar]
- PROTACs suppression of GSK-3β, A crucial kinase in neurodegenerative diseases. Eur. J. Med. Chem.. 2021;210
- [CrossRef] [Google Scholar]
- PROTAC compounds targeting α-synuclein protein for treating neurogenerative disorders: Alzheimer's and Parkinson's diseases. ACS Med. Chem. Lett.. 2020;11(6):1086-1087.
- [CrossRef] [Google Scholar]
- The tale of proteolysis targeting chimeras (PROTACs) for leucine-rich repeat kinase 2 (LRRK2) ChemMedChem. 2021;16(6):959-965.
- [CrossRef] [Google Scholar]
- Liu, J., Plewe, M.B., Wang, J., Han, X., Chen, L, 2020. Tropomyosin Receptor Kinase (Trk) Degradation Compounds and Methods of Use. Patent, WO2020/038415A1
- Discovery of XL01126: a potent, fast, cooperative, selective, orally bioavailable, and blood-brain barrier penetrant PROTAC degrader of leucine-rich repeat kinase 2. J. Am. Chem. Soc.. 2022;144(37):16930-16952.
- [CrossRef] [Google Scholar]
- Discovery of A Keap1-dependent peptide PROTAC to knockdown tau by ubiquitination-proteasome degradation pathway. Eur. J. Med. Chem.. 2018;146:251-259.
- [CrossRef] [Google Scholar]
- Ma, M., Li, C., D, Y., W, Z., East China Normal University, 2022. Tau Protein Visualization PROTAC Degradation Compound and Its Preparation Method and Application. Patent, CN114736264 A
- Proteolysis targeting chimera technology: a novel strategy for treating diseases of the central nervous system. Neural Regen. Res.. 2021;16(10):1944-1949.
- [CrossRef] [Google Scholar]
- Specific non-genetic IAP-based protein erasers (SNIPERs) as a potential therapeutic strategy. Eur. J. Med. Chem.. 2021;216
- [CrossRef] [Google Scholar]
- The potential of flavonoids for the treatment of neurodegenerative diseases. Int. J. Mol. Sci.. 2019;20(12):3056.
- [CrossRef] [Google Scholar]
- PROTACs to address the challenges facing small molecule inhibitors. Eur. J. Med. Chem.. 2021;210
- [CrossRef] [Google Scholar]
- A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature. 1986;319(6056):743-748.
- [CrossRef] [Google Scholar]
- Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov.. 2021;20(4):247-250.
- [CrossRef] [Google Scholar]
- Nichols, W.C., Pankratz, N., Hernandez, D., Paisán-Ruíz, C., Jain, S., Halter, C.A., Michaels, V.E., Reed, T., Rudolph, A., Shults, C.W., Singleton, A., Foroud, T., 2005. Genetic Screening for A Single Common LRRK2 Mutation in Familial Parkinson's Disease. Lancet (London, England) 365 (9457), 410-412. dio:10.1016/S0140-6736(05)17828-3 .
- In vivo knockdown of pathogenic proteins via specific and nongenetic inhibitor of apoptosis protein (IAP)-dependent protein erasers (SNIPERs) J. Biol. Chem.. 2017;292:4556-4570.
- [CrossRef] [Google Scholar]
- From inhibition to degradation: targeting the antiapoptotic protein myeloid cell Leukemia 1 (MCL1) J. Med. Chem.. 2019;62:5522-5540.
- [CrossRef] [Google Scholar]
- Pohl, C., Dikic, I., 2019. Cellular Quality Control by the Ubiquitin-Proteasome System and Autophagy. Science (New York, N.Y.) 366 (6467), 818-822. doi:10.1126/science.aax3769.
- Discovery of PT-65 as a highly potent and selective proteolysis-targeting chimera degrader of GSK3 for treating Alzheimer's disease. Eur. J. Med. Chem.. 2021;226
- [CrossRef] [Google Scholar]
- Raina, K., Lu, J., Qian, Y., Altieri, M., Gordon, D., Rossi, A.M.K., et al., 2016. PROTAC-induced BET Protein Degradation as a Therapy for CastrationResistant Prostate Cancer. Proceedings of the National Academy of Sciences of the United States of America 113, 7124-7129. doi:10.1073/pnas.1521738113.
