

Translate this page into:
Triazolopyridine, a leitmotif of synthetic methods and pharmacological attributes: An extensive review
⁎Corresponding author. iftikharali@uswat.edu.pk (Iftikhar Ali) iftikharali@cuhk.edu.hk (Iftikhar Ali) ia2531@cumc.columbia.edu (Iftikhar Ali)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The interest in Triazolopyridine and its derivatives has grown exponentially. The latest literature and knowledge on synthetic procedures have been described. Novel and efficient methodologies for synthesizing Triazolopyridine and its derivatives have been described. Newer findings on biological and pharmacological activities have been reviewed. This review may be helpful for medicinal chemists and future researchers to develop novel and potent drug candidates.
Abstract
Background
Famous synthetic pharmacophores like Filgotinib, Dapiprazole, and Trazodone have Triazolopyridine as their primary building element. It has become more well-known in medicinal chemistry, to which its broad-spectrum impact may be attributed.
Objective
The derivatization of the triazolopyridine molecule has been widely discussed in several scientific articles, focusing on its biological and pharmacological properties. As lead compounds for developing new drugs, many analogues of Triazolopyridine have been found. This article summarizes and discusses the literature surrounding the synthesis and biological evaluation of the triazolopyridine framework.
Methods
Relevant keywords were used to identify relevant published literature from all relevant databases, including SciFinder, PubMed, and Google Scholar. Supplementary relevant literature bibliographies were also searched to locate linked reports.
Results
Step-by-step explanations of the synthetic approaches to Triazolopyridine-based ring systems were found in the literature. All Triazolopyridine derivative's pharmacological activities were enumerated according to their targets, and a detailed structure–activity link was created.
Conclusion
The current review highlights various synthetic techniques for fabricating the triazolopyridine framework and its uses across several medical chemistry fields. The wide range of biological effects of compounds with the triazolopyridine skeleton, including those that are antibacterial, antifungal, anticancer, anti-inflammatory, antitubercular, analgesic, anticancer, and antidepressant, make them crucial in the process of developing new drugs and so on. Despite extensive research, the structure-based drug design technique can create new, effective pharmacophores.
Keywords
Triazolopyridine
1,2,3-triazoles
Biological activities
1,2,4-triazoles
Synthesis

- T.P
-
Triazolopyridine
- CAN
-
Ceric ammonium nitrate
- NaNO
-
Sodium Nitrite
- EDCI
-
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- ADME
-
Adsorption, Distribution, Metabolism and Distribution
- CoMFA
-
Comparative molecular field analysis
Abbreviations
1 Introduction
Five different heterocyclic systems fall under the umbrella category of triazolopyridines. There are three 1,2,3-triazole structures here and 1,2,4-triazole structures (Abarca-González, 2002). Two do not have bridgehead nitrogen, while the other three do. As building blocks of essential biomolecules like amino acids and nucleotides, nitrogen-containing heterocyclic compounds are indispensable to life's sustenance (East et al., 2009a) (Khaleghi Abbasabadi et al., 2020) (Khaleghi Abbasabadi and Azarifar, 2020) (Khaleghi-Abbasabadi and Azarifar, 2019; Khodabakhshi et al., 2015). Numerous fields, including molecular recognition, medicine, chemical synthesis, and biology, use triazolopyridines, among the most crucial nitrogen-containing five-membered heterocycles (Ribeiro et al., 2019a) (Mahmoudi‐Gom Yek et al., 2020). Triazolopyridines are a class of heterocyclic chemical compounds characterized by a triazole ring connected to a pyridine ring (Abdel-Megid et al., 2013). Isomers can be differentiated based on the arrangement of their nitrogen atoms and the type of ring fusion that occurs (Adam et al., 2014a). In addition, “triazolopyridine” can refer to a class of antidepressant medications with a ring system derived from Triazolopyridine. These medications were developed in the 1950 s. Trazodone is a medication that fits this description. There has been a resurgence of interest in triazolopyridines with the discovery and approval of trazodone, a [1, 2,4]-triazolopyridine, as a selective serotonin reuptake inhibitor (Chea and Giorgi, 2017); “Trazadone, A Triazolopyridine Antidepressant” 1980. Within the larger triazolopyridines category, five different heterocyclic systems are included. These consist of two 1,2,3-triazoles and three 1,2,4-triazoles. Three of these are 1,2,3-triazoles. Only two bridgeheads lack bridgehead nitrogen, in contrast to the three that have it (Fig. 1). In general, this last category reflects differences in the synthesis or characteristics of the compounds(Jones, 2003a). The chemistry of triazolopyridine derivatives and the possible uses of these compounds 5 piques our attention. Our findings add to the exploration of previously undiscovered reactions of Triazolopyridine, a small but versatile substance that led us to discover a fascinating field of research.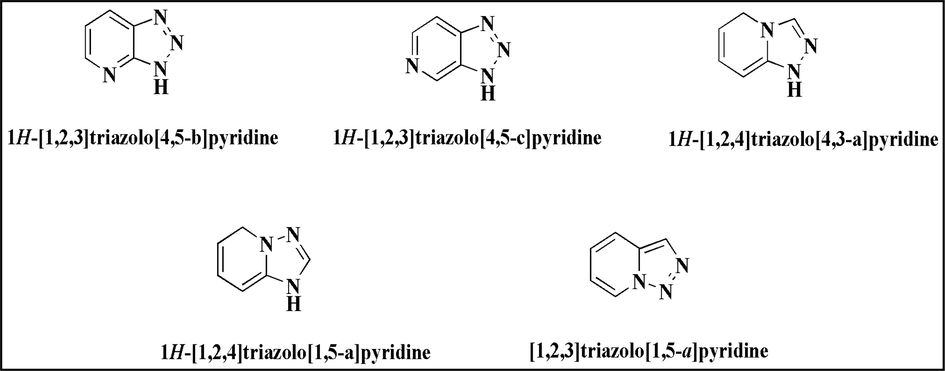
Chemical structures of triazolopyridine isomers.
Our findings contribute to the exploration of previously undiscovered reactions of Triazolopyridine. It summarises their chemistry succinctly (Jones et al., 1985). Synthetic compounds make up the vast bulk of physiologically active T.P.s., as are the analogues of dihydro [1, 2,4] triazolo [1, 5-a] pyrimidine and [1, 2,4] triazolo [1, 5-a] pyrimidinone that has been described so far (Frick et al., 2015). Derivatives of Triazolopyridine constitute a major group of organic compounds that find application in various biochemical, medicinal, and pharmacological processes. Nitrogen-containing heterocycles like triazolopyridines typically emerge as bi- or multiple complexing ligands (Dymińska et al., 2022a; Frick et al., 2015). This is because of lone electron pairs on the nitrogen atoms in the nitrogen-containing heterocycles. They can coordinate metal ions, generating complex compounds that can be used as catalysts, chemosensors, and fluorescence sensors. These isomeric systems of [1, 2,4] triazolo[1, 5-a], [1,2,4] triazolo[4,3-a], [1,2,3] triazolo[1,5-a], [1,2,3] triazolo[4,5-b], and [1,2,3] triazolo[4,5-c] pyridine are especially desirable. This group also contains [1,2,4] triazolo[4,3-a]. According to Dymiska et al., numerous isomeric triazolopyridines each have a specific role in pharmaceutical and medical applications (Dymińska et al., 2022a). Because of the extensive array of uses of triazolopyridines in various fields, the authors of this study concluded that a collection of synthetic protocols geared toward these motifs would be beneficial for younger researchers interested in concentrating on synthesizing triazolopyridines and their applications. In the present review, an effort was made to compile various synthesis developments throughout the last 15 years (2005–2022 up to the present) and the biological activities of triazolopyridines and their derivatives. Young researchers can now work on triazolopyridine motifs to construct several extremely effective lead molecules thanks to the chronological improvements in the synthesis of triazolopyridines and a quick summary of their therapeutic applications.
2 Chemistry
Five heterocyclic systems are included in the broad category of triazolopyridines. These systems appear to number five, with three 1, 2, 3 and two 1, 2, 4 systems.
2.1 Chemical properties
It undergoes oxidation and reduction reactions with ring and ring substituents. It shows reactions with electrophiles as well as nucleophiles. Sometimes undergoes homolytic and ring-opening reactions (Jones, 2003b; Singh et al., 2019).
2.2 Synthesis of triazolopyridines
Triazolopyridines and fused triazoles are two types of effective scaffolds that can be utilized to synthesize therapeutically intriguing compounds. The traditional method for synthesizing triazolopyridines begins with the dehydration of a 2-hydrazidopyridine. The reaction can be accomplished by refluxing phosphorus oxychloride, concentrated hydrochloric acid, and acetic acid(Dymińska et al., 2022b).
3 Synthetic routes of triazolopyridines
3.1 Synthetic route of 1H- [1,2,3] triazolo [4,5-b] pyridine
(E)-3-(2-(4-chlorophenyl) diazinyl)-4-(benzyloxy) pyridine-2,6-diamine (Diazene) 3 was formed by interacting 4-(benzyloxy) pyridine-2,6-diamine 1 with 4-chlorobenzenediazonium chloride 2, produced from diazotization of 4-chloroaniline at 0 °C. Cleavage of the zinc-diazene bond to produce the diamine, which is then cyclized with isoamyl nitrite, was noted by Wurtz et al. and produced 7-(benzyloxy)-1H-[1,2,3] triazolo[4,5-b]pyridin-5-amine 4 by enabling late-stage diversification (Fig. 2) (Wurtz et al., 2018).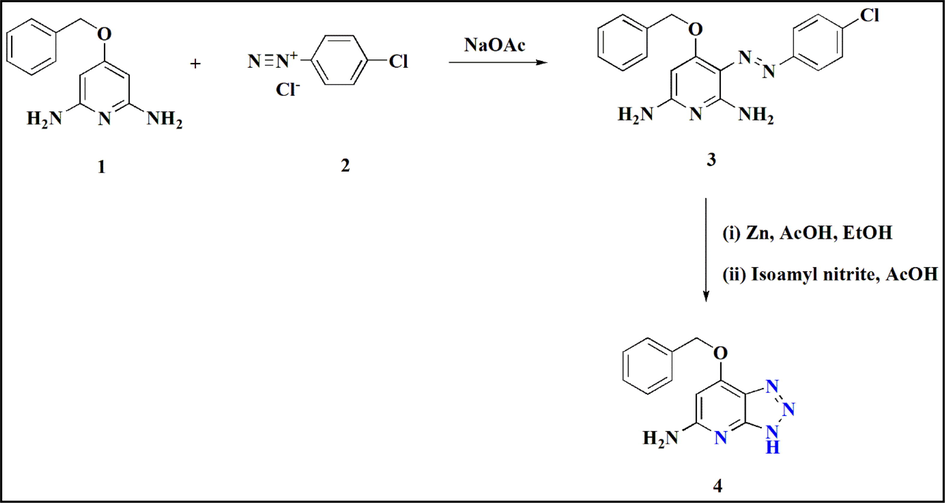
Synthesis of Triazolopyridine via late-stage diversification.
