

Translate this page into:
Recent progress in cycloaddition reactions of cyclopropenone
⁎Corresponding author. lin0741028@yeah.net (Yongqin Ye)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Cyclopropenone has emerged as a highly versatile precursor in organic synthesis due to its diverse reactivity. The cyclopropenone derivatives could undergo various transformations such as cycloaddition reactions, ring-opening reactions, and isomerization reactions in the presence of different chemical reagents. Over the years, significant progress has been made in the manipulation of cyclopropenones for the construction of various carbocycles, heterocycles, and other useful organic compounds. Despite several reviews that have concerned the chemistry and reactions of cyclopropenones, the utility of such motifs in the construction of carbocycles and heterocycles has rarely been reported. Herein, we report recent advancements in the cycloaddition reactions of cyclopropenones as versatile synthetic precursors towards the synthesis of compounds with synthetic and biological significance.
Keywords
Cyclopropenone
Cycloaddition
Heterocycles
Organocatalysis
Transition-metal catalysis

1 Introduction
The cyclopropenone motif is a cyclic olefin skeleton with the chemical formula of C3H2O. The small ring widely exists in natural sources such as penitricin (A) and decalin derivatives (B-D), which could be isolated from Penicillium aculeatum and Telekia speciosa, respectively (Scheme 1) (Bohlmann et al., 1981, Okuda T Fau - Yokose et al., 1984). Notably, penitricin could not only retard bacterial growth, but also inhibit the plasma transglutaminase (factor VIII) (Toru Okuda, 1984, Toru Okuda, 1984, Toru Okuda, 1984, Iwata et al., 2000, Kogen et al., 2000). Breslow and Vol'pin independently reported the first access to cyclopropenone as early as 1959 (Breslow et al., 1959, Vol'pin et al., 1959). Cyclopropenone is of unique significance since it shows a wide range of applications in the fields ranging from ligands, polymer cross-linking agents, dendrimers, and devices (Tobe et al., 1997, Rivard et al., 2004). As shown in Scheme 2, cyclopropenone possesses a Hückel aromatic property and the negative charge mainly concentrates on oxygen atom in the resonance structures. Moreover, its small ring size is characterized by the high ring strain energy, which makes it more likely to ring-opening and participates in various reactions, affording efficient access to a broad range of highly functional molecules under controlled conditions (Komatsu and Kitagawa, 2003). This property facilitates the design of irreversible inhibitors by taking advantage of the reaction with a cysteine or serine residue at the active site of the enzyme or that of antitumor agents by the reaction with a nucleic base in DNA (Istrate et al., Cohen et al., 2013, Veverka et al., 2018, Row et al., 2020). Cyclopropenone derivatives, with substantial ring tensile force and potential chemical transformations, have gained considerable attention, notably in the area of organic and pharmaceutical chemistry (Upitis and Krol, 2002, Row et al., 2017, Robles-Planells et al., 2019, Heiss et al., 2022).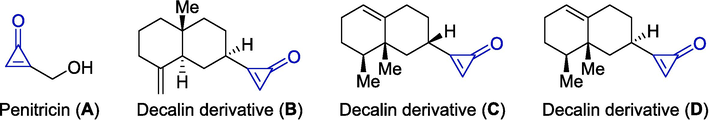
Cyclopropenone derivatives originated from natural sources.
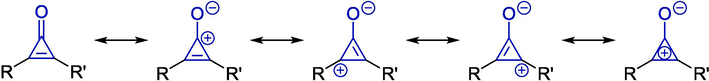
Structural formulas of cyclopropenone and its resonances.
The amphiphilic feature of cyclopropenone bearing a reactive π-system supports numerous reactions with both electrophilic and nucleophilic reagents (Breslow et al., 1965, Werz et al., 2001, Schuster-Haberhauer et al., 2008, Werz and Düfert, 2008). Due to their high strain, cyclopropenones easily participate in cycloaddition, ring-opening, and ring-enlargement reactions (Hollis et al., 2006, Kondo, 2016, Prasad Raiguru et al., 2020, Wang et al., 2021). Among the versatile chemical activities in which the cyclopropenone unit could participate, cycloaddition is one of the most practical and powerful reactions. In general, cycloaddition modes of cyclopropenones can be divided into three types: [2 + n] annulation with C = C double bond, [2 + n] annulation with carbonyl groups, and [3 + n] annulation with broken C–C bond. Typically, this variant of cycloaddition is the most widely used method for the synthesis of heterocyclic compounds. In the past few decades, the cyclopropenone framework has gradually developed into a privileged reaction partner in the regio- and stereoselective cycloadditions, aiming to the construction of highly functionalized heterocycles (Nakamura et al., 2003, Li et al., 2023, Zhou et al., 2024). In light of recent achievements in the synthetic method of heterocyclic compounds, this context mainly concentrated on the diversified annulation reactions of cyclopropenones, which were treated as the integrated platform, and particular concerns on the proposed mechanisms were also depicted. In the presence of metal- and organo-catalytic systems, cyclopropenones could rearrange to the intermediates with C–C bond cleavage and further transform to the corresponding heterocycles. Due to a lack of comprehensive review regarding the cycloaddition chemistry of cyclopropenones, we intended to critically describe the achievements made over the past decade. For a clear and orderly discussion, reactions were classified based on catalytic systems, including metal-free and metal-catalyzed reactions.
2 Transition-metal-free C–C bond activation
Transition-metal-free catalysis has proven to be the most practical and economical strategy to activate the C–C bond of cyclopropenones. A variety of reagents, such as Lewis bases, could be employed as organocatalysts, and cyclopropenones could be transformed into Lewis base-bound intermediates. Coordination of the intermediates and other reaction partners makes delivering a wide range of new heterocyclic compounds possible under certain conditions.
Werz reported the first formal cycloaddition between cyclopropenones 1 with two aryne units 2 in the presence of cesium fluoride and polar solvent acetonitrile (Wallbaum et al., 2015). The strategy served as a convenient approach to synthesize spirocyclic xanthene-cyclopropene frameworks 3 (Schemes 3). It was assumed that the carbon–oxygen double bond cleavage, simultaneous nucleophilic attack of cyclopropenones, and coupling with various arynes delivered the desired products. In addition, the procedure utilized a wide range of cyclopropenone substrates bearing both electron-rich and electron-deficient residues on the aryl moieties to afford the spirocyclic compounds in satisfactory yields. However, when the asymmetric electron-rich arynes were employed as precursors, it would lead to regioisomeric mixtures with decreased yields. This practical protocol demanded only slightly higher temperature to promote the cycloaddition reactions, therefore being convenient and providing simple operation conditions.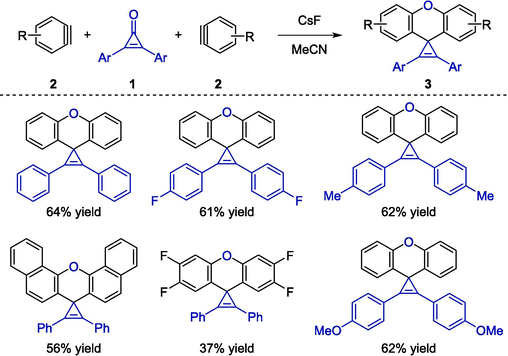
Organocatalytic cycloaddition reactions between cyclopropenones and arynes.
The first strategy for cycloaddition between cyclopropenones 1 and isatins 4 catalyzed by organocatalysts was reported by Shi, Tang, and co-workers (Zhao et al., 2014). The method presented a feasible and efficient way to synthesize multisubstituted 2H-pyran-2-ones 5 (Schemes 4a). In the light of systematic screening on the base catalysts and solvents, 1,4-diazabicyclo[2.2.2]octane (DABCO) as a catalyst in acetonitrile at room temperature were regarded as the optimum reaction conditions. Substituent effects on the aromatic rings did not significantly affect the outcomes, furnishing the desired products in good yields. In addition, a plausible mechanism was proposed to clarify the high reactivity of 1 catalyzed by amine.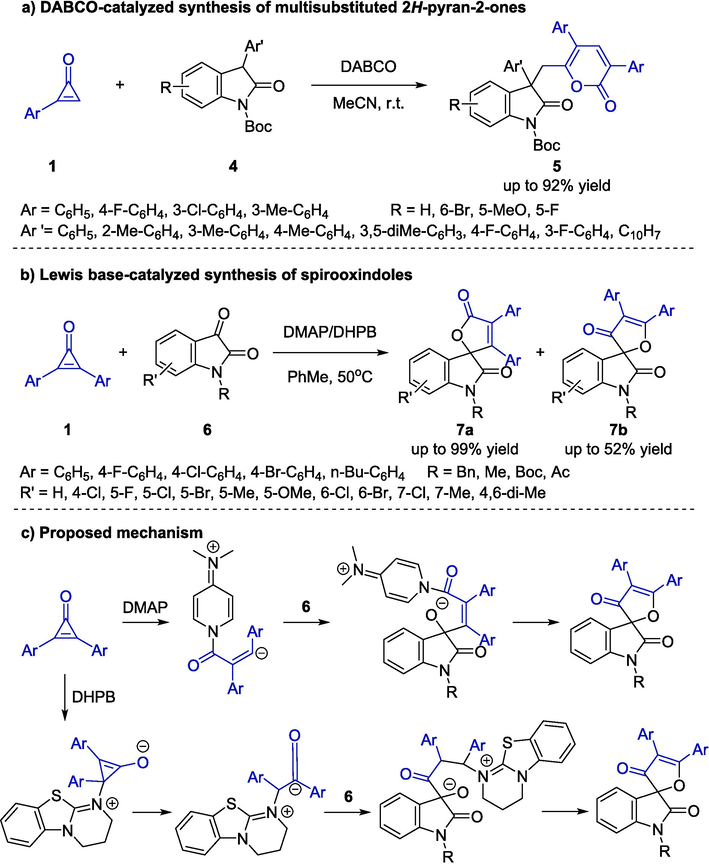
Organocatalytic cycloaddition reactions between cyclopropenones and isatins.
Lu, Du, and co-workers, in 2017, explored the cyclopropenones 1 as synthons for the enantioselective Lewis base-catalyzed [3 + 2] cyclization with isatins 6, and a wide range of spirooxindole furan-one derivatives 7 were generated with generally high reaction efficiency (Schemes 4b) (Xu et al., 2017). Among diverse organocatalysts, the 4-(dimethylamino) pyridine (DMAP) catalyst exhibited the optimal catalytic effects with toluene as solvent under air, providing products 7a in good yields with high regioselectivity. Meanwhile, the protocol catalyzed by 3,4-dihydro-2H-pyrimido[2,1-b] benzothiazole (DHPB) offered the desired products 7b regioselectively in acceptable yields. Concerning the substrate generality of the method, N-protecting isatins bearing electron-donating substituents exhibited better performance than the electron-withdrawing ones in this reaction. As depicted in Scheme 4c, a plausible mechanism of the regioselective reaction was suggested, which underwent three steps, including 1,4-addition, ring-opening process, and lactonization. Notably, this was the first organocatalytic C–C bond activation strategy that applied cyclopropenones as potential 3C synthons in a ring-opening formal cycloaddition.
1,2-Dichalcogen heterocycles represent an essential class due to their significant pharmacological activities (Kensler et al., 1993). Traditional synthetic strategies using elemental sulfur commonly suffered the limitations of harsh reaction conditions, poor yields and selectivity, and low atom economy. In 2020, a selective [3 + 2] annulation reaction of cyclopropenones 1 and elemental chalcogens 8 was explored by the Zhou and Liu group (Scheme 5a) (Wu et al., 2020). Without the help of transition metals, a large number of 1,2-dithiole-3-one heterocycles 9a could be generated in the presence of sulfur powder 8a and potassium fluoride in DMF at room temperature. Furthermore, treating 9a with m-CPBA in acetone for 5 h would provide the oxidized product simultaneously. In addition, elemental selenium 8b also worked well in this protocol and afforded 1,2-diselenol-3-one derivatives 9b in good yields. A proposed mechanism was described in Scheme 5b, including the ring-opening and intramolecular cyclization cascade.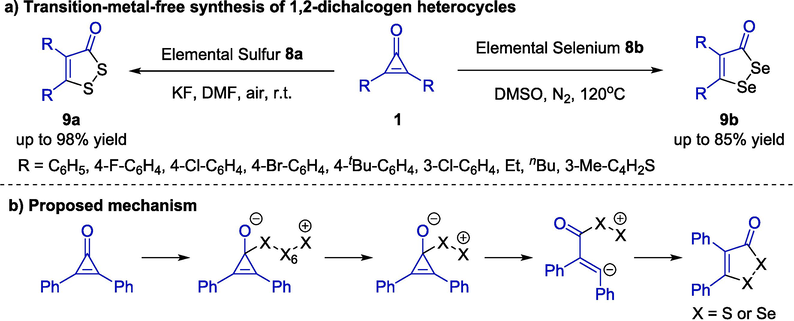
Transition-metal-free cycloaddition reactions between cyclopropenones and elemental chalcogens.
