

Translate this page into:
An overview of ion channels therapeutics in the treatment of pain
⁎Corresponding author at: Department of Pharmaceutical Chemistry, College of Pharmacy, King Khalid University, Abha, Saudi Arabia. drzahin@gmail.com (Mohd. Zaheen Hassan) zaheen@kku.edu.sa (Mohd. Zaheen Hassan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Significance of the study
The clinical management of severe and chronic pain relies heavily on opioids that cause serious side-effects. There therefore exists an urgent need to develop safer and effective analgesics. Moreover, there has been significant progress in the understanding of pain physiology, especially the role of some ion channels in the pain process. Thus, the immense potential of ion channel therapeutics in pain management is a subject of current interest.
Aim of the study
This study is a comprehensive review, focused on ion channels as potential therapeutic targets for the treatment of pain.
Research methodology
A systemic search of available literature on ion channels analgesics was performed. Articles related to the drug discovery and clinical trials on relevant topics were extracted from PubMed and other databases.
Major conclusion of the study
Small molecules targeted at ion channels pathways hold great promise for creating a new approach to pain treatment. Several molecules targeting TRPV1, TRPV3, TRPV4, TRPA1, TRPM8, Nav1.7, Nav1.8, CaV2.2, CaV3.2, ASIC, and P2X3 have demonstrated potential clinical benefits. However, till date US FDA has approved capsaicin, ziconotide, and pregabalin for the treatment of different pain conditions. This review highlights the possibilities for discovery and research on ion channels and their potential for pain treatment.
Keywords
Analgesics
Ion Channels
Transient receptor potential channels
Sodium channels
And calcium channels

1 Introduction
Pain is a serious healthcare problem that can adversely affect human health and quality of life. Chronic pain affects around 17 million people worldwide and among these > 75% experience moderate to severe pain (WHO guidelines, 2018). According to GSK global pain index, globally more than half of people claim to have experienced body pain on a weekly basis (Hunter et al., 2008). Around, 20% of the world’s population experience some sort of enduring pain, and the older patients are more likely to experience several conditions such as arthritis, bone-joint disorders, and other chronic illnesses associated with pain (Thomas, 2013). According to recent surveys, approximately 70% of the medical care in emergency departments of hospitals is being carried out for the management of acute pain and acute exacerbations of chronic pain (Wu et al., 2019). Moreover, the Global Burden of Disease study estimated that the pain and the pain-related syndrome is the leading cause of disability and disease burden worldwide (Vos et al., 2017).
Pain is a traumatic experience often processed by a specific high-threshold sensory receptor called “nociceptor” (Osterweis et al., 1987). The International Association for the Study of Pain (IASP) has defined pain as “an obnoxious sensory and emotional sense due to actual or likely tissue damage” (IASP Terminology, 1979). Pain is a subjective or emotional experience that arises from potentially damaging noxious stimuli, and the release of pain mediators from the injured or inflamed tissues, and dysesthesia due to sensory nerve damage (Yaksh et al., 2018). The neuronal event of pain sensation proceeds through four phases, viz. transduction, transmission, pain modulation, and perception (Yam et al., 2018). Transductions occurs at the end of sensory nerve cells when noxious stimuli such as intense thermal, mechanical, and chemical stimuli is converted into a nerve signal or action potential. The pain information is then transmitted from the peripheral to the central nervous system. Pain modulation refers to up or down regulation of pain signals throughout the spinal cord and the brain. Finally, the signal is processed in the brain for the perception of pain as uncomfortable awareness (Belmonte and Viana, 2007). Sometimes, alteration in these steps due to tissue or nerve damage, as in the case of inflammation and cancer, may cause sensory hypersensitivity, a condition of pain without any stimuli (Cummins et al., 2007). These threshold reduction does not serve as a warning and, over a period, often turn into chronic pain characterized by persistent somatic disorders associated with psychological anguishes such as tension, anxiety, and depression (Katz et al., 2015).
Recent progress in the discovery of analgesics was only possible because of several advanced animal models that mimic many aspects of human pain (Abboud et al., 2021). In the last sixty years, around 140 analgesics belonging to different chemical classes have been approved by the U.S. Food and Drug Administration (FDA) (Igor, 2010). Some important class of approved analgesics includes opiates, phenylacetic acid, propionic acids, salicylates, oxicams, fenamates, diarylpyrazoles etc. However, majority of these drugs are associated with intolerable adverse effects, including nephrotoxicity, cardiotoxicity, constipation, respiratory depression as well as stomach ulcers that can lead to internal bleeding and anaemia (Hill, 2006).
Moreover, several promising clinical candidates failed in the late stages of clinical studies due to their poor drug-like properties. Thus, research focusing on deciphering the underlying mechanisms of chronic pain and developing novel non-addictive and efficacious analgesics is the need of the hour. Taking benefits of exciting new advances in discovery and research on analgesics, we provided an overview of promising investigational molecules that have been evaluated clinically for their analgesic potential. We also highlighted the potential ion channels as novel molecular targets, and their modulators' development, focusing mainly on lead optimization and their progress as clinical candidates.
2 Ion channels as a therapeutic target for pain management
Modulation of ion channel signalling has received much attention from big pharma companies due to their potential to effectively treat pain. The top analgesic companies are now developing more selective, safer, and effective drugs to improve treatment outcomes and patient experience. According to an estimate, ion channels account for around 21% of the total analgesic pipeline. Of the ion channels, Na+ and Ca2+, which play a vital role in pain, lead the ion channel pipeline. Therefore, we have reviewed the current research trends and lists of pipelines with approved drugs of the top pharma companies focusing research on ion channels therapeutics. (Table 1). ® Registered trademark, *Structure not disclosed, $ Orphan-drug designation by the FDA.
S.No.
Drug
Developer
Target channel
Indications
CT Identifier
Progress
1.
QUTENZA®
(8% capsaicin TD patch)Averitas Pharma
TRPV1
PHN, DPN
–
FDA approval-2009
2.
Civamide/Zucapsaicin
Winston Pharmaceuticals
TRPV1
Episodic cluster headache
NCT00033839
Phase III
3.
Vocacapsaicin
Concentric Analgesics
TRPV1
Total knee arthroplasty
NCT03731364
Phase II
4.
Resiniferatoxin
Sorrento Therapeutics
TRPV1
Cancer pain
NCT00804154
Phase I
5.
NEO-6860*
NEOMED Institute
TRPV1
OA
NCT02712957
Phase II
6.
AMG-517
Amgen Inc.
TRPV1
Pain due to tooth extraction
–
Phase I
7.
ABT-102
Abbott
TRPV1
–
NCT00854659
Phase I
8.
AZD-1386
AstraZeneca
TRPV1
Pain due to tooth extraction
NCT00672646
Phase II
9.
DWP-05195
Daewoong
TRPV1
PHN
NCT01557010
Phase II
10.
MK-2295
Merck
TRPV1
Pain due to tooth extraction
NCT00387140
Phase II
11.
SB-705498
GlaxoSmithKline
TRPV1
Pain due to tooth extraction
NCT00281684
Phase II
12.
GRC-15300*
Glenmark
TRPV3
Chronic neuropathic pain
NCT01463397
Phase II
13.
GSK-2798745
GlaxoSmithKline
TRPV4
Diabetic macular edema
NCT04292912
Phase I
14.
GRC-17536
Glenmark Pharmaceuticals
TRPA1
DPN
NCT01726413
Phase II
15.
CB-625*
Cubist Pharmaceuticals Inc.
TRPA1
Acute pain
–
Phase I
16.
HX-100*
Hydra Biosciences
TRPA1
DPN
–
Phase I
17.
ODM-108*
Orion Pharma
TRPA1
–
NCT02432664
Phase I
18.
Menthol
University of Brit. Columbia
TRPM8
DPN
NCT02728687
Phase I/II
19.
PF-05105679
Pfizer
TRPM8
Pain
NCT01393652
Phase I
20.
AMG-333
Amgen
TRPM8
Migraine
NCT01953341
Phase I
21.
Halneuron® (TTX)
WEX Pharmaceuticals
Nav1.7
CRP, CINP
NCT00725114
Phase III
22.
NeoSTX
Grunenthal
Nav1.7
LA
NCT01786655
Phase I
23.
ST-2427*
SiteOne Therapeutics
Nav1.7
Moderate-to-severe pain
NCT04475198
Phase I
24.
Ralfinamide
Newron Pharmaceuticals
Nav1.7
NLBP
NCT01019824
Phase III
25.
PF-05089771
Pfizer Inc
Nav1.7
DPN, Dental pain
NCT02215252
Phase II
26.
Vixotrigine/Raxatrigine $
Convergence Pharmaceuticals
Nav1.7
TN, LR
NCT03637387
Phase III
27.
Funapide$
Flexion Therapeutics
Nav1.7
PHN, OA
NCT02068599
Phase II
28.
DSP-2230/ANP-230
Alphanavi Pharma
Nav1.7
Peripheral neuropathy
IRAS ID 103329
Phase I
29.
GDC-0276
Genentech
Nav1.7
Pain
–
Phase I
30.
GDC-0310
Genentech
Nav1.7
Pain
–
Phase I
31.
AZD-3161
AstraZeneca
Nav1.7
UVC exposed Skin
NCT01240148
Phase I
32.
GSK-2339345
GlaxoSmithKline
Nav1.7
Refractory chronic cough
NCT01899768
Phase II
32.
PF-04531083
Pfizer
Nav1.8
UVB exposed skin, dental pain
NCT01127906
NCT01512160Phase I/II
34.
PF-06305591
Pfizer
Nav1.8
Pain
NCT01776619
Phase I
35.
VX-150
Vertex Pharmaceuticals Inc
Nav1.8
Pain due to SFN
NCT03304522
Phase II
33.
Leconotide/CNSB004
Zenyth Therapeutics
CaV2.2
Intractable pains
–
Phase IIa
34.
Prialt® (Ziconotide)
(Intrathecal infusion)Elan Pharmaceuticals
CaV2.2
Severe chronic pain
–
FDA approval-2004
35.
Z-160
Neuromed Pharmaceuticals
CaV2.2
LR
WO2009146540
Phase II
36.
CNV-2197944
Convergence Pharmaceuticals
CaV2.2
PHN, DPN
NCT01848730
Phase II
37.
ABT-639
AbbVie
CaV3.2
DPN
NCT01345045
Phase II
38.
Z-944
Zalicus Inc
CaV3.2
Neuropathic pain
–
Phase I
39.
Amiloride
University of California
ASIC
Migraine
–
Pilot study
40.
Gefapixant
Merck Sharp & Dohme Corp.
P2X3
Endometriosis-related pain,
OANCT03654326
NCT01554579Phase II
41.
Minodronate
Astellas Pharma
P2X3
Back pain
–
Pilot study
42.
Eliapixant
Bayer
P2X3
DPN
NCT04641273
Phase II
43.
Sivopixant
Shionogi
P2X3
Neuropathic pain
–
Phase I
2.1 Transient receptor potential (TRP) channels modulators
Transient receptor potential (TRP) channels are important nociceptive ion channels of thermal and chemical stimuli that activate the somatosensory system to produce acute or persistent pain (Patapoutian et al., 2009). In the last two decades, appreciable efforts have been made to identify the common pain pathways using natural products as probes. Many of these compounds such as proalgesic capsaicin, cooling menthol and pungent isothiocyanate have been shown to elicit discomfort and pain through a shared molecular mechanism in which they excite sensory, nociceptive neurons by activating TRP ion channels (Maliszewska et al., 2008). Based on these natural probes, three distinct members of the TRP ion channel family have been characterized viz. TRPV1 (the capsaicin receptor) (Menigoz and Boudes, 2011), TRPM8 (the menthol receptor) (Bautista et al., 2007), and TRPA1 (the wasabi receptor) (Guimaraes and Jordt, 2007) as molecular detectors of pain.
The TRPs ligands can be classified according to their pharmacological profiles, the two main classes being agonist and antagonist ligands. Both classes appeared to be extremely useful novel therapeutics for inflammatory and neuropathic pain syndromes. TRP agonists generally elicit pain but also desensitize the channel. This inactivation reduces sensitivity to heat and other ligands, which can be therapeutically utilized as analgesics with limited efficacy. Whereas the latter, TRP channels antagonists reduce pain by preventing transduction in the periphery, acting on channels expressed in keratinocytes or nociceptors, reducing ectopic activity generated by TRP channels along the axon and by reducing transmitter release as well as possibly acting on central neurons (Patapoutian et al., 2009). This section provides a brief overview of promising leads and clinical drug candidates of the TRP channels that have been postulated for pain management.
Cloning of the capsaicin receptor and its identification as the cation channel TRPV1 was one of the breakthrough pain research discoveries in the past 20 years. TRPV1 is a thermosensitive TRP channels that acts as a sensor for noxious heat (greater than ∼ 42 °C) by producing thermal hyperalgesia and pain hypersensitivity due to injury. The channels are activated by some endogenous lipid-derived molecules, acidic solutions (pH < 6.5) and some pungent chemicals and food ingredients such as capsaicin, as well as by toxins such as resiniferatoxin and vanillotoxins (Fig. 1) (Story, 2006). The endogenous lipids which activate the TRPV1 includes anandamide, N-arachidonoyldopamine, and various metabolic products of lipoxygenases (Ross, 2003).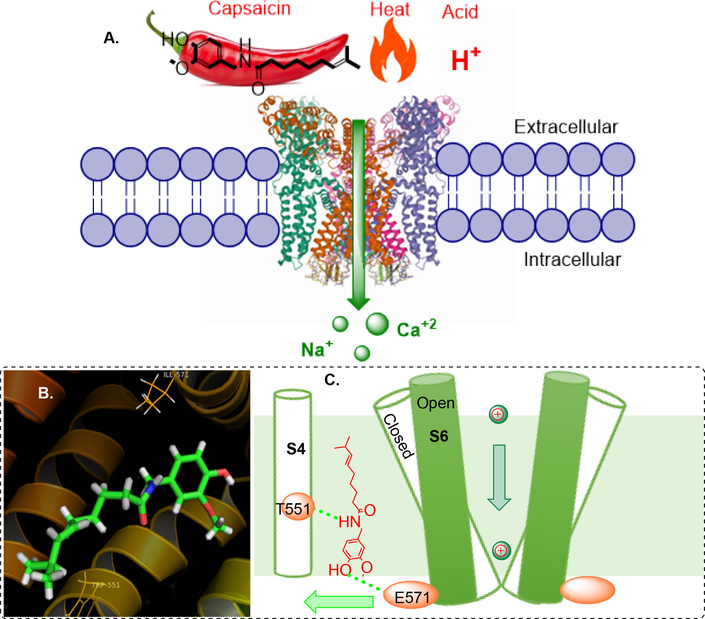
Overall mechanism of TRPV1 activation by capsaicin A. Activation of TRPV1 channel, B. Docked pose of capsaicin at the binding pocket of open state TRPV1 structure (PDB ID: 7LR0), C. Cartoon summarizing capsaicin binding and opening of TRPV1 channel.
