

Translate this page into:
ADMET profiling and molecular docking of pyrazole and pyrazolines derivatives as antimicrobial agents
⁎Corresponding author. f.ennahli@edu.umi.ac.ma (Fatima EN-NAHLI)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
In the present study, a Molecular Docking and in silico ADMET analysis were performed to identify the possible inhibitory effect of 23 molecules, pyrazole and pyrazolines derivatives, on Escherichia coli and to predict the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of all compounds. According to the results, every compound examined might bind to this bacterium's active site (PDB: 1FJ4). The results obtained in silico demonstrated that Only 4 (M6, M17, M19 and M20) of the 23 compounds were selected due to their inhibitory action and proximity to the important catalytic residues Thr302, Thr300, Val270, and His298 of the major protease and could be considered as orally active drug candidates due to their physical and chemical properties. The compounds M6, M17, M19 and M20 were subjected to Lipinski’s rule of five because it has the best binding affinity score in the binding study of the compound with the protein (-9.6, −9.3, −9.5, −10.3 Kcal/mol) successively. Pyrazole derivatives and the structure of pyrazolines are also effectively discussed in this paper for potential application as antibacterial agents due to their significant inhibitory activity. We were also able to predict a new potential inhibitor against a target of interest because to the result that we obtained.
Keywords
ADMET
Escherichia coli
Molecular Docking
Lipinski’s rule
Pyrazole and pyrazolines derivatives

1 Introduction
Escherichia coli continues to stand as a prevalent etiological agent responsible for a variety of prevalent bacterial infections in both human and animal populations (Metelytsia et al. 2020).
Escherichia coli are a bacterium that commonly colonizes the gastrointestinal tracts of humans and warm-blooded animals. It is generally considered to be harmless can cause a variety of intestinal diseases because of their ability to produce shigatoxins (Desvaux et al. 2020). It is one of the most studied microorganisms in the world. The Infection can occur from consuming food, water or meat contaminated with feces and insufficiently cooked (Brooks et al. 2005). There are several highly adapted E.Coli clones, Most strains of this bacterium are harmless, however Enterotoxigenic E. coli (ETEC), Enteropathogenic E. coli (EPEC), Enteroaggregative E. coli (EAEC) Entero-invasive E. coli (EIEC) and Enterohemorrhagic E. coli (EHEC) are pathogenic (Welinder-Olsson and Kaijser 2005). In the last two decade the (EHEC) was considered as the most important group responsible for food-borne illnesses because of the production of several verocytotoxin encoded phages (VT1 and VT2). The infection causes severe and bloody diarrhea as well as fever, vomiting hemorrhagic colitis (HC) and the production of a potent toxin that causes hemolytic uremic syndrome (HUS) which is fatal in 3 to 5% of cases (Brooks et al. 2005). Patients with (HUS) can develop impaired kidney function, a drop in blood cell concentration (red blood cells and platelets) and neurological complications that can lead to a state of coma. The incubation period ranges from 3 to 8 days with a median duration of 3 to 4 days. Despite this, most patients recover within 10 days (Azam, Thathan, and Jupudi 2020).
It was reported that the incidence of ECEH infections varied across age and gender; children under 3 years old and older patients developed severe symptoms compared to young people. Otherwise, back to the European epidemic in 2011, out of 10 affected people, 7 were adult females. This unusual distribution of cases among the population could be related to the differences in the alimentary regime (Garretto et al. 2020)(Luna-Guevara et al. 2019).
According to the World Health Organization (WHO), Escherichia coli is recognized as one of the most resistant bacteria to antibiotics, highlighting the importance of continued investigation for new antimicrobial agents. Therefore, much research is required nowadays, particularly in the design and development of new antimicrobial agents.
Widely recognized compounds incorporating heterocyclic ring structures are of major importance in industrial and medicinal fields. Among them, pyrazole derivatives stand out as an essential five-membered heterocyclic compound, attracting considerable attention in the fields of pharmaceuticals and agriculture. Heterocyclic structures incorporating pyrazole rings serve as versatile fundamental molecules in the medical field, displaying a diverse range of biological activities such as anticancer, antifungal, antimicrobial, antituberculosis, antibacterial, antidepressant, antidiabetic and antioxidant properties (Chinnamanyakar and Ramanathan 2021).
