

Translate this page into:
Advances in synthesis and biological activities of quinazoline scaffold analogues: A review
⁎Corresponding author at: School of Chemistry & Physics, University of KwaZulu-Natal, Westville Campus, P Bag X 54001, Durban 4000, South Africa (S.B. Jonnalagadda). Department of Chemistry, Acharya Nagarjuna University, Guntur, A.P, India (H.B. Bollikolla). dr.b.haribabu@gmail.com (Hari Babu Bollikolla), Jonnalagaddas@ukzn.ac.za (Sreekantha Babu Jonnalagadda)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
This review comprises recently investigated synthetic protocols towards the construction of Quinazolines and its derivatives (from 2017 to 2023 till date). Moreover, the syntheses and significant therapeutical activities of numerous quinazoline based drugs were briefed and assessed.
Abstract
Creating effective, ecologically friendly, and commercially viable synthetic routes is crucial in the design and synthesis of organic substances. Quinazoline, a heterocyclic compound with nitrogen, is one of the most significant heterocyclic motifs with diverse chemical reactivities and many biological applications. Its derivatives comprise a family of fused heterocycles in over 200 naturally occurring alkaloids. Over the past few decades, newer, more complex drugs containing quinazolinone structures have been discovered, with enormous progress in designing various efficient protocols to construct these pharmacologically active scaffolds. This review evaluated the recently investigated protocols for synthesizing quinazolines and their derivatives (from 2017 to 2023 till date). The current review paper provides an up-to-date description of recent advancements in straightforward synthetic procedures that result in the creation of quinazoline molecules. In addition, the significant therapeutical activities of numerous quinazoline-based drugs were briefed and assessed in this review. We envisage that this information would assist researchers in designing and synthesizing novel quinazoline analogues as lead compounds.
Keywords
Quinazoline
Synthesis
Biological activity
Drug
Heterocyclic

- BOP
-
Benzotriazole-1-yl-oxy-tris-(dimethylamino)-phosphonium
- CAR
-
constitutive androstane receptor
- DBU
-
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DCE
-
1,2-Dichloroethene
- DMSO
-
Dimethylsulphoxide
- DMF
-
Dimethylformamide
- DPPA
-
Diphenylphosphorylazide
- DPP-4
-
Dipeptidyl Peptidase IV (DPP IV)
- Me
-
Methyl
- PDE
-
Phosphodiesterase
- PTSA
-
para- Toluenesulfonic acid
- TBHP
-
tert-Butyl hydroperoxide (tBuOOH)
- THF
-
Tetrahydrofuran
Abbreviations
1 Introduction
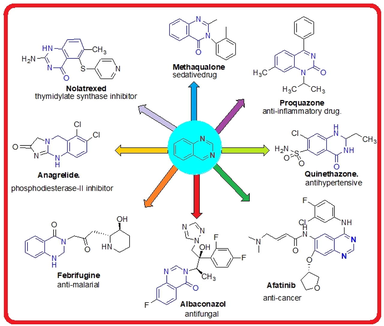
Nitrogen-containing heterocyclic molecules are vital in medical chemistry due to their extensive applications in various fields. Quinazolines are a valuable class of nitrogen-containing heterocyclics with a wide range of biological properties, particularly as anti-bacterial (Ghorab et al., 2013), anti-psychotic (Alvarado et al., 2006), anti-fungal (Al-Amiery et al., 2014), anti-inflammatory (Giri et al., 2009), anti-diabetic (Karan et al., 2021) anti-tuberculosis (Kunes et al., 2000); anti-malarial (Chugh et al., 2020), anti-obesity (Sasmal et al., 2012), anti-viral (Mermer et al., 2021); A2A adenosine receptor antagonists (Bolteau et al., 2022), nematicidal (Wang et al., 2023) agents. Additionally, poly-(ADP-ribose) polymerase (PARP) (Guiles et al., 2009), thymidylate synthase (Liu et al., 2006), tyrosine kinase (Conconi et al., 2013) inhibitors, anti-hepatocellular carcinoma and Radio-sensitizers (Ghorab et al., 2023) characteristics of quinazoline analogues are well documented. Some quinazoline derivatives (Jafari et al., 2016) with diverse biological activities were presented in figure1.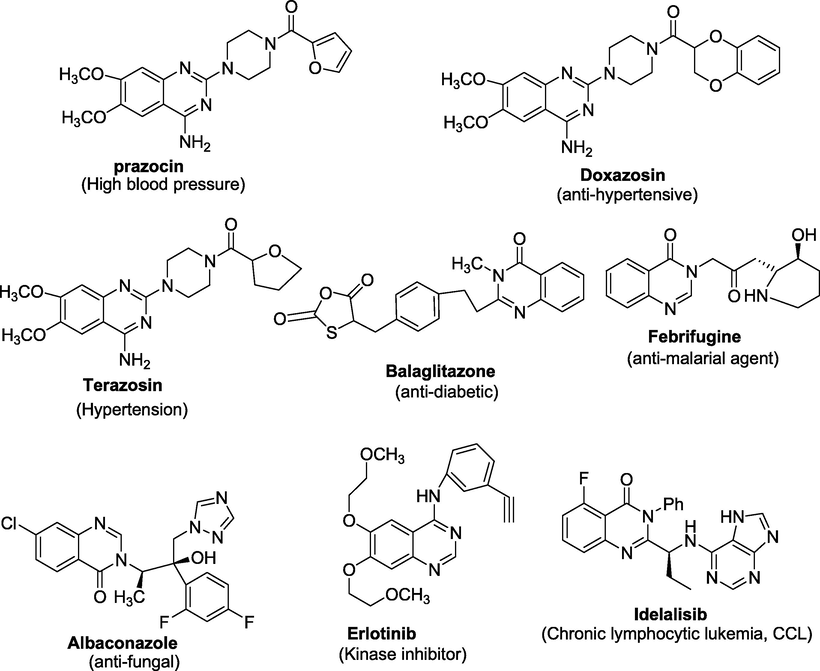
Some bioactive qunazoline sscaffolds.
Several quinazoline derivatives are approved drugs, such as Terazosin hydrochloride, Prazosin hydrochloride and Doxazosin mesylate (Kyprianou, 2003). Moreover, due to the promising therapeutic efficacy against human cancers, various quinazoline derivatives like Erlotinib (Moyer et al., 1997), Gefitinib (Wakeling et al., 2002), Lapatinib (Rusnak et al., 2001); Vandetanib (Wedge et al., 2002) and Afatinib (Li et al., 2008) have been approved for cancer therapy in the clinic (figure 2) by the United States Food and Drug Administration (USFDA). Thus, with significant biological activities, quinazoline derivatives have received the utmost importance in organic synthesis and medicinal chemistry research.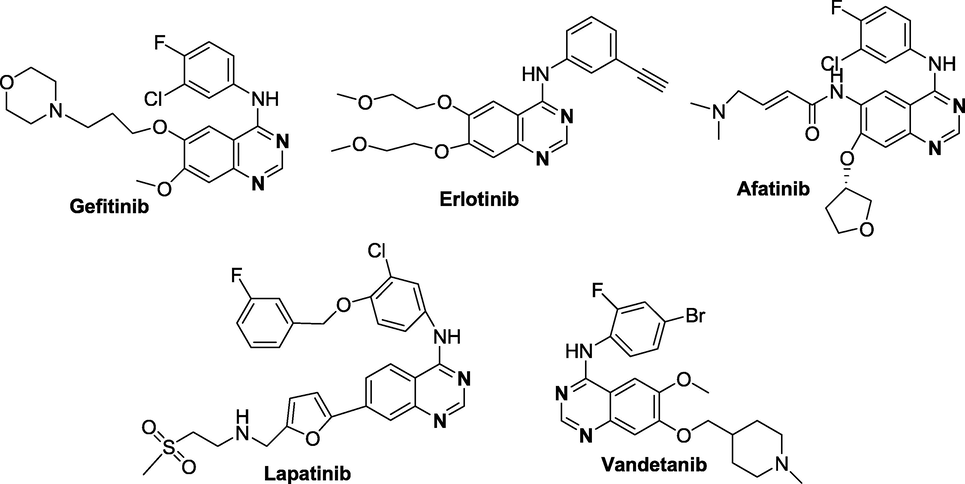
FDA Approved quinazoline motifs for cancer therapy.
Griess produced the first quinazoline moiety, 2-cyano-3,4-dihydro-4-oxoquinazoline (Figure 3), 1869 by reacting cyanogens with 2-aminobenzoic acid. Griess was the first to notice the product's bicyclic character, naming it bicyanoamido benzoyl (Griess, 1869) and using it until 1885.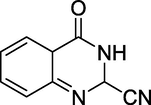
Structure of 2-Cyano-3,4-dihydro-4-oxoquinazoline.
Bichler and Lang made the parent quinazoline (Figure 4) molecule via decarboxylation of the 2-carboxy quinazoline derivative (Bischler and Lang, 1895). Gabriel(1903) developed an improved method for synthesising quinazolines and investigated their characteristics.
Structure of quinazoline.
Widdege proposed the name quinazoline, which comprises two rings, one of which is benzene and the other one is a pyrimidine. Quinazoline has also been referred to by the names phenamiazine, benzyleneamidine, benzo-1,3-diazine-5,6-benzopyrimidine, and 1,3-diazanapthaline. Adding a fused benzene ring changes the characteristics of the pyrimidine ring expressively. The extent of conjugation and substituents on the benzene and pyrimidine rings significantly impact the quinazoline derivative properties.
Despite the remarkable progress in quinazoline chemistry, there is still a shortage of terse and efficient protocols for constructing these medicinally significant quinazoline derivatives. Very few reviews addressing the synthesis and properties of these motifs have appeared. For instance, Paul and co-authors (Prashant et al., 2020) described the advances in lead compounds of quinazoline hybrids and their related heterocycles in medicinal chemistry that help to exaggerate the drug development practice. Chen et al. (Chen et al., 2022) provided a comprehensive depiction with emphasis on the structure–activity relationships (SAR) mechanism, agricultural biological activities, and mechanism of action of quinazoline derivatives in the past decade systematically. Yar et al. (Haider et al., 2022) summarised the recent advances of quinazoline/quinazolinone scaffold-bearing derivatives as anticancer agents acting through vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR) and dual VEGFR/EGFR inhibitors. Neelima and co-workers (Karan et al., 2021) provided information regarding the latest developments in quinazoline analogues with a broad range of pharmacological activities. Al-Salahi and Abuelizz (Abuelizz and Al-Salahi, 2023) provided an up-to-date account of research findings on the pharmacological activities of benzoquinazoline derivatives and suggested directions for future discoveries. Emami et al. (Haghighijoo et al., 2022) presented the relevant information regarding the anti-Alzheimer agents having quinazoline core structure which can promote the discovery and development of novel anti-AD agents. Ionutet al. (Șandor et al., 2023) outlined the general principles for developing (2017–2023) potent EGFR TKIs, exploring the impact of structural modulations in critical positions of the quinazoline core on the anticancer effect. Bhagat and coworkers (Bhagat et al., 2022) disclosed patents of synthetics and natural DHFR inhibitors with quinazoline and diaminopyrimidine nuclei during 2001–2021. The review also emphasised the clinical evolution of various DHFR inhibitors established during 2016–21.Gheidariet al. (Gheidari et al., 2022) described the multiple methods for synthesising quinazoline-2,4(1H,3H)-diones, such as green methods and heterogeneous or homogeneous catalysts from various precursors under different conditions. Joshi and co-authors (Parveen et al., 2022) described the biological significance of quinazoline and its derivatives. They used the diverse nano catalysts (2015–2022) to synthesise various quinazoline derivatives with emphasis on the catalytic performances and synthetic protocols that afford an idea for the coherent designing of efficient, novel and efficient nanocatalysts. Quinazoline and its derivatives have been incessantly evaluated as potent bioactive compounds in recent decades. Table 1 summarises some of the most effective bioactive compounds (Giang et al., 2020; Zhang et al., 2021; Qin et al., 2022; Ghorab et al., 2021; Malasala et al., 2020; Romero et al., 2019; Moreira et al., 2023; Zuzana et al., 2023) with quinazoline nuclei reported in recent years.
Entry
Compound
Biological Activity
Ref. No
1
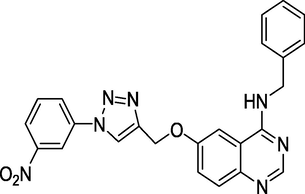
Anti-Alzheimer's activity
(Giang et al., 2020)
2
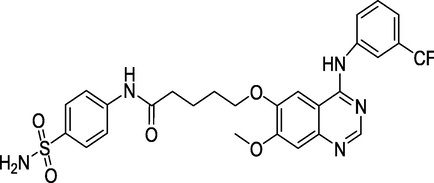
Dual EGFR/CAIX inhibitor
(Zhang et al., 2021)
3
Anti-fungal agent
(Qin et al., 2022)
4
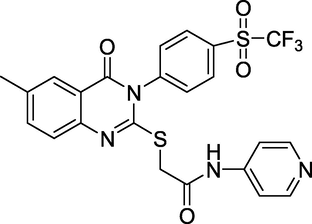
Human carbonic anhydrases IX/XII inhibitors
(Ghorab et al., 2021)
5
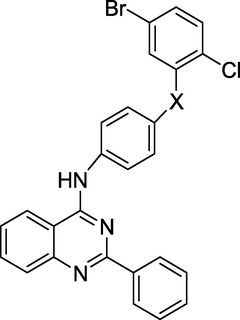
Anti-tubercular agent
(Malasala et al., 2020)
6

Antileishmanial activity
(Romero et al., 2019)
7
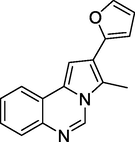
Antiplasmodial activity
(Moreira et al., 2023)
8
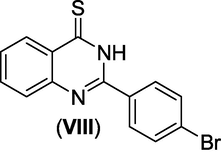
CAR antagonistic activity
(Zuzana et al., 2023)
Gianget al. (Giang et al., 2020) prepared and evaluated the quinazoline-triazole hybrid motifs as acetylcholinesterase inhibitors to treat Alzheimer's disease (AD). The biological assay outcomes revealed that 6-((1-(3-nitrophenyl)-1H-1,2,3-triazol-4-yl)methoxy)-N-benzylquinazolin-4-amine (Compound I, Table 1) is the most potent with IC50 (AChE) value 2.6±0.19 µM. Zhang et al. (Zhang et al., 2021) synthesised a series of quinazoline derivatives containing substituted anilide andsulfamoylphenyl fragments as dual EGFR/CAIX inhibitors. In the obtained compounds, 5-((7-methoxy-4-((3-(trifluoromethyl)phenyl)amino)quinazolin-6-yl)oxy)-N-(4-sulfamoylphenyl)pentanamide (Compound II, Table 1) was identified as the most potent with EGFRWT IC50= 27.0 nM, EGFRT790M = 9.2 nM, and CAIX IC50= 115 nM. Qin et al. (Qin et al., 2022) prepared a library of 2–substituted–4–amino-quinazolines and evaluated their anti-fungal activity against four invasive fungal strains. The antimicrobial screening revealed that compound N-((1E,3E)-penta-1,3-dienyl)-2-p-tolylquinolin-4-amine hydrochloride (Compound III, Table 1) have good anti-fungal activity with MICs 4–16 µg/mL. Ghorab et al., (Ghorab et al., 2021) synthesised a series of iodoquinazolinones with benzenesulfonamide moiety as human carbonic anhydrase (hCA) inhibitors. The screening outcome revealed that 2-(3,4-dihydro-6-iodo-4-oxo-3-(4-(trifluoromethylsulfonyl)phenyl)quinazolin-2-ylthio)-N-(pyridin-4-yl)acetamide (Compound IV, Table 1) is identified as the most potent candidate against hCA IX and hCA XII., on MTT assay, it exhibited potent activity against HepG-2 and MCF-7 cells with IC50 values of 2.53 µM and 4.58 µM. Satyaveni et al. (Malasala et al., 2020) synthesised 2-arylquinazoline benzamide derivatives and screened against Mycobacterium tuberculosis H37RV strain, the Compound N1-(5-bromo-2-chlorophenyl)-N4-(2-phenylquinazolin-4-yl)benzene-1,4-diamine (Compound V, Table 1) exhibits selective and potent antimycobacterial activity with MIC value 4 mg/mL. Romeo et al., (Romero et al., 2019) prepared a series of 2-aryl-quinazolin-4(3H)-ones as leishmania folate inhibitors. The biological screening result revealed that 2-(3-methoxyphenyl)quinazolin-4(3H)-one (Compound VI, Table 1) is identified as a potent antileishmanial agent with ID50 vales 11.04–29.34 μM. Moreira group (Moreira et al., 2023) described the synthesis of Pyrrolo[1,2-c]quinazolines and Pyrrolo[2,1-a]isoquinolines via a C(sp3)–H functionalisation of 4-methylquinazolines and 1-benzylisoquinolines with α-substituted β-nitrostyrenes mediated by inexpensive Cu(OAc)2. The biological activity of the 2-(furan-2-yl)-3-methylH-pyrrolo[1,2-c]quinazoline (Compound VII, Table 1)was found to have promising antiplasmodial action against Plasmodium falciparum. Zuzana and group (Zuzana et al., 2023) reported various 2-Substituted quinazolines as partial agonistic and antagonistic ligands of the constitutive androstane receptor (CAR) among the prepared compounds 2-(4-bromophenyl)quinazoline-4-thione (Compound VIII, Table 1) exhibited significant CAR antagonistic activity with EC50 value of 0.055 μM.
Due to the extensive range of applications of Quinazolines in different fields, the authors believed that the collection of the recent advances in synthetic methods for the construction of Quinazoline scaffolds and reported synthetic protocols of quinazoline-based drug molecules along with its therapeutic application would be exceptionally helpful for the young researchers to spotlight on Quinazoline construction and its applications. In this review, we afforded to collect the recent progressions in synthesizing different quinazoline derivatives from 2017 to till date in 2023 and the pharmacological significance of new quinazoline-based drug moieties along with the developed synthetic pathways towards those drugs. The information presented in this manuscript is beneficial for medicinal chemistry researchers to open inventive channels towards constructing several highly efficient lead quinazoline molecules.
2 Synthesis of quinazoline
The first quinazoline compound was generated in 1869 using anthranilic acid and cyanogens (Griess, 1869). Since then, the field attracted various researchers developing multiple conventional techniques for synthesising quinazoline analogues (Sharma and Kaur, 1989; Moore et al., 1969; Cai et al., 2002; Asakawa and Matano, 1979; Connolly et al., ,2005; Su and Yang, 2002).Most of these approaches use the Niementowski reaction, which fuses the analogues of anthranilic acid with amides (Scheme 1) at temperatures between 130 and 150 °C via the formation of an o-amidobenzamide intermediate (Niementowski, 1895).However, this approach results in low yields and contaminants that are generally difficult to remove by column chromatography or recrystallisation. Besson et al., (Alexandre et al., 2002) used microwave irradiation to re examine the Niementowski synthesis of the 4(3H)- quinazolinones to improve yields and speed up response times. The condensation of anthranilamide (2-aminobenzamide) derivatives with carbonyl compounds and the one-pot three-component reaction of aldehydes, isatoic anhydride and amines (Kim and Cheon, 2014; Zhang et al., 2015; Li et al., 2015; Lobo et al., 2012; Mekala et al., 2017) are two additional significant procedures for the synthesis of quinazolines (Scheme 1).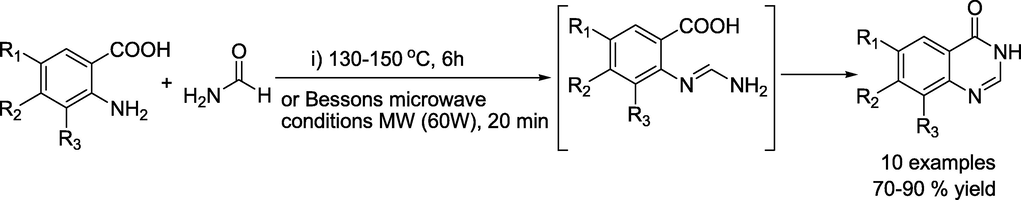
Synthesis of quinazoline by Niementowski reaction or by Besson's microwave conditions.
Transition metal-catalysed C–H activation and functionalisation routes are the most promising approaches. The transition metal-catalysed C–H activation processes arepromising and display an excellent future for organometallic chemistry. Compared to the traditional method, the transition metal-catalysed C–H activation and functionalisation is simple. With no requirement for pre-functionalising the starting material, waste generation is minimised. Significant progress has been achieved in creating heterocycles via C-N and C–C bonds in the last five years. The monograph provides a concise summary and review of the recent literature on the metal-catalysed generation of quinazolines viaC–H activation and C–N coupling reactions over the past fiveyears.
