

Translate this page into:
An overview on generation and general properties of N-heterocyclic carbenes: Applications of 1,2,4-triazolium carbenes as metal free organocatalysts
⁎Corresponding author. annacunha@id.uff.br (Anna Claudia Cunha)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
N-Heterocyclic carbenes (NHCs) have been widely explored in the areas of catalysis, organometallic and medicinal chemistry. In particular, NHCs catalyze the formation of C–C and C-heteroatom bounds with high regioselectivity and stereoselectivity through umpolung and non-umpolung processes for unconventional access to create a vast scope of heterocycle molecules, including β-lactones, tetrahydroquinolines and pyrroloquinoles. Here we summarized the generation and general properties of NHC and important NHC-catalyzed reactions, which typically involve species such as Breslow intermediates, homoenolates, enolates and α, β-unsaturated acyl azoliums, among others. Examples of aerobic oxidative/oxygenative carbene catalysis, specifically highlighting NHC-catalyzed reactions involving oxygen and strategies employed for activation of oxygen in NHC-catalytic processes are discussed in detail.
The catalytic use of NHCs has been one of the most powerful tools in the chemist's arsenal, with applications to commercially important protocols. Given this context, the relevance of N-heterocyclic carbene chemistry is described in this review.
Keywords
1,2,4-triazolium carbene
Organocatalysis
Cascade reaction

- 2,6-DTBP
-
2,6-di-tert-butylphenol
- CAN
-
acetonitrile
- Bn
-
benzyl
- Boc
-
tert-butyloxycarbonyl
- BPZ
-
4,4′-cyclohexane-1,1-diyldiphenol
- CDC
-
cross dehydrogenative coupling
- DABCO
-
1,4-diazabicyclo[2.2.2]octane
- DBU
-
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
-
dichloromethane
- DIEA
-
N,N’-diisopropylethylamine
- DFT
-
Density Functional Theory
- Dipp
-
2,6-diisopropylphenyl
- DMAc
-
N,N-dimethylacetamide
- DMAP
-
4-dimethylaminopyridine
- DME
-
dimethyl ether
- DMF
-
dimethylformamide
- DMSO
-
dimethyl sulfoxide
- DQ
-
3,3′,5,5′-tetra-tert-butyldiphenoquinone
- dr
-
diastereomeric ratio
- ee
-
enantiomeric excess
- Epoxi
-
experimental oxidation potential
- Epred
-
experimental reduction potential
- er
-
enantiomeric ratio
- ETM
-
electron transfer mediator
- EWG
-
electron withdrawing group
- FePc
-
iron(II)phthalocyanine
- FG
-
functional group
- HFIP
-
hexafluoro-2-propanol
- HPLC
-
high-performance liquid chromatography
- LG
-
leaving group
- LiHMDS
-
lithium bis(trimethylsilyl)amide
- MCA
-
methyl cation affinity
- Mes
-
mesityl
- NFSI
-
N-fluorobenzenesulfonimide
- NHC
-
N-heterocyclic carbene
- NMR
-
nuclear magnetic resonance
- o-QDM
-
ortho‐quinodimethanes
- PA
-
proton affinity
- PA10F
-
poly(decamethylene 2,5-furandicarboxylamide)
- PA10T
-
poly(decamethylene terephthalamide)
- PA6T
-
poly(hexamethylene terephthalamide)
- PAM
-
polyamide
- PETA
-
poly(p-ethylene terephthalamide)
- p-QM
-
para-quinone methide
- PIDA
-
phenyliodine (III) diacetate
- Pre-NHC
-
N-heterocyclic carbene precursor
- RC
-
Rauhut-Currier
- SCE
-
saturated calomel electrode
- SET
-
single electron transfer
- SFC
-
supercritical fluid chromatography
- TBD
-
triazabicyclodecene
- TBDPS
-
tert-butyldiphenylsilyl
- TBHP
-
tert-butyl hydroperoxide
- TBS
-
tert-butyldimethylsilyl
- TEA
-
triethylamine
- TEMPO
-
2,2,6,6-tetramethylpiperidinyloxy
- THF
-
tetrahydrofuran
- UV/Vis
-
ultraviolet/visible
Abbreviations
1 Introduction
N-Heterocyclic carbenes (NHCs) (Fèvre et al., 2013; Huynh, 2018) represent a class of neutral heterocyclic molecules containing a neutral carbon species with two coordinately unsaturated electrons, and at least one nitrogen atom is positioned adjacent to the carbene donor within the heterocyclic ring (Shen et al., 2022).
Contrary to classical carbenes, which exist in a triplet state, N-Heterocyclic carbenes (NHCs) exist in a singlet ground state (Fig. 1a). In the cyclic structure, mesomeric effects due to π-electron donation from adjacent heteroatoms into the empty p orbital of the carbene and the inductive effect due to the σ-electron withdrawal of the heteroatoms help to favor the singlet state (Hopkinson et al., 2014). NHCs can be represented as an ylide or as a carbene species, as shown in Fig. 1b (Feroci et al., 2016b; Hopkinson et al., 2014; Okano, 2013).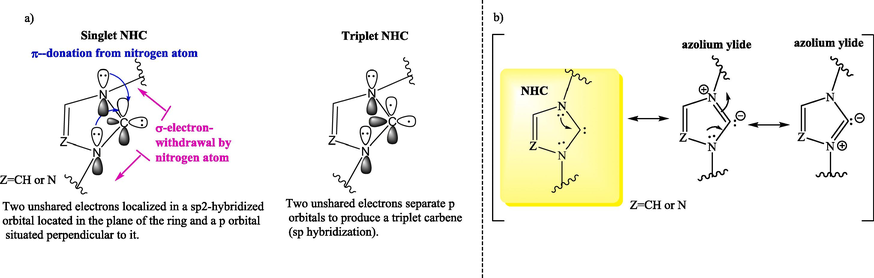
a) Structures of the singlet and triplet ground states; b) Mesomeric structures of NHCs.
Because they do not respect the “octet rule”, general alkyl based carbenes have long been recognized as an unstable species; however, in 1991, Arduengo et al. (1991) reported the synthesis and structural characterization of 1,3-di(adamantyl)imidazol-2-ylidene 2 (Fig. 2a), a stable N-heterocyclic carbene obtained from deprotonation of the corresponding imidazolium chloride 1. The nitrogen atoms adjacent to the carbene serves to make the NHCs more stable than the general alkyl based carbene (Arduengo et al., 1991; Feroci et al., 2016b; Hopkinson et al., 2014).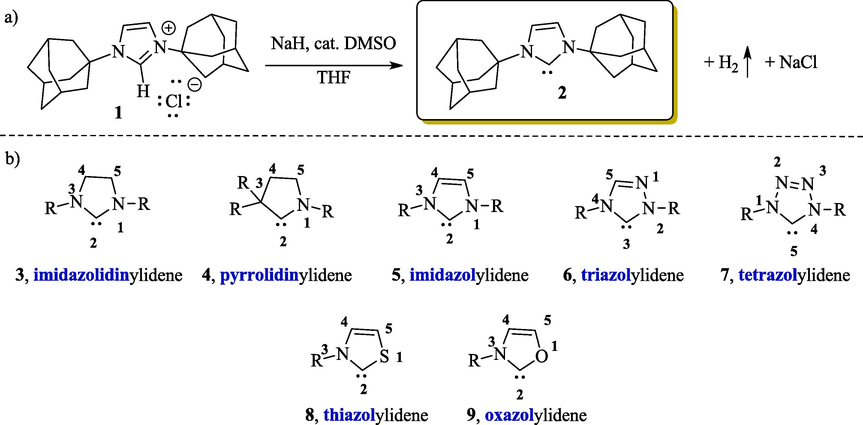
a) Synthesis of the first crystalline carbene; b) Some examples of five-membered NHCs.
Most of the known NHCs are derived from five-membered rings with additional oxygen, sulfur or phosphorus heteroatoms. Fig. 2b summarizes some of the more common examples of NHCs 3–9 and their associated names (Cavallo and Cazin, 2010; Jahnkea and Hahn, 2017).
The origin of the abundance of publications on NHCs lies in the structural diversity of the air-stable azolium salt precursors (Pre-NHCs), as shown in Fig. 3 for the 1,2,4-triazolium salts 10–65 (Flanigan et al., 2015).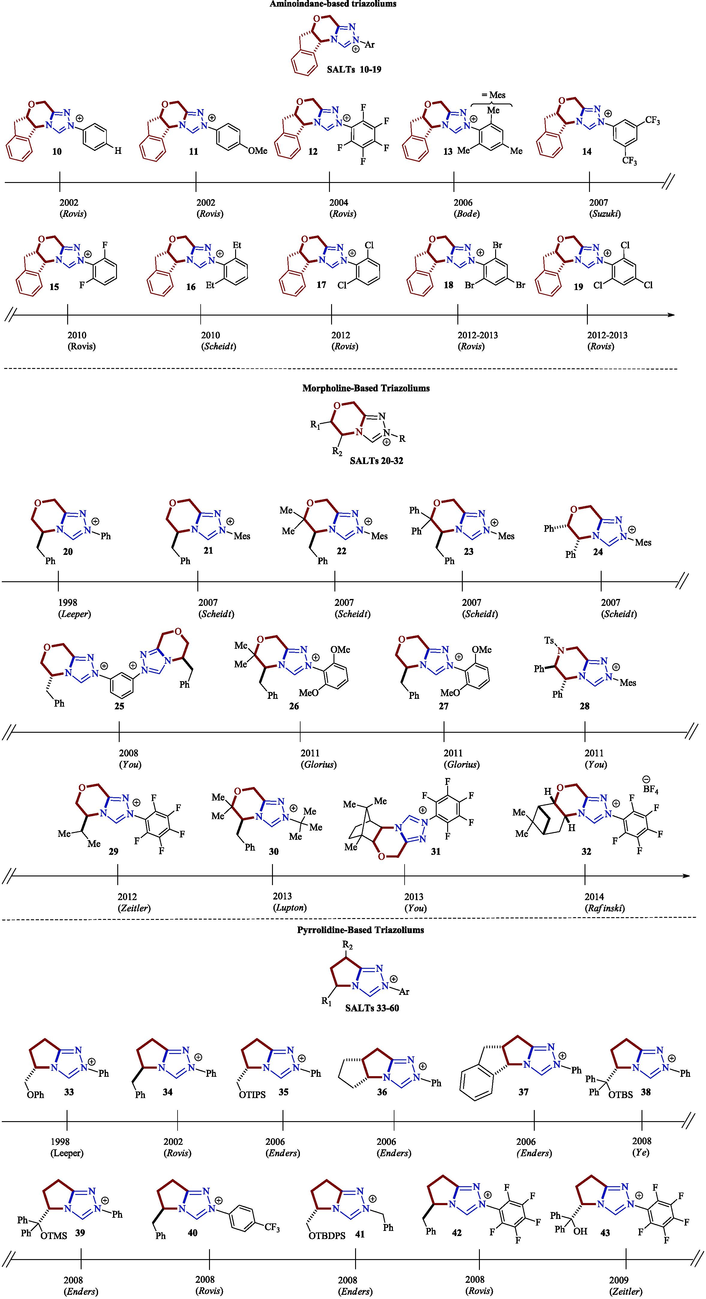
Some examples of 1,2,4-triazolium salt structures extracted from the literature (Flanigan et al., 2015).
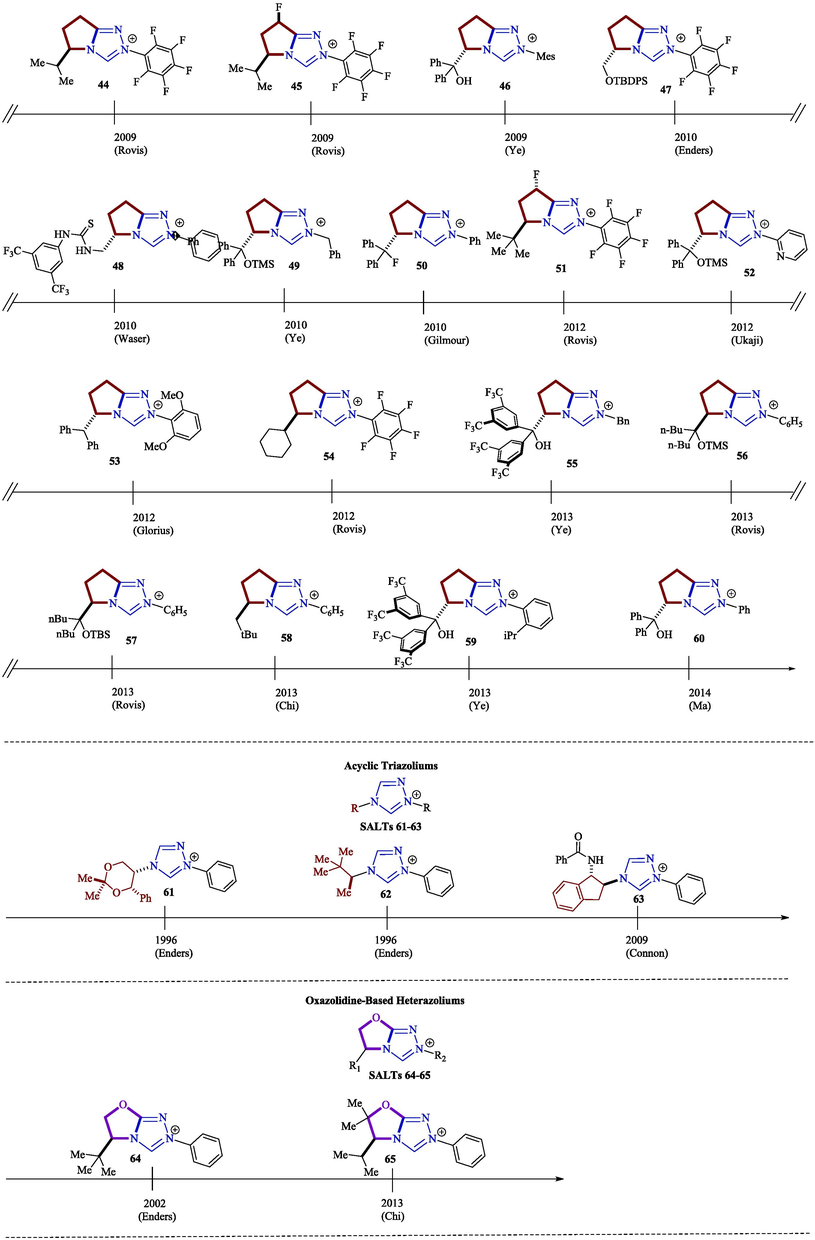
Some examples of 1,2,4-triazolium salt structures extracted from the literature (Flanigan et al., 2015).
The synthesis of Pre-NHCs has been reported in the literature by several research groups (Benhamou et al., 2011; Grieco et al., 2015; Hutchinson et al., 2019).
1.1 Generation of free NHCs
Free NHCs are most frequently generated in situ via the deprotonation of the corresponding Pre-NHCs (Scheme 1). Several bases, including NaH, KH, TEA, tBuOK, DBU, NaOH, K2CO3 and NaOEt, and solvents, such as THF, ACN and DCM, among others, have been used for this purpose (Vetica et al., 2021). Free NHCs (Dong et al., 2022; Mondal et al., 2019; Bhatia et al., 2013) and a wide range of metal-NHCs (NHCMLn-1) (Zhao et al., 2020) have been found to be useful as catalysts in various organic reactions. The free form of NHCs 67 can usually be achieved through the reductive desulfurization of imidazol-2-thione derivatives 66 with strong reducing metals (Scheme 2) (Bhatia et al., 2013) or via various thermally labile (Fèvre et al., 2013; Naumann and Buchmeiser, 2014) NHC precursors (Fig. 4), which can be divided into typical categories such as betaine adducts, azolium hydrogen carbonate, azolium carboxylate salts, neutral NHC adducts and NHC-metal adducts. These protected NHCs are versatile tools in organocatalysis and polymerization catalysis (Fèvre et al., 2013; Naumann and Dove, 2016).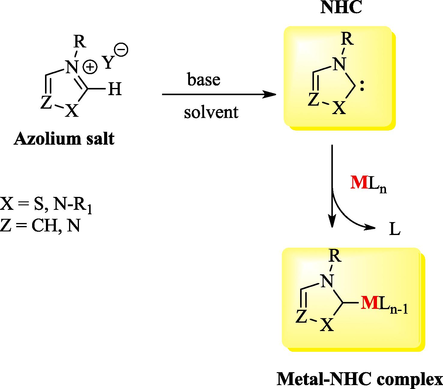
Chemical generation of NHCs and metal-NHC complex.
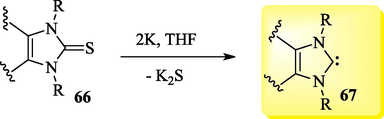
Reduction of imidazole-2-thiones to obtain NHCs.