Capsaicin (1) and (and its synthetic cis-isomer Zucapsaicin) induces a sensation of irritation and burning pain by activating a TRPV1 (EC50 40 nM) (Smutzer and Devassy, 2016), on sensory nerve endings in mammals however, birds are indifferent to the pain-producing effects of vanilloid compounds capsaicin. It is also currently under investigation for the relief of severe pain in adults suffering from episodic cluster headache (Jordt and Julius, 2002). This interesting fact led to the discovery of molecular basis of capsaicin sensitivity through the vanilloid receptor. Vanilloid sensitivity reside within the residues of transmembrane TM2 and TM3 regions of the capsaicin receptor (Voets et al., 2005). Capsaicin is a topical analgesic (orally not active), approved by the FDA as an orphan drug for the treatment of neuropathic pain associated with peripheral neuropathy (PHN) (Ausín-Crespo et al., 2022; Hong et al., 2019). It has since become an attractive lead molecule for the development of safer and effective analgesics and subsequently several capsaicinoids have reached preclinical or clinical development stages. (Fig. 2). One of such small molecules is a capsaicin-prodrug vocacapsaicin (CA-008) (2) which is a non-opioid, water-soluble drug developed by Concentric Analgesics, Inc. The prodrug was a first-in-class TRPV1 agonist successfully going for Phase III trial for the management of postsurgical pain. Results of its Phase II trial showed that patients administered with 36 mg of vocacapsaicin during surgery had significantly reduced pain and opioid consumption (NCT04203537). Nevertheless, capsaicin and related analogues has very strong pungency, therefore several non-pungent analogues have also been discovered (Huang et al., 2013).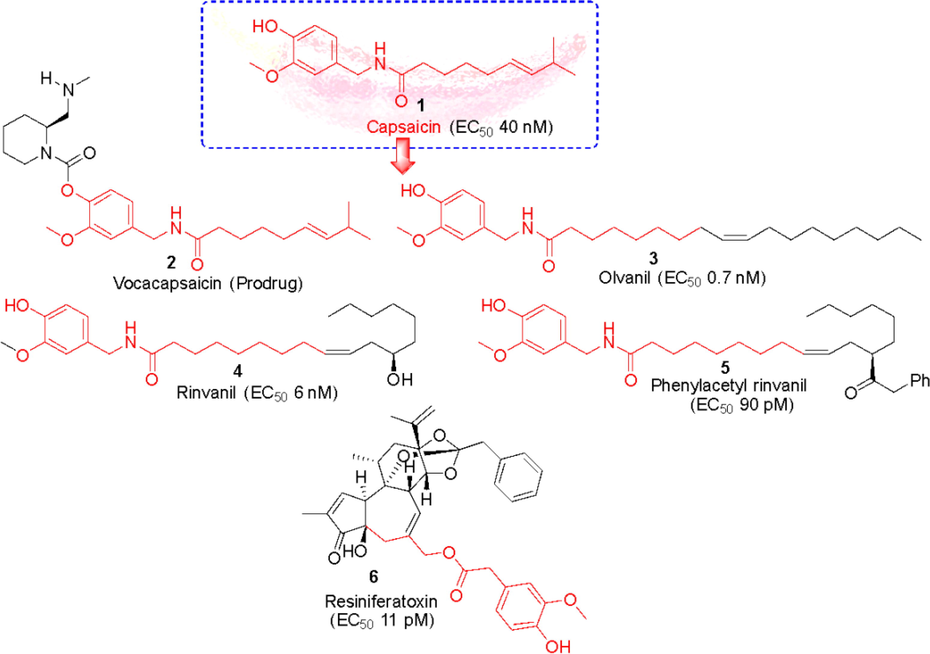
Capsaicin based TRPV1 agonists under clinical development.
In pursuit of this, a non-pungent, synthetic, and orally active capsaicin derivative, olvanil (N-9-Z-octadecenoyl-vanillamide, NE-19550) (3) was developed as a potent agonist (TRPV1 EC50 0.7 nM). Appendino et al., 2005 further modified the fatty acyl chain of olvanil and introduced a hydroxyl group at C-12 yielding a compound rinvanil (4) (EC50 6.0 nM) named after after ricinoleic acid. Phenylacetylation of rinvanil dramatically enhanced the potency of vanillamide with two-digit picomolar EC50 of 90 pM yielding phenylacetylrinvanil (5) (IDN5890). IDN 5890 was the first ultra-potent capsaicinoid agonist at TRPV1 channels. At present several capsaicin-based formulations are also being gauged. For example, phase III clinical trial has established the efficacy of 8% capsaicin patch (Qutenza®) in treating peripheral neuropathic pain and subsequently FDA has approved Qutenza® for the treatment of neuropathic pain associated with diabetic peripheral neuropathy (DPN) in adults (NCT01533428). In early 90s, scientists at the National Cancer Institute (NCI), Bethesda discovered a natural ultrapotent TRPV1 agonist resiniferatoxin (6) (RTX) (EC50, 11 pM) from Euphorbia cactus (Szallasi and Blumberg, 1990). RTX is currently undergoing clinical phase I and II trials for the treatment of severe pain in patients with advanced cancer (NCT00804154). Further, iodination of its vanillyl moiety, led to the most potent TRPV1 antagonist available to date, 5́-iodoresiniferatoxin (7) (Wahl et al., 2001).
The advent of radioligand [3H]RTX binding assay and TRPV1 gene cloning has led to a new concept of design and development of small molecule analgesics (Pearce et al., 2017). Since then, many pharmaceutical companies have started drug screening and lead optimization programs for the discovery of clinically useful small molecule TRPV1 antagonists. And this endeavour has led to the discovery of several TRPV1 antagonists in the different stages of clinical phases (Fig. 3). In the early 90 s, collaborative endeavours of scientists at the Sandoz Institute of medical research, UK (Novartis) resulted in the development of a first competitive vanilloid antagonist, capsazepine (8) which is a conformationally constraint analogue of capsaicin. Capsazepine competitively blocks the painful sensation of heat produced by capsaicin and resiniferatoxin with moderate potency in a variety of in vitro and in vivo bioassays (Walpole et al., 1994). Later, many promising compounds in preclinical studies have successfully moved forward and their clinical trials progressed at unprecedented speed. A number of TRPV1 antagonists belonging to different chemotypes, viz., cinnamides (AMG0347 (9) and AMG9810 (10)), pyrimidines (AMG-517 (11)), ureas (JYL-1421 (12) and A-425619 (13)), and piperazines (BCTC (14)), ABT-102 (15), AZD-1386 (16), DWP-05195 (17), JTS-653 (18), MK-2295 (19), and SB-705498 (20) have already entered clinical trials (Garami et al., 2020). However, clinical trials for some of these first generation TRPV1 antagonists are being discontinued due to their inherent deleterious side effects such as hyperthermia and long-lasting alteration of the noxious heat sensation. For example,
-
AstraZeneca discontinued the phase II clinical studies of AZD-1386 (16) because it causes a moderate increase in core body temperature. AstraZeneca was developing this benzimidazole based orally active TRPV cation channel inhibitor for the treatment of gastro-oesophageal reflux disease (GERD) and pain (Quiding et al., 2013).
-
Another novel 4-oxopyrimidine derivative, AMG-517 (11) developed by Amgen Inc. was found to be potent (IC50 < 10 nM) orally bio-available TRPV1 antagonist. AMG-517 reverses inflammation induced pain in rats with ED50 of 0.33 mg/kg p.o. Based on its promising selectivity, pharmacokinetic, and safety profile, the drug candidate was selected for three independent phase I clinical trials on healthy adults. In one of the studies, AMG-517 showed emergence of marked and persistent hyperthermia in subjects undergoing molar extraction. Due to these, clinical studies of AMG-517 were discontinued and very few subjects were evaluated for the potential analgesic effect of AMG 517 (Doherty et al., 2007; Gavva et al., 2008).
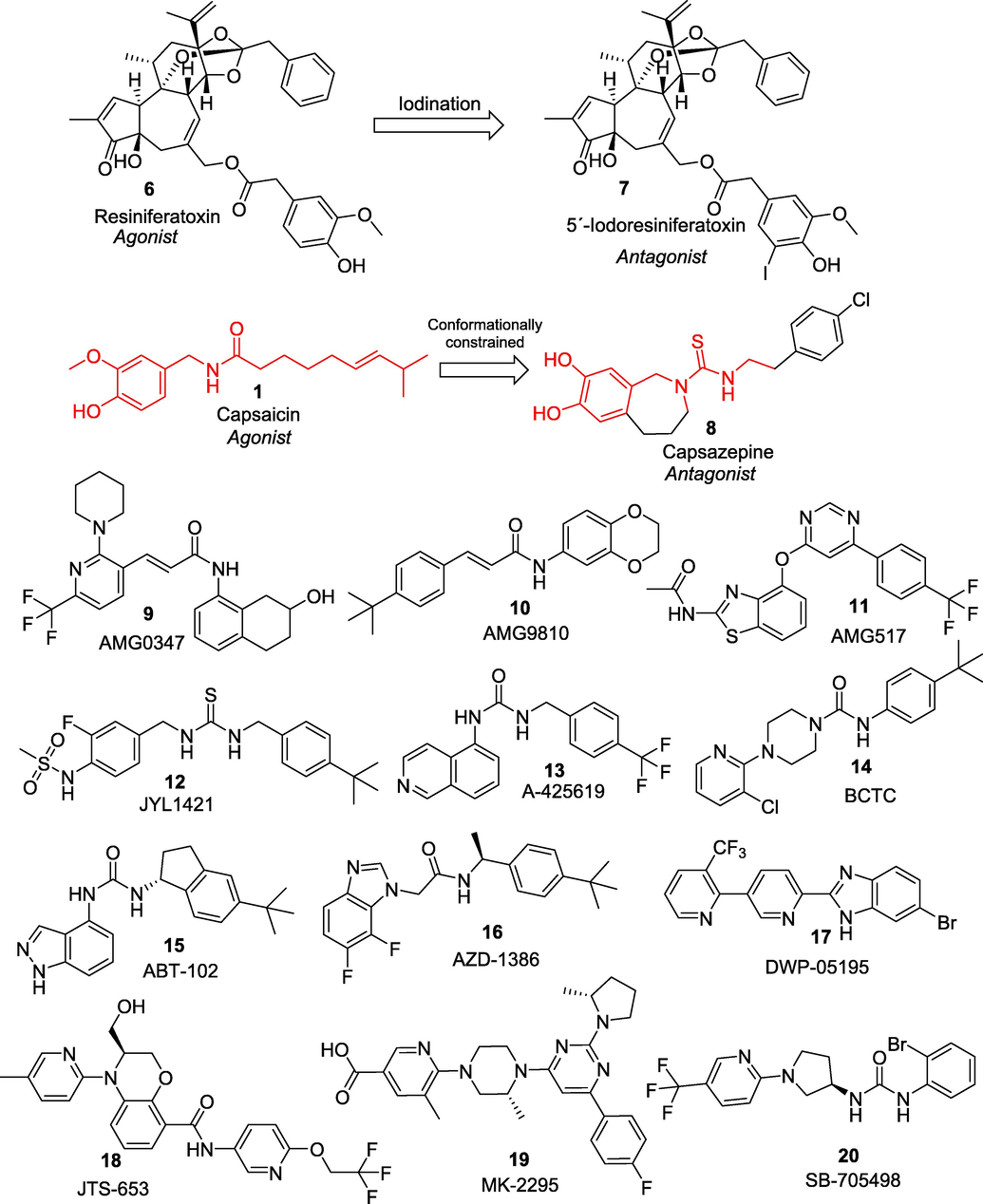
- Selective blockers of TRPV1 ion channel developed as a potential treatment for chronic pain.
Customarily, TRPV1 has been considered the most important TRP channel involved in nociceptive transduction. However, some other subfamilies of TRP channels are also most prominently expressed in nociceptors and have been extensively studied. Here, we summarize the clinical updates of the other TRP channels modulators as a miscellaneous class. Fig. 4 summarizes the structures of the reported miscellaneous TRP-modulators progressed to the clinical trials.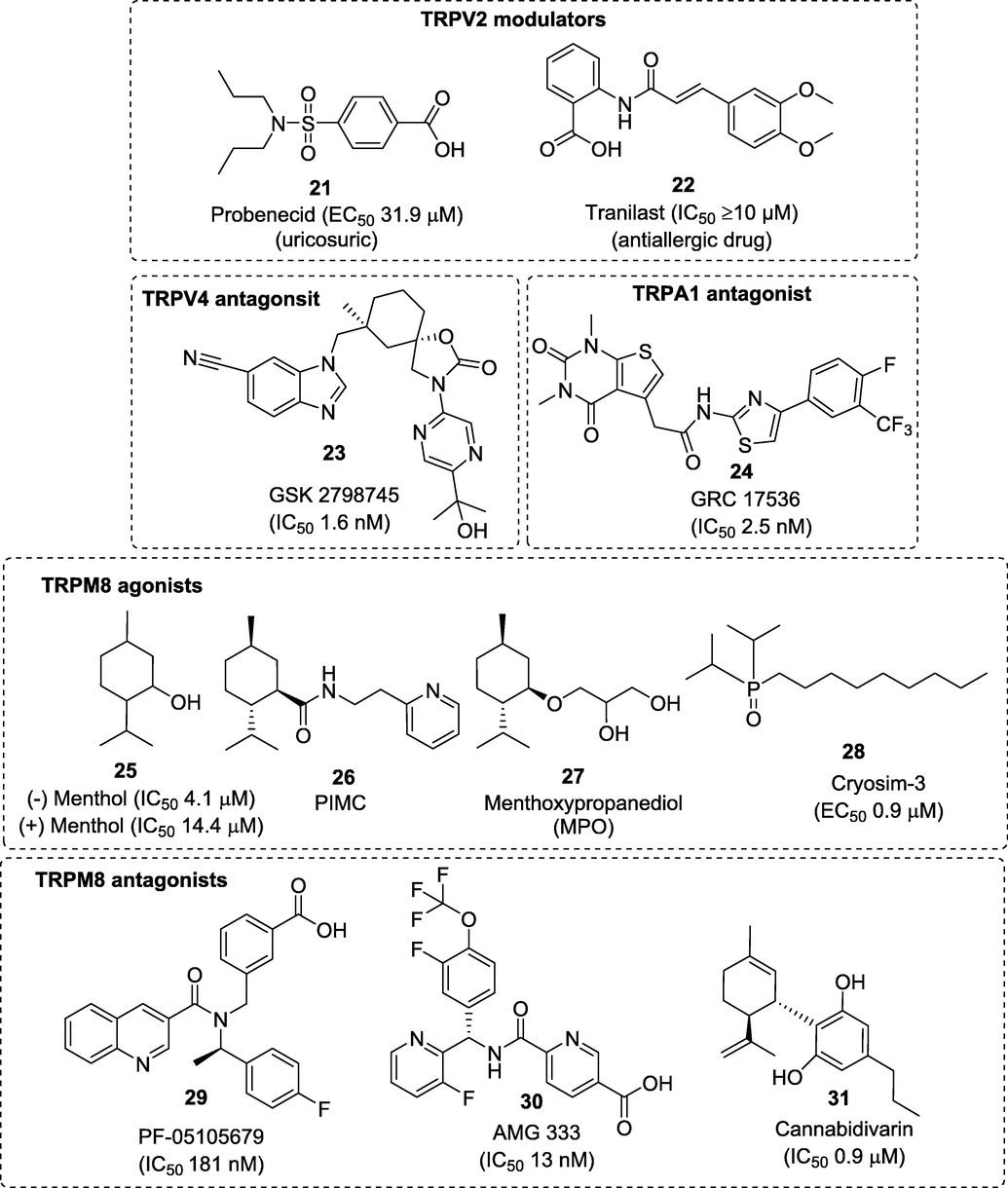
Structures of the reported miscellaneous TRP-modulators progressed to the clinical trails.