3.2 Synthetic route of 3H– [1,2,3] triazolo[4,5-c] pyridine
Matsuda and co-workers successfully created synthetic 3H–[1,2,3] triazolo[4,5-c]pyridine derivatives using 2-Bromo-5-fluoropyridine 5 N-oxidized utilizing trifluoroacetic anhydride, hydrogen peroxide, and urea with constant stirring at room temperature for 15 hrs followed by quenching with saturated solution of sodium sulfite in ice cold condition. The resultant compound 6 was nitrated utilizing strong sulfuric acid and fuming nitric acid by stirring at 100 °C for 4 hrs to get 7. Furthermore, compound 8 was produced by the ArNS reaction of 7 with the appropriate amine, and Fe then reduced the nitro groups that resulted from this reaction to produce compounds 9, which underwent cyclization using Trifluoroacetic acid with NaNO2 to produce derivatives of the 3H–[1,2,3] triazolo[4,5-c] pyridine 10 through continuous stiring at room temperature for 1hr followed by concentrating the reaction mixture in vacuo. The resultant residue was diluted with aqueous sodium bicarbonate solution and extracted with chloroform. (Fig. 3) (Matsuda et al., 2017).![Synthesis of 3H–[1,2,3]triazolo [4,5-c] pyridine analogues.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig3.png)
Synthesis of 3H–[1,2,3]triazolo [4,5-c] pyridine analogues.
3.3 Synthetic route of 1H- [1,2,4] triazolo[4,3-a] pyridine
According to Nakka et al., by Ceric ammonium nitrate (CAN) (5 mol%), a reusable reaction medium, accelerated oxidative amidrazones of 1-(pyridin-2-yl) hydrazine undergo cyclization 11 and aldehydes 12 using reusable (consecutive three times) polyethylene glycol as solvent were carried out to produce a variety of 3-substituted-[1,2,4] triazolo[4,3-a]pyridine 13 (Fig. 4) that are economical and may find usage in commercial applications (Nakka et al., 2014).![CAN catalysed synthesis of 3-substituted-[1,2,4] triazolo[4,3-a]pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig4.png)
CAN catalysed synthesis of 3-substituted-[1,2,4] triazolo[4,3-a]pyridine.
Baucom et al. developed an operationally efficient 1,1′carbonyldiimidazole (CDI) mediated method for synthesizing [1,2,4] triazolo[4,3a] pyridines 16. This research involved a single flask tandem coupling of substituted carboxylic acids 14 and 1-(pyridin-2-yl) hydrazine 11 in presence of acetonitrile at ambient temperature for 30 min followed by Cyclization of intermediate formed, substituted N'-(pyridin-2-yl) benzo hydrazide 15 using CDI (2.0 equiv) as a gentle and effective reagent to produce 16 (Fig. 5). The methodology suits batch mode and continuous process (Baucom et al., 2016).![CDI catalysed synthesis of [1,2,4] triazolo[4,3-a] pyridine via Tandem coupling and cyclization.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig5.png)
CDI catalysed synthesis of [1,2,4] triazolo[4,3-a] pyridine via Tandem coupling and cyclization.
Reichelt and co-workers established a quick and easy method for making 3-substituted-[1,2,4] triazolo[4,3-a]pyridines 13, which on addition of alkyl or aryl hydrazides 18 to 2-chloropyridine 17 in the presence of 2–5 mol% Pd2(dba)3 and a phosphine ligand, Josiphos as a catalyst to form an intermediate 19 on heating in sealed vial at 100 °C for 15 hr and separated as brown oil. It happens chemo-selectively at the hydrazide's terminal nitrogen. Product 19, brown oil, which was filtered through, was microwave irradiated at 180 °C for 30 min in presence of glaial acetic acid. SiO2 undergoes dehydrative Cyclization when acetic acid is present to form desired product 13 (Fig. 6) (Reichelt et al., 2010).![Palladium-Catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig6.png)
Palladium-Catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.
For the synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine 21, Jatangi et al. discovered iodine-mediated oxidative C-N and N-S bond forms in water, which allow for a simple, ecologically friendly, metal-free method. Through the interaction of 1-(pyridin-2-yl) hydrazine 11 and isothiocyanates 20 when magnetically stirred at room temperature for 3hr using water as solvent, the desired compound 21 was formed. The reaction mixture was further quenched with sodium thisulphate to remove unreacted iodine. (Fig. 7) (Jatangi et al., 2018).![Synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine via iodine mediated oxidative reaction.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig7.png)
Synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine via iodine mediated oxidative reaction.
According to Krylov et al., when the equimolar concentration of chloroethynylphosphonates 23 reacts with substituted 2-hydrazinylpyridines 22 in the presence of anhydrous potassium carbonate (K2CO3), and acetonitrile at room temperature for 4 hr afforded [1,2,4] triazolo[4,3-a] pyridines phosphonate derivatives 24. 31P NMR spectroscopy was used to monitor the reaction. (Fig. 8) (Krylov et al., 2019).![Synthesis of [1,2,4] triazolo[4,3-a] pyridines phosphonate derivatives using chloroethynylphosphonates.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig8.png)
Synthesis of [1,2,4] triazolo[4,3-a] pyridines phosphonate derivatives using chloroethynylphosphonates.
Flefel and co-workers have synthesized [1,2,4] triazolo[4,3-a] pyridine-8-carbonitrile derivatives 26–29 by cyclo condensation of hydrazine compound 25. During the research, 2-hydrazinonicotinonitrile was prepared by chlorinating naphthalen-2-pyridone using phosphorous oxychloride (POCl3) or phosphorus pentachloride (PCl5). When intermediate 25 was refluxed for 2–8 hrs with benzoyl chloride, acetic acid/acetic anhydride, formic acid, or carbon disulfide using ethanol as a solvent, the corresponding triazolopyridine derivatives 26–29 were produced. (Fig. 9) (Flefel et al., 2018; Kotb et al., 2017a).![Synthesis of [1,2,4] triazolo[4,3-a] pyridine-8-carbonitrile derivatives via Cyclocondensation.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig9.png)
Synthesis of [1,2,4] triazolo[4,3-a] pyridine-8-carbonitrile derivatives via Cyclocondensation.
Higgins and co-workers discovered method for synthesis of 3-(cyclopropyl methyl)-8-(trifluoromethyl)-7-alkoxy-[1,2,4] triazolo[4,3-a]pyridine 34 which includes 4 steps. During the research, chlorine is displaced with alkoxy groups on constant stirring for 1 hr in presence of strong base sodium hydride at 0 °C from 2,4-dichloro-3-trifluoromethyl pyridine 30 to form ethers 31, followed by displacement of second chlorine with hydrazine to generate hydrazine adducts 32. 2-cyclopropyl-N'-(3-(trifluoromethyl)-4-alkoxypyridin-2-yl) acetohydrazide 33 was formed by acylating adduct 32 with cyclopropyl acetyl chloride, which underwent mild cyclodehydration using methyl N-(triethylammonium sulfonyl) carbamate known as Burgess reagent using dioxane and acetonitrile solvent to form the desired products 34 (Fig. 10) (Higgins et al., 2017).![Synthesis of 3-(cyclopropyl methyl)-8-(trifluoromethyl)-7-alkoxy- [1,2,4] triazolo[4,3-a] pyridine using Burgess reagent.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig10.png)
Synthesis of 3-(cyclopropyl methyl)-8-(trifluoromethyl)-7-alkoxy- [1,2,4] triazolo[4,3-a] pyridine using Burgess reagent.
Gandikota et al. efficiently synthesized 3-chloromethyl-[1,2,4] triazolo[4,3-a]- pyridine 36 by treating 2-Chloro pyridine 17 with hydrazine hydrochloride at 100 °C for 48 hrs to give 1-(pyridin-2-yl)hydrazine 11 followed by selective mono acylation with chloro acetyl chloride through continuous stiring at ambient temperature for 12 hrs to form 2-chloro-N'-(pyridin-2-yl)acetohydrazide 35 which underwent dehydrative Cyclization using phosphorous oxychloride POCl3 to afford desired compound 36 with constant stiring at 120 °C for 12 hrs. (Fig. 11) (Gandikota et al., 2017).![Synthesis of 3-chloromethyl- [1,2,4] triazolo[4,3-a]- pyridine by dehydrative cyclization using POCl3.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig11.png)
Synthesis of 3-chloromethyl- [1,2,4] triazolo[4,3-a]- pyridine by dehydrative cyclization using POCl3.
Srinivasan et al. have established a workable one-pot synthesis method of 3-substituted-[1,2,4] triazolo[4,3-a]- pyridine 13. This research involved the CuBr2 (10 mol%) catalyzed oxidative Cyclization of hydrazone 37 under mild conditions, formed by interacting various aldehydes and 1-(pyridin-2-yl) hydrazine 11 in presence of oxone at room temperature for 3 hrs. (Fig. 12) (Srinivasan et al., 2016).![Copper-Catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig12.png)
Copper-Catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.
Ramesha et al. have developed a useful one-pot synthetic strategy for the synthesis of 3-substituted-[1,2,4] triazolo[4,3-a]- pyridine 13, which involved a novel approach by treating 1-(pyridin-2-yl)hydrazine 11 with thioesters 38 in the presence of propyl phosphonic anhydride (T3P) and DMSO. The reaction was completed within 2–4 hrs using THF as a solvent. (Fig. 13) (Ramesha et al., 2016).![T3P-DMSO catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig13.png)
T3P-DMSO catalysed synthesis of 3-substituted- [1,2,4] triazolo[4,3-a] pyridine.
