

Translate this page into:
A suitable drug structure for interaction with SARS‐CoV‐2 main protease between boceprevir, masitinib and rupintrivir; a molecular dynamics study
⁎Corresponding author. m.yoosefian@kgut.ac.ir (Mehdi Yoosefian)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
In recent years, more than 200 countries of the world have faced a health crisis due to the epidemiological disease of COVID-19 caused by the SARS-CoV-2 virus. It had a huge impact on the world economy and the global health sector. Researchers are studying the design and discovery of drugs that can inhibit SARS‐CoV‐2. The main protease of SARS‐CoV‐2 is an attractive target for the study of antiviral drugs against coronavirus diseases. According to the docking results, binding energy for boceprevir, masitinib and rupintrivir with CMP are −10.80, −9.39, and −9.51 kcal/mol respectively. Also, for all investigated systems, van der Waals and electrostatic interactions are quite favorable for binding the drugs to SARS-CoV-2 coronavirus main protease, indicating confirmation of the complex stability.
Keywords
SARS‐CoV‐2 main protease
Boceprevir
Masitinib
Rupintrivir
Protease inhibitor

1 Introduction
An acute respiratory illness (COVID-19) that accelerated across the world has been found to be caused by a completely unique strain of coronaviruses (SARS-CoV-2) (Hu et al., 2021). In 2019, a serious respiratory infectious disease appeared in China and quickly in other countries, such as Spain, the United States of America, Britain, Russia, Brazil, and France. The disease continued to worsen until January 30, 2020, when the World Health Organization (WHO) named the infectious disease the new 2019 coronavirus (2019-nCoV) (World Health Organization, 2021). On January 30, 2020, the WHO announced the prevalence of the Coronavirus in a globalized dangerous situation. And on March 11, 2020, the WHO described Covid-19 as an epidemic. No specific antiviral drug is currently available for the treatment or prevention of coronavirus diseases, so there is still a serious need for treatment and prevention against coronavirus infection (Hafeez et al., 2020). Covid-19 is a highly contagious viral disease with a very high transmission rate, and given the dangers and lethality of the virus, the best way to control the disease is to control the disease and prevent new cases, or in other words, cut the transmission chain. As of March 23, 2022, the disease has spread to more than 474,424,710 confirmed worldwide (https://coronavirus.jhu.edu/). The structure of the newest type of coronavirus has a single-stranded RNAs genome with positive polarity and is also surrounded by a lipid bilayer membrane with a diameter of approximately 50–200 nm (Tolksdorf et al., 2021; Ewert et al., 2021; Dhar, 2022). The viral particles of the coronaviruses have four important structural proteins: spike (S) proteins, envelope (E), membrane (M), and nucleocapsid (N) proteins (Satarker and Nampoothiri, 2020; Yadav et al., 2021). Protein N is located in the structure of the virus genome, and the other three proteins together form the coat of the virus (Boson et al., 2021). Of these four proteins, the most important is the protein (Spike), which is located on the membrane of the virus. S membrane protein is a glycoprotein and forms homotrimers on the viral surface. Protein E may be a passing protein that's important to pathogenesis. Protein M is a small membrane protein with three passing areas that may be important for virion shape protein N plays a very important role in the packaging of RNA and also the release of viral particles. Several non-structural proteins have an enzyme activity such as protease and RNA polymerase activity with RNA, so an important and effective strategy for antiviral development against SARS–COV-2 is blocking enzymatic activity (Li and Kang, 2020). The proliferation cycle of SARS-COV-2 is based on the binding of the S protein to the angiotensin 2 receptor at the human cell surface, which changes the cell membrane and the viral membrane with the change of the protein spike S (Delpino and Quarleri, 2020; Gheblawi et al., 2020). After this stage, viral genes can enter the host cell and cause more virus replication. Human infection with SARS-COV-2 and its proliferation in human cells depends on different proteins, including (i) structural proteins corresponding to the glycosylated spike (S) protein, that mediates host cell receptor recognition and host cell entry, and induces host immune responses; and (ii) non-structural proteins such as polymer-dependent RNA enzyme (RdRp), (iii) main proteolytic enzyme Mpro. Mpro protease plays a key role in transcription and replication of the genome of coronavirus 2 (SARS-Cov-2). Understanding proteases to identify and develop specific antiviral drugs can effectively prevent or treat Covid-19 (Luan et al., 2020; Jankun, 2020). One of the important points about finding drugs is to use the available drug screening method with the help of software and predefined algorithms in the form of computer-aided drug design (Yoosefian et al., 2022; Yoosefian et al., 2018; Yu and MacKerell, 2017; Ziaadini et al., 2019; Afzali et al., 2015). The mechanism of action and knowledge of the qualitative nature of the interactions, in addition to the quantitative nature and amounts of energy exchanged, is important in deciding whether a drug combination is effective for a specific purpose.