Without doing experimental tests, computer models can provide information about the potential effects of the chemicals on metabolism and if they are suitable for use as drugs. With the help of cheminformatics, scientists may deduce a molecule's pharmacokinetics, physical, chemical, solubility, adsorption, and other properties from its chemical structure. Every year, numerous new compounds are created worldwide. Check these chemicals bioactivity, but be aware that in-vivo and in-vitro testing are highly expensive.
Consequently, ADME and molecule target prediction have become essential for the sectors in which these molecules can be used. Structure-based drug design techniques and ligand-based drug design techniques are employed as important drug discovery tools in the rational drug discovery process. Docking studies are advanced computational methods in structure-based drug design to obtain an optimized conformation of the ligand-receptor interaction and study their relative orientation through a minimized energy-free system. Computer-aided drug design is a fast and economically modern technique that enables valuable, precise and in-depth understanding of experimental results and new suggestions for molecular structures to be synthesized (Ouassaf, Belaidi, Khamouli, et al. 2021).
For this, the current work aimed to perform in silico study to propose an effective drug by using a series of 23 molecules of pyrazoline and pyrazole derivatives and studying their affinity and their interaction with this bacterium by molecular docking method, the binding mode between active ligand and the target have been explored based on the extra-precision molecular docking and to identify more potent molecules active against E.Coli strains. As well as to study the toxicity of all compounds by ADMET and Lipinski’s rules methods.
2 Methods and computational details
2.1 Biological data set
For the present study, twenty-three molecules of new pyrazoline and pyrazole derivatives were selected from the literature and were docked with the organism: Escherichia coli (Hassan 2013). Initially, the 2D structure of the compounds was designed using the ChemDraw professional 15.0 software, Chem3D 15.0 was then used to convert all compounds from 2D to 3D structures and they are optimized by this software (Ouabane et al. 2022). Finally, the molecules were saved in PDBQT file format by using Autodock tools. The structure of the studied molecules represented in Table 1.
molecules
structures
molecules
Structure
M1
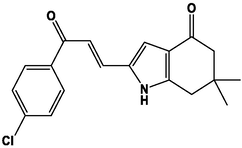
M13
M2
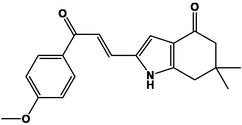
M14
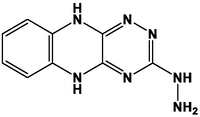
M3
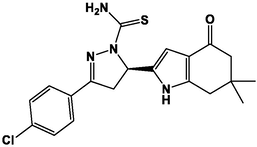
M15
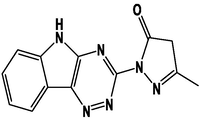
M4
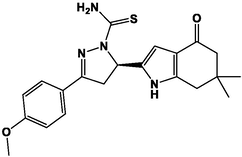
M16
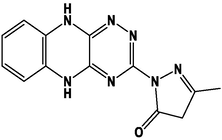
M5
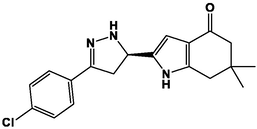
M17
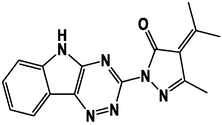
M6
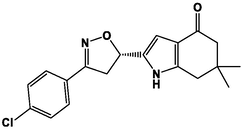
M18
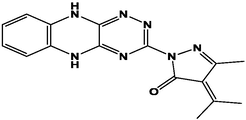
M7
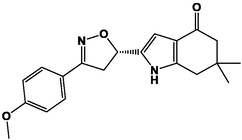
M19
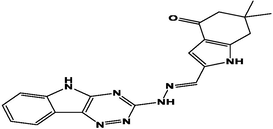
M8
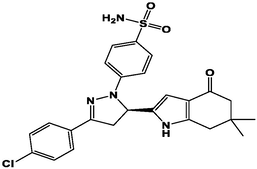
M20
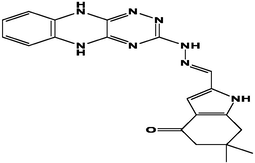
M9
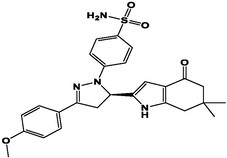
M21
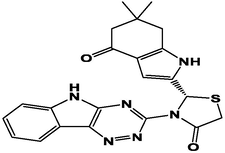
M10
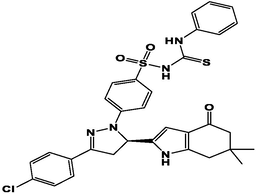
M22
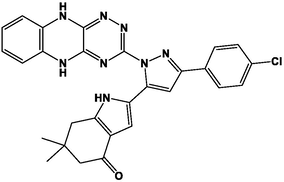
M11
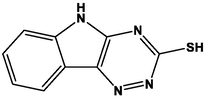
M23
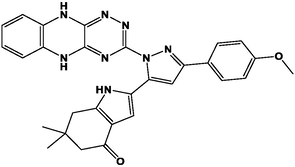
M12
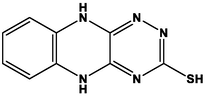
2.2 Molecular docking