In 2018, Xu and co-workers utilized cyclopropenones 1 and o-hydroxy aromatic aldimines 10 for the synthesis of enantiopure heterocyclic products 11a through a catalyst-substrates intermediate (Cao et al., 2018). In the presence of bifunctional squaramide Ⅳ, the cascade reactions proceeded well in mesitylene at ambient temperature. The investigation on substrate scope indicated that both aromatic aldimines and cyclopropenones bearing various electronic properties of substituents were appropriated to the protocol (Scheme 6a). In addition, due to the multiple electrophilic sites of key intermediate, cascade reactions between methylphenylcyclopropenone 1b and 10 would offer the different cyclization products 11b. Diverse substituents on the aryl group of 10 were generally well tolerated, delivering the target compounds in excellent yields with satisfactory ee and dr values (Scheme 6b). Very recently, Ravikumar and co-workers reported a divergent synthesis of azetidinones and benzoxazepines via N-Heterocyclic carbene-catalyzed formal [3 + 1] or [3 + 4] cyclization of diphenylcyclopropenones and ortho-aminophenols (Scheme 6c) (Nanda et al., 2023). The selectivity was based on the N-substituted groups of aminophenol substrates. The formation of [3 + 1] addition products involved the generation of carbenes from NHC salts, coordination with cyclopropenones, 1,4-nucleophilic addition, NHC regeneration, nucleophilic attack of the amino groups, and proton transfer. By comparison, the formation of [3 + 4] addition products involved nucleophilic attack of the hydroxy groups. This strategy provided a facile access to the structural diversification of biologically active azetidinones and benzoxazepines.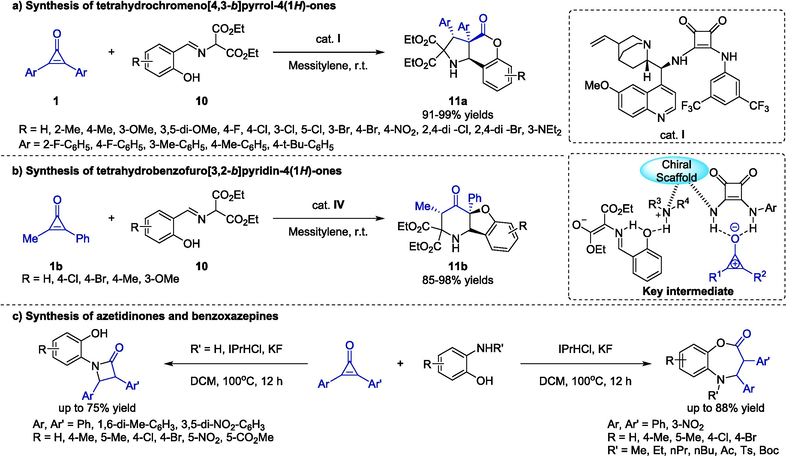
Organocatalytic cascade reactions between cyclopropenones and o-substituted phenols.
To further investigate the reactivity of cyclopropenone, Alya and co-workers utilized diphenylcyclopropenones as electrophiles for the [3 + 3] cycloaddition with amidrazones 12. (Scheme 7a) (Aly et al., 2016). With the triethylamine as a catalyst, various substituents bearing electron-donating or withdrawing groups on the aryl groups of amidrazone substrates 12 were compatible, serving a wide variety of desired pyrimidine-4-ones 13 in moderate to good yields (67–87 % yields). The rational mechanism, involving hydrazine addition, ring-opening reaction, intramolecular nucleophilic addition, and ammonia elimination, was also illustrated in Scheme 7b.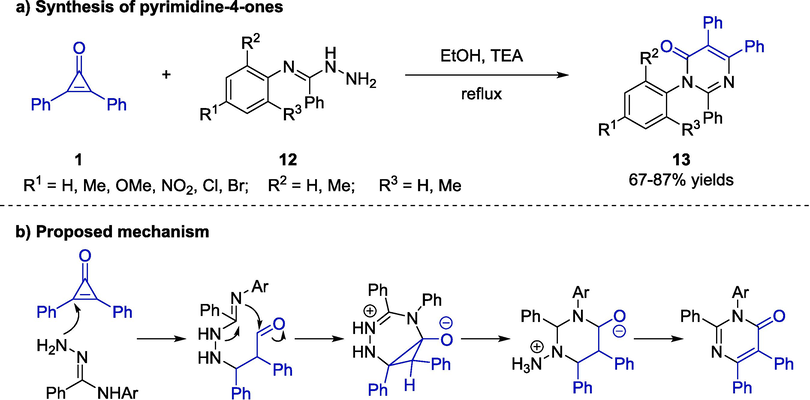
Triethylamine-catalyzed cycloaddition reactions between cyclopropenone and amidrazones.
1,3-Thiazine rings have attracted tremendous attention owing to their powerful antifungal, antibacterial, anti-inflammatory, and anti-tubercular biological activities (Koketsu et al., 2002, Nagaraj and Sanjeeva Reddy, 2008). In 2017, El-Sheref disclosed a transition-metal free catalyzed [3 + 3] cycloaddition reaction between 2,3-diphenyl cyclopropenone 1 and pyrazolylthioureas 14 to generate thiazinan-4-ones 15a in absolute ethanol (Scheme 8a) (El-Sheref, 2017). In addition, the author improved reaction conditions for the sake of a complete conversion of 15a, and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) which served as an effective hydrogen acceptor was added to achieve further dehydrogenation. As a result, a series of 1,3-thiazin-4(3H)-ones 15b could be furnished efficiently. Notably, there were four potential reactive sites (thione of thioamide motif, pyrazol-NH thiourea-N1, and thiourea-N3) of substrate 14, and the isolated product demonstrated that the reaction was more likely to occur via the nucleophilic attack of thione on C2-site of cyclopropenone.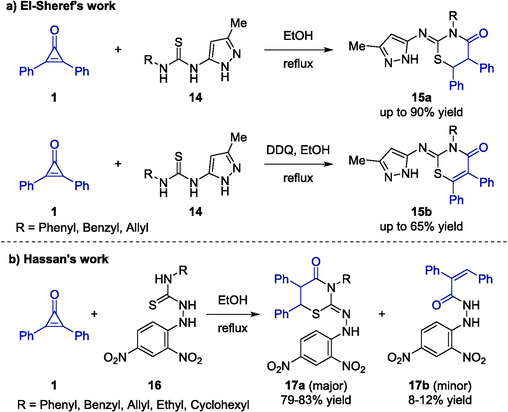
Organocatalytic cycloaddition reactions between cyclopropenones and thioureas.
In addition, Hassan and co-workers described a similar transition-metal-free catalytic system (Hassan et al., 2019). In this study, a highly efficient method for the synthesis of 1,3-thiazinan-4-one scaffold 17a through a [3 + 3] annulation reaction starting from 2,3-diphenyl cyclopropenone 1 and carbothioamides 16 in refluxing ethanol (Scheme 8b). With high atom economy, various functional substituent groups on the carbothioamide substrates were generally well tolerated, generating the target compounds 17a as the major products in good yields (79 %-83 % yields). Moreover, under the established condition, acrylohydrazides 17b were also isolated as minor products presenting the (E)-configuration which had been confirmed by X-ray analysis.
Multi-substituted oxazinones have long been employed as an important building block in medicinal and agrochemical materials (Li et al., 2018). In 2018, Shi and co-workers reported a base-promoted [3 + 3] cycloaddition of cyclopropenones 1 and benzamides 18, which afforded a convenient access to 1,3-oxazin-6-ones 19 (Scheme 9a) (Niu et al., 2018). Under the established reaction conditions (CsOAc as base, DCE as solvent at ambient temperature in the air), the target compounds were delivered in good to excellent yields. Additionally, the benzamides bearing electron-donating groups provided lower yields than those bearing electron-withdrawing groups. In this reaction, the use of cyclopropenones with other substituents such as two different aryl groups would provide the cycloadduct mixtures. The plausible mechanism for the base-induced cyclization reaction was outlined in Scheme 9b, which involved a series of isomerization, ring-opening, intramolecular nucleophilic attack reaction, and Newman-Kwart rearrangement to generate the desired products 19.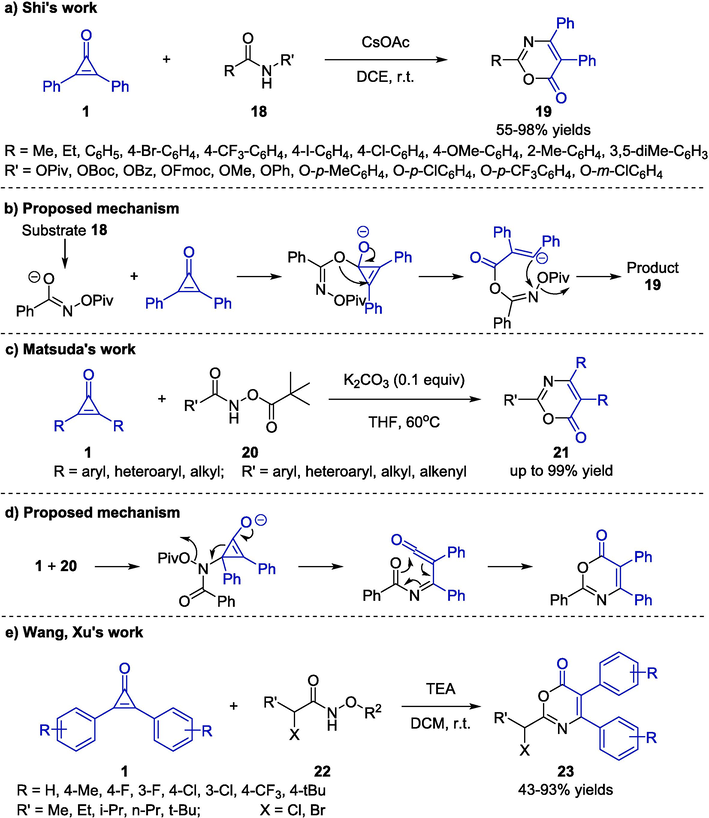
Base-promoted cycloaddition reactions between cyclopropenones and amides.
In the same year, Matsuda and co-workers further extended the application of amides, and they employed N-(pivaloyloxy)amides 20 as N,O-dipoles to react with cyclopropenones 1 (Matsuda et al., 2018). The six-membered azalactone scaffolds 21 could be obtained via a [3 + 3]-type cycloaddition reaction catalyzed by K2CO3 in THF (Scheme 9c). Notably, when the catalytic dosage of K2CO3 was decreased from 0.5 equiv to 0.1 equiv under the established condition, similar outcomes were obtained. Furthermore, the products would be barely isolated by reducing the amounts of base continuously. In this transformation, a wide range of aryl substituted benzamides and cyclopropenones with functional groups regardless of steric or electronic effects were well tolerated and furnished the desired products 21 in excellent yields. A proposed mechanism for the reaction comprised three steps: conjugate addition, ring opening, and electrocyclization (Scheme 9d). In 2021, Wang, Xu, and co-workers reported a highly effective synthetic method for the synthesis of 6H-1,3-Oxazin-6-one framework 23 through a triethyl amine-induced [3 + 3] annulation reaction originating from cyclopropenones 1 and hydroxamates 22 (Scheme 9e) (Sizhan Liu, 2021). Under the metal-free and ambient conditions, the reactions exhibited good functional group tolerance and broad substrate scope, providing the title compounds 23 in good to excellent yields. In addition, the strategy could be successfully scaled up to the gram level.
In 2022, Nechaev and colleagues reported a simple synthetic procedure to access indolizin-1-ol derivatives 25 starting from cyclopropenones 1 and pyridines 24 bearing electron acceptors at the γ-positions (Scheme 10a) (Nechaev and Cherkaev, 2022). This immediate transformation permitted MeOH as the solvent under an inert atmosphere. Brightly colored products were afforded and almost no byproducts were monitored. In addition, investigations on the stability showed that indolizin-1-ols were stable in the solid state but exhibited a short lifetime in solution. The treatment of compound 25a with air as an oxidant at 100 °C could provide a dimeric indolizin-dione 26 in a moderate yield (Scheme 10b). Compared with the previously reported similar reactions, the introduction of pyridinium salts improved the reaction efficiency with no chromatographic purification required. Similar to pyridines, isoquinolines are also prevalent and readily available. In this regard, Cao and co-workers developed a dearomative [3 + 2] cyclization of cyclopropenones with isoquinolines or pyridines to provide fused indolizinones (Scheme 10c) (Liu et al., 2023). Notably, DFT calculations indicated that the substituents at the 2-position of pyridine substrates played an important role in the dearomatization process. This synthetic protocol was attractive because of simple conditions and excellent reaction regioselectivity.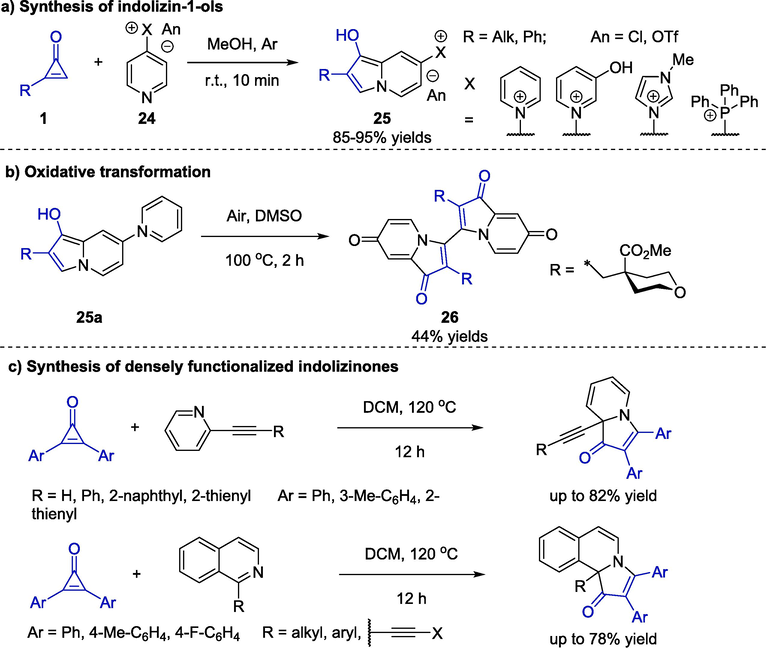
Transition-metal-free catalytic reactions to construct indolizinones.