Paul and coauthors reported (Chakraborty et al., 2019) a simple atom-efficient reaction of nitriles with 2-aminobenzyl alcohol to obtain quinazolines in good yields via the biomimetic dehydrogenative condensation/coupling process (Scheme 2). Singlet diradical Ni(II) with two anti-ferromagnetically connected singlet diradical diamine type ligands catalyses this reaction.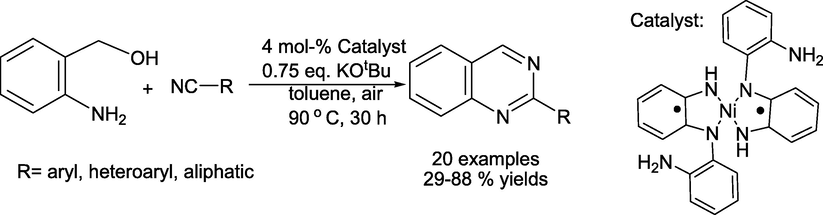
Singlet diradical diamine Ni(II) catalysed the synthesis of arylquinazolines.
Catalytic system from the cationic ruthenium−hydride complex [(C6H6)- (PCy3)(CO)RuH]+ BF4− (precatalyst) with 4-(1,1-dimethylethyl)-1,2- benzenediol (Ligand) was found to give the highest activity and selectivity for the coupling of 2-(aminophenyl)ethanone with 4-methoxybenzylamine in yielding the quinazoline product (Scheme 3) by Yi and group (Arachchige and Yi, 2019). The reactions work without using any reactive reagents or forming any toxic byproducts.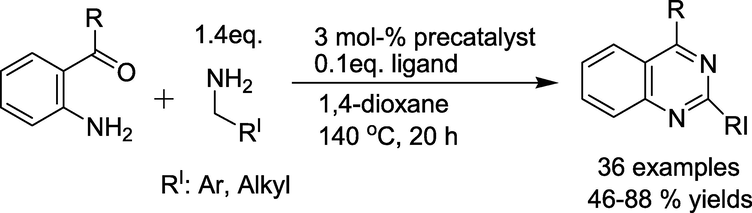
Cationic ruthenium−hydride complex catalysed synthesis of quinazolines.
Chen and coworkers (Chen et al., 2018) described a competent synthesis of quinazolines through a Fe(II) catalysed C(sp3)-H oxidation and intramolecular C−N bond formation (Scheme 4). Initially, the reaction of 2-alkylamino benzonitriles with various organometallic reagents led to 2-alkylamino N–H ketimine species. Next, the FeCl2-catalysed C(sp3)-H oxidation of the alkyl group employing tert-BuOOH, intramolecular C−N bond formation and aromatisation afforded a wide variety of 2,4-disubstituted quinazolines in good to excellent (43–86 %) yields.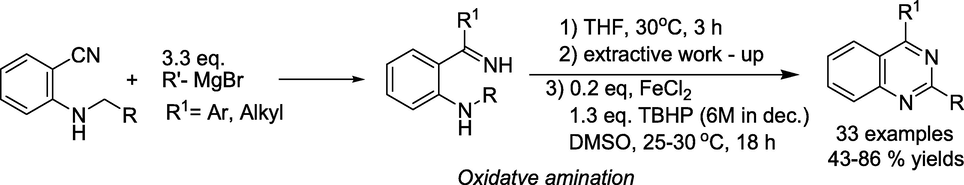
Ferrous chloride catalysed synthesis of quinazolines.
Chen et al. reported (Hu et al., 2018) an efficient Pd-catalysed one-pot three-component tandem reaction of 2-aminobenzonitriles, aldehydes, and arylboronic acids. The reaction proceeds through the cyano group's carbopalladation, generating a wide range of quinazolines (Scheme 5). The significant feature of this protocol is tolerance of bromo and iodo groups, broad substrate scope with remarkable chemoselectivity.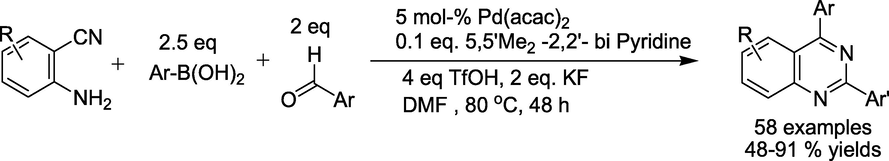
Palladium-catalysed catalysed synthesis of arylquinazolines.
Bhanageet al. (Raut et al., 2017)produced cuprous oxide nanocubes as a heterogeneous nano-catalytic system and used one-pot tandem cyclisation of 2-bromobenzaldehyde derivatives with amidine hydrochlorides without the use of ligands. This simple, eco-friendly method can produce quinazoline frameworks in excellent isolated 81–95 % yields Within a few minutes (Scheme 6). The cuprous oxide nano catalytic system could also be recycled and regenerated up to four times without significantly losing its catalytic effectiveness.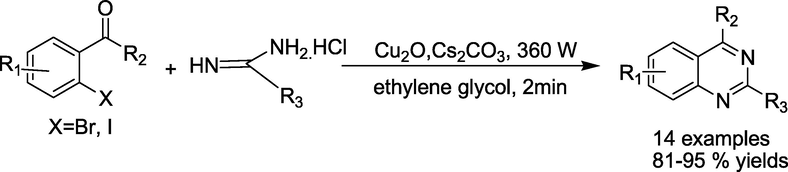
Cu2O–catalysed synthesis of quinazolinone derivatives.
Fan et al. recently described (Guo et al., 2018) the production of quinazolines in moderate to good isolated yields (40–72%) using the Cu-catalysed aerobic oxygenation of 2-(2-amidoaryl)-1Hindoles, followed by intramolecular cyclisation under acidic conditions (Scheme 7). This method features controlled selectivity and a simple operational procedure.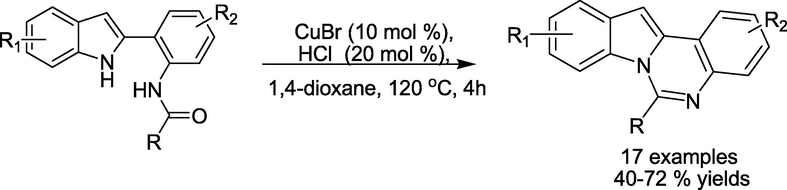
CuI -catalysed synthesis of quinazoline derivatives.
The construction of quinazoline derivatives from I2 catalyzed reaction of 2-aminobenzaldehydes or 2-aminobenzophenones with benzyl-amines was recently described by Bhanageet al. (Deshmukh and Bhanage, 2018) With a sample range of functionalised 2-aminobenzaldehydes or 2-aminobenzophenones, many functionalised hetero-aryl or aryl amines were examined to produce quinazolines in 49–92% yields (Scheme 8). The method is more environmentally benign and cost-effective due to using O2 as a safe oxidant without transition metals, additives, and solvents. The absence of an aqueous workup also increases the efficiency of this process.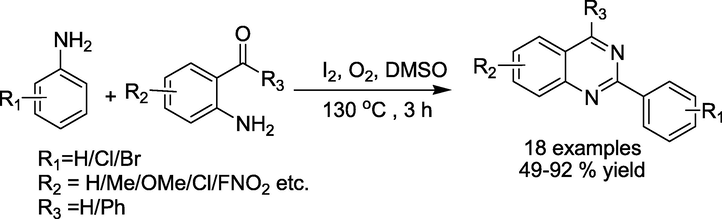
Molecular iodine catalysed reaction of benzyl-amines with 2-amino benzophenones or 2-aminobenzaldehydes.
Wu et al. (Wu et al., 2018) presented a rhodium-catalysed direct and distinctive process through annulation of ketones and 2-arylimidazoles,for a library of 5- arylimidazo[1,2-c]quinazolines in 49–95 % yields (Scheme 9). This approach is distinguished by (i) being devoid of halo functionalisation handles and (ii) using readily available starting materials.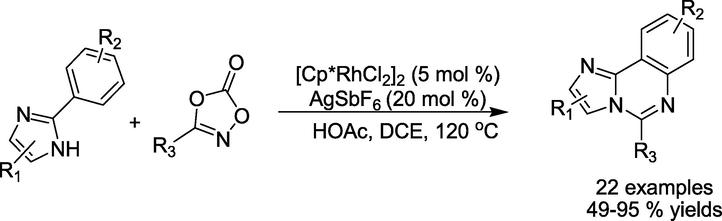
Rhodium-catalysed annulation of C–H bonds.
Wang and Jiao documented (Wang and Jiao, 2016) the synthesis of biologically active quinazolines by reacting imidate derivatives with alkyl azides. The novel [4 + 2] carbon-hydrogen bond activation and annulation was co-catalysed by Cu and Rh. Simple alkyl azide derivatives are usefully used in this aerobic oxidative process to produce N-heterocycles, with water and nitrogen as byproducts(Scheme 10).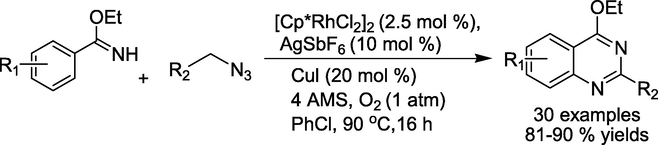
Rh-catalyzed protocol for the formation of quinazoline skeletons.
While employing inexpensive 2-aminobenzonitriles, aryl boronic acids and aldehydes, Chen et al. (Kun et al., 2018) demonstrated Pd-catalysed three-component, one-pot tandem assembly for quinazolines. The technique exhibits astounding chemoselectivity and a broad substrate range (Scheme11). This method stands out for its tolerance of iodo and bromo moieties allowing flexibility for future modifications.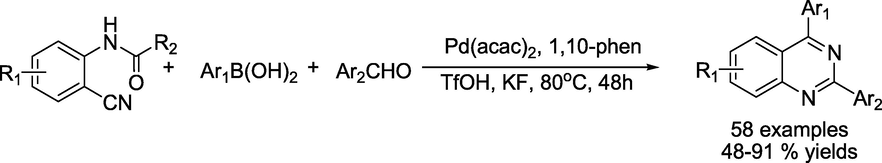
Pd-catalyzed synthesis of quinazolinones.
The same team reported the reaction of aryl boronic acids with 2-(quinazolinone-3(4H)-yl)benzonitriles (Zhang et al., 2018) in another approach. This tandem synthesis produced the quinazoline scaffolds in good to exceptional isolated yields (31–93%) through nucleophilic addition, intramolecular cyclisation, and subsequent ring-opening (Scheme 12).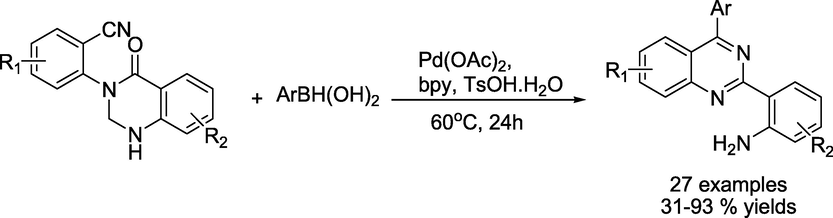
Pd-catalysed synthesis of multi-substituted quinazolines.
Another interesting study by Chen et al. (Zhu et al., 2018) describes the synthesis of 2,4-disubstituted quinazoline derivatives using 1,10-phen and trifluoroacetic acid in THF at 80 °C in the Pd-catalyzed reaction of aryl boronic acidswith N-(2-cyanoaryl)benzamides (Scheme 13). With N-(2-cyanoaryl)benzamides, the reaction showed a wide range of functional group tolerance, encompassing electron-deficient and electron-rich aryl boronic acids.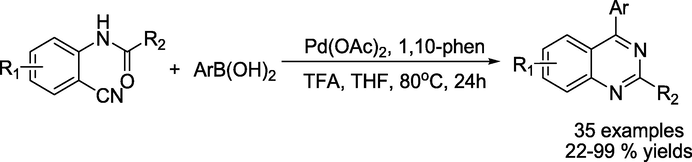
Pd-catalysed synthesis of quinazolinones.
In 2021, Senadiet al. (Philips et al., 2021) reported an atom economical and environmentally benign Cu-promoted oxidative coupling strategy towards the construction of 4(3H)-quinazolinones and benzoimidazoquinazoline utilising 0.25–0.5 equivalent D-glucose as renewable C1 synthon in the open air for 12– 24 h at 120 °C (Scheme 14). Biomass-derived platform chemical as carbon synthon; synthesis of naturally occurring alkaloids and precursors are the significant features of this method.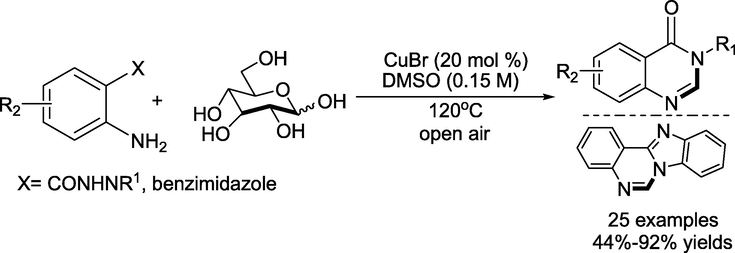
Cu promoted synthesis of 4(3H)-quinazolinones and benzoimidazoquinazoline.
To find a new fungicide to combat Rhizoctoniasolani, Lei and coworkers reported (Lei et al., 2022) a library of novel quinazolinone scaffold-containing pyrazole carbamide derivatives (Scheme 15). Initially, the substituted quinazolinone wasobtained from anthranilic acid and formamidine acetate in ethylene glycol monomethyl ether at 95–130 °C to get the quinazolin-4-ones (Scheme 16). The key intermediate N-(2-(chloromethyl)phenyl)-3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxamide was obtained in 75% yield by dissolving 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic acid in DMF followed by the addition of triethylamine and thionyl chloride at room temperature (Scheme 17). Next, the targeted quinazolinone-scaffold-containing pyrazole carboxamides were achieved in 41–83 % yield by the reaction of quinazolin-4-ones and N-(2-(chloromethyl)phenyl)-3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxamide in DMF under alkaline conditions (Scheme 15).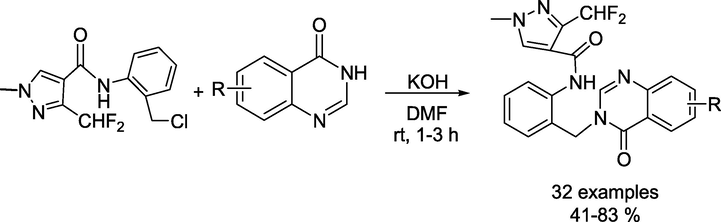
Synthesis of quinazolinone containing pyrazole carbamide derivatives.
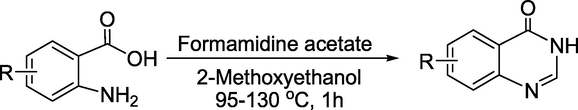
Synthesis of quinazolin-4-one derivatives.
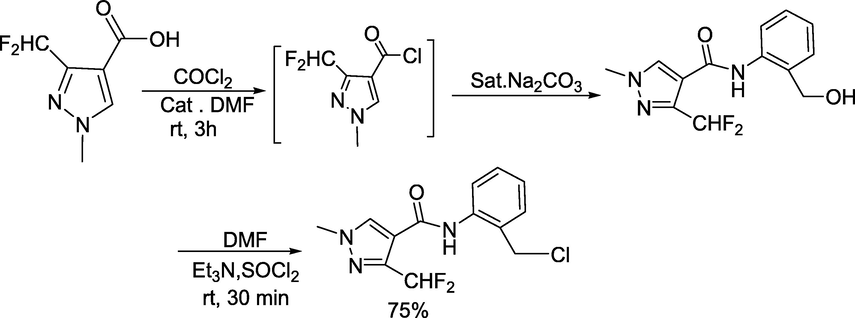
Synthesis of N-(2-(chloromethyl)phenyl)-3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxamide.
Tskhovrebov and group demonstrated (Osmanov et al., 2022) an efficient method for the synthesis of 2-selenoxo-1,2,3,4-tetrahydro-4-quinazolinone via cyclisation reaction between methyl anthranilate and isoselenocyanates (Scheme 18). The latter compounds readily undergo oxidation with H2O2 to produce the appropriate diselenides in high yields (Scheme 19). Additionally, all chalcogen bonding (ChB) interactions were established by DFT calculations, followed by the topological analysis of the electron density distribution.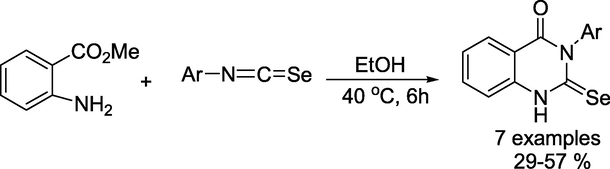
Synthetic path for2-selenoxo-1,2,3,4-tetrahydro-4-quinazolinones.
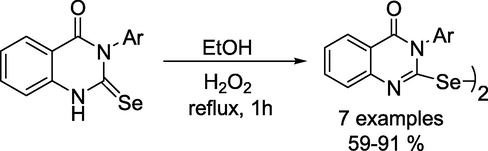
Synthetic route for2,2′-diselane-1,2-diylbis(3-arylquinazolin-4(3H)-ones.
Al-Tel' group (Ravi et al., 2023) reported a metal-free, stereo-divergent build/couple/pair strategy that allows access to a unique collection of benzo[5,6][1,4]) (oxazino[4,3-a]quinazoline, quinolino[1,2-a]quinazoline scaffolds (Scheme 20) with total diastereo control and wide distribution of molecular architectures. This metal-free protocol proceeds through the desymmetrisation of phenol derivatives. The cascade connects Mannich with aza-Michael addition reactions, providing hasty entries to different classes of molecular shapes in a single operation.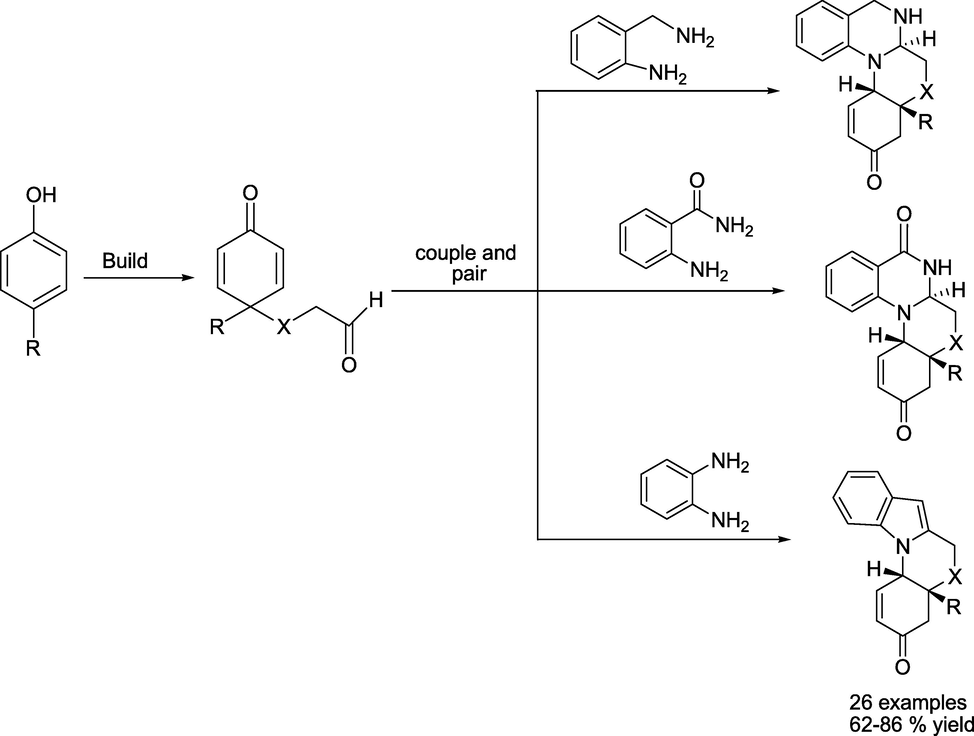
Synthetic protocols for functionalised Quinazoline Scaffolds.