The docking study contains three steps: the first step is Preparation/Optimization of the protein, the second step is Preparation of ligands and the last step is Molecular docking analysis (Belghalia et al. 2023).
In this work, we have focused on a bacterium that is found in the digestive tract of humans and warm-blooded animals and that causes several risks, this bacterium is Escherichia coli. The crystal structures of Escherichia coli (PDB ID: 1FJ4; Resolution: 2.35 Å), was obtained from protein data bank (PDB) (https://www.rcsb.org/pdb/home/home.do) (See Fig. 1. Moreover, the software AutoDockTools was used to adjust gasteiger charges, add polar hydrogen, and save the protein structure in the PDBQT file format (Flamandita et al., 2020). The enzymes are constituted of two chains; we chose one chain and delete the water molecules and ligand to allow easier use of the enzyme with a free active site during molecular docking (Abchir et al. 2022). The next step is the preparation of the grid, the grid plane should be in the center near the binding site, and it should be large enough to contain all the residues that interact with the ligand and large enough to put the ligand to have a full rotation, then the enzyme was placed in the center of the grid box while the number of grid points in the X, Y and Z directions was kept to the maximum (Belhassan et al. 2022). For the second step, the ligand library was collected from the literature, the 2D structures of the ligands were converted to.pdb format using autodock tools, Gasteiger charges were added, non-polar hydrogen was fused, and rotational bonds were detected and defined (Ouabane et al. 2023). Then, the docking was performed using autodock vina. The last step is devoted to the analysis of the docking results; we focus on the results of the simulation of the protein–ligand interaction that allowed us to obtain the best conformations with the binding energy, the conformation of the ligand with the protein that had a minimum energy present the best result for docking.
Co-crystal structure of protein 1FJ4.pdb.
In this work, docking analysis was performed using the AutoDock Vina software. Using a number of points in the (x,y,z) = (20, 20, 20) Å box, Autodock Tools performed the GRID box, after docking we used Discovery Studio Visualizer for examined the structures and analyzed the interaction between receptor and the protein.
2.3 Prediction of ADME/Tox and bioavailability
Chemical database contain 23 molecules in the SMILES format are submitted one by one to each web server pkCSM provides information about Absorption, Distribution, Metabolism, Elimination, and Toxicity (ADMET). The most stable molecules that have the best interactions with the protein of Escherichia coli are also studied by calculating the Lipinski’s rule of five by using SwissADME considering molecular weight, log P, H-bond acceptors, H-bond donors and rotatable bonds, the Lipinski’s rule of five was used to evaluate the drug likeness of the potential inhibitor. Lipinski noted that molecules that comply with these 5 rules remain bioavailable (Stitou et al. 2021). According to the RO5, a drug-like compound should have a molecular weight (MW) < 500 g/mol, a log P < 5, hydrogen bond donors (HBDs) < 5, hydrogen bond acceptor (HBA) < 10 and rotatable bonds < 10 (En-nahli et al. 2022). A substance will have improved pharmacokinetic characteristics and greater bioavailability in the organism's metabolic process if it complies with the five principles (Ouassaf, Belaidi, Mogren Al Mogren, et al. 2021).