Jerome and co-workers have explored two approaches for synthesizing 5-Bromo 3-substituted- [1,2,4] triazolo[4,3-a] pyridine 42. In both methods, initially, 2,5-dibromopyridine 39 was converted to 1-(5-bromopyridin-2-yl) hydrazine 40. In Approach 1, one-pot synthesis of product 42 was achieved by acylation of 40 using acyl chloride or oxalyl dichloride, followed by dehydrative Cyclization using POCl3. Approach 2 involves an additional step of formation of hydrazide 41 by coupling 40 with various carboxylic acids when there was 1-Ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDCI). The hydrazide formed then undergo dehydrative Cyclization in thionyl chloride's presence to get the desired product 42 (Fig. 14) (Jerome et al., 2010).![Synthesis of 6-Bromo-3-substituted- [1,2,4] triazolo[4,3-a] pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig14.png)
Synthesis of 6-Bromo-3-substituted- [1,2,4] triazolo[4,3-a] pyridine.
Roberge et al. synthesized 6-Bromo-3-isopropyl-[1,2,4] triazolo[4,3-a]pyridine 44 using a modified Mitsunobu reaction. The research showed the conversion of N'-(5-bromopyridin-2-yl) isobutyrohydrazide 43 in presence of Triphenylphosphine (Ph3P) and Diethyl azodicarboxylate (DEAD) to product 44 by using the trimethylsilyl-azide (TMS-N3) as an additive for 72 hrs. (Fig. 15) (Roberge et al., 2007).![TMS-N3 catalysed synthesis of 6-Bromo-3-isopropyl- [1,2,4] triazolo[4,3-a] pyridine using Mitsunobu reaction.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig15.png)
TMS-N3 catalysed synthesis of 6-Bromo-3-isopropyl- [1,2,4] triazolo[4,3-a] pyridine using Mitsunobu reaction.
Wang et al. created a successful method for the microwave-irradiated synthesis of substituted-[1,2,4] triazolo[4,3-a]pyridine 46. The desired product was synthesized by treating carboxylic acid 45 with substituted 2-hydrazinylpyridines 22 in the presence of solid phase reagent PS-PPh3/CCl3CN, which played an important role in acylation and dehydration. The reaction mixture was irradiated in dichloromethane at 150 °C for 15 min. (Fig. 16) (Wang et al., 2007).![Synthesis of substituted- [1,2,4] triazolo[4,3-a] pyridine using solid phase reagent, PS-PPh3/CCl3CN.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig16.png)
Synthesis of substituted- [1,2,4] triazolo[4,3-a] pyridine using solid phase reagent, PS-PPh3/CCl3CN.
3.4 Synthetic route of 1H- [1,2,4] triazolo[1,5-a] pyridine
Methyl ester of 1H- [1,2,4] triazolo[1,5-a] pyridine 49 was synthesized by Akiu et al. via chemical modification of the pyridine atom of methyl 4-aminobenzoate 47 with ethyl O-mesitylsulfonyl acetohydroxamate 48, following by Condensation with the suitable carboxylic acid or Condensation with the appropriate acyl chlorides at 80 °C using triethylamine as a base and EDCI catalyst. (Fig. 17)(Akiu et al., 2021a).![Synthesis of methyl ester of 1H- [1,2,4] triazolo[1,5-a] pyridine using EDCI.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig17.png)
Synthesis of methyl ester of 1H- [1,2,4] triazolo[1,5-a] pyridine using EDCI.
In Fig. 18, the equivalent N[4-(ethylthio)-2-oxopyridin 6 was made by reacting N-cyanoacetohydrazide 53 with 2,2-dicyanoethene-1,1-bis(ethylthiolate) 52 for 24 h at room temperature under the presence of KOH-dioxane. Additionally, by reacting 2 with N'-[(aryl)-methylene]-2cyanoacetohydrazides, 7a-f were treated in KOH-dioxane for 24 h at room temperature to obtain the corresponding 7,5-(ethylthio)-3,2,4 triazolo[1,5-a]pyridines 55.(Azzam and Elgemeie, 2019a).![Synthesis of 7-(ethylthio)-3,5-dihydro [1,2,4] triazolo[1,5-a] pyridines.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig18.png)
Synthesis of 7-(ethylthio)-3,5-dihydro [1,2,4] triazolo[1,5-a] pyridines.
A copper-catalyzed tandem addition for N-C bond formation and oxidative Cyclization through an N-N coupling procedure was established by Ueda et al. to produce 2-substituted 1H-[1,2,4] triazolo[1,5-a]pyridine 58. To explore the synthetic strategy, they substituted 2-aminopyridine 56 and aryl nitriles 57 in the presence of CuBr (5 mol%) and 1,10-Phen (5 mol%). The reaction efficacy was improved by adding ZnI2 (10 mol%) and refluxing at 130 °C for 24 hrs in dichlorobenzene (Fig. 19) (Ueda and Nagasawa, 2009).![2-substituted 1H- [1,2,4] triazolo[1,5-a] pyridine synthesis via Tandem Addition-Oxidative Cyclization Catalysed by Copper.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig19.png)
2-substituted 1H- [1,2,4] triazolo[1,5-a] pyridine synthesis via Tandem Addition-Oxidative Cyclization Catalysed by Copper.
Guba and co-workers have synthesized 8-amino-2-substituted- [1,2,4] triazolo[1,5-a] Pyridine methyl ester 60 by regioselective N-amination of methyl 5,6-aminopyridine-3-carboxylate 59 using O-mesitylene sulfonyl hydroxylamine followed by Condensation with different aldehyde 12 and oxidative Cyclization in alkaline medium (Fig. 20) (Guba et al., 2004a).![Synthesis of 8-amino-2-substituted- [1,2,4] triazolo[1,5-a] pyridine methyl ester.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig20.png)
Synthesis of 8-amino-2-substituted- [1,2,4] triazolo[1,5-a] pyridine methyl ester.
3.5 Synthetic route of 1H- [1,2,3] triazolo[1,5-a] pyridine
Landman et al. developed the method for synthesizing 1H- [1,2,3] triazolo[1,5-a] pyridine derivatives were formed utilizing nitrous oxide as a diazo transfer reagent. Nitrous oxide and metalized 2-alkyl pyridines combined to generate triazolopyridines. The reactions can be carried out in a friendly environment and produce moderate to good yields of the desired product.
Researchers had first lithiated substituted 2-benzyl pyridine 61 using butyl lithium (n-BuLi) in the presence of diethyl ether as a solvent to form substituted lithiated 2-benzyl Pyridine which, on treatment with nitrous oxide (N2O) in the presence of THF at 50 °C for 2 hrs to synthesize 1H-[1,2,3] triazolo[1,5-a]pyridine derivatives 62 (Fig. 21). Similarly they also synthesized 3-(1-(pyridin-2-yl) pentyl)- [1,2,3] triazolo[1,5-a] pyridine 64 using lithiated trans-1,2di(2-pyridyl) ethylene as an intermediate which is formed by lithiation of trans-1,2di(2-pyridyl) ethylene 63 using butyl lithium with constant stirring at −78 °C to RT for 1 hr followed by treatment with 0.05 M THF. (Fig. 22). Furthermore, they modified the approach for the synthesis of 3-substituted 1H- [1,2,3] triazolo[1,5-a] pyridines 66 (Fig. 23) (Landman et al., 2021).![1H- [1,2,3] triazolo[1,5-a] pyridine derivatives.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig21.png)
1H- [1,2,3] triazolo[1,5-a] pyridine derivatives.
![Synthesized 3-(1-(pyridin-2-yl) pentyl)- [1,2,3] triazolo[1,5-a] pyridine.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig22.png)
Synthesized 3-(1-(pyridin-2-yl) pentyl)- [1,2,3] triazolo[1,5-a] pyridine.
![3-substituted 1H- [1,2,3] triazolo[1,5-a] pyridines.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig23.png)
3-substituted 1H- [1,2,3] triazolo[1,5-a] pyridines.
Metalated methyl 2-(pyridine-2-yl) acetate was solubilized by adding 18-Crown-6 as an additive and deprotonated by using potassium tert-butoxide (KOtBu). Subsequent addition of nitrous oxide in the presence of 0.1 M DMSO The spotless emergence of potassium salt has been accomplished via exposure to heat (Fig. 24). Acidic workup gave carboxylic acid (Lapier et al., 2019a).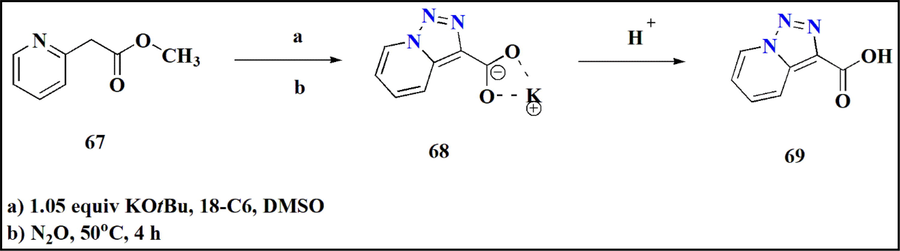
Triazolopyridine carboxylic acid.
Lapier and co-workers have used the methodology for synthesizing 3-substituted- [1,2,3] triazolo[1,5-a] pyridine derivatives 72. The research involved manganese dioxide (MnO2) catalyzed Cyclization of 1-(substituted(pyridin-2-yl) methylene) hydrazine 71, which was formed by treating pyridin-2-yl ketone 70 with methanolic hydrazine monohydrate to form desired compound 72. This compound 72 was then subjected to lithiation using n-BuLi in presence of toluene at −40 °C to form intermediate 73, which, treated with various electrophilic reagents, afforded 7-keto-(3-substituted-[1,2,3] triazolo[1,5-a] Pyridine derivatives 74 (Fig. 25)(Lapier et al., 2019b).![[1,2,3] triazolo[1,5-a] pyridine derivatives.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig25.png)
[1,2,3] triazolo[1,5-a] pyridine derivatives.
Cai et al. have developed an efficient, free, one-step synthesis method for synthesizing substituted [1,2,3] triazolo[1,5-a] Pyridine derivatives 76 at room temperature. In this method, substituted pyridin-2-ylmethanamines 75 were diazotized using tert-butylnitrite and undergo 1,3-H migration followed by Cyclization and dehydration to afford desired product 76 (Fig. 26).![[1,2,3] triazolo[1,5-a] pyridine derivatives.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig26.png)
[1,2,3] triazolo[1,5-a] pyridine derivatives.