Gil and team (Victor et al., 2020) described a simple method for the synthesis of substituted 3-benzyl-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-diones (Scheme 21). Initially, 3-benzylquinazolinediones were obtained by reacting 2-aminoaryl esters and aryl isocyanates via method A or B. Further, the obtained 3-benzylquinazolinedione in DMF combines with 4-(trifluoromethyl) benzyl chloride in the presence of NaHCO3 and the reaction mixture is heated to 130 °C overnight to produce the consecutive 3-benzyl-1-(4-(trifluoromethyl)benzyl)- quinazolin-2,4(1H,3H)-diones in 10–71 % yields.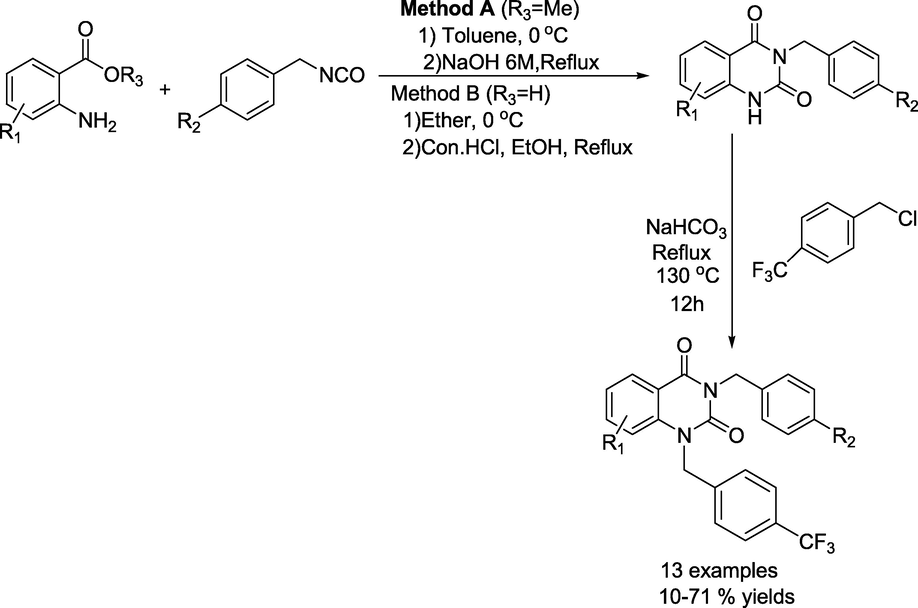
Synthesis of 3-benzyl-1-(4-(trifluoromethyl)benzyl)quinazolin-2,4(1H,3H)-diones.
Chang and coauthors presented (Wang et al., 2023) the metal-free synthesis of quinazolinone-fused polycyclic skeletons from 2-aminobenzamide precursors via an iodine-promoted intramolecular sp3 C–H amination reaction (Scheme 22). The reaction progressed well with crude 2-aminobenzamide derivatives, permitting the synthesis of the products from tetrahydroisoquinolines and simple 2-aminobenzoic acids without purification of the 2-aminobenzamide intermediates. This reaction has a broad substrate scope and can be used on a gram scale under optimal reaction conditions.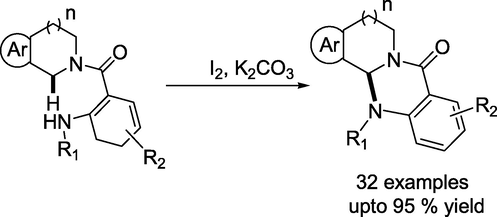
I2-promoted synthesis of Quinazolinone-Fused Tetrahydro isoquinolines.
Tabatabai group reported (Hejazi et al., 2020) a series of new quinazoline-4(3H)- one derivatives with varying steric and electronic properties synthesised from 4-nitrobenzaldehyde and anthranilamide (Scheme 23). Initially, the mixture of 4-nitrobenzaldehyde and anthranilamide in DMSO was irradiated under ultrasound irradiation for 3h in an open flask to produce the 2-(4-nitrophenyl)quinazoline-4(3H)-one that reacts with sodium dithionite in 1:9 H2O-DMF at 90 °C with stirring to obtain the 2-(4-aminophenyl)quinazoline-4(3H)-one. This intermediate, stirring with consequent anhydrides or acyl chlorides under an argon atmosphere at 0 °C to room temperature, followed by adding water, produced the target quinazoline-4(3H)-one derivative in 62–95 % yields.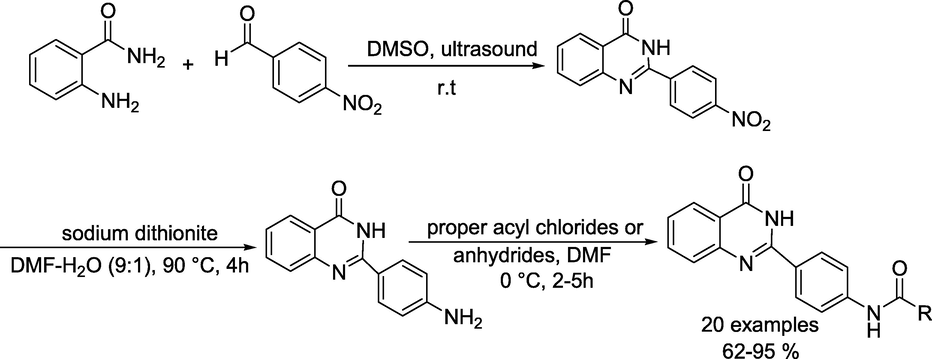
Synthesis of quinazoline-4(3H)- one derivatives.
Chang and coauthors (Wu et al., 2023) reported a mild metal-free protocol towards the construction of quinazolines scaffolds from 2,2,2–trichloroethyl imidates hydrochloride as nitrogen source and 2-aminophenyl ketones in the presence of sodium acetate (Scheme 24). This protocol afforded several quinazoline derivatives, especially 2-substituted quinazolines, in moderate to high yields. This approach has advantages like readily available starting materials and gram-scale synthesis.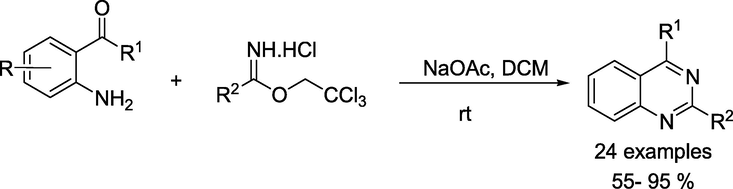
Synthesis of quinazolines from 2-aminophenyl ketones and 2,2,2–trichloroethyl imidates hydrochloride.
Ding and coauthors reported an efficient metal-/solvent-free protocol (Tan et al., 2022) towards the construction of quinazolines by elemental sulfur-promoted oxidative condensation of 2-(aminomethyl)anilines and nitriles (Scheme 25). The method tolerates a broad range of functional groups to obtain the consecutive products in 56–91% yield and can also be achieved on a gram scale.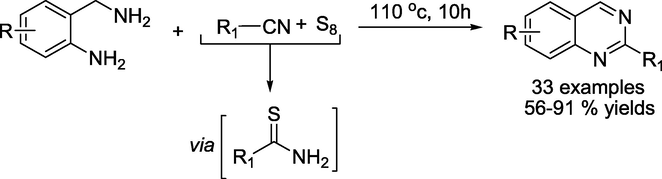
Synthesis of Quinazolines by elemental sulphur promoted oxidative condensation.
Lu and group (Pan et al., 2022) prepared a series of novel 4-aminoquinazoline derivatives. Initially, 7-methoxy-4-oxo-3,4-dihydroquinazolin-6-yl acetate and sulfuryl dichloride were added into N, N-dimethylformamide dropwise under nitrogen atmosphere to obtain 4-chloro-7-methoxyquinazolin-6-yl acetate, that reacts with ammonia in CH3OH, produced 4-chloro-7-methoxyquinazolin-6-ol. Next, the obtained 4-chloro-7-methoxyquinazolin-6-ol reacts with 2-(pyrrolidin-1-yl)ethan-1-ol in THF under DTAD and PPh3 produced 6-(2-(pyrrolidin-1-yl)ethoxy)-4-chloro-7-methoxyquinazoline. Finally, in the presence of 4-methylbenzene-1-sulfonic acid and propan-2-ol, the compounds 6-(2-(pyrrolidin-1-yl)ethoxy)-4-chloro-7-methoxyquinazoline reacts with various N1-benzylbenzene-1,4-diamines and 4-(aryloxy)benzenamine to obtain the consecutive target compounds N1-(6-(2-(pyrrolidin-1-yl)ethoxy)-7-methoxyquinazolin-4-yl)-N4-arylbenzene-1,4-diamines and 6-(2-(pyrrolidin-1-yl)ethoxy)-N-(4-(benzyloxy)phenyl)-7-methoxyquinazolin-4-amine(Scheme 26). Similarly, N1-(6-(2-(1H-pyrrol-2-yl)ethoxy)-7-methoxyquinazolin-4-yl)-N4-arylbenzene-1,4-diamines were obtained by the reaction of 6-(3-(1H-pyrrol-2-yl)butyl)-4-chloro-7-methoxyquinazoline with N1-benzylbenzene-1,4-diamines (Scheme 27).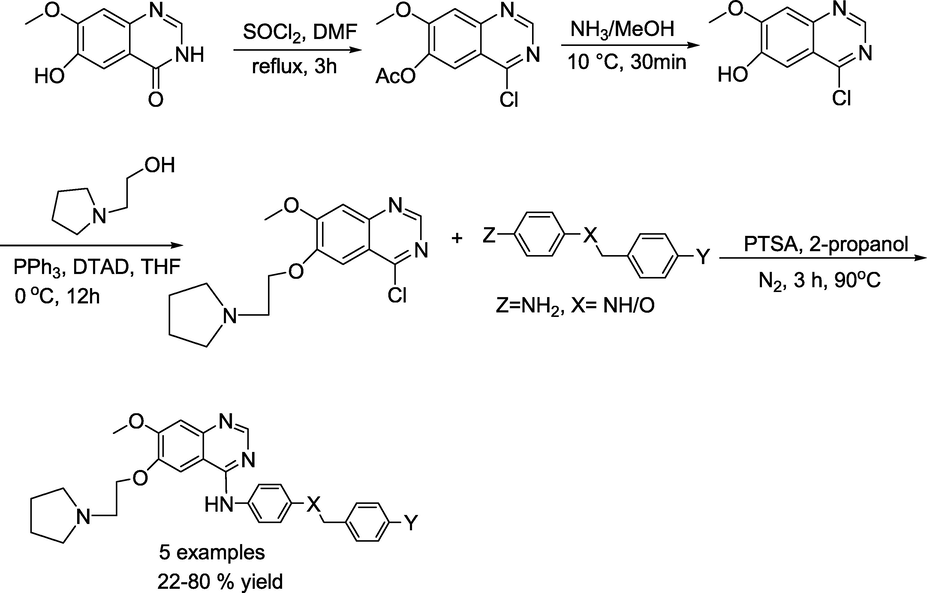
Synthetic procedure for quinazoline derivatives.
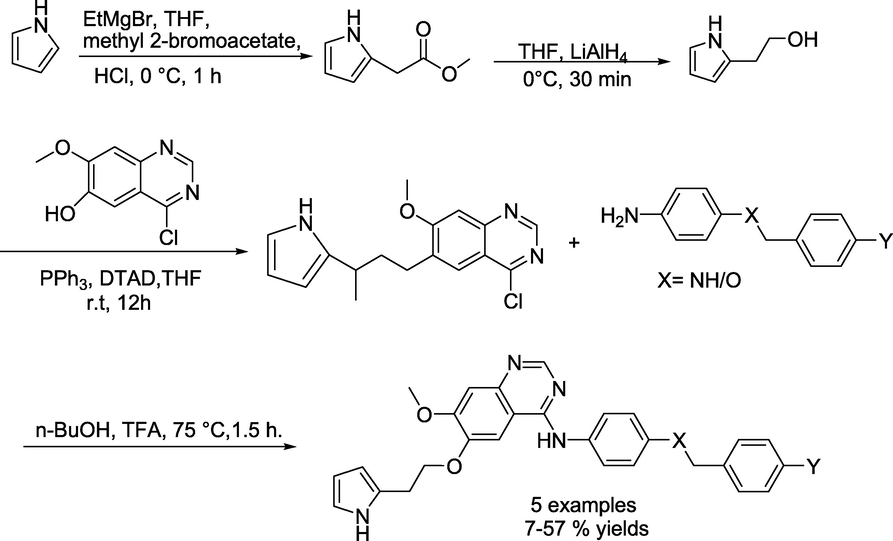
Synthetic procedure for quinazoline analogues.
Raju Dey et al. (Hima et al., 2023) described a transition metal-free protocol towards constructing quinazoline derivatives starting from aryl amides and 2-aminobenzyl alcohols through a potassium tertiary butoxide-promoted alcohol dehydrogenation strategy (Scheme 28). This method tolerates a range of functional groups affording simple access to diverse quinazolines with good to excellent yields.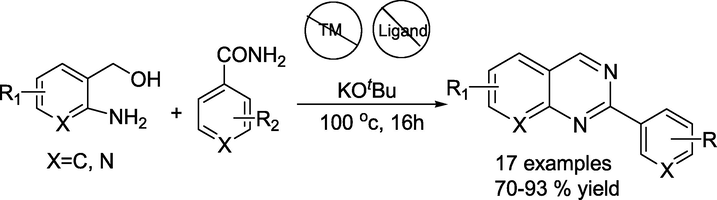
Metal-free synthesis of quinazoline analogues.
3 Drugs having quinazoline and its derivatives as a scaffold
In 1968, only two quinazoline derivatives, Methaqualone (soporific and anticonvulsant) and Quinethazone (diuretic), by 1980, over 50 derivatives with varied biological properties such as sedative, tranquillising, analgesic, anticonvulsant, antitussive, myorelexant, antirheumatic, hypotensive, antiallergic, bronchodilating, antidiabetic, cholagogue, diuretic, cystatic, anti-malarial and spermicidal have been reported.
3.1 Methaqualone (1954)
Methaqualone (Fig. 5) is a hypnotic and sedative medicine. It is also used as a muscle relaxant. The drug was marketed under the brand names Quaalude and Sopor. A combination medication Mandrax, which had 250 mg methaqualone and 25 mg diphenhydramine in one tablet, was also promoted (Kacker and Zaheer, 1951). In 1984, commercial methaqualone production was prohibited due to extensive usage.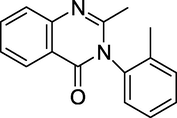
Structure of Methaqualone.
Synthesis: There are two primary synthetic routes (Van, 2001; Angelos and Meyer, 1985) for manufacturing Methaqualone
Route-1: Two-step preparation- In the first step, N-acetylanthranilic acid (3) is synthesised from anthranilic acid (1) and acetic anhydride (2), followed by condensation of (3) with o-toluidine (4) in the presence of phosphorus trichloride in the second step (Scheme 29).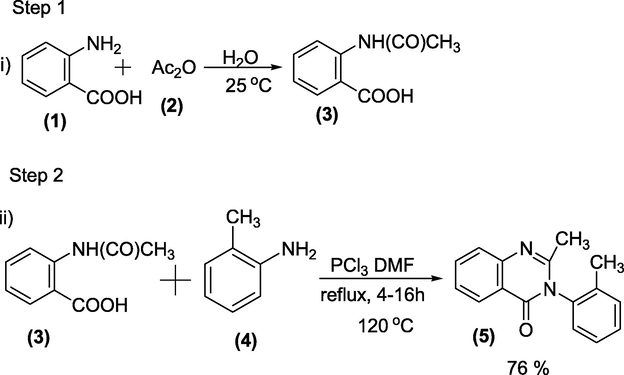
Phosphorous trichloride mediated synthesis of Methaqualone.
Route-2: One-step process- It is carried out by refluxing a mixture of 2-aminobenzoic acid (1), acetic anhydride (2), and o-toluidine (4) to give Methaqualone (5). Poly Phosphoric acid may be added to remove water (Scheme 30).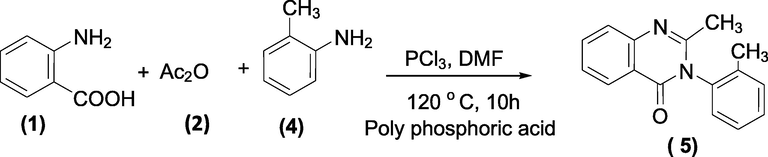
Phosphoric acid mediated synthesis of Methaqualone.
3.2 Diproqualone (1958)
Diproqualone (Fig. 6) belongs to the quinazolinone class of GABAergics and is an analogue of Methaqualone developed by a team at Nogentaise de ProduitsChimiques in the late 1950's, as a result of its agonist activity at the GABAA subtype, antagonist activity at all histamine receptors, inhibition of the cyclooxygenase-1 enzyme, and possibly agonist activity at both the sigma 1 and sigma 2 receptors. Due to its beneficial anti-inflammatory and analgesic effects and the sedative and anxiolytic effects common to other medicines in this family, diproqualone is the only analogue of Methaqualone that is still widespread in clinical use. Its many benefits are sedative, anxiolytic, antihistamine, and analgesic effects. Also valuable for the treatment of osteo arthritis and rheumatoid arthritis-related inflammatory discomfort. Moreover, it is also used to treat insomnia, anxiety and neuralgia, although not common practice.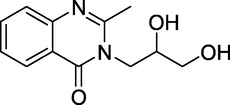
Structure of Diproqualone.
Synthesis: Diproqualone is synthesised through a one-pot synthesis of 2-iodobenzoic acid (6) and acetamidine hydrochloride (7) employing a copper-glucose catalysed quinazolinone reaction as a crucial step (Scheme 31). After the completion of the reaction, the N-arylated molecule was obtained by adding 3-chloro-1,2-propanediol to the same flask under reflux conditions. (±)-Diproqualone (8) is the end product (Dubey and Kumar, 2018).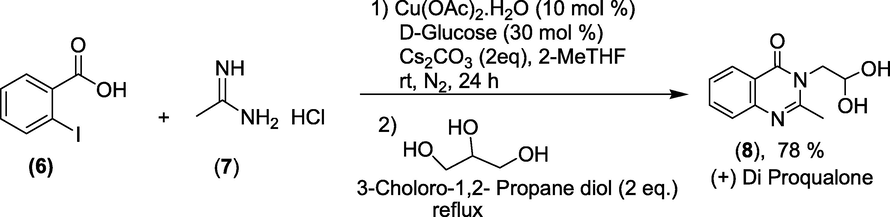
Phosphoric acid mediated synthesis of Methaqualone.
3.3 Etaqualone (1960)
Etaqualone (Fig. 7) is a quinazolinone-class GABAergic that was created in the 1960 s as an equivalent of Methaqualone. The dosage and effects are similar to Methaqualone but have a shorter half-life and milder effect. Aolan, Ethinazone, and Athinazone are some of their other names (Pflegel and Wagner, 1967; GB patent, 1963). It treats insomnia because it possesses sedative, hypnotic, muscle relaxant, and central nervous system depressant characteristics.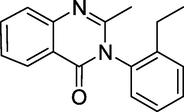
Structure of Etaqualone.
Synthesis: N-Acetylanthranilic acid (9) is refluxed at 120 °C with 2-ethylaniline (10) in the presence of PCl3 to give Etaqualone (11) as presented in Scheme 32 (Casale and Hays, 2012).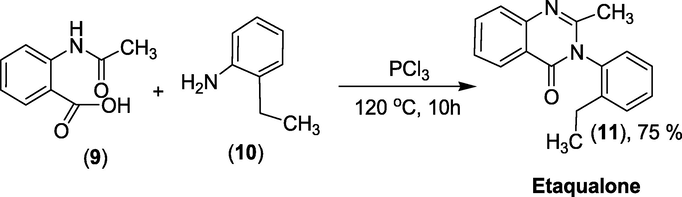
Synthesis of Etaqualone.
3.4 Prazosin (1974)
2-[4-(2-furoyl)piperazin-1-yl]-6,7-dimethoxyquinazolin-4amine is the chemical formula for prazosin.(Fig. 8). It's an alpha-adrenergic blocker, which works by relaxing blood arteries to reduce blood pressure. Prazosin is specific for alpha-1 receptors in vascular smooth muscle (Howard, 2008). These receptors mediate the vasoconstrictive action of norepinephrine, which typically elevates blood pressure. Prazosin lowers blood pressure by inhibiting these receptors, a sympatholytic medicine used to treat high blood pressure. It helps to lower blood pressure. Minipress, Vasoflex, Pressin, and Hypovase are some other names.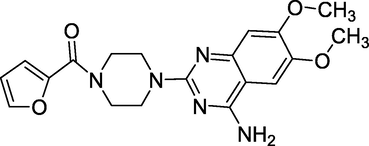
Structure of Prazosin.