Transient receptor potential vanilloid 2 (TRPV2) belongs to the TRPV subfamily of TRP channels (TRPV1-TRPV4) however, TRPV2 does not contribute to in vivo thermal nociception and is insensitive to vanilloids (Iwata et al., 2020). The analgesic potential of TRPV2 has been least studied however, it has a proven track record of success mainly in treating heart failure and cardiomyopathy. The literature revealed that only two FDA approved drugs having TRPV2 modulatory effects such as probenecid (21) (uricosuric) and tranilast (22) (antiallergic) entered clinical trials for the treatment of heart disorders due to its positive ionotropic effects (Robbins et al., 2018; Matsumura et al., 2018). Recently, a cell-based calcium mobilization and electrophysiological assays to identify novel cannabinoid TRPV2 agonists was carried out. Results showed that cannabidiol (CBD) which is a non-psychotropic therapeutically active ingredient of Cannabis sativa, is an activator of TRPV2 (EC50 3.7 μm) and modulator of other TRP channels (Quin et al., 2008).
Another member of the TRPV family is a heat-sensitive TRPV3 ion channel, which shares around 43% sequence similarity with TRPV1. TRPV3 is highly expressed in the skin, where it is involved in skin barrier function, keratinocyte proliferation, skin homeostasis, wound healing, and nociception (Nilius et al., 2014). TRPV3 can be activated by chemical agonists that include, 2-aminoethoxydiphenyl borate (2APB), farnesyl pyrophosphate, and various natural compounds such as camphor, carvacrol, eugenol, and thymol (Colton and Zhu, 2007; Xu et al., 2008). Recent reports suggest that TRPV3 plays significant roles in inflammatory skin disorders, itch, and pain sensations. Despite several compounds have shown high potentiality towards TRPV3, GRC-15300 (structure not disclosed) is the one and only first-in-class TRPV3 inhibitor developed by Glenmark that entered clinical trials globally for the treatment of neuropathic pain (NCT01463397). Unfortunately, the trial was dropped in 2014 when the compound failed in Phase II proof of concept trial (WO2007056124; NCT01463397).
TRPV4 channels, as an important sensor for osmotic and mechanical stimuli, play a central role in the essential hallmarks of nociception. Several small molecules such as, anandamide, 5,6-epoxyeicosatrienoic acid, GSK 1016790A, and heat (27–34 °C) are known to activate TRPV4 channel (Lawhorn et al., 2020). Many selective TRPV4 antagonists have been evaluated in recent years however, the only TRPV4 ligand to have entered clinical trials is GSK-2798745 (23). Compound 23 demonstrated excellent in vivo efficacy (hTRPV4 IC50 1.8 ± 0.2 nM, rTRPV4 IC50 1.6 ± 0.2 nM) and satisfactory preclinical safety profile that supported its subsequent development to clinical studies. Compound 23 from GlaxoSmithKline turned out to be highly potent and orally active TRPV4 inhibitor, that is currently being investigated for the treatment of chronic cough (NCT03372603) (NCT03372603) and diabetic macular oedema (NCT04292912).
The wasabi receptor, TRPA1, represents another TRP subfamily as a detector of chemical irritants. TRPA1 is a non-selective cationic channel generally co-expressed with TRPV1 channels in the CNS and PNS and can be activated by several chemical, thermal, mechanical, and osmotic stimuli. TRPA1 is activated by isothiocyanates and thiosulfinates that constitute pungent agents from mustard (e.g., wasabi) and allium (e.g., garlic and shallot) plants, respectively. Pharmacological and genetic studies have shown that TRPA1 plays an essential role in the nociceptive response to these and other environmental irritants (King et al., 2019). Therefore, TRPA1 has become a validated target for the development of analgesic drugs. Recent literature showed that many promising TRPA1 modulators have been postulated to have potential role in pain management. TRPA1 antagonists such as 6-Methyl-5-(2-(trifluoromethyl)phenyl)-1H-indazole, CMP1, AZ868, HC-030031, and A-967079 were found to be effective in reversing the chemically-induced hyperalgesia and allodynia in mice without changing the core body temperature (Giorgi et al., 2019). To date, four TRPA1 modulators including GRC-17536 (24) (Glenmark) (NCT01726413), CB-625 (structure not disclosed) (Cubist Pharmaceuticals Inc.) (Giorgi et al., 2019), HX-100 (structure not disclosed) (Hydra Biosciences) (Giorgi et al., 2019), and ODM-108 (structure not disclosed) (Orion Pharma) (NCT02432664) have entered clinical trials for the treatment of neuropathic pains but latter discontinued mainly due to their low oral bioavailability.
Transient receptor potential melastatin-8 (TRPM8) is a non-selective cation channel activated by cold temperature and by cooling agents such as menthol. Several novel TRPM8 modulators have shown promising effects in reducing both acute and chronic pain (Perez de Vega et al., 2016). As menthol (25) acts by selectively activating TRPM8 channel therefore, widely used as a topical analgesic (up to 16%) to relieve acute, inflammatory, and neuropathic pain. >200 different topical and oral formulations of menthol are being evaluated under phase I/II clinical trials for cosmetic and medical indications including carpal tunnel syndrome, knee osteoarthritis, chemotherapy-induced peripheral neuropathy, migraine, and cancer pain (Fernández-Carvajal et al., 2020). Inspired by this, many pharma companies have begun drug development program on menthol-based carboxamide and ester derivatives as TRPM8 agonists (González-Muñiz et al., 2019). In 2016, scientists at Beiersdorf AG, Germany assessed two similar type of compounds, (1R,2S,5R)-N-(2-(2-pyridinyl)ethyl)-2-ispropyl-5-methylcyclohexancarboxamide (26) and menthoxypropanediol (MPO) (27) in a randomized, double-blind, pilot study in dry skin patients with pruritus (NCT00669708). Combined applications of these two cooling compounds have shown stronger activation of TRPM8 with significant improvement in skin roughness, dryness and hydration conditions (Stander et al., 2017). Lately, a group of scientists at Chonnam National University Hospital, South Korea also found that administration of water-soluble, non-menthol derivative, cryosim-3 (28) (TRPM8 agonist) significantly cured dry eye disease when tested on human subjects (ISRCTN24802609/ISRCTN13359367) (Yang et al., 2017). TRPM8 antagonists have also been implicated in pain, inflammation, and cancer. Till now, only three TRPM8 antagonists have progressed to clinical studies. The first two promising compounds viz. PF-05105679 (29) and AMG-333 (30) were precluded after completing phase I clinical trials due to its severe side effects (Andrews et al., 2015; Horne et al., 2018) however the third compound, cannabidivarin (31) is currently being evaluated in the phase II clinical trial for autism spectrum disorder (ASD) (NCT03202303). As cannabidivarin, is effective in treating paediatric epilepsy therefore, the drug demonstrates potential mechanisms for treating ASD (NCT03202303).
2.2 Sodium channel blockers
The afferent neurons transmit pain signals of noxious stimuli from the periphery to the CNS (von Hehn et al., 2012). These pain signals are conducted as electrical excitability or action potential (AP) across axonal membrane due to sequential opening and closing of voltage-gated ion channels (Lodish et al., 2000). Several studies have shown that many different types of ion channels play significant roles in nociception and altered pain sensitivity (Matzner et al., 1994; Baker et al., 2001; Du et al., 2013). Among these, voltage-gated sodium (Nav) channels are vital for AP electrogenesis and transmission of painful stimuli (Cummins et al., 2019). The central role of Nav channels in regulating nociception was confirmed by many studies one of which showed that local anaesthetics relieve pain through Nav blocking actions and have potential therapeutic applications for the treatment of pain (Clare et al., 2000; Holmdahl et al., 1998). Gene cloning studies showed that around nine distinct Nav isoforms viz. Nav1.1-Nav1.9 are known to be expressed in humans (Levinson et al., 2012). Preclinical studies involving gain-of-mutations research identified only three isoforms, Nav1.7, Nav1.8 and Nav1.9 as validated targets for pain management as these isoforms are preferentially expressed in the peripheral nervous system (Cregg et al., 2010). Many non-selective Nav channel blockers belonging to antiarrhythmic drugs (ADs), antiepileptic drugs (AEDs) and local anaesthetics (LAs) has also widely been prescribed as “off-label drugs” for the treatment of various pain conditions. Some of the examples of these drugs include carbamazepine (32), oxcarbazepine (33), eslicarbazepine (34), phenytoin (35), lacosamide (36), rufinamide (37), and mexiletine (38) that hold promise for treating migraine, trigeminal neuralgia (TN), DPN, fibromyalgia, chronic post-thoracotomy pain syndrome, cryptogenic sensory polyneuropathy, erythromelalgia etc.
The clinical benefits of some of these approved drugs are continuously being verified and consequential additional clinical indications are also being approved by the FDA through supplemental biologic licensing (Fig. 5). However, these drugs have narrow therapeutic margins due to their associated adverse effects like cardiac arrhythmia, ataxia, sedation, and because of their non-selective actions on CNS, cardiac and skeletal muscle tissues (Fischer et al., 2009; Sheets et al., 2011, Ryder and Stannard, 2005).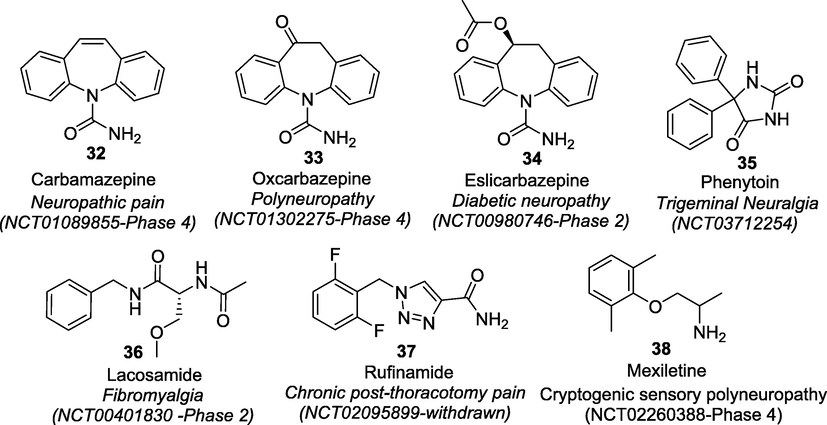
Clinical trials of non-specific Nav blockers in pain management (https://clinicaltrials.gov/).
Thus, the development of selective Nav channel blockers targeting peripheral sensory neurons has gained tremendous interest in research on ion channel therapeutics for pain. Towards this end, Nav1.7 channel has become a potential pharmacotherapeutic target for the treatment of pain because of its remarkable validation from human genetics and preclinical studies. Many clinical and preclinical genetic studies including knockout mice clearly implicate a major role for Nav1.7 in acute and inflammatory pain as loss of Nav1.7 function leads to complete insensitivity to pain (Yeomans et al., 2005). These findings were also confirmed by Geoff Woods who noticed that some Pakistani children are insensitive to pain, a rare condition known as congenital insensitivity to pain (CIP) due to loss-of-function Nav1.7 mutations (Cox et al., 2006). Thus, Nav1.7 is an ideal target for the development of novel analgesics. Genomic analyses, together with molecular modelling studies have tremendously helped researchers understand the sodium ion channel structure (Baker and Nassar, 2020; Namadurai et al., 2014). The ion channel is consisting of a gene SCN9A with a large pore forming α-subunit and one or more smaller β-subunits. The α-subunit consists of four homologous domains (DI-IV) with six subdomains (S1-S6). The segments S1-S4 form the voltage-sensing domain (VSD) whereas, the S5 and S6 domains forms the ion pore domain (PD) made up of amino acid sequence Asp-Glu-Lys-Ala (DEKA) which selectively allows the flux of hydrated Na + through the ion pore. The sequences between S5 and S6 also comprise the selectivity filter (SF) (Fig. 6) (Shen et al., 2019).[81].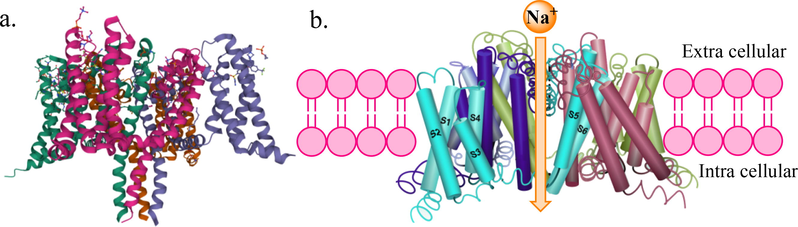
(a) Crystal structure of human voltage-gated sodium channel Nav1.7 showing the α subunit which folds to four homologous repeats, each containing six transmembrane helices designated S1-S6 (PDB ID: 6J8J). (b) Side view of the open-channel conformation showing channel pores.