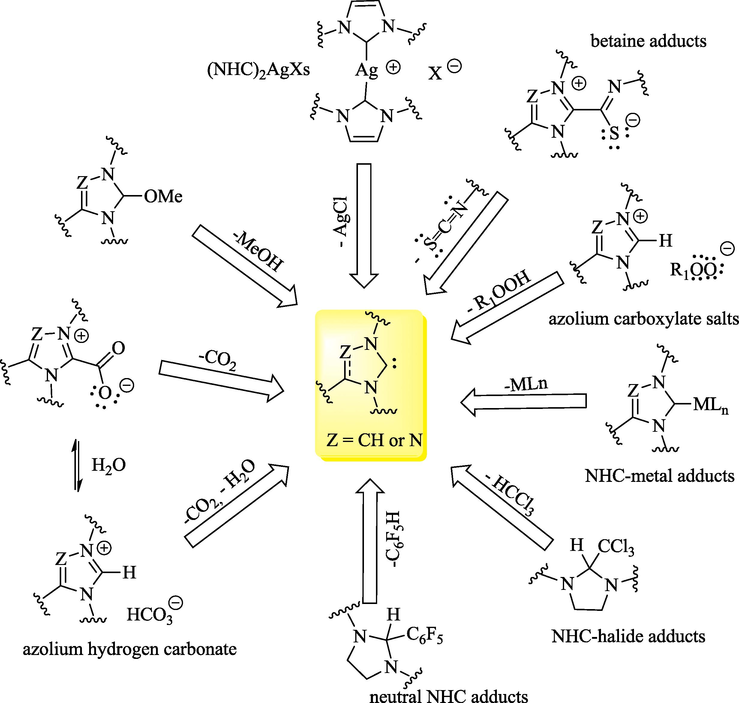
Some examples of thermo-latent NHC progenitors.
According to Scheme 3, another method that can be used to obtain free NHCs is the electron cathodic reduction of the C–H bond between the heteroatoms of Pre-NHCs (Schotten et al., 2021a, 2021b). An electrochemically synthesized NHC is called electrogenerated. In the electrochemical reaction, the Pre-NHC is reduced to a free NHC at the cathode with the evolution of hydrogen. At the anode, the NHC can posteriorly react with a substrate (Schotten et al., 2021a) or with a metal cation (Schotten et al., 2021b), which is produced from the sacrificial anode via oxidation, to provide a reaction product or metallo-complex, respectively. Still, other reactions can occur, including solvent decomposition, counterion oxidation or the oxidation of a sacrificial anode to release metal ions. This method represents a green and advantageous alternative to traditional synthetic methods that often rely on strong bases and air-sensitive materials (Schotten et al., 2021a).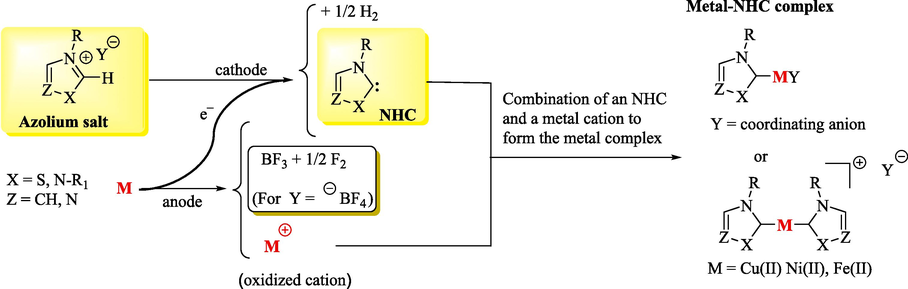
Electrochemical generation of NHCs and metal-NHC complex.
The literature shows that the limited lifetime of various NHCs can be related to the formation of dimeric structures. Steric and electronic effects, among other factors, can affect the dimerization process. For example, saturated NHCs have a stronger tendency to dimer than unsaturated NHCs (Feroci et al., 2016b). The irreversible capture of the NHC by the metallic cation prevents its oxidation and degradation (Schotten et al., 2021a).
As previously shown, free NHCs are most often prepared via the deprotonation of Pre-NHCs, and knowing the pKa values of these species is an important factor, mainly in reactions involving NHCs as Brønsted base catalysts (N. Wang et al., 2018).
1.2 Quantifying the electronic properties of NHCs
1.2.1 pKa measurements of Pre-NHCs
The pKa values reflect how easily Pre-NHC can form an NHC precursor (Hopkinson et al., 2014; Konstandaras et al., 2020; Quinn et al., 2021; Zhu et al., 2022). The –NC(H)N- proton in Pre-NHCs is acidic due to its connection to the carbon atom bonded to the two adjacent electron-withdrawing nitrogen atoms. The reactivity of the NHCs is highly sensitive to the azolium nucleus and structural changes in their cyclic carbon skeletons (Zhu et al., 2022), but the nature of the solvent and the counter-ion Y− also plays a significant role (Feroci et al., 2016b). The Brønsted basicity of NHCs has been investigated by combining computational and experimental methodologies to determine the pKa values of the Pre-NHCs, both in solution and in the gas phase, and the relative proton affinities (PAs) of the NHCs (Fèvre et al., 2013). For example, the equilibrium acidities of the most commonly used NHC were measured in DMSO using an overlapping indicator method via UV/vis spectra. The pKa values range from 12.8 to 23.4 (Fig. 5a) (Li et al., 2017). Triazolium salts 68a–71 are generally more acidic, with pKa values ranging from 12.8 to 14.7, than thiazolydene 72 and imidazolydene 73–75 salts due the inclusion of a third electronegative nitrogen atom in their azole ring, leading to a lowering of the pKa value. In the triazolylidene series, the change from a pentafluorophenyl group (electron-withdrawing substituent) 68a to a mesityl group 70 (Ar = Mes, electron-donating substituent) results in an increased pKa (15.52) (Li et al., 2017). Fig. 5b shows pKa values determined in water for some select Pre-NHCs 76–80. In general, imidazolium and imidazolinium salts are less acidic (pKa ≈ 19.8–23.4) in aqueous solution than 1,2,4-triazoliums (pKa ≈ 17.2–21.5) (Flanigan et al., 2015; Higgins et al., 2011; Massey et al., 2012). The pKa value of a Pre-NHC 73 in DMSO is lower than the pKa value determined in aqueous media (compound 76a), because neutral carbene is insensitive to solvent stabilization, while the azolium cation can be stabilized by hydrogen bonding in water to a greater extent than in an aprotic solvent (Konstandaras et al., 2020).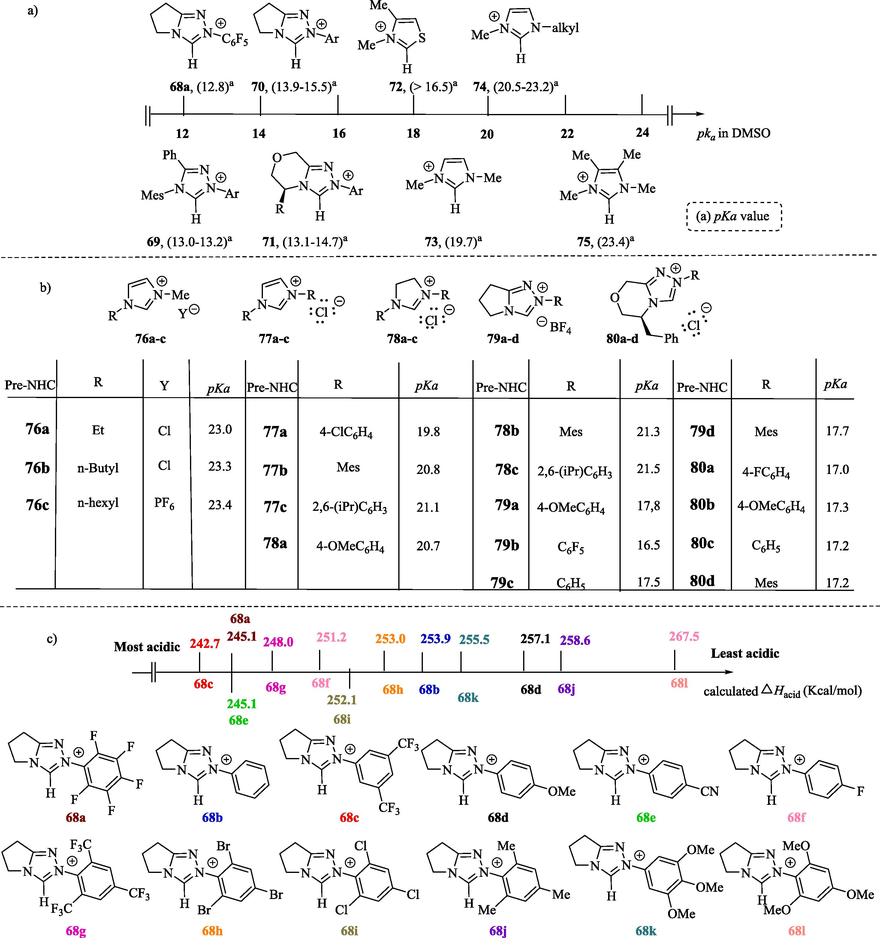
pKa values of the Pre-NHCs in a) DMSO solution and b) H2O; c) Acidities (ΔHacid) of the triazolium salts calculated using computational method.
The experimental and computational gas-phase acidities of triazolylidene salts 68a–l (Fig. 5c) in the gas phase were investigated by Niu et al. (2017). The calculated acidities (ΔHacid) of the triazolium salts, which correspond to the proton affinity of their corresponding carbene species, were determined using the B3LYP method at a 6–31 + G* basis (Niu et al., 2017). The higher the value of the proton affinity, the stronger the base and the weaker the conjugate acid in the gas phase. In general, the electron-withdrawing N-aryl substituent favors the destabilization of the cationic triazolium carbon acid in relation to the neutral NHC conjugate base, thus favoring the deprotonation process, while the electron-donating N-aryl substituent has the opposite effect on the acidity (pKa increase), which will stabilize the cation and shift the equilibrium towards the protonated form. Comparing 68c with 68 g, the ortho-substituted N-aryl triazolium salt forces the phenyl ring out of planarity relative to the azolium nucleus. This steric restriction present in salt 68 g does not allow the conjugation between the electron withdrawing phenyl ring and the azole ring, resulting in the stabilization of the acidic proton and, consequently, reducing the acidity of the ortho-substituted triazolium salt.
The basicity and nucleophilicity of NHCs have also been reported using the electrochemical reduction potential of the corresponding Pre-NHCs in different solvents by several research groups (Feroci et al., 2016a, 2016b; Green et al., 2015; Ogawa and Boydston, 2014; Petersen et al., 2022; Rocco et al., 2022; Schotten et al., 2021a; Zhou et al., 2022). The researchers identified a linear relationship between the pKa value and the experimental reduction potential (Epred) of Pre-NHCs, while the oxidation potential (Epoxi) of NHCs is closely related to their nucleophilicity. The cathodic process is concerned with breaking the C–H bond between the heteroatoms of the Pre-NHCs to form the corresponding azolium-based NHCs (reduction of the NHCH+ azolium cations to NHCs) and H2 (Scheme 4), while the anodic pathway is related to the oxidation of the electrogenerated NHCs. NHC formation is usually connected with the acidity of the corresponding Pre-NHC, while the carbene reactivity is usually connected with its lone-pair availability (Feroci et al., 2016b). The reduction potential of Pre-NHCs correlates with their acidity. Larger positive values of the reduction potential are indicative of a greater tendency to be reduced. According to Fig. 6, thiazolium salts 83a–c are easier to reduce to the corresponding NHCs than triazolium salt 82 (lower reduction potential), which in turn has a more positive E1/2 (-2.76 V) compared to imidazolium-based cations 81a E1/2 (-3.0 V) and 77b E1/2 (-2.87 V). Data from the literature show that imidazolium salts are less acidic (pKa = 22–24 in water and 22 in DMSO) than thiazolium and triazolium cations (pKa = 16–19 in water and 14 in DMSO) (Feroci et al., 2016a). There is an essential difference between the pKa values for the triazolium and imidazolium salts, as the presence of an additional electronegative nitrogen atom in the triazolium ring destabilizes the Pre-NHC with respect to the corresponding NHC, thus increasing its acidity.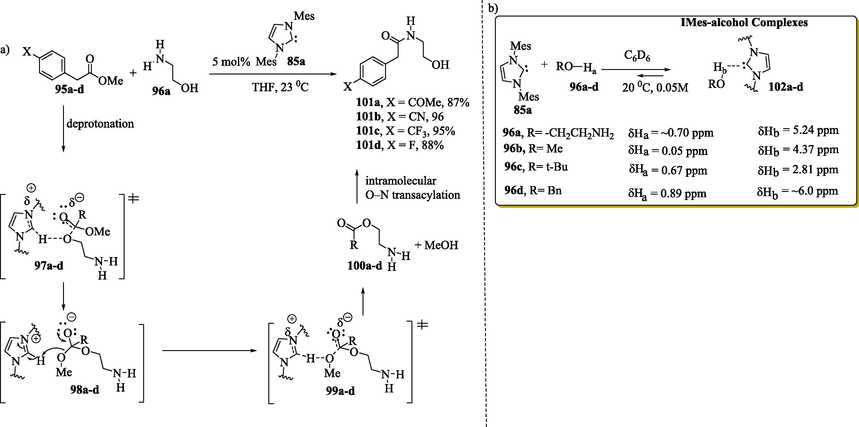
a) NHC-catalyzed amidation of esters; b) Evidence by NMR of the involvement of Brønsted base catalysis.
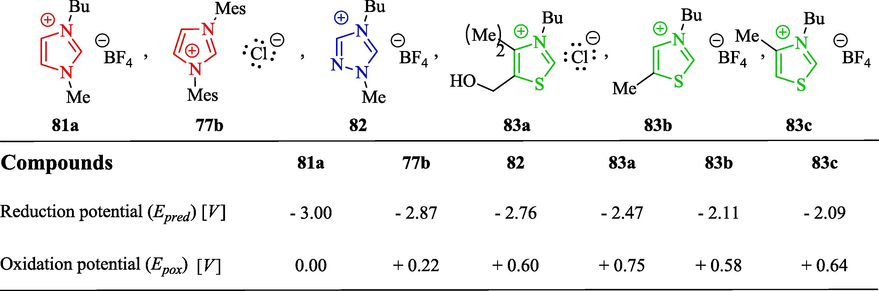
Reduction and oxidation potentials of the Pre-NHCs and NHC parents, respectively, (0.10 M in DMF), in V vs saturated calomel electrode (SCE) according to data from the literature (Feroci et al., 2016a).
The anodic process is related to the electroactivity of the electrogenerated carbene. From electrochemical studies, the authors suggest that Epox values decrease with increasing NHC nucleophilicity, which reflects its ability to attack an electrophilic reactant. Imidazolium salts 81a and 77b (Epox = 0.00 and + 0.22) are three orders of magnitude more nucleophilic than the triazolium salt 82 (Epox = +0.60) (Fig. 6).
1.2.2 Nucleophilicity and Lewis basicity of NHCs
In addition to the use of NHCs as Lewis base catalysts, they are also nucleophiles. In general, NHCs are of moderate nucleophilicity but high Lewis basicity. However, most of the organocatalytic transformation NHCs act as nucleophilic species attacking an electrophilic reactant to form umpolung species, such as Breslow intermediates, this being the first and crucial step (Gudimetla et al., 2022). Nucleophilicity is also a crucial factor for the coordination of NHCs to transition metal complexes (Nesterov et al., 2018). The nature of the azolium ring and N-substituent plays an important role in defining the nucleophilicity of the NHC as a catalyst. The nucleophilicity of several NHCs has been quantified according to the Mayr-Patz equation, log k (20 0C) = sN(N + E) (Eq. 1), which is described by three parameters: the electrophile-dependent reactivity parameter E (electrophilicity), nucleophilicity parameter (solvent dependent) and nucleophile-specific sensitivity parameter (sN), which is sensitive to changes in the electrophilicity of the reaction partner (solvent dependent). From the above equation, Maji et al. (2011) determined the nucleophilicity parameters (N) of NHCs 84–86 and other nucleophiles 87–89 (Fig. 7a) through reaction, with structurally related quinone methide 90a or benzhydrylium ion 92 used as reference electrophiles (Fèvre et al., 2013; Levens et al., 2016; Maji et al., 2012, 2011; Mayr et al., 2012; Wei et al., 2008) to form adducts 91 and 93.
The results of these studies showed that imidazolidinylidene 84a and imidazolylidene 85a carbenes were significantly more nucleophilic than Enders carbene (TPP, 86) (Fig. 7a). This fact can be explained on the basis of the inductive electron withdrawal exerted by the extra nitrogen in the triazolium carbene. In general, the number, identity and position of ring heteroatoms and backbone substitution play a role in influencing the NHC stability and reactivity. In addition to these considerations, compounds with unsaturated backbones such as imidazolylidene 85a and triazolylidene 86 benefit from increased stability because of their partial aromatic character. The saturated imidazolinylidene 84a reacted at a slightly faster rate than the unsaturated analog 85a. The less nucleophilic triazolium carbene 86 is moderately more nucleophilic than PPh3 87, 4-(dimethylamino)pyridine (88, DMAP) and 1,8-diazabicyclo[5.4.0]undec-7-ene (89, DBU) (Maji et al., 2011).![a) Nucleophilicity parameters for NHCs in THF; b) MCAs (kJ/mol) [MP2/6–31 + G(2d,p)//B98/6-31G(d)].](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105527-fig12.png)
a) Nucleophilicity parameters for NHCs in THF; b) MCAs (kJ/mol) [MP2/6–31 + G(2d,p)//B98/6-31G(d)].
Maji et al. (2011) also determined the Lewis basicity of the NHCs 84a, 85a and 86 and of their related azolium carbenes 84b–d, 85b–d and 94a–d (Fig. 7b) by using the methyl cation affinity (MCA), which corresponds to ΔH298 to generate the naked NHC and +CH3 from the covalent adduct NHC–CH3+ (Maji et al., 2011).