In 2020, Wu and co-workers established a base-catalyzed cycloaddition reaction of cyclopropenones 1 and sulfoxonium ylides 27 to furnish 2-pyrone derivatives 28 (Scheme 11a) (He et al., 2020). After investigating the reaction conditions, the employment of NaOAc in DCE at 95 °C for 24 h afforded cyclic products through [3 + 3] annulation. Sulfoxonium ylides 27 with electron-donating and electron-withdraw groups on aromatic rings, as well as alkyl substituted ylides were well tolerated in this synthetic approach, and a wide range of 2-pyrones 28 were synthesized in moderate to good yields. A proposed mechanism was outlined in Scheme 11b, which involved a cascade process of nucleophilic addition, ring-opening process, and 6-exo-trig cyclization. Wang and co-workers further extended the Lewis-base catalyst system by developing a mild [3 + 3] annulation reaction between cyclopropenones 1 and sulfur ylides 29 (Scheme 11c) (Yuan et al., 2023). The 2H-pyran-2-ones 30 were generated smoothly under the catalysis of triethylamine (TEA) in 1,2-dichloroethane (DCE) at 60 °C. The investigation on substrate scope indicated that the aromatic groups on both cyclopropenones and sulfur ylides containing various electron-donating or electron-withdrawing substituents showed good compatibility for the synthetic protocol. Of note, alkyl groups attached to the sulfur ylides 29 could also give corresponding products in excellent yields. However, cyclopropenones bearing alkyl groups were not investigated using this method.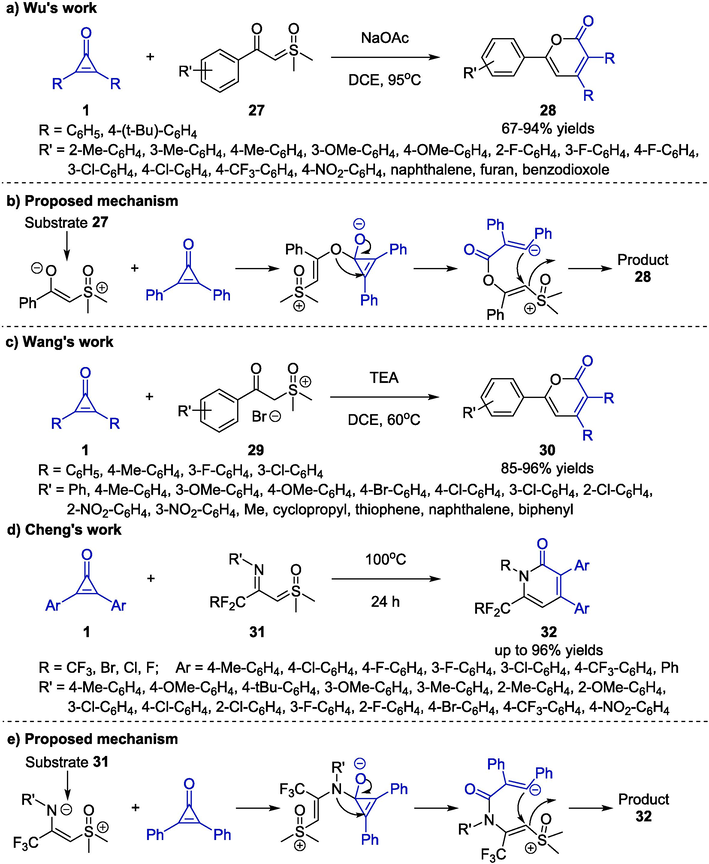
Base-promoted cycloaddition reactions between cyclopropenones and ylides.
A similar achievement for the construction of CF2R-containing pyridones 32 through stepwise [3 + 3] cycloaddition of cyclopropenones 1 and imidoyl sulfoxonium ylides 31 was developed by the Cheng group (Scheme 11d) (Wen et al., 2022). In terms of substrate scope, N-arenes bearing electron-rich or electron-deficient species at ortho-, meta-, and para-positions could be well tolerated in this protocol. It was worth noting that the eco-friendly transformation proceeded efficiently in the absence of transition-metal, additive and solvent, affording a series of the target products 32 with good yields (up to 96 % yield). The possible mechanism, involving nucleophilic addition, ring-opening process, and intramolecular nucleophilic annulation, was depicted in Scheme 11e.
The synthetic method of 2-pyrones has been further advanced in various ring-opening/cyclization tandem processes by applying different nucleophiles. Among diverse strategies transforming β-ketosulfoxonium ylides to 2-pyrones, the use of more readily available α-bromoketones or β-ketoethers significantly upgraded the synthetic method. In 2022, Xu and colleagues employed bromoketones 33 to react with cyclopropenones 1 and furnished substituted 2-pyrones 34 in high efficiency (Scheme 12a) (Qiao et al., 2022). Under the DMAP-catalyzed conditions, the reaction could proceed well and finish within 12 h, using potassium hydroxide as a base, 1,4-dioxane as a solvent in nitrogen atmosphere. Substrates including cyclopropenones and bromoketones bearing various alkyl and aryl groups could be conveniently transformed to the target products 34 in good yields with notable regioselectivity. A mechanism was proposed encompassing sequentially ring cleavage of 1, elimination of DMAP and intramolecular electrocyclization which was further clarified by DFT calculations. Bai et al. also discovered that 2-pyrones 36 could be easily afforded by the [3 + 3] annulation reaction of cyclopropenones 1 and β-ketoethers 35 (Scheme 12b) (Bai et al., 2021). By using t-BuOK as a catalytic base and 1,2-dimethoxyethane (DME) as solvent under argon, a straightforward method was proposed to construct 2-pyrone frameworks via the cleavage process of C-O bond of keto alkyl ethers and C–C bond of cyclopropenones. In this approach, the annulation reaction exhibited good functional group tolerance and excellent chemoselectivity, delivering a series of desired cyclization products 36 with satisfactory yields and simple operation.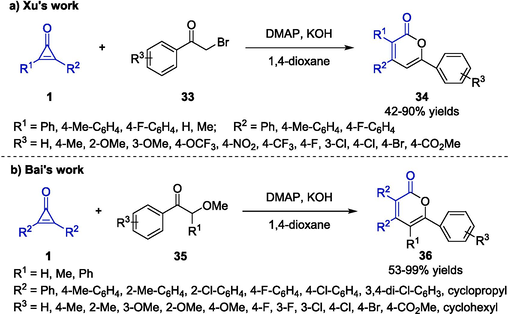
Synthesis of 2-pyrone derivatives through annulation reaction.
In 2016, Lin and co-workers provided an unprecedented one-pot [3 + 2] cyclization reaction between cyclopropenones 1 and β-ketoesters 37 to construct a butenolide scaffold (Scheme 13a) (Li et al., 2017). The protocol eliminated the demand for transition metal catalysts and provided a straightforward method to a series of substituted butenolides 38 bearing a quaternary center under mild conditions. Under the catalysis of DBU, the reaction tolerated various functional substituents at different positions of the substrates including diverse aromatic disubstituted cyclopropenones or ketoesters. As a result, the corresponding substituted butenolides 38 were generated in moderate to good yields and excellent chemoselectivity. Furthermore, reaction mechanism exploration validated the formation of an enol intermediate exerted a great impact on the reaction results. Subsequently, Kanger's group developed a similar approach for the synthesis of butenolide 39 via asymmetric catalyzed [3 + 2] cyclization of cyclopropenone 1 with β- keto ester 37a (Scheme 13b) (Reitel et al., 2018). In the presence of chiral catalyst VIIId, the transformation proceeded smoothly in a biphasic system of DCM/aq KOH by merging a ring-opening process with a classic intramolecular annulation pattern in a cascade reaction, affording the product 39 in a moderate yield with low enantioselectivity. Disappointingly, the substrate scope was proved only by one example.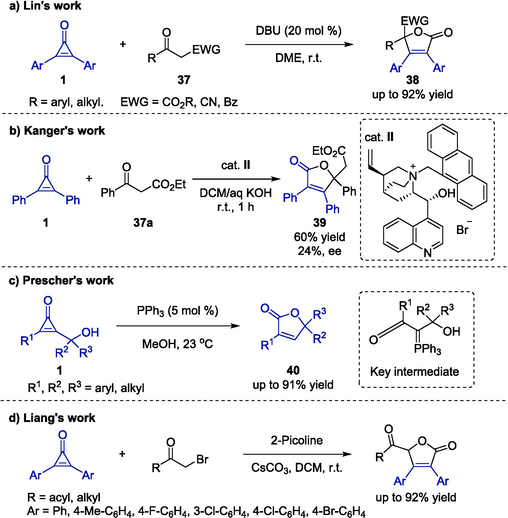
Organocatalyzed synthesis of substituted butenolides derived from cyclopropenones.
In 2019, Prescher and co-workers developed a general methodology to construct butenolide derivatives 40 from cyclopropenones 1 (Scheme 13c) (Nguyen et al., 2019). This reaction could be finished in a short time under the catalysis of triphenyl phosphine in methanol, and it would provide substituted butenolides 40 in good yields. The cyclization protocol exhibited excellent functional group tolerance, and various substrates bearing electron-donating or electron-withdrawing substituents on the aromatic ring could be efficiently converted. In addition, the authors proposed a plausible mechanism involving a cascade process that underwent ring-opening reaction and phosphine cycloaddition successively. It was worth noting that the protocol featuring good generality and practicability provided a complementary access to butenolides.
In addition to β-ketoesters, bromomethyl carbonyl compounds were also found to be viable substrates to react with cyclopropenones to afford functionally useful furan-2(5H)-ones (Scheme 13d) (Liang et al., 2024). Notably, Cs2CO3 serving as the base as well as the oxygen source, played an important role in promoting this [3 + 2] annulative reaction. The formation of products involved C–H activation of methyl bromoacetate to afford methoxycarbonylmethylpyridinium followed by the intermolecular Michael addition with cyclopropenone, ring-opening, carboxylation, decarboxylation, intramolecular SN2 reaction, and oxidation process. This protocol provided a facile access to valuable furan-2(5H)-one frameworks.
In 2017, Czarnocki and co-workers successfully developed a cascade reaction between cyclopropenones 1 and guanidine 41 (Scheme 14a) (Ahmad et al., 2017). This practical reaction proceeded under mild conditions in a mixed solvent system of benzene and ethanol. 1H-pyrimidine-4-one 42 was afforded as a single diastereomer and the structure was further confirmed by X-ray analysis. In addition to the guanidine group, the thiourea group in thiosemicarbazones could also serve as an effective synthon. Very recently, Aly and co-workers disclosed a formal [3 + 3] annulation of hydrazinecarbothioamides with diphenylcyclopropenone by using another convenient method (Scheme 14b) (Aly et al., 2024). This reaction was performed in refluxing ethanol in the presence of triethylamine to give the substituted thiazinanone derivatives in good yields. The suggested mechanism was illustrated as follows: the conjugate double bond of diphenylcyclopropenone was attacked by the sulfur atom to generate Zwitter salt, which subsequently underwent 1,3-H shift, rearrangement, ring opening, annulation, and another 1,3-H shift, resulting in the formation of the final product.![Formal [3 + 3] annulation reactions between cyclopropenones with guanidine or thiourea group.](/content/184/2024/17/8/img/10.1016_j.arabjc.2024.105845-fig14.png)
Formal [3 + 3] annulation reactions between cyclopropenones with guanidine or thiourea group.
In 2020, Cui et al. reported a convenient synthesis of indolizinoindoles 44 from cyclopropenones 1 and unactivated dihydro-β-carbolines 43 through catalyst-free [3 + 2] annulation (Scheme 15a) (Liu et al., 2020). The initial reaction occurred in DCM for 17 h at room temperature, and the product was generated in low yield. Subsequently, the reaction efficiency was improved significantly by changing the solvent to ethanol and increasing the temperature to 50 °C, giving rise to a higher yield of 43 in a short time. Moreover, the established strategy could be further applied to furnish dihydro-pyrrolo-1(5H)-ones 46 and dihydro-benzo-pyrrolo-1(5H)-ones 48 by using dihydroisoquinolines 45 and 3,4-dihydrobenzo[b][1,7]naphthyridine 47 to react with cyclopropenones 1, respectively. This work enabled the preparation of more structurally diverse indolizinoindole derivatives for potential biomedical research.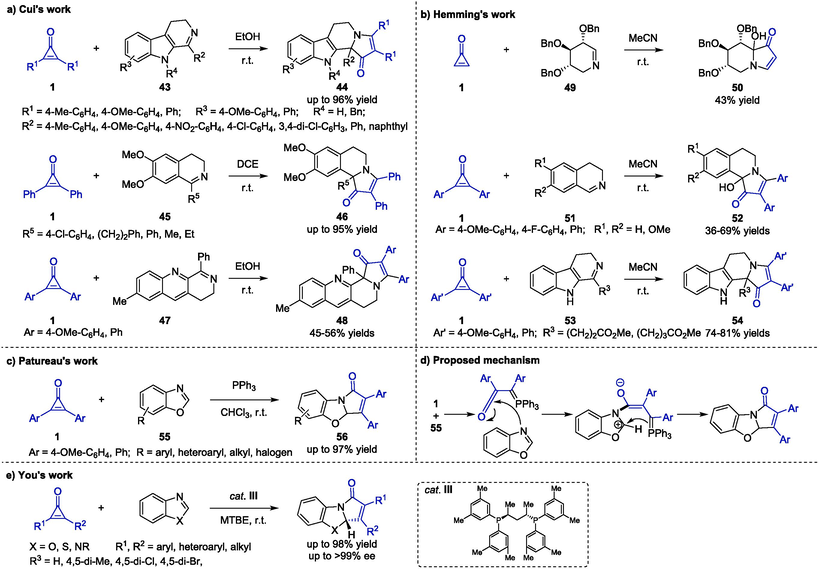
Organocatalyzed cycloaddition reactions between cyclopropenones and imines.