3 Results and discussion
3.1 Docking results
In order to define theoretically the antibacterial mechanism of twenty-three Pyrazoline and Pyrazole Derivatives, a molecular docking study was carried out using AUTODOCK Vina. All the study compounds were docked into the crystal structure of Escherichia coli (PDB ID: 1FJ4) with a resolution of 2.35 Å. The first step in docking protocol is validation by redocking the reference ligand. Indeed, it was found that such ligand was attached to the binding site of the bacteria, through the same interactions i.e H-bond with His 298 and His 333 as shown in Fig. 2.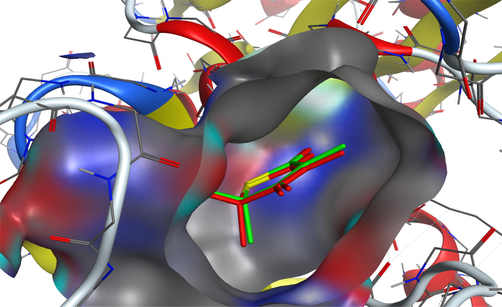
3D view Superposition of the reference ligand on the 1FJ4 pocket (the red stick represents the reference ligand; the green stick represents the redocked ligand).
The results shown in Table 2 indicate that compounds M6, M17, M19 and M20 are stabilized in 1FJ4 receptor pocket by various interactions with very low binding affinities of −9.6, −9.3, −9.5 and −10.3 (Kcal/mol) respectively. And the predicted docking results of these four compounds are shown in Fig. 3. We noted that a combination of hydrogen bonding and hydrophobic interaction maintained these three molecules in the active pocket. The interaction of the hydrogen bond was observed with the amino acid residues Thr (3 0 2), Thr (3 0 0), Val (2 7 0), and His (A: 298). And the major hydrophobic contacts between protein and compounds Ala (2 7 1), Pro (3 0 3), Ala (2 0 6), Pro (2 7 2), Phe (2 2 9), Cys (1 6 3), Val (3 0 4), His (3 3 3), Phe (3 9 2), His (2 9 8). These findings demonstrated the significance of hydrophobic interactions and hydrogen bonds in the antibacterial activity of pyrazoline and pyrazolone derivatives.
Molecules
Affinity (Kcal/mol)
Hydrophobic interactions
Hydrogen Bond interactions
Unfavorable interactions
M1
−9.3
Ala (A:271); Pro(A:203); Ala (A:206); Pro (A:272).
Thr (A:302)
M2
−9.2
Ala (A:271); Pro(A:303); Ala (A:206); Pro (A:272).
Asn (A:396); Asp (A:265)
M3
−8.7
Phe (A:392); Met(A:204); Ala (A:206); Pro (A:272).
Val (A:270); Thr (A:302)
M4
−8.3
Ala (A:206); Met(A:204); Val (A:270); Phe (A:392); Pro (A:272).
Thr (A:302)
M5
−9.0
Pro (A:303); Ala (A:271); Pro (A:272).
Gly (A:305); Val (A:304).
M6
−9.6
Ala (A:271); Pro(A:303); Ala (A:206); Pro (A:272).
Thr (A:302)
M7
−8.9
Ala (A:271); Pro(A:303); Ala (A:206); Pro (A:272).
Thr (A:302); Asn (A:396);
M8
−6.9
Met (A:204); Val (A:270); Ala (A:271); His (A:298); Val (A:304).
Thr (A:302).
M9
−7.0
Val (A:270); Val (A:304); Ala (A:271).
Met (A:204); Thr (A;302)
M10
−6.7
Met (A:204); Val (A:270); Ala (A:271); His (A:298); Val (A:304).
Thr (A:302);
M11
−7.4
Phe (A:392); Cys (A:163); Pro (A:272).
Phe (A:390).
M12
−8.0
Ala (A:271); Pro (A:272).
Thr (A:300); Glu (A:309).
M13
−8.8
Ala (A:271); Pro (A:272); Gly (A:305)
Thr (A:300).