Synthesis: Prazosin is synthesised from 2-amino-4,5-dimethoxybenzoic acid (12), which undergoes hetero-cyclisation into 2,4-dihydroxy-6,7-dimethoxyquinazoline (13) when it reacts with sodium cyanate. 2,4-dichloro-6,7-dimethoxyquinazoline (14) is obtained by replacing the hydroxyl groups with chlorine via a reaction with thionyl chloride or a mixture of phosphorous oxychloride and phosphorous pentachloride. The chlorine atom at C4 of the pyrimidine ring is replaced with an amino group in a subsequent reaction with ammonia, resulting in 4-amino-2-chloro-6,7-dimethoxyquinazoline (15). Finally, by combining (15) with 1-(2-furoyl)piperazine (16) is produced Prazosin (17) as presented in Scheme 33(Vardanyan and Hruby, 2006).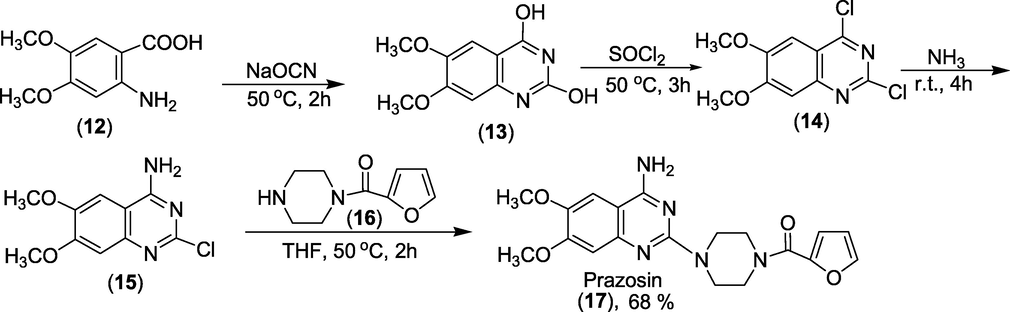
Synthesis of Prazosin.
3.5 Cloroqualone (1980)
Cloroqualone (Fig. 9) is a quinazolinone-class GABAergic that was established in the 1980's as an analogue of Methaqualone and marketed mostly in France and a few other European nations. Its agonist activity at the subtype of the GABAA receptor and the sigma-1 receptor gives it soothing and antitussive qualities. It was advertised as a cough treatment alone or in combination with other components. Cloroqualone has a weaker sedative effect than Methaqualone and was recognised for its cough-suppressing characteristics. 3-(2,6-dichlorophenyl)-2-ethylquinazolin-4-one is its IUPAC name. It possesses sedative and antitussive properties and acts as a cough-suppressing agent.
Stucture of Cloroqualone.
3.6 Alfuzosin (1988)
UroXatral; Urion; Xatral; Alfetim are brand names for alfuzosin, which is chemically known as N[3-[(4-amino-6,7-dimethoxy-quinazolin-2-yl)-methyl-amino]propyl] tetrahydrofuran-2-carboxamide (Fig. 10). It makes it easier to urinate by relaxing the muscles in the prostate and bladder neck. In patients with severe renal insufficiency, Alfuzosin should be taken with caution. It is not advised for individuals with a QT prolongation history or taking drugs extending the QT interval (Herbert, 2016; Fischer and Ganellin, 2006). It is an α1 receptor antagonist used to treat benign prostatic hyperplasia (BPH). Additionally, it aids in relaxing the muscles in the prostate and bladder neck, making urination easier.
Structure of Alfuzosin.
Synthesis: Initially, 4-amino-2-chloro-6,7-dimethoxyquinazoline (18) is condensed with 3-methylaminopropionitrile (19) in the presence of a polar aprotic solvent chosen from the group consisting of diglyme, dimethyl formamide, t-butanol, hexamethylphosphoramide, or mixtures there of to form -(4-amino-6,7-dimethoxyquinazol-2-yl)-N-methyl-2-cyanoethylamine (20). Using a hydrogenating agent and a pressure of less than 10 kg/cm2, hydrogenating the N-(4-amino-6,7-dimethoxyquinazol-2-yl)-N-methyl-2-cyanoethylamine (20) to generate N-(4-amino-6,7-dimethoxyquinazol-2-yl)-N-methylpropylenediamine (21). Moreover, tetrahydrofuran-2-carbonyl chloride (23) was obtained by chlorinating the tetrahydrofuran-2-carboxylic acid (22) with POCl3 in the presence of a base. Condensing the intermediate (23) with the N-(4-amino-6,7-dimethoxyquinazol-2-yl)-N-methylpropylenediamine (21) or with the acid addition salt thereof to generate alfuzosin base (24) and optionally converting alfuzosin base to a salt of alfuzosin as presented in scheme 34 (Kankan et al., n.d.).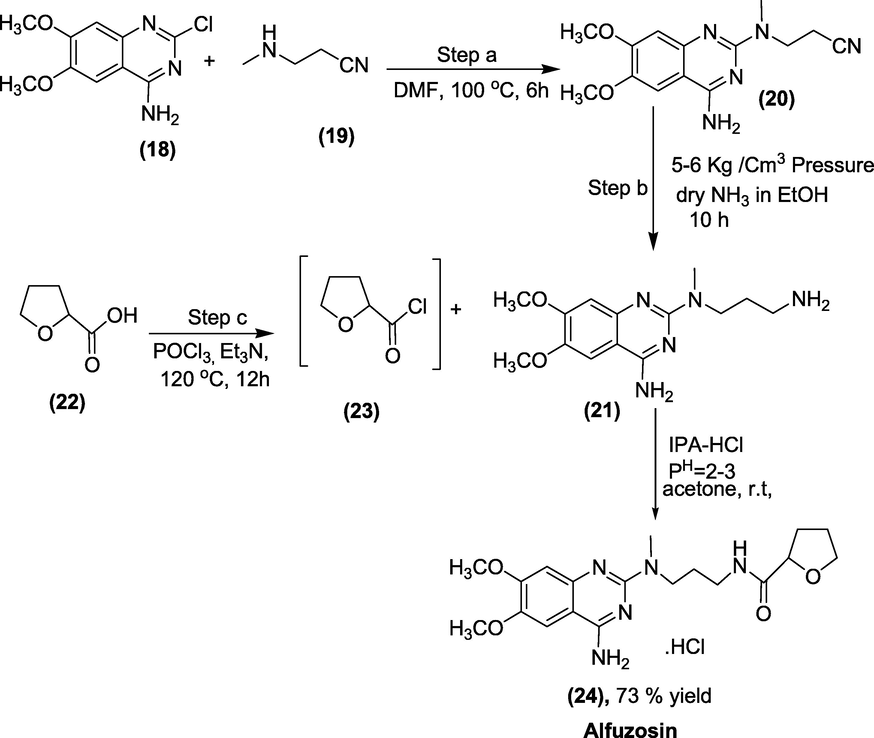
Synthesis of Alfuzosin.
3.7 Bunazosin (1988)
Bunazosin (Fig. 11), also known as andante, is an alpha-1 antagonist with the chemical formula 1-(4-(4-amino-6,7-dimethoxyquinazolin-2-yl)-1,4-diazepan-1-yl)butan-1-one. Bunazosin was created to treat benign prostatic hyperplasia (BPH). The mechanism of action involves lowering intraocular pressure by reducing aqueous outflow through the uveoscleral pathway. Intraoperative Floppy Iris Syndrome was linked to systemic Alpha-1 adrenergic receptor antagonists (IFIS). Bunazosin may have the same effect, but no research proved it as a cataract surgery risk. Its applications include the treatment of benign prostatic hyperplasia (BPH), the treatment of glaucoma, and the improvement of blood flow to the optic nerve.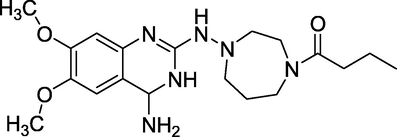
Structure of Bunazosin.
Synthesis: It is made from 2-amino-4,5-dimethoxybenzoic acid (25), which undergoes hetero-cyclisation, producing 2,4-dihydroxy-6,7-dimethoxyquinazoline (26) when reacting with sodium cyanate. Next, 2,4-dichloro-6,7-dimethoxyquinazoline is generated by replacing the hydroxyl groups of (26) with chlorine using thionyl chloride. The chlorine atom at C4 of the pyrimidine ring is replaced with an amino group in a subsequent reaction with ammonia, resulting in 4-amino-2-chloro-6,7-dimethoxyquinazoline (28). Finally, Bunazosin (30) is produced by coupling (28) with 1-(1,4-diazepan-1-yl)butan-1-one (29) as in scheme 35(Manoury et al., 1986).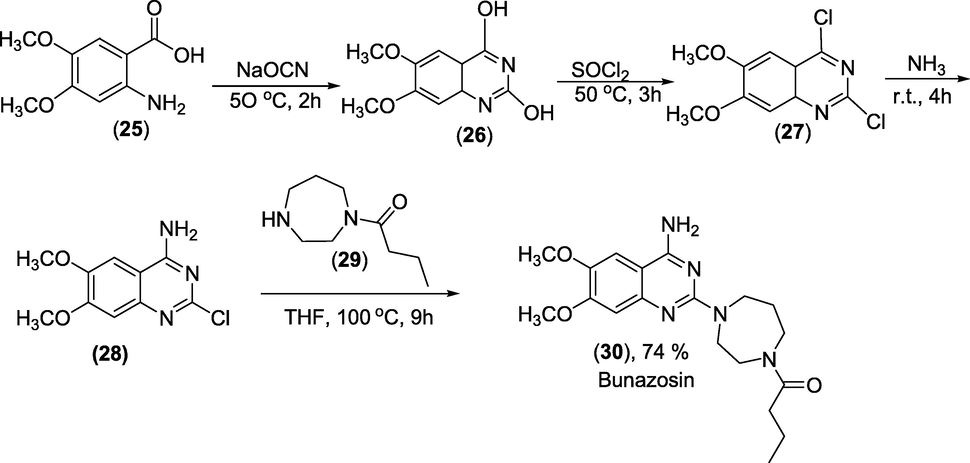
Synthesis of Bunazosin.
3.8 Trimetrexate (December 1993)
Trimetrexate (Fig. 12), also known as 5-methyl-6-[(3,4,5-trimethoxyphenyl) aminomethyl]quinazoline-2,4-diamine, is a nonclassical folic acid inhibitor that works by inhibiting the dihydrofolate reductase enzyme. It treats pneumocystis pneumonia alongside leucovorin and is researched for treating leiomyosarcoma. Trimetrexateisa methotrexate (MTX) derivative that works against transport-deficient MTX-resistant tumor cells, overcoming methotrexate resistance both acquired and natural (Wong et al., 1990; Smith et al., 2002). It is an antineoplastic and antiparasitic agent to treat pneumocystis pneumonia in AIDS patients and skin lymphoma.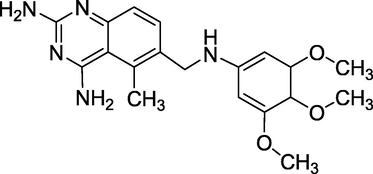
Structure of Trimetrexate.
Synthesis: To obtain Trimetrexate (39), the 2,4-diamino-6-quinazolinecarbonitrile (37) is reduced over raney nickel in the presence of benzneamine (38) at a pressure of 50 psig (Elslager et al., 1983) as represented in below scheme 36.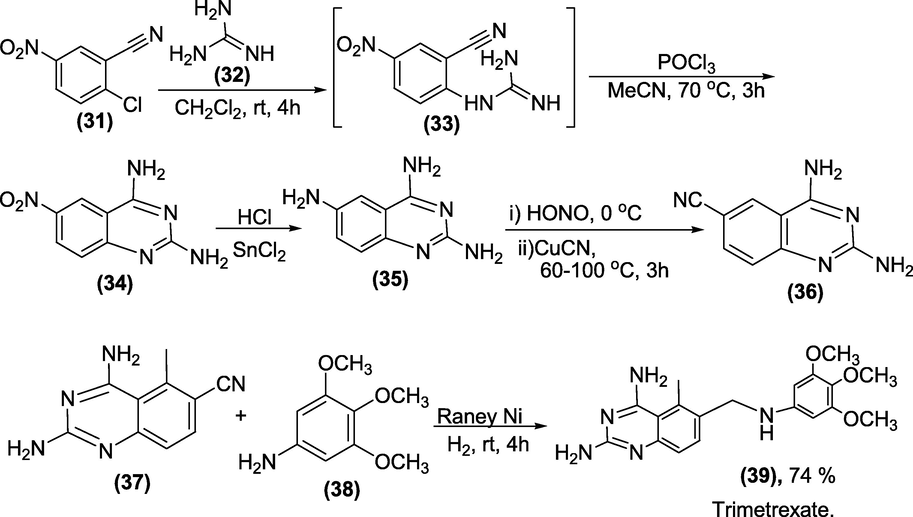
Synthesis of Trimetrexate.
3.9 Anagrelide (1997)
Agrylin/Xagrid, Shire, and anagrelide are brand names, and the chemical name is 6,7-dichloro1,5-dihydroimidazo(2,1-b)quinazolin-2(3H)-one (Fig. 13). Anagrelide works by preventing platelets from megakaryocytes from maturing. Although it is known to be a PDE inhibitor, the specific mechanism of action is unknown. It is a strong phosphodiesterase-II inhibitor (IC50 = 36 nM). PDE-3 and phospholipase A2 are both inhibited by it. According to a randomised experiment conducted by the Medical Research Council in 2005, hydroxyurea with aspirin is superior to anagrelide and aspirin for treating essential thrombocytosis (ET). Myelofibrosis, arterial thrombosis, and bleeding decreased in the hydroxyurea group, although venous thrombosis was somewhat higher (Voglova et al., 2006). It is used to treat ET, or excessive blood platelet production. It also has been used in the treatment of chronic myeloid leukemia.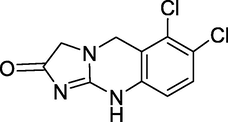
Structure of Anagrelide.
Synthesis: When 1,2-dichloro-4-nitro-3-propylbenzene (40) is combined with the ethyl ester of glycine to form an alkylated product (41). The nitro group is reduced to form aniline and converted to cyanamide (42) by reacting it with cyanogen bromide. The aliphatic would then undergo cyclisation, resulting in the development of a quinazoline ring (44). Whatever the intricacies of the sequence, amide creation between the newly generated imide and the ester would next suffice to construct the imidazolone ring, yielding anagrelide (45).Scheme 37 represents the total synthetic route (Hitoshi and Fumiyoshi, 1981).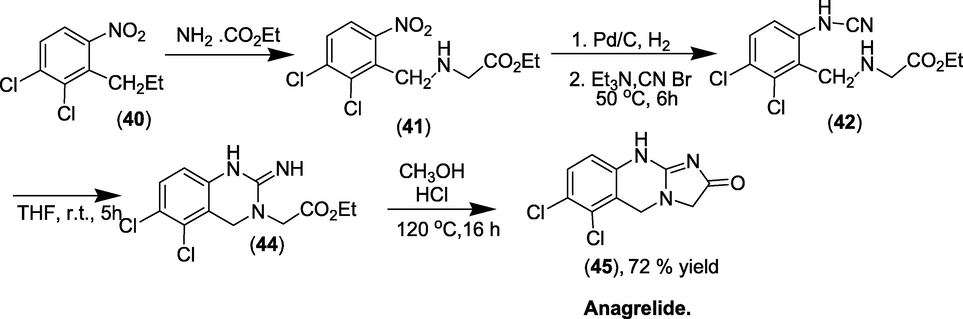
Synthesis of Anagrelide.
3.10 Gefitinib (May 2003)
Gefitinib (Fig. 14), a quinazoline-containing medication, has been approved by the US Food and Drug Administration (FDA). AstraZeneca's medication is an inhibitor of the protein kinase of the epidermal growth factor receptor (EGFR). It binds to the EGFR ATP-binding site, inactivating the anti-apoptotic ras signal transduction cascade and stopping cancer cells from growing further (Sordella et al., 2004). The drug is used to treat non-small-cell lung cancer if platinum- and taxane-based chemotherapies have failed.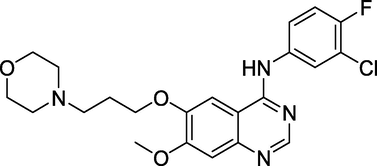
Structure of Gefitinib.
Synthesis: Methyl 3-hydroxy-4-methoxybenzoate (46) on alkylation with 1-bromo-3-chloropropane (47) produced the intermediate (48) in 94.7 percent yield. Compound (49) was obtained by nitrating the intermediate (48) with nitric acid in acetic acid, which was then reduced by powdered iron in acetic acid to provide Compound (50) with an acceptable yield of 77 %. Even after long reaction periods, catalytic hydrogenation using Raney/Ni or 5% Pd/C yielded partial conversions. Compound (53) is obtained by cyclising (50) with formamidine acetate and chlorinating it with thionyl chloride after two reactions with various amines, Gefitinib (54) was created (Scheme 38) (Li et al., 2007).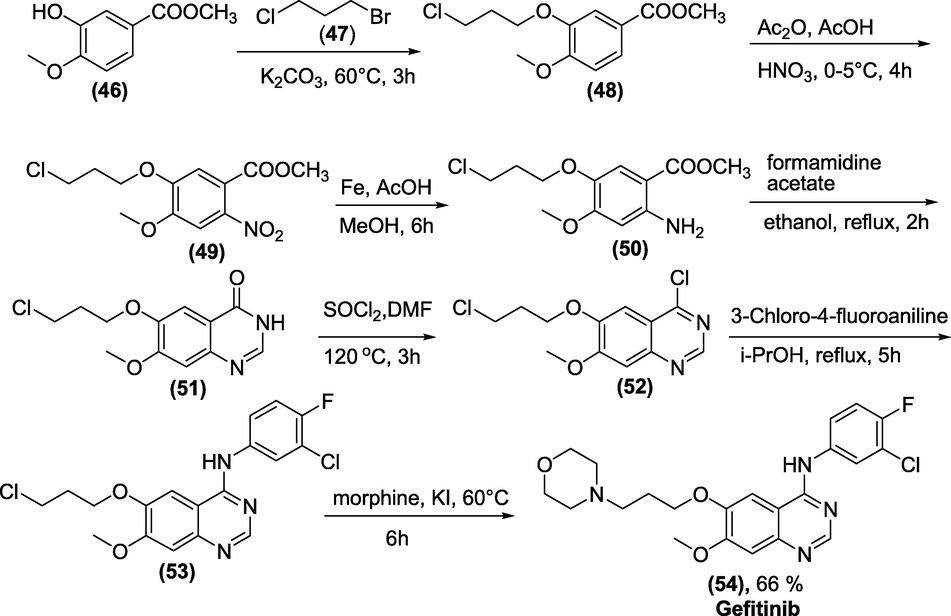
Synthesis of Gefitinib.
3.11 Erlotinib (2004)
Tarceva is the brand name for erlotinib (Fig. 15). N-(3-ethynylphenyl)6,7-bis(2-methoxyethoxy)quinazolin-4-amine is its chemical name. It is used to treat NSCLC that has progressed to other parts of the body and contains mutations in the epidermal growth factor receptor (EGFR), either an exon 19 deletion (del19) or an exon 21 (L858R) substitution mutation (Martin et al., 2015). It treats non-small cell lung cancer, advanced unresectable metastatic prostate cancer, and pancreatic cancer.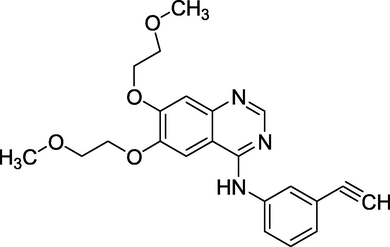
Structure of Erlotinib.
Synthesis: Erlotinib hydrochloride (63) was manufactured in seven stages, starting with 3,4-dihydroxy benzoic acid (55), as shown in Scheme 39. One essential modification in this work is reducing the 6-nitrobenzoic acid derivative (59) to the 6-aminobenzoic acid derivative (60). In the presence of palladium/charcoal (Pd/C), a cheap reagent, ammonium formate was utilised as an in situ hydrogen donor instead of hydrogen gas at high pressure to obtain intermediate (61), that on cyclisation and chlorination produced the precursor (62), that couples with 3-ethynylaniline to afford the Erlotinib hydrochloride (63) as a final product (Zhang et al., 2015).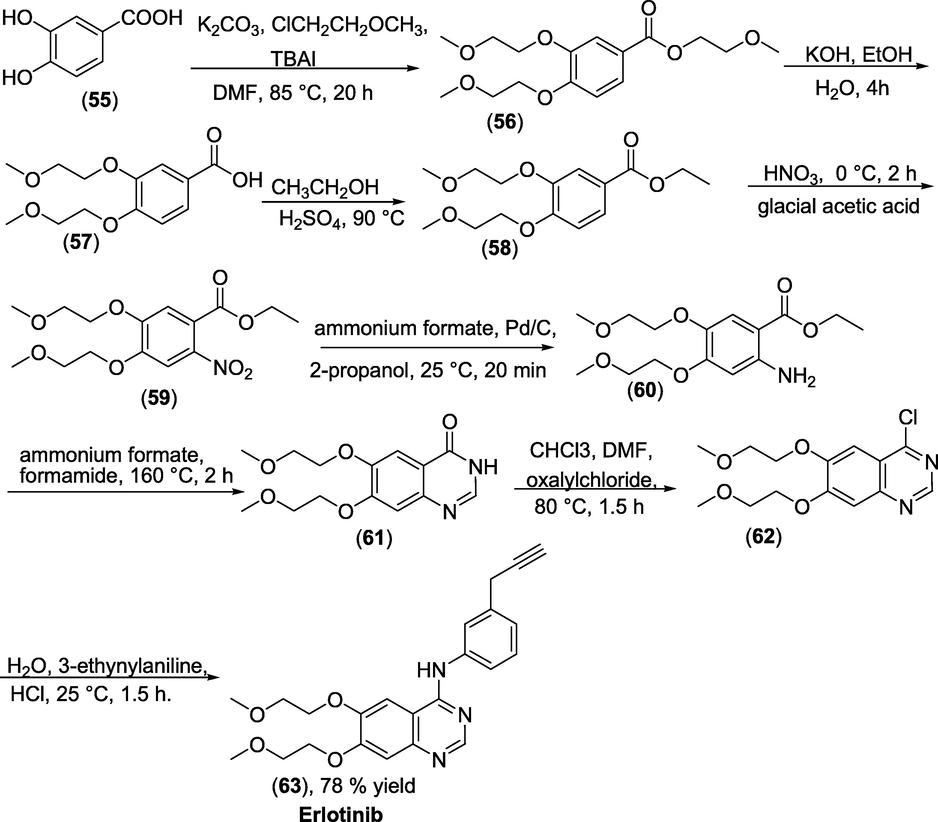
Synthesis of Erlotinib.