Recently, use of toxins to probe various ion channel structures and functions has contributed tremendously on our understanding of voltage-gated sodium ion channels (VGSCs) physiology. Moreover, the ensuing potential natural and synthetic peptide-based toxins turned out to be promising lead compounds in developing novel pain killers. Some small molecules neurotoxins such as tetrodotoxin (TTX) (39) and saxitoxin (STX) (40) are also a relatively new class of compounds acting as pore blockers (Yasumoto and Murata, 1993). These neurotoxins interact with VGSCs either through physically obstructing the pores and inhibiting the sodium ion conductance or modifying the gating kinetics (Marijke et al., 2011). These inhibitors can bind at the six different binding sites in the channels based on their physicochemical properties and resulting into different mechanism of actions (Fig. 7). TTX (IC50 18.6 nM, Nav1.7) and STX (IC50 702 nM, Nav1.7) are the hydrophillic secondary metabolites causing paralysis and produced by puffer fish and molluscs, respectively (Walker et al., 2012). Both are chemically alkaloidal guanidinium class of compounds containing protonated guanidino group which is required for efficient interaction with the putative binding site 1 present at the P-loops connecting S5 and S6 domains (Suppiramaniam et al., 2010; Marijke et al., 2011). The protonated guanidinium group of TTX forms ionic interactions with the negatively charged amino acids Asp384 and Glu387 of DI and Glu942 of DII, whereas the hydroxyl groups at C9, C10 and C11 form hydrogen bonds with Glu945 of DII and Asp1532 in DIV. TTX block the pore sterically and occludes sodium ion permeation, thereby inhibiting the AP generation and propagation and consequently blocking nerve conduction and paralysis (Lipkind and Fozzard, 1994; Lee and Ruben, 2008). STX is chemically similar to TTX. It belongs to the water-soluble neurotoxin containing positively charged guanidinium groups which bind with the negatively charged carboxyl groups in the outer pore loops of Nav. However, STX structure has an additional positive guanidinium group that leads to slightly different binding interactions as that of TTX (Penzotti et al., 1998). Apart from these nonpeptidic guanidinium toxins, venom peptides such as μ-conotoxins from the venoms of predatory cone snails have been shown to act at Site 1 and preferentially block skeletal muscle voltage-gated sodium channels. The μ-conotoxin has long been used as a paralyzing agent for fish-hunting until recently, studies recognized that it preferentially blocked Nav channels (Green et al., 2014). Several similar peptidic venoms belonging to conotoxin families have also been characterized recently e.g. μO-, δ-, and ι-conotoxins, and μO§-conotoxins (Gajewiak et al., 2014). Site 2 is mainly targeted by lipophilic toxins such as batrachotoxin (BTX) (41) and its analogues produced by frogs (Phyllobates spp.). These toxins are also known as activators as they modulate Nav channel to open it more easily and stay open for longer duration. BTX binding modulates the voltage-dependent movement of the DIV S4 voltage sensor and thereby alters channel activation and its coupling to inactivation (Linford et al., 1998). Site 3 neurotoxins are mainly produced from scorpions, sea anemones and spiders. Scorpion toxins targeting the Nav channels are classified into two categories viz. α- and β-toxins. The α-toxins generally inhibit the inactivation of VGSCs, whereas β-toxins produce a strong hyperpolarizing shift in the voltage dependence of activation at the neurotoxin site 4 (Gordon et al., 1996). Site 5 is targeted by highly lipophilic, cyclic polyether compounds such as brevetoxins (PbTxs) (42) and ciguatoxins (CTXs) (43) produced by marine dinoflagellates Karenia brevis and Gambierdiscus toxicus, respectively (Schreibmayer and Jeglitsch, 1992). Bindings of PbTx at the site 5 produces distinct alteration in channel gating and stabilize the conductance level, whereas CTXs shift the activation of channels towards more negative potential and suppress the fast inactivation (Schreibmayer and Jeglitsch, 1992; Hogg et al., 2002).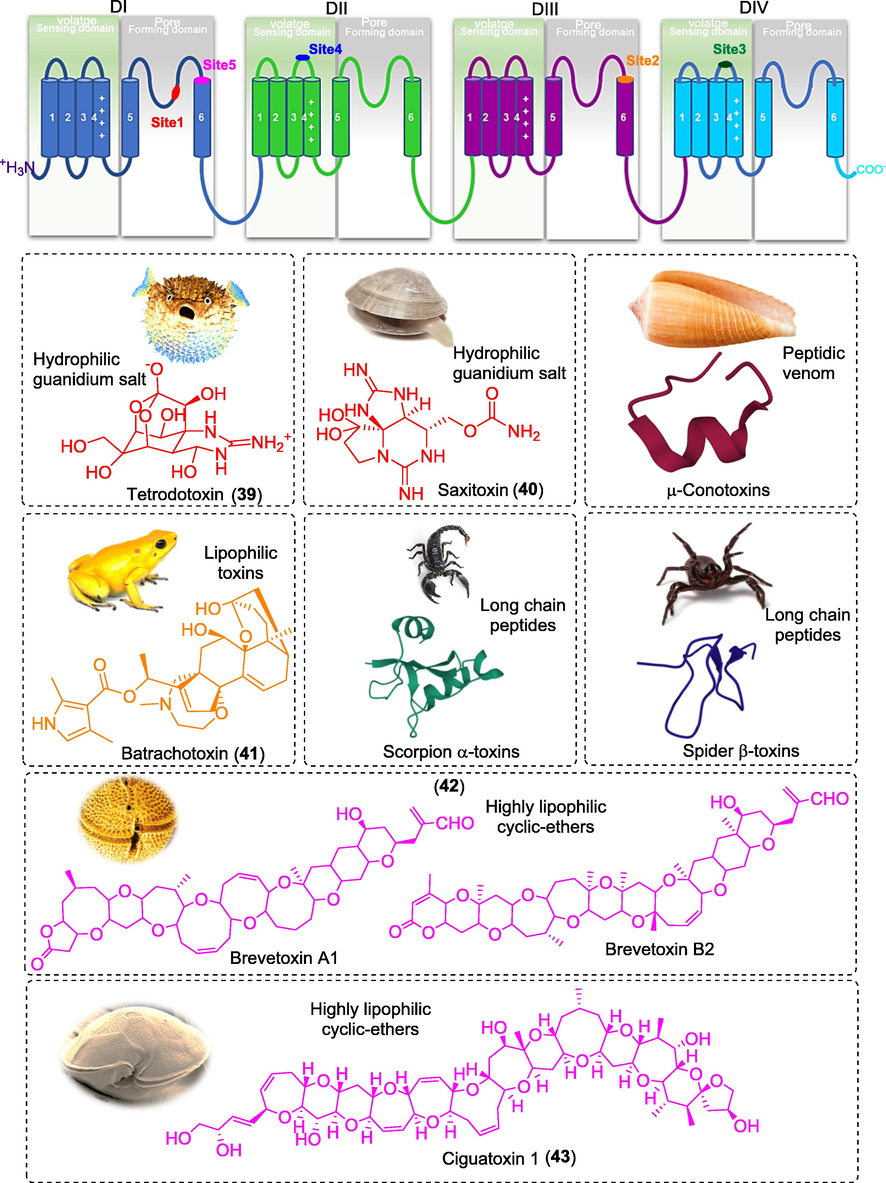
Figure showing the trans-membrane diagram of α-subunit of Nav channel with their binding sites ligands.
Considering the promising selectivity of these neurotoxins towards Nav1.7, several small molecules and peptides are in pre-clinical and clinical development. Many of these molecules, from Pfizer, Convergence, Xenon, Genetech, Teva, Sumitomo Dainippon Pharma, Nektar Therapeutics, Wex Pharmaceuticals and AstraZeneca, have demonstrated significant results in the preclinical and clinical studies. However, some of these molecules failed to achieve end-results in early clinical studies and therefore, dropped from their pipelines. Therefore, in this section we are pursuing on the investigator’s investigational new drugs (INDs) that are being developed to manage acute and chronic pain (Fig. 8).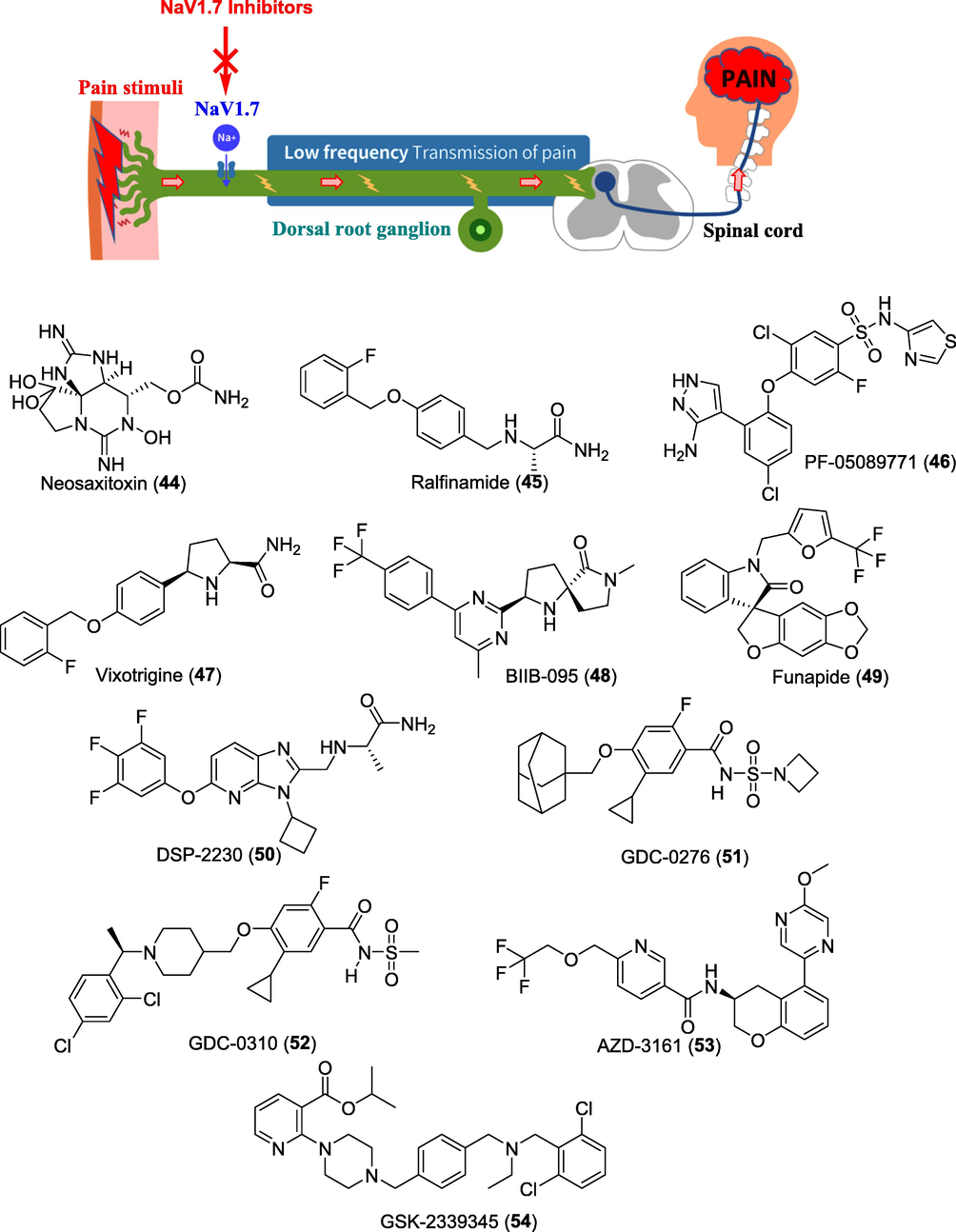
Figure showing role of Nav1.7 inhibitors in treating pain and potential clinical candidates.
WEX Pharmaceuticals Inc. is a biopharmaceutical company based in Canada focused on the therapeutic fields of non-opioid analgesics. The company has completed open-label, phase III trial for Halneuron® (Ingredient: TTX) to treat cancer related pain (CRP) and chemotherapy-induced neuropathic pain (CINP) in the United States and Canada (NCT00725114, NCT00726011). The study demonstrated that patients receiving a subcutaneous 30 μg b.i.d. dose of TTX for 4-days had effectively reduced pain outcomes and improved quality of life (NCT00725114; NCT00726011). Prof. Charles Berde at the Boston Children's Hospital, US had finished phase I trial of neosaxitoxin (NeoSTX) (44), an analogue of STX as subcutaneous injection in combination with the commonly used local anaesthetic, bupivacaine, and epinephrine (NCT01786655). SiteOne Therapeutics scientists have consistently used this template to benchmark their synthetic compounds based on toxins. Recently they have initiated a phase I study of its saxitoxin-based synthetic molecule ST-2427, for managing moderate to severe pain. ST-2427 acts as a selective inhibitor of Nav1.7. To date, structure of the molecule ST-2427 has not been disclosed by the company (NCT04475198).
Newron Pharmaceuticals is a fully integrated biopharmaceutical company focused on the development of novel therapies for patients with diseases of the central and peripheral nervous system. Currently, there is one drug in its analgesic portfolio acting through Nav1.7 mechanism. The drug is an orally active, selective blocker of the Nav1.7 known as ralfinamide (45) and has been progressed into phase IIb/III study in patients with moderate neuropathic low back pain (NLBP) to evaluate its safety and efficacy of two dose regimens compared to placebo. Results demonstrated that patients with neuropathic pain due to nerve compression showed a response to ralfinamide treatment (NCT00736151; NCT01019824).
Researchers at Pfizer Inc. are also working tirelessly to design new molecules acting as Nav1.7-selective inhibitors. One of these molecules is a PF-05089771 (46), that has successfully completed phase I studies to assess its safety and tolerability in healthy subjects. Efficacy of PF-05089771 in treating postoperative dental pain and DPN were also completed in phase II clinical trials in 2018 (NCT01529346; NCT02215252). Biogene is conducting numerous clinical trials in neuropathic pain to evaluate the efficacy and safety of two orally administered Nav1.7 inhibitors viz. BIIB074 (vixotrigine/raxatrigine) (47) and BIIB-095 (48). Vixotrigine developed by Convergence Pharmaceuticals (now acquired by Biogene), has been evaluated in phase II clinical studies for their efficacy of in treating pain with lumbosacral radiculopathy (LR) (NCT02935608). Vixotrigine (formerly known as CNV1014802) was previously granted orphan-drug designation by the FDA for the treatment of TN. Results of its phase IIa demonstrated that, patients treated with vixotrigine showed a significant reduction in TN paroxysms compared with those receiving placebo (Zakrzewska et al., 2017). Recently, phase III studies evaluating efficacy and safety study of BIIB074 in participants with TN has been registered (NCT03637387). BIIB095 has also completed phase I studies for safety, tolerability, and pharmacokinetics in healthy participants (NCT03454126).
In 2021, Flexion Therapeutics, Inc. announced the FDA clearance of the IND application for FX301, a preclinical program for the extended-release thermosensitive hydrogel of funapide (49) (XEN402) as peripheral nerve blocking agent for controlling post-operative pain. The molecule funapide was earlier developed by Xenon Pharmaceuticals as a Nav1.7 and Nav1.8 blocker and had orphan drug designation from the FDA for treating pain associated with erythromelalgia (EM) (Price et al., 2017). Funapide also completed phase II clinical studies in patients with PHN (NCT02365636) and osteoarthritis (OA) (NCT02068599).
A Japan based company Sumitomo Dainippon Pharma Co. Ltd. and its venture AlphaNavi Pharma Co. are also continuing the development of imidazo-pyridine derivative DSP-2230/ANP-230 (50) for the treatment of voltage-gated sodium channel (Nav)-mutated rare pain diseases. The molecule is currently in phase I clinical trial for its safety, tolerability, and pharmacokinetic studies (Wulff et al., 2019).
Stumpf et al. at the Genentech, Inc. discovered acylsulfonylurea GDC-0276 (51) as a potent and efficacious, orally bioavailable, inhibitor of Nav1.7 and was chosen for clinical development. Phase I studies demonstrated that GDC-0276 exhibited a safety and pharmacokinetic profile that supports its future investigation as a potential therapeutic for pain (Stumpf et al., 2019). Further, optimization through blocking the labile benzylic position led to the discovery of GDC-0310 (52). GDC-0310 exhibited improved metabolic stability, Nav selectivity and pharmacokinetic profile as compared to GDC-0276 in the phase I trial (Safina et al., 2021).
AstraZeneca, Sweden developed first of its kind chromane derivative AZD-3161 as a potent Nav1.7 inhibitor (IC50 Nav1.7 is 66 nM). AZD-3161 (53) was investigated in the phase I study for its effect on mechanical pain sensitivity in ultraviolet C irradiated skin. Unfortunately, further development of this compound was halted as it failed in the clinical proof-of-mechanism study (NCT01240148).
Scientists at GlaxoSmithKline have worked extensively on the discovery of Nav1.7 inhibitors. They disclosed a pan-Nav channel inhibitor, GSK-2339345 (54), that was evaluated in phase II clinical trials for patients with chronic refractory cough. The effect of GSK2339345 on cough responses during cough challenges was inconclusive, and further progress of this molecule was stopped (WO2013006596) (Boehm et al., 2013).