The five-membered ring is of a planar nature for most NHCs; the di-tert-butyl imidazoline carbene 84d adopts a twist conformation with an N-C–C-N interplanar angle of 18°. In the NHCs 84b, 85b and 94b, the phenyl groups are coplanar with the heterocycle ring or only slightly distorted out of the plane, while the mesityl groups in 84a, 85a and 94a are almost perpendicular to the plane of the aza-heterocycle moiety. Phenyl groups can stabilize the NHCs by π-conjugation, unlike 2,6-(OMe)2C6H3-group, which cannot operate through mesomeric effect. Regarding the products (NHCs–CH3+) obtained through the methylation of N-mesityl-substituted NHCs 84a, 85a and 94a and N-phenyl-substituted NHCs 84b, 85b and 94b, in both series, the aryl groups are distorted out of the plane to minimize the steric hindrance effect, with the methyl group at the former carbene center. This means that the phenyl-substituted series are weaker Lewis bases than mesityl analogues, because the phenyl group can stabilize the NHCs by π-conjugation but not their corresponding adducts NHCs–CH3+. The electron-withdrawing effect of the additional nitrogen in series 94a–d makes these NHCs less basic than other NHCs. The NHC 86 is more basic than 94b due to the mesomeric effect of the additional phenyl group. Imidazoline 84a (768.9 kJ mol−1) and imidazole-type 85a (767.2 kJ mol−1) NHCs, respectively, are thus stronger Lewis bases than the triazolium carbene 94a (728.4 kJ mol−1); the latter is stronger than other neutral Lewis bases that have been studied with PPh3 (618.4 kJ mol−1) and DMAP (581.2 kJ mol−1).
Other methods for quantifying the properties of NHCs using a variety of different parameters have been described (Ando et al., 2020; Dröge and Glorius, 2010; Falivene and Cavallo, 2017; Gómez-Suárez et al., 2017; Huynh, 2018; Nelson et al., 2013).
1.3 Some examples of NHC-Brønsted base-catalyzed reactions
In 2005, Movassaghi and Schmidt (2005) described the NHC Brønsted base-catalyzed amidation of esters 95a–d with amino alcohol 96a (Scheme 4a). They proposed that NHC 85a acts as a Brønsted base catalyst based on evidence of hydrogen-bonding interactions between NHC 85a and alcohols 96a–d (Scheme 4b) supported by 1H NMR, 13C NMR and a single-crystal X-ray of 102b (Chen and Huang, 2019; Movassaghi and Schmidt, 2005; N. Wang et al., 2018). Initially, the carbene 85a acts as a Brønsted base interacting with the alcohol 96a through hydrogen bonding. Upon the activation of the hydroxyl group of the amino alcohol by the NHC catalyst, the oxygen atom is nucleophilic enough to attack the carbon carbonyl of the esters (transition states 97a–d), followed by the liberation of the alcohol (transition states 99a–d). Movassaghi and Schmidt (2005) observed that the products 101a–d were obtained from 100a–d through an intramolecular O-N acyl transfer (Movassaghi and Schmidt, 2005).
In the NMR analysis, it was observed that the proton of the hydroxyl group in the complexes 102a–d (Scheme 4b) becomes more deshielded than alcohols 96a–d (Movassaghi and Schmidt, 2005). The X-ray structure of a stable hydrogen-bonded carbene-alcohol complex 102b and NMR study support a carbene-base nucleophile activation mechanism.
Another example that involved an NHC acting as a Brønsted base catalyst was reported by Lai et al. (2005) (Fèvre et al., 2013; Lai et al., 2005; Ryan et al., 2013) in their theoretical studies on the mechanism of transesterification reactions. They investigated two types of mechanisms: an NHC acting as a nucleophilic catalyst (Path I) (Scheme 5) and an NHC acting as a Brønsted base catalyst (Path II). In the first case, the nucleophilic attack of the NHC 85c on carbonyl ester 103 leads to the formation of a tetrahedral zwitterion 105, which releases an alkoxide anion, transforming into acyl azolium intermediate 106. In the second case, NHC 85c assists in the proton transfer from alcohol 104 to the carbonyl group of ester 103, forming a concerted four-member-ring transition state 108, followed by a stable tetrahedral intermediate 109, which transfers a proton to the –OR1 group via transition state 110, increasing its ability to act as a leaving group. Reforming the carbonyl double bond causes the elimination of an alcohol (HOR1) and the formation of the desired product. In the study, it was observed that neutral intermediate 109 (Path II) was significantly more stable than tetrahedral zwitterionic intermediate 105 and the acyl azolium 106 (Path I), suggesting that the mechanism involved an NHC acting as a Brønsted base and not as a nucleophilic catalyst.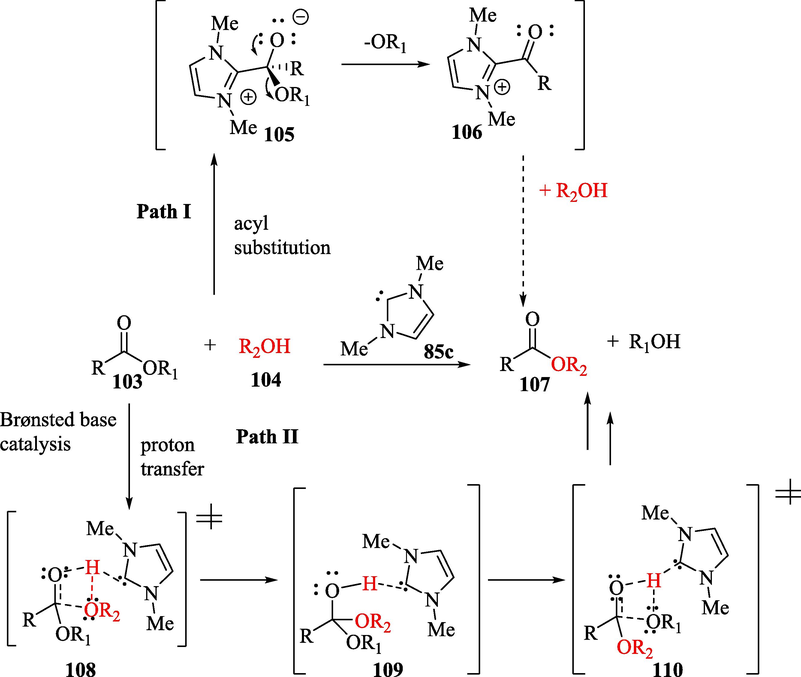
Proposed mechanism of NHC-catalyzed transesterification.
A highly stereoselective 1,6-addition reaction of 1,3-ketoamides 111a–b with acidic N–H to p-quinone methides (p-QMs) 90b–g using an NHC as a Brønsted base catalyst was reported by Santra et al. (2018) to obtain the compounds 114a–f with excellent stereoselectivity (ee and dr) and yields. Among these compounds, the substance 114f stands out; it was used as a synthetic precursor in the design and synthesis of chiral γ-lactam 116 and β-lactam 118, bearing three stereocenters in good yields with excellent stereocontrol (Scheme 6). Based on mechanistic studies, the authors suggest that, after the deprotonation of the N–H acid of the amides by the NHC Brønsted base, the reaction proceeds through an intermediate pair composed of the enolate ion and the azolium cation 113. The chemistry reduction of 114f led to the attainment of 115 as the major product in good yield and with excellent ee; it can subsequently be transformed into spirocyclic γ-lactam 116 via oxidation with phenyliodine(III) diacetate (PIDA). Substance 115 was also subjected to the mesylation reaction to form compound 117, which later, under basic conditions, can be converted into the enantioenriched β-lactam 118 in good yield with excellent stereocontrol. The absolute configuration of the stereocenters of compound 114a was unambiguously determined by single-crystal X-ray analysis. (Santra et al., 2018).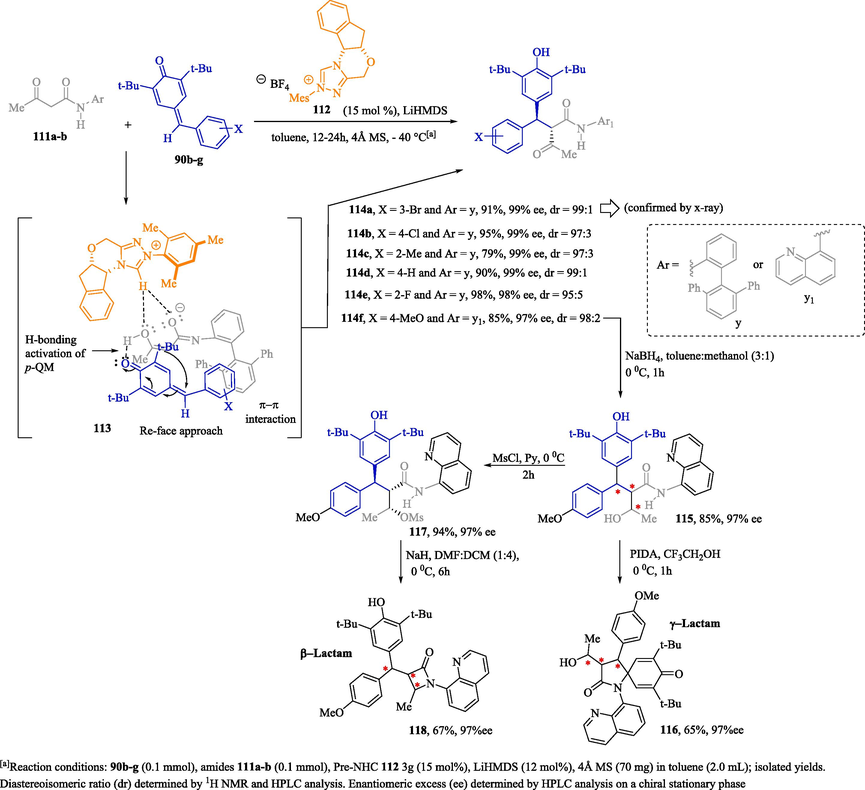
Synthesis of chiral amides 114a–f and γ- and β-lactams 116 and 118, respectively, mediated by NHCs acting as chiral Brønsted base catalysts.
1.4 Application of 1,2,4-triazolium carbenes as nucleophilic organocatalysts
Since vitamin B1, a naturally occurring thiazolium salt, was utilized by Ukai et al. (Ukai et al., 1943) as an organic catalyst to promote a class of umpulong reactions that induce aldehydes to undergo nucleophilic addition reactions, a wide range of related chemical transformations catalyzed by different Pre-NHCs has been investigated intensively (Zhu et al., 2022).
Free NHCs, as nucleophilic species, have shown significant importance as organocatalysts and ligands for transition metals (Riederer et al., 2011), and they have been used in metal-free polymer synthesis (Fèvre et al., 2013; Kohsaka et al., 2021), ionic liquids (Alpers et al., 2018) and metallodrugs (Vellé et al., 2017). Particularly in the field of organocatalysis, chiral NHC-catalyzed reactions have been a powerful strategy for the stereoselective formation of C–C and C-heteroatom bonds, leading to the construction of interesting cyclic systems (Ni et al., 2015). Specifically, 1,2,4-triazolium carbene catalysts have proved to be more effective at inducing enantioselectivity with systems that have asymmetric centers incorporated into a rigid structure (Mahatthananchai and Bode, 2014; Zhu et al., 2022). Other examples of chemical transformations involving 1,2,4-triazolylidenes include the oxidative formation of esters and carboxylic acids from aldehydes and the oxidation of alcohols, among others (Harnying et al., 2021).
In order to understand the reactions mediated by NHCs acting as nucleophilic catalysts, this review will initially present the different types of NHC intermediates (subsection 1.4.1), followed by a discussion of the chemical part of some of the synthetic steps (subsections 1.4.2 and 1.4.3) that are common in the different types of catalytic cycles demonstrated throughout this work.
1.4.1 NHC catalyzed umpolung reactions: Generation of NHC intermediates
N-Heterocyclic carbenes are known to confer umpolung reactivity to saturated 120 and α,β-unsaturated 121 aldehydes, which are then exploited in the generation of important reactive species (Stenkvist et al., 2023; Chen et al., 2018a, 2018b; Li et al., 2020; Mahatthananchai and Bode, 2014; Pareek et al., 2021; Que and He, 2020; Zhao et al., 2021) (Scheme 7a–b) such as Breslow intermediates 124, acyl azoliums 125, azolium enolates 126, homoenolate equivalents 127, unsaturated Breslow intermediates 128, γ-unsaturated acyl azolium intermediates 129 and azolium dienolates 130. The in situ activation of carboxylic acid derivatives 131 and 132, followed by the addition of NHC 119, provides the acyl azoliums 125 and 129, respectively, which have been used for the preparation of esters 133 and amides 134. At this point, the azolium dienolate intermediates 130 (Scheme 7b) can be generated through the addition of NHC 119 to a carbonyl compound with an acidic α,β-hydrogen atom 139–141. The most commonly used methods for obtaining NHC-derived azolium dienolate intermediates rely on the redox activation of α,β-unsaturated aldehydes 139 and 140, α,β-unsaturated acid derivatives 141 and cyclobutenones 142 (Chen et al., 2020, 2018a). The reactivity of the aforementioned intermediates has been extensively exploited in various chemical synthetic methods, including annulation, benzoin, Stetter, Claisen rearrangement, cycloaddition, and C–C and C–heteroatom bond functionalization reactions (Mahatthananchai and Bode, 2014), to obtain different classes of organic compounds 133–138 (Scheme 7a) and 143–145 (Scheme 7b). Examples of these reactions are shown throughout this review.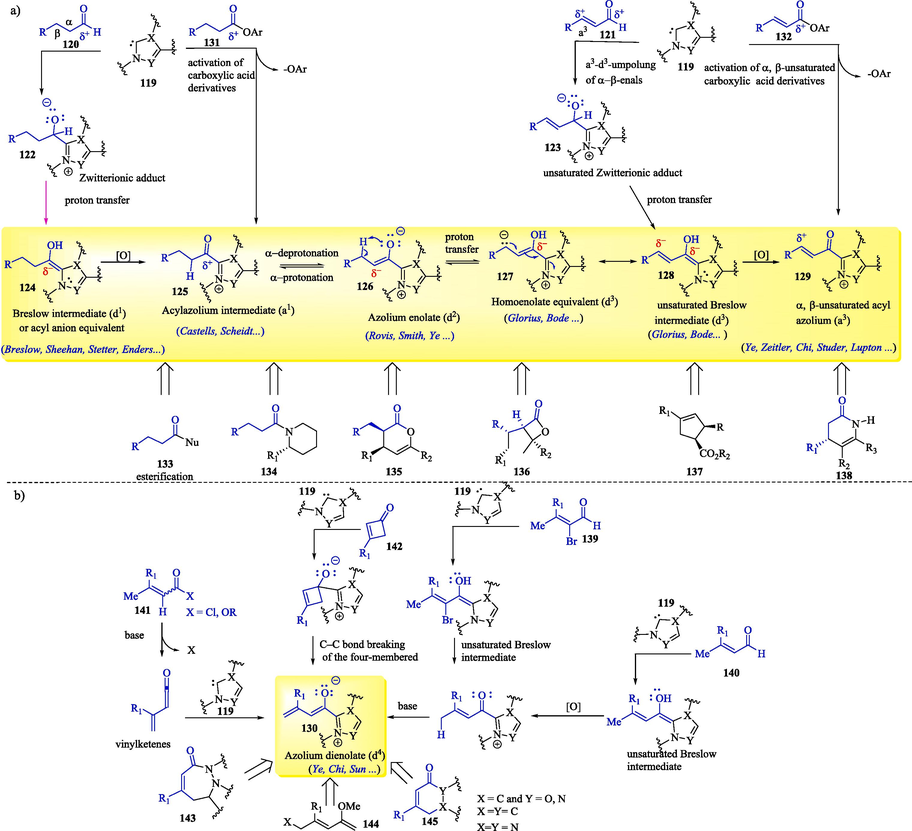
Examples of reactive intermediates and different classes of compounds from NHC catalysis.
In addition to NHCs, metallophosphites and phosphines are organocatalysts that can provide access to the umpolung of organic compounds (Bugaut and Glorius, 2012). The concept of umpolung, or reversed polarity, introduced by D. Seebach and E. J. Corey (Rezayee et al., 2022), can be better understood by considering Scheme 7. The carbon atom (C-3 or Cβ) of the α,β-unsaturated aldehyde 121, a potential electrophilic Michael acceptor, undergoes a polarity inversion and becomes a nucleophilic site in an a3 to d3 umpolung (also called a conjugate umpolung) when the nucleophilic catalyst transiently transforms it into homoenolate equivalent 127 through an addition − proton transfer sequence (Scheme 8a). If the carbon atom (C-3) is a positively polarized synthon (acceptor synthon), it is denoted by the symbol a3, while if the carbon atom (C-3) is a negatively polarized synthon (donor synthon), it is denoted by the symbol d3. Synthons a and d are numbered with respect to the relative positions of a functional group (FG) and the reaction site. Activation of the carbonyl group of an electrophilic aldehyde 120 via the umpolung strategy to form a nucleophilic Breslow intermediate is shown in Scheme 8b (Bugaut and Glorius, 2012; Vetica et al., 2021).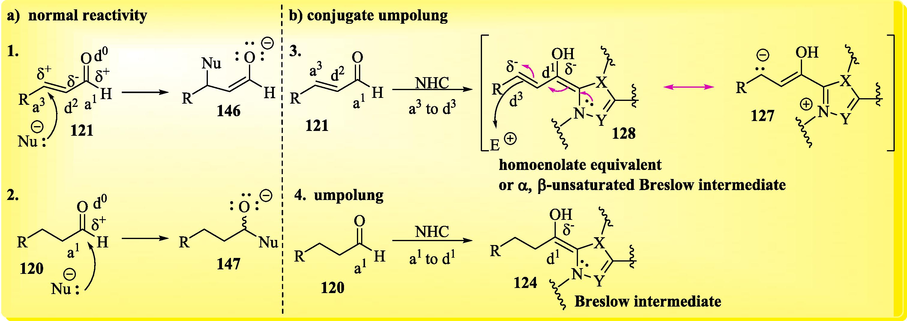
a) Normal reactivity of aldehydes with nucleophiles (e.g., amines); b) a3-d3 and a1-d1 umpolungs of aldehydes with NHCs.