In the same year, the Hemming group reported a similar annulation strategy with the attempt to construct a castanospermine framework by using cyclopropenones and cyclic imines (Scheme 15b) (Jamshaid et al., 2020). Initially, piperideine 49 was employed in the reaction with cyclopropenone 1 to give the desired indolizidine 50 smoothly under aerial oxidation in moderate yield. However, the corresponding non-oxidized compound failed to form even in the absence of oxygen. Moreover, a series of novel pyrroloisoquinoline derivatives 52 were formed via a [3 + 2] annulation reaction of dihydroisoquinolines 51 and cyclopropenones 1. In addition, further exploration on generality of the method was conducted by using indolizino[8,7-b]indole alkaloids 53 and cyclopropenones 1 as reaction partners, affording the expected indolizinoindoles 54 in good yields.
In 2022, Patureau and co-workers developed a phosphine-catalyzed [3 + 2] annulation of cyclopropenones 1 and benzoxazoles 55 (Scheme 15c) (Wang et al., 2022). The optimal reaction condition was confirmed by the screening of catalysts, solvents and substrates ratios, and triphenylphosphine utilized as the organocatalyst with an excess of 55 (1/55 M ratio = 1/3) in chloroform was proved to be the best. A wide range of cyclopropenones 1 and benzoxazoles 55 were well compatible with this protocol, offering the corresponding benzopyrrolo-oxazolone derivatives 56 in acceptable to good yields. The plausible mechanism proposed to undergo three crucial steps including ring-opening reaction, nucleophilic attack and intramolecular cycloaddition (Scheme 15d). Another organocatalyzed cycloaddition reaction between cyclopropenones and imines was achieved by You and co-workers. (Zhang et al., 2024) As the chiral cat. III was used as a catalyst in the reaction between cyclopropenones and benzimidazoles, a series of dearomatized heterocycles were furnished in excellent yields and enantioselectivity (Scheme 15e). Mechanistically, the formation of product involved C–C bond cleavage of cyclopropenone to form a ketene ylide intermediate, coordination with benzimidazole followed by intramolecular nucleophilic addition and reductive elimination.
Regarding the N2-substituted bis(1,2,3-triazolyl) compounds synthesis, strategies involving radical coupling reactions, transition-metal catalyzed cross coupling reactions, and direct nucleophilic substitutions were classical synthetic methods (Katritzky et al., 1995, Chen et al., 2008, Liu et al., 2008). Since it was proposed, some variations on the reaction conditions have been developed. In 2017, Li et al. reported a one-pot ditriazolylation reaction of cyclopropenones 1 and triazoles 57 in reflux 1,2-dichloroethane (DCE), yielding the desired bis(1,2,3-triazolyl) compounds 58 in moderate to excellent yields (Scheme 16a) (Li et al., 2017). Notably, trace amounts of water were proved to be critical for the protocol. The reaction began with the hydration of triazole in the presence of residual water to generate the corresponding intermediate, which would further react with two molecules of cyclopropenone in sequence to yield the target 1,1-ditriazolylation product and Ts2O as the only byproduct. In contrast to previous synthetic strategies, this work provided a N2-selective autocatalytic chemical reactions with more economical and user-friendly conditions.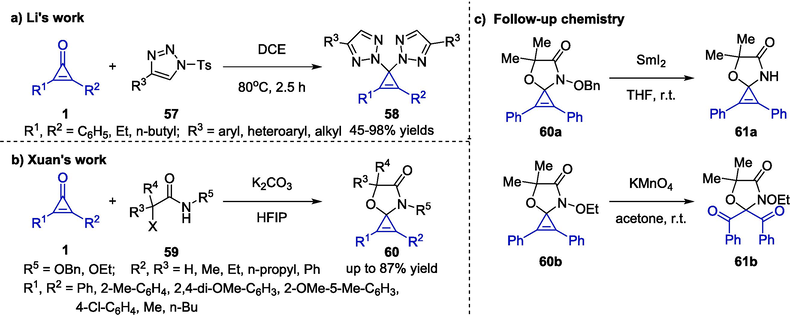
Organocatalyzed synthesis of cyclopropenes bearing a quaternary center.
In 2019, Xuan and co-workers reported a base-induced one-pot construction of spirocyclic oxazoles 60 via a convenient [3 + 2] cycloaddition reaction between cyclopropenones 1 and halohydroxamate analogues 59 in a polar medium (Scheme 16b) (Zhou et al., 2019). After evaluating the efficiency of various bases and solvents, the use of potassium carbonate as a base and hexafluoroisopropanol (HFIP) exhibited the best results. Under the optimal condition, attentions were turned to exploring the scope and limitations. A wide variety of cyclopropenones and halohydroxamate analogues were well tolerated, and the target oxazoles could be generated in moderate to high yields. However, halohydroxamate 59 with N-benzyl protecting group could not take part in this reaction. In addition, oxazoles 60 were separately treated with samarium(Ⅱ) iodide and potassium permanganate, resulting in unprotected oxazole compounds 61a and and diketone scaffold 61b, respectively (Scheme 16c).
N,O-Bidentate ligands are significant organic compounds due to their good catalytic activity and chemoselectivity (Clevenger et al., 2020, Rej et al., 2020). In 2021, Liu, Cao and co-workers reported an efficient deconstructive cycloaromatization reaction between cyclopropenones 1 and indolizines 62 to construct novel N,O-bidentate derivatives 63 (Scheme 17a) (Zhou et al., 2022). The protocol proceeded effectively in dichloromethane at 120 °C in the absence of any catalysts, offering a series of polyaryl N,O-bidentates that contain diverse substituents in moderate to high yields. In addition, the one-pot reaction also proceeded smoothly with the addition of BF3·OEt2 and triethylamine, and the target N,O-bidentate-BF2 complexes 64 were produced in moderate yields (Scheme 17b). Furthermore, the authors successfully assembled various polyaryl phenolic esters 65 by using acetic acid as an additive (Scheme 17c). This work provided new perspectives to assemble polyaryl N,O-bidentate ligands and accelerate the pace of N,O-bidentate complexes discovery.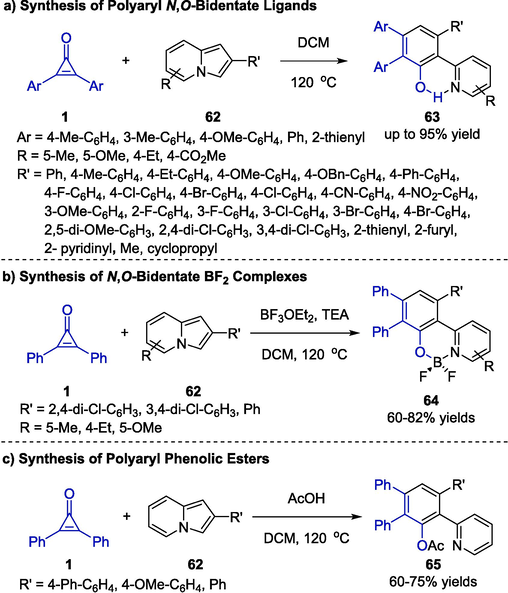
Organocatalyzed synthesis of polyaryl N,O-bidentate frameworks.
Hu and co-workers discovered a novel cyclization reaction for the synthesis of conjugate benzofurans 67 through multiyne tandem coupling between cyclopropenones 1 and N-tetrayne substrates 66 in reflux acetonitrile under an oxygen atmosphere (Scheme 18) (Yao et al., 2021). Substrate scope investigation showed that tetraynes 66 bearing electron-deficient groups on the aryl rings generally exhibited better reactivity than the electron-donating groups, the functionalized benzofuran derivatives 67 were obtained in good yields with excellent regioselectivity. In addition, DFT calculations revealed the unprecedented homolytic cleavage of C = C double bond and the significant role of molecular oxygen in promoting the protocol. Being different from general strategies which featured harsh reaction conditions and expensive catalyst systems, this protocol provided a mild and metal-free strategy toward benzofurans by employing oxygen as the green and economical oxidant.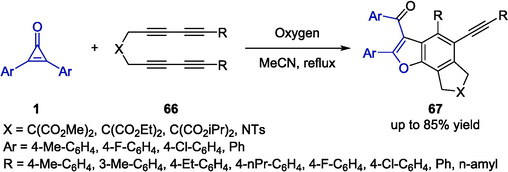
Cycloaddition reactions between cyclopropenones and tetraynes.
3 Rhodium-catalyzed C–C bond activation
Rhodium-catalyzed C–C bond activation reactions have developed rapidly in the past decades, and become a powerful tool to solve considerable problems especially in the field of modern organic synthesis (Li et al., 2016). Wender et al. reported the first approach to cyclopentadienones 69 through the rhodium-catalyzed C − C bond activation strategy in 2006 (Scheme 19a) (Wender et al., 2006). In this seminal study, the construction of cyclopentadienone framework could be accomplished by a [3 + 2] annulation reaction between cyclopropenones 1 and alkynes 68 in the presence of [RhCl(CO)2]2 in toluene. Moreover, substitutions of cyclopropenones or alkynes generally exerted little influence on the reaction results.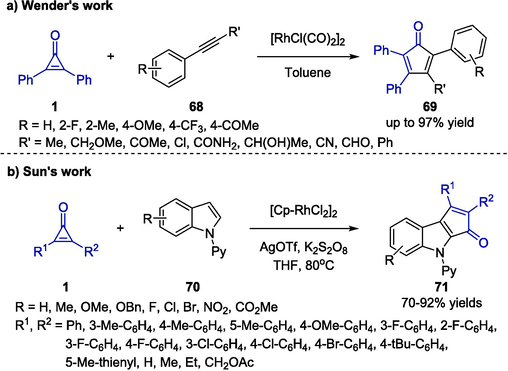
Rh-catalyzed synthesis of cyclopentadienone frameworks.
Quite recently, Sun and co-workers reported the utilization of indoles 69 instead of alkynes as the reaction partners for assembling cyclopentadienone frameworks (Scheme 19b) (Zhang et al., 2023). A versatile Rh-catalyzed [3 + 2] C–H functionalization of 69 with cyclopropenones 1 was established. In the presence of AgOTf as an additive and K2S2O8 as the oxidant, [Cp*RhCl2]2 was proved to be the most effective catalyst for the transformation and afforded the desired cyclopenta[b]indole products 70 in good yields with excellent regioselectivity. Both cyclopropenones 1 and indoles 69 bearing electron-rich or electron-deficient groups were compatible with the reaction system. This strategy provided an efficient tool for accessing biologically and pharmaceutically indole-fused molecules.
In 2019, the group of Chen reported a convenient approach for Rh-catalyzed coupling annulation of cyclopropenones 1 and N-nitrosoanilines 71 (Scheme 20a) (Liu et al., 2019). With N-nitroso as the directing group, 4-quinolones 72 were delivered with good regioselectivity through intermolecular [3 + 3] cycloaddition. Various reaction conditions were also evaluated, indicating that the use of [Cp*RhCl2]2, Ag2SO4 and NaF in 2,2,2-trifluoroethanol (TFE) was proved to be the optimal manner. With this protocol, a range of substitutions were tolerated and the desired products 72 were obtained in good yields. In addition, the authors proposed that the rate-limiting step was the reversible C–H activation which was induced by a concerted metallation-deprotonation mechanism.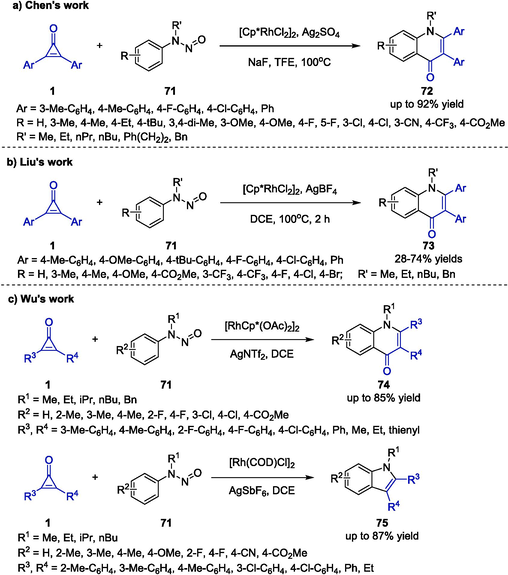
Rh-catalyzed annulation between cyclopropenones and N-nitrosoanilines.