His (A:298); Glu (A: 309); Asp (A:306).
M14
−9.0
Ala (A:271); Pro (A:272); Gly (A:305)
Asp (A:306); Glu (A:309).
M15
−9.1
Ala (A:206); Val (A:304).
Thr (A:302).
M16
−9.1
Ala (A:271); Pro (A:272);
Thr (A:302).
M17
−9.3
Phe (A:229); Cys (A:163); Pro (A:303); Val (A:304); His (A:333); Phe (A: 392) His (A:298).
Thr (A:302).
M18
−9.3
Phe (A:392); Pro (A:303); Pro (A:272); Val (A:304); His (A:333); Phe (A: 392) His (A:298).
M19
−9.5
Pro (A:272); Ala (A:271)
Thr (A:300); Val (A:270).
M20
−10.3
Ala (A:206); Val (A:304); Pro (A:272); Ala (A:271).
His (A: 298).
Thr (A:307)
M21
−8.3
Phe (A:392); Val (A:304); Pro (A:272); Ala (A:271). Val (A: 270); Ala (A:206).
Thr (A:302), Glu (A:228).
M22
−8.0
Phe (A:229); Ala (A:271); Ala (A:206); Val (A:304); His (A:333); Val (A: 270) Met (A:204); Thr (A: 302).
M23
−8.7
Phe (A:392); Pro (A:272); Ala (A:206); Val (A:304); Pro (A:303); Val (A: 270) Met (A:204).
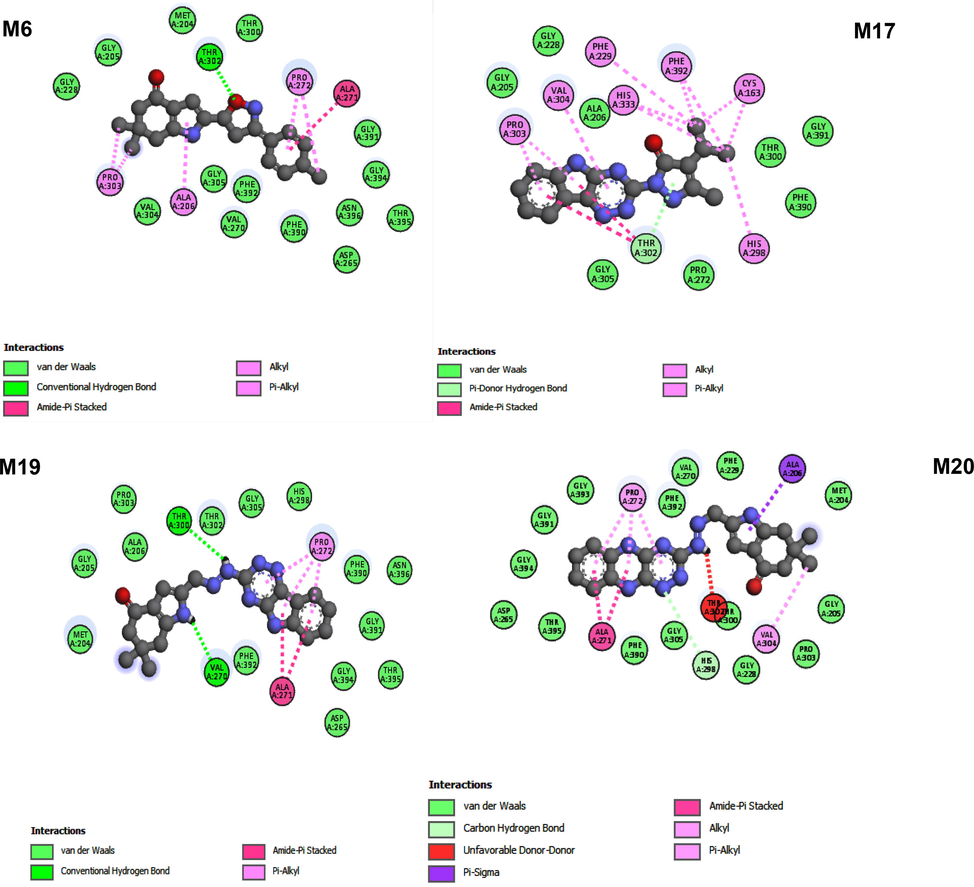
2D view of the binding conformations and hydrogen bond interactions of the four inhibitors at the active site of 1FJ4.