This polarity inversion makes it possible to explore new synthetic possibilities that cannot be realized from the normal reactivity of functional groups. From the reactive species (Scheme 7), depending on the reaction conditions and catalyst choice, different pathways can be taken, resulting in a diverse array of products that can be formed under NHC catalysis.
1.4.2 Proposed mechanisms for the transformation of a zwitterionic adduct into a Breslow intermediate
In NHC-catalyzed reactions, the most crucial step in the formation of the Breslow intermediate is the deprotonation of the carbon attached to the negatively charged oxygen atom in the zwitterionic adducts 122 and 123 (Scheme 7a) (Reddi and Sunoj, 2017). The pathway to the formation of the Breslow intermediates 124 and 128 from zwitterionic adducts 122 and 123, respectively, has been explicated computationally and experimentally under different conditions, including direct [1,2] proton shift, acid/base catalysis and the assistance of protic media (Huang et al., 2022; Nandi et al., 2021; Pareek et al., 2021; Wessels et al., 2022).
Recently, based on quantum chemistry calculations using density-functional theory (DFT), Huang et al. (2022) proposed a bimolecular mechanism to explain the formation of the Breslow intermediates 158 from the zwitterionic adducts 150 under aprotic conditions. These last intermediates were obtained via the reaction between benzaldehyde 148a and an imidazolinylidene 149 (Scheme 9). According to researchers, the conversion of the zwitterionic adducts 150 into the Breslow intermediates 158 takes place via hemiacetals 152. This step involved the oxy-addition of the zwitterionic adduct 150 to another benzaldehyde molecule 148a, followed by the proton migration of 151 to form hemiacetal 152. The hydrogen bond interaction between the zwitterionic adducts 150 and 152 results in the complex 153, which advances the hemiacetal decomposition to form Breslow intermediate 158 with the assistance of the zwitterionic adduct 150. The hydrogen bond interaction between the zwitterionic adduct and the hemiacetal (complex 153) / Breslow intermediate (complex 157) plays an important role (Huang et al., 2022; Wessels et al., 2022).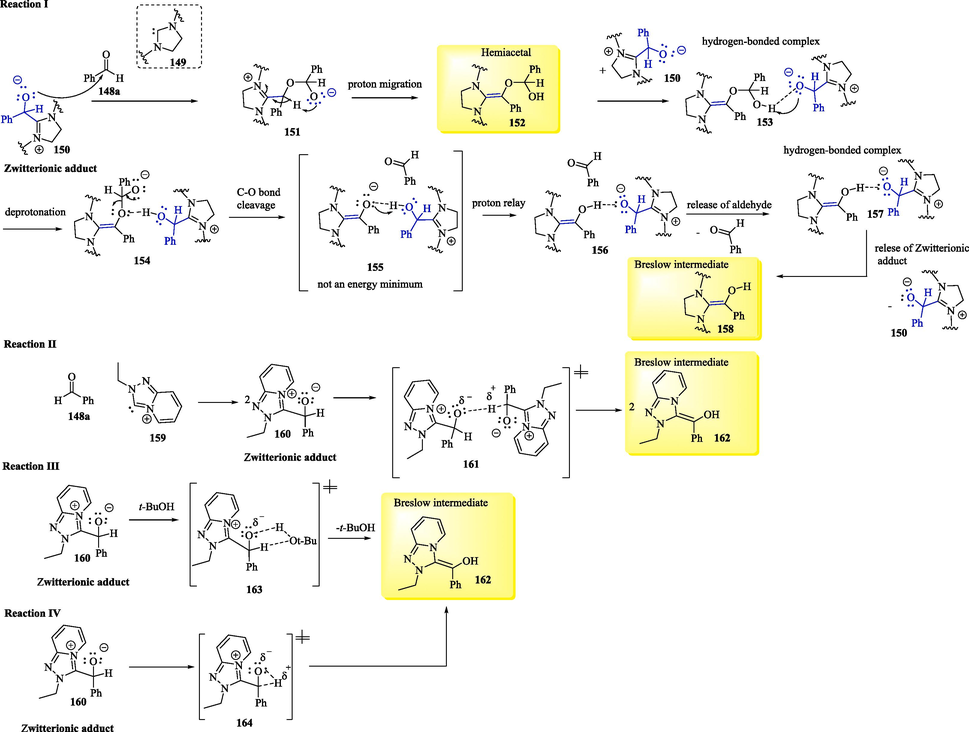
Proton transfer mechanisms to provide Breslow intermediate.
Theoretical investigations of the mechanism of formation of Breslow intermediates were also proposed (He and Xue, 2011). In Scheme 9 (Equation II), the Breslow intermediate 162 is formed via two sequent intermolecular proton transfers between two NHC/benzaldehyde coupled intermediates. The theoretical results show that this intermolecular proton transfer to form a tertiary alcohol and enolate anion is rate-determining, with a barrier of 30.93 kcal/mol. In Scheme 9 (Equation III), the protic solvent-assisted hydrogen transfer furnishing Breslow intermediate 162 is rate-determining, with a barrier of 28.84 kcal/mol. These studies and other research reports suggest that both pathways (Scheme 9, Equations II and III) are competitive for their similar barriers. Furthermore, it was reported that the great tension associated with the three-membered transition state (Scheme 9, Equation IV) results in the high intramolecular barrier (45.23 kcal/mol) that leads to the formation of Breslow intermediate 162, which is an unfavorable process.
1.4.3 Generation of acylazoliums by internal and external oxidation of Breslow intermediates
The scope of NHC chemistry can be expanded with protocols for the oxidative transformations of Breslow intermediates into electrophilic acyl azolium intermediates via an internal redox process or through the addition of an external oxidant. Electrophilic acyl azolium has been generated from internal redox processes, such as redox isomerization, the loss of a leaving group or ring-opening, with several α-oxidizable aldehydes (e.g., ynals 165, α-haloaldehydes 166 and epoxyaldehydes 167) (Dzieszkowski et al., 2020; Flanigan et al., 2015; Ni et al., 2015) (Scheme 10, Equation I). Carboxylic acid derivatives (Biswas et al., 2021) 168 and 169 (Scheme 10, Equation II) are other examples of compounds that lead to the formation of the acylazonium intermediates without the use of an external oxidant agent.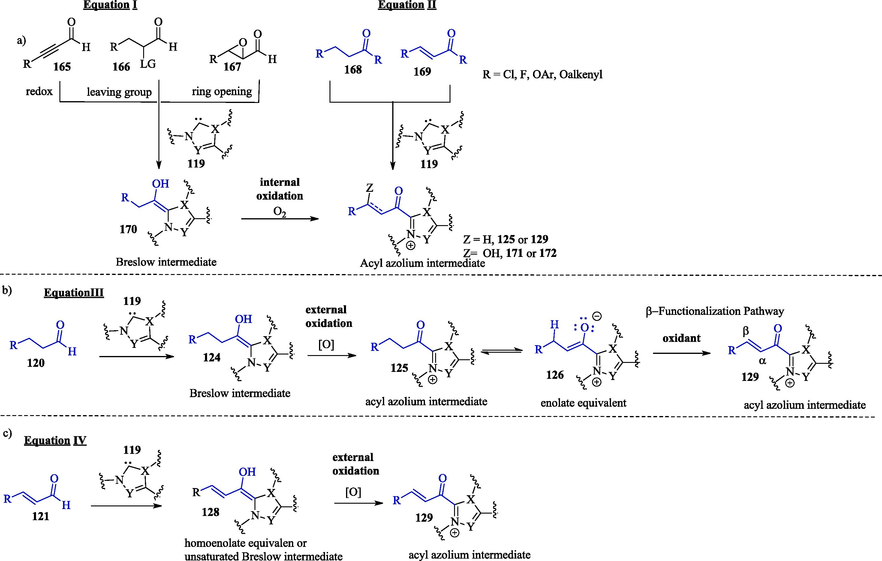
Oxidative pathways towards acylazolium intermediates.
The addition of an external oxidant for the oxidation of the Breslow intermediates 124 and 128 into the corresponding acyl azolium intermediates 125 and 129 paves the way for the use of other raw materials, including saturated 120 and ,β-unsaturated 121 aldehydes (Scheme 10, Equations III–IV). Some examples of external inorganic/organic oxidants that can be used for the oxidation of the Breslow intermediate to the corresponding reactive acylazolium ion intermediate include 3,3′,5,5′-tetra-tert-butyldiphenoquinone (Kharasch-reagent, DQ), nitrobenzene, MnO2, ferricyanide, 2,2,6,6-Tetramethylpiperidinyloxy (TEMPO), N-fluorobenzenesulfonimide (NFSI), riboflavin, phenazine, azabenzene, polyhalides and electrochemical (Finney et al., 2012; Ishii et al., 2020; Wu et al., 2017). It should be noted that normally, the use of substrates capable of undergoing redox processes using external oxidants requires a stoichiometric amount and has cost, toxicity and sustainability problems (Wu et al., 2019). Cooperative systems that employ atmospheric oxygen as a terminal oxidant, such as organocatalysis with NHCs and a transition metal-based catalyst in combination with O2, would be more desirable (Zhao et al., 2013). Molecular oxygen (O2) has been considered as an ideal oxidant due to its natural, inexpensive, and environmentally friendly characters. (Stenkvist et al., 2023).
Some examples of NHC-catalyzed reactions are shown below.
1.5 Oxidative/oxygenative pathways for the aerobic oxidation of aldehydes through NHC catalysis
The aerobic oxidation of the Breslow intermediate 170 generated in situ from NHC 119 and aldehyde 173 can occur through two different pathways (Scheme 11): two-electron transfer to the oxidant species (O2 or inorganic/organic oxidants in stoichiometric quantities) (oxidative route) and/or oxygen atom transfer from the oxidant (O2) (oxygenative route). In most cases, the Breslow intermediate is transformed into an electrophilic acylazolium ion, which may be attacked by nucleophiles, but not all oxidations necessarily occur in this direction. For example, when oxygen is used as the oxidant agent, a complex ion between the Breslow-derived radical cation 174 and superoxide radical anion [O2].- is formed via single electron transfer (SET) from the Breslow intermediate to the molecular oxygen.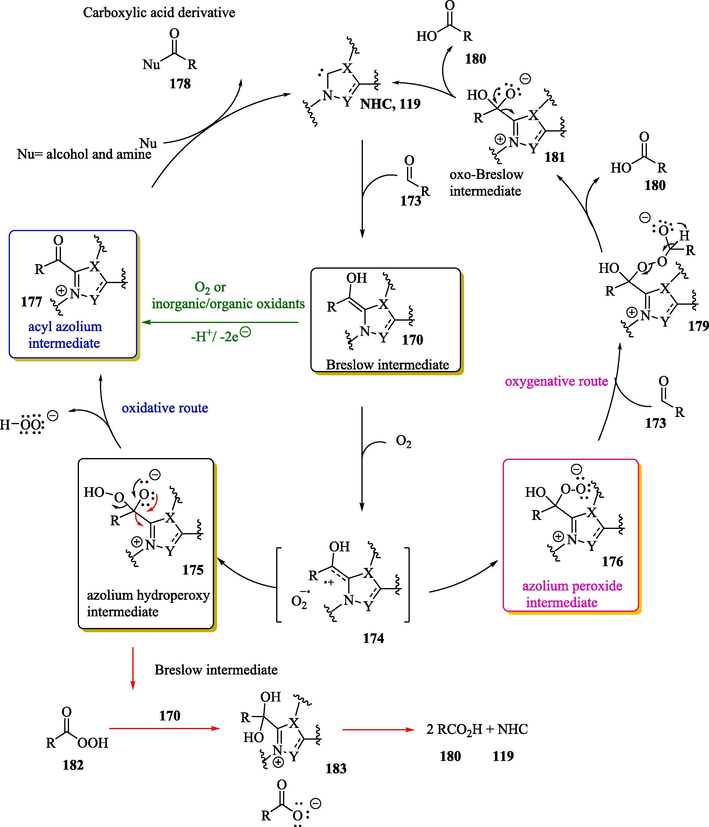
Possible mechanisms for the aerobic oxidation of aldehydes to carboxylic acid derivatives (oxidative path) and carboxylic acid (oxygenative path).
Subsequently, the complex 174 recombines to provide the hydroperoxy derivative 175 and the peroxide anion. Zwitterionic hydroperoxy 175 forms the acyl azolium ion 177 after the liberation of the hydroperoxy anion in the oxidative pathway, which can then react with further nucleophiles according to a nucleophilic acyl substitution mechanism, with the regeneration of the NHC catalyst (oxidative route, Scheme 11). Alternatively, the peroxide 176 reacts with a second molecule of aldehyde 173 to form intermediate 179 (oxygenative route, Scheme 11), which, through a process reminiscent of Baeyer-Villiger oxidation, forms the first molecule of acid 180. The final step is the release of an additional molecule of acid 180 from intermediate 181, with the regeneration of the catalyst. As reported by Bortolini et al., (2014) all intermediates of both catalytic pathways were identified under mass spectrometry conditions (Bortolini et al., 2017, 2014; De Risi et al., 2023). It has also been reported in the literature (Zhao et al., 2013) that azolium hydroperoxy intermediate 175 can undergo fragmentation, with the release of the peracid compound 182. This later, in turn, reacts with aldehyde 173 to provide two equivalents of the carboxylic acid 180. According to Studer, another possible way of generating the carboxylic acid 180 involves the fragmentation of the salt 183 obtained from the reaction between the Breslow intermediate 170 and the peracid 182 (Zhao et al., 2013).
1.6 Aerobic oxidative NHC-catalysis
1.6.1 Oxidative NHC-catalyzed esterification of aldehydes
Within the previously discussed concept, Delany et al. (2013) described NHC-catalyzed aerobic aldehyde-esterifications with methanol without additives or cocatalysts. Benzaldehyde 148a and several aromatic aldehydes 148b–d and 184–185 containing electronically different substituents were converted into the corresponding methyl esters 187a–f in excellent yields in the presence of DBU, methanol and air (Scheme 12). (Delany et al., 2013).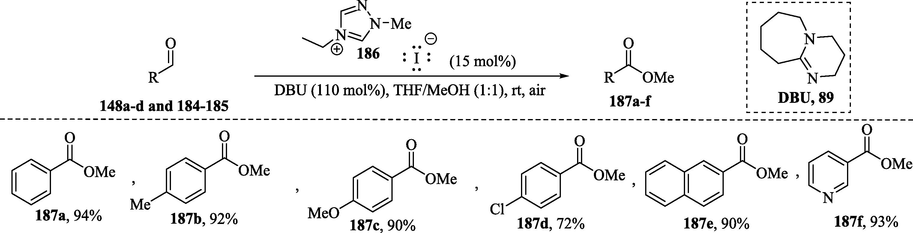
Oxidative carbene-catalyzed esterification of aldehydes.
1.6.2 NHC-catalyzed ester synthesis from organic halides
Tan et al. (2021) described the NHC-catalyzed synthesis of esters in moderate to excellent yields via the incorporation of oxygen atoms from O2 into organic halides (Scheme 13a). Aliphatic bromide 189 k (n = 3), compared to benzylic halides 189a–n, failed to provide the compound 190 k. It should be noted that halides can be transformed into aldehydes and ketones under several conditions, with the need for excessive additives and stoichiometric strong oxidant agents, including NaIO4, H5IO6/V2O5, pyridine N-oxide in Ag2O, o-iodoxybenzoic acid and tert-butyl hydroperoxide (TBHP), while the NHC-catalyzed oxidation of organic halides provides a green strategy for ester synthesis. In Scheme 13b, the NHC-catalyzed oxygenation cross-esterification of benzyl bromides 189a–d with aliphatic alcohols 191 produced the desired esters 187a–b and 193a–i in lower to good yields (Tan et al., 2021).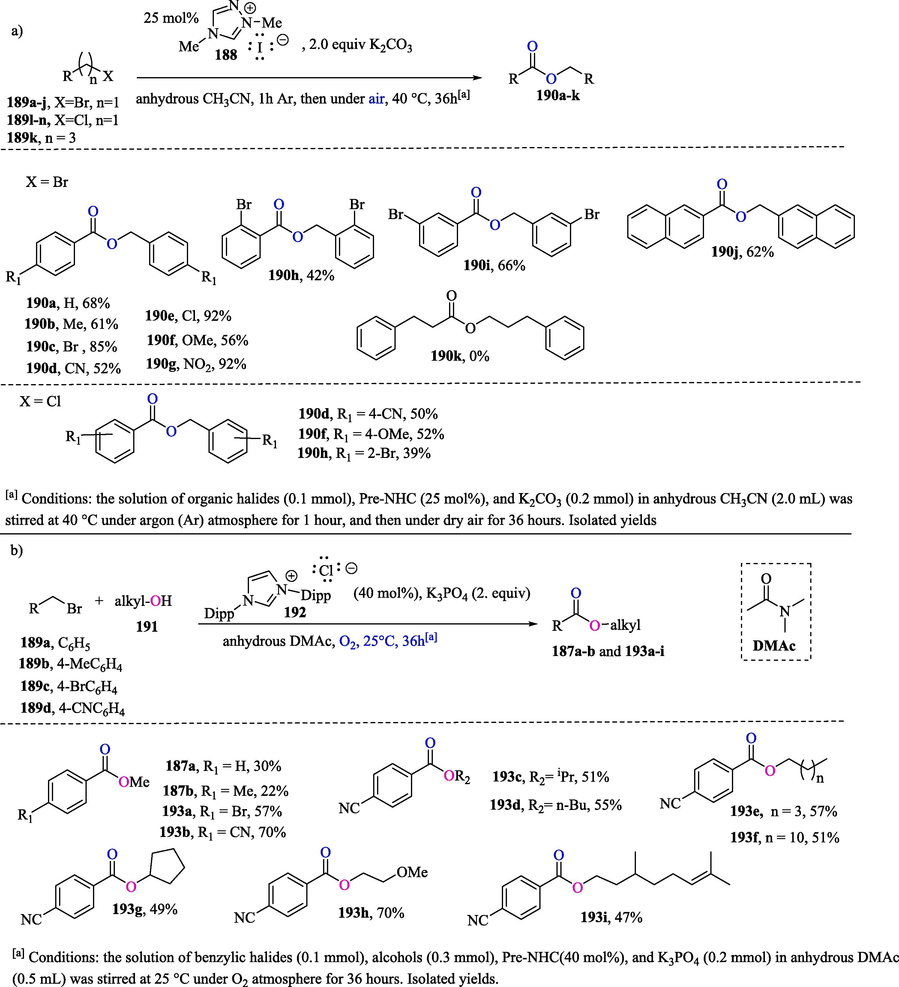
a) Esterification of organic halides 189a-j;189 l-n and 189 k b) Cross-esterification of organic halides 189a-d with aliphatic alcohols 191.