With a similar redox-neutral strategy, Liu and colleagues achieved an Rh-catalyzed [3 + 3] cyclization between cyclopropenones 1 and N-nitrosoanilines 71 to afford quinolin-4(1H)-one scaffolds 73 in moderate to good yields (Scheme 20b) (Liu et al., 2020). Compared with the previous reports, this transformation proceeded smoothly under the [Cp*RhCl2]2/AgBF4 catalytic system without the extra additives. Furthermore, the method was characterized with short reaction time and high atom economy offering a promising route for the synthesis of bioactive heterocyclic compounds. However, only aryl substituted cyclopropenones were studied, which limited their application in synthesizing functionalized quinolones.
A series of C–H activation reactions catalyzed by rhodium were reported by the group of Wu (Shi et al., 2020). The authors utilized cyclopropenones 1 and N-nitrosoanilines 71 as substrates to furnish divergent cyclization products including quinolone 74 and indole 75 scaffolds, respectively (Scheme 20c). By simply switching the catalysts, regiodivergent C–H activations were realized to enable [3 + 3] and [3 + 2] cycloadditions. To be specific, [RhCp*(OAc)2]2 promoted the formation of quinolones 74 while [Rh(COD)Cl]2 rendered the indole products 75. The protocols showed excellent functional group compatibility and amenability to scale-up synthesis. These discoveries shed light on the development of new catalytic domino reactions for challenging C–H activation regiodivergent and regioselectivity via catalyst-controlled methodologies.
As the cyclopropenone substrates were highly vulnerable to occurring ring-opening reactions under the catalysis of transition metals, Wu and co-workers employed N-arylamidines 76 to participate in Rh-catalyzed [5 + 1] cycloaddition reactions with cyclopropenones 1 in 2020 (Scheme 21a) (Xing et al., 2020). The resulting 4-ethenyl quinazolines 77 bearing diverse aryl, alkyl and various other functional groups were generated in good yields. It was noteworthy that the arylamidine substrates 76 with substituted benzyl moiety underwent further oxidation under the established protocol to afford 2-benzoyl quinazoline products 78. This protocol overcome traditional restrictions such as the utilization of highly functionalized aniline derivatives or stoichiometric strong oxidants. Shortly afterwards, the same group discovered a Rh-catalyzed multi-step construction of indenol derivatives 80 via a one-pot tandem reaction of cyclopropenone 1 and N-hydroxybenzamidines 79 (Scheme 21b) (Wu et al., 2022). In the presence of [RhCp*(OAc)2]2 catalyst, the functionalized indenol products 80 were provided in moderate yields. Moreover, 80 could react with another molecule of cyclopropenone under base-promoted conditions, and transform to the corresponding products 81 in acceptable yields.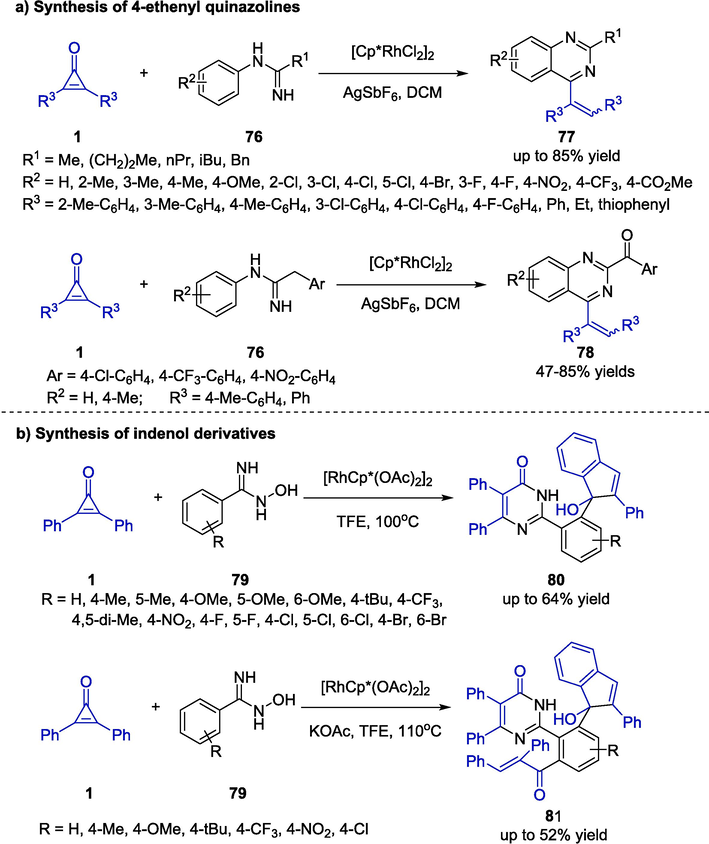
Synthesis of 4-ethenyl quinazolines and indenol derivatives.
In 2019, Chatani and colleagues developed a straightforward strategy for the construction of pyrrol-2-ones 83 via Rh-catalyzed [3 + 2] cycloaddition reactions between cyclopropenones 1 and amides 82 (Scheme 22a) (Haito and Chatani, 2019). After screening various reaction conditions, employing [Rh(OAc)(cod)]2 in toluene under carbon monoxide atmosphere was proved to be the optimal condition to furnish desired products 83 bearing a wide range of functional groups in good yields. The authors discovered that the reaction proceeded smoothly in the presence of sp2-hybridized nitrogen atom attached to the alkyl substituent of the amide. In addition, 83 would further undergo dehydration to provide the corresponding α,β-unsaturated γ-lactam products 84 under acidic conditions (Scheme 22b).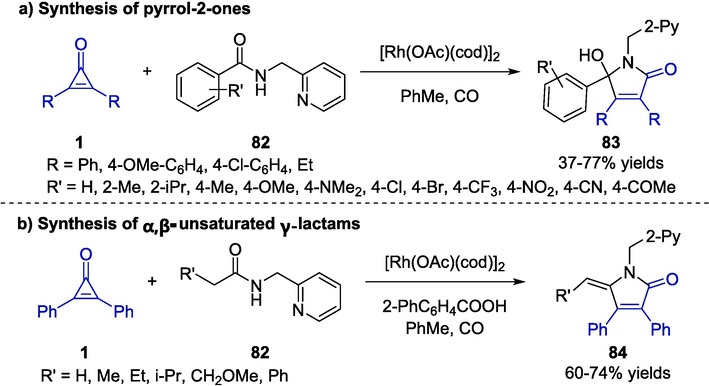
Rh-catalyzed annulation between cyclopropenones and amides.
In 2022, Hu et al. developed Ru-catalyzed [3 + 3] cascade annulation of cyclopropenones 1 and ketimines 85, affording the desired spirodienones 86 via successive C–H/C–C bond activation (Scheme 23a) (Hu et al., 2022). The catalytic system of [Cp*RhCl2]2/AgNTf2 exhibited excellent conversion efficiency, thereby generating the target products 86 in good yields. Additionally, no desired products were observed in the absence of a catalyst or additive, demonstrating their significance in this reaction. Moreover, a plausible mechanism involving C–H metalation, migratory insertion, β-carbon elimination, and nucleophilic addition, was depicted in Scheme 23b. In addition, Zhou et al. utilized cyclopropenones 1 and ketosulfoxonium ylides 87 as substrates to furnish a Ru-catalyzed one-pot [3 + 3] annulation for the construction of highly substituted 2-pyrone derivatives 88 under mild conditions, in which sodium acetate was employed as the promoting agent of elimination (Scheme 23c) (Zhou et al., 2020).![Rh-catalyzed synthesis of spiro[4,5]dienones and 2-pyrones.](/content/184/2024/17/8/img/10.1016_j.arabjc.2024.105845-fig23.png)
Rh-catalyzed synthesis of spiro[4,5]dienones and 2-pyrones.
4 Ruthenium-catalyzed C–C bond activation
Driven by ring-strain release of C–C bond cleavage, the group of Mitsudo pioneered the ruthenium catalytic system by using cyclopropenones 1 to react with alkynes 89. The target pyranopyrandiones 90 were generated with high selectivity via a cascade process of Ru-catalyzed C–C bond cleavage and triethylamine-induced intramolecular annulation reaction (Scheme 24a) (Kondo et al., 2002). However, this method could only be applied in specific examples. In 2020, Wu and co-workers developed a facile strategy for the synthesis of 6-ethenyl phenanthridine derivatives 92 through a cyclization reaction of cyclopropenones 1 and 2-arylanilines 91 (Scheme 24b) (Chen et al., 2020). The free-amine substituent of 91 which acted as a directing group was proved to be extremely critical for the reaction. The 2-arylaniline substrates bearing both electron-donating and electron-withdrawing groups on the aromatic group were well tolerated. In addition, cyclopropenones with various substituents could participate in the transformation effectively. In general, the protocol proceeded smoothly regardless of significant steric or electronic influence, and afforded a variety of 6-ethenyl phenanthridines with high atom-economy.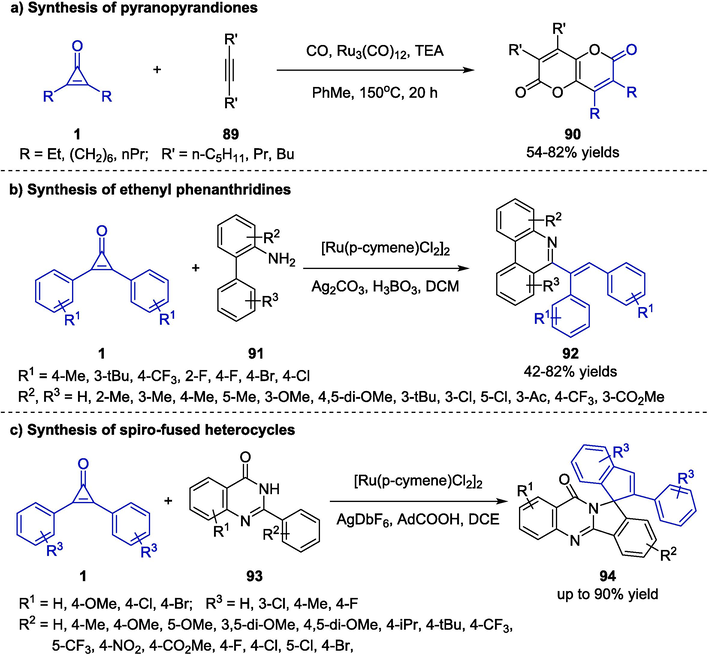
Ru-catalyzed synthesis of pyranopyrandiones, ethenyl phenanthridines and spiro-fused heterocycles.
Subsequently, a similar strategy was further developed for the construction of spiro-fused heterocycles 94 by the same group (Shi et al., 2021). The authors employed aquinazolinones 93 as the reaction partners in a ruthenium catalyst system for the annulation reaction (Scheme 24c). The 1-adamantane carboxylic acid was screened as the optimal acid additive to achieve C–H activation cascades under mild conditions. In addition, the protocol was scalable, proceeded efficiently with good regioselectivity and functional group tolerance.
5 Silver-catalyzed C–C bond activation
Silver could also promote the cycloaddition of cyclopropenones to furnish diversely substituted heterocyclic compounds. Matsuda and co-workers presented a [3 + 2] annulation reaction between cyclopropenones 1 and amide derivatives 95 to afford highly substituted 5-amino-2-furanone products 96, which was promoted by AgOTf (Scheme 25a) (Matsuda et al., 2018). A wide range of electron-donating and electron-withdrawing diaryl cyclopropenones were well compatible with the novel method. Additionally, amides bearing various substituents were also proved to be amenable substrates. The proposed mechanism was illustrated in Scheme 25b, which underwent a nucleophilic attack followed by an annulation reaction to generate desired compounds and release the Ag catalyst.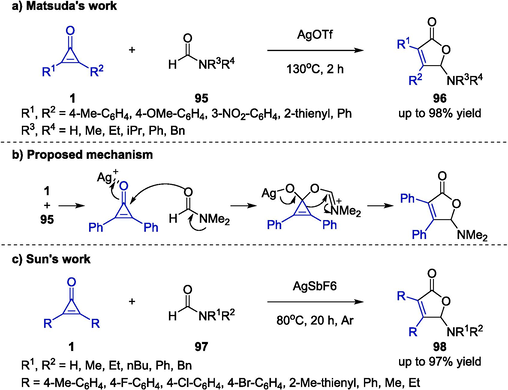
Ag-catalyzed synthesis of γ-aminobutenolide derivatives.
Based on the above-mentioned powerful strategies, Sun and co-workers further extended the C–C bond cleavage approach of cyclopropenones to construct a diverse range of γ-aminobutenolide derivatives 98 (Scheme 25c) (Nanda et al., 2022). In this work, a facile and efficient protocol has been reported wherein cyclopropenones 1 were coupled with formamides 97 serving as available C = O bond in the presence of AgSbF6 under argon atmosphere. The transformation could be achieved smoothly in a short time at 80 °C, forming a series of target products with good chemo- and regio-selectivity.