3.2 Lipinski’s rule and ADMET prediction
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) play key roles in drug discovery. In our study, prediction of the properties of ADMET was carried out using pKCSM web server; Prediction of ADMET parameters is listed in Table 3. This method is very important to select best molecules we can use as a drug candidate.
Molecules
Absorption
Distribution
Metabolism
Excretion
Toxicity
Intestinal absorption (human)
Blood Brain Barrier Permeability
CYP
Renal OCT2 substrate
AMES toxicity
Hepatotoxicity
2D6
3A4
2D6
3A4
Substrate
Inhibitor
Numeric (% Absorbed)
Numeric (log BB)
Numeric (log PS)
Categorical (Yes/No)
Categorical (Yes/No)
Categorical (Yes/No)
M1
92.44
0.106
−1.609
No
Yes
No
Yes
No
No
Yes
M2
94.74
0.134
1.902
No
Yes
No
Yes
No
No
Yes
M3
91.45
−0.179
−2.771
No
Yes
No
Yes
No
No
Yes
M4
93.90
−0.215
−2.857
No
Yes
No
Yes
No
No
Yes
M5
90.98
0.206
−1.62
No
Yes
No
No
No
No
Yes
M6
91.57
0.213
−1.599
No
Yes
No
Yes
No
No
Yes
M7
94.09
−0.038
−2.766
No
Yes
No
Yes
No
No
Yes
M8
98.66
−0.805
−1.878
No
Yes
No
Yes
Yes
No
Yes
M9
95.99
−0.858
−2.208
No
Yes
No
Yes
No
No
Yes
M10
100
−1.203
−1.539
Yes
Yes
No
Yes
No
Yes
Yes
M11
89.93
0.835
−2.981
No
No
No
No
No
Yes
No
M12
88.56
0.458
−2.28
No
No
No
No
No
No
No
M13
74.06
−0.909
−3.456
No
No
No
No
No
Yes
No
M14
73.11
−0.962
−2.666
No
No
No
No
No
No
No
M15
93.89
−0.402
−3.049
No
No
No
No
No
Yes
Yes
M16
86.64
−0.568
−2.491
No
No
No
No
No
No
Yes
M17
92.9
−0.276
−2.932
No
No
No
Yes
No
No
Yes
M18
88.55
−0.443
−2.175
No
No
No
No
No
No
Yes
M19
85.81
−1.293
−3.388
No
Yes
No
Yes
Yes
Yes
Yes
M20
84.59
−1.34
−2.321
No
Yes
No
Yes
No
No
Yes
M21
96.57
−1.217
−3.109
No
Yes
No
Yes
Yes
Yes
Yes
M22
100
−1.758
−1.603
No
Yes
No
Yes
No
No
Yes
M23
100
−1.777
−3.032
No
Yes
No
Yes
No
No
Yes
We are interested in M6, M17, M19 and M20 the most potent inhibitor in the dataset. As a result, we can observe that these compounds have high calculated values of intestinal absorption by humans, which are larger than 80%., compared to the criterion, an intestinal absorption value of<30% indicates poor absorbance. This indicates that these molecules can be easily absorbed by the intestine and circulate in the bloodstream (Norinder and Bergström 2006).
The distribution analysis shows that the molecules M19 and M20 are poorly distributed in the brain with values less than −1 (Abdelrheem et al. 2021), on the other hand, the molecules M6 and M17 present values higher than −1 which indicates that these two molecules are easily distributed in the brain.
Measures such as the permeability of the blood-surface area of the central nervous system (CNS) permeability (log PS) can be obtained from in situ brain perfusions with a direct injection of the compound into the carotid artery, which create a lakes of systemic distribution effects that could distort brain penetration. In this case compounds that present log PS > -2 are able to penetrate the CNS. However, compounds with log PS < -3 are unable to penetrate the CNS.