The proposed mechanism for ester synthesis illustrated in Scheme 14 was based on 18O labeling experiments, such as the cross-esterification of 4-nitrobenzyl bromide and hexyl bromide performed under 18O-labeled O2. From this study, it was established that the two oxygen atoms of the ester originated from 18O2. In Scheme 14, the deoxy Breslow intermediate 195 generated in situ from NHC 94c or 194 and organic halide 189 undergoes an oxidative reaction with O2, transforming into peroxide anion 196. The reaction of this latter compound with 195 leads to the formation of intermediate 197, which subsequently reacts with an electrophilic halide 189 to form 198 (Path I). The release of a hydrogen halide from 198 culminates in the formation of 199, which then provides 200 via an oxidative process with molecular oxygen. Finally, the reaction between species 199 and 200 produces intermediate 201, which is converted into an ester product 190, with the release of the NHC. In Path II, when an alcohol 191 is employed as one of the starting materials, the Breslow intermediate 202 is oxidized by O2 to form acyl azolium compound 203, which is subsequently attacked by the nucleophilic oxygenated compound to provide esters 187a-b and 193a-i and the NHC.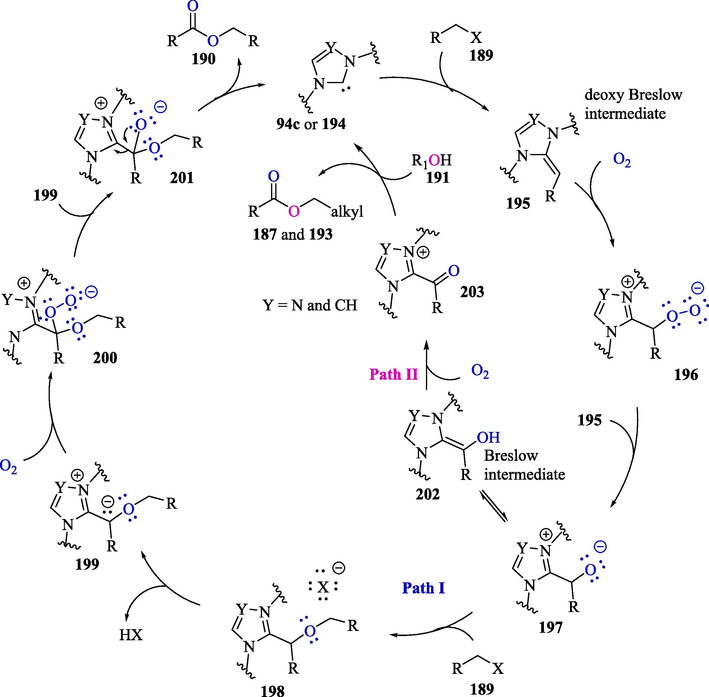
Proposed mechanism for NHC-catalyzed esterification of organic halides (Path I) and cross-esterification of organic halides with aliphatic alcohols (Path II).
1.7 Examples of NHC-catalyzed reactions using external oxidizing agents
1.7.1 Aldehyde esterification in the presence of an external oxidant and with molecular oxygen as the terminal oxidant
Based on the mechanistic study of the NHC-catalyzed aerobic oxidation of aldehydes to esters, as demonstrated in Scheme 11, Bortolini et al. (2023) found that the oxidation of a Breslow intermediate with O2 in the presence of alcohol led to the formation of the carboxylic acid 208 and esters 190a and 209 as a side product (Schemes 15 and 16a). According to researchers, the ratio of the compounds 208 and 190a and 209 is related to the substitution pattern of the R-group (De Risi et al., 2023; Zhao et al., 2013). An alternative method for the preparation of esters 190a and 209 with good yields involves an efficient cooperative NHC– and metal-based redox catalysis of aldehydes 148a, 148c–e, 184, 204a, 205–206 or 207 in the presence of alcohols (Schemes 16a-b) using aerobic oxygen as the terminal oxidant for the regeneration of the metal cocatalyst (Zhao et al., 2013).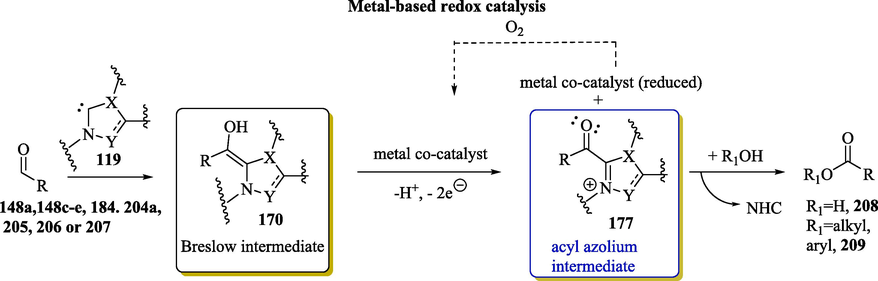
NHC-catalyzed oxidative esterification of aldehydes via cooperative NHC/metal-based catalysis.
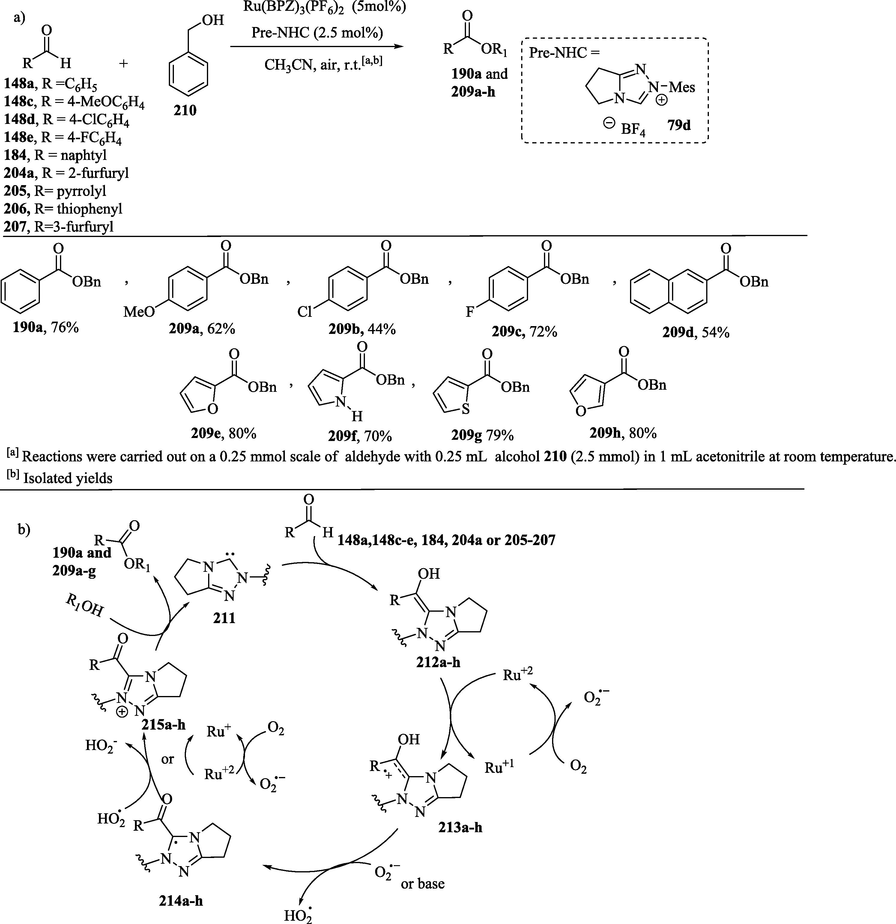
a) Cooperative NHC– and ruthenium-based redox catalysis for oxidative esterification of aldehyde; b) Proposed mechanism for oxidative esterification.
In this regard, several aromatic and heteroaromatic aldehydes 148a, 148c–e, 184, 204a, 205–206 or 207 could be esterified in moderate to good yields through cooperative NHC and Ru(II)-based redox catalysis, with aerobic oxygen being used as the terminal oxidant (Scheme 16a) (Zhao et al., 2013).
In the proposed mechanism (Scheme 16b), the Breslow intermediates 212a–h are oxidized by Ru(BPZ)3(PF6)2 through a single electron transfer (SET) process to obtain the radical cations 213a–h. Molecular oxygen acts as the terminal oxidant, restoring the catalytic activity of the catalyst through the oxidation of the intermediate Ru(I) complex to provide the Ru(BPZ)3+2 complex, along with the superoxide radical anion (O2⋅-). Subsequently, the deprotonation reaction of 213a–h with a base or with a superoxide radical anion leads to the formation of tertiary radicals 214a–h, which then undergo oxidation, yielding the acylazolium ions 215a–h. This oxidation step can be carried out with Ru(BPZ)3+2 or with the hydroperoxyl radical (HO2⋅). The attack of the nucleophile on the electrophilic center of acylazolium intermediate culminates in the formation of the esters 190a and 209a–g and the release of the NHC. Zhao et al. report (Zhao et al., 2013) that the major side product identified in this esterification reaction was the carboxylic acid resulting from the oxidation of the Breslow intermediate with O2, without the involvement of Ru catalysis. Zhao et al. (2013) studies also show that in the absence of the Ru(II) complex and alcohol, aldehydes were oxidized under aerobic conditions to the corresponding acids in good to excellent yields (Scheme 17, and see Scheme 11—oxygenative route). (Zhao et al., 2013).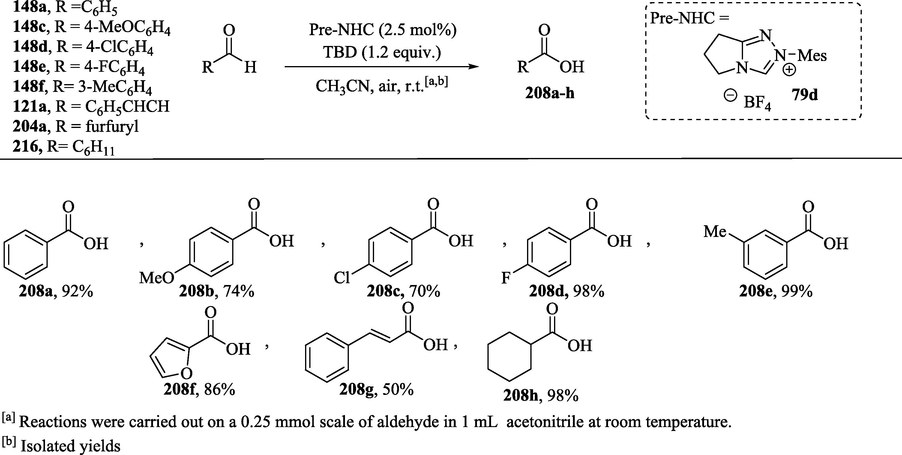
NHC-catalyzed aerobic oxidation of aldehydes.
1.7.2 NHC-catalyzed aerobic oxidation coupled to ETM system for ester synthesis
The oxidative NHC-catalyzed aerobic esterification of α, β-unsaturated aldehydes 121a-b with alcohols 96b and 96e in the presence of a coupled system of electron transfer mediators (ETMs/ETMs’) 2,6-di-tert-butylphenol (2,6-DTBP, 218) and iron(II)phthalocyanine (FePc, 217) in a basic medium provided ester derivatives in excellent yields (Scheme 18a). In the proposed mechanism, the 2,6-DTBP catalyst 218 could be introduced as a precursor of DQ 220 through in situ chemical oxidation with molecular oxygen. Posteriorly, the electron-transfer process between Breslow intermediates 222a–b and DQ 220 leads to the formation of the acyl azolium ions 223a–b and the reduced diol DQH 221, which is posteriorly re-oxidized by FePc and air (O2) acting as the terminal oxidant (Scheme 18b) (De Risi et al., 2023; Liu et al., 2021; Ta et al., 2016). It should be noted that ETMs provide a low-energy path that the electrons can use to flow from the Breslow intermediates 222a–b to a suitable terminal oxidant such as O2 (Axelsson et al., 2016; Ta et al., 2016). In the last step of the catalytic cycle, the esters 219a–b are formed through the nucleophilic attack of alcohol on the electrophilic carbonyl carbon of 223a–b, regenerating the catalyst.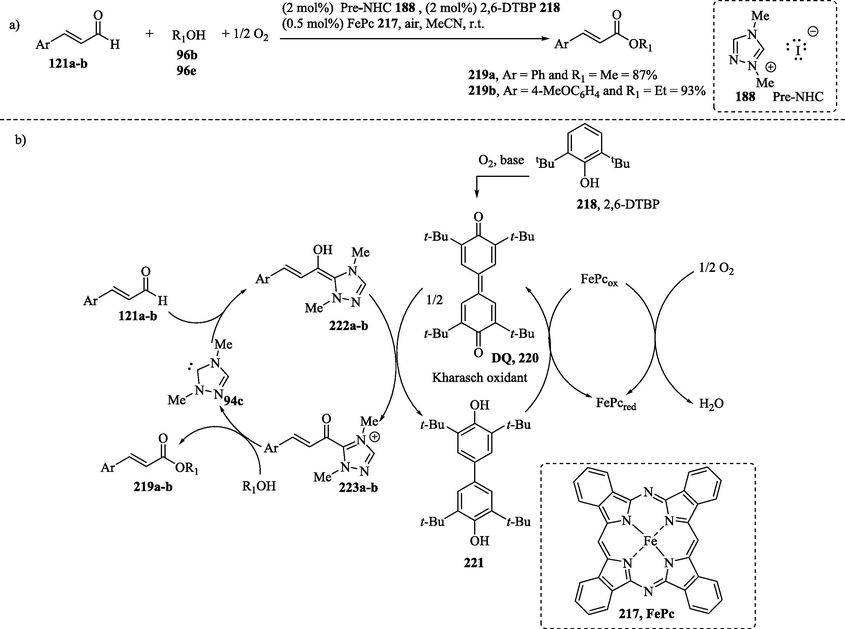
NHC-catalyzed ester synthesis; b) Proposed mechanism of aerobic oxidation via multistep electron transfer.
The oxidative coupling of 2-chlorobenzaldehyde with methanol for the preparation of methyl ester promoted by polystyrene-supported 1,2,4-triazolium carbene offers practical advantages in terms of purification and reusability compared with the homogeneous catalytic process. However, NMR relaxation studies of the solvent effect in the oxidation reaction of 2-chlorobenzaldehyde into the methyl ester promoted by heterogeneous NHC catalysis in the presence of a quinone-based redox mediator such as 2,6-di-tert-butylphenol show that the solvent interaction with the surface of the organocatalyst immobilized over a solid support (polystyrene) plays a key role in determining the catalyst activity. Solvents with a high surface affinity lead to a decrease in catalyst activity, possibly by hindering the access of reactant molecules to the catalytic sites over the surface (Di Carmine et al., 2020).