Palepu et al. have synthesized 3-(pyridin-2-yl)-[1,2,3] triazolo[1,5-a]pyridine 79 by nucleophilic substitution reaction in which p-tosyl hydrazine 77 was condensed with dipyridyl ketone 78 by refluxing for 6 hrs in presence of acetic acid. In this reaction. The tosyl group is nucleophilically substituted with the nitrogen of the pyridyl group (Fig. 27)(Palepu and Kollipara, 2017).![Synthesis of 3-(pyridin-2-yl)- [1,2,3] triazolo[1,5-a] pyridine by nucleophilic substitution reaction.](/content/184/2023/16/10/img/10.1016_j.arabjc.2023.105181-fig27.png)
Synthesis of 3-(pyridin-2-yl)- [1,2,3] triazolo[1,5-a] pyridine by nucleophilic substitution reaction.
4 Biological applications of triazolopyridines
In synthesizing [1,2,4] triazolo[1,5-a] pyrimidines, crucial for medicinal chemistry, the heterocyclic compound is a possible bio-isostere for purines, carboxylic acid, and N-acetylated lysine. Antibacterial(Kotb et al., 2017b), antiviral(Ribeiro et al., 2019b), antifungal(Kuroyanagi et al., 2010a), anti-breast cancer(Sachdeva et al., 2019a), Leishmanicidal(Martín-Montes et al., 2017a), and neurotropic(Paronikyan et al., 2019a), anti-tumour(Ryu et al., 2021) properties have been observed in several T.P.s and analogues, highlighting their relevance in various medical contexts. Many enzyme inhibitors, including those that block the action of the mGlu2 receptor, DNA gyrase, and the HIV-1 allosteric integrase.
4.1 Anti-breast cancer activity
Tanisha Sachdeva et al. reported a new [1,2,4] triazolo[4, 3-a] pyridine scaffolds based on phenothiazine that was synthesized and produced in significant quantities. All chemicals were tested on cancer cells from humans (MDA-MB-231, MDA-MB-468, MCF7, SKBR3, and T47D) and non-tumorigenic breast cells (MCF10 A). The cytotoxic and apoptotic effects of a molecule with a pendent phenyl group on phenothiazine were tested against a breast cancer cell line, and the IC50 ranged from 10.2 to 17.6 M. Importantly, cancer cell cytotoxicity was 3-fold higher than that of normal cells, suggesting that breast cancer might be managed with minimal side effects. The G2/M blockade of breast cancer cells from humans was confirmed in a cell-cycle investigation. Compound 80a, which has a pendant phenyl ring on phenothiazine, significantly inhibited the growth of breast cancer cell types and induced apoptosis, with IC50 values ranging from 10.2 to 17.6 M compared to 80b (Fig. 28).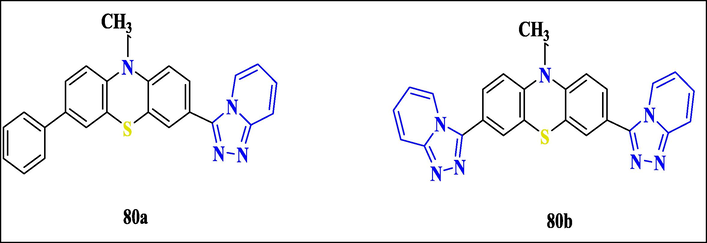
Structure of compound 80 with anti-Breast Cancer Activity.
The anticancer activity on cancer cells was notable since it was three times more likely to be robust than on non-cancer cells, suggesting the prospect of controlling breast cancer with fewer side effects. The molecular docking results, which demonstrated hydrophobic interaction between the linked phenyl ring, Triazolopyridine, and phenothiazine, confirmed this assessment(Sachdeva et al., 2019b).
Volker K. Schulze et al. discussed cancer treatment by removing the checkpoint for the spindle assembly. Inhibition of monopolar spindle 1 (MPS1) kinase induces mitosis regardless of DNA damage in cancerous cells instead of halting the cell cycle. It results in aneuploidy and cell death. One HTS hit from the “triazolopyridines” was a starting point for Schulze et al., optimized to identify MPS1 inhibitors. The preclinical analysis of these molecules in a few in vivo models, their production, development, and ability to bind the ATP site of MPS1 kinase and various binding interactions led to the evolution of Clinical candidate 81(Fig. 29) (Schulze et al., 2020).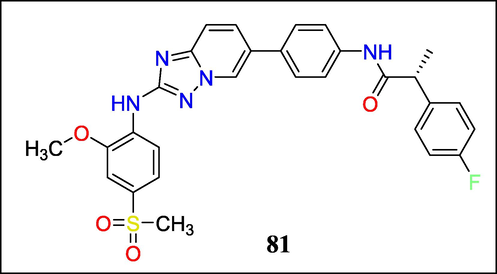
Structure of compound 81 with anti-Breast Cancer Activity.
4.2 Leishmanicidal activity
Leishmaniasis remains a significant global problem, and new drugs are intensely interested in combating it. Montes et al. described triazole [1,2,3] pyridine compounds with potent Leishmanicidal activity. Synthesis of novel [1,2,3] triazolo[1,5-a] pyridinium salts from triazolopyridines and five of the tested compounds (82, 83, 84, 85, 86) displayed selectivity indices that were higher than those of the standard medication Glucantime (Fig. 30). Furthermore, these compounds inhibit Fe-SOD in all three parasite species studied but have little effect on human CuZn-SOD. These compounds are attractive prospects for developing affordable leishmanicidal medications because of their low toxicities, stability and solubility, low cost of raw materials, and ease of production. These substances have a considerable impact on the parasites' gluconeogenesis as well as inhibiting Fe-SOD (Martín-Montes et al., 2017b).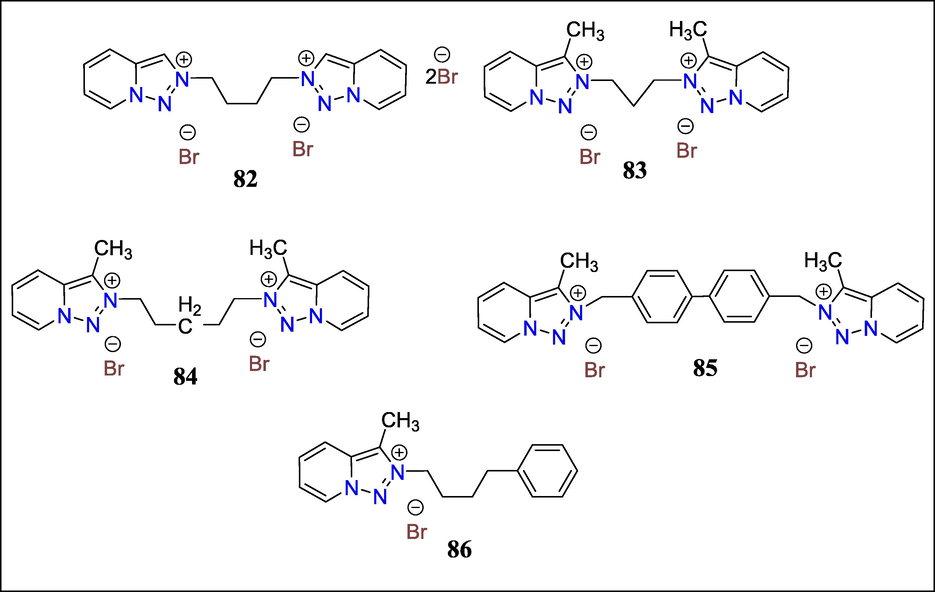
Structures of compounds 82, 83, 84, 85 and 86 with Leishmanicidal activity.
The most effective leishmanicidal agents were discovered to be compounds 87 and 88 (Fig. 31). Both exhibited efficacy versus cultured Leishmania spp. Promastigotes at micromolar doses (IC50 = 99.8–26.8 M), without toxicity toward J774 macrophage cells. Furthermore, a mouse model of L. infantum infection was used to assess these two substances in vivo. Triazolopyridine derivative 87 was efficient versus parasites of the spleen and liver; however, derivative 88 was inefficient. Through DNA binding experiments, mechanistic features of the antileishmanial action were examined. The active ligands are capable of interacting effectively with DNA, according to the results [Kb = 1.14 105 M−1 (87) and 3.26 105 M−1 (88)]. Additionally, it has been suggested that both 87 and 88 bind to DNA grooves. Fluorescence titrations and UV–visible spectrophotometer were used to track the association of compounds 8 and 9 with bovine serum albumin (BSA) to shed further light on the mechanism of action of these substances under natural settings. The binding coefficients and quenching constants were identified. Strong affinity has been shown for BSA by triazolopyridine ketones 87 and 88 [Kb = 2.5 104 M−1 (87) and 1.9 104 M−1 (88)] (Adam et al., 2014b).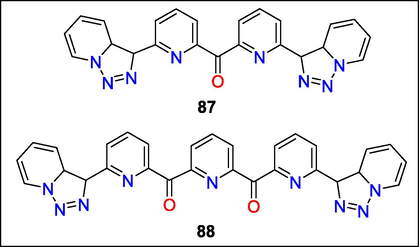
Structures of compounds 87 and 88 with Leishmanicidal activity.
4.3 Neurotropic activity
Ervand G. Paronikyan et al. synthesized 8-Hydrazino pyranoij3,4-c] pyridine derivatives and all compounds' neurotropic activity was tested. Six compounds (89, 90, 91, 92, 93 and 94) anticonvulsant testing with pentylene tetrazole and maximal electroshock seizure results indicated activity (MES) (Fig. 32). Additionally, open field, elevated plus maze (EPM), and forced swimming tests were used to assess the anxiolytic and antidepressant effects of these drugs. The “rotating rod” test showed no effect of the substances on neuromuscular synchronization. Substance 91 proved to be the most effective across all testing. The Inhibitory GABA Receptor, Serotonin Reuptake Blocker, and 5-Hydroxy receptor were paired with the most effective molecules (Paronikyan et al., 2019b).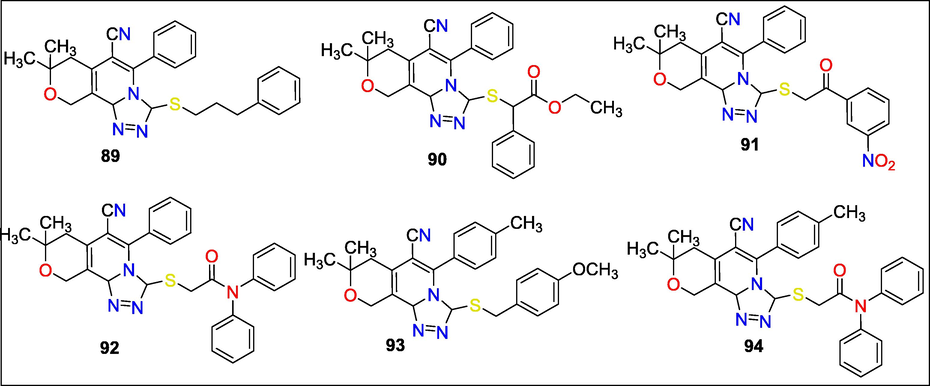
Structure of compounds 89 to 94 with Neurotropic activity.
