

Translate this page into:
A spin-coated TiOx/Pt nanolayered anodic catalyst for the direct formic acid fuel cells
⁎Corresponding author. ammohammad@cu.edu.eg (Ahmad M. Mohammad)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The CO poisoning of the platinum anodic catalyst which typically functions the catalytic deterioration of the direct formic acid fuel cells could be minimized with a simple modification for Pt with titanium oxide. The fabrication scheme involved the spin-coating of a Ti precursor onto a Pt thin layer that was physically sputtered onto a Si substrate. The whole assembly was subjected to a post-annealing processing to produce the TiOx layer (60 nm) in a porous structure (mostly Anatase) atop of the Pt surface. The porous nature of the TiOx layer permitted the participation of Pt in the electrocatalysis of the formic acid electro–oxidation (FAO). The annealing temperature was critical in identifying the catalytic efficiency and durability of the catalyst toward the FAO. Interestingly, if compared to bare-Pt substrates, the TiOx-modified catalysts could successfully steer the FAO toward the direct dehydrogenation (favorable and less energetic) pathway with more than an order of magnitude increase in the catalytic activity. It also provided a great opportunity for the mitigation of poisoning CO; concurrently with a lowering (∼0.3 V) in the onset potential of the FAO. The scanning electron microscopy (SEM), energy dispersive X-ray spectroscopy (EDS), X-ray diffraction spectroscopy (XRD), cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) techniques were all combined to evaluate, respectively, the catalyst’s morphology, composition, crystal structure and activity and further to understand the role of the TiOx in the catalytic enhancement.
Keywords
Direct formic acid fuel cells
Carbon monoxide
Poisoning
Transition metal oxides
Electrocatalysis

1 Introduction
The demand to sustain renewable, energy-efficient and eco-friendly power sources has recently spurred a tremendous interest in fuel cells (FCs) (Al-Akraa, 2017; Al-Akraa et al., 2015; Boddien et al., 2011; Cheng et al., 2010a, 2010b; Li et al., 2018; Xia et al., 2011). In fact, FCs together with water-based electrolyzers succeeded to represent a green, harmless and efficient saving/restoring mechanism for excess electricity that renewable power plants may deliver under certain conditions (plenty time) to be used later under the conditions of deficiency (Dunn, 2002; Gahleitner, 2013). They could also promote the movement into the era of transferring the power generators rather than the power (electricity) itself (Bayod-Rújula, 2009). That will definitely save the huge energy losses over the long transmission and distribution of electricity. Besides, FCs with their greenness (higher than 90% reduction in major contaminations exhausted from the combustion of fossil fuels), improved efficiency (up to 60% in electrical energy conversion), robustness, reliability, safety and moving flexibility have become a decent alternative for batteries in several stationary and portable electronic devices (Borup et al., 2007; Debe, 2012; Steele and Heinzel, 2001; Wang et al., 2011). Nevertheless, for FCs to replace batteries, they must demonstrate an economic feasibility with a remarkable increase in their delivered power density. The amount of precious metals (mostly Pt) employed for the catalysis of the involved electrochemical reactions has to be minimized and secured from expected poisoning (Baschuk and Li, 2001; Gottesfeld and Pafford, 1988). The rates of electrochemical reactions involved therein have to be boosted together with a significant reduction in the area-specific resistivity (ASR) of the FCs' cell components to attain an improvement for the power density. Ideally, the ASR of the electrolyte, anode and cathode should lie close to 0.1 Ω cm2 to support the 1 kW dm−3 and 1 kW kg−1 required for transport applications (Steele and Heinzel, 2011).
From another perspective, poisoning (with CO principally) the Pt catalysts that are typically suggested for the electro-oxidation of liquid carbon-containing fuels still represents a major dilemma for the industry of liquid FCs. The problem resides in the gradual marginalization of a huge portion of the Pt surface from the participation in the targeted electrochemical reactions which ultimately deteriorates the overall performance of the FCs. This poisoning was not terrible for the H2/O2 FCs which employed H2; the smallest and cleanest (carbon-free) fuel (Camara et al., 2002; Dhar et al., 1987). Nevertheless, the use, transport and storage of H2 were challenging from safety and economic perspectives which obliged the industry to switch into carbon-containing liquid fuels (Shinnar, 2003). Of the least affected liquid fuels by CO-poisoning is formic acid (FA) which, moreover, features an incomparable safety (a typical food-additive) and a minor crossover through the through Nafion® membranes that typically represent the electrolyte in liquid FCs (Joó, 2008; Markoví and Ross, 2002; Zhu et al., 2005). Yet, effort is still running to minimize/overcome the CO poisoning to improve the kinetics of the formic acid electro–oxidation (FAO) at Pt-based catalysts.
In this regard, three major strategies were adapted. The first manipulated the surface composition of the catalyst by doping with foreign metals (e.g., Ru, Fe, Cu, Au, Pd) to enforce the adsorption of FA at the Pt surface in a favorable orientation impeding the release of poisoning CO (Ge et al., 2013; Scofield et al., 2015). Under certain conditions, scattering the Pt surface with minute amount of nano-Au succeeded to disturb the contiguity of active Pt sites that is required to stabilize CO; hence, mitigated the poisoning (Al-Akraa et al., 2019a). Recently, supporting the Pt catalyst onto a conducting polymer (e.g., poly-aniline) could perform similarly as nano-Au to yield an improved FAO (Abd El-Moghny et al., 2017). The second approach employed the surface modification of the Pt surface with oxide nanostructures of transition metals as Ni, Mn, Co and Cu which enriched the Pt surface with oxygen moieties that facilitated the oxidative removal of poisoning CO at relatively lower potentials (if compared to that required at the bare-Pt surface) (Mohammad et al., 2018). These oxides could moreover mediate the oxidation scheme of the FAO with their multiple oxidation states and their available d-orbital vacancies to facilitate the electron transfer and provide better kinetics. The modification procedure (sequential layer-by-layer or simultaneous co-deposition) of the Pt catalyst with nano-Au or transition metal oxide nanostructures can intensively impact the catalytic activity of the catalysts toward FAO (Asal et al., 2018, 2019). The third approach involved tuning the surface electronic properties of Pt in a way weakening the Pt–CO bonding; hence, facilitating the release of CO and overcoming poisoning (Mohammad et al., 2018).