1.7.3 NHC-catalyzed synthesis of dihydropyranones utilizing ETMs
Asymmetric aerobic oxidative NHC-catalyzed annulation between enals and 1,3-dicarbonyl compounds was investigated using Kharasch-reagent 220 with a catalytic amount of NaBF4 (Scheme 19a) or in combination with another redox mediator such as the phthalocyanine of iron (II) (FePc, 217) (Scheme 19b). In the first case, the desired dihydropyranones 226a–f were provided in modest to good yields and with moderate to excellent enantioselectivities (Flanigan et al., 2015). In Scheme 19b, the combination of two electron transfer mediators (DT/FePc) led to the formation of dihydropyranones 227a–g in good to high yields with ee values of 81–91 % (Axelsson et al., 2016).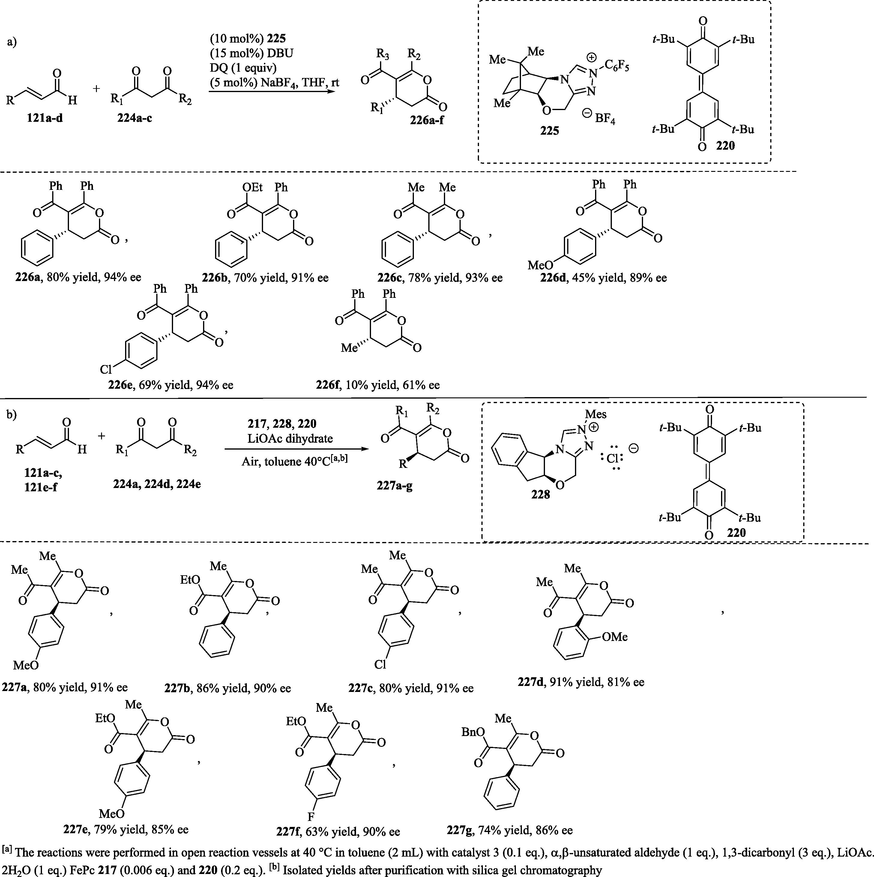
NHC-catalyzed enantioselective annulation of enals with 1,3-dicarbonyls (Shen et al., 2011).
1.7.4 NHC-catalyzed [4 + 2] annulation of ,β-disubstituted enals with trifluoromethyl ketones
In situ-generated 1,2,4-azolium dienolate intermediates 236 (Scheme 20) from α,β-unsaturated aldehyde 229a were used by Chen et al. (2013) for the synthesis of γ-lactones 232a–f via the enantioselective γ-addition of enals to activated ketones under oxidative conditions using an NHC / Lewis acid cocatalysts [Sc(OTf)3 and Mg(OTf)2] / quinone as an external oxidant agent (Chen et al., 2018b; Curti et al., 2020; Mo et al., 2012). In the proposed mechanism (Scheme 20b), the α, β-unsaturated-Breslow intermediates 234 are oxidized by quinone, generating the unsaturated acyl azolium intermediate 235, which subsequently undergoes γ-deprotonation in the presence of a base, transforming into reactive vinyl enolate intermediate 236. The coordination of the Lewis acid Sc(III) or combined Sc-(OTf)3/Mg(OTf)2 with trifluoromethyl ketones 230a–f and azolium dienolate 236 produces the complexes 237a-f. The Lewis acid easily coordinates with the carbonyl oxygen of trifluoromethyl ketone, increasing its electrophilicity. In addition, the oxidative γ-functionalization of the enal under NHC catalysis exhibits lower enantioselectivity (Scheme 20b), and the Lewis acid is necessary for obtaining the desired lactones 232a–f with good to high enantioselectivities. The chelating ketone-azolium dienolate complexes 237a-f amplify chiral induction by the chiral NHC catalyst because the Lewis acid brings the ketone into proximity with the azolium dienolate-containing chiral NHC catalyst. Furthermore, in this reaction, an enal containing steric hindrance at the β-position was utilized as a substrate, so that its reactivity was smoothly switched from the β-carbon to the γ-position upon oxidation (X. Chen et al., 2013b).![1,2,4-triazolium-catalyzed oxidative [4 + 2] cycloaddition of an enal with trifluoromethyl ketones; b) Proposed mechanism.](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105527-fig28.png)
1,2,4-triazolium-catalyzed oxidative [4 + 2] cycloaddition of an enal with trifluoromethyl ketones; b) Proposed mechanism.
1.7.5 NHC-catalyzed oxidative [4 + 2] cycloaddition via ortho-quinodimethane intermediate
The asymmetric functionalization of the benzylic C − H bonds of heteroaryl aldehydes via NHC organocatalysis has been developed by Chen et al. (2020). This strategy involves the in situ generation of heterocyclic ortho‐quinodimethanes (o-QDM, 242), which undergo highly enantioselective formal [4 + 2] cycloaddition with trifluoromethyl ketones 230a-c or isatins 244a-d to form polycyclic lactones 243 and 245a–f, which contain a quaternary carbon center, in good to excellent yields and with over 90 % ee (Scheme 21) (Chen et al., 2020; X. Chen et al., 2013b).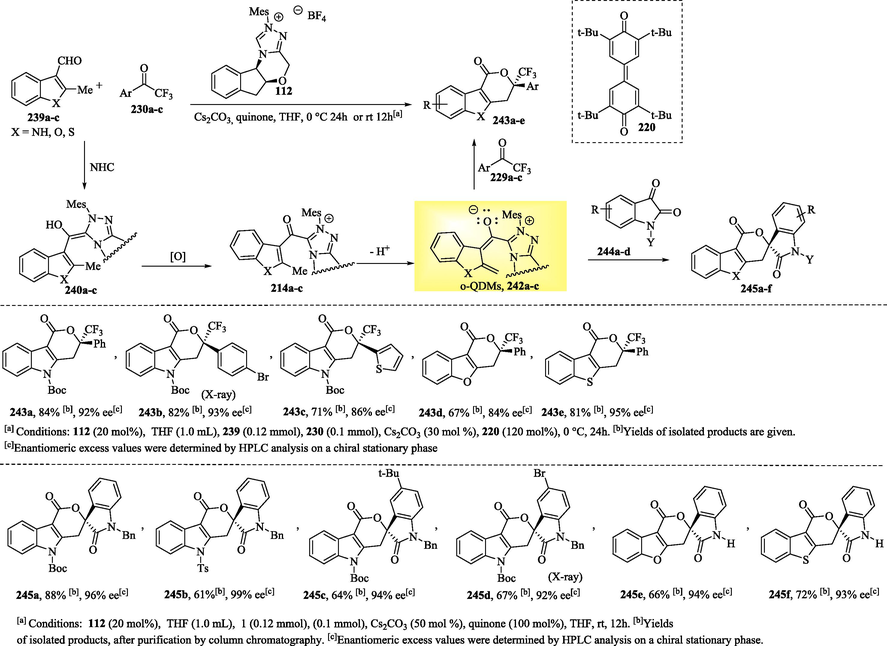
NHC-catalyzed oxidative cycloaddition via ortho-quinodimethane intermediate.
Other examples of the annulation of o-quinodimethanes through NHC catalysis are reported in the literature (Janssen-muller et al., 2016).
1.7.6 Synthesis of oligomeric polyamides mediated by NHC catalysis
The synthesis of oligomeric polyamides (PAs) 248a–d (Scheme 22) via the polycondensation of diamines 246a-c and dialdehydes 247a-b mediated by a 1,2,4-triazolium carbene catalyst in the presence of a quinone oxidant and hexafluoro-2-propanol (HFIP), a nucleophilic additive that guarantees oligomer growth, was proposed by Ragno et al. (2021) (Ragno et al., 2021). The polyamides were obtained with a number-average molecular weight (Mn) in the range from 1.7 to 3.6 kg mol−1, as determined by NMR experiments. In the catalytic process, the deprotonation reaction of the 1,2,4-triazolium salt with DBU, followed by the nucleophilic attack of the carbene 94c on dialdehydes 247a-b, provides the Breslow intermediates 249a-b, which undergo oxidation with quinone, transforming into the corresponding acyl azolium intermediates 250a-b. Posteriorly, the nucleophilic attack of hexafluoro-2-propanol (HFIP) on the carbonyl carbon of 250a-b results in the formation of the hexafluoroisopropyl monoester 251a-b and the release of the NHC catalyst. According to the authors, the hexafluoroisopropyl diester 252a-b can be obtained via the functionalization of monoesteres 251a-b in a second catalytic cycle. They suggest that the concurrent oxidative esterification of the carbonyl function of dialdehyde 247a-b seems to be less likely; however, it cannot be ruled out. The reaction of diesteres 252a-b with diamine culminates in the formation of the amide and the release of HFIP. (Ragno et al., 2021).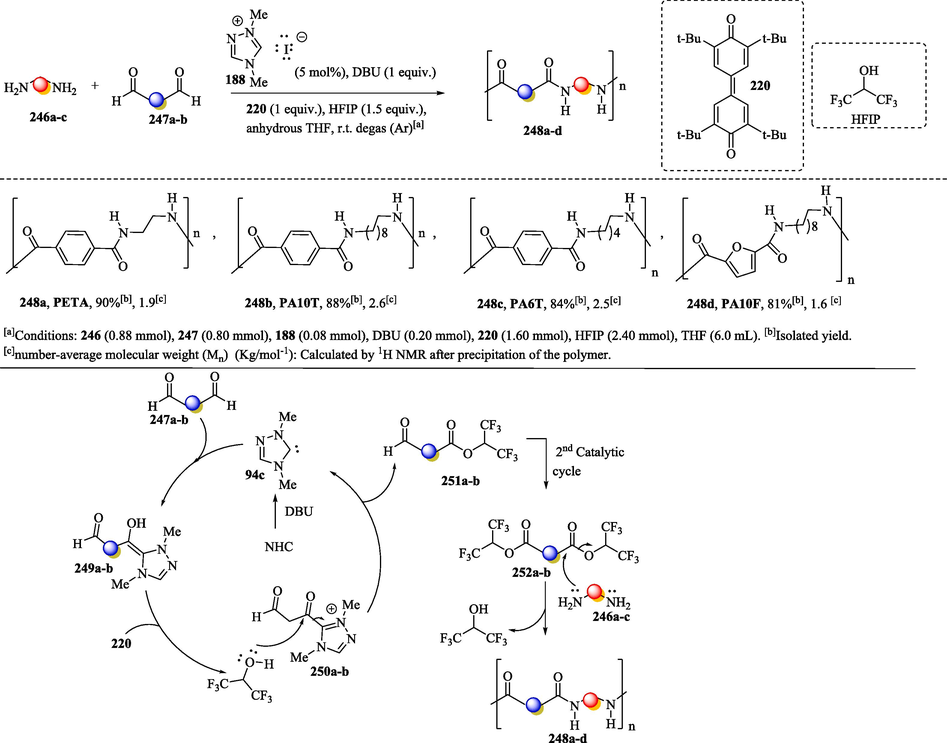
a) Synthesis of polyamide oligomers via oxidative nhc catalysis; b) Proposed mechanism.
Several reviews of polymers obtained from NHC-mediated polymerization reactions are reported in the literature (Fèvre et al., 2013; Naik et al., 2012; Ou et al., 2022; Wei et al., 2022).
1.7.7 NHC-catalyzed reactions via single electron transfer (SET) process
In 2008, Studer et al reported(Guin et al., 2008; Ishii et al., 2019a; Song and Chi, 2019) the pioneering example of NHC-catalyzed conversion of aldehydes 173 to carboxylic esters 253 through two single-electron oxidation steps in the presence 2,2,6,6-tetramethylpiperidine N-oxyl radical (TEMPO) as the oxidizing agent (Scheme 23). For this methodology, cinnamaldehyde, benzaldehyde and electron-donating and –withdrawing para-substituted benzaldehyde derivatives provided the corresponding ester products in excellent yields (Guin et al., 2008). Mechanistically, the process involves two continuous single-electron oxidations of the Breslow intermediates by TEMPO to form the azolium ketones 203, which react with TEMPO- to give the desired TEMPO-ester derivatives 253 and regenerate the N-heterocyclic carbene.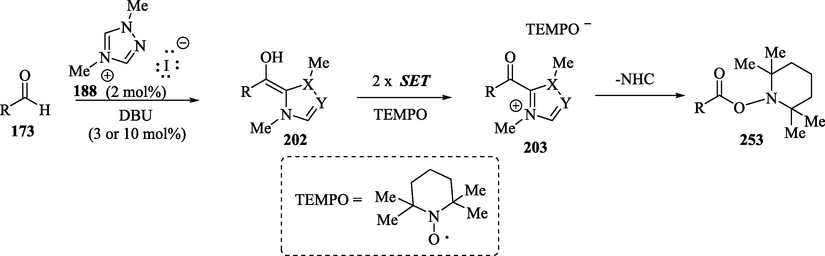
NHC-catalyzed SET reaction.
N–Heterocyclic carbene-catalyzed decarboxylative alkylation between aldehydes 148a, 148d-e, 148 g-h, 184 and redox ester 254a-e (Scheme 24a) was reported by Ohmiya et al. (Ishii et al., 2019a, 2020; Song and Chi, 2019). This work constitutes an important example for generating reactive radical intermediates in NHC catalysis. In proposed mechanism, the reaction of 148a, 148d-e, 148 g-h, 184 with NHC 257 produces a neutral Breslow intermediate 258a-f, which undergoes a deprotonation reaction generating the reducing enolate form of the Breslow intermediate 259a-f. The SET event between enolate 259a-f and 254a-e gives Breslow intermediate type radical 260a-g and alkyl radical 261a-e, respectively, (Scheme 24b). This oxidation involves a decarboxylation step to release CO2 and form two radical species. The authors suggested that Lewis acidic activation of the phthalimide moiety with the counterion (Cs+) favored the oxidation of the enolate 259a-f to the radical adduct 260a-f. Radical-radical recombination between 260a-f and 261a-e followed by elimination of NHC 257 led to the formation of the ketone compound 255a-i.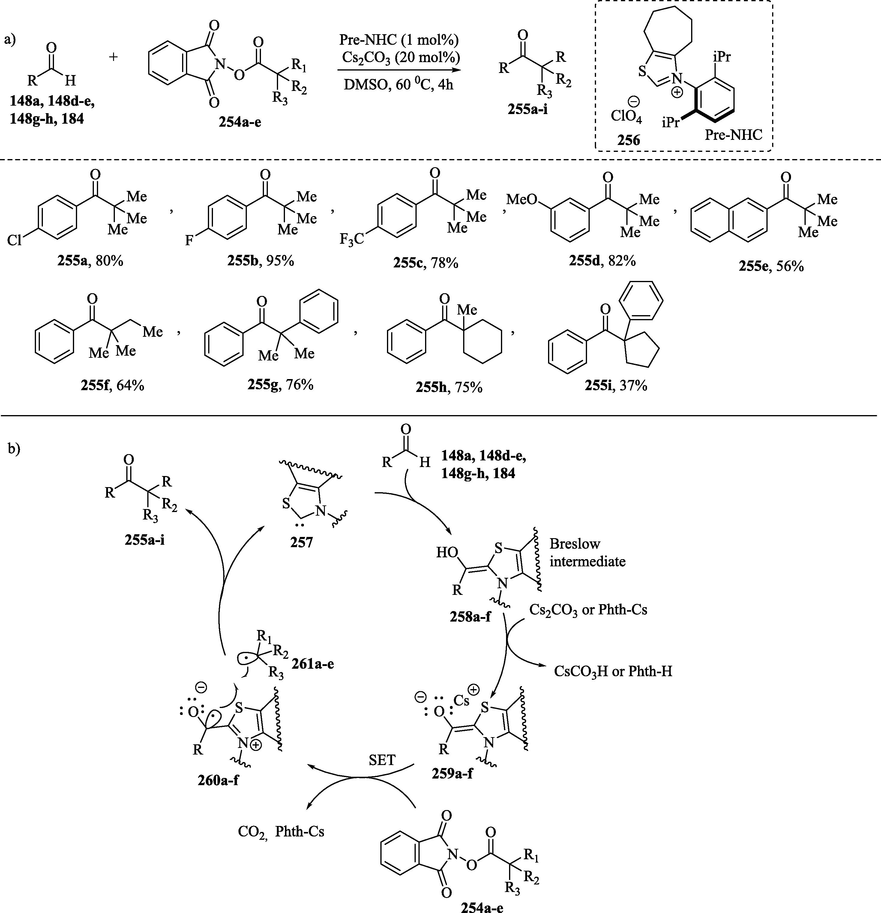
a)NHC– catalyzed decarboxylative alkylation of aldehydes; b) Proposed mechanism.
N-Heterocyclic carbene-catalyzed radical cross-coupling between aldehydes and α-bromoesters or α-bromoamides was also reported in the literature by Ohmiya and colleagues in 2021 (Ishii et al., 2020).