In compound 95, Mayuko Akiu discovered a potent nicotinamide phosphoribosyltransferase stimulator with decreased CYP inhibition. NAMPT catalyzes the rate-limiting step in the NAD + salvage pathway. As NAD + is involved in multiple biochemical functions, including metabolism and ageing, stimulating NAMPT is a possible treatment method for various illnesses. Compound 95, a triazolopyridine derivative, was the initial HTS hit optimized. To develop 1-[2-(1-methyl-1H-pyrazol-5-yl)-[1,2,4] triazolo[1,5-a] pyridin-6-yl], researchers had to address concerns about CYP direct inhibition (DI), which they did by modifying the compound's lipophilicity(Fig. 33). NAMPT solid activity was seen with the chemical 3-(pyridin-4-ylmethyl) urea (96), and CYP DI was reduced concerning many CYP isoforms (Akiu et al., 2021b).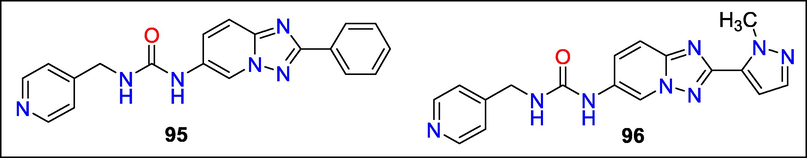
Structure of compounds 95 and 96 with Neurotropic activity.
Novel N-substituted triazolopyridines were synthesized and checked for antibacterial activity by Rasha Azzam et al. The compounds were evaluated for their antibacterial properties versus 4 fungi and eight bacterial species. Among the three Gram-negative bacteria examined, compounds 97 and 98 of the triazolo[1,5-a] pyridine family showed the highest antibacterial activity (Fig. 34)(Azzam and Elgemeie, 2019b).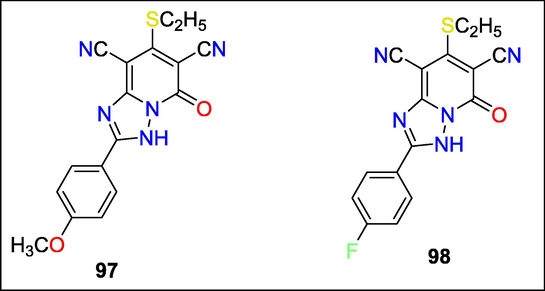
Structure of compounds 97 and 98 with Neurotropic activity.
Kathryn Bell recently revealed the SAR of a novel class of 6-aryl-2-amino-triazolopyridines, selective and potent PI3Kc inhibitors. Chemoproteomic examination of a kinase-specific database uncovered the 6-aryl-triazolopyridine core. Fast chemical expansion around a bifunctional core allowed the identification of the critical elements needed for PI3Kc activity and selectivity. The compound 99 was created as a highly bioavailable, potent PI3Kc inhibitor with selectivity over PI3Ka and PI3Kd. Compound 100 showed improved selectivity over the whole kinome, especially over PI3Kb, due to a core alteration(Fig. 35) (Bell et al., 2012).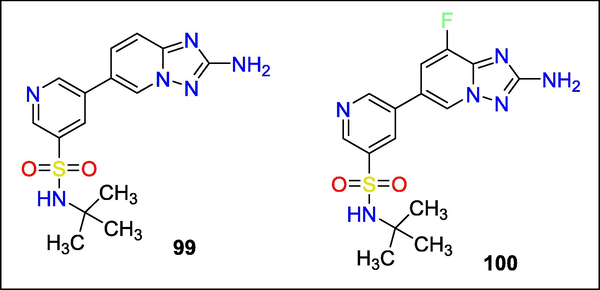
Structure of compounds 99 and 100 with Neurotropic activity.
The discovery of 99 potent, orally accessible small-molecule inhibitors of PI3Kc, stems from the work of Mihiro Sunose et al., who identified a family of 7-substituted triazolopyridines. Oral delivery of Compound 99 results in excellent kinome specificity, significant efficacy for cell-based tests, and in vivo testing in a mouse collagen-induced osteoarthritis model (Sunose et al., 2012).
4.4 Kinase inhibition
The ongoing investigation of the Triazolopyridine scaffolds as a basis for p38 MAP kinase inhibition was described by Kevin D. Jerome et al. structure-based drug development discusses the production and SAR of several C6 sulphur-linked triazolopyridines-based p38 inhibitors. Using a structure-based design, sulphur-linked molecules like 101 were simulated and proved effective p38a inhibitors; however, they limited metabolic stability (Fig. 36). Modelling demonstrated that methylene-linked antagonists like 102 are biologically stable in p38a and bioisosteric. The triazolopyridine series binding models were validated by an X-ray of the ethylene-related molecule 103 (Jerome et al., 2010b).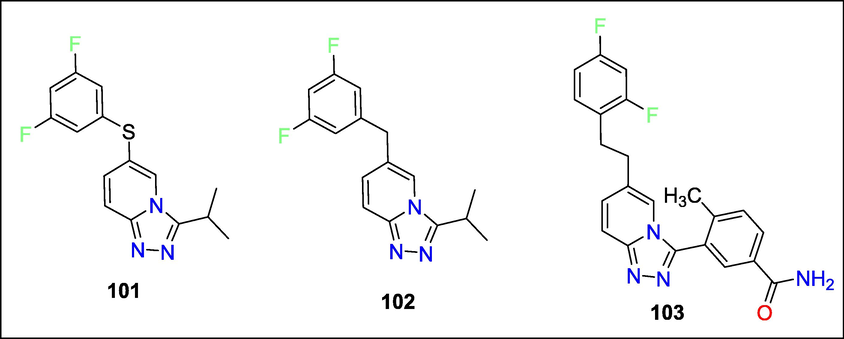
Structure of compounds with kinase inhibition activity.
Triazolopyridine oxazole compounds were examined as antagonists of p38 MAP alpha kinases by M. Ravi Shashi Nayana et al. utilizing 3D-QSAR and Integrated Molecular Field Analysis to find new physiologically chemical ingredients with enhanced inhibitory therapeutic potential (CoMFA). The simulation result for CoMFA produced q2 values of 0.707 and r2 values of 0.942. The discoveries may potentially help in the creation of new, more powerful inhibitors, in addition to improving our knowledge of the structural requirements for p38 alpha inhibitors. The 104 interaction mechanisms of the drugs at the p38 MAP alpha kinase active site were examined using the Glide docking tool (Fig. 37). Hydrogen-bonding relationships between the inhibitors and the target were found. The contour plots are connected with a brief discussion of the specifics of the amino acid linkages at the active site (Shashi Nayana et al., 2008).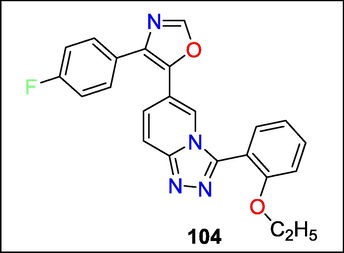
Structure of compound 104 with kinase inhibition activity.
4.5 DNA gyrase
Inhibitors of DNA gyrase were created by Stephen P. East et al. three DNA supercoiling blockers (105, 106, and 107) chemotypes based on [1,2,4] triazolo[1,5-a] pyridine scaffolds are synthesized and tested for their antimicrobial properties (Fig. 38). These inhibitors target the DNA gyrase and topoisomerase IV (GyrB/ParE) ATPase subunits. Triazolopyridine scaffold molecules 105, 106, and 107 have potent antibacterial action through GyrB/ParE suppression (East et al., 2009b).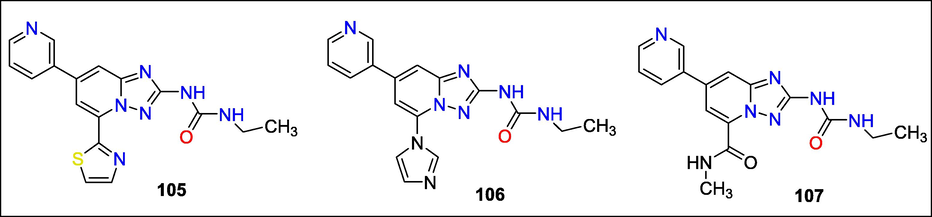
Structure of compounds with DNA gyrase inhibition activity.
4.6 β-Hydroxysteroid dehydrogenase type 1 (11β-HSD-1) inhibitors
According to research by Jun Li et al., a 1,2,4-triazolopyridine (TP) core was shown to have several effective and selective inhibitors of the human 11-hydroxysteroid dehydrogenase type 1 (11-HSD-1) enzyme. Notable lead compounds like 108 were among the most promising (Fig. 39). This finding shows that the TP core might be a perfect, scalable substrate for synthesizing novel chemicals (Li et al., 2014).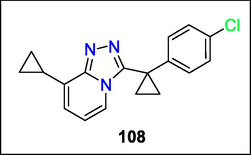
Structure of compound 108 with (11-HSD-1) enzyme inhibition activity.
Human 11b-hydroxysteroid dehydrogenase type 1 was effectively inhibited by 3,8-substituted 1,2,4-triazolopyridines that Haixia Wang and colleagues created. Following modification of substituents at the 3 and 8 sites of the TZP core, molecule 109 was discovered to be a strong and metabolically stable blocker of the enzyme (Fig. 40). Future research on this chemotype focuses on refining 109 to identify a candidate that can be tested in vivo (Li et al., 2014).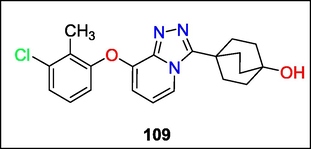
Structure of compound 109 with (11-HSD-1) enzyme inhibition activity.
4.7 Adenosine 2A receptor inhibitors
A study conducted by Wolfgang Guba et al. showed that the amino group in the 5-amino series 110 is a significant indicator for enhanced affinity and hA2a/hA1 specificity in this series, favouring generally triazolopyridine derivatives 110. This is based on the relationship between the hydrogen bond donor and acceptor capabilities of the nitrogen atoms in both scaffold series. (Fig. 41). Despite the structural similarity of the two scaffold series, the bioactivity of the two differs noticeably due to variations in electron density (Guba et al., 2004b).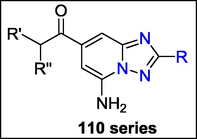
Structure of compound 110 with Adenosine 2A Receptor Inhibitors.