3.12 Nolatrexed (2005)
Nolatrexed (Fig. 16) has the chemical name 2-Amino-6-methyl-5-(4-pyridylthio)-1H-quinazolin-4-one. Nolatrexed is a thymidylate synthase inhibitor. Used for the treatment of liver cancer (Hughes et al., 1999).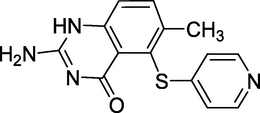
Structure of Nolatrexed.
Synthesis: The synthesis was carried out in three phases. The first step included converting 4-bromo-5-methylisatin(64) to methyl anthranilate(65) using potassium peroxydisulfate/sodium methoxide. In the final Ullmann reaction, potassium carbonate was used instead of sodium hydride, and copper catalysts were used in much smaller amounts. Furthermore, instead of using hydrogen sulfide/methanol under very acidic conditions, sodium sulphide solution was used to extract copper efficiently in almost neutral conditions (Scheme 40). These adjustments prepared nolatrexed dihydrochloride (69) in high yield and purity (Zhao et al., 2010).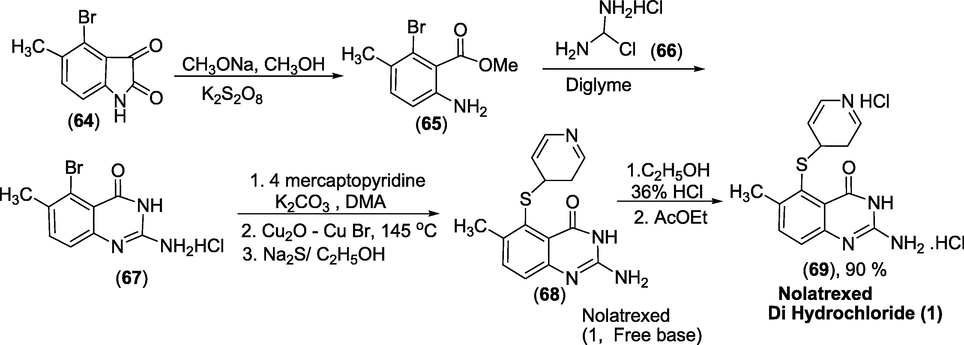
Synthesis of Nolatrexed.
3.13 Proquazone (2005)
Its trade name is Biarison is chemically known as 1-isopropyl-7-methyl-4phenylquinazolin-2(1H)-one. Proquazone (Fig. 17) is a non-steroidal anti-inflammatory drug. Its uses are cyclooxygenase inhibitors and analgesics.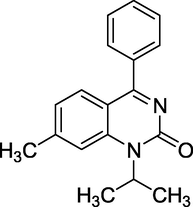
Structure of Proquazone.
Synthesis: Proquazone is prepared (https://pharmaceutical-substances.thieme.com/ps/searchresults?docUri=KD-16-0230, n.d.) as presented in scheme 41. 2- amino-4-methyl benzophenone (71) undergoes reductive coupling with acetone in the presence of sodiumborohydride to form 2-(isopropylamino)-4-methylphenyl)(phenyl)methanone (72) that, on cyclisation with urea produced the proquazone (73) in good yield.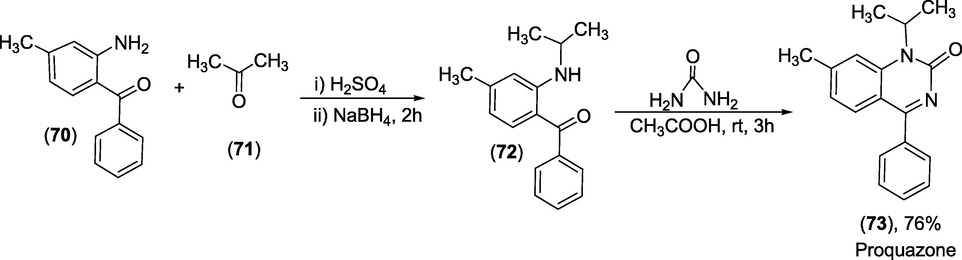
Synthesis of Proquazone.
3.14 Albaconazole (2005)
Albaconazole (Fig. 18) is an anti-fungal triazole with a wide range of effects. Its IUPAC name is 7-Chloro-3-[ (2R,3R) -3-(2,4-difluorophenyl)-3-hydroxy-4-(1,2,4-triazol-1-yl)butan-2-yl] quinazolin-4-one (Karsdal, 2009). Its anti-fungal properties include toenail fungus, distal onychomycosis, and subungual onychomycosis. It has also completed early clinical trials for the treatment of tinea pedis.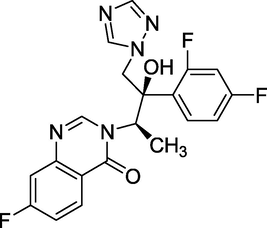
Structure of Albaconazol.
Synthesis: The Albaconazole (84) is made from 1,3-thiazolo[4,5-g]quinazolin-8(7H)-one (83). Intially,7-bromo-3H-quinazolin-4-one(75) is obtained by interacting 4-bromo-2-nitrobenzoic acid (74) with formamide under microwave irradiation. Further, the intermediate (75) from appropriate chemical transformations afford the thiazolo[4,5-g]quinazolin-8(7H)-one(82) that couples with 1-(((2R,3S)-2-(2,4-difluorophenyl)-3-methyloxiran-2-yl)methyl)-1H-1,2,4-triazole (83) to obtain the Albaconazol(84) as shown in Scheme 42(Guillon et al., 2013).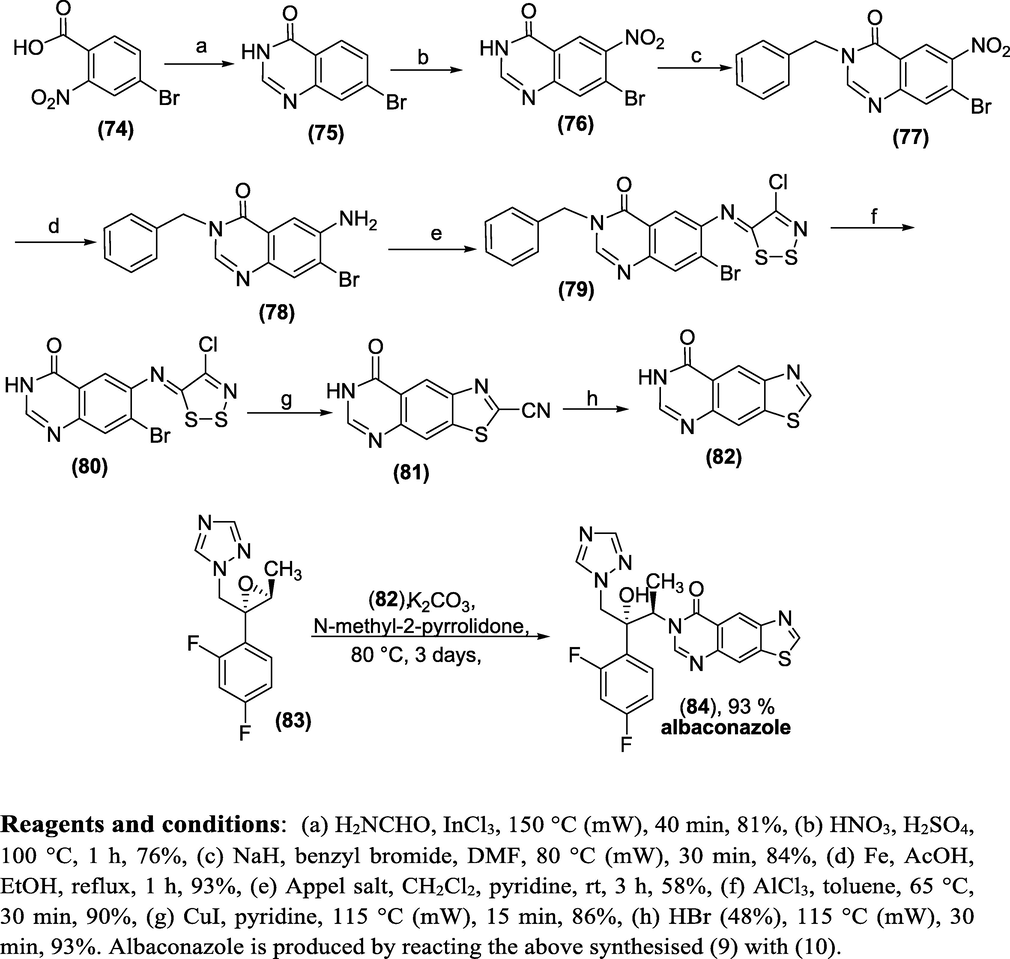
Synthesis of Albaconazole.
Reagents and conditions: (a) H2NCHO, InCl3, 150 °C (mW), 40 min, 81%, (b) HNO3, H2SO4, 100 °C, 1 h, 76%, (c) NaH, benzyl bromide, DMF, 80 °C (mW), 30 min, 84%, (d) Fe, AcOH, EtOH, reflux, 1 h, 93%, (e) Appel salt, CH2Cl2, pyridine, rt, 3 h, 58%, (f) AlCl3, toluene, 65 °C, 30 min, 90%, (g) CuI, pyridine, 115 °C (mW), 15 min, 86%, (h) HBr (48%), 115 °C (mW), 30 min, 93%. Albaconazole is produced by reacting the above synthesised (82) with (83).
3.15 Ispinesib (2006)
Ispinesib (Fig. 19) inhibits the mitotic motor protein kinesin spindle protein (KSP), causing mitotic spindle construction to be hindered, cell cycle arrest during the mitotic phase to be induced, and cell death in actively dividing tumour cells. Ispinesib may be less likely to cause peripheral neuropathy than tubulin-targeting drugs since KSP is not engaged in nonmitotic activities such as neuronal transport. IUPAC name- N-(3-aminopropyl)-N-[(1R)-1-[7-chloro-4-oxo-3-(phenylmethyl)-2-quinazolinyl]-2-methylpropyl] -4-methylbenzamide (Wang et al., 2013). It has anti-cancer characteristics and is used to treat breast cancer.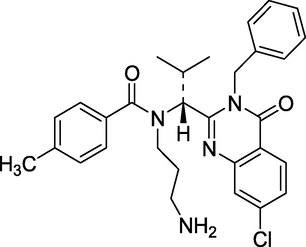
Structure of Ispinesib.
Synthesis: Swern oxidation was used to convert optically active alcohol (85) from L-valine to R-phthalimidyl aldehyde (86). The aldehyde (86) converted to oxime (87) was reacted with NCS to obtain C-chlorooxime. C-chlorooxime was then coupled with aldimine (88) by [3+2]-cycloaddition in the presence of Et3N to yield 4,5-dihydro-1,2,4-oxadiazole (89) as a 1:1 diastereomixture. Aerobic treatment of each isomer of (89) produced quinazolinone (90) in 62% and 50% yields, respectively, with partial racemisation (87 % ee each). The phthalimido moiety of (90) was further deprotected with hydrazine, resulting in the essential precursor amine (91), which couples with p-methylbenzoyl chloride to form the targeted drug Ispinesib(92) in good yield (Scheme 43) (Agrawal et al., 2012).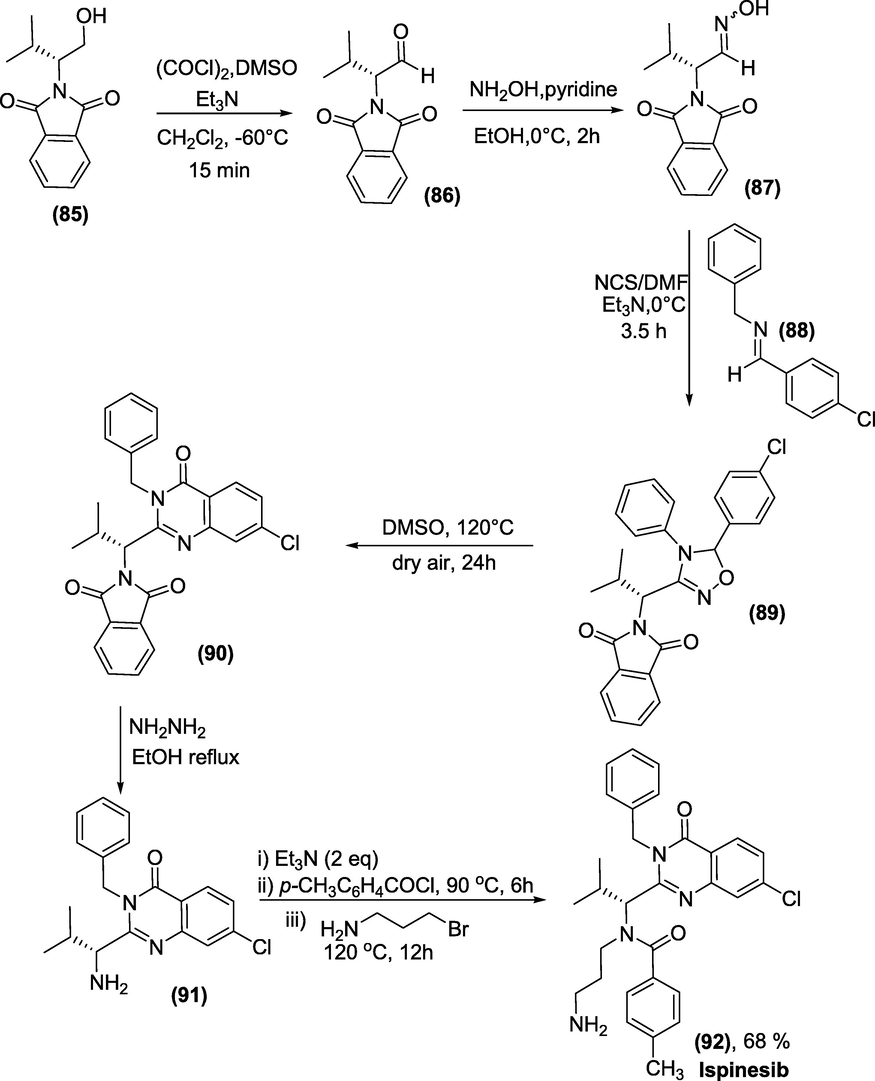
Synthesis of Ispinesib.
3.16 Balaglitazone (October 2006)
The antihyperglycemic drug balaglitazone (Fig. 20) is a thiazolidinedione derivative. Balaglitazone is a partial agonist for the peroxisome proliferator-activated receptor (PPAR) gamma and appears to have fewer negative effects than complete PPAR gamma agonists. 5-[[4-[(3-methyl-4-oxoquinazolin-2-yl)methoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione is its IUPAC name (Su and Yang, 2002). Among the applications it plays a key role in controlling insulin, triglycerides, and lipid metabolism; it's a promising target for Type 2 diabetes treatment; and Balaglitazone is a partial agonist of PPAR. Balaglitazone inhibits PPAR activity to a maximum of 52 %. As a result, Balaglitazone is considered to have fewer negative effects. In numerous animal models, it is effective at lowering blood glucose levels.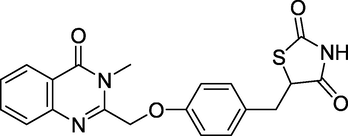
Structure of Balaglitazone.
Synthesis: In the presence of a base, 2-(chloromethyl)-3-methylquinazolin-4(3H)-one(93) combines with p-Hydroxybenzaldehyde (94) to produce ether compound (95). Balaglitazone is formed when ether (95) connects with thiazolidine-2,4-dione(96) to generate precursor(97), which is then reduced with hydrogen on palladium carbon to form Balaglitazone(98) as presented in scheme 44(Lohray et al., n.d.).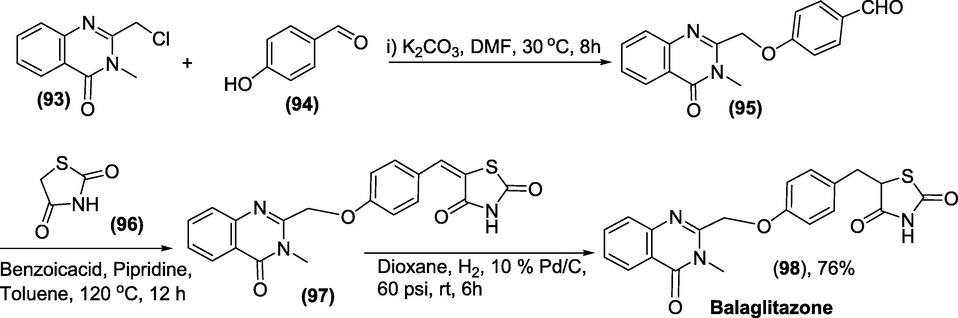
Synthesis of Balaglitazone.
3.17 Lapatinib (March 2007)
The FDA has approved Lapatinib (Fig. 21) in combination therapy for breast cancer patients already taking capecitabine (Xeloda). The medicine is marketed by GlaxoSmithKline (GSK) under the brand names Tykerb (primarily in the United States) and Tyverb (mainly in Europe) (mostly Europe and Russia). The tyrosine kinase activity of two oncogenes, EGFR (epidermal growth factor receptor) and HER2/neu, is inhibited by Lapatinib (human EGFR type 2). Overexpression of the HER2/neu gene in women has been linked to some forms of high-risk breast cancers. Lapatinib treats a specific form of breast cancer (HER2-positive). It acts by slowing or preventing cancer cell proliferation (Erickson et al., 2014).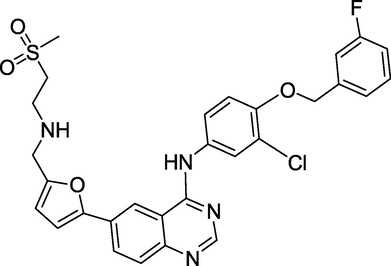
Structure of Lapatinib.
Synthesis: The total synthesis of Lapatanib was presented in scheme 45(Erickson et al., 2014).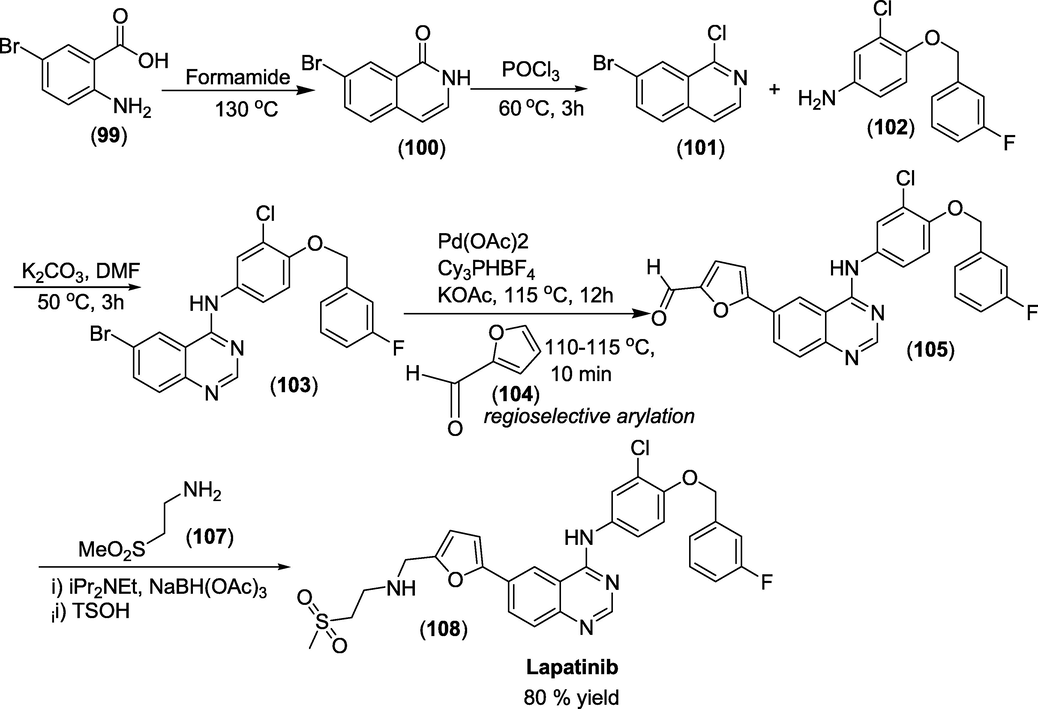
Synthesis of Lapatinib.