NHC-catalyzed radical reaction relay facilitates the vicinal alkylacylation of styrenes, acrylates and acrylonitriles by employing aldehydes and redox-active esters derived from tertiary alkyl carbonic acids (Scheme 25) (Dai and Ye, 2021; Ishii et al., 2019b). The following chemical transformation has a similar mechanism to that illustrated in Scheme 24b up to the stage of formation of the enolate ion 259. The SET process between enolate 259a-b and redox ester 254f-g gives a ketyl radical 260a-b and tertiary alkyl radical 261a-b, respectively. This last radical species reacts with alkene 262a-d to form the secondary radical 264a-e. Radical-radical combination between 260a-b and 264a-e followed by elimination of the NHC catalyst 257 produces the alkylacylation compound 263a-f.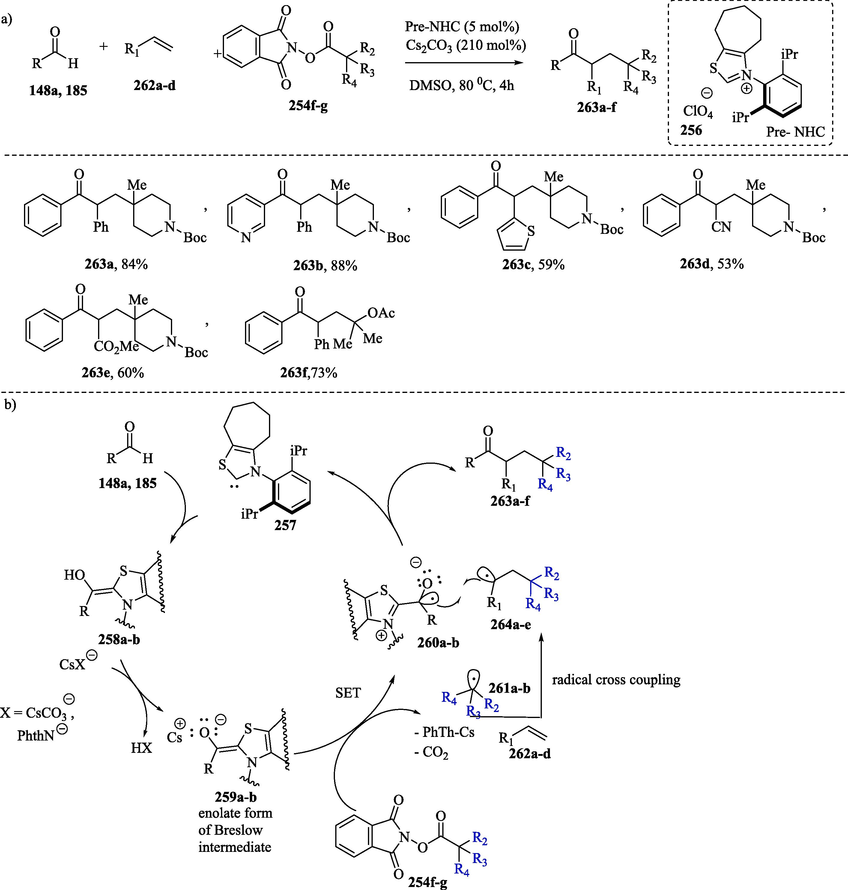
NHC-catalyzed alkylacylation of alkenes via radical process.
Other examples of reactions based on NHC-catalyzed radical pathways can be found in the literature (Dai and Ye, 2021; Dong et al., 2020; Ishii et al., 2020; Li et al., 2022, 2021; K. Liu et al., 2022; Liu and Chen, 2020; Man et al., 2023; Matsuki et al., 2021; Mulks et al., 2023).
1.8 Other examples of NHC mediated reactions
1.8.1 NHCs as organocatalysts for cross-benzoin condensation
Benzoin and cross-benzoin condensations represent an important strategy for creating new C–C bonds, leading to the formation of α-functionalized carbonyl compounds (Menon et al., 2016). In the following example, 1,2,4-triazolium carbene-catalyzed asymmetric cross-benzoin reactions of heteroaromatic aldehydes 204a–c with trifluoromethyl ketone 230a–b and 230 g-h led to the formation of α-hydroxy ketones 266a–f in good to excellent yields (71–96 %) and moderate to good enantioselectivities (ee = 52–84 %) (Scheme 26a). After recrystallization, these compounds were obtained with an enantiomeric excess of up to 99 % (Enders et al., 2010).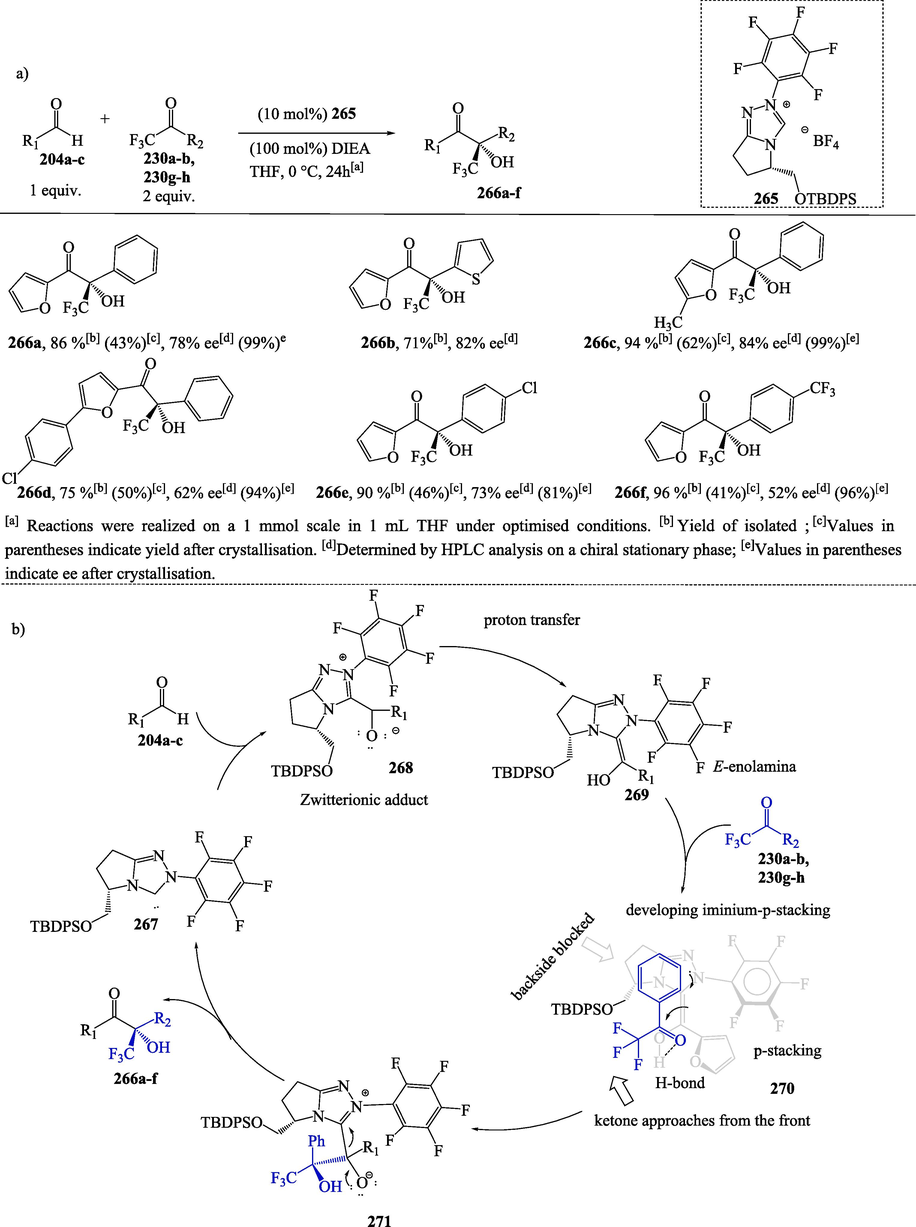
a) Asymmetric cross-benzoin-type reaction of heteroaromatic aldehydes and various trifluoromethyl ketones; b) Proposed mechanism.
In the proposed mechanism, the nucleophilic addition of the NHC 267 to aldehydes 204a-c generates a zwitterionic intermediate 268, which subsequently undergoes a critical proton transfer through some of the pathways described in Scheme 26b, furnishing Breslow intermediate 269 (Enders et al., 2010). DFT studies have suggested the (E) stereochemistry to be favored in Breslow intermediates due to steric and stereoelectronic interaction (Hawkes and Yates, 2008). Leeper (1997) postulated that restricting the rotation of a chiral NHC through the formation of a conformationally restricted chiral N-substituted bicyclic structure would more effectively block only one face of the triazolydene-derived acyl anion equivalent (Breslow intermediate) and therefore improve the enantioselectivity of the nucleophilic addition step (Enders and Kallfass, 2002). In the catalytic cycle illustrated in Scheme 26b, other factors may contribute to the selectivity results for the formation of an excess of one enantiomer over the other from a chiral Breslow-intermediate-derived zwitterionic adduct and trifluoromethyl ketone. The sterically demanding silyl protection group attached to the bicyclic 1,2,4-triazolium catalyst plays an important role in the stereochemistry of the product. Ketone should approach from the side opposite to the relief of steric interactions with the silyl protecting group attached to the Breslow intermediate (2 7 0) formed in a catalyzed reaction of the NHC and aldehyde. Other additional factors for enantioselectivity involve a hydrogen bond between the Breslow intermediate and the ketone and a π-π interaction between the triazolium ring and the aromatic substituent of the ketone. The release of catalytically active NHC from 271 provides the desired product 266a-f.
Other examples of benzoin reactions are reported in the literature (Menon et al., 2016).
1.8.2 Chiral NHC-catalyzed Stetter reaction
The Stetter reaction is a 1,4-addition of an aldehyde 173 to an α,β-unsaturated compound 274, catalyzed by cyanide or a Pre-NHC (Heravi et al., 2020). In this case, the NHC reaction involves a Michael acceptor 274 as the electrophile for the Breslow intermediate 170, resulting in 1,4-bifunctional compounds 275 bearing quaternary centers (Scheme 27).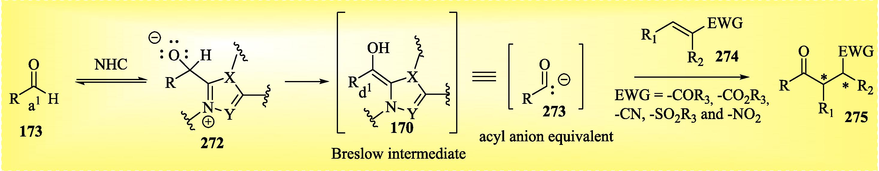
NHC-catalyzed Stetter reaction.
In the chiral 1,2,4-triazolium-catalyzed Stetter reaction between a variety of aldehydes 148d, 148 g, 148i–k and N-acylamido acrylates acting as the Michael acceptor α-amino acid derivatives 276 (Scheme 28a), compounds 277a–e were obtained in good yields and with excellent stereoinduction (Biju et al., 2011; Jousseaume et al., 2011). On the basis of computational calculations, Dudding and Houk (2004) suggested that Breslow intermediate 278 derived from aldehyde and triazolylidene Pre-NHC 80d are electronically suited to preferentially form the E-isomer. (Dudding and Houk, 2004) In the catalytic asymmetric intermolecular Stetter reaction, Jousseaume et al. (2011) suggest that the stepwise addition of chiral Breslow intermediate 278 to the Michael acceptor 276 from the Si face is supported by an intramolecular protonation involving a hydrogen bond between the enol hydrogen atom and the carbonyl oxygen of the Michael acceptor 279 (Scheme 28b). The protonation of 280 and subsequent regeneration of the carbonyl group of 281 provides α-amino acid derivative 277a–e. Jousseaume et al. (2011) also proposed the synthesis of 277a–e through an alternative concerted mechanism, which proceeds via cyclic five-membered transition state 282 (Biju et al., 2011; Jousseaume et al., 2011; Mondal et al., 2014).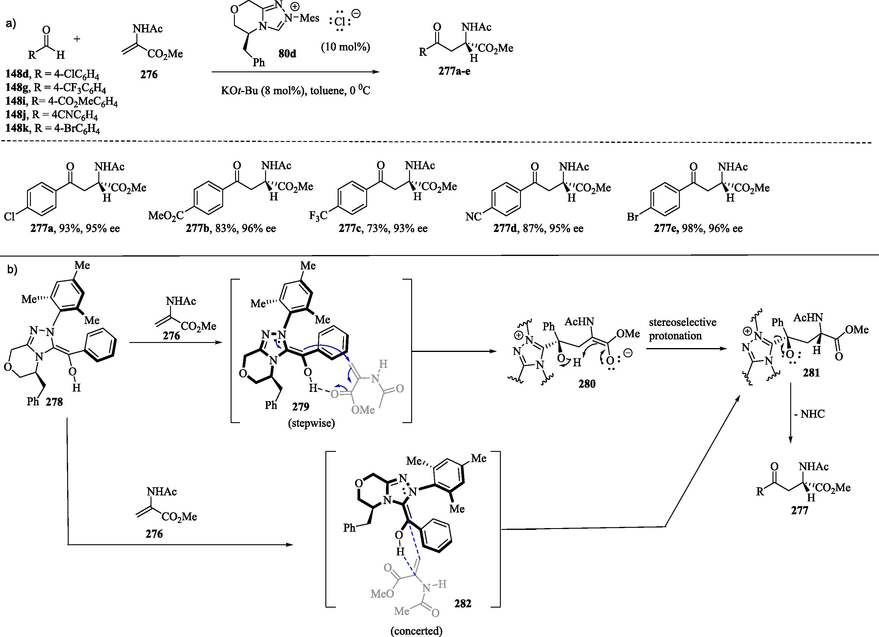
a) Preparation of α-amino acid derivatives through the Stetter reaction; b) Proposed mechanistic pathways for the enantioselective stetter reaction.
1.8.3 Intramolecular Stetter reaction
Ghosh et al. (2019) described a method of preparation of 2-aryl-naphthoquinone derivatives 284a–q in good yields via cross dehydrogenative coupling (CDC) involving an NHC-catalyzed endo Stetter reaction (Scheme 29a) (Ghosh et al., 2019). The 1,2-addition of free carbene 285 to carbonyl compounds 283a–q leads to the formation of intermediate 286, which then undergoes a proton transfer to produce the Breslow intermediate 287 (Scheme 29b). The intramolecular addition of the nucleophilic acyl anion equivalent to the Michael acceptor/electron-poor double bond of 287 occurs through the 6-endo-trig pathway (Gilmore and Alabugin, 2011) to provide enolate intermediate 288, which subsequently undergoes proton exchange, leading to alkoxide intermediate 289. This alkoxide intermediate eliminates free carbene to produce the dihydronaphthoquinone derivative 290, which through atmospheric oxidation is converted into the desired product 284.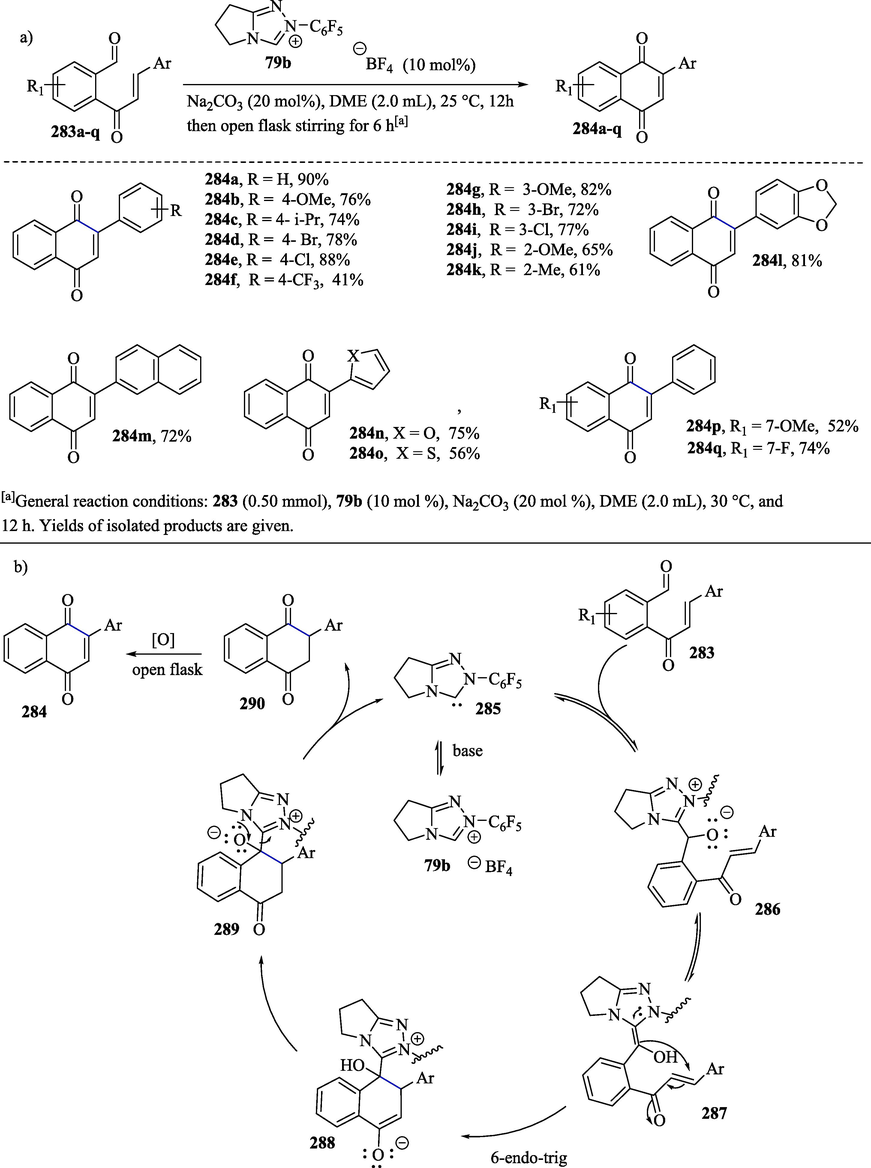
a) Synthesis of 2-aryl-naphthoquinone through NHC-catalyzed intramolecular Stetter reaction; b) Proposed mechanism.