4.8 Antibacterial activity
Anil K. Sadana et al. produced 1-aryl/hetryl-1,2,4-trizolo-[4,3-a] pyridines and 1-aryl/hetryl-5-methyl-1,2,4-trizolo[4,3-a] quinolones by oxidising 2-pyridyl and 2-quinylhydrazones in dichloromethane. In vitro tests for antibacterial on seven substances were conducted. S. typhi was much more resistant to 1-(5′-Nitro-2-furyl)-5-methyl-1,2,4-triazolo[4,3-a] quinoline 111 at the MIC of 10 g/ml than to several commercialized antibiotics (Fig. 42) (Sadana et al., 2003).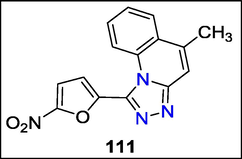
Structure of compound 111 with antibacterial activity.
Triazolopyridines were identified by Jun-ichi Kuroyanagi et al. as antagonists of the production of β-1,6-glucan. Triazolopyridines, imidazopyridines, and pyrazolopyridine were produced for in vitro assessment of their antifungal activity. A selective blocker β-1,6-glucan, a necessary constituent of the fungal cell wall, and significant antifungal activity against numerous Candida species were shown to exist in the Triazolopyridine compounds 112, 113, 114, 115, and 116 (Fig. 43) (Kuroyanagi et al., 2010b).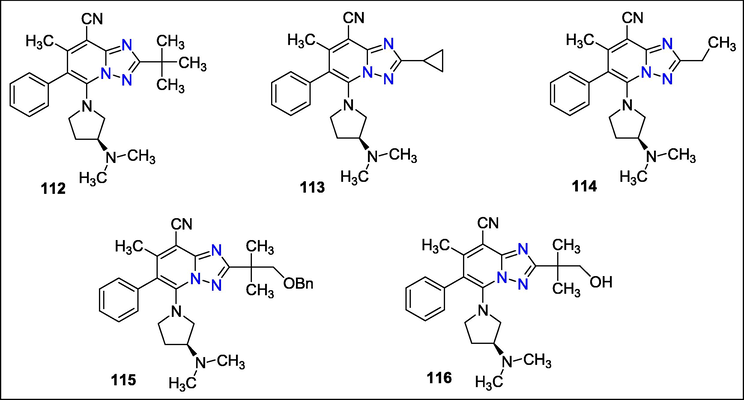
Structure of compounds with antibacterial activity.
Shaw et al. reported that chronic MPO stimulation has also been associated with random protein alteration that damages tissue, atherosclerosis, acute cardiovascular events, and chronic inflammatory disorders. Triazolopyrimidine 117 (Fig. 44). is a DNA repair protein called methyl guanine methyl transferase inhibitor and a reversible MPO inhibitor with low acid stability (MGMT). Using structure-based drug design, benzyl triazolopyridines with enhanced MPO activity, acid stability, no interaction with MGMT, and specificity against thyroid peroxidase were found (TPO) (Shaw et al., 2020).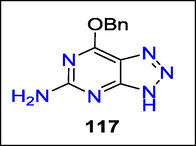
Structure of compound 117 with antibacterial activity.
4.9 Inhibitors of the HIV-1 allosteric integrase
The work by Kyle Parcella et al. describes fundamental modifications to the 1,2,4 triazolopyridine class of HIV-1 allosteric integrase inhibitors by adding a benzofuropyrimidine substituted tetrahydroisoquinoline at the C-5 position. Regarding rat P.K. characteristics, compound 119 performed better than the prior lead compound 118 while retaining outstanding antiviral efficacy and spectrum against the initial polymorphic screening panel (Fig. 45). Nevertheless, an in vitro lipid droplet production study showed that 119 profile was comparable to 118′s. Due to an unfavourable in vivo vacuolation risk, the advancement of 118 was stopped, and 119 was likewise stopped (Parcella et al., 2022).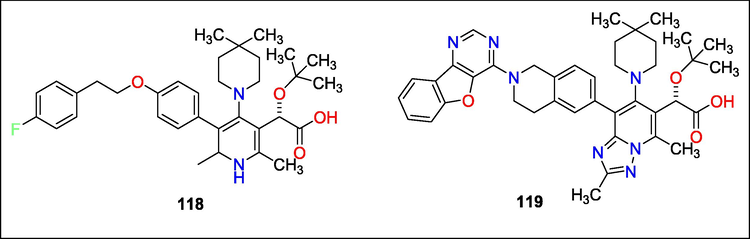
Structure of compounds with HIV-1 allosteric integrase inhibitors.
4.10 Allosteric modulators of mGlu2 receptors
José Mara Cid et al. demonstrate that 1,2,4-triazole-[4,3-a] pyridines have PAM activity at the mGlu2 receptor. The presented leads 120 showed improved activity, in vitro ADMET and hERG, and a reason in vivo P.K. profile by modifying the analogous imidazo[1,2-a] pyridine series (Fig. 46). Modifications to the scaffold substituents improved metabolic stability while maintaining lipophilicity.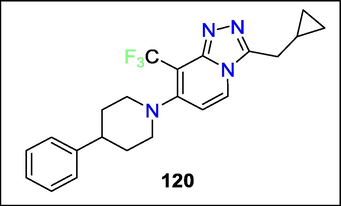
Structure of compound 120 with mGlu2 receptors activity.
The combination of this series with previously described mGlu2 receptor PAMs demonstrates an increase in the efficiency of lipophilic ligands throughout the programme. At a dose of 3 mg/kg P.O., 120 of the best compounds demonstrated central in vivo efficacy in the rat sleep-wake EEG paradigm by decreasing the REM sleep state, a characteristic proven to be mGlu2 mediated. The antipsychotic ED50 of Compound 120 was 5.4 mg/kg sc, and it blocked PCP-induced hyperlocomotion in mice (Cid et al., 2012).
4.11 JAK1 inhibitors
The first documented selective JAK1 inhibitor in clinical trials, 121, was found by Christel Jeanne Menet et al., who also developed a powerful and unique class of JAK1 inhibitors (Fig. 47).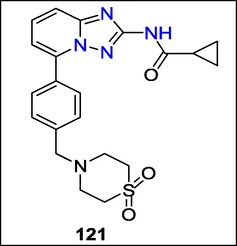
Structure of compound 121 with JAK1 inhibitors activity.
121 excellent has in vitro ADME qualities, high effectiveness in enzyme and cellular systems, specificity against JAK2 in whole blood assays, oral absorption in animal species, and dose-dependent activity in CIA when administered orally (Menet et al., 2014; Ribeiro et al., 2019c).
5 Drugs containing triazolopyridines
Filgotinib (1 2 2), marketed under the trade name Jyseleca, is employed to combat rheumatoid arthritis (R.A.). It act by inhibiting Janus Kinase enzyme also called as second generation JAK1 inhibitor. (Tarrant et al., 2020). It is used in treatment of rheumatoid arthritis alone or in combination with Methotrexate. A small molecule HER2 inhibitor, tucatinib (1 2 3), marketed under the trade name Tukysa, is used to combat breast and colorectal cancer that is HER2-positive (Kulukian et al., 2020). Tucatinib, is an oral tyrosine kinase inhibitor. A medication called enarodustat (development code JTZ-951; brand name Energy) is prescribed to treat anaemia, particularly when it is brought on by chronic renal disease (CKD). The action of enarodustat (1 2 4) is to block proly hydroxylase, a component of hypoxia-inducible factor (Markham, 2021). A familiar brand name for the antidepressant drug trazodone (1 2 5) is a first FDA approved drug for depression, mental illnesses and sleep issues are all treated with it. It belongs to the serotonin antagonist and reuptake inhibitor (SARI) class of phenylpiperazine compounds (Khouzam, 2017). It can be utilized as a component of combination therapy with other drugs or psychotherapies, or employed independently for the treatment of depression. Nefazodone is a triazolopyridine antidepressant that is metabolized by CYP3A4 and also inhibits the activity of this enzyme, which can lead to drug-drug interactions(Larrey and Ripault, 2013). An alpha-1 blocker is Dapiprazole (1 2 6), also known as Rev-Eyes. After an inspection of the eyes, it is employed to reverse mydriasis (Giakoumaki et al., 2005). It produces miosis by blocking the alpha-adrenergic receptors on the dilator muscle of the iris. The contraction of the ciliary muscle is not significantly affected by dapiprazole. As a result, there are no modifications to the depth of the anterior chamber or the thickness of the lens. A triazolinone herbicide called azafenidin (1 2 7) is utilized to manage grasses and weeds with broad leaves (Fig. 48) (Daugrois et al., 2005). Chemically, is 2-[2,4-dichloro-5-(prop-2-yn-1-yloxy)phenyl]-5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-a]pyridin-3(2H)-one.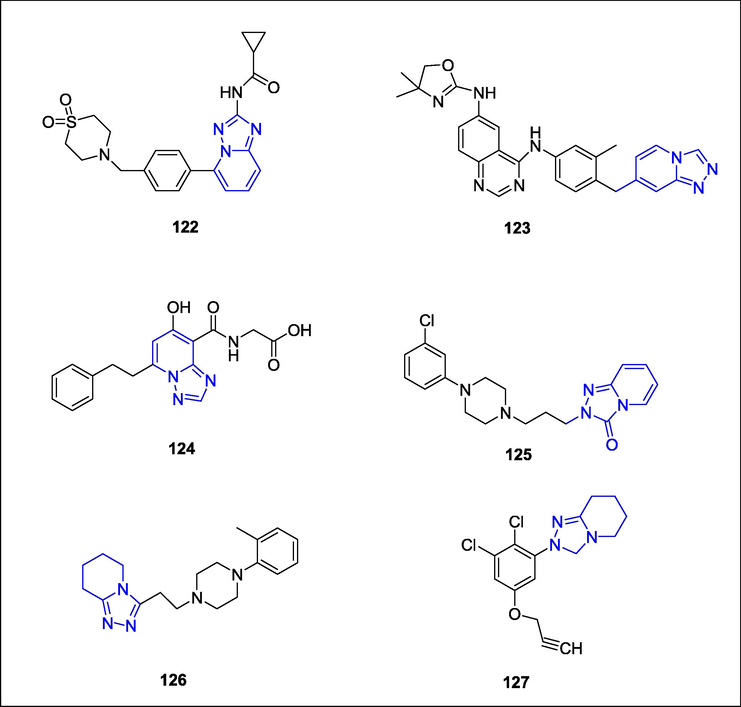
Structure of drugs containing triazolopyridine ring.