The key intermediate 6-bromo-N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)quinazolin-4-amine(1 0 3)was obtained from 2-amino-5-bromobenzoic acid (99) through suitable chemical transformations shown in scheme 45. Next, the palladium catalysed regioselective arylation of furfural(1 0 4) with 6-bromo-N-(3-chloro-4-((3-fluorobenzyl)oxy)phenyl)quinazolin-4-amine(1 0 3) yield the Lapatanib (1 0 8) (Scheme 45). This crucial step is an atom-inefficient Suzuki cross-coupling reaction, resulting in a considerable reduction in synthesis time.
3.18 Vandetanib (April 2011)
Vandetanib's (Fig. 22) marketing name is Zactima, and its chemical name is N-(4bromo-2-fluorophenyl)-6-methoxy-7-[(1-methylpiperidin-4-yl)methoxy] quinazolin-4-amine. It is a vascular endothelial growth factor receptor (VEGFR) and epidermal growth factor receptor (EGFR) antagonist (EGFR). It works by inhibiting tyrosine kinases. The drug also has a third target: it inhibits the activity of RET-tyrosine kinase, a crucial growth driver in particular thyroid cancer (Selvam and Kumar, 2011). It treats patients with unresectable locally advanced or metastatic illnesses with symptomatic or progressing medullary thyroid carcinoma.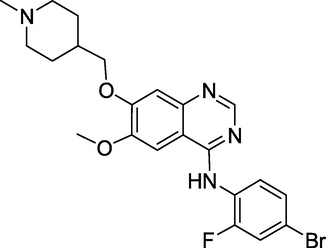
Structure of Vandetanib.
Synthesis: Synthesis is carried out utilising an unstable 4-chloroquinazoline scaffold, which necessitates the use of strong chemicals as well as sequential protection and deprotection processes. 7-(benzyloxy)-4-chloro-6-methoxyquinazoline hydrochloride (1 0 9) on appropriate chemical transformation produce the precursor (1 1 5) that on N-methylation affords the target Vandetanib in good yield. Vandetanib was synthesised using the Dimroth rearrangement in the primary quinazoline-producing step. The precursor7-((piperidin-4-yl)methoxy)-N-(4-bromo-2-fluorophenyl)-6-methoxyquinazolin-4-amine was obtained. The total synthesis for Vandetanib was presented in Scheme 46(Brocklesby et al., 2017).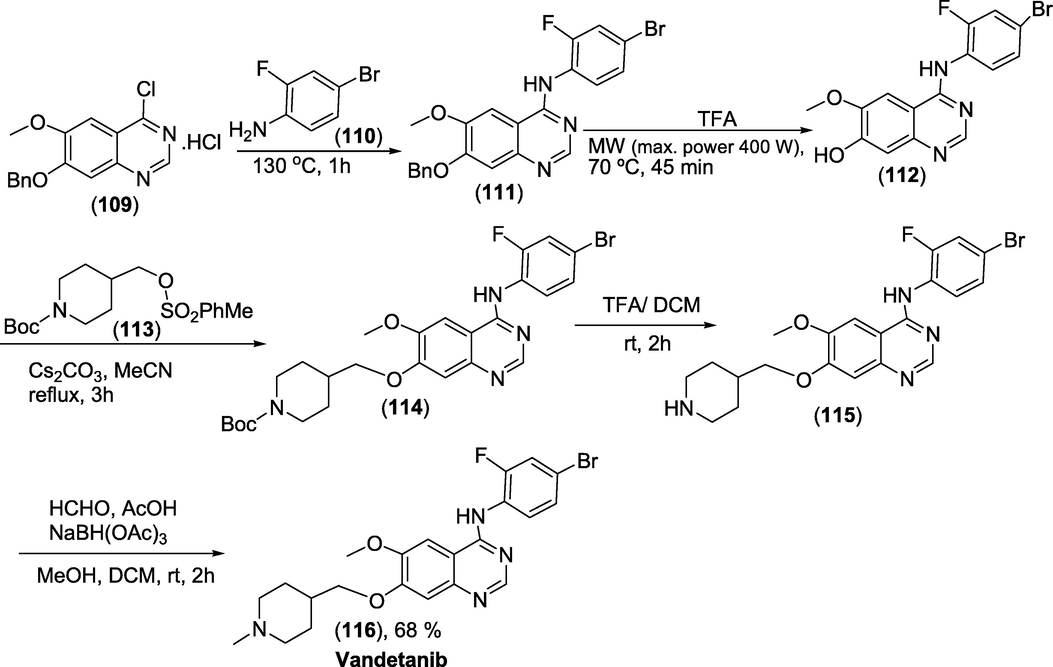
Synthesis of Vandetanib.
3.19 Afatinib (2013)
Afatinib (Fig. 23) treats non-small cell lung cancer. It is available under the trade names Gilotrif and others (NSCLC) (Ghorab et al., 2013; Alvarado et al., 2006; Al-Amiery et al., 2014). It belongs to the class of drugs known as tyrosine kinase inhibitors. Afatinib, like Lapatinib and Neratinib, is a protein kinase inhibitor that inhibits human epidermal growth factor receptor 2 (Her2) and epidermal growth factor receptor (EGFR) kinases irreversibly. Afatinib is effective against EGFR mutations targeted by first-generation TKIs like Erlotinib or Gefitinib and against less prevalent mutations resistant to these medicines (Selvam and Kumar, 2011). It treats non-small cell lung, breast cancer, and cancers resistant to Gefitinib and Erlotinib.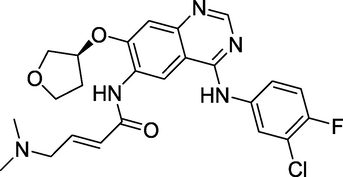
Structure of Afatinib.
Synthesis: Scheme 47 illustrates the total synthesis of Afatanib (Kovacevic et al., 2018). The desired intermediate (1 2 1)was synthesised from commercially available 4-fluoro anthranilic acid (1 1 7), which was iodinated with potassium iodide and sodium periodate to yield (1 1 8), and the 4-quinazolinone ring was closed conventionally using formamide acetate to produce intermediate (1 1 9). Next, the etherification and introduction of 3-chloro-4-fluoroaniline to intermediate (1 1 9) afforded the key intermediate (1 2 1), which on Cu promoted C-N coupling with trans-N,N’-dimethyl-1,2-cyclohexanediamine procured the desired Afatanib (1 2 3) in 56 % yield.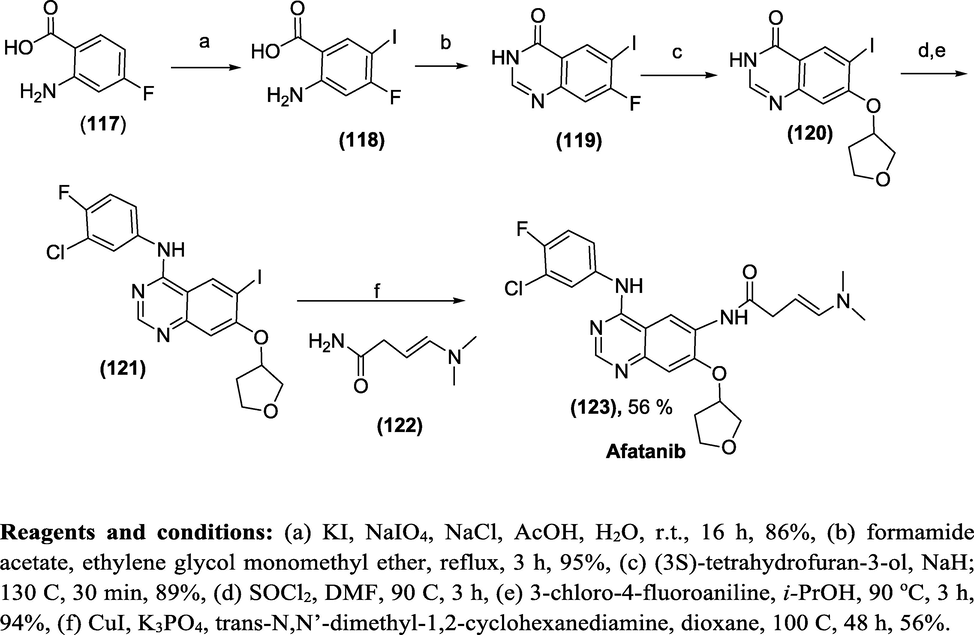
Synthesis of Afatinib.
Reagents and conditions: (a) KI, NaIO4, NaCl, AcOH, H2O, r.t., 16 h, 86%, (b) formamide acetate, ethylene glycol monomethyl ether, reflux, 3 h, 95%, (c) (3S)-tetrahydrofuran-3-ol, NaH; 130 oC, 30 min, 89%, (d) SOCl2, DMF, 90 oC, 3 h, (e) 3-chloro-4-fluoroaniline, i-PrOH, 90 °C, 3 h, 94%, (f) CuI, K3PO4, trans-N,N’-dimethyl-1,2-cyclohexanediamine, dioxane, 100 oC, 48 h, 56%.
3.20 Evodiamine (1915)
21-methyl-3,13,21-triazapentacyclo [11.8.0.02,10.04, 9.015,20]henicosa-2(10),4,6,8,15,17,19-heptaen-14-one is its chemical name. Evodiamine (Fig. 24) is made from the Evodia spp. plant family, and proved to lower fat intake in the mouse trials. This substance has been found in various over-the-counter bodybuilding supplements, but neither its fat-burning benefits nor possible hazards and side effects have been empirically established (Viola, 2016). Its properties include increasing body warmth and inhibiting the growth of some cancer cells. Furthermore, when taken with certain medicines, it alters your metabolism and impacts catecholamine output from your adrenal glands.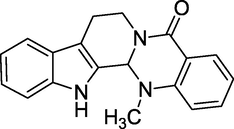
Structure of Evodiamine.
Synthesis: The synthetic path for constructing the drug Evodiamine(1 2 6) was shown in scheme 48(Wang et al., 2018), with triethoxymethane as a cosolvent, one-pot synthesis of evodiamine from tryptamine (1 2 4) and N-methylisatoic anhydride (1 2 5). The transformation proceeded efficiently in dimethylacetamide (DMA) at 100 °C with the addition of 1.0 equiv of TFAA and 1.5 equiv of 1,4-diazabicyclo[2.2.2]octane (DABCO) as shown in below scheme 48.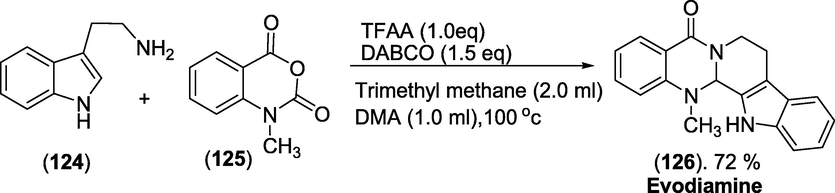
Synthesis of Evodiamine.
3.21 Quinethazone (1960)
The drug chemically known as 7-chloro-2-ethyl-4-oxo-1,2,3,4-etrahydroquinazoline-6-sulfonamide is marketed as Hydromox (Fig. 25). Common side effects include dizziness, dry mouth, nausea, and low potassium levels (Kobayashi et al., 2001). It is used as a thiazide diuretic to treat hypertension.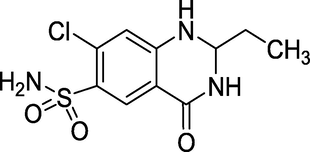
Structure of Quinethazone.
Synthesis:The total synthesis of quinethazone is shown in Scheme 49 (Cohen et al., 1960). The 5-chloro-o-acetotoluide (1 2 8) was produced by the acetylation of 5-chlorotoluidine (1 2 7). Then it was chlorosulfonated and then treated with aqueous ammonia to generate 5-chloro-4-sulfamyl-o-acetotoluidide (1 2 9), which on oxidation with permanganate,afford the 4-chloro-5-sulfamyl-N-acetylanthranilic acid (1 3 0). Next, intermediate (1 3 0)on deacetylation with a base or an acid, 4- chloro-5-sulfamylanthranilic acid (1 3 1) is produced. Finally, fusing(1 3 1) with propionamideand cyclising with urethane give the target quinethazone(1 3 3) in good yield.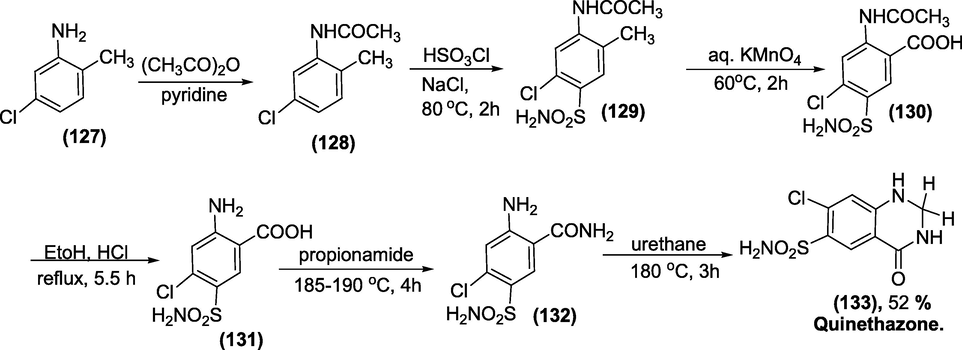
Synthesis of quinethazone.
3.22 Febrifugine (1949)
3–3-[(2S,3R)-3-Hydroxypiperidin-2-yl]-2-oxo propyl quinazolin-4(3H)-one is the chemical name for Febrifugine (Fig. 26). The quinazolinone alkaloid was discovered from the Chinese herb Dichroa febrifuge. However, it is also present in the garden plant Hydrangea (McLaughlin and Evans, 2010). It possesses anti-malarial effects, and it's halogenated derivative, halofuginone, is utilised as a coccidiostat in veterinary medicine.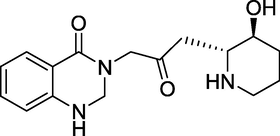
Structure of Febrifugine.
Synthesis: Scheme 50 presents the total synthesis of Febrifugine (Smullen et al., 2018). The 2-acetyl furan (1 3 4) was transformed to the pyridine (1 3 5), which on treatment with NH2OH and methylation, produced the intermediate (1 3 5), which combined with acetaldehyde and hydrogenated with PtO2 to afford cis-piperidine (1 3 6). Next, it was oxidised to the ketone, alpha- brominated, N-protected and reacted with 4-(3H)-quinazolinone to furnish the N,O protected Febrifugine. Finally,the N,O-deprotection gave access to Febrifugine (1 3 8).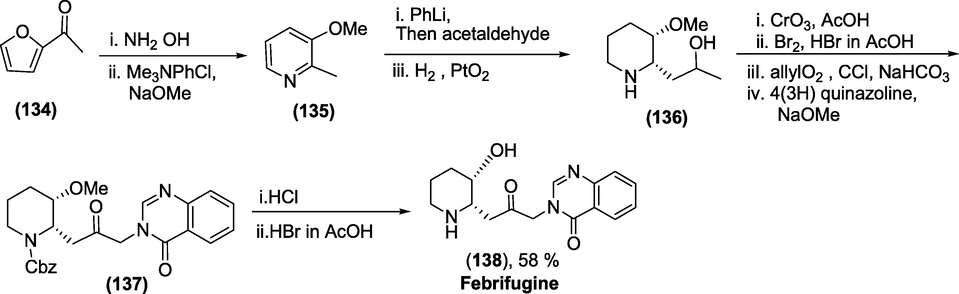
Synthesis ofFebrifugine.
3.23 Fenquizone (1969)
Idrolone is a diuretic that belongs to the low-ceiling sulfonamide diuretic class. Its chemical name is 7-chloro-4-oxo-2-phenyl-1,2,3,4 tetrahydroquinazoline-6-sulfonamide. Fenquizone (Fig. 27) is most commonly used to treat oedema and hypertension.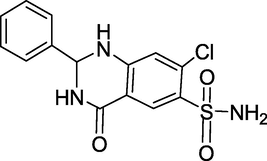
Structure of Fenquizone.
Synthesis: Fenquizone(1 4 1) was generated in good yields in water under neutral conditions by the reaction of anthranilamide (1 3 9)withbenzaldehyde (1 4 0) mediated by β-cyclodextrin (Scheme 51). With a slight loss of enzymatic activity, β -cyclodextrin can be recovered and reused (Ramesh et al., 2012).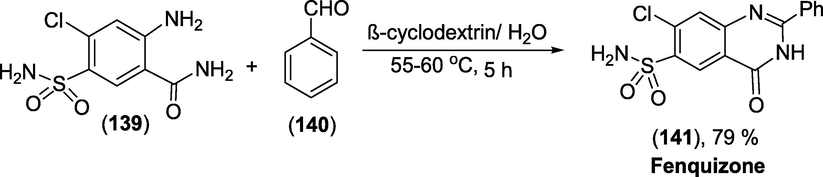
Synthesis of Fenquizone.
3.24 Metolazone (1970)
It is sold under the brand names Zytanix, Metoz, Zaroxolyn, and Mykrox and is a thiazide-like diuretic. Metolazone (Fig. 28) reduces the quantity of water reabsorbed into the bloodstream by the kidney in an indirect manner, resulting in a drop in blood volume and an increase in urine volume. Metolazone reduces blood pressure and prevents extra fluid from building up in heart failure patients. Metolazone is sometimes used with loop diuretics like furosemide or bumetanide, although these combinations can cause dehydration and electrolyte imbalances (Fischer and Ganellin, 2006). Furthermore, it is primarily utilised to treat congestive heart failure and hypertension.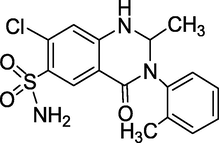
Structure of Metolazone.
Synthesis: Metolazone is produced from5-chloro-2-methylaniline (1 4 2), which on treatment with Ethyl chloroformate, acylates the amino group, yielding 5-chloro-N-ethoxycarbonyl-2-methylaniline (1 4 3). Following a reaction with chlorosulfonic acid and ammonia, the intermediate (1 4 3) is converted into 4-sulfonamido-5-chloro-N-ethoxycarbonyl-2-methylaniline (1 4 4). On oxidation with potassium permanganate, the methyl group of this molecule yields 5-sulfonamido-4-chloro-N-ethoxycarbonyl anthranilic acid (1 4 5). It cycles into the corresponding anhydride (1 4 6) after being treated with thionyl chloride. When this comes into contact with o-toluidine, it forms 2-amino-5-aminosulfonyl-4-chloro-o-toluolbenzamide (1 4 7). Finally, metolazone (1 4 8) is produced by reacting the intermediate with dimethylacetal acetic acid (Scheme 52) (Vardanyan and Hruby, 2006).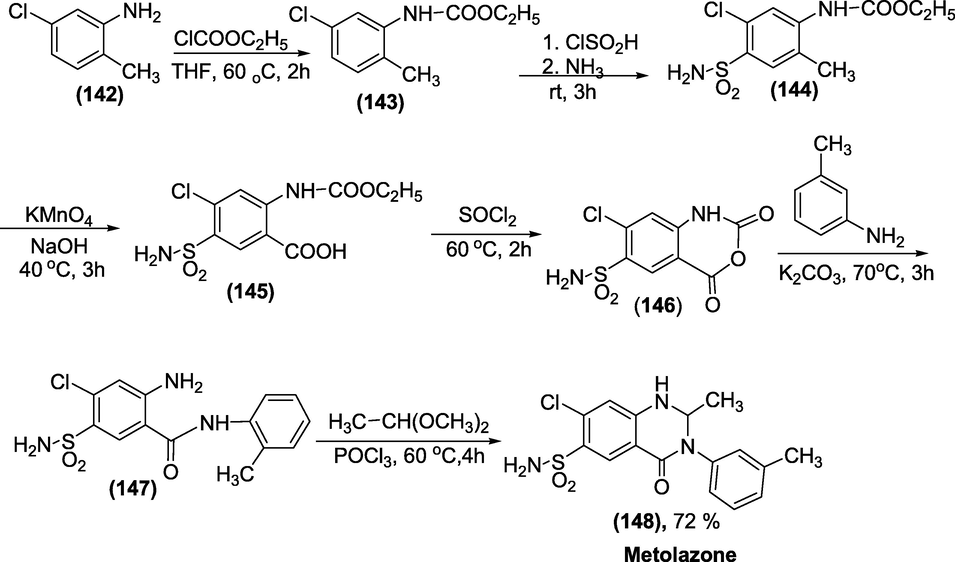
Synthesis of Metolazone.