Nav1.8 has also gained much attention as the inhibition of Nav1.8 has been associated with hyperalgesia, neuropathic pain, and reduced functionality of opioid receptors. Studies on Nav1.8 knockout mice showed deficits in nociception with inflammation, advocating the association of Nav1.8 in inflammatory pain. Nav1.8 turned out to be the first tetrodotoxin-resistant (TTX-r) VGSC mainly expressed in DRG neurons and sensory fibres; therefore, Nav is a potential target for pain (Alsaloum et al., 2020). Despite the promising therapeutic effects of Nav1.8 inhibitors in animal models of inflammatory and neuropathic pain, there have been a lack of studies into the efficacy of Nav1.8-specific inhibitors in humans. Currently, three Nav1.8 inhibitors are undergoing clinical studies for treating different pain conditions (Fig. 9). Molecule PF-04531083 (55), is an orally active, small molecule Nav1.8 VGSC blocker (hNav1.8 IC50 0.7 μM) developed by Pfizer Neusentis, UK. Compound 55 was efficacious in preclinical models of neuropathic pain and tibial nerve transection (TNT) induced mechanical allodynia model in rats (Bagal et al., 2015). The clinical efficacy of compound 55 was established by assessing its effect on heat pain in healthy volunteers in Phase I with ultraviolet light sensitized skin (NCT01127906) and post-surgical dental pain in Phase II (NCT01512160). Pfizer reported another clinical candidate PF-06305591 (56), as selective inhibitor of Nav1.8. Compound 56 was found to be potent, highly selective Nav1.8 blocker (IC50 15 nM) with an excellent preclinical in vitro ADME and safety profile. Compound 56 has completed the investigation in Phase I clinical trial for its safety and tolerability (NCT01776619). However, Pfizer is not listing this compound in its present-day pipeline and might be keeping this as a backup molecule. The third small molecule VX-150 (57), is a highly selective inhibitor of Nav1.8 (>400-fold). Compound 57 was developed as an orally bioavailable prodrug that rapidly converts into its active metabolite (Anderson et al., 2016). In 2018, Vertex Pharmaceuticals Inc. revealed the promising Phase II results of its investigational Nav1.8 inhibitor VX-150 in patients with pain caused by small fibre neuropathy. VX-150 was well tolerated in this study and demonstrated statistically significant and clinically meaningful pain reductions. Moreover, this study was the third consecutive proof-of-concept study for VX-150 and validated the potential role of Nav1.8 inhibitors in treating several pain disorders (NCT03304522).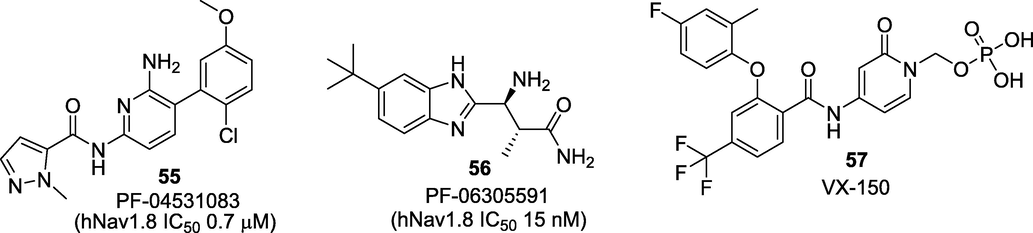
Small molecule Nav1.8 inhibitors that progressed to clinical trials.
2.3 Calcium channel blockers
Ca2+ channels play a critical role in controlling sensory functions associated with the transduction, transmission, processing, and modulation of pain signals (Yaksh et al., 2006). An increase in intracellular Ca2+ ion through influx of Ca2+ ion due to opening of membrane channels causes depolarization of membrane current and contributes to neuronal firing. Calcium channels are classified based on their activation characteristics as high- and low-voltage activated channels, structural subunit composition as CaV1, CaV2, CaV3, and their pharmacology as L, P/Q, N.R, T-type (Bourinet et al., 2005). L-type Ca2+ channels are high-voltage activated channels containing four subunits CaV1.1, CaV1.2, CaV1.3, CaV1.4. L-type Ca2 + channels function in the excitation-secretion coupling of endocrine cells and some neurons. The L-type channels are blocked by a phenylalkylamines, benzothiazepines and dihydropyridines (Striessnig, 1999). Research involving the intrathecal administration of L-type Ca2+ channel blockers have shown that they do not offer satisfactory pain relief (Lee, 2013). N-type calcium channel contains a single α1 subunit gene (also known as α1B or CaV2.2) restricted to primary afferent neurons, the dorsal horn, and the synaptic connections of nociceptive afferent neurons. Studies showed that indirect inhibition of N-type Ca2+ channels via morphine-induced activation produces potent analgesic actions (Westenbroek et al., 1992). Several peptides from the snail’s venoms were reported as potent N-type calcium channel blockers. ω-Conotoxin is a peptide containing 24–30 amino acids with three disulphide bonds obtained from Conus geographus, C. magus and C. catus. ω-Conotoxin mainly acting through inhibit synaptic transmission by blocking N-type Ca2 + channels (Ellinor et al., 1994). Leconotide (ω-conotoxin CVID, AM-336) has been investigated in phase I/IIa trials as a drug under the patronage of Zenyth Therapeutics for the treatment of intractable pains. Despite its selectivity and stability, the development of leconotide is now on hold due to the sluggish market scenario (Schroeder et al., 2012). Currently, the only FDA approved ω-conotoxin is ziconotide (Prialt®) (58) which is the first-in-class drug acting as CaV2.2 channel blocker. Ziconotide is a synthetic peptide of ω-conotoxin MVIIA [H-Cys-Lys-Gly-Lys-Gly-Ala-Lys-Cys-Ser-Arg-Leu-Met-Tyr-Asp-Cys-Cys-Thr-Gly-Ser-Cys-Arg-Ser-Gly-Lys-Cys-NH2] indicated for the treatment of severe chronic pain in patients for whom intrathecal therapy is necessary (Pope et al., 2013) (Fig. 10). Till date, only two drugs, morphine and ziconotide have been approved by the FDA for targeted intrathecal drug delivery administration for chronic pain as monotherapy (Van Zundert and Rauck, 2023). A small molecule NMED-160 (Z-160) (59) acting as a potent N-type Ca2+ channel blocker was developed by Neuromed Pharmaceuticals. The compound was evaluated in phase II trials for the treatment of lumbosacral radiculopathy (LR) and PHN but further study was discontinued due its poor pharmaceutical characteristics (Pajouhesh et al., 2009). Another small molecule benzenesulfonamide derivative, CNV-2197944 (60) is being developed by Convergence Pharmaceuticals (acquired from GSK in 2010) as CaV2.2 selective blocker. The molecule has completed its phase II trials for both PHN (NCT01848730) and DPN (NCT01893125). Scientists have been investigating a plethora of AEDs that may be repurposed to combat pain. One of these drugs, levetiracetam (61) acting via blocking N-type Ca2+ channel has been evaluated in phase IV trial for painful polyneuropathy (NCT00286260), PHN (NCT00160511) and fibromyalgia (NCT00254657).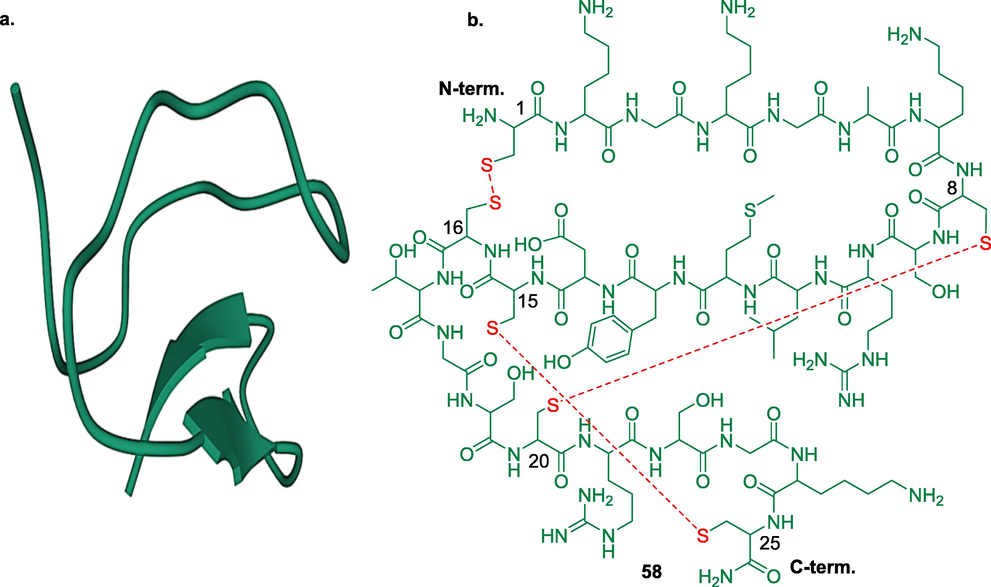
a. crystal structure of ω-conotoxin from Conus geographus (PDB ID: 1TTL) and b. ziconotide showing three disulphide bonds.
P-type Ca2+ channels are distributed to Purkinje cells where they facilitate depolarization-induced repetitive spikes. These channels are blocked by ω-agatoxin IVA isolated from the funnel web spider venom which could be useful as a therapeutic lead for pain treatment. Unfortunately, none of the P-type Ca2 + channels blockers progressed into clinical studies (Olivera et al., 1994).
T-type Ca2+ channels (CaV3) are low voltage activated channels that open in response to small membrane depolarization and can contribute to secretory processes (Snutch et al., 2018). Clinically available AEDs, ethosuximide (62) and zonisamide (63) are the molecules known to act through blocking T-type Ca2+ channels (Kostyuk et al., 1992; Kawata et al., 1999). Currently, ethosuximide is being evaluated in phase II clinical trials in the treatment of peripheral neuropathic pain (NCT04431778) and phase III trial in abdominal pain (NCT04217733). Zonisamide is being investigated in phase II trial for its safety and effectiveness in subjects with migraine headache (NCT00055484). ABT-639 (64), a benzenesulfonamide derivative was also developed as a selective T-type CaV blocker. The compound has been shown to be efficient in reducing nociceptive and neuropathic pain in various preclinical models but failed in achieving significant pain-attenuating actions in phase II trial (Ziegler et al., 2015). Zalicus Inc., a biopharmaceutical company focused on developing novel treatments for pain has recently completed phase I and Ib trials of novel, orally active T-type Ca2+ channel blocker Z-944 (65). The results demonstrated that Z-944 was well tolerated and efficacious in human models measuring laser-evoked potentials (LEP) from skin irritated by topical application of capsaicin (Lee, 2014).
Recently, gabapentinoid blockbuster drugs such as gabapentin (66) and pregabalin (67) acting through modulation via binding to the α2δ-1 and α2δ-2 Ca2 + channel subunit received much attention for their analgesic profile. These drugs have been approved by the FDA for the treatment of PN, PHN and fibromyalgia apart from the indication of epilepsy (Patel et al., 2018; Gee et al., 1996; Arnold et al., 2017; Najam et al., 2022) (Fig. 11).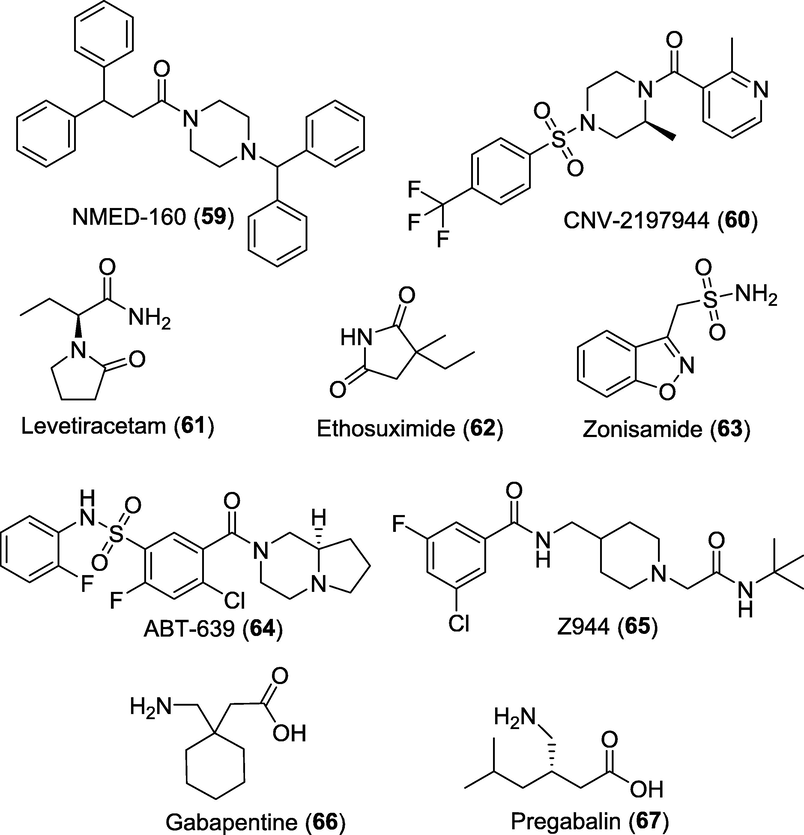
Promising pipeline analgesics acting as calcium channel blockers.
2.4 Acid-sensing ion channel modulators
Acid-sensing ion channels (ASICs) are crucial acid sensors implicated in neural modulation in the CNS and pain-associated tissue acidosis in the peripheral system. Several pain-causing stimuli, including inflammation, lower extracellular pH and local tissue acidosis (pH < 6) due to tissue damage activate the TRPV1 through protons. ASICs are also activated by the decrease in extracellular pH and initiate the proton-evoked currents in the sensory neurons. ASICs at the extracellular amino‐acid residues E600 and E648, open the channel (at low pH < 6) and lower the threshold for TRPV1 activation, by inflammatory meditators (at high pH > 6) (Holzer, 2009; Jordt et al., 2000). Four acid-sensing ion channel (ASIC) genes (ASIC1, ASIC2, ASIC3 and ASIC4) and six ASIC subunits (ASIC1A, ASIC1B, ASIC2A, ASIC2B, ASIC3 and ASIC4) have been identified. Vanillotoxins (VaTxs 1, 2 and 3) are potent peptidic neurotoxins found in the venom of the Trinidad chevron tarantula (Psalmopoeus cambridgei) inhibiting ASIC1A homomultimeric channel activity. These spider toxins are also potent agonists of TRPV1 and antagonists of Kv2-type voltage-gated potassium channels (Siemens et al, 2006).