Relatively, most of these mechanisms succeeded to boost FAO but the catalyst’s durability remained undesirable where most of these Pt modifiers underwent either a phase change or peeling off under the continuous operation in acidic environment for long time. Herein, a much more stable modifier (titanium oxide) is recommended for Pt surfaces to boost the FAO. The synthetic scheme involved the spin-coating of Ti precursors over a Pt layer that was pre-sputtered on a Si wafer. The whole sample was next subjected to annealing before testing the catalyst’s activity toward the FAO.
2 Experimental
2.1 Chemicals
Chemicals of analytical grades were used to prepare the aqueous solutions with deionized water. The high purity of these chemicals permitted no prior treatment before the preparation. Alcohol-dissolved Ti (3%) precursors were purchased from Koujundo Kagaku. Co., Ltd., Japan.
2.2 Catalyst’s preparation
The same catalyst was recommended previously for the electro-generation of ozone and water splitting; hence, more details about the catalyst’s preparation can be retrieved (Al-Akraa et al., 2019b; Mohammad et al., 2007, 2009). Shortly, the catalyst had the sequential Si–TiOx/Pt/TiOx structure. The Si–TiOx substrate was prepared by radio-frequencies (RF) sputtering (ULVAC, Inc.) of titanium oxide (TiOx) on a Si substrate for 10 min at room temperature (RT) under a total gas pressure of 0.6 Pa (Ar/O2 ratio = 0.48/0.52) and an RF power density of 6.4 W cm−2. Platinum was next deposited on the Si–TiOx substrate for 1 min at RT under an Ar gas pressure of 0.7 Pa and RF power density of 4.8 W cm−2. The sputtered TiOx layer was primarily deposited to reinforce the adhesion of the Pt layer to the Si substrate and to mitigate the mutual diffusion of Si and Pt. The Si–TiOx/Pt was next moved to a spin-coater (Kyowariken, K-359 S-1, Japan) where a Ti layer was spun from alcohol-dissolved Ti (3%) precursors onto the Pt layer in two consecutive steps (1000 rpm for 10 s then at 3000 rpm for 30 s). After that, the electrode was dried in air at RT for 10 min and next at 200 °C for another 10 min. The average thickness of the spin-coated Ti layer was ca. 60 nm. Before moving into materials and electrochemical inspection, the electrode was annealed at three different temperatures 500 °C, 600 °C and 700 °C for 10 min in air. Based on the annealing temperature, the catalysts acquired the corresponding (Ti500, Ti600 and Ti700) abbreviations.
2.3 Electrochemical measurements and materials characterization
The electrochemical measurements were performed at RT (25 ± 1 °C) in a two-compartment three-electrode Pyrex cell. The Ti500, Ti600 and Ti700 electrodes were used as the working electrode while a saturated calomel electrode (SCE) and a spiral Pt wire were used as the reference and counter electrodes, respectively. All potentials, even if not mentioned, will be reported in reference to SCE. The measurements were performed using a Bio-Logic SAS potentiostat (model SP-150) operated with EC-Lab software. Current densities were calculated on the basis of the real surface areas of the working electrodes that were determined based on the charge associated the reduction of the PtO layer (420 µC/cm2) (Trasatti and Petrii, 1991). The electrocatalytic activity of the modified electrodes toward FAO was examined in an aqueous solution 0.3 M FA at a scan rate of 100 mV s−1. The electrochemical impedance spectroscopy (EIS) assisted in revealing the role of TiOx in the catalytic enhancement of the Ti500, Ti600 and Ti700 catalysts toward FAO.
The diffraction patterns of the electrodes were obtained by grazing incidence X–ray diffraction (XRD) spectroscope operated with Cu Kα (λ = 1.54056 Å) radiation at 45 kV and 360 mA (D8-DISCOVER, Bruker AXS). The morphology of the electrode was inspected using a field-emission scanning electron microscope (FE-SEM, QUANTA FEG 250) which was equipped with an energy dispersive X-ray spectrometer (EDS).