To provide antiviral drugs that can be quickly used to fight the COVID-19 pandemic, a study was conducted to identify existing drugs that could potentially be used as inhibitors of the SARS-CoV-2 virus that causes the disease COVID-19. Eight HCV drugs (Simeprevir, grazoprevir, Telaprevir, Paritaprevir, Narlaprevir, Vaniprevir, Boceprevir, Asunaprevir) have been identified to inhibit viral replication in human Vero and/or 293T cells expressing the ACE2 receptor (Bafna et al., 2021). Another study suggests the reuse of three chemicals, dextromethorphan, prednisolone, and dexamethasone, as anti-covid agents. The tertiary structure of the major protease of Coronavirus (Mpro) with PDB ID 6LU7 was used as the target protein in this analysis. Molecular docking and further dynamics study revealed their synergistic effect against the protease protein of COVID-19 (Sarkar and Sen, 2022). Hydroxychloroquine (HCQ) and remdesivir were effective in treating COVID-19 patients. Docking and molecular dynamics (MD) simulations of HCQ and remdesivir with Mpro protein have been performed, which provided promising results for the inhibition of Mpro protein in SARS-CoV-2. On the basis of results obtained we designed structurally modified 18 novel derivatives of HCQ, remdesivir and tetrahydrocannabinol (THC) and carried out docking studies of all the derivatives. Further, molecular dynamics simulation of one derivative of HCQ and one derivative of tetrahydrocannabinol showing excellent docking score was performed along with the respective parent molecules. The two derivatives gave excellent docking score and higher stability than the parent molecule as validated with molecular dynamics (MD) simulation for the binding affinities towards Mpro of SARS-CoV-2 thus represented as strong inhibitors at very low concentration. (Mishra et al., 2021). In this study, with the help of molecular dynamics simulations, we investigated three drugs as inhibitors of SARS-CoV-2 coronavirus main protease (CMP), which are:
-
Boceprevir: a protease inhibitor used to treat hepatitis caused by hepatitis C infection (HCV) genotype 1. It binds to the HCV non-structural protein 3 dynamic site (Rizza et al., 2011; Bacon et al., 2011).
-
Masitinib: a tyrosine kinase inhibitor that inhibits the stem-cell factor receptor, KIT (kinase inhibitor targeting) in both biochemical and cellular assays (Dubreuil et al., 2009; Marech et al., 2014).
-
Rupintrivir: an enterovirus protease inhibitor, which has been reported to have anti–human norovirus activity in vitro. It is a peptidomimetic inhibitor of rhinovirus 3Cpro and an analogue (AG7404) have advanced to clinical trials, but failed due to limited efficacy (Rocha-Pereira et al., 2014; Hung et al., 2011).
2 Method
2.1 Molecular dynamics simulations:
The complex structure of CMP and the candidate drugs (i) boceprevir (PDB code: 7C6S, BCV/CMP), masitinib (PDB code: 7JU7, MSB/CMP) and rupintrivir (PDB code: 7L8J, RPV/CMP) were prepared to simulate the molecular dynamics simulation. The crystal structure of SARS-CoV-2 main protease in complex with a ligand has been presented in Fig. 1A. Furthermore, the active sites for each of these three complexes i.e. BCV/CMP, MSB/CMP and RPV/CMP are shown in Fig. 1B–D. In this region of an enzyme ligand molecules bind and undergo a chemical reaction.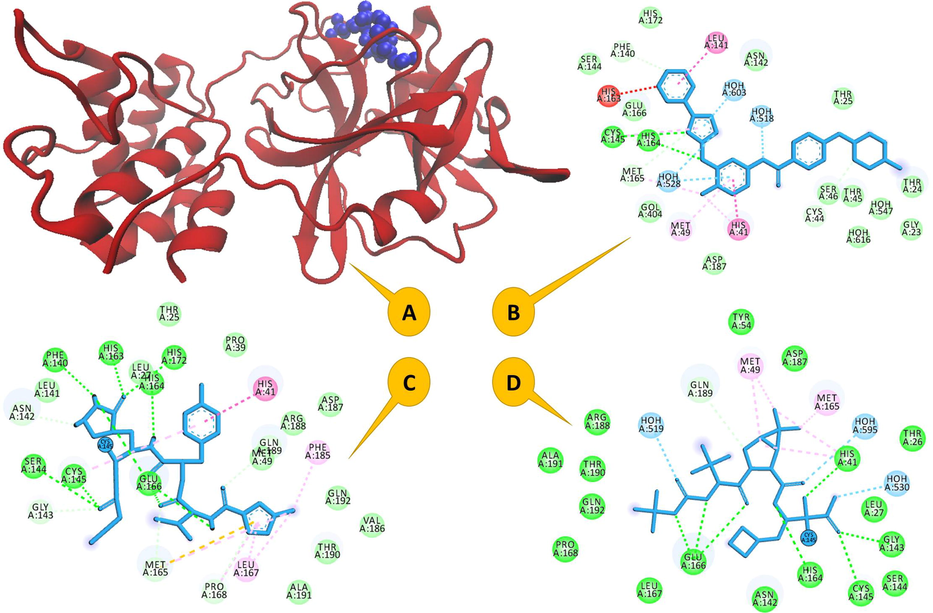
(A) The crystal structure of SARS-CoV-2 main protease in complex with a drug (drug in blue and CMP in red), the important resediues in the active site of (B) MSB/CMP complex, (C) RPV/CMP complex and (D) BCV/CMP complex.