Recent studies employing intrathecal injection of VaTx1 had significantly reduced thermal, mechanical, chemical, inflammatory and neuropathic pain in mice (Mazzuca et al., 2007; Baron et al., 2013). The potential roles of ASIC inhibitors amiloride (68) (a K + -sparing diuretic, IC50 5–100 μM) and benzamil (69) (benzyl amiloride) in nociception have recently been investigated (Kleyman and Cragoe et al., 1988). The clinical efficacy of amiloride in alleviating aura and headache has also been proved successful in an open-labelled pilot study consisting of 4–7 patients (Holland et al., 2012). Several synthetic and natural compounds modulating the ASICs such as GMQ, A-317567, NSAIDs, tetraethylammonium, 4-aminopyridine, aminoglycosides and local anaesthetics have been identified (Fig. 12). GMQ (70) (2-guanidine-4-methylquinazoline) is amiloride like compound belonging to the guanidinium class that activates ASIC3 at physiological pH7.4 and induce pain in an ASIC3-dependent manner, when injected into the paw of a mouse (EC50 2 mM) (Alijevic and Kellenberger et al., 2012).[156] Abbott Neuroscience Research Laboratory discovered a more potent amidine analogue A-317567 (71), as ASIC3 blocker (IC501025 nM). Compound 71 was found to be effective in the rat CFA model of inflammatory pain and in the skin incision model of postoperative pain (Dubé et al., 2005). Multiple studies have also demonstrated the ASIC inhibitory potential of NSAIDs such as flurbiprofen (72) and ibuprofen (73) against ASIC1a (IC50 ∼ 350 mM) whereas, salicylic acid (74), aspirin (75) and diclofenac (76) against ASIC3 (IC50 90–260 mM) (Dorofeeva et al., 2008). The analgesic effects of local anaesthetics (LAs) mainly due to voltage-gated Na + ion channel inhibition is well known however, some evidence for their ASIC inhibition activity was recently observed. The peak current of ASIC3 can be blocked by tetracaine (77) (IC50 10 mM) whereas, ASIC1a by lidocaine (78) (IC50 ∼ 12 mM) (Leng et al., 2013; Lin et al., 2011). Despite these significant developments, ASICs modulators has not yet produced compounds that are suitable for clinical use (Baron et al., 2015).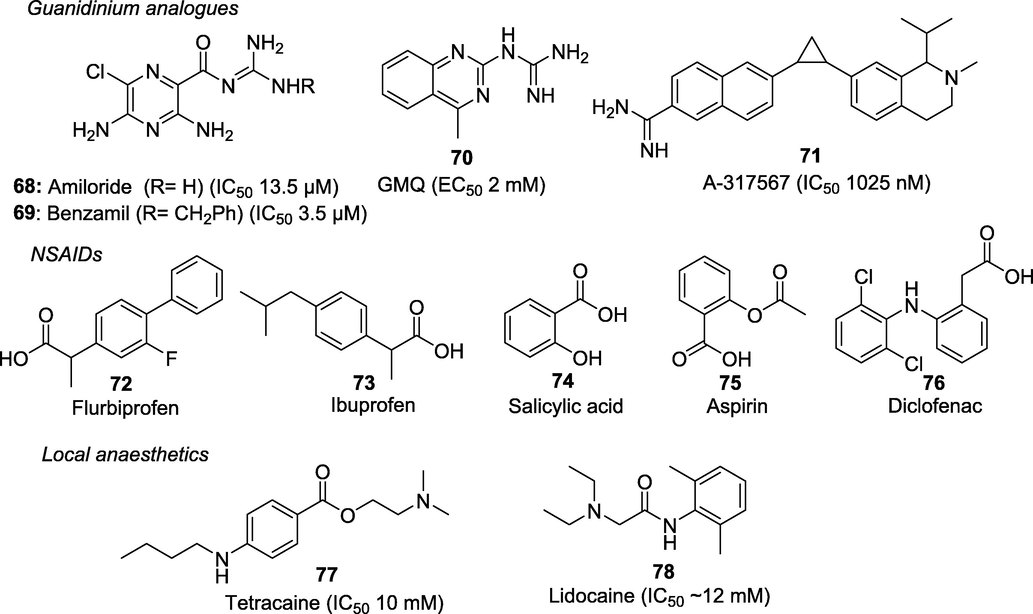
Acid-sensing ion channel modulators.
2.5 Piezo channel modulators
Scientists at the Scripps Research Institute, USA have characterized a revolutionary mechanosensitive PIEZO ion channel in a mouse neuroblastoma cell line. These are the family of mechanotransducers composed by two nonselective cationic channels known as Piezo1 and Piezo2 with a relatively homologous structure (Coste et al., 2010). Among the recently discovered mechanically sensitive ion channels, Piezo2, has been implicated in mediating proprioception and detecting light touch on the skin, as well as mechanical allodynia in a model of nerve injury. Studies have found that inflammatory signals also enhance Piezo2-mediated mechanosensitive currents in vitro and produces mechanical hyperalgesia (Dubin et al., 2012). Till now, very few modulators such as Yoda1 (79), Jedi1 (80) and Jedi2 (81) have been identified through high-throughput screening assays (Fig. 13). These small molecules activators utilize the key mechanotransduction components to activate Piezo1 (Wang et al., 2018).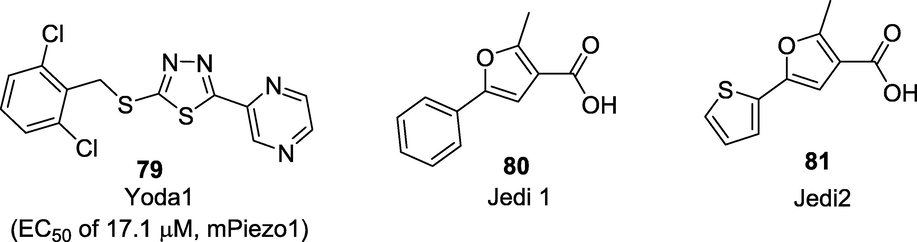
Human and mouse Piezo1 activators.
2.6 P2X receptor ligands
Purinergic receptors are ion gated channels distributed in the CNS and immune system. They are classified into two subfamilies: G-protein-coupled metabotropic (P2Y) and ligand-gated ionotropic (P2X) receptors (Burnstock, 2012). Among these, P2X which consists of seven members P2X1-7 have shown to play significant roles in the pathogenesis of pain and accumulating evidence indicates that activation of P2X3 receptors mainly mediates this effect. P2X3 receptor channels are chiefly expressed in sensory neurons and are activated by the extracellular ATP and thus play significant roles in nociception and sensory hypersensitization (Chen et al., 1995). Significant progress has been made to discover selective and more potent P2X antagonists for the treatment of several diseases associated with hypersensitive nerve fibres, including chronic pain, neurogenic inflammation, overactive bladder (OAB), and refractory and/or unexplained chronic cough (RUCC), as evidenced by the patents and publications. Several compounds belonging to diverse classes such as nucleotide ATP analogues, and non-nucleotide molecules comprising of diaminopyrimidines, imidazo-pyridine, arylamides, pyrrolinones etc. have been reported (Shieh et al, 2006). Many of these first generation P2X3 antagonists had very poor drug-like properties with unfavourable pharmacokinetic and pharmacodynamic characteristics. Currently, only four small molecules viz. gefapixant (82) (Merck), BLU-5937 (83) (Bellus Health), eliapixant (84) (Bayer), and sivopixant (85) (Shionogi) are being evaluated in different phases of clinical trials (Fig. 14). Gefapixant (82) (AF-219/MK-7264) is a first-in-class, orally active P2X3 antagonist (IC50 0.03 μM), that is being evaluated by Afferent Pharmaceuticals in phase II trials (NCT01554579) against patients with moderate to severe pain associated with osteoarthritis (OA) of the knee and cystitis/bladder pain syndrome (Richards et al., 2019). Despite the report of its unpleasant side effects including loss of taste, this small molecule has successfully progressed to Phase III trials for RUCC (NCT03449134). Another molecule is an oral imidazopyridine based bisphosphonate known as minodronate (86) which is approved in Japan for the oral treatment of osteoporosis (Kubo et al., 2010). Recently, the analgesic effects of minodronate mediated by the purinergic P2X2/3 receptor have been confirmed by its ability to reduce low back pain in patients (Yoshioka et al., 2013). BLU-5937 (83) is another small molecule of the imidazopyridine chemical class acting as a P2X3 antagonist with high selectivity (>1500 fold) and no taste alteration adverse effect (Garceau et al., 2019). The molecule has shown good drug and pharmacokinetic properties in healthy volunteers and currently being studied in phase II trial for the treatment of chronic cough and chronic pruritus (NCT03979638). Eliapixant (84) (BAY-1817080) a P2X3 antagonist belonging to arylamide class, is being developed by Bayer for the treatment of persistent chronic cough, diabetic neuropathic pain in Phase II trial (NCT04641273) and endometriosis-associated pelvic pain in Phase II (NCT04614246). Sivopixant (85) (S-600918), a dioxotriazine analgesic for the treatment of neuropathic pain and cough is being developed by Shionogi. Compound 82 was found to reduces chronic cough by selectively antagonizing the P2X3 receptors (Kai et al., 2021; Dicpinigaitis et al., 2020).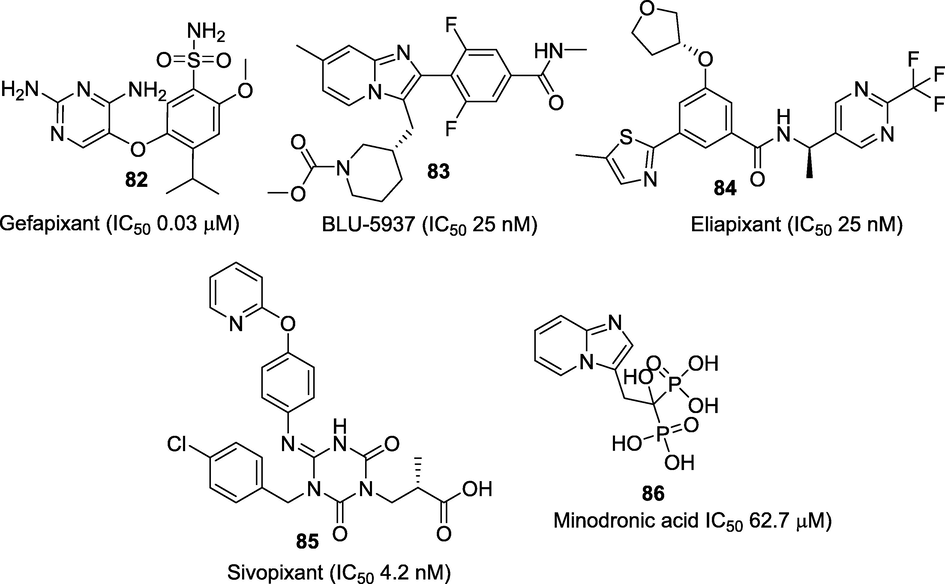
Selective P2X3 receptor antagonists under clinical studies.
3 Conclusion, authors comments and future perspective
Pain affects the physical and mental health of patients and has a tremendous financial impact globally. Despite the strong consensus that urgent pursuit required to combat the opioid crisis, there has been insufficient development of analgesics in the past for treating acute pains, such as trauma and surgery. Moreover, the global analgesics market size is expected to reach $42.6 billion in the coming few years. Therefore, the big pharma companies have now lately started focusing on development of novel non-opioid pain killers. As a result, around nine hundred preclinical analgesics in the pipeline are now rapidly advancing towards the clinical trials. Among these, ion channels are the second-largest target family, accounting >21% of the total drug candidates. Ion channels offer great potential for developing analgesics due to their high level of genomic diversity. Furthermore, technical advancements in the protein crystallography, high throughput screening and structure-guided drug discovery efforts have identified several classes of ion channels modulators. In several instances, these novel pain relievers have demonstrated good efficacies in their early and late stages of clinical trials. One such promising molecule is capsaicin from pepper which holds great promise for the treatment of pain. Development of its newer analogues may result not only in the discovery of safer and effective analgesics but also in better understanding of pain mechanisms. Similarly, toxins from spiders, snakes and cone snails etc. are a treasure trove of ligands that may aid in the development of new analgesics with potential therapeutic values. Moreover, several clinically available drugs that have lately proven to be acting as ion channels modulators (e.g. AEDs) can be repurposed to accelerate the development of newer analgesics. Future research involving SCN9A gene will potentially lead to entirely new avenues of novel antibody-based drugs and gene therapy, as a significant number of therapeutic opportunities are still unexplored.
CRediT authorship contribution statement
Yahya I. Asiri: Supervision, Conceptualization.
Acknowledgements
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under grant number RGP2/108/44.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Animal models of pain: Diversity and benefits. J. Neuro. Met.. 2021;348:108997
- [CrossRef] [Google Scholar]
- Subtype-specific modulation of acid-sensing ion channel (ASIC) function by 2-guanidine-4-methylquinazoline. J. Biol. Chem.. 2012;287:36059-36070.
- [CrossRef] [Google Scholar]
- Alsaloum, M., G.P. Higerd, P.R. Effraim, S.G., 2020. Waxman, Status of peripheral sodium channel blockers for non-addictive pain treatment. Nat. Rev. Neurol. 16, 689-705. https://doi.org/10.1038/s41582-020-00415-2.
- Anderson, C., Ruah, S.S.H., Golec, J.M.C., Zhang, B., Littler, B.J., Keshavarz-Shokri, A., Alcacio, T.E., Belmont, D.T., 2016. Prodrugs of pyridone amides useful as modulators of sodium channels. US patent 9464102.
- Discovery of a selective TRPM8 antagonist with clinical efficacy in cold-related pain. ACS Med. Chem. Lett.. 2015;6:419-424.
- [CrossRef] [Google Scholar]
- Development of the first ultra-potent “capsaicinoid” agonist at transient receptor potential vanilloid type 1 (TRPV1) channels and its therapeutic potential. J. Pharmacol. Exp. Ther.. 2005;312:561-570.
- [CrossRef] [Google Scholar]
- Dose-response of pregabalin for diabetic peripheral neuropathy, postherpetic neuralgia, and fibromyalgia. Postgrad. Med.. 2017;129:921-933.
- [CrossRef] [Google Scholar]
- Capsaicin 8% dermal patch for neuropathic pain in a pain unit. Pain Manag. Nurs.. 2022;23:452-457.
- [CrossRef] [Google Scholar]
- Discovery and optimization of selective nav1.8 modulator series that demonstrate efficacy in preclinical models of pain. ACS Med. Chem. Lett.. 2015;6:650-654.
- [CrossRef] [Google Scholar]
- Painful and painless mutations of SCN9A and SCN11A voltage-gated sodium channels. Pflugers. Arch. Eur. J. Physiol.. 2020;472:865-880.
- [CrossRef] [Google Scholar]
- Involvement of Na+ channels in pain pathways. Trends Pharmacol. Sci.. 2001;22:27-31.
- [CrossRef] [Google Scholar]
- Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon. 2013;75:187-204.
- [CrossRef] [Google Scholar]
- Pharmacology of acid-sensing ion channels e Physiological and therapeutical perspectives. Neuropharmacol.. 2015;94:19-35.
- [CrossRef] [Google Scholar]
- The menthol receptor TRPM8 is the principal detector of environmental cold. Nature. 2007;448:204-208.
- [CrossRef] [Google Scholar]
- Transduction and Encoding of Noxious Stimuli. In: Schmidt R., Willis W., eds. Encyclopedia of Pain. Berlin, Heidelberg: Springer; 2007. p. :229-246.
- [Google Scholar]
- Boehm, J.C., Davis, R.S. Kerns, J. Lin, G., 2013. Murdoch, H. Nie, Voltage-gated sodium channel blockers. PCT Int. Appl. WO2013006596.
- Voltage gated calcium channels as targets for analgesics. Curr. Top. Med. Chem.. 2005;5:539-546.
- [CrossRef] [Google Scholar]
- Discovery of purinergic signalling, the initial resistance and Current explosion of interest. Br. J. Pharmacol.. 2012;167:238-255.
- [CrossRef] [Google Scholar]
- A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428-431.
- [CrossRef] [Google Scholar]
- Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today. 2000;5:506-520.
- [CrossRef] [Google Scholar]
- 2-Aminoethoxydiphenyl borate as a common activator of TRPV1, TRPV2, and TRPV3 channels. Handb. Exp. Pharmacol.. 2007;179:173-187.
- [Google Scholar]
- Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. 2010;330:55-60.
- [CrossRef] [Google Scholar]
- An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894-898.
- [CrossRef] [Google Scholar]
- The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131:243-257.
- [CrossRef] [Google Scholar]
- Sodium Channels and Pain. In: Wood J.N., ed. the Oxford Handbook of the Neurobiology of Pain. UK: Oxford Handbooks Online; 2019. p. :1-35.
- [Google Scholar]
- P2X3-Receptor antagonists as potential antitussives: summary of current clinical trials in chronic cough. Lung. 2020;198:609-616.