3 Results and discussion
3.1 Electrochemical preliminary testing
Investigation of the cyclic voltammograms (CVs) has been employed to reveal the electrochemical performance of the catalysts and to confirm the deposition of catalyst's constituting ingredients. Fig. 1 shows a series of CVs measured in 0.5 M H2SO4 solution under N2 atmosphere at a potential scan rate of 0.1 V s−1 for a bare Pt electrode (Fig. 1A), Ti500 (Fig. 1B), Ti600 (Fig. 1C) and Ti700 (Fig. 1D). The characteristic behavior of a polycrystalline Pt electrode appeared in all the CVs with the oxidation of Pt to PtO (from ca. 0.6 to 1.2 V) and its subsequent reduction (at ca. 0.4 V). This accompanied the appearance of well-defined split hydrogen adsorption/desorption (Hads/des) peaks in the potential range from 0.0 to −0.2 V . The splitting of the Hads/des peaks infers the exposure of different crystal facets (Pt (1 0 0) at a lower potential and Pt (1 1 1) at a higher potential) of the bare polycrystalline Pt substrate to the electrolyte. In fact, all the major characteristic (Pt/PtO and Hads/des) peaks of poly-crystalline Pt surfaces retained in the CVs of Ti500 (Fig. 1B), Ti600 (Fig. 1C) and Ti700 (Fig. 1D) catalysts which highlighted the incomplete coverage of the Pt surface with the spun TiOx modifier. This cracked-like structure is most likely behind the little distortion of the CV response of the spin-coated Ti500, Ti600 and Ti700 catalysts (Fig. 1B–D) which may get enlarged with H2 spillover (Malevich et al., 1997). In addition, the potential of the PtO/Pt reduction peak which was located at ca. 0.43 V at the bare Pt electrode (Fig. 1A) was negatively shifted to ca. 0.32 V at the TiOx modified catalysts (Fig. 1B–D). It worth mentioning that the onset potential (ca. 0.75 V) of the PtO/Pt reduction which is a substrate-dependent thermodynamic function retained unchanged at all catalysts. This might result because of the large increase of the Pt surface area of the Ti500, Ti600 and Ti700 electrodes if compared to that of the bare Pt electrode. This was also obvious in the large increase of the PtO/Pt peak currents of the TiOx modified catalysts comparatively to that of the bare Pt electrode. One more important observation in Fig. 1 was the increase in the double-layer charging current (at ca. 0.0 to 0.2 V) of the Ti500, Ti600 and Ti700 electrodes if compared to that of the bare Pt electrode. This indicates certainly the successful deposition of the capacitive TiOx film.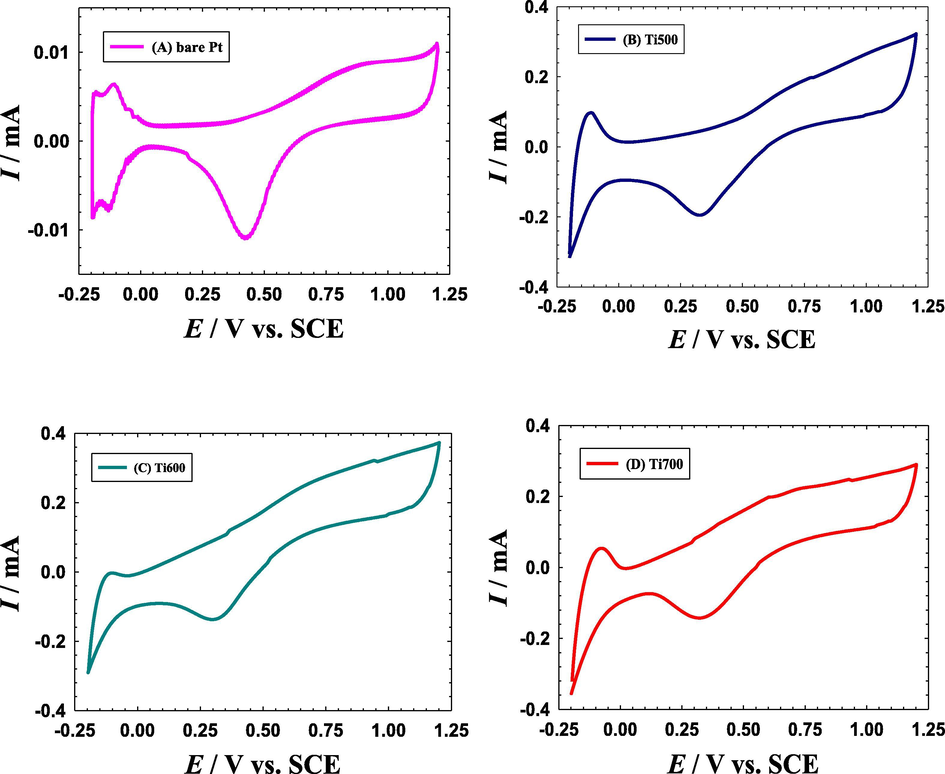
The CVs obtained in N2–saturated 0.5 M H2SO4 solution at a potential scan rate of 0.1 V s−1 for a bare-Pt electrode (A), Ti500 (B), Ti600 (C) and Ti700 (D) electrodes.