ITP and PDB files of drugs (BCV, MSB and RPV) were generated based on Charm 36 force field. Calculations of molecular dynamics simulation were performed using GROMACS program version 1 (2019) (Abraham et al., 2015). VMD was used to visualize the whole process (Humphrey et al., 1996). First, the protein was placed in the center of the simulated box; then the drug molecule was added to the simulated box. The distance between the protein-drug complex and the simulation box wall was determined as 1 nm. TIP3P was chosen as the water model in this force field, and chlorine ions were added to neutralize the net load of the system. The algorithm of “steepest” was used for integration in 50,000 steps to perform simulations in the system neutralization steps and minimize energy. Before simulating molecular dynamics, temperature equilibration was achieved for 10 ns at 300 K using a V-rescale thermostat during NVT. In addition, the pressure was balanced for 10 ns using NPT and 1 bar using parrinello-rahman barostat. In this process, the LINCS algorithm, along with the Particle Mesh Ewald (PME) method, was used to calculate long-range electrostatic forces (Hess et al., 1997). During the simulation, the Fourier grid spacing and Coulomb radius were set at 0.16 and 1.2 nm, respectively, and the van der Waals interactions were limited to 1.2 nm. The MD trajectories were saved at every 10 ps for energy stabilization, and the root means square deviation (RMSD) calculations. Furthermore, the equations of motion were integrated using the leap-frog algorithm, with a time step of 2 fs and a total time of 100 ns.
3 Results and discussion
3.1 Molecular dynamics simulation
In this study, in order to evaluate the stability and equilibrium of the system, various parameters such as potential energy and total system energy are calculated and the goal of minimizing energy is to bring the system to the most stable state. Potential energy and total energy for all three systems (BCV/CMP, MSB/CMP and RPV/CMP) was calculated and shown in Fig. 2.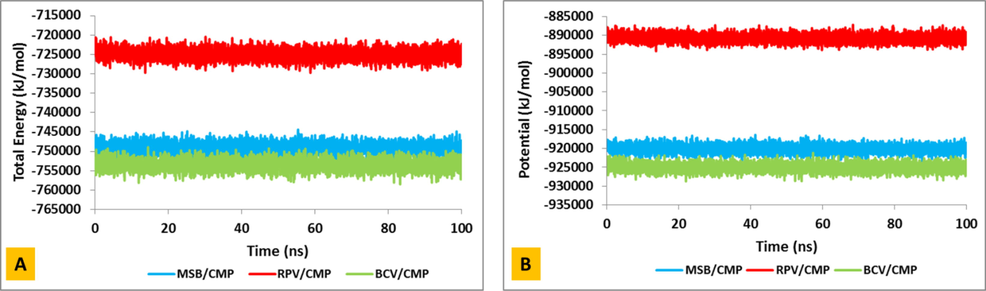
Potential and total energies of the BCV/CMP (green), MSB/CMP (blue) and RPV/CMP (red) complexes during 100 ns simulation.