3.25 Linagliptin (2011)
Ondero is its brand name, and its chemical name is 8-[(3R)-3-aminopiperidin-1-yl]-7-(but-2yn-1-yl)-3-methyl-1-[(4-methylquinazolin-2-yl)methyl]-3,7-dihydro-1H-purine-2,6-dione. It's a DPP-4 inhibitor produced by Boehringer Ingelheim for type 2 diabetes research. A Phase III clinical trial of linagliptin (Fig. 29) found that the medicine effectively lowers blood sugar levels (Huang et al., 2016).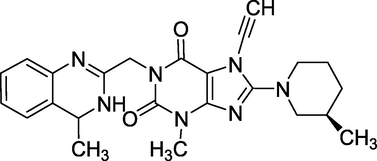
Structure of Linagliptin.
Synthesis: The synthesis of Linagliptin (1 5 5)involves(Scheme 53) cyclisation of 1-(2-aminophenyl)ethanone (1 4 9) with 2-chloroacetonitrile in the presence of HCl to afford 2-(chloromethyl)-4-methylquinazoline (1 5 0). The compound (1 5 0) is condensed with 8-bromo-7-(but-2-yn-1-yl)-3-methyl-1H-purine-2,6(3H,7H)-dione (1 5 1) in the presence of Na2CO3 as a basic reagent, giving 8-bromo-7-(but-2-yn-1-yl)-3-methyl-1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione (1 5 2).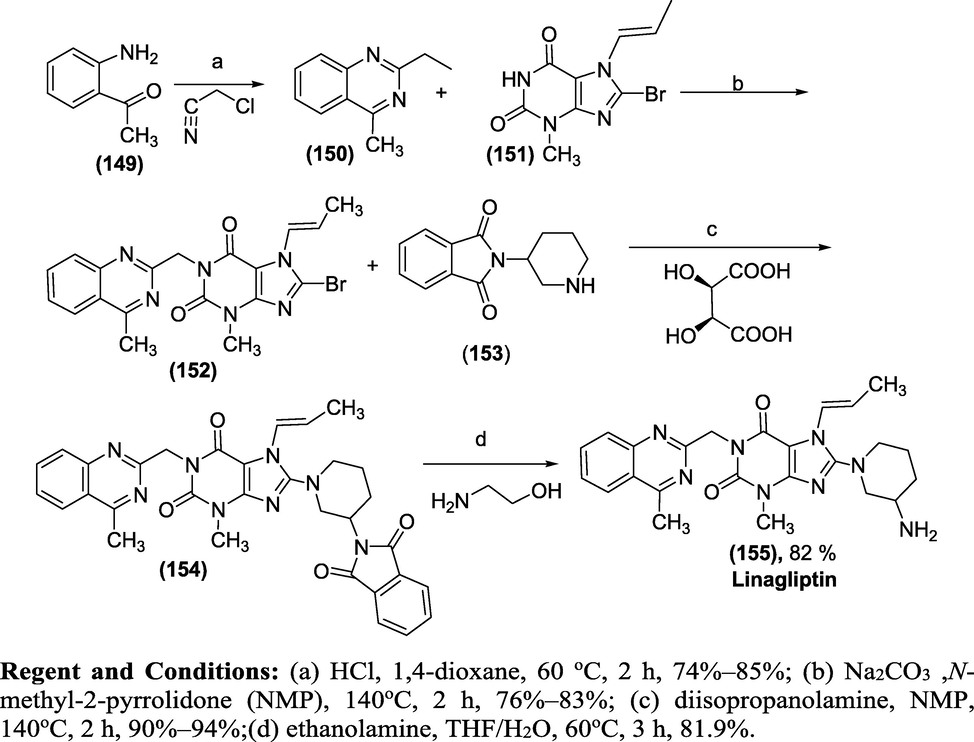
Synthesis of Linagliptin.
Regent and Conditions: (a) HCl, 1,4-dioxane, 60 °C, 2 h, 74%–85%; (b) Na2CO3, N-methyl-2-pyrrolidone (NMP), 140 °C, 2 h, 76%–83%; (c) diisopropanolamine, NMP, 140 °C, 2 h, 90%–94%;(d) ethanolamine, THF/H2O, 60 °C, 3 h, 81.9%.
Subsequently, the condensation of (1 5 2) with (R)-2-(piperidin-3-yl)isoindoline-1,3-dione (1 5 3) and D-tartaric acid using diisopropylethylamine as a basic reagent provided (R)-7-(but-2-yn-1-yl)-8-(3-(1,3-dioxoisoindolin-2-yl) piperidin-1-yl)-3-methyl1-((4-methylquinazolin-2-yl)methyl)-1H-purine-2,6(3H,7H)-dione(1 5 4). Finally, Compound (1 5 4) was converted to the desired Linagliptin (1 5 5) in 82 % yield via aminolysis using ethanolamine as the aminolysis agent(Scheme 53).
3.26 Quazinone (1980)
Quinazone (Fig. 30 was developed and marketed in the 1980's for the treatment of heart disease under the brand name Dozonone, which is chemically known as (3R)-6-chloro-3-methyl-5,10dihydroimidazo[2,1-b] quinazolin-2(3H)-one. It works as a selective PDE3 inhibitor (Daly et al.,1985; Belz et al., 1985). It's a cardiotonic and vasodilator medication.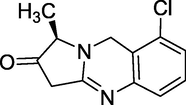
Structure of Quazinone.
Synthesis: Quinazone was prepared by combining 1-chloro-(2-chloromethyl)nitrobenzene (1 5 6) with (S)-ethyl-2-aminopropanoate (1 5 7) to form (1 5 8), then hydrogenating the nitro group to form NH2 to form intermediate (1 5 9). The intermediate reacts with cyanogen bromide followed by cyclisation resulting in the formation of the product Quazinone (1 6 1) (Scheme 54) (Chodnekar and Kaiser, n.d.).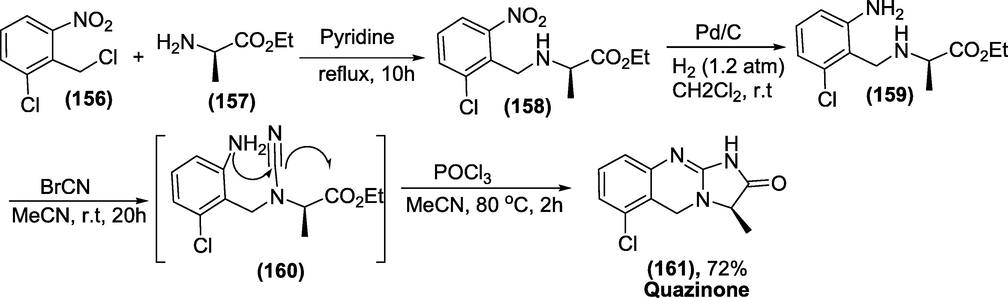
Synthesis of Quazinone.
3.27 Raltitrexed (1996)
Tomudex is the commercial name for N-[(5-{methyl[(2-methyl-4-oxo-1,4dihydroquinazolin-6-yl)methyl]amino}-2-thienyl)carbonyl]-L-glutamic acid, and is manufactured by AstraZeneca. Raltitrexed (Fig. 31) is a chemotherapeutic medication chemically related to folic acid and belongs to the folate antimetabolites class. It prevents the formation of thymidylate by inhibiting dihydrofolate reductase, an enzyme involved in generating tetrahydrofolate. After polyglutamylation, Raltitrexed is fully functional. Raltitrexed hinders the production of DNA and RNA, which are necessary for the growth and survival of both normal and malignant cells, by blocking the formation of precursor pyrimidine nucleotides. It is an antimetabolite medicine used in cancer chemotherapy; it is a thymidylate synthase inhibitor. Raltitrexed has been employed in treating colorectal cancer since 1998.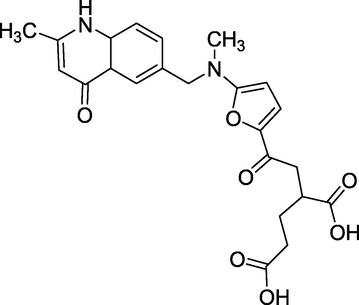
Structure of Raltitrexed.
Synthesis: The synthesis of Raltitrexed (1 6 9) was presented in Scheme 55(Cao et al., 2003). Initially, monocoupling 2,5-thiophenedicarboxylic acid (1 6 2) with diethyl L-glutamate. Next, the intermediate (1 6 2)on consecutive modified Curtius reaction,N-methylation, deprotection of BOC, condensation with (bromomethyl)quinazolinone, and saponification reactions afford the targeted drug molecule Raltitrexed (1 6 9).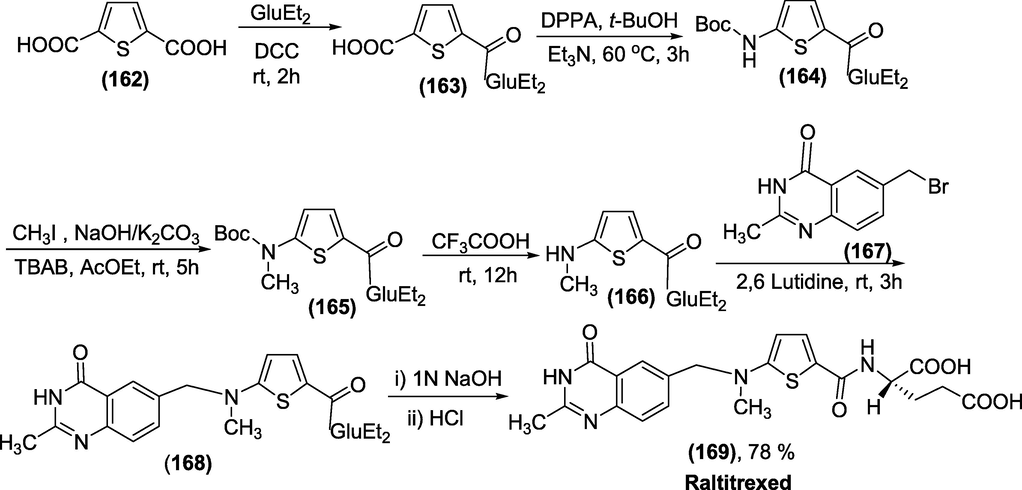
Synthesis of Raltitrexed.
3.28 EBE-A22 (2011)
PD153035 is another name for EBE-A22 (Fig. 32), which is chemically known as N-(3-bromophenyl)-6,7-dimethoxyquinazolin-4-amine (Selvam and Kumar, 2011). It is an antineoplastic and intercalating agent (capable of insertingitself between DNA bases). These are employed in DNA research.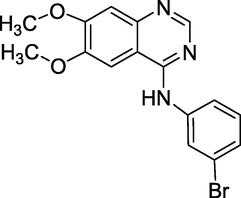
Structure of EBE-A22.
Synthesis: The synthetic path for EBE-A22 was shown in scheme 56 (Johnström et al., 1998). Initially, formamide was heated with 2-amino-4,5-dimethoxybenzoic acid(1 7 0) to 150 °C for 7 h. The obtained brown solid (1 7 1) was refluxed with POCl3 for 4 h. Excess POCl3 should be vaporised before adding CHCl3 and NaOH to the wash residue. Compound (1 7 2) was produced by heating 3-bromoaniline and a minor amount of 3-bromoaniline hydrochloride with CH2Cl2 and 2-propanol for 2 h at 60 °C (Scheme 56).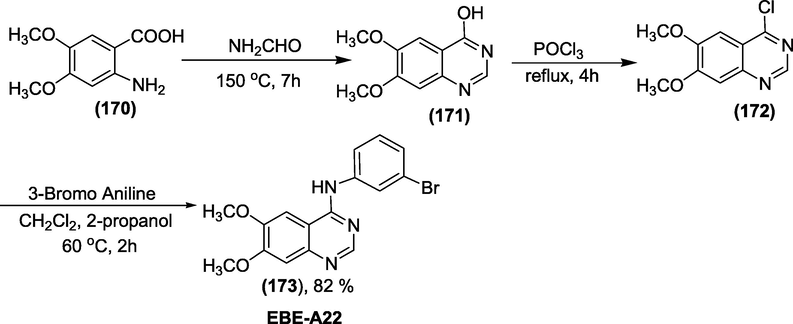
Synthesis of EBE-A22.
3.29 NSC127213 (2015)
Tetrazolo [1,5-c]quinazoline is the chemical name for NSC127213. NSC127213 (Fig. 33) can treat or prevent inflammatory, autoimmune, allergy, and ophthalmic illnesses by inhibiting H1R and H4R (Selvam and Kumar, 2011).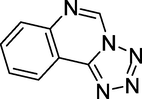
Structure of NSC127213.
Synthesis: The synthesis of the fused tetrazole NSC127213 (Wan et al., 2010) was achieved by treating quinazolin-4(3H)-one (1 7 4) with 0.5 equivalents of BOP reagent in the presence of DBU and sodium azide as the nitrogen nucleophile(Scheme 57). The reaction proceeds via BOP-promoted homo-dimerisation of 3,4-dihydro-4-methylenequinazoline to obtain the targeted fused tetrazole NSC127213(1 7 5).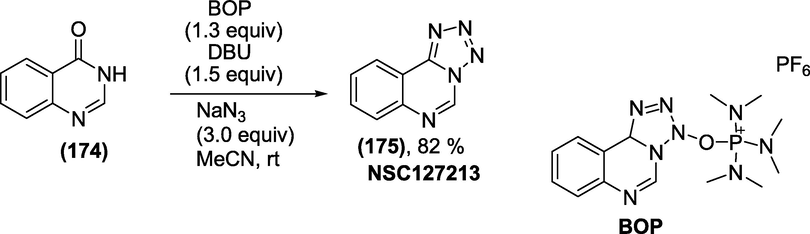
Synthesis of NSC127213.
4 Conclusions
Quinazoline and its derivatives play a crucial role in medicinal chemistry, evident in the chemical makeup of a wide range of FDA-approved medications, clinical candidates, and bioactive compounds. The findings state that several strategies have been designed to create these scaffolds. However, there is still an opportunity to develop highly effective protocols for synthesising and biological assessment of various quinazoline derivatives. The development of molecular hybrids of quinazolines with a wide range of therapeutic values is receiving a lot of interest these days. Additionally, understanding presently prescribed quinazoline-based medications, their therapeutic value, and the methods for developing them is crucial for any researcher in medicinal chemistry. Substantial synthetic advancements have been made in meeting this requirement. In this review, we presented a precise account of recent advancements and noteworthy developments in synthesising different novel quinazoline analogues (2017–2023) focusing on design and therapeutic activity. We briefed the synthetic protocols of significant quinazoline-based drugs currently in use. We have also demonstrated how these skeletons can serve as artificial substrates for further alterations. Considering that quinazoline and quinazolinone compounds exhibit various biologically significant characteristics, this review will be beneficial for medicinal chemistry research and future drug development from the straightforward and varied nature of synthetic methods for producing quinazoline- and quinazolinone-based heterocycles.
Author contributions
The authors S.N.M.B. and H.B.B. designed the project; R.K.V.P., G.B.K., H.S.S., and N.R. helped prepare the manuscript, while S.N.M.B, H.B.B. and S.B.J. provided scientific guidance and helped to draft the manuscript. All authors have read and agreed to the submission of the manuscript.
Acknowledgements
Dr. S. N. Murthy Boddapati thanks the University of KwaZulu- Natal authorities for the fellowship. All the authors thank the Acharya Nagarjuna University, for the constant support and motivation. H. S. Saini expresses his gratitude to CSIR for its SRTP program 2020.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Significant pharmacological activities of benzoquinazolines scaffold. Pharmacol. Rep.. 2023;75:223-235.
- [CrossRef] [Google Scholar]
- Balaglitazone: a second generation peroxisome proliferator-activated receptor (PPAR) gamma (γ) agonist. Mini Rev. Med. Chem.. 2012;12:87-97.
- [CrossRef] [Google Scholar]
- Antioxidant and antimicrobial activities of novel quinazolinones. Med. Chem. Res.. 2014;23:236-242.
- [CrossRef] [Google Scholar]
- Microwave-assisted Niementowski reaction. Back to the roots. Tetrahedron Lett.. 2002;43:3911-3913.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of new quinazoline and cinnoline derivatives as potential atypical antipsychotics. Chem. Biodivers.. 2006;1:106-117.
- [CrossRef] [Google Scholar]
- The isolation and identification of the precursors ad reaction products in the clandestine manufacturing of Methaqualone and mecloqualone. J. Forensic Sci.. 1985;30:1022-1047.
- [Google Scholar]
- Synthesis of quinazoline and quinazolinone derivatives via ligand-promoted ruthenium-catalyzed dehydrogenative and deaminative coupling reaction of 2-aminophenyl ketones and 2-aminobenzamides with amines. Org. Lett.. 2019;21:3337-3341.
- [CrossRef] [Google Scholar]
- Synthesis of 6-Chloro-5-sulfamoyl-and 6-Chloro-3- sulfamoylanthranilic acid derivatives. Chem. Pharm. Bull.. 1979;27:1287-1298.
- [CrossRef] [Google Scholar]
- Dose-response following single administrations of a new cardiac performance enhancer RO 13–6438 in normal volunteers. J. Cardiovas. Pharmacol.. 1985;7:86-90.
- [CrossRef] [Google Scholar]
- Preet Mohinder Singh Bedi. Dihydrofolate reductase inhibitors: patent landscape and phases of clinical development (2001–2021) Expert Opin. Therap. Patents. 2022;32:1079-1095.
- [CrossRef] [Google Scholar]
- ZurKenntniss der phenmiazinderivative. Ber. Dtsch. Chem. Ges.. 1895;28:279-293.
- [CrossRef] [Google Scholar]
- High ligand efficiency quinazoline compounds as novel A2A adenosine receptor antagonists. Eur. J. Med. Chem.. 2022;241:114620
- [CrossRef] [Google Scholar]
- An alternative synthesis of Vandetanib (Caprelsa™) via a microwave accelerated Dimroth rearrangement. Tetrahedron Lett.. 2017;58:1467-1469.
- [CrossRef] [Google Scholar]
- A. one-pot synthesis of 2-aryl-2, 3-dihydro-4 (1H)-quinazolinones by use of samarium iodide. J Heterocycl Chem.. 2002;39:1271-1272.
- [CrossRef] [Google Scholar]
- New synthesis of thymidylate synthase inhibitor raltitrexed. Synth. Commun.. 2003;33:3519-3526.
- [CrossRef] [Google Scholar]
- The characterization of etaqualone and differentiation from its 3- and 4-ethyl analogues. Microgram J.. 2012;9:47-51.
- [Google Scholar]
- Dehydrogenative synthesis of quinolines, 2-aminoquinolines, and quinazolines using singlet diradical Ni(II)-catalysts. J. Org. Chem.. 2019;84:2626-2641.
- [CrossRef] [Google Scholar]
- Chen,C.yi., He, F., Tang, G., Yuan, H., Li, N., Wang, J., Faessler, R., 2018. Synthesis of Quinazolines via an Iron-Catalyzed Oxidative Amination of N−H Ketimines. J. Org. Chem. 83, 2395-2401. 10.1021/acs.joc.7b02943
- Recent research progress and outlook in agricultural chemical discovery based on quinazoline scaffold. Pest. Biochem. Physiol.. 2022;184:105122
- [CrossRef] [Google Scholar]
- Chodnekar, M.S., Kaiser, A.,Imidazo[2,1-b]quinazolin-2(3H)-ones and pharmaceutical compositions for treatment and prophylaxis of cardiac insufficiency and cardiac failure. US 4256748.
- A review of anti-malarial activity of two or three nitrogen atoms containing heterocyclic compounds. Med. Chem. Res.. 2020;29:1723-17500.
- [CrossRef] [Google Scholar]
- Conconi, M.T., Marzaro, G., Urbani, L., Zanusso, I., Ferrarese, A., Guiotto, Chilin,A., 2013.Quinazoline-based multityrosine kinase inhibitors: synthesis, modeling, antitumor and antiangiogenic properties. Eur. J. Med. Chem. 67 373–383. 10.1016/j.ejmech.2013.06.057.
- Connolly, D.J., Cusack, D., O'sullivan, T.P., Guiry, P.J.,2005. Synthesis of quinazolinones and quinazolines. Tetrahedron, 61, 10153–10202.http://dx.doi.org/10.1016/j.tet.2005.07.010.
- Daly, P.A., Chatterjee, K., Viquerat, C.E.,1985.R013-6438, A new inotrope-vasodilator: systemic and coronary hemodynamic effects in congestive heart failure. Am. J. Cardio. 55, 1539-1544. 10.1016/0002-9149(85)90969-5.