3.2 Morphology and structure
In fact, the inspection of the surface morphology was not so descriptive; the typical imaging of thin films. However, the appearance of dark and bright areas in Fig. 2A matches the sequential multilayer structuring of the catalyst. The dark areas can likely correspond to the topmost cracked TiOx layer while the brighter areas correspond to the underneath Pt layer. This visualization harmonizes with a previous inspection for the same catalyst after a surface modification with MnOx nanorods (Al-Akraa et al., 2019b). Fig. 2B provides a Ti-mapping for the same catalyst (Ti700) which indicated the homogeneous distribution of TiOx in a porous nanotexture atop of the Si–TiOx /Pt substrate. This will likely permit the accessibility of the electrolyte to the Pt surface, in agreement with the electrochemical evidences.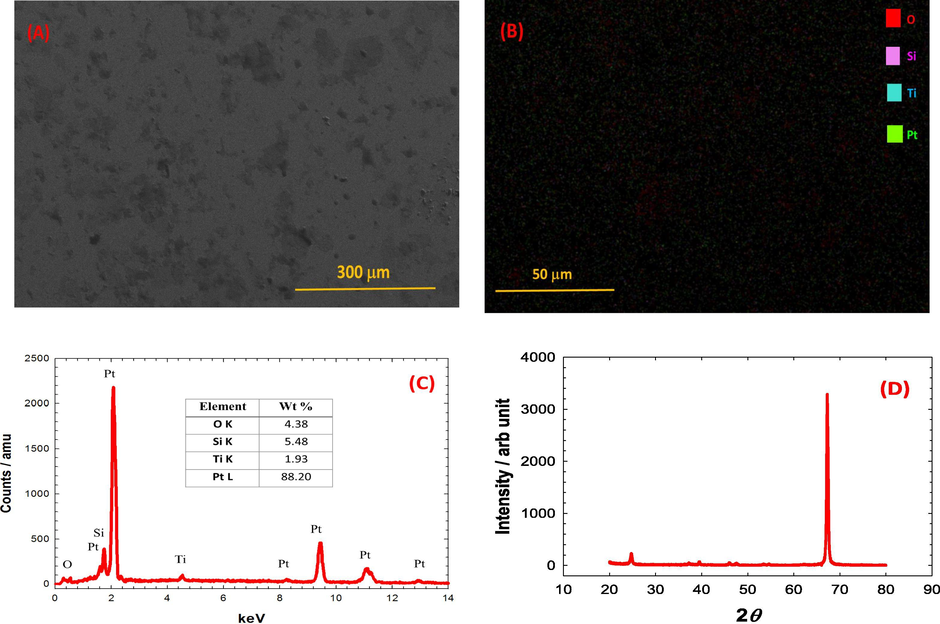
SEM image (A), elemental mapping analysis (B), EDS analysis (C) and XRD analysis (D) of the Ti700 electrode.
The EDS analysis of the catalyst (Ti700) provided a confirmation for the deposition of all constituting ingredients in the catalyst (Si, Ti, Pt and O) and further assessed their relative compositions (see Fig. 2C).
Additionally, the XRD analysis in Fig. 2D assisted in revealing the crystal structure of the Si–TiOx/Pt/TiOx catalyst where TiOx was crystallized mostly in Anatase structure. The diffraction peaks at 2θ of 24.74, 37.24 and 46.04° corresponded to the (1 0 1), (0 0 4) and (2 0 0) planes of TiO2 (anatase) (Kim et al., 2007). On the other hand, the diffraction peaks at 2θ of 39.48, 47.58 and 67.45 were assigned to the (1 1 1), (2 0 0) and (2 2 0) planes of Pt (Mohammad et al., 2018).
3.3 Formic acid electro-oxidation (FAO): Activity assessment
Fig. 3 shows the CVs of the FAO at a bare Pt (Fig. 3A), Ti500 (Fig. 3B), Ti600 (Fig. 3C) and Ti700 (Fig. 3D) electrodes in 0.3 M FA (pH = 3.5). As expected, the FAO proceeded at all catalysts with different activities and tolerances against poisoning. To understand the assessment of the catalytic activity toward FAO as a consequence of a given structural/compositional amendment in the catalyst, the inherent mechanism of FAO on Pt-based materials should be elaborated. Generally, a ‘dual pathway’ mechanism is adapted for this reaction (Larsen et al., 2006; Lović et al., 2005) with the direct favorable dehydrogenation (FA → CO2) and the indirect unfavorable dehydration (FA → CO → CO2). As obviously seen, both avenues end up with CO2 but the consequences of both are really different, particularly economically. In reality, the direct FAO pathway proceeds with no poisoning intermediates at low overvoltage, which eventually immune the Pt surface against deactivation and avoid unnecessary energy loses. Eq. (1) represents the oxidation half-reaction involved in this direct pathway where the first anodic peak in the forward potential scan (at ca. 0.2 V in Fig. 3A) depicts its corresponding electrochemical response in an acidic (0.3 M FA) medium. The highest (peak) current, Ipd, of this direct oxidation pathway probes the density of the free active (non-poisoned) Pt sites available to participate in this direct pathway.
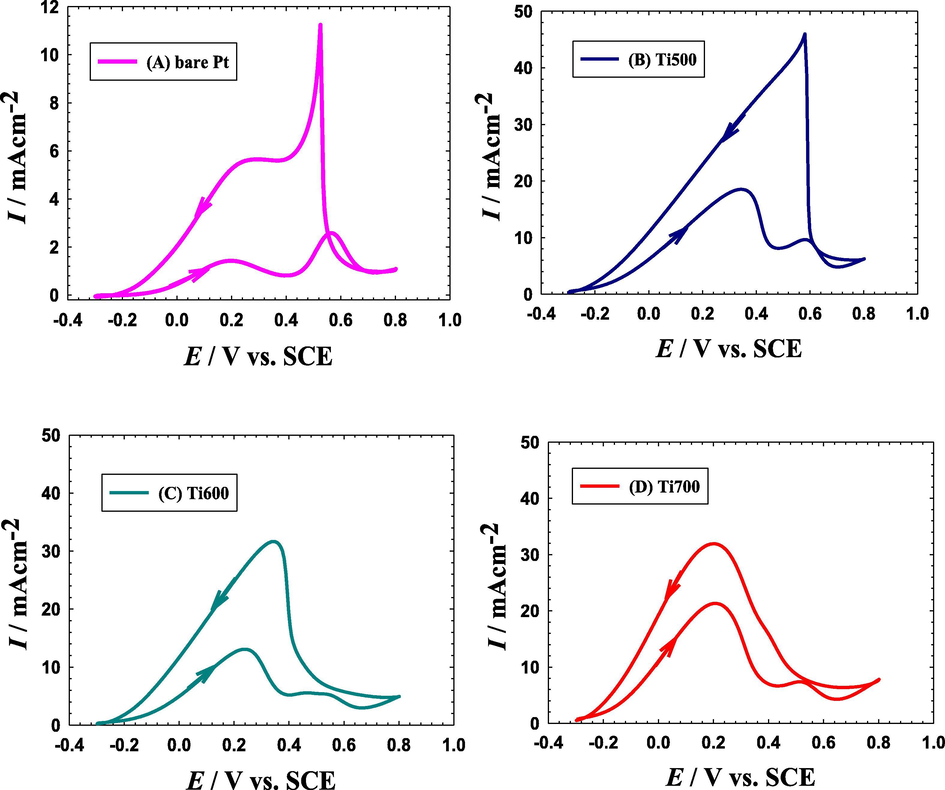
The CVs obtained in 0.3 M FA (pH = 3.5) at a potential scan rate of 0.1 V s−1 for a bare-Pt electrode (A), Ti500 (B), Ti600 (C) and Ti700 (D) electrodes.