From Table 3 it can be seen that the compound M19 is unable to penetrate the CNS, while the other (M6, M17, M20) are capable to penetrate the CNS (Larik et al., 2019). In addition, metabolizing enzymes is the major study in phase I in discovery of drug, as indicate in Table 3, cytochrome P450 (CYP) includes substrate and inhibitor enzyme, the most important cytochromes P450 are CYP 2D6 and CYP 3A4 and it is involved in the metabolism of approximately half of the drugs currently in use, the results obtained indicate that M6, M17, M19 and M20 are non-substrate and non-inhibitor of 2D6 CYP enzyme. On the other hand, they are substrate and inhibitor of 3A4 CYP except for M17 is no substrate of the last enzyme (Moroy et al. 2012).
The Lipinski’s rule including molecular weight, number of hydrogen bonds donor, number hydrogen bonds acceptor, number of rotatable bonds and log P were shown in Table 4.
Compounds
Molecular weight (g/mol)
LogP
H-bond acceptors
H-bond donors
Rotatable bonds
M6
342.82
2.97
3
1
2
M17
306.32
2.63
5
1
1
M19
373.41
2.48
5
3
3
M20
388.43
2.59
5
4
3
In the filter that combines the drug-likeness criteria of Lipinski's rule of five (MW ≤ 500, log P ≤ 5, H-bond donors ≤ 5 and H-bond acceptors ≤ 10) and rotatable bonds ≤ 10) (da Cunha Xavier et al. 2021). Lipinski's rule of five is strictly adhered to by the four compounds, which indicates that they exhibit strong drug-like properties. For excretion parameters, the molecule M19 is considered an OCT2 renal substrate. For toxicity parameters, the AMES test results suggest that only molecule M11 is active as AMES toxicity, but the four molecules’ studies are also predicted to be hepatotoxic.
4 Conclusions
Throughout this study, AutoDock tools were utilized to verify 23 of pyrazole and pyrazolines derivatives inhibitors targeting Escherichia coli using bacterial protein 1FJ4. From this docking result, it is found these compounds, namely M6, M17, M19, and M20, four derivatives show high binding affinity score, established stable interactions. The in-silico ADME property was carried out to these four compounds. From this result of ADME property and Lipinski’s role, it is established that selected compounds have a good drug-likeness score.
Such parameters hold significant value in facilitating the design of novel inhibitors that possess substantial potential for pharmaceutical development.
Acknowledgments
The authors want to thank the Moroccan Association of Theoretical Chemestry (MATC) for its relevant help regarding to the software.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Design of novel benzimidazole derivatives as potential α-amylase inhibitors using QSAR, pharmacokinetics, molecular docking, and molecular dynamics simulation studies. J. Mol. Model.. 2022;28(4)
- [CrossRef] [Google Scholar]
- Molecular docking, bioactivity score, drug-likeness and admet studies of eight phytoconstituents from brown alga Sargassum Platycarpum. J. Mol. Struct.. 2021;1225:129245
- [CrossRef] [Google Scholar]
- Azam, Mohammed Afzal, Janarthanan Thathan, and Srikanth Jupudi. 2020. “Pharmacophore Modeling, Atom Based 3D-QSAR, Molecular Docking and Molecular Dynamics Studies on Escherichia Coli ParE Inhibitors.” Computational Biology and Chemistry 84 (September 2018): 107197. https://doi.org/10.1016/j.compbiolchem.2019.107197.
- In silico research on new sulfonamide derivatives as BRD4 inhibitors targeting acute myeloid leukemia using various computational techniques including 3D-QSAR, HQSAR, molecular docking, ADME / Tox, and molecular dynamics. J. Biomol. Struct. Dyn. 2023:1-19.
- [CrossRef] [Google Scholar]
- In silico detection of potential inhibitors from vitamins and their derivatives compounds against SARS-CoV-2 main protease by using molecular docking, molecular dynamic simulation and ADMET profiling. J. Mol. Struct.. 2022;1258:132652
- [CrossRef] [Google Scholar]
- Non-O157 Shiga toxin-producing Escherichia coli infections in the United States, 1983–2002. J. Infect. Dis.. 2005;192(8):1422-2149.