- Genetics and neuropathology of Huntington's disease. Int. Rev. Neurobiol.. 2011;98:325-372.
- [CrossRef] [Google Scholar]
- Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC) Chem. Commun.. 2017;53:7577-7580.
- [CrossRef] [Google Scholar]
- Expression of mRNA for the neurotrophin receptor Trkc in neuroblastomas with favourable tumour stage and good prognosis. Br. J. Cancer. 1996;74(5):773-779.
- [CrossRef] [Google Scholar]
- Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421(6921):373-379.
- [CrossRef] [Google Scholar]
- Alzheimer's disease. Lancet (London, England). 2021;397(10284):1577-1590.
- [CrossRef] [Google Scholar]
- Discriminating α-Synuclein strains in Parkinson's disease and multiple system atrophy. Nature. 2020;578(7794):273-277.
- [CrossRef] [Google Scholar]
- Targeted degradation of aberrant Tau in frontotemporal dementia patient-derived neuronal cell models. Elife. 2019;8
- [CrossRef] [Google Scholar]
- Homo-PROTACs for the chemical knockdown of cereblon. ACS Chem. Biol.. 2018;13(9):2771-2782.
- [CrossRef] [Google Scholar]
- PROTACs: great opportunities for academia and industry. Signal Transduct. Targ. Ther.. 2019;4:64.
- [CrossRef] [Google Scholar]
- Thaler, A., Ash, E., Gan-Or, Z., Orr-Urtreger, A., Giladi, N., 2009. The LRRK2 G2019S Mutation as the Cause of Parkinson's Disease in Ashkenazi Jews. Journal of Neural Transmission (Vienna, Austria: 1996) 116 (11), 1473-1482. dio:10.1007/s00702-009-0303-0.
- Ibrutinib combinations in CLL therapy: scientific rationale and clinical results. Blood Cancer J.. 2021;11(4):79.
- [CrossRef] [Google Scholar]
- Tomoshige, S., Nomura, S., Ohgane, K., Hashimoto, Y., Ishikawa, M., 2017. Discovery of Small Molecules that Induce the Degradation of Huntingtin. Angewandte Chemie (International ed. in English) 56 (38), 11530-11533. dio:10.1002/anie.201706529.
- Degradation of Huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett.. 2018;28(4):707-710.
- [CrossRef] [Google Scholar]
- Toure, M., Crews, C.M., 2016. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angewandte Chemie (International ed. in English) 55 (6), 1966-1973. dio:10.1002/anie.201507978.
- Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1α protein-protein interface. Chem. Biol.. 2012;19:1300-1312.
- [CrossRef] [Google Scholar]
- Developments of CRBN-based PROTACs as potential therapeutic agents. Eur. J. Med. Chem.. 2021;225
- [CrossRef] [Google Scholar]
- PROTACs technology for targeting non-oncoproteins: advances and perspectives. Bioorg. Chem.. 2021;114
- [CrossRef] [Google Scholar]
- A novel small-molecule PROTAC selectively promotes tau clearance to improve cognitive functions in Alzheimer-like models. Theranostics. 2021;11(11):5279-5295.
- [CrossRef] [Google Scholar]
- Recent advances of degradation technologies based on PROTAC mechanism. Biomolecules. 2022;12(9):1257.
- [CrossRef] [Google Scholar]
- Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: recent progress and future challenges. Eur. J. Med. Chem.. 2021;210
- [CrossRef] [Google Scholar]
- TrkC-targeted kinase inhibitors and PROTACs. Mol. Pharm.. 2019;16(10):4313-4318.
- [CrossRef] [Google Scholar]
- Cereblon expression is required for the antimyeloma activity of Lenalidomide and Pomalidomide. Blood. 2011;118(18):4771-4779.
- [CrossRef] [Google Scholar]