On the other hand, the indirect dehydration pathway of FAO involves the release of CO intermediate that has a strong adsorption affinity at the Pt surface. As long as the Pt surface is kept protected from hydroxylation (typically occurs if overpotential exceeded ca. 0.4 V), the CO remains adsorbed and blocking the active Pt sites from participating in the FAO; a phenomenon known as “poisoning of the Pt surface”. If overpotential higher than 0.4 V is supplied, the Pt surface gets hydroxylated and, hence, CO can be oxidized to CO2. The second peak in the forward potential scan of Fig. 3A corresponds to this indirect oxidation (CO → CO2) with a peak current (Ipind) at ca. 0.6 V. Equations 2–4 describe the mechanism of the indirect pathway of FAO. It becomes now manifested the energy loss (corresponding to an increase of “0.6–0.2 = 0.4 V” in overpotential) associating the poisoning avenue of FAO. The same result can be obtained if the onset potential (Eonset) of FAO and CO oxidation are compared. Fortunately, the indirect oxidation of CO retrieves again the activity of the Pt surface to participate again in the FAO. This explains the high current (Ib) associating the backward cathodic scan peak.
The modification of the Pt catalyst was inspired from the incredible interest in using TiOx supports in the catalytic oxidation of small organic molecules. This was not only because of the large surface area they imparted particularly if used in nanostructures but also for their potential to mediate the oxidation reaction via the multiple oxidation states and the vacant d-orbital of Ti (Li et al., 2017; Song et al., 2007; Wang and Xia, 2010). They possessed, moreover, a short charge transport distance and little carrier transport losses, which undoubtedly presages enhanced electron transfer kinetics for many electrocatalytic applications (Li et al., 2017; Roy et al., 2011). Nevertheless, it remained to justify a more quantitative index to compare the impact of the modification on the performance of different catalysts toward FAO. It was really comfortable to employ the Ipd/Ipind and Ipd/Ib in this assessment. A relatively higher value of Ipd/Ipind indicates certainly the availability of more free active Pt sites for the direct FAO while a higher Ipd/Ib value designates a lower CO poisoning level for the Pt surface. These two indices along with Eonset of the direct FAO will be evaluated to sort the different catalysts based on their activity toward the FAO.
First of all, these indices (Ipd/Ipind and Ipd/Ib) read ca. 0.6 and 0.2, respectively at the bare Pt electrode (Fig. 3A). Surprisingly, the Ti500, Ti600 and Ti700 electrodes (Fig. 3B-D) surpassed the bare Pt electrode in the catalytic performance toward FAO with Ipd/Ipind values of 6 (tenfold increase), 7.6 (∼13 times increase) and 10 (∼17 times increase) and Ipd/Ib values of 0.4, 0.45 and 0.67, respectively. In addition, they all offered a significant (ca. 250–300 mV) lowering in Eonset if compared to that of the bare Pt electrode. This indicated the improved catalytic activity and associated tolerance against CO poisoning of the Ti500, Ti600 and Ti700 catalysts toward FAO.
3.4 Role of TiOx in the catalytic enhancement
The electrochemical impedance spectroscopy (EIS) technique was used to probe the charge transfer resistance (Rct) of the different catalysts during FAO. Fundamentally, the EIS technique visualizes the electrode/electrolyte interface as a combination of passive electrical circuit elements, i.e., resistance, capacitance and inductance. If an alternating current is applied to this interface, Ohm's law can predict the resulting current. The representation of EIS measurements appears in one of two plots called “Nyquist and Bode diagrams”. Fig. 4 shows the Nyquist plots obtained at the bare Pt (Fig. 4a), Ti500 (Fig. 4b), Ti600 (Fig. 4c) and Ti700 (Fig. 4d) catalysts in a 0.3 M aqueous solution of FA (pH = 3.5) at a potential of 0.2 V in the frequency range (10 mHz to 100 kHz). Principally, the diameter of the extrapolated semicircle in the Nyquist diagram represents the charge transfer resistance (Rct) that is equivalent to the polarization resistance of the electrochemical system (RIBEIRO et al., 2015). Hence, the larger the diameter of the semicircle the higher Rct, and hence, the slower kinetics of the reaction (Yavuz et al., 2015). As clearly seen in Fig. 4, the Rct of the Ti700 (0.1 kΩ) is the smallest if compared to those of the Ti600 (0.37 kΩ) and Ti500 (0.96 kΩ) catalysts. Promisingly, all these Rct values were very low if compared to that of the bare Pt electrode (18 kΩ). This finding agrees perfectly with the data of Fig. 3 to confirm undoubtedly a facilitated charge transfer and boosted catalytic activity of the spin-coated Si–TiOx/Pt/TiOx catalysts toward FAO.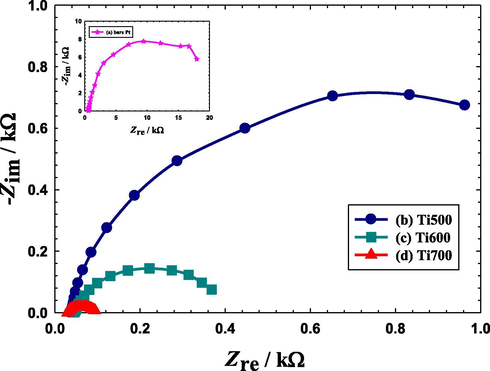
Nyquist plots obtained in 0.3 M FA (pH = 3.5) at a potential of 0.2 V for a bare-Pt electrode (a, inset), Ti500 (b), Ti600 (c) and Ti700 (d) electrodes. Frequency range 10: mHz-100 kHz.