6 Conclusion and future perspective
The investigation of [1,2,4] triazolo[1,5-a], Pyridine, a part of a significant chemical class known as triazole, is now ongoing and has been intensified by scientists working in the fields of agricultural and medicinal chemistry. Extensive research proved that molecules containing this scaffold had unusual biological activities in various fields. These biological activities included antibacterial, antifungal, anti-breast cancer, Leishmanicidal neurotropic, allosteric modulators of mGlu2 receptors, DNA gyrase and Kinase inhibitors, HIV-1 allosteric integrase inhibitors, JAK1, and inhibitors of various enzymes. Other biological activities included JAK1 inhibitors. Many strategies that inhibit a wide variety of enzymes can be mined for information that can help develop triazolopyridine-based drugs that are more clinically and economically effective. [1,2,4] triazolo[1,5-a] Pyridine have significant biological activities in various circumstances and plays an essential role as an agrochemical and in developing medicinal applications. New synthetic techniques that allow quick access to an extensive range of functionalized heterocyclic compounds have several advantages, including the capacity to expand the accessible drug-like chemical space and to facilitate more effective delivery of drug development programmes. Another factor that helps speed up the medication development process is the creation of effective synthetic routes for mass-producing the target molecule. While drug discovery programmes often employ tried and true synthetic methods, recent advances in a heterocyclic synthesis that allows for a wide range of bond formation methods have profoundly affected the pharmaceutical industry. Triazolopyridine's structural characteristics, recent synthetic advances, and new biological uses are highlighted in the review to promote a more profound comprehension and potential future research into or discovery of these substances.
7 Future perspective
Investigations to enhance the triazolopyridine scaffold's biological and pharmacological capabilities and its synthesis and modification have been ongoing. Today, scientists are concentrating on creating and implementing environmentally friendly reaction processes and producing Triazolopyridine and its derivatives. Green protocols often use ionic liquid-catalyzed processes in aqueous media without using metals or solvents. Triazolopyridine and its derivatives have shown promise in treating various human infections, including cancer, malaria, fungal infections, and bacterial infections. The synthesis processes stated above that are based on green synthesis techniques are typically recommended for preparing this noble organic molecule and its analogues. In future the in-silico studies and green synthesis of triazolopyridines is the area of study for our team.
CRediT authorship contribution statement
Popat Mohite: Conceptualization, Formal analysis, Data curation. Deepali Nahar: Conceptualization. Rahul Pawara: Formal analysis, Data curation. Taha Alqahtani: Formal analysis, Data curation. Sayed M. Eldin: Formal analysis, Data curation. Nabendo Mukherje: Formal analysis, Data curation. Abdel Rahman Mohammad Said Al-Tawaha: Formal analysis, Data curation. Rashid Iqbal: Formal analysis, Data curation. Sami Bawazeer: Formal analysis, Data curation. Iftikhar Ali: Conceptualization, Formal analysis, Funding Acquisition, Writing Original draft.
Acknowledgement
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under grant number RGP2/410/44.
Funding
The authors extend their appreciation to the Deanship of Scientific Research at King Khalid University for funding this work through large group Research Project under grant number RGP2/410/44.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The chemistry of [1,2,3]triazolo[1,5- a] pyridines. J. Enzyme Inhib. Med. Chem.. 2002;17:359-367.
- [CrossRef] [Google Scholar]
- Synthesis of some new nitrogen bridge-head triazolopyridines, pyridotriazines, and pyridotriazepines incorporating 6-methylchromone moiety. J. Heterocycl. Chem.. 2013;50:615-624.
- [CrossRef] [Google Scholar]
- Triazolopyridyl ketones as a novel class of antileishmanial agents. DNA binding and BSA interaction. Bioorg. Med. Chem.. 2014;22:4018-4027.
- [CrossRef] [Google Scholar]
- Triazolopyridyl ketones as a novel class of antileishmanial agents. DNA binding and BSA interaction. Bioorg. Med. Chem.. 2014;22:4018-4027.
- [CrossRef] [Google Scholar]
- Discovery of 1-[2-(1-methyl-1H-pyrazol-5-yl)-[1,2,4]triazolo[1,5-a]pyridin-6-yl]-3-(pyridin-4-ylmethyl)urea as a potent NAMPT (nicotinamide phosphoribosyltransferase) activator with attenuated CYP inhibition. Bioorg. Med. Chem. Lett.. 2021;43:128048
- [CrossRef] [Google Scholar]
- Discovery of 1-[2-(1-methyl-1H-pyrazol-5-yl)-[1,2,4]triazolo[1,5-a]pyridin-6-yl]-3-(pyridin-4-ylmethyl)urea as a potent NAMPT (nicotinamide phosphoribosyltransferase) activator with attenuated CYP inhibition. Bioorg. Med. Chem. Lett.. 2021;43
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial evaluation of novel N-substituted 4-ethylsulfanyl-2-pyridones and triazolopyridines. Med. Chem. Res.. 2019;28:62-70.
- [CrossRef] [Google Scholar]
- Synthesis and antimicrobial evaluation of novel N-substituted 4-ethylsulfanyl-2-pyridones and triazolopyridines. Med. Chem. Res.. 2019;28:62-70.
- [CrossRef] [Google Scholar]
- 1,1′-carbonyldiimidazole (CDI) mediated coupling and cyclization to generate [1,2,4]triazolo[4,3- a ]pyridines. Org. Lett.. 2016;18:560-563.
- [CrossRef] [Google Scholar]
- SAR studies around a series of triazolopyridines as potent and selective PI3Kγ inhibitors. Bioorg. Med. Chem. Lett.. 2012;22:5257-5263.
- [CrossRef] [Google Scholar]
- Trazodone: A review of its pharmacological properties and its off-label use in dogs and cats. Am. J. Anim. Vet. Sci.. 2017;12:188-194.
- [CrossRef] [Google Scholar]
- Discovery of 3-cyclopropylmethyl-7-(4-phenylpiperidin-1-yl)-8-trifluoromethyl[1,2,4]triazolo[4,3- a ]pyridine (JNJ-42153605): A positive allosteric modulator of the metabotropic glutamate 2 receptor. J. Med. Chem.. 2012;55:8770-8789.
- [CrossRef] [Google Scholar]
- Protoporphyrinogen oxidase inhibitor herbicide effects on pythium root rot of sugarcane, pythium species, and the soil microbial community. Phytopathology. 2005;95:220-226.
- [CrossRef] [Google Scholar]
- The structural and optical properties of 1,2,4-triazolo[4,3-a]pyridine-3-amine. Molecules. 2022;27:721.
- [CrossRef] [Google Scholar]
- The structural and optical properties of 1,2,4-triazolo[4,3-a]pyridine-3-amine. Molecules. 2022;27:721.
- [CrossRef] [Google Scholar]
- DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: Synthesis and antibacterial activity. Bioorg. Med. Chem. Lett.. 2009;19:894-899.
- [CrossRef] [Google Scholar]
- DNA gyrase (GyrB)/topoisomerase IV (ParE) inhibitors: Synthesis and antibacterial activity. Bioorg. Med. Chem. Lett.. 2009;19:894-899.
- [CrossRef] [Google Scholar]
- Synthesis, molecular docking and in vitro screening of some newly synthesized triazolopyridine, pyridotriazine and pyridine-pyrazole hybrid derivatives. Molecules. 2018;23:2548.
- [CrossRef] [Google Scholar]
- Synthesis of tricyclic 1,2,3-triazolopyridines. Synthesis (Stuttg). 2015;47:2593-2598.
- [CrossRef] [Google Scholar]
- A facile synthesis of amide derivatives of [1,2,4]triazolo[4,3-a]pyridine. Asian J. Chem.. 2017;29:1920-1924.
- [CrossRef] [Google Scholar]
- Effects of peripheral sympathetic blockade with dapiprazole on the fear-inhibited light reflex. J. Psychopharmacol.. 2005;19:139-148.
- [CrossRef] [Google Scholar]
- Comparison of inhibitory activity of isomeric triazolopyridine derivatives towards adenosine receptor subtypes or do similar structures reveal similar bioactivities? Bioorg. Med. Chem. Lett.. 2004;14:3307-3312.
- [CrossRef] [Google Scholar]
- Comparison of inhibitory activity of isomeric triazolopyridine derivatives towards adenosine receptor subtypes or do similar structures reveal similar bioactivities? Bioorg. Med. Chem. Lett.. 2004;14:3307-3312.
- [CrossRef] [Google Scholar]
- Triazolopyridine ethers as potent, orally active mGlu2 positive allosteric modulators for treating schizophrenia. Bioorg. Med. Chem.. 2017;25:496-513.
- [CrossRef] [Google Scholar]
- I 2 -mediated oxidative C-N and N–S bond formation in water: A metal-free synthesis of 4,5-disubstituted/N-fused 3-amino-1,2,4-triazoles and 3-substituted 5-amino-1,2,4-thiadiazoles. J. Org. Chem.. 2018;83:5715-5723.
- [CrossRef] [Google Scholar]
- Continued exploration of the triazolopyridine scaffold as a platform for p38 MAP kinase inhibition. Bioorg. Med. Chem. Lett.. 2010;20:469-473.
- [CrossRef] [Google Scholar]
- Continued exploration of the triazolopyridine scaffold as a platform for p38 MAP kinase inhibition. Bioorg. Med. Chem. Lett.. 2010;20:469-473.
- [CrossRef] [Google Scholar]
- The chemistry of the triazolopyridines: An update. Chem. Inform.. 2003;34
- [CrossRef] [Google Scholar]
- The chemistry of the triazolopyridines: An update. Chem. Inform.. 2003;34
- [CrossRef] [Google Scholar]
- Triazolopyridines. Part 6. Ring opening reactions of triazolopyridines. J. Chem. Soc. Perkin. 1985;1:2719.