- [CrossRef] [Google Scholar]
- Anti-cancer and antimicrobial activity, in-silico adme and docking studies of biphenyl pyrazoline derivatives. Biointerface Res. Appl. Chem.. 2021;11(1):8266-8282.
- [CrossRef] [Google Scholar]
- Pathogenicity factors of genomic Islands in intestinal and extraintestinal Escherichia coli. Front. Microbiol.. 2020;11(September)
- [CrossRef] [Google Scholar]
- High-throughput virtual screening approach of natural compounds as target inhibitors of Plasmepsin-II. J. Biomol. Struct. Dyn. 2022:1-11.
- [CrossRef] [Google Scholar]
- Garretto, Andrea, Taylor Miller-Ensminger, Adriana Ene, Zubia Merchant, Aashaka Shah, Athina Gerodias, Anthony Biancofiori, et al. 2020. “Genomic Survey of E. Coli From the Bladders of Women With and Without Lower Urinary Tract Symptoms.” Frontiers in Microbiology 11 (September). https://doi.org/10.3389/fmicb.2020.02094.
- Synthesis, antibacterial and antifungal activity of some new pyrazoline and pyrazole derivatives. Molecules. 2013;18(3):2683-2711.
- [CrossRef] [Google Scholar]
- The role of pathogenic E. coli in fresh vegetables: Behavior, contamination factors, and preventive measures. Int. J. Microbiol.. 2019;2019
- [CrossRef] [Google Scholar]
- Design of (Quinolin-4-Ylthio)carboxylic acids as new Escherichia coli DNA gyrase B inhibitors: Machine learning studies, molecular docking, synthesis and biological testing. Comput. Biol. Chem.. 2020;85(January):107224
- [CrossRef] [Google Scholar]
- Toward in silico structure-based ADMET prediction in drug discovery. Drug Discov. Today. 2012;17(1–2):44-55.
- [CrossRef] [Google Scholar]
- Ouabane, Mohamed, Halima Hajji, Assia Belhassan, Yassine Koubi, Mhamed Elbouhi, Hassan Badaoui, Chakib Sekkat, and Tahar Lakhlifi. 2022. “2D-QSPR of the Retention/Release Property for Odorant Molecules in Pectin Gels of Different Concentration.” RHAZES: Green and Applied Chemistry 14: 15–35.
- Structure-odor relationship in pyrazines and derivatives : A physicochemical study using 3D-QSPR, HQSPR, Monte Carlo, molecular docking, ADME-Tox and. Arab. J. Chem. 2023105207
- [CrossRef] [Google Scholar]
- Combined 3D-QSAR and molecular docking analysis of thienopyrimidine derivatives as staphylococcus aureus inhibitors. Acta Chim. Sloven.. 2021;68(2):289-303.
- [CrossRef] [Google Scholar]
- Combined docking methods and molecular dynamics to identify effective antiviral 2, 5-diaminobenzophenonederivatives against SARS-CoV-2. J. King Saud Univ. - Sci.. 2021;33(2):101352
- [CrossRef] [Google Scholar]
- Quantitative structure-activity relationships analysis, homology modeling, docking and molecular dynamics studies of triterpenoid saponins as kirsten rat sarcoma inhibitors. J. Biomol. Struct. Dyn.. 2021;39(1):152-170.
- [CrossRef] [Google Scholar]
- Enterohemorrhagic Escherichia coli (EHEC) Scand. J. Infect. Dis.. 2005;37(6–7):405-416.
- [CrossRef] [Google Scholar]
- Cunha Xavier, Jayze da, Francisco Wagner de Queiroz Almeida-Neto, Priscila Teixeira da Silva, Amanda Pereira de Sousa, Emmanuel Silva Marinho, Márcia Machado Marinho, Janaina Esmeraldo Rocha, et al. 2021. “Structural Characterization, DFT Calculations, ADMET Studies, Antibiotic Potentiating Activity, Evaluation of Efflux Pump Inhibition and Molecular Docking of Chalcone (E)-1-(2-Hydroxy-3,4,6-Trimethoxyphenyl)-3-(4-Methoxyphenyl)Prop-2-En-1-One.” Journal of Molecular Structure 1227: 129692. https://doi.org/10.1016/j.molstruc.2020.129692.