RMSD is the most reliable method of molecular dynamics simulation analysis. It is also used to analyze time-dependent particles, especially to determine the stability of the structure on a time scale and the degree of deviation from the original coordinates during simulation. Deviation from the original coordinates indicates an imbalance of the system during the simulation. To investigate the exact structural behavior and test the stability of the inhibitors, the RMSD of the systems was calculated within 100 ns. As shown in Fig. 3A, the average values of RMSD are 6 for MSB/CMP and RPV/CMP systems and 6.4 for BCV/CMP system indicating the stability and equilibrium of the systems during simulation.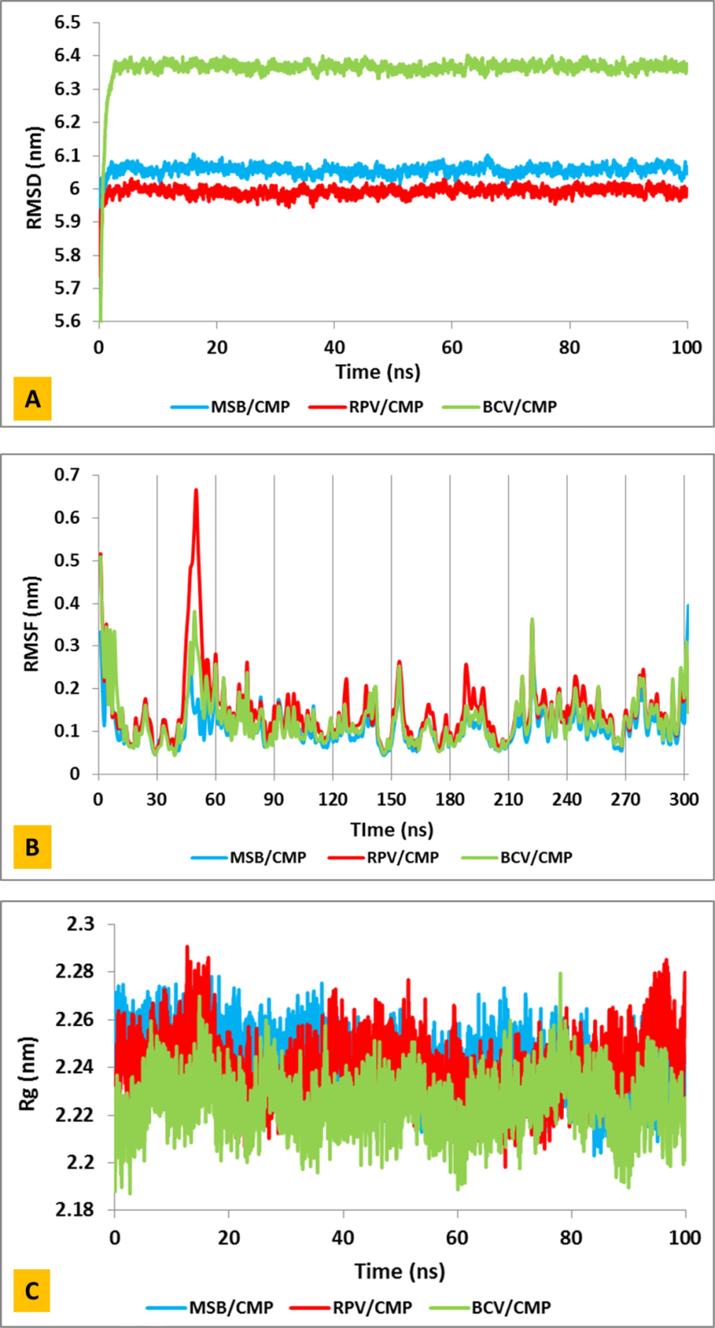
(A) Modifications RMSD, (B) Root-mean-square fluctuation (RMSF) plots and (C) Radius of gyration (Rg) of Covid-19 virus main protease complexed by Masitinib (blue) and Boceprevir (green) and Rupinrivir (red) system during 100 ns simulation.
The amino acid motions of CMP were analyzed using the square root of the fluctuation squares for all three systems (BCV/CMP, MSB/CMP and RPV/CMP). This calculation is a measure of system flexibility. Fig. 3B of the RMSF diagram shows that many residues are below 0.2 nm, indicating that the residues were stable during molecular dynamics simulation.
Radius of gyration (Rg) is one of the useful analyzes obtained from the results of molecular dynamics simulations and has been selected as the first criterion to study the three-dimensional structure of the studied systems. Because it can be directly compared to laboratory results, for large molecules such as proteins, the radius of gyration is a good measure of the folding of proteins. The amount of Rg in the secondary structure of the protein is always constant and any change in this structure leads to a change in the amount of Rg. As shown in Fig. 3C, the gyration radius fluctuates over 100 ns with an almost horizontal slope around the mean value of 2.24 ± 0.02 until the end of the simulation. The small standard deviation, i.e. the small amplitude of the oscillation around the mean value, indicates the high stability of the second and third protein structures in all three systems, within 100 ns of the simulation.