- [CrossRef] [Google Scholar]
- β-Alanine-functionalized magnetic graphene oxide quantum dots: an efficient and recyclable heterogeneous basic catalyst for the synthesis of 1 H -pyrazolo[1,2- b ]phthalazine-5,10-dione and 2,3-dihydroquinazolin-4(1 H)-one derivatives. Appl. Organomet. Chem.. 2020;34
- [CrossRef] [Google Scholar]
- Sulfonic acid-functionalized Fe 3 O 4 -supported magnetized graphene oxide quantum dots: A novel organic-inorganic nanocomposite as an efficient and recyclable nanocatalyst for the synthesis of dihydropyrano[2,3- c ]pyrazole and 4 H -chromene derivatives. Appl. Organomet. Chem.. 2020;34
- [CrossRef] [Google Scholar]
- Magnetic Fe3O4-supported sulfonic acid-functionalized graphene oxide (Fe3O4@GO-naphthalene-SO3H): a novel and recyclable nanocatalyst for green one-pot synthesis of 5-oxo-dihydropyrano[3,2-c]chromenes and 2-amino-3-cyano-1,4,5,6-tetrahydropyrano[3,2-c]quinolin-5-ones. Res. Chem. Intermed.. 2019;45:2095-2118.
- [CrossRef] [Google Scholar]
- A green strategy to prepare warfarin-like compounds catalyzed by zirconium oxychloride. J. Chinese Chem. Soc.. 2015;62:9-12.
- [CrossRef] [Google Scholar]
- A review of trazodone use in psychiatric and medical conditions. Postgrad. Med.. 2017;129:140-148.
- [CrossRef] [Google Scholar]
- New pyridine and triazolopyridine derivatives: Synthesis, antimicrobial and antioxidant evaluation. Acta Pol. Pharm.. 2017;74:861-872.
- [Google Scholar]
- New pyridine and triazolopyridine derivatives: synthesis, antimicrobial and antioxidant evaluation. Acta Pol. Pharm.. 2017;74:861-872.
- [Google Scholar]
- Synthesis of ([1,2,4]triazolo[4,3- a ]pyridin-3-ylmethyl)phosphonates and their benzo derivatives via 5- exo - dig cyclization. Beilstein J. Org. Chem.. 2019;15:1563-1568.
- [CrossRef] [Google Scholar]
- Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol. Cancer Ther.. 2020;19:976-987.
- [CrossRef] [Google Scholar]
- Novel antifungal agents: Triazolopyridines as inhibitors of β-1,6-glucan synthesis. Bioorg. Med. Chem.. 2010;18:5845-5854.
- [CrossRef] [Google Scholar]
- Novel antifungal agents: Triazolopyridines as inhibitors of β-1,6-glucan synthesis. Bioorg. Med. Chem.. 2010;18:5845-5854.
- [CrossRef] [Google Scholar]
- Nitrous oxide as a diazo transfer reagent: the synthesis of triazolopyridines. Chem. Commun.. 2021;57:11537-11540.
- [CrossRef] [Google Scholar]
- Novel [1,2,3]triazolo[1,5-a]pyridine derivatives are trypanocidal by sterol biosynthesis pathway alteration. Future Med. Chem.. 2019;11:1137-1155.
- [CrossRef] [Google Scholar]
- Novel [1,2,3]triazolo[1,5-a]pyridine derivatives are trypanocidal by sterol biosynthesis pathway alteration. Future Med. Chem.. 2019;11:1137-1155.
- [CrossRef] [Google Scholar]
- Larrey, D., Ripault, M.-P., 2013. Hepatotoxicity of Psychotropic Drugs and Drugs of Abuse, in: Drug-Induced Liver Disease. Elsevier, pp. 443–462. https://doi.org/10.1016/B978-0-12-387817-5.00025-X.
- Optimization of 1,2,4-triazolopyridines as inhibitors of human 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD-1) ACS Med. Chem. Lett.. 2014;5:803-808.
- [CrossRef] [Google Scholar]
- Heterogenized magnetic graphene oxide-supported N 6 -Schiff base Cu (II) complex as an exclusive nanocatalyst for synthesis of new pyrido[2,3- d ]pyrimidine-7-carbonitrile derivatives. Appl. Organomet. Chem.. 2020;34
- [CrossRef] [Google Scholar]
- Synthesis and in vitro leishmanicidal activity of novel [1,2,3]triazolo[1,5-a]pyridine salts. RSC Adv.. 2017;7:15715-15726.
- [CrossRef] [Google Scholar]
- Synthesis and in vitro leishmanicidal activity of novel [1,2,3]triazolo[1,5-a]pyridine salts. RSC Adv.. 2017;7:15715-15726.
- [CrossRef] [Google Scholar]
- Novel 3 H -[1,2,3]triazolo[4,5- c ]pyridine derivatives as GPR119 agonists: Synthesis and structure-activity/solubility relationships. Bioorg. Med. Chem.. 2017;25:4339-4354.
- [CrossRef] [Google Scholar]
- Triazolopyridines as selective JAK1 inhibitors: From hit identification to GLPG0634. J. Med. Chem.. 2014;57:9323-9342.
- [CrossRef] [Google Scholar]
- A simple and efficient synthesis of 3,4,5-trisubstituted/N-fused 1,2,4-triazoles via ceric ammonium nitrate catalyzed oxidative cyclization of amidrazones with aldehydes using polyethylene glycol as a recyclable reaction medium. Synthesis (Stuttg). 2014;47:517-525.
- [CrossRef] [Google Scholar]
- Half-sandwich ruthenium, rhodium and iridium complexes of triazolopyridine ligand: Synthesis and structural studies. J. Chem. Sci.. 2017;129:177-184.
- [CrossRef] [Google Scholar]
- Core modifications to the 4-(4,4-dimethylpiperidinyl) 2,6-dimethylpyridinyl class of Hiv-1 allosteric integrase inhibitors. SSRN Electron. J. 2022
- [CrossRef] [Google Scholar]
- Derivatives of a new heterocyclic system – pyrano[3,4- c ][1,2,4]triazolo[4,3- a ]pyridines: synthesis, docking analysis and neurotropic activity. Medchemcomm. 2019;10:1399-1411.
- [CrossRef] [Google Scholar]
- Derivatives of a new heterocyclic system – pyrano[3,4- c ][1,2,4]triazolo[4,3- a ]pyridines: synthesis, docking analysis and neurotropic activity. Medchemcomm. 2019;10:1399-1411.
- [CrossRef] [Google Scholar]
- A novel approach for the synthesis of imidazo and triazolopyridines from dithioesters. New J. Chem.. 2016;40:7637-7642.
- [CrossRef] [Google Scholar]
- Palladium-catalyzed chemoselective monoarylation of hydrazides for the synthesis of [1,2,4]triazolo[4,3- a ]pyridines. Org. Lett.. 2010;12:792-795.
- [CrossRef] [Google Scholar]
- Triazolopyrimidine and triazolopyridine scaffolds as TDP2 inhibitors. Bioorg. Med. Chem. Lett.. 2019;29:257-261.
- [CrossRef] [Google Scholar]
- Triazolopyrimidine and triazolopyridine scaffolds as TDP2 inhibitors. Bioorg. Med. Chem. Lett.. 2019;29:257-261.
- [CrossRef] [Google Scholar]
- Synthesis of triazolopyridines and triazolopyrimidines using a modified Mitsunobu reaction. Arkivoc. 2007;2007:132-147.
- [CrossRef] [Google Scholar]
- Discovery of a novel triazolopyridine derivative as a tankyrase inhibitor. Int. J. Mol. Sci.. 2021;22:7330.
- [CrossRef] [Google Scholar]
- Design, synthesis and characterisation of novel phenothiazine-based triazolopyridine derivatives: Evaluation of anti-breast cancer activity on human breast carcinoma. ChemistrySelect. 2019;4:12701-12707.
- [CrossRef] [Google Scholar]
- Design, synthesis and characterisation of novel phenothiazine-based triazolopyridine derivatives: Evaluation of anti-breast cancer activity on human breast carcinoma. ChemistrySelect. 2019;4:12701-12707.
- [CrossRef] [Google Scholar]
- Hypervalent iodine mediated synthesis of 1-aryl/hetryl-1,2,4-triazolo[4,3-a] pyridines and 1-aryl/hetryl 5-methyl-1,2,4-triazolo[4,3-a]quinolines as antibacterial agents. Eur. J. Med. Chem.. 2003;38:533-536.
- [CrossRef] [Google Scholar]
- Treating cancer by spindle assembly checkpoint abrogation: discovery of two clinical candidates, BAY 1161909 and BAY 1217389, targeting MPS1 Kinase. J. Med. Chem.. 2020;63:8025-8042.
- [CrossRef] [Google Scholar]
- CoMFA and docking studies on triazolopyridine oxazole derivatives as p38 MAP kinase inhibitors. Eur. J. Med. Chem.. 2008;43:1261-1269.
- [CrossRef] [Google Scholar]
- Discovery and structure activity relationships of 7-benzyl triazolopyridines as stable, selective, and reversible inhibitors of myeloperoxidase. Bioorg. Med. Chem.. 2020;28:115723
- [CrossRef] [Google Scholar]
- An exhaustive compilation on chemistry of triazolopyrimidine: A journey through decades. Bioorg. Chem.. 2019;88:102919
- [CrossRef] [Google Scholar]
- A Convenient one-pot synthesis of triazolopyridine and related heterocycle fused-triazole analogs through copper catalyzed oxidative cyclization strategy. J. Heterocycl. Chem.. 2016;53:606-614.
- [CrossRef] [Google Scholar]
- Discovery of 5-(2-amino-[1,2,4]triazolo[1,5-a]pyridin-7-yl)-N-(tert-butyl)pyridine-3-sulfonamide (CZC24758), as a potent, orally bioavailable and selective inhibitor of PI3K for the treatment of inflammatory disease. Bioorg. Med. Chem. Lett.. 2012;22:4613-4618.
- [CrossRef] [Google Scholar]
- Filgotinib, a JAK1 inhibitor, modulates disease-related biomarkers in rheumatoid arthritis: Results from two randomized, controlled phase 2b trials. Rheumatol. Ther.. 2020;7:173-190.
- [CrossRef] [Google Scholar]
- Facile synthesis of 1,2,4-triazoles via a copper-catalyzed tandem addition−Oxidative cyclization. J. Am. Chem. Soc.. 2009;131:15080-15081.
- [CrossRef] [Google Scholar]
- A simple and efficient automatable one step synthesis of triazolopyridines from carboxylic acids. Tetrahedron Lett.. 2007;48:2237-2240.
- [CrossRef] [Google Scholar]
- Potent triazolopyridine myeloperoxidase inhibitors. ACS Med. Chem. Lett.. 2018;9:1175-1180.
- [CrossRef] [Google Scholar]