A practical silver-promoted ring-opening/annulation cascade reaction was reported by Liu and co-workers in 2020, which created a notable route for the regiospecific construction of oxazinone derivatives 100 (Yang et al., 2020). Through a formal [4 + 2] cycloaddition of cyclopropenones 1 and oximes 99, the procedure provided a direct access to 1,3-oxazinone framework (Scheme 26a). The optimal reaction conditions identified were 15 mol % Ag2O in cyclohexane solvent at 80 ℃ under nitrogen atmosphere for the acquisition of desired products. The transformation was screened with various oxime substrates and the corresponding oxazinones were generated with good efficiency. A range of cyclopropenones were also screened and most presented good yields except for 1 bearing electron-donating groups or heterocyclic rings. Overall, this methodology had several desirable features involving a simple work-up process and atom-economical, which allowed the efficient and economical synthesis of oxazinones under mild conditions.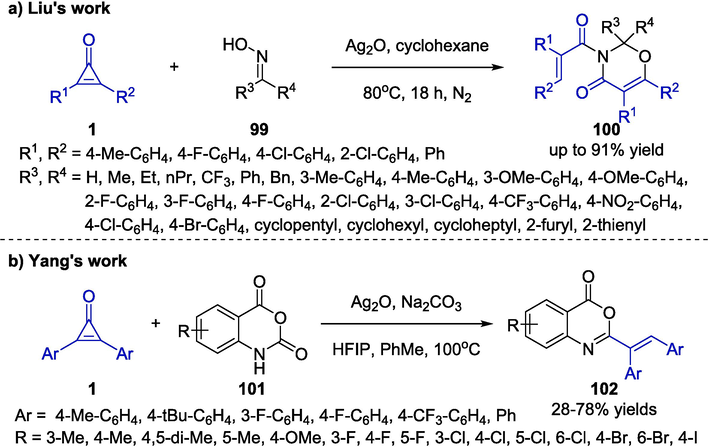
Ag-catalyzed synthesis of oxazinones and benzoxazin-4-ones.
In 2021, an efficient silver-catalyzed HFIP-mediated [4 + 2] cycloaddition reaction was reported by Yang et al. for the rapid assembly of benzoxazin-4-one derivatives 102 from cyclopropenones 1 and isatoic anhydrides 101 (Scheme 26b) (Yang et al., 2021). The authors developed a cascade reaction involving decarboxylation, intermolecular addition, nucleophilic cyclization, ring-opening and isomerization. In this strategy, hexafluoroisopropanol (HFIP) was used as the additive, which was extremely critical for promoting the process of decarboxylative cycloaddition. This available protocol featured with a wide substrate scope, as well as excellent functional group compatibility.
6 Other transition-metal-catalyzed C–C bond activation
Apart from the aforementioned strategies, a variety of explorations have dealt with cyclopropenone chemistry for the construction of heterocyclic ring frameworks which were promoted by various transition metals such as nickel, copper, palladium, gold, iron, scandium, lanthanum, bismuth, and et al. In 2020, Bai et al. reported a nickel-catalyzed [3 + 2] annulation process of cyclopropenones 1 and ketones 103 (Scheme 27a) (Bai et al., 2020). The transformation was discovered to generate the highest yield and excellent enantioselectivity in the presence of Ni(cod)2 (4.0 mol %), chiral ligands (7.0 mol %) in toluene (2.0 mL). A variety of substrates were well compatible with the standard conditions, generating the corresponding butenolides 104 with a tetrasubstituted carbon in good yields. In addition, the authors utilized α,β-unsaturated imines 105 as the reaction partners to construct γ-alkenyl lactam derivatives 106 efficiently under the same catalytic system (Scheme 27b). Furthermore, in 2021, the same group extended the strategy for the asymmetric synthesis of γ-butenolide derivatives 109 via Ni-catalyzed [3 + 2] annulation using α-CF3 enones 107 or 1,2-diones 108 as reaction partners (Scheme 27c) (Bai et al., 2021). The transformation proceeded smoothly through C–C activation of cyclopropenones 1 and further cycloaddition in the catalytic system of Ni(cod)2 (5.0 mol %) and chiral ligands (6.0 mol %), providing the corresponding products in good yields with remarkable ee values. Additionally, detailed DFT studies were performed to verify the observed chemo- and regioselectivity.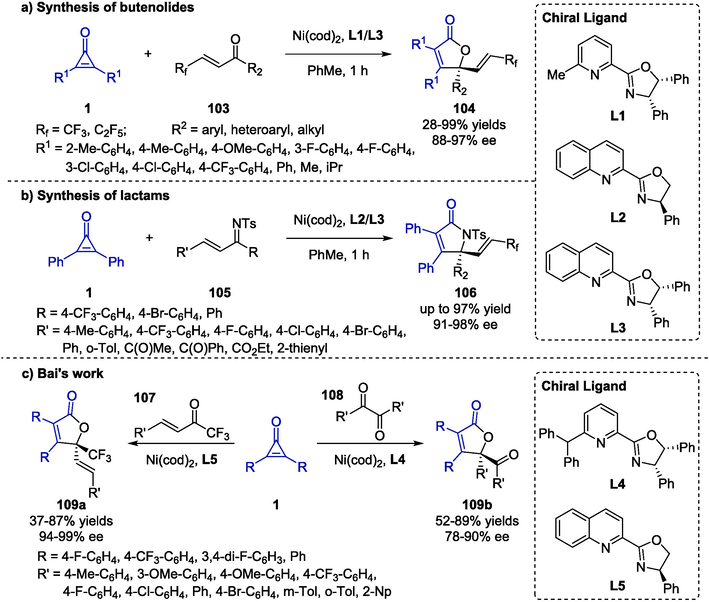
Ni-catalyzed synthesis of butenolides and lactams.
Despite the significant role of transition metal-catalyzed σ-bond cross exchange reaction, some formidable challenges remained in the reactivity, selectivity and substrate scope. The group of Zhao developed an unprecedented ring expansion protocol for the creation of sila-(benzo)cycloheptenone derivatives 111 by treating cyclopropenones 1 with (benzo)silacyclobutanes 110 (Scheme 28a) (Zhao et al., 2018). An exchange reaction of cross-metathesis between C–C bond and C-Si bond occurred in the presence of Pd(OAc)2 or Ni(COD)2 catalyst and led to a variety of sila-(benzo)suberones bearing diverse functional groups. The facile protocol promoted the discovery of novel candidate compounds as a result of the specificity of such frameworks in medicinal chemistry. In addition, the key intermediates that took part in the transformation were shown in Scheme 28b.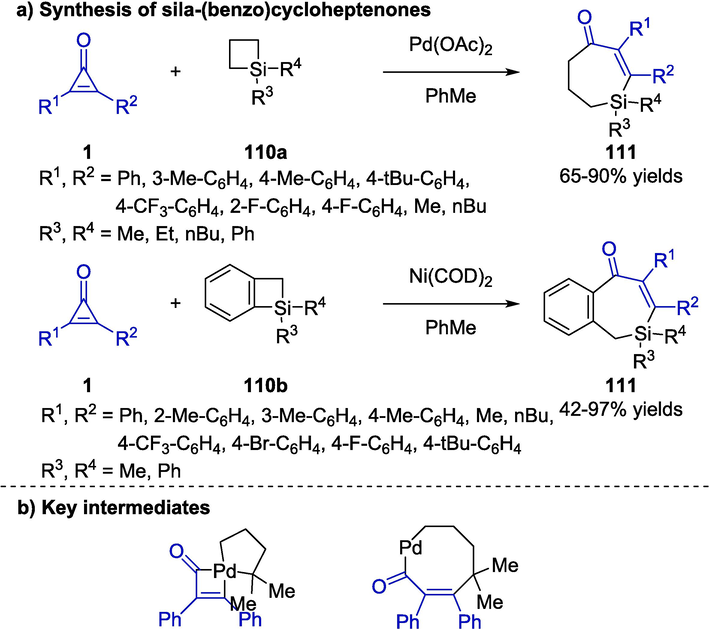
Transition metal-catalyzed synthesis of sila-(benzo)cycloheptenones.
Yang and co-workers reported an efficient route to synthesize a series of spirolactone derivatives 113 via the coupling reaction between two different molecules of cyclopropenones (Scheme 29a) (Syu et al., 2014). In the presence of a catalytic amount of copper bromide in 1,2-dichloroethane, cyclopropenones bearing electron-withdrawing or electron-donating groups could conveniently transform to the target products in good yields. Although the use of copper catalyst improved the efficiency, only several novel benzofuran-containing spirolactones could be synthesized, thus the substrate scope was limited. Unlike traditional α-keto sulfoxonium ylides, vinyl sulfoxonium ylides were first utilized as powerful building blocks to construct phenol skeletons by Zhang and co-workers (Chen et al., 2023). The authors developed a straightforward one-pot [3 + 3] annulation strategy of cyclopropenone and vinyl sulfoxonium ylides 115 for the construction of tetrasubstituted phenol derivatives 116 (Scheme 29b). The transformation proceeded smoothly via a copper carbenoid intermediate in the presence of NHC-Cu catalyst in dioxane. Therefore, a series of tetrasubstituted phenols were furnished in good to excellent yields by applying various ylides.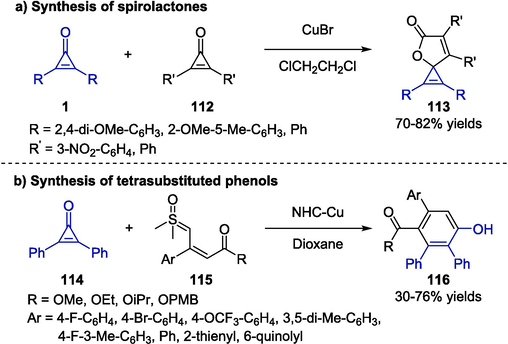
Copper-catalyzed synthesis of spirolactones and phenols.
Interestingly, cyclopropenone could serve as a nontoxic source of carbon monoxide under the catalysis of palladium which was first discovered by the Ravikumar group in 2020 (Nanda and Ravikumar, 2020). The authors reported a cascade carbonylative amination of cyclopropenones 1 and anilines 117 to provide highly substituted maleimides 118 (Scheme 30a). A wide range of anilines bearing electron-donating or −withdrawing as well as cyclopropenones bearing various substituents were compatible with the protocol, highlighting the great significance and application value of the method. Additionally, the key intermediate that participated in the conversion was a four-membered pallada-cyclobutenone which was depicted in Scheme 30a. Similarly, Zhu et al. further developed the utilization of cyclopropenones as the carbon monoxide source in the presence of a palladium catalyst (Zhu et al., 2021). Under mild conditions, a formal [2 + 2 + 1] cascade cycloaddition among cyclopropenone 1, iodochromones 119 and bridged olefins 120 was disclosed, which provided access to chromone fused cyclopentanone derivatives (Scheme 30b). The diversification of protocol was discovered to be compatible with a wide range of substrates bearing various electronic and sterical properties, furnishing the target compounds in good yields with excellent chemoselectivity. Furthermore, a possible mechanism was proposed to undergo three key sequences involving the Heck reaction, C–H activation, and carbonylation reaction.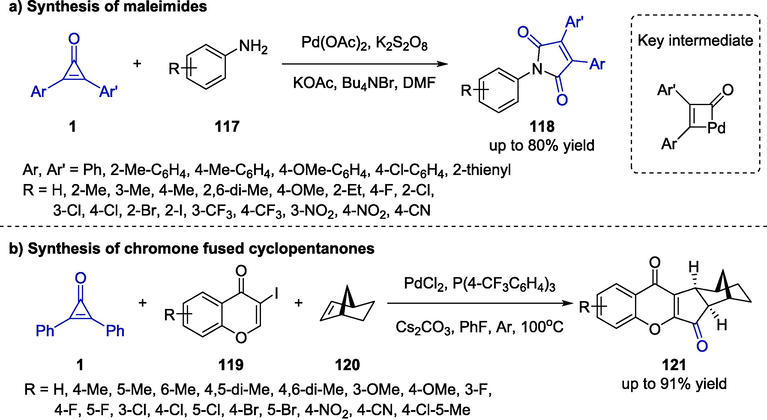
Palladium-catalyzed synthesis of maleimides and chromone fused cyclopentanones.
In 2014, Matsuda and co-workers developed a ring-expanding reaction of cyclopropenones 1 with enynes 122, which constructed functional spirocyclic cyclopentenone derivatives 123 effectively (Scheme 31a) (Matsuda and Sakurai, 2014). The transformation proceeded smoothly under the catalysis of (IPr)AuNTf2 with gentle reaction conditions, possessing excellent functional group tolerance. The final spirocyclic products could undergo further conversions, such as a dehydration reaction in the presence of p-toluenesulfonic acid. In the same year, an efficient scandium-catalyzed [3 + 2] cycloaddition reaction of cyclopropenones 1 and cyclopropanes 124 was reported for the construction of 4-oxaspiro[2.4]hept-1-ene derivatives 125 by Sierra and co-workers (Scheme 31b) (Rivero et al., 2015). In this protocol, the authors utilized Sc(OTf)3 as the catalyst, which played a significant role in stabilizing the cyclopropenyl cation. In addition, the protocol featured with a wide substrate scope as well as good functional group tolerance, delivering a variety of spirocyclic products other than oxabicyclic species which was further confirmed by DFT calculations. Cunha et al. disclosed a formal bismuth-catalyzed [3 + 2] annulation of cyclopropenone 1 and acyclic enaminones 126 as a convenient access to pyrrolinone derivatives 127 (Scheme 31c) (Cunha et al., 2017). Microwave irradiation was applied in a solvent-free system, delivering a series of polysubstituted 2-pyrrolinones with good selectivity and the formation of two new bonds. This protocol was attractive because it provided a structural scaffold with potential cytotoxic activities against glioblastoma cells.![Transition metal-catalyzed synthesis of cyclopentenones, 4-oxaspiro[2.4]hept-1-enes and pyrrolinones.](/content/184/2024/17/8/img/10.1016_j.arabjc.2024.105845-fig31.png)
Transition metal-catalyzed synthesis of cyclopentenones, 4-oxaspiro[2.4]hept-1-enes and pyrrolinones.