In order to investigate the interaction between BCV, MSB and RPV drugs and CMP, the relative distances between the CMP and drugs were measured. Fig. 4A shows the calculated simulations of the distances between CMP and drugs during 100 ns. The distance between CMP with inhibitors shows normal fluctuations. The average distance for BCV/CMP, MSB/CMP and RPV/CMP systems are 1.88, 2.04 and 1.93 Å, respectively. The results of this analysis show that among the three studied drugs, drug BCV has a smaller distance with the CMP and shows that it is better placed in the active site of the CMP.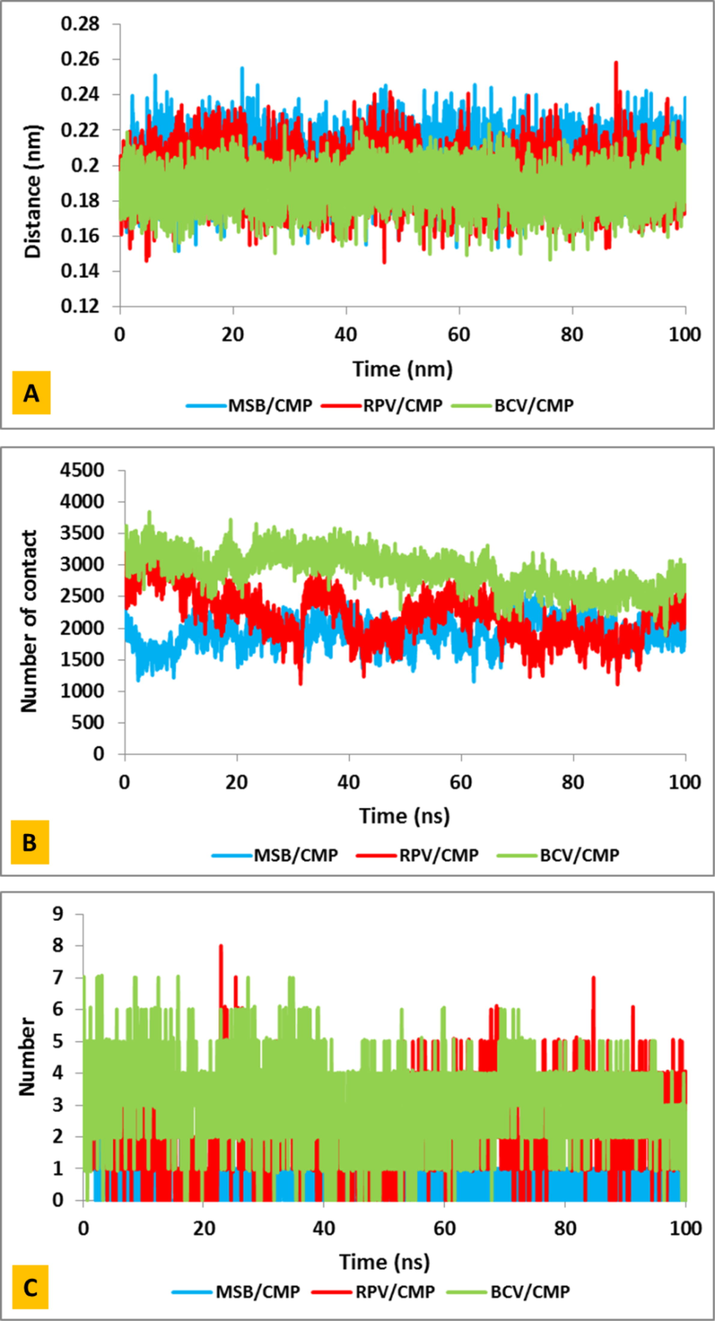
(A) distance, (B) number of contacts and (C) number of hydrogen bond between Covid-19 virus main protease and Masitinib (blue) and Boceprevir (green) and Rupinrivir (red) drugs during 100 ns simulation.
To better understand of the interaction between CMP and investigated drugs, the number of contact between CMP and drugs was calculated for all three systems (BCV/CMP, MSB/CMP and RPV/CMP). Fig. 4B shows the close relationship and interaction between these three complexes over the simulation time. According to the results, the average number of contacts for BCV/CMP is 2922 and for RPV/CMP 2221 and finally the average number of contacts for MSB/CMP is 1939 and these results are consistent with the distance data.
Analysis of hydrogen bonds provides useful information about protein–ligand interactions. Hydrogen bonding, interaction involving a hydrogen atom located between a pair of other atoms having a high affinity for electrons; such a bond is weaker than an ionic bond or covalent bond but stronger than van der Waals forces (Yoosefian et al., 2011; Raissi et al., 2010; Mirhaji et al., 2018; Afzali et al., 2014; Ghanbarian et al., 2013). Hydrogen bonds contribute favorably to protein stability and the contribution of hydrogen bonds to protein stability is strongly context dependent. Hydrogen bonds by side chains and peptide groups make similar contributions to protein stability. The greater the number of hydrogen bonds, the stronger the bond between BCV/CMP, MSB/CMP and RPV/CMP protein–ligand complexes. As shown in Fig. 4C, the behavior of the hydrogen bonds between these three complexes is quite different. The average hydrogen bonds number obtained in BCV/CMP, MSB/CMP and RPV/CMP complexes are 3.19, 0.66 and 2.08, respectively. The higher number of hydrogen bonds in the BCV/CMP complex shows that there is a good connection between the BCV drug and the CMP, and it can be concluded that among these three drugs, BCV is the best candidate for inhibiting the CMP.