To understand the role of TiOx in the catalytic enhancement of FAO, CO was allowed to be chemisorbed from 0.5 M FA at an open circuit potential at the four investigated electrodes for 10 min. Then this adsorbed CO layer was stripped electrochemically in 0.5 M Na2SO4 (pH = 3.5), as shown in Fig. 5. The CO oxidation occurred at 0.72 V with a peak current intensity of ca. 1.8 mA cm−2 at the bare Pt electrode (see Fig. 5a). After the modification with TiOx (Fig. 5b–d), two interesting features appeared. The peak current intensity was lowered dramatically to 0.53 mA cm−2 at the Ti700 electrode; inferring a lower opportunity for CO poisoning, exactly as the data of Fig. 3 informed. This lowering in the peak current density was concurrent with a negative shift (ca. 0.4 V) in the peak potential of CO oxidation, which again highlights the favorable modification in the electronic properties of the Pt surface in the way facilitating the oxidative removal of poisoning CO, in agreement with the data of Fig. 4. Based on the data of Figs. 4 and 5, one can safely assume a potential role for TiOx in facilitating the charge transfer during FAO at Pt surfaces, mitigating the CO adsorption and facilitating the oxidative desorption of CO at relatively much lower potentials that ultimately favor the direct pathway of FAO.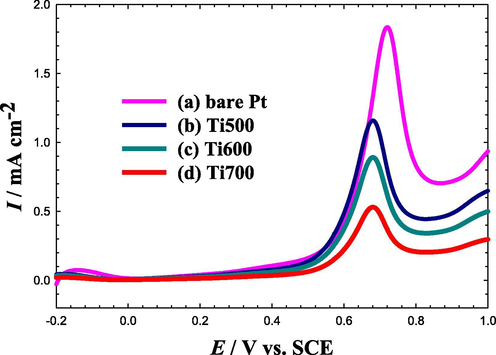
LSVs for oxidative CO stripping obtained at the bare Pt (a), Ti500 (b), Ti600 (c) and Ti700 (d) electrodes in 0.5 M Na2SO4 (pH = 3.5). Potential scan rate: 50 mVs−1. Before measurements, CO was adsorbed from 0.5 M FA at the open circuit potential for 10 min.
3.5 Stability measurements
Furthermore, the catalytic stabilities of the spin-coated Si–TiOx/Pt/TiOx catalysts were inspected and compared with that obtained at the bare Pt electrode. Fig. 6 shows the chronoamperometric (i-t) curves obtained at the bare Pt (Fig. 6a), Ti500 (Fig. 6b), Ti600 (Fig. 6c) and Ti700 (Fig. 6d) catalysts in a 0.3 M aqueous solution of FA (pH = 3.5) at a potential of 0.2 V for 2000 s. A quick inspection of Fig. 6 again sorts ascendingly the bare Pt, Ti500, Ti600 and Ti700 catalysts according to their activities toward FAO. However, a poor catalytic stability is obviously seen at the bare Pt electrode which owned a fast chronic decay in current; in agreement with previous investigations (Mohammad et al., 2018). Fascinatingly, this undesirable decay disappeared at the spin-coated Si–TiOx/Pt/TiOx electrodes (Fig. 6b–d). The maximum stability, in terms of the highest and steady-state current density, was obtained at the Ti700 catalyst. It worth mentioning that the Ti700 catalyst exhibited the highest activity (see Fig. 3), the lowest Rct (Fig. 4) and the minimum CO poisoning degree (Fig. 5). All these findings pointed out the importance of the modification of Pt-based catalysts with the spin-coated TiOx layer in minimizing the CO poisoning and enhancing the charge transfer at the Pt surface.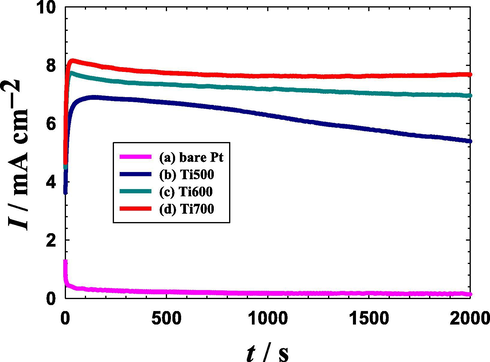
Chronoamperometric (i-t) curves obtained in 0.3 M FA (pH = 3.5) at a potential of 0.2 V for a bare-Pt electrode (a), Ti500 (b), Ti600 (c) and Ti700 (d) electrodes.