Based on the C–C bond activation of cyclopropenone, in 2022, Yan and co-workers developed an iron-catalyzed cascade reaction to synthesize isothiazolone derivatives 129 via the sequential formation of C-N, N-S and C-S bonds from cyclopropenones 1, anilines 128, and sulfur powder (Scheme 32a) (Wang and Yan, 2022). This conversion was promoted to be compatible with a range of anilines bearing both electron-donating and −withdrawing groups irrespective of substituted positions, delivering the expected products 129 effectively. Subsequently, selenium powder was selected as the model substrate to perform tandem reactions, thereby preparing a series of the corresponding 1,2-selenazolone derivatives 130 in acceptable yields (Scheme 32b). Xu and co-workers reported a facile synthetic strategy for the construction of pyrano[2,3-b]indol-2-one derivatives 132 under a tandem process (Scheme 32c) (Chen et al., 2021). Initially, the authors applied cyclopropenone 1 and N-ethyl isatin 131 as original materials, and [La]-N(SiMe3)2/ligand L6 were proved to be the optimal catalytic system. As a result, a variety of target products were provided in good yields with high efficiency. The protocol served a straightforward synthetic route to pyrano[2,3-b]indole scaffolds, which were flexible motifs possessing a feature with extensive application in pharmaceutical chemistry.![Transition metal-catalyzed synthesis of isothiazolones, 1,2-selenazolones and pyrano[2,3-b]indol-2-ones.](/content/184/2024/17/8/img/10.1016_j.arabjc.2024.105845-fig32.png)
Transition metal-catalyzed synthesis of isothiazolones, 1,2-selenazolones and pyrano[2,3-b]indol-2-ones.
7 Summary and outlook
Herein we have summarized and discussed recent studies of the cyclopropenone motif which serves as a flexible building unit for the advancement of synthetic chemistry towards bioactive molecules. Since its small ring size with high ring strain energy, the three-membered carbocycle is inclined to undergo a ring-opening process and subsequential cycloaddition reactions. These transformations could be promoted by a variety of efficient catalysts, such as Lewis bases as well as various transition metals involving rhodium, ruthenium, silver, nickel, copper, palladium, gold, scandium, bismuth, iron, lanthanum, and so on, delivering a wide range of corresponding heterocyclic products with good chemoselectivity, regioselectivity and stereoselectivity. Despite the great achievements have been made, the exploration of novel protocols that facilitate synthetic strategies based on C–C bond activation of cyclopropenone is still of great interest to chemists. Furthermore, one of the common phenomena concerning synthetic methodologies lies in the narrow substrate scope. Some methods had limited generality because they were applied in the construction of special substrates involving complex natural products. While others could only provide access to simple structures but not complex or functional cycloadducts. Thus, the development of this field should concentrate on discovering more general strategies that could facilitate the synthesis of both simple and complex structures. Meanwhile, the highly challenging enantioselective cycloaddition methods are still very limited so far and need to be enriched. In addition, most mechanistic studies are not particularly in-depth and only stay in the proposal stage. Additional experimental and computational investigation should be adopted to reveal the reaction mechanism. We are convinced that significant progress on catalytic transformations will be made in the area of cyclopropenone chemistry, providing powerful and straightforward tools to accelerate its development.
CRediT authorship contribution statement
Shibo Lin: Funding acquisition, Writing – original draft. Lan Xiang: Conceptualization. Chun Liu: Investigation. Xi Chen: Supervision. Yongqin Ye: Validation.
Acknowledgments
We greatly appreciate the financial support from China International Medical Foundation (No. Z-2021-46-2101), the Medical Research Youth Innovation Project of Sichuan Province (Q22025), and the Open Project of Antibiotics Research and Re-evaluation Key Laboratory of Sichuan Province (ARRLKF23-01).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis of novel chiral guanidine catalyst and its application in the asymmetric Pictet-Spengler reaction. Catal. Commun.. 2017;89:44-47.
- [CrossRef] [Google Scholar]
- Reaction of amidrazones with 2,3-diphenylcyclopropenone: Synthesis of 3-(aryl)-2,5,6-triphenylpyrimidin-4(3H)-ones. J. Chem. Res.. 2016;40:637-639.
- [CrossRef] [Google Scholar]
- Facile synthesis of new thiazinanones derived by acenaphythylenone. J. Sulfur Chem.. 2024;45:173-183.
- [CrossRef] [Google Scholar]
- Nickel(0)-catalyzed enantioselective [3+2] annulation of cyclopropenones and α, β-unsaturated ketones/imines. Angew. Chem. Int. Ed.. 2020;59:2740-2744.
- [CrossRef] [Google Scholar]
- Catalytic [3+3] annulation of β-ketoethers and cyclopropenones via C(sp3)—O/C—C bond cleavage under transition-metal free conditions. Chin. J. Chem.. 2021;39:2769-2773.
- [CrossRef] [Google Scholar]
- Mechanistic studies on nickel-catalyzed enantioselective [3 + 2] annulation for γ-butenolide synthesis via C-C activation of diarylcyclopropenones. Org. Chem. Front.. 2021;8:3023-3031.
- [CrossRef] [Google Scholar]
- Naturally occurring cyclopropenone derivatives. Angew. Chem. Int. Ed. Eng.. 1981;20:292-293.
- [CrossRef] [Google Scholar]
- The synthesis of diphenylcyclopropenone. J. Am. Chem. Soc.. 1959;81:247-248.
- [CrossRef] [Google Scholar]
- Organocatalytic regiodivergent C−C bond cleavage of cyclopropenones: A highly efficient cascade approach to enantiopure heterocyclic frameworks. Chem. Eur. J.. 2018;24:18863-18867.
- [CrossRef] [Google Scholar]
- Chen, Y., Y. Liu, J. L. Petersen, et al., 2008. Conformational control in the regioselective synthesis of N-2-substituted-1,2,3-triazoles. Chemical Communications. 3254-3256. doi: 10.1039/B805328F.
- Ruthenium(ii)-catalyzed [5 + 1] annulation reaction: a facile and efficient approach to construct 6-ethenyl phenanthridines utilizing a primary amine as a directing group. Org. Chem. Front.. 2020;7:2944-2949.
- [CrossRef] [Google Scholar]
- Lanthanide silylamide-catalyzed synthesis of pyrano[2,3-b]indol-2-ones. Org. Lett.. 2021;23:4785-4790.
- [CrossRef] [Google Scholar]
- Divergent synthesis of tetrasubstituted phenols via [3 + 3] cycloaddition reaction of vinyl sulfoxonnium ylides with cyclopropenones. Org. Lett.. 2023;25:4286-4291.
- [CrossRef] [Google Scholar]
- Trends in the usage of bidentate phosphines as ligands in nickel catalysis. Chem. Rev.. 2020;120:6124-6196.
- [CrossRef] [Google Scholar]
- Peptidyl cyclopropenones: Reversible inhibitors, irreversible inhibitors, or substrates of cysteine proteases? Protein Sci.. 2013;22:788-799.
- [CrossRef] [Google Scholar]
- One-step synthesis of 3,4-diphenyl-2-pyrrolinones by solvent-free and Bi2O3-catalyzed approaches and cytotoxicity screening against glioma cells. J. Heterocycl. Chem.. 2017;54:3700-3710.
- [CrossRef] [Google Scholar]
- One-pot synthesis of 3-substituted-2-[(3-methyl-1H-pyrazol-5-yl)imino]-5,6-diphenyl-1,3-thiazin-4-ones. J. Sulfur Chem.. 2017;38:625-634.
- [CrossRef] [Google Scholar]
- Rh(i)-Catalyzed [3+2] annulation reactions of cyclopropenones with amides. Chem. Commun.. 2019;55:5740-5742.
- [CrossRef] [Google Scholar]
- Convenient diastereoselective synthesis of annulated 3-substituted-(5S*,6S*, Z)-2-(2-(2,4-dinitrophenyl)hydrazono)-5,6-diphenyl-1,3-thiazinan-4-ones. Mol. Divers.. 2019;23:821-828.
- [CrossRef] [Google Scholar]
- Transition-metal-free [3+3] annulation reaction of sulfoxonium ylides with cyclopropenones for the synthesis of 2-pyrones. Green Synth. Catal.. 2020;1:180-182.
- [CrossRef] [Google Scholar]
- Fluorogenic cyclopropenones for multicomponent, real-time imaging. J. Am. Chem. Soc.. 2022;144:7871-7880.
- [CrossRef] [Google Scholar]
- Cyclopropenone (c-H2C3O): A new interstellar ring molecule. Astrophys. J.. 2006;642:933.
- [CrossRef] [Google Scholar]
- Rh(iii)-Catalyzed spiroannulation of ketimines with cyclopropenones via sequential C-H/C–C bond activation. Chem. Commun.. 2022;58:4743-4746.
- [CrossRef] [Google Scholar]
- Istrate, A., M. A.-O. Geeson, C. A.-O. X. Navo, et al., Platform for Orthogonal N-Cysteine-Specific Protein Modification Enabled by Cyclopropenone Reagents.
- Iwata, Y., K. Tago, T. Kiho, et al., 2000. Conformational analysis and docking study of potent factor XIIIa inhibitors having a cyclopropenone ring1 1Color Plate 1, Color Plate 2, Color Plate 3, Color Plate 4 and color plate 5 for this article are on pages 602–604. Journal of Molecular Graphics and Modelling. 18, 591-599. doi: doi: 10.1016/S1093-3263(00)00054-1.
- Cyclopropenones in the synthesis of indolizidine, pyrrolo[2,1-a]isoquinoline and indolizino[8,7-b]indole alkaloids. Tetrahedron. 2020;76:131570
- [CrossRef] [Google Scholar]
- Study of the orientation of lithiation of 1- and 2-alkylbenzotriazoles. J. Org. Chem.. 1995;60:1244-1249.
- [CrossRef] [Google Scholar]
- Kensler, T. W., J. D. Groopman, B. D. Roebuck, et al., 1993. Chemoprotection by l,2-Dithiole-3-thiones. Food Phytochemicals for Cancer Prevention I, American Chemical Society. 546: 154-163.
- Alutacenoic acids A and B, rare naturally occurring cyclopropenone derivatives isolated from fungi: Potent non-peptide factor XIIIa inhibitors. J. Am. Chem. Soc.. 2000;122:1842-1843.
- [CrossRef] [Google Scholar]
- Synthesis of 1,3-thiazine derivatives and their evaluation as potential antimycobacterial agents. Eur. J. Pharm. Sci.. 2002;15:307-310.
- [CrossRef] [Google Scholar]
- Cyclopropenylium cations, cyclopropenones, and heteroanalogues recent advances. Chem. Rev.. 2003;103:1371-1428.
- [CrossRef] [Google Scholar]
- Ruthenium- and rhodium-catalyzed strain-driven cleavage and reconstruction of the C-C bond. Eur. J. Org. Chem.. 2016;2016:1232-1242.
- [CrossRef] [Google Scholar]
- Rapid ruthenium-catalyzed synthesis of pyranopyrandiones by reconstructive carbonylation of cyclopropenones involving C−C bond cleavage. J. Am. Chem. Soc.. 2002;124:6824-6825.
- [CrossRef] [Google Scholar]
- Organocatalyzed [3 + 2] annulation of cyclopropenones and β-ketoesters: An approach to substituted butenolides with a quaternary center. Org. Lett.. 2017;19:778-781.
- [CrossRef] [Google Scholar]
- N2-selective autocatalytic ditriazolylation reactions of cyclopropenones and tropone with N1-sulfonyl-1,2,3-triazoles. Adv. Synth. Catal.. 2017;359:3304-3310.
- [CrossRef] [Google Scholar]
- Recent advances in nucleophilic Lewis base-catalyzed cycloadditions for synthesis of spirooxindoles. Adv. Synth. Catal. 2023
- [CrossRef] [Google Scholar]
- Rhodium-catalyzed C-C coupling reactions via double C–H activation. Org. Biomol. Chem.. 2016;14:4554-4570.
- [CrossRef] [Google Scholar]
- Synthesis and bio-inspired optimization of drimenal: Discovery of chiral drimane fused oxazinones as promising antifungal and antibacterial candidates. Eur. J. Med. Chem.. 2018;143:558-567.
- [CrossRef] [Google Scholar]
- Pyridine-catalyzed ring-opening reaction of cyclopropenone with bromomethyl carbonyl compounds toward furan-2(5H)-ones. Chem. Commun.. 2024;60:1900-1903.
- [CrossRef] [Google Scholar]
- Rhodium(III)-catalyzed redox-neutral [3+3] annulation of N-nitrosoanilines with cyclopropenones: A traceless approach to quinolin-4(1H)-one Scaffolds. Journal. 2020;25
- [CrossRef] [Google Scholar]
- Dearomative cyclization of pyridines/isoquinolines with cyclopropenones: Access to indolizinones and benzo-fused indolizinones. Chem. Commun.. 2023;59:4051-4054.
- [CrossRef] [Google Scholar]
- Efficient synthesis of N-2-aryl-1,2,3-triazole fluorophores via post-triazole arylation. Org. Lett.. 2008;10:5389-5392.