PCA analysis helps us to better understand the behavior of proteins when binding to ligands. The PCA representation is a reflection of the specific vector of the location of each carbon atom during the simulation. Fig. 5A shows the eigenvalues versus the eigenvectors and shows that the three PCAs initially cover the highest motion reflectance, and therefore the two-dimensional shape of the PCA diagram for these systems is a good representation of protein motions. As shown in Fig. 5B–D, the BCV diagram, which takes up less space, shows that the protein moves less when bound to the compound, and therefore takes up less space. MSB and RPV also take up more space than MSB. This also suggests that BCV carbon atoms tend to reduce protein compression when bond to this ligand.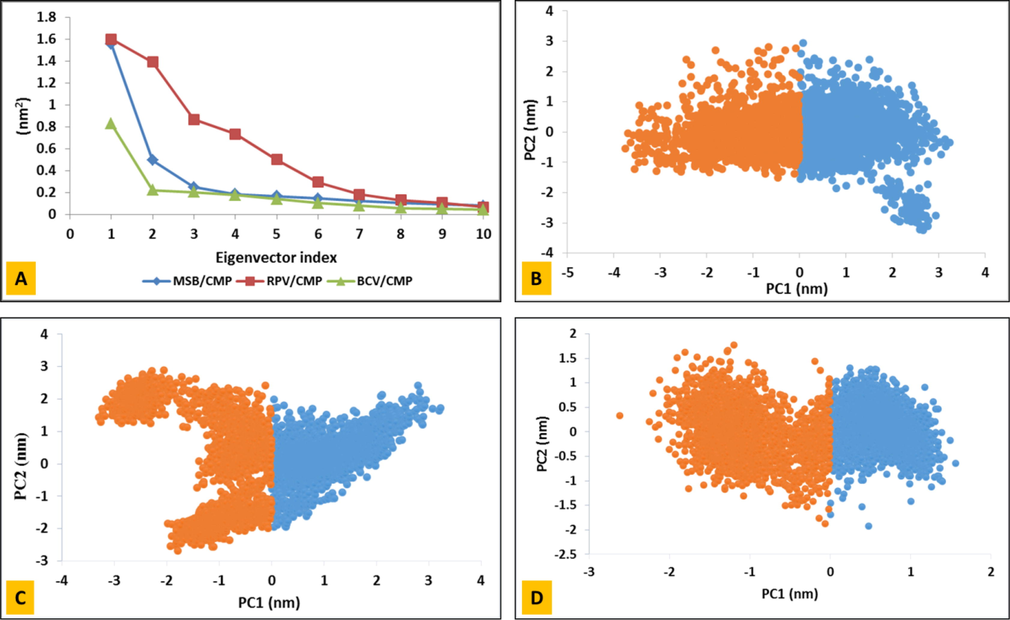
Principal component analysis (PCA) of SARS-CoV-2 main protease in complex with a drug was performed using gmx covar and gmx anaeig utility toolkits of GROMACS. (A) The eigenvalues plotted against the corresponding eigenvector indices obtained from the Cα covariance matrix constructed from the 100 ns MD trajectory. Projection of the motion of the structures of the backbone atoms of (B) MSB/CMP (C) RPV/CMP and (D) BCV/CMP complexes along the first two principal eigenvectors (EV1 and EV2).
As a final analysis, free binding energies were investigated at molecular mechanics/Poisson Boltzmann surface area (MM-PBSA) methodology implemented in the g_mmpbsa apparatus of GROMACS was utilized in Poisson Boltzmann (MM-PBSA) (Kumari et al., 2014). After the complexes are fully balanced, 10 frames are separated from the last 20 ns of molecular dynamics simulation and these frames are used for the free binding energies calculation. As presented in Table 1, for all investigated systems BCV/CMP, MSB/CMP and RPV/CMP, van der Waals and electrostatic interactions are quite favorable for binding the BCV, MSB and RPV to CMP, indicating confirmation of the complex stability.
RPV/CMP
BCV/CMP
MSB/CMP
Van der Waals energy
−185.45
−192.77
−166.58
Electrostatic energy
−29.39
−15.67
−49.93
Polar solvation energy
148.45
126.03
180.62
SASA energy
−21.39
−19.57
−21.26
Binding energy
−87.77
−101.98
−57.14
3.2 ADMET result
Pharmacokinetic and toxicological data were analyzed and supported by the server from the ADMET lab 2.0 website (https://admetmesh.scbdd.com/) (Xiong et al., 2021) and (https://tox-new.charite.de/protox_II). In the present study, ADMET properties and biological activities of different drugs are observed; all three drugs are probably metabolized by CYP3A4. MSB and BCV have the ability to act as P-glycoprotein (Pgp) substrate; also MSB has the ability to cross the blood–brain barrier (BBB) (see Table2). The toxicity profiles of compounds such as their ability to cause liver damage, mutation and cancer, cytotoxicity and immunotoxicity were predicted. Table 4 predicts the toxicity of compounds in silico. LD stands for “Lethal Dose”. LD50 is the amount of a material, given all at once, which causes the death of 50% (one half) of a group of test animals. As seen in the Table 3, the LD50 for BCV, MSB and RPV are 1500, 1000 and 4000 (mg/kg), respectively. The higher the LD50, the less toxic the substance. Also, the results show that the hepatotoxicity of MSB is active.