To test the catalyst's durability, the Ti700 electrode was morphologically, compositionally and structurally inspected once again after the electrochemical inspections (Figs. 1, 3–6). The SEM image (Fig. 7A), elemental mapping analysis (Fig. 7B), EDS analysis (Fig. 7C) and XRD analysis (Fig. 7D) did not show a significant variation from the data in Fig. 2 (for the same catalyst before the electrochemical investigations). This once again confirms the high stability of the proposed Ti700 catalyst toward the FAO.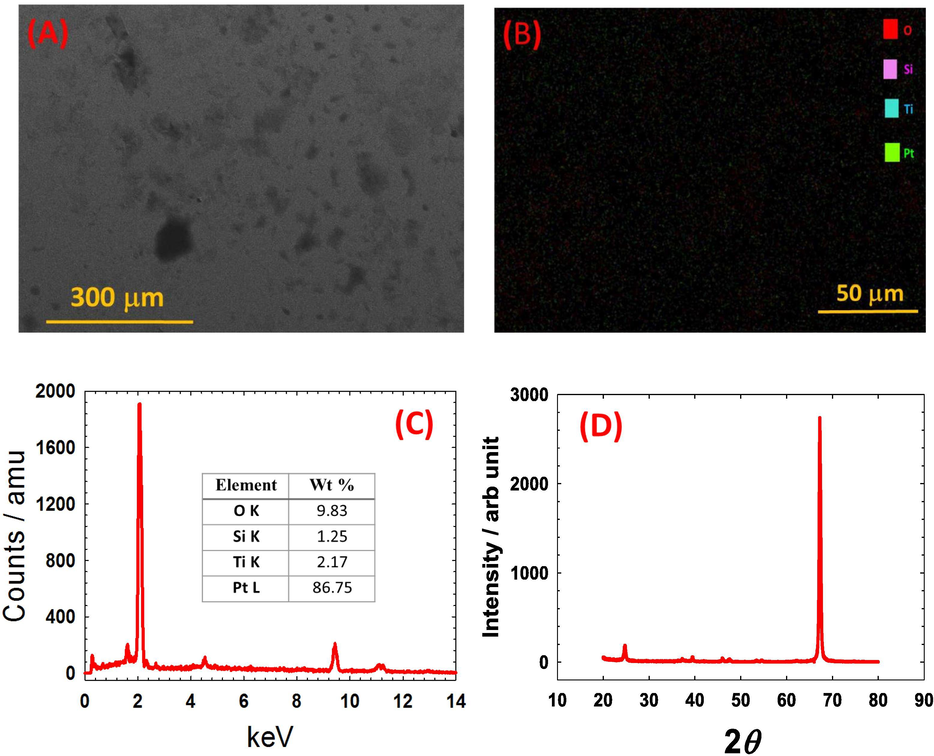
SEM image (A), elemental mapping analysis (B), EDS analysis (C) and XRD analysis (D) of the Ti700 electrode after the electrochemical investigations of Figs. 1, 3–6.
4 Conclusion
A sequential compilation of physical sputtering, spin coating and a post-annealing processing was employed in the synthesis of a Si–TiOx/Pt/TiOx catalyst for the formic acid electro–oxidation (FAO). The annealing temperature could influence the catalytic performance of the catalyst where 700 °C was proved optimum in terms of the observed catalytic efficiency (17-fold more than that of the bare Pt catalyst) and the largest negative shift (∼300 mV) in the onset potential of FAO. The EIS and stability measurements agreed consistently with the electrocatalytic data for the superiority of the Ti700 catalyst. The role of TiOx in the catalytic enhancement appeared in facilitating the charge transfer during FAO perhaps with the vacant d-orbital and the multiple oxidation states of Ti. It might further tailor geometrically the Pt surface against the adsorption of poisoning CO that ultimately favor the direct pathway of FAO.
Funding statement
This research was supported from the General Scientific Research Department at Cairo University (Grant 77/2016). Dr. Al-Akraa appreciate the financial support given from the British University in Egypt (Young Investigator Research Grant/YIRG2017-03).
References
- Conducting polymers inducing catalysis: enhanced formic acid electro-oxidation at a Pt/polyaniline nanocatalyst. Int. J. Hydrogen Energy. 2017;42:11166-11176.
- [Google Scholar]
- Efficient electro-oxidation of formic acid at Pd-MnOx binary nanocatalyst: optimization of deposition strategy. Int. J. Hydrogen Energy. 2017;42:4660-4666.
- [Google Scholar]
- Facile synthesis of a tailored-designed Au/Pt nanoanode for enhanced formic acid, methanol, and ethylene glycol electrooxidation. J. Nanomater.. 2019;2019:1-9.
- [CrossRef] [Google Scholar]
- Advances in direct formic acid fuel cells: Fabrication of efficient Ir/Pd nanocatalysts for formic acid electro-oxidation. Int. J. Electrochem. Sci.. 2015;10:3282-3290.
- [Google Scholar]
- A promising amendment for water splitters: Boosted oxygen evolution at a platinum, titanium oxide and manganese oxide hybrid catalyst. Arab. J. Chem.. 2019;12:897-907.
- [Google Scholar]
- A competent simultaneously co-electrodeposited Pt-MnOx nanocatalyst for enhanced formic acid electro-oxidation. J. Taiwan. Inst. Chem. Eng.. 2018;96:169-175.
- [Google Scholar]
- Design of efficient bimetallic Pt–Au nanoparticle-based anodes for direct formic acid fuel cells. Int. J. Hydrogen Energy. 2019;44:3615-3624.
- [Google Scholar]
- Carbon monoxide poisoning of proton exchange membrane fuel cells. Int. J. Energy Res.. 2001;25:695-713.
- [Google Scholar]
- Future development of the electricity systems with distributed generation. Energy. 2009;34:377-383.
- [Google Scholar]
- Efficient dehydrogenation of formic acid using an iron catalyst. Science. 2011;333:1733.
- [Google Scholar]
- Scientific aspects of polymer electrolyte fuel cell durability and degradation. Chem. Rev.. 2007;107:3904-3951.