- [CrossRef] [Google Scholar]
- Rhodium(iii)-catalyzed [3 + 3] annulation reactions of N-nitrosoanilines and cyclopropenones: an approach to functionalized 4-quinolones. Org. Chem. Front.. 2019;6:3973-3977.
- [CrossRef] [Google Scholar]
- Catalyst-free [3+2] cycloaddition of unactivated imines with cyclopropenones. Asian J. Org. Chem.. 2020;9:82-85.
- [CrossRef] [Google Scholar]
- Gold(I)-catalyzed ring-expanding spiroannulation of cyclopropenones with enynes. J. Org. Chem.. 2014;79:2739-2745.
- [CrossRef] [Google Scholar]
- Silver-catalyzed ring-opening [3+2] annulation of cyclopropenones with amides. New J. Chem.. 2018;42:19178-19182.
- [CrossRef] [Google Scholar]
- Synthesis of trisubstituted 1,3-oxazin-6-ones via base-catalyzed ring-opening annulation of cyclopropenones with N-(pivaloyloxy)amides. Tetrahedron Lett.. 2018;59:1458-1460.
- [CrossRef] [Google Scholar]
- Synthesis and biological study of novel bis-chalcones, bis-thiazines and bis-pyrimidines. J. Iran. Chem. Soc.. 2008;5:262-267.
- [CrossRef] [Google Scholar]
- Cyclopropenone acetals synthesis and reactions. Chem. Rev.. 2003;103:1295-1326.
- [CrossRef] [Google Scholar]
- A palladium-catalyzed cascade C-C activation of cyclopropenone and carbonylative amination: Easy access to highly functionalized maleimide derivatives. Org. Lett.. 2020;22:1368-1374.
- [CrossRef] [Google Scholar]
- Recent advancement in palladium-catalyzed C-C bond activation of strained ring systems: Three- and four-membered carbocycles as prominent C3/C4 building blocks. ACS Catal.. 2022;12:13247-13281.
- [CrossRef] [Google Scholar]
- N-Heterocyclic carbene catalyzed base assisted C-C bond cleavage of cyclopropenones: An approach towards diastereoselective synthesis of azetidinones and benzoxazepines. Org. Chem. Front.. 2023;10:6166-6171.
- [CrossRef] [Google Scholar]
- Indolizin-1-ols with charged electron acceptors: A direct way to 3H-indolizinium-1-olates with donor functions. J. Org. Chem.. 2022;87:14137-14154.
- [CrossRef] [Google Scholar]
- Butenolide synthesis from functionalized cyclopropenones. Org. Lett.. 2019;21:8695-8699.
- [CrossRef] [Google Scholar]
- Base-promoted [3 + 3] cyclization of cyclopropenones and cyclopropenethiones with amides for the synthesis of 6H–1,3-oxazin-6-ones and 6H–1,3-thiazin-6-ones. Org. Chem. Front.. 2018;5:1267-1271.
- [CrossRef] [Google Scholar]
- Okuda T Fau - Yokose, K., T. Yokose K Fau - Furumai, H. B. Furumai T Fau - Maruyama, et al., 1984. Penitricin, a new class of antibiotic produced by Penicillium aculeatum. II. Isolation and characterization. J Antibiot. 37, 718-722. doi: doi: 10.7164/antibiotics.37.718.
- Synthetic applications of cyclopropene and cyclopropenone: Recent progress and developments. Asian J. Org. Chem.. 2020;9:1088-1132.
- [CrossRef] [Google Scholar]
- DMAP-catalyzed [3+3] annulation of cyclopropenones with α-bromoketones for synthesis of 2-pyrones. Eur. J. Org. Chem.. 2022;2022:e202200243.
- [CrossRef] [Google Scholar]
- Study of the asymmetric organocatalyzed [3+2] annulation of cyclopropenone and β-keto ester. Chem. Heterocycl. Compd.. 2018;54:929-933.
- [CrossRef] [Google Scholar]
- Bidentate directing groups: An efficient tool in C-H bond functionalization chemistry for the expedient construction of C–C bonds. Chem. Rev.. 2020;120:1788-1887.
- [CrossRef] [Google Scholar]
- Donor-stabilized cations and imine transfer from N-silylphosphoranimines. J. Am. Chem. Soc.. 2004;126:2286-2287.
- [CrossRef] [Google Scholar]
- Synthesis of oxaspiranic compounds through [3 + 2] annulation of cyclopropenones and donor-acceptor cyclopropanes. J. Org. Chem.. 2015;80:1207-1213.
- [CrossRef] [Google Scholar]
- Lithraea caustic (Litre) extract promotes an antitumor response against B16 melanoma. Front. Pharmacol.. 2019;10
- [Google Scholar]
- Cyclopropenones for metabolic targeting and sequential bioorthogonal labeling. J. Am. Chem. Soc.. 2017;139:7370-7375.
- [CrossRef] [Google Scholar]
- Chemically triggered crosslinking with bioorthogonal cyclopropenones. Chem. Commun.. 2020;56:10883-10886.
- [CrossRef] [Google Scholar]
- CpCo-mediated reactions of cyclopropenones: Access to CpCo-capped benzoquinone complexes. Organometallics. 2008;27:1361-1366.
- [CrossRef] [Google Scholar]
- Divergent C-H activation synthesis of chalcones, quinolones and indoles. Chem. Commun.. 2020;56:1585-1588.
- [CrossRef] [Google Scholar]
- Divergent construction of diverse Scaffolds through catalyst-controlled C−H activation cascades of quinazolinones and cyclopropenones. Chem. Eur. J.. 2021;27:13346-13351.
- [CrossRef] [Google Scholar]
- Sizhan Liu, M. C. B. W. C. H. Y. Z. J. L. X. X. Z. W. S. W., 2021. Triethyl Amine-Promoted Cyclization Reaction between Cyclopropenone and <i>α</i>-Halogenated Hydroxamate for the Synthesis of Polysubstituted 6<i>H</i>-1,3-Oxazin-6-one. Chinese Journal of Organic Chemistry. 41, 1622-1630.
- Synthesis of benzofuran-containing spirolactones from diarylcyclopropenones. Tetrahedron Lett.. 2014;55:1207-1211.
- [CrossRef] [Google Scholar]
- Diels−alder reactions of tetraethynylcyclopentadienones. An approach to differentially substituted hexaethynylbenzenes of C2v symmetry. J. Org. Chem.. 1997;62:3430-3431.
- [CrossRef] [Google Scholar]
- Penitricin, a new class of antibiotic produced bypenicillium aculeatum. J. Antibiot.. 1984;37:712-717.
- [CrossRef] [Google Scholar]
- Penitricin, a new class of antibiotic produced by penicillium aculeatum. J. Antibiot.. 1984;37:723-727.
- [CrossRef] [Google Scholar]
- Penitricin, a new class of antibiotic produced by penicillium aculeatum. J. Antibiot.. 1984;37:718-722.
- [CrossRef] [Google Scholar]
- The use of diphenylcyclopropenone in the treatment of recalcitrant warts. JCMS. 2002;6:214-217.
- [CrossRef] [Google Scholar]
- Responses to topical diphenylcyclopropenone as an adjunct treatment for in-transit melanoma: A tertiary referral center experience. Dermatol. Surg.. 2018;44
- [Google Scholar]
- Vol'pin, M. E., Y. D. Koreshkov and D. N. Kursanov, 1959. Diphenylcyclopropenone, a three-membered analog of tropone. Bulletin of the Academy of Sciences of the USSR, Division of chemical science. 8, 535-536. doi: 10.1007/BF00917720.
- Reacting cyclopropenones with arynes: Access to spirocyclic xanthene-cyclopropene motifs. J. Org. Chem.. 2015;80:3730-3734.
- [CrossRef] [Google Scholar]
- Wang, D., L. Désaubry, G. Li, et al., 2021. Front Cover Picture: Recent Advances in the Synthesis of C2-Functionalized Pyridines and Quinolines Using N-Oxide Chemistry (Adv. Synth. Catal. 1/2021). Advanced Synthesis & Catalysis. 363, 1-1. doi: doi: 10.1002/adsc.202001427.
- Iron-catalyzed one-step synthesis of isothiazolone/1,2-selenazolone derivatives via [3+1+1] annulation of cyclopropenones, anilines, and elemental chalcogens. Adv. Synth. Catal.. 2022;364:715-719.
- [CrossRef] [Google Scholar]
- Phosphine-catalyzed dearomative [3 + 2] cycloaddition of benzoxazoles with a cyclopropenone. Org. Lett.. 2022;24:1127-1131.
- [CrossRef] [Google Scholar]
- Transition-metal-, additive-, and solvent-free [3 + 3] annulation of RCF2-imidoyl sulfoxonium ylides with cyclopropenones to give multifunctionalized CF3-pyridones. J. Org. Chem.. 2022;87:1124-1132.
- [CrossRef] [Google Scholar]
- Cyclopentadienone synthesis by rhodium(I)-catalyzed [3 + 2] cycloaddition reactions of cyclopropenones and alkynes. J. Am. Chem. Soc.. 2006;128:14814-14815.
- [CrossRef] [Google Scholar]
- σ/π versus π/π conjugation: DFT studies on oligocyclopropenones and related systems. Org. Lett.. 2008;10:5231-5234.
- [CrossRef] [Google Scholar]
- Tethered dithiacyclopropenones. Syntheses and structural properties of tetrathiacyclopropenonophanes. J. Org. Chem.. 2001;66:3416-3422.
- [CrossRef] [Google Scholar]
- Selective [3 + 2] cycloaddition of cyclopropenone derivatives and elemental chalcogens. Org. Lett.. 2020;22:5555-5560.
- [CrossRef] [Google Scholar]
- Construction of indenols and derivatives through Rh(III) catalyzed CH activation in a one-pot manner. Tetrahedron Lett.. 2022;107:154110
- [CrossRef] [Google Scholar]
- Synthesis of 4-ethenyl quinazolines via rhodium(iii)-catalyzed [5 + 1] annulation reaction of N-arylamidines with cyclopropenones. Org. Chem. Front.. 2020;7:672-677.
- [CrossRef] [Google Scholar]
- Organocatalytic C-C bond activation of cyclopropenones for ring-opening formal [3 + 2] cycloaddition with isatins. Org. Chem. Front.. 2017;4:560-564.
- [CrossRef] [Google Scholar]
- Ag2O-promoted ring-opening reactions of cyclopropenones with oximes. Org. Biomol. Chem.. 2020;18:5822-5825.
- [CrossRef] [Google Scholar]
- Synthesis of 2-alkenyl-4H-3,1-benzoxazin-4-ones through HFIP-mediated decarboxylative [4+2]-annulation of isatoic anhydrides with cyclopropenones under silver catalysis. Adv. Synth. Catal.. 2021;363:4085-4090.
- [CrossRef] [Google Scholar]
- Fully substituted conjugate benzofuran core: Multiyne cascade coupling and oxidation of cyclopropenone. Org. Lett.. 2021;23:4971-4975.
- [CrossRef] [Google Scholar]
- Transition-metal free synthesis of 2-pyrones by [3 + 3] annulation of cyclopropenones and sulfur ylides. J. Heterocycl. Chem.. 2023;60:1458-1463.
- [CrossRef] [Google Scholar]
- Rh(III)-catalyzed double C-H functionalization of indoles with cyclopropenones via sequential C–H/C–C/C–H bond activation. Org. Lett.. 2023;25:3922-3926.
- [CrossRef] [Google Scholar]
- Phosphine-catalyzed asymmetric dearomative [3+2] annulation reaction of benzimidazoles with cyclopropenones. CCS Chemistry. 2024:1-11.
- [CrossRef] [Google Scholar]
- Phosphane- and amine-catalyzed ring-opening reactions of cyclopropenones with isatin derivatives: Synthesis of carboxylated 1H-indoles and multisubstituted 2H-pyran-2-ones. Eur. J. Org. Chem.. 2014;2014:2672-2676.
- [CrossRef] [Google Scholar]
- Intermolecular σ-bond cross-exchange reaction between cyclopropenones and (Benzo)silacyclobutanes: Straightforward access towards sila(benzo)cycloheptenones. Angew. Chem. Int. Ed.. 2018;57:6329-6332.
- [CrossRef] [Google Scholar]
- [3+2]-cycloaddition of azaoxyallyl cations with cyclopropenones and cyclopropenethiones: Synthesis of spirocyclic oxazole and thiazole derivatives. Asian J. Org. Chem.. 2019;8:1376-1379.
- [CrossRef] [Google Scholar]
- C-H activation-initiated spiroannulation reactions and their applications in the synthesis of spirocyclic compounds. Org. Biomol. Chem.. 2024;22:2324-2338.
- [CrossRef] [Google Scholar]
- Deconstructive cycloaromatization strategy toward N, O-bidentate ligands from indolizines and cyclopropenones. Org. Lett.. 2022;24:3238-3243.
- [CrossRef] [Google Scholar]
- Rh(III)-catalyzed [3 + 3] annulation reaction of cyclopropenones and sulfoxonium ylides toward trisubstituted 2-pyrones. J. Org. Chem.. 2020;85:360-366.
- [CrossRef] [Google Scholar]
- Palladium-catalyzed [2 + 2 + 1] annulation: access to chromone fused cyclopentanones with cyclopropenone as the CO source. Org. Chem. Front.. 2021;8:3082-3090.
- [CrossRef] [Google Scholar]