- [CrossRef] [Google Scholar]
- Novel vanilloid receptor-1 antagonists: 2. Structure-activity relationships of 4-oxopyrimidines leading to the selection of a clinical candidate. J. Med. Chem.. 2007;50:3515-3527.
- [CrossRef] [Google Scholar]
- Mechanisms of non-steroid anti-inflammatory drugs action on ASICs expressed in hippocampal interneurons. J. Neurochem.. 2008;106:429-441.
- [CrossRef] [Google Scholar]
- Potassium channels in peripheral pain pathways: expression, function and therapeutic potential. Curr. Neuropharmacol.. 2013;11:621-640.
- [CrossRef] [Google Scholar]
- Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain. 2005;117:88-96.
- [CrossRef] [Google Scholar]
- Inflammatory signals enhance piezo2-mediated mechanosensitive currents. Cell. Rep.. 2012;2:511-517.
- [CrossRef] [Google Scholar]
- Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 1994;372:272-275.
- [CrossRef] [Google Scholar]
- Investigational drugs in early phase clinical trials targeting thermotransient receptor potential (thermoTRP) channels. Exp. Opin. Investig. Drugs. 2020;29:1209-1222.
- [CrossRef] [Google Scholar]
- A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann. Neurol.. 2009;65:733-741.
- [CrossRef] [Google Scholar]
- A disulfide tether stabilizes the block of sodium channels by the conotoxin μO§-GVIIJ. Proc. Natl. Acad. Sci. USA. 2014;111:2758-2763.
- [CrossRef] [Google Scholar]
- Hyperthermia induced by transient receptor potential vanilloid-1 (TRPV1) antagonists in human clinical trials: Insights from mathematical modeling and meta-analysis. Pharmacol. Therap.. 2020;208:107474
- [CrossRef] [Google Scholar]
- BLU-5937: A selective P2X3 antagonist with potent anti-tussive effect and no taste alteration. Pulm. Pharmacol. Ther.. 2019;56:56-62.
- [CrossRef] [Google Scholar]
- Pharmacological blockade of the vanilloid receptor TRPV1 elicits marked hyperthermia in humans. Pain. 2008;136:202-210.
- [CrossRef] [Google Scholar]
- The novel anticonvulsant drug, gabapentin (Neurontin), binds to the alpha2delta subunit of a calcium channel. J. Biol. Chem.. 1996;271:5768-5776.
- [CrossRef] [Google Scholar]
- Is TRPA1 burning down TRPV1 as druggable target for the treatment of chronic pain? Int. J. Mol. Sci.. 2019;20:2906.
- [CrossRef] [Google Scholar]
- Recent progress in TRPM8 modulation: An update. Int. J. Mol. Sci.. 2019;20:2618.
- [CrossRef] [Google Scholar]
- Scorpion toxins affecting sodium current inactivation bind to distinct homologous receptor sites on rat brain and insect sodium channels. J. Biol. Chem.. 1996;271:8034-8045.
- [CrossRef] [Google Scholar]
- Structure and function of μ-conotoxins, peptide-based sodium channel blockers with analgesic activity. Fut. Med. Chem.. 2014;6:1677-1698.
- [CrossRef] [Google Scholar]
- Guimaraes, M.Z.P., Jordt, S.E., 2007. TRPA1: A Sensory Channel of Many Talents, in: Liedtke, W.B., Heller, S. (Eds.), TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis. https://www.ncbi.nlm.nih.gov/books/NBK5237/ (accessed 11 January 2023).
- Hill, R.G., 2006. Analgesic drugs in development, in: McMahon, S.B., Koltzenburg M. (Eds.), Wall and Melzack's Textbook of Pain. Elsevier Churchill Livingstone, 5th ed., Philadelphia, pp-541–552.
- Ciguatoxin-induced oscillations in membrane potential and action potential firing in rat parasympathetic neurons. Eur. J. Neurosci.. 2002;16:242-248.
- [CrossRef] [Google Scholar]
- Acid-sensing ion channel 1: a novel therapeutic target for migraine with aura. Ann. Neurol.. 2012;72:559-563.
- [CrossRef] [Google Scholar]
- Xylocain (lidocaine, lignocaine), its discovery and Gordh’s contribution to its clinical use. Acta Anaesth. Scand. Suppl.. 1998;113:8-12.
- [Google Scholar]
- Acid-sensitive ion channels and receptors. Handb. Exp. Pharmacol.. 2009;194:283-332.
- [Google Scholar]
- Patient-reported outcomes in orphan drug labels approved by the US food and drug administration. Value Health. 2019;22:925-930.
- [Google Scholar]
- Discovery of TRPM8 antagonist (S)-6-(((3-Fluoro-4-(trifluoromethoxy)phenyl)(3-fluoropyridin-2-yl)methyl)carbamoyl) nicotinic Acid (AMG 333), a clinical candidate for the treatment of migraine. J. Med. Chem.. 2018;61:8186-8201.
- [CrossRef] [Google Scholar]
- Capsaicin and its analogues: structure-activity relationship study. Curr. Med. Chem.. 2013;20:2661-2672.
- [CrossRef] [Google Scholar]
- The symptoms of osteoarthritis and the genesis of pain. Rheum. Dis. Clin. North. Am.. 2008;34:623-643.
- [CrossRef] [Google Scholar]
- IASP Terminology, 1979. https://www.iasp-pain.org/Education/Content.aspx?ItemNumber=1698 (accessed 11 January 2023).
- The development of new analgesics over the past 50 years: A lack of real breakthrough drugs. Anesth. Analg.. 2010;110:780-789.
- [Google Scholar]
- TRPV2 channel as a possible drug target for the treatment of heart failure. Lab. Invest.. 2020;100:207-217.
- [Google Scholar]
- Molecular basis for species-specific sensitivity to “Hot” chili peppers. Cell. 2002;108:421-430.
- [Google Scholar]
- Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc. Natl. Acad. Sci. USA. 2000;97:8134-8139.
- [Google Scholar]
- Kai, T., Horiguchi, T., Kameyma, N., Onodera, N., Itoh, Y., Fujii, Y., Ichihashi, K., Hirai, T., Shintani, K., Nakamura, K., Minami, E., Kasai, S., Yoneda, Y., Murakami, H., Ogawa, R., Sekimoto, S., Shinohara, O., Yoshida, N., Kurose., 2021. Discovery of clinical candidate Sivopixant (S-600918): Lead optimization of dioxotriazine derivatives as selective P2X3 receptor antagonists. Bioorg. Med. Chem. Lett. 52, 128384. https://doi.org/10.1016/j.bmcl.2021.128384.
- Chronic pain, psychopathology, and DSM-5 somatic symptom disorder. Can. J. Psychiatry.. 2015;60:160-167.
- [Google Scholar]
- Effects of zonisamide on K+ and Ca2+ evoked release of monoamine as well as K evoked intracellular Ca2+ mobilization in rat hippocampus. Epilep. Res.. 1999;35:173-182.
- [CrossRef] [Google Scholar]
- A cell-penetrating scorpion toxin enables mode-specific modulation of TRPA1 and pain. Cell. 2019;178:1362-1374.
- [CrossRef] [Google Scholar]
- Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol.. 1988;105:1-21.
- [CrossRef] [Google Scholar]
- Different action of ethosuximide on low- and high-threshold calcium currents in rat sensory neurons. Neurosc.. 1992;51:755-758.
- [CrossRef] [Google Scholar]
- Minodronate for the treatment of osteoporosis. Drugs Today (Barc). 2010;46:33-37.
- [CrossRef] [Google Scholar]
- Recent advances in TRPV4 agonists and antagonists. Bioorg. Med. Chem. Lett.. 2020;30:1-6.
- [CrossRef] [Google Scholar]
- Pharmacological inhibition of voltage-gated Ca(2+) channels for chronic pain relief. Curr. Neuropharmacol.. 2013;11:606-620.
- [CrossRef] [Google Scholar]
- Z944: A first in class T-type calcium channel modulator for the treatment of pain. J. Peripher. Nerv. Syst.. 2014;19:S10-S14.
- [CrossRef] [Google Scholar]
- Interaction between voltage gated sodium channels and the neurotoxin, tetrodotoxin. Channels. 2008;2:407-412.
- [CrossRef] [Google Scholar]
- Subunit and frequency-dependent inhibition of Acid Sensing Ion Channels by local anesthetic tetracaine. Mol. Pain. 2013;9:27.
- [CrossRef] [Google Scholar]
- The role of sodium channels in chronic pain. Muscle & Nerve. 2012;46:155-165.
- [CrossRef] [Google Scholar]
- Inhibition of acid sensing ion channel currents by lidocaine in cultured mouse cortical neurons. Anesth. Analg.. 2011;112:977-981.
- [CrossRef] [Google Scholar]
- Interaction of batrachotoxin with the local anesthetic receptor site in transmembrane segment IVS6 of the voltage-gated sodium channnel. Proc. Natl. Acad. Sci. USA. 1998;95:13947-13952.
- [CrossRef] [Google Scholar]
- A structural model of the tetrodotoxin and saxitoxin binding site of the Na channel. Biophys. J.. 1994;66:1-13.
- [CrossRef] [Google Scholar]
- Lodish, H., Berk, A., Zipursky, S.L., 2000. Molecular Cell Biology, fourth ed. Freeman, W.H., New York. https://www.ncbi.nlm.nih.gov/books/NBK21668/, 2000 (accessed 5 February 2023).
- Effect of capsaicin and other thermo-TRP agonists on thermoregulatory processes in the American cockroach. Molecules. 2008;23:3360.
- [CrossRef] [Google Scholar]
- Neurotoxins and their binding areas on voltage-gated sodium channels. Front. Pharmacol.. 2011;2:71.
- [CrossRef] [Google Scholar]
- A pilot study of tranilast for cardiomyopathy of muscular dystrophy. Intern. Med.. 2018;57:311-318.
- [CrossRef] [Google Scholar]
- Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J. Neurophysiol.. 1994;72:349-359.
- [CrossRef] [Google Scholar]
- A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci.. 2007;10:943-945.
- [CrossRef] [Google Scholar]
- The expression pattern of TRPV1 in brain. J. Neurosci.. 2011;31:13025-13027.
- [CrossRef] [Google Scholar]
- Najam, F., Jafri, N., Khan, M.N., 2022. Daraz U. Reduction of Acute Postoperative Pain With Pre-Emptive Pregabalin Following Laparoscopic Cholecystectomy. Cureus. 14, e27783. doi: 10.7759/cureus.27783.
- Namadurai, S., Balasuriya, D., Rajappa, R., Wiemhöfer, M., Stott, K., Klingauf, J., Edwardson, J.M., Chirgadze, D.Y., Jackson, A.P., 2014. Crystal structure and molecular imaging of the Nav channel β3 subunit indicates a trimeric assembly. J. Biol. Chem. 289, 10797−10811. https://doi.org/10.1074/jbc.M113.527994.
- NCT00055484: A Study to Measure the Safety and Effectiveness of Zonisamide in Subjects With Migraine Headache. https://clinicaltrials.gov/ct2/show/NCT00055484?term=zonisamide&cond=Pain&draw=2&rank=2 (accessed 25 Jun 2022).
- NCT00160511: A 16 Week Study Evaluating Levetiracetam in the Treatment of Post Herpetic Neuralgia (PHN) https://clinicaltrials.gov/ct2/show/NCT00160511?term=Levetiracetam&cond=Pain&draw=2&rank=6 (accessed 20 June 2022).
- NCT00254657: Levetiracetam for Treatment of Pain Associated With Fibromyalgia. https://clinicaltrials.gov/ct2/show/NCT00254657?term=Levetiracetam&cond=Pain&draw=2&rank=4 (accessed 20 June 2022).
- NCT00286260: Levetiracetam for Painful Polyneuropathy. https://clinicaltrials.gov/ct2/show/NCT00286260?term=Levetiracetam&cond=Pain&draw=2&rank=2 (accessed 20 Jun 2022).
- NCT00725114: Safety & Efficacy Study of Subcutaneous Tetrodotoxin for Moderate to Severe Inadequately Controlled Cancer-related Pain (TEC-006). https://clinicaltrials.gov/ct2/show/NCT00725114?term=Tetrodotoxin&cond=Pain&draw=2&rank=1 (accessed 5 February 2023).
- NCT00726011:Tetrodotoxin Open-label Efficacy and Safety Continuation Study (TEC-006OL). https://clinicaltrials.gov/ct2/show/NCT00726011?term=Tetrodotoxin&cond=Pain&draw=2&rank=3 (accessed 5 February 2023).
- NCT00736151: Phase II Dose Titration Study in Patients With Neuropathic Pain. https://clinicaltrials.gov/ct2/show/NCT00736151?term=Ralfinamide&cond=Pain&draw=2&rank=1 (accessed 5 February 2023).
- NCT00804154: Resiniferatoxin to Treat Severe Pain Associated with Advanced Cancer. https://www.clinicaltrials.gov/ct2/show/NCT00804154 (accessed 20 January 2023).
- NCT01019824: Efficacy and Safety of Ralfinamide in Patients With Chronic Neuropathic Low Back Pain. https://clinicaltrials.gov/ct2/show/NCT01019824?term=Ralfinamide&cond=Pain&draw=2&rank=2 (accessed 5 February 2023).
- NCT01127906: Study to assess the effect of PF-04531083 on heat pain in healthy volunteers with ultraviolet light sensitized skin. https://clinicaltrials.gov/ct2/show/NCT01127906 (accessed 04 March 2023).
- NCT01240148: Determine the Effect of AZD3161, Injected Intradermally, on Quantitative Sensory Testing Variables in Normal and Ultraviolet-C (UVC) Exposed Skin in Healthy Volunteers. ClinicalTrials.gov, U.S. National Institutes of Health: Bethesda, MD, Nov 10, 2010, http:// clinicaltrials.gov/show/NCT01240148 (accessed 20 February 2023).
- NCT01463397: Efficacy and Safety of SAR292833 Administration for 4 Weeks in Patients with Chronic Peripheral Neuropathic Pain (Alchemilla). https://clinicaltrials.gov/ct2/show/NCT01463397 (accessed 5 February 2023).
- NCT01512160: Efficacy Of Pf-04531083 In Treating Post-Surgical Dental Pain. https://clinicaltrials.gov/ct2/show/NCT01512160. (accessed 04 March 2023).
- NCT01529346: Efficacy Of PF-05089771 In Treating Postoperative Dental Pain. https://clinicaltrials.gov/ct2/show/NCT01529346?term=PF-05089771&cond=Pain&phase=1&draw=2&rank=1 (accessed 5 February 2023).
- NCT01533428: A Study to Evaluate Efficacy and Safety of a Single Application of Capsaicin 8%Transdermal Delivery System Compared to Placebo in Reducing Pain Intensity in Subjects With Painful Diabetic Peripheral Neuropathy (PDPN) (STEP). https://clinicaltrials.gov/ct2/show/NCT01533428 (accessed 20 January 2023).
- NCT01554579: A Four-Week Multicenter Study Evaluating the Safety and Efficacy of Gefapixant (AF-219/MK-7264) in Subjects with Osteoarthritis of the Knee. https://clinicaltrials.gov/ct2/show/NCT01554579 (accessed 04 Nov 2022).
- NCT01726413: A Clinical Trial to Study the Effects GRC 17536 in Patients With Painful Diabetic Peripheral Neuropathy (Painful Extremities Due to Peripheral Nerve Damage in Diabetic Patients). https://clinicaltrials.gov/ct2/show/NCT01726413 (accessed 5 February 2023).