DRUG
LogP
LogS
BBB
Pgp-inhibitor
Pgp-substrate
CYP1A2 inhibitor
CYP2C19 inhibitor
CYP2C9 inhibitor
CYP2D6 inhibitor
CYP3A4 inhibitor
BOSEPERIVIR
3.979
−5.066
0.328
0.994
0.991
0.003
0.296
0.206
0.263
0.891
MASITINIB
4.768
−4.185
0.854
1
0.98
0.52
0.658
0.581
0.835
0.906
RUPINTRIVIR
3.28
−4.189
0.236
0.291
0.985
0.035
0.394
0.843
0.01
0.931
DRUG
LD50
CLASS
Hepatotoxicity
Carcinogenicity
Immunotoxicity
Mutagenicity
Cytotoxicity
BOSEPERIVIR
1500
4
Inactive
Inactive
Inactive
Inactive
Inactive
MASITINIB
1000
4
Active
Inactive
Inactive
Inactive
Inactive
RUPINTRIVIR
4000
5
Inactive
Inactive
Inactive
Inactive
Inactive
DRUG
Target
PDB
Ki
Binding Energy
Boceprevir
HCV protease
3SV6
912.24 nM
−8.24
Masitinib
tyrosine kinase
5MQL
16.08 nM
−10.63
Rupintrivir
3C protease
7L8H
1.63 mM
−7.9
Boceprevir
CMP
7C6S
12.08 nM
−10.8
Masitinib
CMP
7JU7
130.36 nM
−9.39
Rupintrivir
CMP
7L8J
107.47 nM
−9.51
3.3 Molecular docking results
In this section, all docking simulations were analyzed using AutoDockTools. The atomic coordinates for the best scoring conformation obtained for each drug-protein complex, were stored in PDB format for analysis. The interactions of these protein–ligand complexes were mapped using Ligplot, a fully automated protein–ligand interaction profiler, for comparative analysis of inhibitor binding simulations. Docking simulations were used to investigate the affinity of BCV, MSB and RPV for their common targets compared to the CMP (see Table 4). Docking scores for BCV, MSB and RPV with CMP are −10.80, −9.39, and −9.51 kcal/mol respectively. According to Fig. 1, more than 4 hydrogen bonds between BCV and RPV with CMP have also interacted with two important residues of the active site (Her 41, cys145). But MSB did not make good interactions with the important residues of the active site of CMP and no favorable binding energy was observed.
4 Conclusions
In this study, the ability of three drugs boceprevir, rupintrivir and masitinib to inhibit the SARS‐CoV‐2 main protease was investigated so that it can be used as a potential drug for the treatment of Coronavirus disease. RMSD results show that all systems are in perfect equilibrium. Furthermore, analyze of RMSF structural parameters and gyrus radius for BCV, MSB and RPV drugs with CMP showed that the protease has a stable structure with high strength. The results showed that all three drugs have the ability to inhibit the CMP. And among the three studied drugs, drug BCV showed the best performance. Overall the docking results show that among these three drugs selected to interact with CMP, the two drugs boceprevir and rupintrivir have a higher docking score may be a successful treatment against COVID-19. MM-PBSA analysis show that, among the three drugs studied, drug BCV has the highest binding energy, although the other two drugs also have good binding energies.
Funding
No funding was received for conducting this study.
CRediT authorship contribution statement
Mehdi Yoosefian: Methodology, Visualization, Writing – review & editing. Razieh Dashti: . Mohamad Mahani: . Leila Montazer: . Amirabbas Mir: .
Acknowledgements
The authors wish to thank Graduate University of Advanced Technology, Kerman, Iran, for their support.
Author contribution
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Razieh Dashti. The first draft of the manuscript was written by Razieh Dashti and all authors commented on previous versions of the manuscript. Methodology, visualization, writing - review & editing were done by Mehdi Yoosefian. All authors read and approved the final manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19-25.
- [Google Scholar]
- Determination of trace amounts of zirconium in real samples after microwave digestion and ternary complex dispersive liquid–liquid microextraction. Environ. Monit. Assess.. 2014;186(6):3523-3529.
- [Google Scholar]
- Determination of trace amounts of antimony (III) based on differential pulse voltammetric method with multi-walled carbon-nanotube-modified carbon paste electrode. Ionics. 2015;21(2):565-570.
- [Google Scholar]
- Boceprevir for previously treated chronic HCV genotype 1 infection. N. Engl. J. Med.. 2011;364(13):1207-1217.
- [Google Scholar]
- Hepatitis C virus drugs that inhibit SARS-CoV-2 papain-like protease synergize with remdesivir to suppress viral replication in cell culture. Cell Rep.. 2021;35(7):109133
- [Google Scholar]
- The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem.. 2021;296
- [Google Scholar]
- SARS-CoV-2 pathogenesis: imbalance in the renin-angiotensin system favors lung fibrosis. Front. Cell. Infect. Microbiol.. 2020;10:340.