- [Google Scholar]
- The CO poisoning mechanism of the hydrogen oxidation reaction in proton exchange membrane fuel cells. J. Electrochem. Soc.. 2002;149:A748-A753.
- [Google Scholar]
- An ambient aqueous synthesis for highly dispersed and active Pd/C catalyst for formic acid electro-oxidation. J. Power Sources. 2010;195:7246-7249.
- [Google Scholar]
- One-step growth of 3–5nm diameter palladium electrocatalyst in a carbon nanoparticle–chitosan host and characterization for formic acid oxidation. Electrochim. Acta. 2010;55:6601-6610.
- [Google Scholar]
- Electrocatalyst approaches and challenges for automotive fuel cells. Nature. 2012;486:43-51.
- [Google Scholar]
- Nature of CO adsorption during H2 oxidation in relation to modeling for CO poisoning of a fuel cell anode. J. Electrochem. Soc.. 1987;134:3021-3026.
- [Google Scholar]
- Hydrogen futures: toward a sustainable energy system. Int. J. Hydrogen Energy. 2002;27:235-264.
- [Google Scholar]
- Hydrogen from renewable electricity: an international review of power-to-gas pilot plants for stationary applications. Int. J. Hydrogen Energy. 2013;38:2039-2061.
- [Google Scholar]
- A core-shell nanoporous Pt-Cu catalyst with tunable composition and high catalytic activity. Adv. Funct. Mater.. 2013;23:4156-4162.
- [Google Scholar]
- A new approach to the problem of carbon monoxide poisoning in fuel cells operating at low temperatures. J. Electrochem. Soc.. 1988;135:2651-2652.
- [Google Scholar]
- Breakthroughs in hydrogen storage\-formic acid as a sustainable storage material for hydrogen. Chem. Sus. Chem.. 2008;1:805-808.
- [Google Scholar]
- The growth and structure of titanium oxide films on Pt(111) investigated by LEED, XPS, ISS, and STM. Electrochim. Acta. 2007;52:2492-2497.
- [Google Scholar]
- Unusually active palladium-based catalysts for the electrooxidation of formic acid. J. Power Sources. 2006;157:78-84.
- [Google Scholar]
- Dendritic defect-rich palladium–copper–cobalt nanoalloys as robust multifunctional non-platinum electrocatalysts for fuel cells. Nat. Commun.. 2018;9:3702.
- [Google Scholar]
- Light-induced deposition of Pd-based nanoalloy on TiO2 nanotubes for formic acid electrooxidation. J. Taiwan. Inst. Chem. Eng.. 2017;76:109-114.
- [Google Scholar]
- Kinetic study of formic acid oxidation on carbon-supported platinum electrocatalyst. J. Electroanal. Chem.. 2005;581:294-302.
- [Google Scholar]
- Studies in surface science and catalysis. Stud. Surf. Sci. Catal.. 1997;112:359-366.
- [Google Scholar]
- Surface science studies of model fuel cell electrocatalysts. Surf. Sci. Rep.. 2002;45:117-229.
- [Google Scholar]
- Superior electrocatalysis of formic acid electro-oxidation on a platinum, gold and manganese oxide nanoparticle-based ternary catalyst. Int. J. Hydrogen Energy. 2018;43:139-149.
- [Google Scholar]
- Development of spin-coated Si/TiOx/Pt/TiOx electrodes for the electrochemical ozone production. Appl. Surf. Sci.. 2009;255:8458-8463.
- [Google Scholar]
- Superior electrocatalysis of spin-coated titanium oxide electrodes for the electrochemical ozone production. Chem. Lett.. 2007;36:1046-1047.
- [Google Scholar]
- Use of Electrochemical Impedance Spectroscopy (EIS) to monitoring the corrosion of reinforced concrete. Rev. IBRACON Estrut. Mater.. 2015;8:529-546.
- [Google Scholar]
- TiO2 nanotubes: synthesis and applications. Angew. Chem. Int. Ed.. 2011;50:2904-2939.
- [Google Scholar]
- Tailoring the composition of ultrathin, ternary alloy PtRuFe nanowires for the methanol oxidation reaction and formic acid oxidation reaction. Energy. Environ. Sci.. 2015;8:350-363.
- [Google Scholar]
- The hydrogen economy, fuel cells, and electric cars. Technol. Soc.. 2003;25:455-476.
- [Google Scholar]
- TiO2 nanotubes promoting Pt/C catalysts for ethanol electro-oxidation in acidic media. J. Power Sources. 2007;170:50-54.
- [Google Scholar]
- Materials for fuel-cell technologies. Materials for Sustainable Energy 2011:224-231.
- [Google Scholar]
- Real surface area measurements in electrochemistry. Pure. Appl. Chem.. 1991;63:711-734.
- [Google Scholar]
- The influence of the crystal structure of TiO2 support material on Pd catalysts for formic acid electrooxidation. Electrochim. Acta. 2010;55:851-856.
- [Google Scholar]
- A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Appl. Energy. 2011;88:981-1007.
- [Google Scholar]
- Electrode processes at gas|salt|Pd nanoparticle|glassy carbon electrode contacts: salt effects on the oxidation of formic acid vapor and the oxidation of hydrogen. New J. Chem.. 2011;35:1855-1860.
- [Google Scholar]
- Electrochemical preparation, characterization of molybdenum-oxide/platinum binary catalysts and its application to oxygen reduction reaction in weakly acidic medium. Electrochim. Acta. 2015;151:72-80.
- [Google Scholar]
- The behavior of palladium catalysts in direct formic acid fuel cells. Electrochim. Acta. 2005;139:15-20.
- [Google Scholar]