- Molecular iodine catalysed benzylic sp3 C-H bond amination for the synthesis of 2-arylquinazolines from 2- aminobenzaldehydes, 2-aminobenzophenones and 2-aminobenzyl alcohols. Synlett. 2018;29:979-985.
- [CrossRef] [Google Scholar]
- Cu(II)-glucose sustainable catalyst for the synthesis of quinazolinones in a bio-mass derived solvent 2- MethylTHF and application for the synthesis of diproqualone. ACS Sustain. Chem. Eng.. 2018;6:14283-14291.
- [CrossRef] [Google Scholar]
- Synthesis and antitumor and anti-malarial properties of trimetrexate and related 6-[(Phenylamino)methyl]-2,4-quinazolinediamines. J. Med. Chem.. 1983;26:1753-1760.
- [CrossRef] [Google Scholar]
- Synthesis of Lapatinib via direct regioselective arylation of furfural. Tetrahedron Lett.. 2014;55:6007-6010.
- [CrossRef] [Google Scholar]
- Fischer, J., Ganellin, C.R.,2006.Analogue-based Drug Discovery. John Wiley & Sons.p. 457.
- Fischer J., Ganellin C.R., 2006. Analogue-based Drug Discovery. John Wiley & Sons, p. 455.
- GB patent 936902, Quinazolinone Derivatives, issued 1963-09-18, assigned to Beiersdorf.
- Recent advances in synthesis of quinazoline-2,4(1H,3H)-diones: Versatile building blocks in N-heterocyclic compounds. Appl. Organomet. Chem.. 2022;36:e6631.
- [Google Scholar]
- Ghorab, W.M., El-Sebaey , S.A., Ghorab, M.M., 2023. Design, synthesis and Molecular modeling study of certain EGFR inhibitors with a quinazolinone scaffold as anti-hepatocellular carcinoma and Radio-sensitizers.Bioorganic Chemistry,131,106310. 10.1016/j.bioorg.2022.106310.
- Synthesis and pharmacophore modeling of novel quinazolines bearing a biologically active sulfonamide moiety. Acta Pharm.. 2013;63:1-18.
- [CrossRef] [Google Scholar]
- Supuran. Biological evaluation, radiosensitising activity and structural insights of novel halogenated quinazoline-sulfonamide conjugates as selective human carbonic anhydrases IX/XII inhibitors. Bioorg. Chem.. 2021;107:104618
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of novel quinazoline-triazole hybrid compounds with potential use in Alzheimer's disease. Bioorg. Med. Chem. Lett.. 2020;30:127404
- [CrossRef] [Google Scholar]
- Design, synthesis and characterisation of novel 2-(2,4-disubstitutedthiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-κB and AP-1 mediated transcription activation and as potential anti-inflammatory agents. Eur. J. Med. Chem.. 2009;44:2184-2189.
- [CrossRef] [Google Scholar]
- Guiles, J., Sun, X., Critchley, L.A., Bertino, J., Green, Dallmann, G., . McHenry, C.S., Janjic, N., 2009.Quinazolin-2-ylamino-quinazolin-4-ols as novel non-nucleoside inhibitors of bacterial DNA polymerase III, Bioorg. Med. Chem. Lett. 19, 800–802. 10.1016/j.bmcl.2008.12.038
- Discovery of a novel broad-spectrum anti-fungal agent derived from albaconazole. ACS Med. Chem. Lett.. 2013;4:288-292.
- [CrossRef] [Google Scholar]
- Solvent dependent copper-catalysed indolyl C3-oxygenation and N1 cyclisation reactions: selective synthesis of 3 H-indol-3-ones and indolo [1, 2-c] quinazolines. J. Org. Chem.. 2018;83:3889-3896.
- [CrossRef] [Google Scholar]
- Therapeutic potential of quinazoline derivatives for Alzheimer's disease: A comprehensive review. Eur. J. Med. Chem.. 2022;227:113949
- [CrossRef] [Google Scholar]
- An appraisal of anticancer activity with structure-activity relationship of quinazoline and quinazolinone analogues through EGFR and VEGFR inhibition: A review. Drug Develop. Res.. 2022;83:859-890.
- [CrossRef] [Google Scholar]
- Quinazoline-4(3H)-one derivatives as novel and potent inhibitors of soluble epoxide hydrolase: Design, synthesis and biological evaluation. Bioorg. Chem.. 2020;99:103736
- [CrossRef] [Google Scholar]
- Alpha-blockers for the Treatment of Benign Prostatic Hyperplasia. Urol. Clin. North Am.. 2016;43:311-323.
- [CrossRef] [Google Scholar]
- Transition metal-free synthesis of 2-aryl quinazolines via alcohol dehydrogenation. Mol. Catal.. 2023;542:113110
- [CrossRef] [Google Scholar]
- Synthesis and reactions of 2-chloro-3,4-dihydrothienopyrimidines and quinazolines. J. Heterocycl. Chem.. 1981;18:67-70.
- [CrossRef] [Google Scholar]
- Howard S.,2008. Illustrated Pharmacology Memory Cards: PharMnemonics. Minireview. p. 13.
- Palladium-catalysed three-component tandem process: One-pot assembly of quinazolines. Org. Lett.. 2018;20:3083-3087.
- [CrossRef] [Google Scholar]
- Synthesis and characterisation of process-related impurities of antidiabetic drug linagliptin. Molecules. 2016;21:1041.
- [CrossRef] [Google Scholar]
- Phase I studies with the nonclassical antifolate nolatrexed dihydrochloride (AG337, THYMITAQ) administered orally for 5 days. Clin. Cancer Res.. 1999;5:111-118.
- [Google Scholar]
- Quinazolinone and quinazoline derivatives: recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci.. 2016;11:1-14.
- [Google Scholar]
- Synthesis of [methoxy-11C]PD153035, a selective EGF receptor tyrosine kinase inhibitor. J. Label. Compd. Radiopharma.. 1998;41:623-629.
- [CrossRef] [Google Scholar]
- Potential analgesics. Part I. Synthesis of substituted 4-quinazolones. J. Ind. Chem. Soc.. 1951;28:344-346.
- [Google Scholar]
- Kankan, R.N., Ramachandra, D., Birari R.D., Process for the Preparation of AlfuzosinHydrochloride.https://patents.google.com/patent/US20100256370A1/en.
- Recent advances on quinazoline derivatives: a potential bioactive scaffold in medicinal chemistry. Chem. Eng.. 2021;5:73.
- [CrossRef] [Google Scholar]
- A comparison of glycemic control, water retention, and musculoskeletal effects of balaglitazone and pioglitazone in diet-induced obese rats. Eur. J. Pharmacol.. 2009;616(1–3):340-345.
- [CrossRef] [Google Scholar]
- Synthesis of quinazolinones from anthranilamides and aldehydes via metal-free aerobic oxidation in DMSO. Tetrahedron Lett.. 2014;55:2340-2344.
- [CrossRef] [Google Scholar]
- Capsaicin-like anti-obese activities of evodiamine from fruits of Evodia rutaecarpa, a vanilloid receptor agonist. Planta Med.. 2001;67(7):628-633.
- [CrossRef] [Google Scholar]
- Kovacevic, T., Mesic, M., Avdagic, A., Zegarac, M., 2018.An alternative synthesis of the non-small cell lung carcinoma drug afatinib. Tetrahedron Lett. 59, 4180-4182. 10.1016/j.tetlet.2018.10.026.
- Palladium-catalysed three-component tandem process: one-pot assembly of quinazolines. Org. Lett.. 2018;20:3083-3087.
- [CrossRef] [Google Scholar]
- Kunes, J., Bazant, J., Pour, M., K. Waisser, K., Sloś arek, M.,Janota, J., 2000. Quinazoline derivatives with antitubercular activity.Farmaco. 55, 725–729. 10.1016/S0014-827X(00)00100-2.
- Doxazosin and terazosin suppress prostate growth by inducing apoptosis: clinical significance. J. Urol.. 2003;169:1520-1525.
- [CrossRef] [Google Scholar]
- Design, synthesis, and bioactivity of novel quinazolinone scaffolds containing pyrazole carbamide derivatives as anti-fungal agents. Curr. Issues Mol. Biol.. 2022;44:5605-5621.
- [CrossRef] [Google Scholar]
- BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702-4711.
- [CrossRef] [Google Scholar]
- One-pot solvent-free synthesis of 2, 3-disubstituted 4(3H)- quinazolinones catalysed by long-chain double SO3H-functionalized Brønsted acidic ionic liquids under microwave irradiation. J. Iran. Chem. Soc.. 2015;12:897-901.
- [CrossRef] [Google Scholar]
- Synthesis of Gefitinib from Methyl 3-Hydroxy-4-methoxy-benzoate. Molecules. 2007;12:2160.
- [CrossRef] [Google Scholar]
- The 3D-QSAR analysis of 4 (3H)-quinazolinone derivatives with dithiocarbamate side chains on thymidylate synthase. Bioorg. Med. Chem.. 2006;14:1425-1430.
- [CrossRef] [Google Scholar]
- Bio-compatible eutectic mixture for multi-component synthesis: a valuable acidic catalyst for synthesis of novel 2,3- dihydroquinazolin-4(1H)-one derivatives. Catal. Commun.. 2012;27:179-183.
- [CrossRef] [Google Scholar]
- Lohray, V.B., Lohray, B.B., Paraselli, R.B., Gurram, R.M., Ramanujam, R.,Chakrabarti, R., Pakala, S.K.S., Novel heterocyclic compounds process for their preparation and pharmaceutical compositions containing them and their use in the treatment of diabetes and related diseases, US patent, WO9741097A2.
- Synthesis, biological evaluation and molecular modelling insights of 2-arylquinazoline benzamide derivatives as anti-tubercular agents. J. Mol. Struct.. 2020;1218:128493
- [CrossRef] [Google Scholar]
- Synthesis and antihypertensive activity of a series of 4-amino-6,7-dimethoxyquinazoline derivatives. J. Med. Chem.. 1986;29:19-25.
- [CrossRef] [Google Scholar]
- FDA drug approval summary erlotinib (Tarceva) tablets. The Oncologist. 2015;10:461-466.
- [CrossRef] [Google Scholar]
- Dihydroxylation of vinyl sulfones: Stereoselective synthesis of (+)- and (−)-febrifugine and halofuginone. J. Org. Chem.. 2010;75:518.
- [CrossRef] [Google Scholar]
- Efficient synthesis of 2,3-dihydroquinazolin-4(1H)-ones catalysed by titanium silicon oxide nanopowderin aqueous media. Synth. Commun.. 2017;47:121-130.
- [CrossRef] [Google Scholar]
- Recent studies of nitrogen containing heterocyclic compounds as novel antiviral agents: a review. Bioorg. Chem.. 2021;114:105076
- [CrossRef] [Google Scholar]
- Reactions of anthranilamide and o-aminoacetophenone with benzil and benzoin. J. Org. Chem.. 1969;34:887-892.
- [CrossRef] [Google Scholar]
- Moreira, N.M., Ingrid T. M.,Jhonathan R. N., Opatz, T., Oliva, G., Rafael V. C. G., Arlene .G.C., 2023. Copper-Catalysed Synthesis of Pyrrolo[1,2-c]quinazolines and Pyrrolo[2,1-a]isoquinolines and Antiplasmodial Evaluation.J. Org. Chem. 88, 8781–8790. 10.1021/acs.joc.3c00616
- Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res.. 1997;57:4838-4848.
- [Google Scholar]
- Facile access to 2-Selenoxo-1,2,3,4-tetrahydro-4-quinazolinone Scaffolds and corresponding diselenides via cyclization between methyl anthranilate and isoselenocyanates: synthesis and structural features. Molecules. 2022;27:5799.
- [CrossRef] [Google Scholar]
- Pan, J., Ma, L., Tang, Y,X., Tian, Y., Lin, Y.H., Zhang, L.J., Feng G.B., Lu, G.M., 2022. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential NF-jb inhibitors. Arabian Journal of Chemistry, 15 , 103908. 10.1016/j.arabjc.2022.103908.
- Parveen K., G., Vijesh T., Joshi, R.K., Meena N.,2022. Nanocatalyzed synthetic approach for quinazoline and quinazolinone derivatives: A review (2015–present). Synth. Commun., 52, 795-826. 10.1080/00397911.2022.2041667.
- On the polarography of 2-methyl-3-(2-methylphenyl)-3,4-dihydroquinazolinone-(4) methaqualone, Dormutil) and 2-methyl-3-(2-ethylphenyl-3,4-dihydroquinazolinone-(4) (ethinazone, Aolan) Die Pharmazie. 1967.On;22:643-650.
- [Google Scholar]
- Philips, A., Raja, D., Arumugam, A., Dr. Lin, W.Y.,Senadi G.C.,2021.Copper-Catalysed Oxidative C−C Cleavage of Carbohydrates: An Efficient Access to Quinazolinone Scaffolds.Asian J. Org. Chem. 10, 1795–1800. 10.1002/ajoc.202100317.
- Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids. RSC Adv.. 2020;10:41353-41392.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of new 2–substituted–4–amino-quinolines and -quinazoline as potential anti-fungal agents. Bioorg. Med. Chem. Lett.. 2022;72:128877
- [CrossRef] [Google Scholar]
- A concise aqueous phase supramolecular synthesis of 2-phenyl-2,3-dihydroquinazolin-4(1H)-one derivatives. Tetrahedron Lett.. 2012;53:6936-6939.
- [CrossRef] [Google Scholar]
- Ultrasoundassisted preparation of copper(I) oxide nanocubes: high catalytic activity in the synthesis of quinazolines. Chem. Cat. Chem.. 2017;9:1292-1297.
- [CrossRef] [Google Scholar]
- Stereodivergent desymmetrization of phenols en route to modular access to densely functionalized quinazoline and oxazine Scaffolds. J. Org. Chem.. 2023;88:1600-1612.
- [CrossRef] [Google Scholar]
- Romero, A.H., Rodríguez, N., Oviedo, H., 2019.2-Aryl-quinazolin-4(3H)-ones as an inhibitor of leishmania folate pathway: In vitro biological evaluation, mechanism studies and molecular docking. Bioorganic Chemistry, 83, 145-153. 10.1016/j.bioorg.2018.10.028,
- The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther.. 2001;1:85-94.
- [Google Scholar]
- Structure-activity relationship studies based on quinazoline derivatives as EGFR kinase inhibitors (2017–Present) Pharmaceuticals. 2023;16:534.
- [CrossRef] [Google Scholar]
- Sasmal, S., Balasubrahmanyam, D., Reddy, H.R.K.,Balaji, G., Srinivas, G., Cheera, S.,. Abbineni, C.,.Sasmal,P.K., Khanna, I., Sebastian, V.J., Jadhav, V.P., Singh, M.P., Talwar, R., Elster, Hogberg, L.Y., 2012. Design and optimisation of quinazoline derivatives as melanin concentrating hormone receptor 1 (MCHR1) antagonists: part 2.Bioorg. Med. Chem. Lett. 22, 3163–3167. 10.1016/j.bmcl.2012.03.049.
- Synthesis of 3-Oxa- and 3-Aza-1-dethiacepham analogs. Synthesis. 1989;677–80
- [CrossRef] [Google Scholar]
- Trimetrexate in the treatment of recurrent or advanced leiomyosarcoma of the uterus: a phase II study of the Gynecologic Oncology Group. Gynecol. Oncol.. 2002;84:140-144.
- [CrossRef] [Google Scholar]
- Chemical synthesis of Febrifugine and analogues. Bioorg. Med. Chem.. 2018;26:2199-2220.
- [CrossRef] [Google Scholar]
- Gefitinib-sensitising EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163-1167.
- [CrossRef] [Google Scholar]
- Reductive cyclisation of nitro and azide compounds with aldehydes and ketones promoted by metallic samarium and catalytic amount of iodine. Aust. J. Chem.. 2002;55:695-697.
- [CrossRef] [Google Scholar]
- One-pot synthesis of quinazolines via elemental sulfur-mediated oxidative condensation of nitriles and 2-(Aminomethyl)anilines. Adv. Synth. Catal.. 2022;364:3600-3606.
- [CrossRef] [Google Scholar]
- A survey of reported synthesis of Methaqualone and some positional and structural isomers. Forensic Sci. Int.. 2001;122:142-149.
- [CrossRef] [Google Scholar]
- Vardanyan, R.S., Hruby, V.J.,2006. Synthesis of Essential Drugs, 1st Edition. 10.1016/B978-044452166-8/50036-4.
- Vardanyan R.S., HrubyV.J..2006. Adrenoblocking Drugs. Synthesis of Essential Drugs, p.161-177.
- Deciphering the enzymatic target of a new family of antischistosomal agents bearing a quinazoline scaffold using complementary computational tools. J. Enzyme Inhib. Med. Chem.. 2020;35:511-523.
- [CrossRef] [Google Scholar]
- Treatment of advanced thyroid cancer with targeted therapies: ten years of experience. Endocrine-Related Cancer. 2016;23(4):R185-R205.
- [CrossRef] [Google Scholar]
- Combination of imatinib and anagrelide in treatment of chronic myeloid leukemia in blastic phase. Vnitr̆níLékar̆ství. 2006;52:819-822.
- [Google Scholar]
- ZD1839 (Iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res.. 2002;62:5749-5754.
- [Google Scholar]
- Phosphonium- and benzotriazolyloxy-mediated bond-forming reactions and their synthetic applications. Synlett 2010:1143-1169.
- [CrossRef] [Google Scholar]
- Rh-and Cu-cocatalyzed aerobic oxidative approach to quinazolines via [4+ 2] C-H annulation with alkyl azides. Org. Lett.. 2016;18:2150-2153.
- [CrossRef] [Google Scholar]
- Discovery of quinazoline compound as a novel nematicidal scaffold. Pest. Biochem. Physiol.. 2023;189:105310
- [CrossRef] [Google Scholar]
- One-pot total synthesis of evodiamine and its analogues through a continuous biscyclization reaction. Org. Lett.. 2018;20:6380-6383.
- [CrossRef] [Google Scholar]
- Synthesis of quinazolinone-fused tetrahydroisoquinolines and related polycyclic scaffolds by iodine-mediated sp3 C-H amination. J. Org. Chem.. 2023;88:1061-1074.
- [CrossRef] [Google Scholar]
- Oxidative radical skeletal rearrangement induced by molecular oxygen: Synthesis of quinazolinones. Org. Lett.. 2013;15:2842-2845.
- [CrossRef] [Google Scholar]
- ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res.. 2002;62:4645-4655.
- [Google Scholar]
- Metabolic disposition of trimetrexate, a nonclassical dihydrofolate reductase inhibitor in rat and dog. Drug Metab. Dispos.. 1990;18(6):980-986.
- [Google Scholar]
- Rh-catalyzed annulation of ortho-C– H bonds of 2-arylimidazoles with 1, 4, 2-dioxazol-5-ones toward 5-arylimidazo [1, 2-c] quinazolines. Adv. Synth. Catal.. 2018;360:1111-1115.
- [CrossRef] [Google Scholar]
- Mild synthesis of quinazolines from 2,2,2-trichloroethyl imidates and 2-aminophenyl ketones. Tetrahedron Lett.. 2023;116:154313
- [CrossRef] [Google Scholar]
- Design, synthesis and biological evaluation of sulfamoylphenyl-quinazoline derivatives as potential EGFR/CAIX dual inhibitors. Eur. J. Med. Chem.. 2021;216:113300
- [CrossRef] [Google Scholar]
- Synthesis and evaluation of novel erlotinib–NSAID conjugates as more comprehensive anticancer agents. ACS Med. Chem. Lett.. 2015;6:1086-1090.
- [CrossRef] [Google Scholar]
- Palladium catalysed tandem reaction of quinazolinone-based nitriles with arylboronic acids: synthesis of 2-(4-arylquinazolin-2-yl) anilines. Adv. Synth. Catal.. 2018;360:3260-3265.
- [CrossRef] [Google Scholar]
- The cascade synthesis of quinazolinones and quinazolines using an α-MnO2 catalyst and tert-butyl hydroperoxide (TBHP) as an oxidant. Chem. Commun.. 2015;51:9205-9207.
- [CrossRef] [Google Scholar]
- A new method for synthesis of nolatrexed dihydrochloride. Org. Process Res. Develop.. 2010;14:346-350.
- [CrossRef] [Google Scholar]
- Pd-Catalyzed tandem reaction of N-(2-cyanoaryl)benzamides with arylboronic acids: synthesis of quinazolines. Org. Biomol. Chem.. 2018;16:8596-8603.
- [CrossRef] [Google Scholar]
- 2-Substituted quinazolines: Partial agonistic and antagonistic ligands of the constitutive androstane receptor (CAR) Eur. J. Med. Chem. 2023:115631.
- [CrossRef] [Google Scholar]