- [Google Scholar]
- Diagnostic assay and technology advancement for detecting SARS-CoV-2 infections causing the COVID-19 pandemic. Anal. Bioanal. Chem. 2022:1-32.
- [Google Scholar]
- Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS One. 2009;4(9):e7258.
- [Google Scholar]
- Cationic liposomes as vectors for nucleic acid and hydrophobic drug therapeutics. Pharmaceutics. 2021;13(9):1365.
- [Google Scholar]
- Displacement-Dispersive Liquid-Liquid Microextraction Based on Solidification of Floating Organic Drop of Trace Amounts of Palladium in Water and Road Dust Samples Prior to Graphite Furnace Atomic Absorption Spectrometry Determination. J. AOAC Int.. 2013;96(4):880-886.
- [Google Scholar]
- Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ. Res.. 2020;126(10):1456-1474.
- [Google Scholar]
- A review of COVID-19 (Coronavirus Disease-2019) diagnosis, treatments and prevention. Ejmo. 2020;4(2):116-125.
- [Google Scholar]
- LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem.. 1997;18(12):1463-1472.
- [Google Scholar]
- Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol.. 2021;19(3):141-154.
- [Google Scholar]
- Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis.. 2011;203(12):1784-1790.
- [Google Scholar]
- COVID-19 pandemic; transmembrane protease serine 2 (TMPRSS2) inhibitors as potential drugs, Translation: The University of Toledo. J. Med. Sci.. 2020;7:1-5.
- [Google Scholar]
- g_mmpbsa□ A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model.. 2014;54(7):1951-1962.
- [Google Scholar]
- Progress in developing inhibitors of SARS-CoV-2 3C-like protease. Microorganisms. 2020;8(8):1250.
- [Google Scholar]
- Targeting proteases for treating COVID-19. J. Proteome Res.. 2020;19(11):4316-4326.
- [Google Scholar]
- Masitinib (AB1010), from canine tumor model to human clinical development: where we are? Crit. Rev. Oncol. Hematol.. 2014;91(1):98-111.
- [Google Scholar]
- Boron nitride nanotubes as a nanotransporter for anti-cancer docetaxel drug in water/ethanol solution. J. Mol. Liq.. 2018;271:151-156.
- [Google Scholar]
- Structurally modified compounds of hydroxychloroquine, remdesivir and tetrahydrocannabinol against main protease of SARS-CoV-2, a possible hope for COVID-19: Docking and molecular dynamics simulation studies. J. Mol. Liq.. 2021;335:116185
- [Google Scholar]
- The effects of substitutions on structure, electron density, resonance and intramolecular hydrogen bonding strength in 3-mercapto-propenethial. J. Mol. Struct. (Thoechem). 2010;960(1):1-9.
- [Google Scholar]
- The enterovirus protease inhibitor rupintrivir exerts cross-genotypic anti-norovirus activity and clears cells from the norovirus replicon. Antimicrob. Agents Chemother.. 2014;58(8):4675-4681.
- [Google Scholar]
- In silico screening predicts common cold drug Dextromethorphan along with Prednisolone and Dexamethasone can be effective against novel Coronavirus disease (COVID-19) J. Biomol. Struct. Dyn.. 2022;40(8):3706-3710.
- [Google Scholar]
- Structural proteins in severe acute respiratory syndrome coronavirus-2. Archiv. Med. Res.. 2020;51(6):482-491.
- [Google Scholar]
- Inhibition of SARS-CoV-2 replication by a small interfering RNA targeting the leader sequence. Viruses. 2021;13(10):2030.
- [Google Scholar]
- World Health Organization, 2021. Coronavirus disease (COVID-19). https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed July 11, 2021).
- ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res.. 2021;49(W1):W5-W14.
- [Google Scholar]
- Role of structural and non-structural proteins and therapeutic targets of SARS-CoV-2 for COVID-19. Cells. 2021;10(4):821.
- [Google Scholar]
- Substituent effect on structure, electron density, and intramolecular hydrogen bonding in nitroso-oxime methane. Int. J. Quantum Chem. 2011;111(14):3505-3516.
- [Google Scholar]
- Nanofilter platform based on functionalized carbon nanotubes for adsorption and elimination of Acrolein, a toxicant in cigarette smoke. Appl. Surf. Sci.. 2018;444:598-603.
- [Google Scholar]
- In silico evaluation of atazanavir as a potential HIV main protease inhibitor and its comparison with new designed analogs. Comput. Biol. Med. 2022:105523.
- [Google Scholar]
- Photocatalytic degradation of methylene blue from aqueous solution using Fe3O4@ SiO2@ CeO2 core-shell magnetic nanostructure as an effective catalyst. Adv. Environ. Technol.. 2019;5(2):127-132.
- [Google Scholar]