- NCT01776619: Safety and Tolerability Study of Multiple Doses of PF-06305591. https://clinicaltrials.gov/ct2/show/NCT01776619. (accessed 04 March 2023).
- NCT01786655: Safety Study of Long-Acting Local Anesthetic. Available online: https://clinicaltrials.gov/ct2/show/NCT01786655 (accessed 5 February 2023).
- NCT01848730: Efficacy and Safety of CNV2197944 Versus Placebo in Patients With Post-herpetic Neuralgia. https://clinicaltrials.gov/ct2/show/NCT01848730?term=CNV-2197944&cond=Pain&draw=2&rank=2 (accessed 15 Jun 2022).
- NCT01893125: Efficacy and Safety of CNV2197944 Versus Placebo in Patients With Diabetic Peripheral Neuropathy. https://clinicaltrials.gov/ct2/show/NCT01893125?term=CNV-2197944&cond=Pain&draw=2&rank=1 (accessed 15 Jun 2022).
- NCT02068599: A Study to Evaluate the Safety and Efficacy of Topically Applied TV-45070 (Ointment) in Participants With Primary Osteoarthritis (OA) Affecting a Single Knee. https://clinicaltrials.gov/ct2/show/NCT02068599?term=funapide&cond=Pain&draw=2&rank=2 (accessed 22 February 2023).
- NCT02215252: A Clinical Trial To Evaluate PF-05089771 On Its Own And As An Add-On Therapy To Pregabalin (Lyrica) For The Treatment Of Pain Due To Diabetic Peripheral Neuropathy (DPN). https://clinicaltrials.gov/ct2/show/NCT02215252?term=PF-05089771&cond=Pain&phase=1&draw=2&rank=2 (accessed 5 February 2023).
- NCT02365636: A Study to Evaluate the Safety and Efficacy of Topically Applied TV 45070 Ointment in Patients With Postherpetic Neuralgia (PHN) ((PHN)). https://clinicaltrials.gov/ct2/show/NCT02365636? term=funapide&cond=Pain&draw=2&rank=3 (accessed 20 February 2023).
- NCT02432664: Safety, Tolerability, Pharmacokinetic and Pharmacodynamic Effects of ODM-108: in Healthy Male Volunteers (FIMTRIP). https://clinicaltrials.gov/ct2/show/NCT02432664 (accessed 5 February 2023).
- NCT02935608: Study to Evaluate the Efficacy and Safety of BIIB074 in Neuropathic Pain From Lumbosacral Radiculopathy (RELAY-1). https://clinicaltrials.gov/ct2/show/NCT02935608?term=BIIB074 &cond=Pain&draw=2&rank=4 (accessed 18 February 2023).
- NCT03202303: Cannabidivarin (CBDV) vs. Placebo in Children With Autism Spectrum Disorder (ASD). https://clinicaltrials.gov/ct2/show/NCT03202303 (accessed 5 February 2023).
- NCT03304522: A Study to Evaluate the Efficacy and Safety of VX-150 in treating subjects with pain caused by small fiber neuropathy. https://clinicaltrials.gov/ct2/show/NCT03304522. (accessed 04 March 2023).
- NCT03372603: A Study to Assess the Effectiveness and Side Effects of GSK2798745 in Participants With Chronic Cough. https://clinicaltrials.gov/ct2/show/NCT03372603 (accessed 5 February 2023).
- NCT03449134: A Study of Gefapixant (MK-7264) in Adult Participants with Chronic Cough (MK-7264-027) https://www.clinicaltrials.gov/ct2/show/NCT03449134 (accessed on 05 Nov 2022).
- NCT03454126: Evaluating the Safety, Tolerability, and Pharmacokinetics of BIIB095 in Healthy Participants. https://clinicaltrials.gov/ct2/show/NCT03454126?term=biib095&cond=Pain&draw=2&rank=2 (accessed 20 February 2023).
- NCT03637387: 802NP302 Efficacy and Safety Study of BIIB074 in Participants With Trigeminal Neuralgia (SURGE-2). https://clinicaltrials.gov/ct2/show/NCT03637387?term=BIIB074&cond=Pain&draw=2&rank=10 (accessed 20 February 2023).
- NCT03979638: A Dose Escalation Study of BLU-5937 in Unexplained or Refractory Chronic Cough (RELIEF). https://clinicaltrials.gov/ct2/show/NCT03979638 (accessed 04 Nov 2022).
- NCT04203537: Study Evaluating the Safety, Efficacy and Pharmacokinetics of CA-008 (Vocacapsaicin). https://www.clinicaltrials.gov/ct2/show/NCT04203537 (accessed 20 January 2023).
- NCT04217733: Ethosuximide and Pentoxifylline in the Treatment of Abdominal Pain Related to Irritable Bowel Syndrome. https://clinicaltrials.gov/ct2/show/NCT04217733?term=Ethosuximide&cond=Pain&draw=2&rank=4 (accessed 22 Jun 2022).
- NCT04292912: Study 212669: A Phase I Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GSK2798745 in Participants With Diabetic Macular Edema. https://clinicaltrials.gov/ct2/show/NCT04292912 (accessed 5 February 2023).
- NCT04431778: Evaluation of the Efficacy and Tolerance of Low Doses of Ethosuximide in the Treatment of Peripheral Neuropathic Pain (E-PENEPA). https://clinicaltrials.gov/ct2/show/NCT04431778?term=Ethosuximide&cond=Pain&draw=2&rank=1 (accessed 22 Jun 2022).
- NCT04475198: Single Ascending Dose Study to Evaluate the Safety, Tolerability, and Pharmacokinetics of ST-2427. https://clinicaltrials.gov/ct2/show/NCT04475198#contacts (accessed 5 February 2023).
- NCT04614246: Study to Gather Information How Well Three Different Doses of BAY1817080 Given Twice Daily Over 12 Weeks Work in Comparison to an Inactive Pill (Placebo) and Elagolix in Women Suffering From Pain Related to a Condition Where the Tissue That Usually Grows Inside the Womb Grows Outside of the Womb (SCHUMANN). https://clinicaltrials.gov/ct2/show/NCT04614246 (accessed 04 Nov 2022).
- NCT04641273: A 2-part Trial to Learn More About How BAY1817080 Works, How Safe it is, and What the Right Dose is for Participants with Diabetic Neuropathic Pain. https://clinicaltrials.gov/ct2/show/NCT04641273 (accessed 04 Nov 2022).
- TRPV3: time to decipher a poorly understood family member! J. Physiol.. 2014;592:295-304.
- [CrossRef] [Google Scholar]
- Calcium channel diversity and neurotransmitter release: the omega-conotoxins and omega-agatoxins. Annu. Rev. Biochem.. 1994;63:823-867.
- [Google Scholar]
- Institute of Medicine (US) Committee on Pain, Disability, and Chronic Illness Behavior. US: National Academies Press; 1987.
- Pajouhesh, H., Kaul, R., Ding, Y., Zhu, Y., Zhang, L., Chakka, N., Grimwood, M., Tan, J., Zhou, Y., 2009. N- Piperidinyl acetamide derivatives as calcium channel blockers Patent number: WO2009146540. US20090420793.
- Transient receptor potential channels: targeting pain at the source. Nat. Rev. Drug. Discov.. 2009;8:55-68.
- [CrossRef] [Google Scholar]
- Calcium channel modulation as a target in chronic pain control. Br. J. Pharmacol.. 2018;175:2173-2184.
- [CrossRef] [Google Scholar]
- Novel radiolabeled vanilloid with enhanced specificity for human transient receptor potential vanilloid 1 (TRPV1) J. Med. Chem.. 2017;60:8246-8252.
- [CrossRef] [Google Scholar]
- Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys. J.. 1998;75:2647-2657.
- [CrossRef] [Google Scholar]
- Pérez de Vega, M.J., Gómez-Monterrey, I., Ferrer-Montiel, A., González-Muñiz, R., 2016. Transient Receptor Potential Melastatin 8 Channel (TRPM8) Modulation: Cool Entryway for Treating Pain and Cancer. J. Med. Chem. 59, 10006-10029. https://doi.org/10006-10029. 10.1021/acs.jmedchem.6b00305.
- Ziconotide: a clinical update and pharmacologic review. Exp. Opin. Pharmacother.. 2013;14:957-966.
- [CrossRef] [Google Scholar]
- Safety and efficacy of a topical sodium channel inhibitor (TV-45070) in patients with postherpetic neuralgia (PHN): A randomized, controlled, proof-of-concept, crossover study, with a subgroup analysis of the Nav1.7 R1150W genotype. Clin. J. Pain. 2017;33:310-318.
- [Google Scholar]
- TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J. Neurosci.. 2008;28:6231-6238.
- [CrossRef] [Google Scholar]
- TRPV1 antagonistic analgesic effect: A randomized study of AZD1386 in pain after third molar extraction. Pain. 2013;154:808-812.
- [CrossRef] [Google Scholar]
- Fountain, S.J. Action of MK-7264 (gefapixant) at human P2X3 and P2X2/3 receptors and in vivo efficacy in models of sensitisation. Br. J. Pharmacol.. 2019;176:2279-2291.
- [CrossRef] [Google Scholar]
- Probenecid improves cardiac function in patients with heart failure with reduced ejection fraction in vivo and cardiomyocyte calcium sensitivity in vitro. J. Am. Heart. Assoc.. 2018;7:1-11.
- [CrossRef] [Google Scholar]
- Anandamide and vanilloid TRPV1 receptors. Br. J. Pharmacol.. 2003;140:790-801.
- [CrossRef] [Google Scholar]
- Treatment of chronic pain: antidepressant, antiepileptic and antiarrhythmic drugs. Cont. Edu. Anaesth. Crit. Care Pain. 2005;5:18-21.
- [CrossRef] [Google Scholar]
- Discovery of Acyl-sulfonamide Nav1.7 Inhibitors GDC-0276 and GDC-0310. J. Med. Chem.. 2021;64:2953-2966.
- [CrossRef] [Google Scholar]
- The sodium channel activator Brevetoxin-3 uncovers a multiplicity of different open states of the cardiac sodium channel. Biochim. Biophys. Acta. 1992;1104:233-242.
- [CrossRef] [Google Scholar]
- Therapeutic potential of conopeptides. Fut. Med. Chem.. 2012;4:1243-1255.
- [CrossRef] [Google Scholar]
- Lidocaine reduces the transition to slow inactivation in Na(v)1.7 voltage-gated sodium channels. Brit. J. Pharmacol.. 2011;164:719-730.
- [CrossRef] [Google Scholar]
- Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303-1308.
- [CrossRef] [Google Scholar]
- P2X receptor ligands and pain. Exp. Opin. Ther. Pat.. 2006;16:1113-1127.
- [CrossRef] [Google Scholar]
- Spider toxins activate the capsaicin receptor to produce inflammatory pain. Nature. 2006;444:208-212.
- [CrossRef] [Google Scholar]
- Integrating TRPV1 receptor function with capsaicin psychophysics. Adv. Pharmacol. Sci. 2016:1512457.
- [CrossRef] [Google Scholar]
- Recent advances in the development of T-typecalcium channel blockers for pain intervention. Br. J. Pharmacol.. 2018;175:2375-2383.
- [CrossRef] [Google Scholar]
- Novel TRPM8 agonist cooling compound against chronic itch: results from a randomized, double-blind, controlled, pilot study in dry skin. J. Eur. Acad. Dermatol. Venereol.. 2017;31:1064-1068.
- [CrossRef] [Google Scholar]
- The emerging role of TRP channels in mechanisms of temperature and pain sensation. Curr. Neuropharmacol.. 2006;4:83-96.
- [CrossRef] [Google Scholar]
- Pharmacology, structure and function of cardiac L-type Ca(2+) channels. Cell Physiol. Biochem.. 1999;9:242-269.
- [CrossRef] [Google Scholar]
- The discovery of the Nav1.7 inhibitor GDC-0276 and development of an efficient large-scale synthesis. ACS Symp.. 2019;1332:107-123.
- [Google Scholar]
- Ion Channels. In: McQueen C.A., ed. Comprehensive Toxicology. USA: Elsevier; 2010. p. :129-171.
- [Google Scholar]
- Resiniferatoxin and analogs provide novel insights into the pharmacology of the vanilloid (capsaicin) receptor. Life Sci.. 1990;47:1399.
- [Google Scholar]
- Management of pain in the Emergency Department. ISRN Emerg. Med.; 2013. p. :583132.
- Intrathecal drug delivery in the management of chronic pain. Best Pract. Res. Clin. Anaesthesiol.. 2023;37:157-169.
- [CrossRef] [Google Scholar]
- Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron. 2012;73:638-652.
- [Google Scholar]
- Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211-1259.
- [Google Scholar]
- Iodo-resiniferatoxin, a new potent vanilloid receptor antagonist. Mol. Pharmacol.. 2001;59:9-15.
- [Google Scholar]
- Marked difference in saxitoxin and tetrodotoxin affinity for the human nociceptive voltage-gated sodium channel (Nav1.7) Proc. Natl. Acad. Sci. USA. 2012;109:18102-18107.
- [Google Scholar]
- The discovery of capsazepine, the first competitive antagonist of the sensory neuron excitants capsaicin and resiniferatoxin. J. Med. Chem.. 1994;37:1942-1954.
- [CrossRef] [Google Scholar]
- A lever-like transduction pathway for long-distance chemical- and mechano-gating of the mechanosensitive Piezo1 channel. Nat. Commun.. 2018;9:1300.
- [Google Scholar]
- Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9:1099-1115.
- [CrossRef] [Google Scholar]
- WHO guidelines for the pharmacological and radiotherapeutic management of cancer pain in adults and adolescents. Geneva: World Health Organization, 2018. http://apps.who.int/iris. (accessed 10 January 2023).
- The prevalence and years lived with disability caused by low back pain in China, 1990 to 2016: findings from the global burden of disease study 2016. Pain. 2019;160:237-245.
- [Google Scholar]
- Antibodies and venom peptides: new modalities for ion channels. Nat. Rev. Drug Discov.. 2019;18:339-357.
- [CrossRef] [Google Scholar]
- Oregano, thyme and clove-derived flavors and skin sensitizers activate specific TRP channels. Nat. Neurosci.. 2008;9:628-635.
- [Google Scholar]
- Calcium channels as therapeutic targets in neuropathic pain. J. Pain. 2006;7:S13-S30.
- [Google Scholar]
- Development of new analgesics: An answer to opioid epidemic. Trends Pharmacol. Sci.. 2018;39:1000-1002.
- [Google Scholar]
- General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int. J. Mol. Sci.. 2018;19:2164.
- [Google Scholar]
- Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Nav1.7 sodium channels in primary afferents. Hum. Gene. Ther.. 2005;16:271-277.
- [Google Scholar]
- A comparative study of the effects of daily minodronate and weekly alendronate on upper gastrointestinal symptoms, bone resorption, and back pain in postmenopausal osteoporosis patients. J. Bone Miner. Metab.. 2013;31:153-160.
- [Google Scholar]
- Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lan. Neurol.. 2017;16:291-300.
- [CrossRef] [Google Scholar]
- randomized double-blind, placebo-, and active-controlled study of T-type calcium channel blocker ABT-639 in patients with diabetic peripheral neuropathic pain. Pain. 2015;156:2013-2020.
- [CrossRef] [Google Scholar]