1.8.4 Chiral NHC-catalyzed annulations of α,β-unsaturated aldehydes (enal) with enols and enamines
The NHC-catalyzed [3 + 3] annulation of α-bromo-α, β-unsaturated aldehydes 291a–c with β-keto enols forms of 1,3-dicarbonyl compounds 223a, 223f–g and enamines 294a–d provides functionalized 3,4-dihydropyranones 292a–e (Scheme 30a) and dihydropyridinones 295a–e, respectively, in good yields and with a high enantiomeric excess (Scheme 30b) (Mondal et al., 2019; Yetra et al., 2013).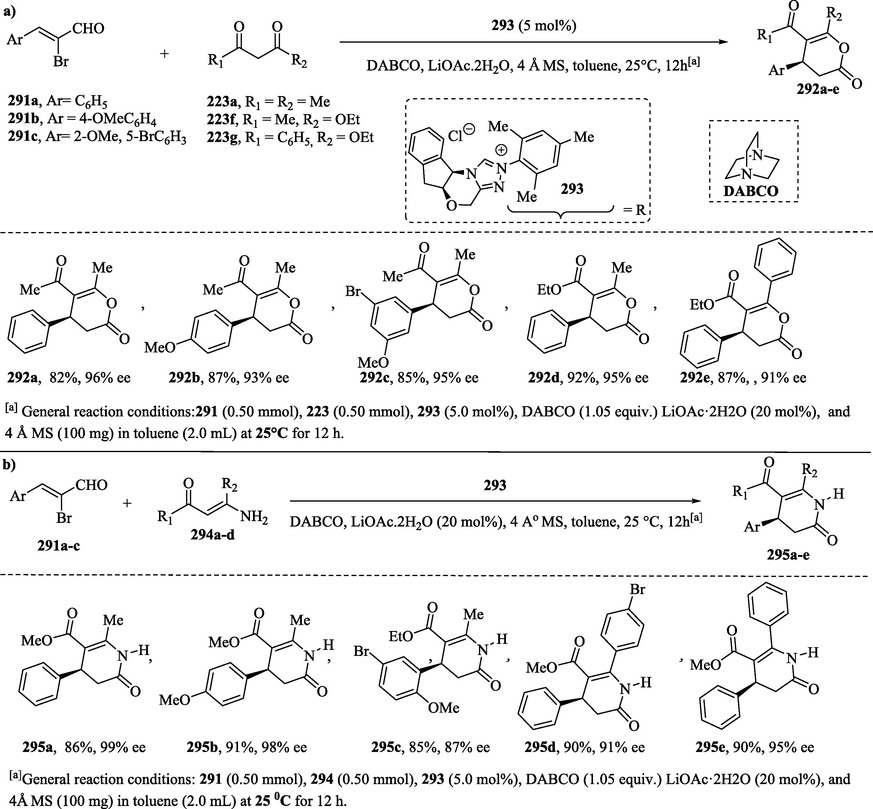
NHC-catalyzed reaction of α-bromo-α, β-unsaturated aldehydes with a) β-keto enols compounds and b) enamines.
In the proposed mechanism (Scheme 31), Breslow intermediates 299 were generated in situ from the reaction of α-Bromo,β-unsaturated aldehydes 291 with the 1,2,4-triazolylidene-derived carbene 296; then, they were converted into acylazolium ions 301 through a3 → d3 umpolung and debromination. The acylazolium ions 301 reacted with the β-keto enol forms of 223 and 302 and enamines 294 in a Michael addition reaction, with subsequent tautomerization and intramolecular nucleophilic acyl substitution to provide the corresponding dihydropyranones 292 and dihydropyridinones 295 and to regenerate the NHC catalyst. Alternatively, the intermediates 301 can undergo 1,2-addition with β-keto enols 302 and enamines 294, followed by a Claisen rearrangement, a [3,3] sigmatropic rearrangement of an allyl vinyl ether 305 (X = O) and ally vinyl enamine 306 (X = NH), respectively, and intramolecular nucleophilic acyl substitution to provide the corresponding products 292 and 295, respectively, and regenerate the NHC catalyst (Mahatthananchai et al., 2012; Mahatthananchai and Bode, 2014; Mondal et al., 2019; Samanta et al., 2012; Yao et al., 2012; Yetra et al., 2013).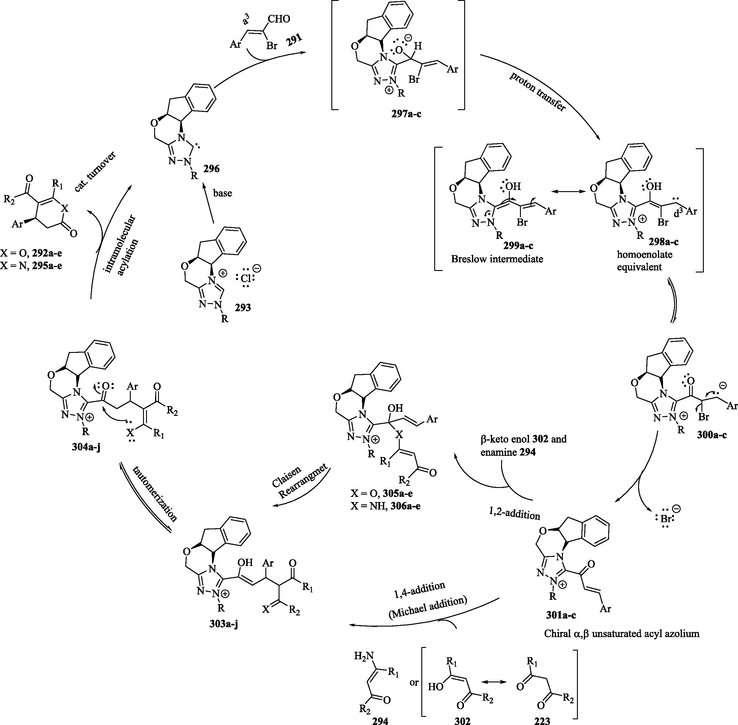
Proposed mechanism of the NHC-catalyzed reaction of α-bromo-α,β-unsaturated aldehydes with the β-keto enol forms of 1,3-dicarbonyl compounds and enamines.
Based on kinetic experiments and theoretical data on the mode of addition of nucleophiles (β-keto enols) to the α, β-unsaturated acyl azolium ions, Samantha et al. (2012) suggest that 1,4-addition is more preferable than 1,2-addition (Samanta et al., 2012). In contrast, Kaeobamrung et al. (2010) proposed that the annulation step-mechanism occurs via a Claisen-type rearrangement rather than via the 1,4-addition of the enol and enamines to the catalytically generated α, β-unsaturated acyl azolium (Kaeobamrung et al., 2010; Lyngvi et al., 2012; Mahatthananchai et al., 2012; Samanta et al., 2012).
1.8.5 Chiral NHC-catalyzed annulations of ynals with enols
Zhu et al. (2010) reported the use of ynals 164a–d as precursors for the NHC-catalyzed generation of α, β-unsaturated acyl azoliums 311 for the preparation of functionalized dihydropyranones 226 g-k (Scheme 32) in moderate yields and with a high enantiomeric excess (Mahatthananchai et al., 2012; Zeitler, 2006; Zhu et al., 2011; Zhu and Xiao, 2010). The use of sterically chiral carbene generated from the 1,2,4-triazolium salt 307 was an important factor for the stereoselectivity of the annulation reaction. The 1,2-addition of the NHC catalyst to ynals 165a–d forms tetrahedral intermediates 308, which undergo proton transfer to provide Breslow intermediates 309. The latter can then generate the α,β-unsaturated acyl azoliums 311 through β-protonation, followed by the tautomerization reaction of the allenol intermediates 310. The generated acyl azoliums 311 are captured by enol derivatives 302d-f to provide dihydropyranone compounds 226 g–k and regenerate the catalyst. As previously reported in Scheme 31, two different mechanisms have been suggested to transform acyl azoliums into dihydropyranones and dihydropyridinones. Zhu and Xiao (2010) (Zhu and Xiao, 2010) and (2012) Samantha (Samanta et al., 2012) suggest that the acylazolium salt reacts with various nucleophiles via the 1,4-addition pathway. The proposal by Kaeobamrung et al. (2010) (Kaeobamrung et al., 2010; Lyngvi et al., 2012) based on kinetic investigations shows that the annulation process does not proceed via the 1,4-addition of the enol to the catalytically generated α, β-unsaturated acyl azolium; instead, a Claisen-type rearrangement is the key catalytic step.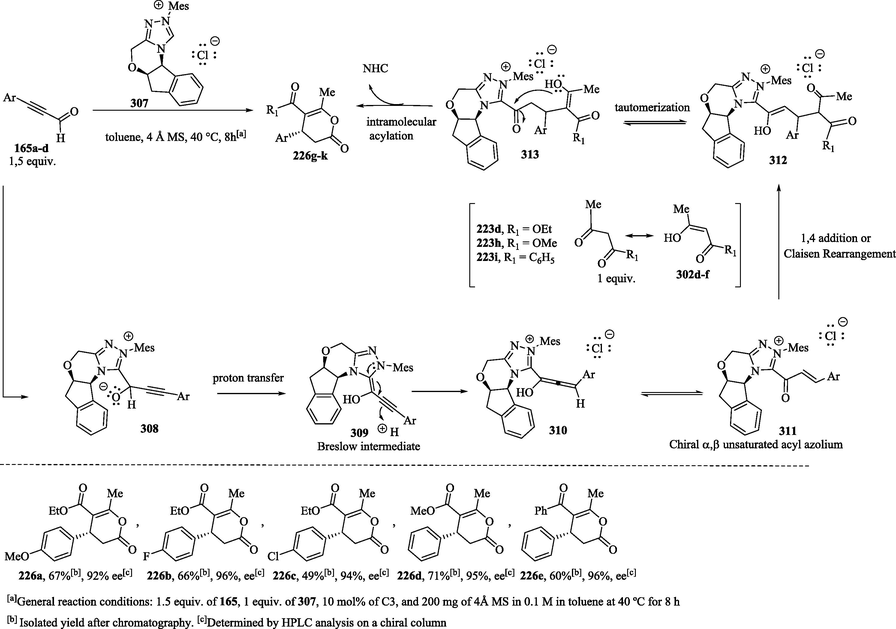
NHC-catalyzed annulations of ynals 165a-d with enols to obtain dihydropyranone derivatives 226a-e.
A literature review (Dong et al., 2022) also focuses on the generation and reactivity of NHC-linked alkynyl acyl azolium intermediates from ynals.
1.8.6 Synthesis of pyrrolo[3,2-c]quinolines via NHC-catalyzed enantioselective addition of benzylic carbon to unsaturated acyl azolium
Wang et al. (2018) (J. Wang et al., 2018) related the synthesis of pyrrolo[3,2-c] quinolines 315a–f (Scheme 33a) via the catalyzed enantioselective addition of benzylic carbon to α,β-unsaturated acyl 1,2,4,-triazolium intermediates 301b and 301d–h generated in situ from α-bromo enals 291b and 291d–h and a chiral triazolium carbene precursor. In the proposed simplified mechanism (Scheme 33b), the reaction proceeds via the formation of Breslow intermediates 299f through the addition of NHC 296 generated in situ from chiral 1,2,4-triazolium salt 112 to α-bromo enal 291f under basic conditions. Subsequently, the intermediate 299f undergoes bromide elimination to provide the α, β-unsaturated acyl azolium intermediate 301f, which then reacts with the nucleophilic intermediate 316 generated by a deprotonation reaction of imine derivative 314. The authors suggest that due to the formation of an intramolecular hydrogen bond and the steric effect provided by the chiral portion of the NHC-based acyl azolium intermediate in complex 317d, the Re face of NHC-bound α, β-unsaturated acyl azolium is blocked by the catalyst, while the Si face is attacked by the Re face of the nucleophilic sp2 carbon of intermediate 316. In complex 317d, the NHC-catalyzed Michael addition between species 301f and 316 provides the chiral adduct 318d, which then undergoes intramolecular cyclization, providing chiral pyrrolidine intermediate 319. The intramolecular nucleophilic acyl substitution of 319 provides the desired pyrrolo[3,2-c]quinoline product 315 and releases the NHC catalyst.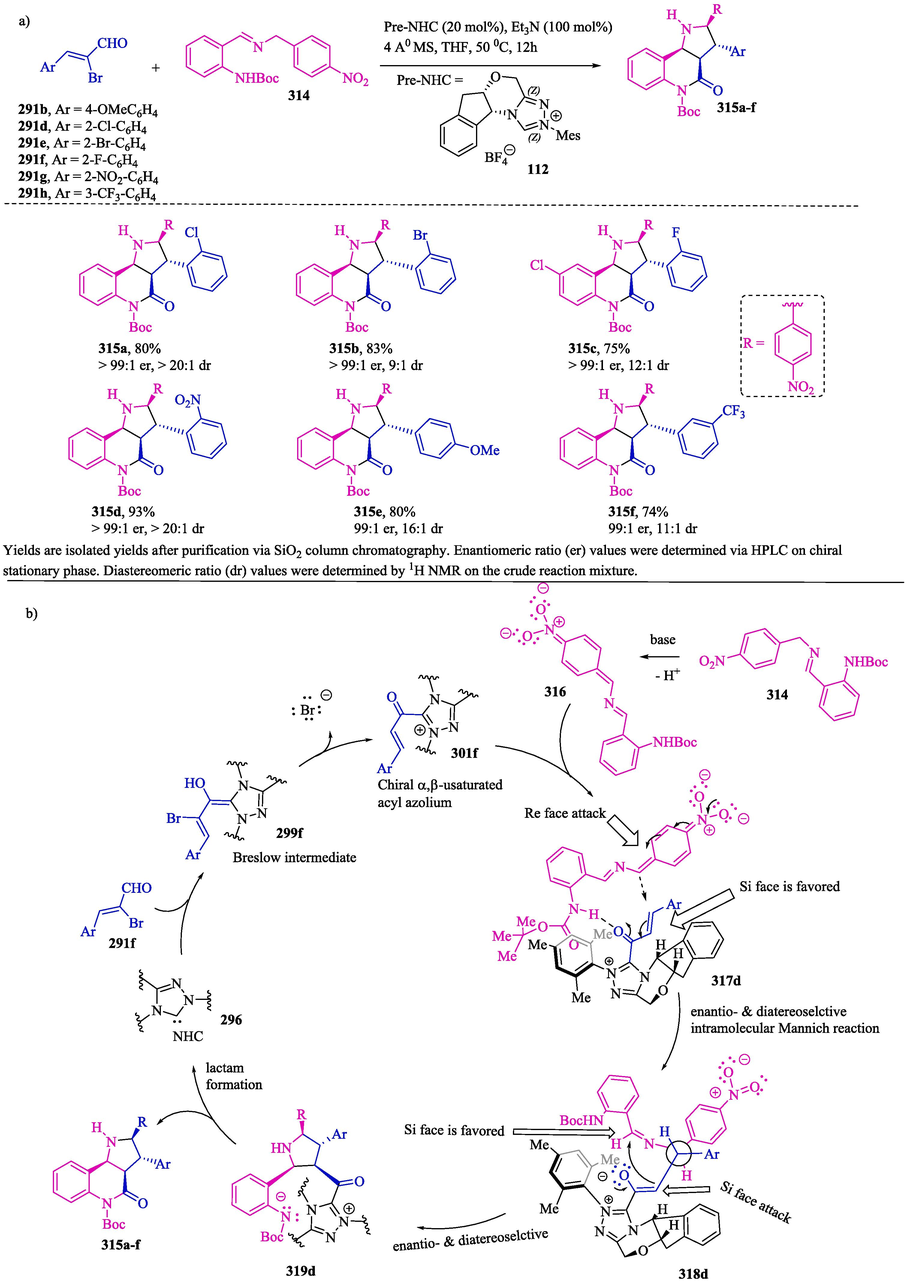
a) NHC-catalyzed enantioselective addition of benzylic carbon to unsaturated acyl azolium; b) Proposed mechanism.
1.8.7 Synthesis of functionalized tetrahydroquinolines through NHC-catalyzed cascade reaction
Zhang et al. (2013) (Zhang et al., 2013) reported the stereoselective aza-Michael-Michael-lactonization cascade synthesis for the construction of chiral functionalized tetrahydroquinolines 321a–e, which contain three consecutive asymmetric centers, in high yields (up to 98 %), with excellent diastereoselectivities (>25:1) and enantioselectivities (up to 98.3 % ee) (Scheme 34a). The synthesis of these compounds involved the reaction between 2-bromoenal derivatives 291a and 291i–k and 2-aminophenylenones 320a–b catalyzed by chiral Pre-NHC 293 (10 mol %). In the proposed mechanism (Scheme 34b), the Breslow intermediates 299 formed by the nucleophilic addition of NHC 296 to the carbonyl group of 2-bromoenal analogs 291a and 291i–k were transformed into compounds 300, which undergo tautomerization and debromination, leading to the formation of compounds 301. The aza-Michael addition of aminophenylenones 320a–b to 301 provided the enolates 322, which were later subjected to intramolecular Michael addition to provide compounds 323. These latter substances were subjected to intramolecular lactonization, transforming into the desired products 321a–e and regenerating the NHC catalyst. The spatial arrangement of atoms in chiral molecules (4aR, 5R, 10bS) was determined through the X-ray crystal analysis of compound 321b.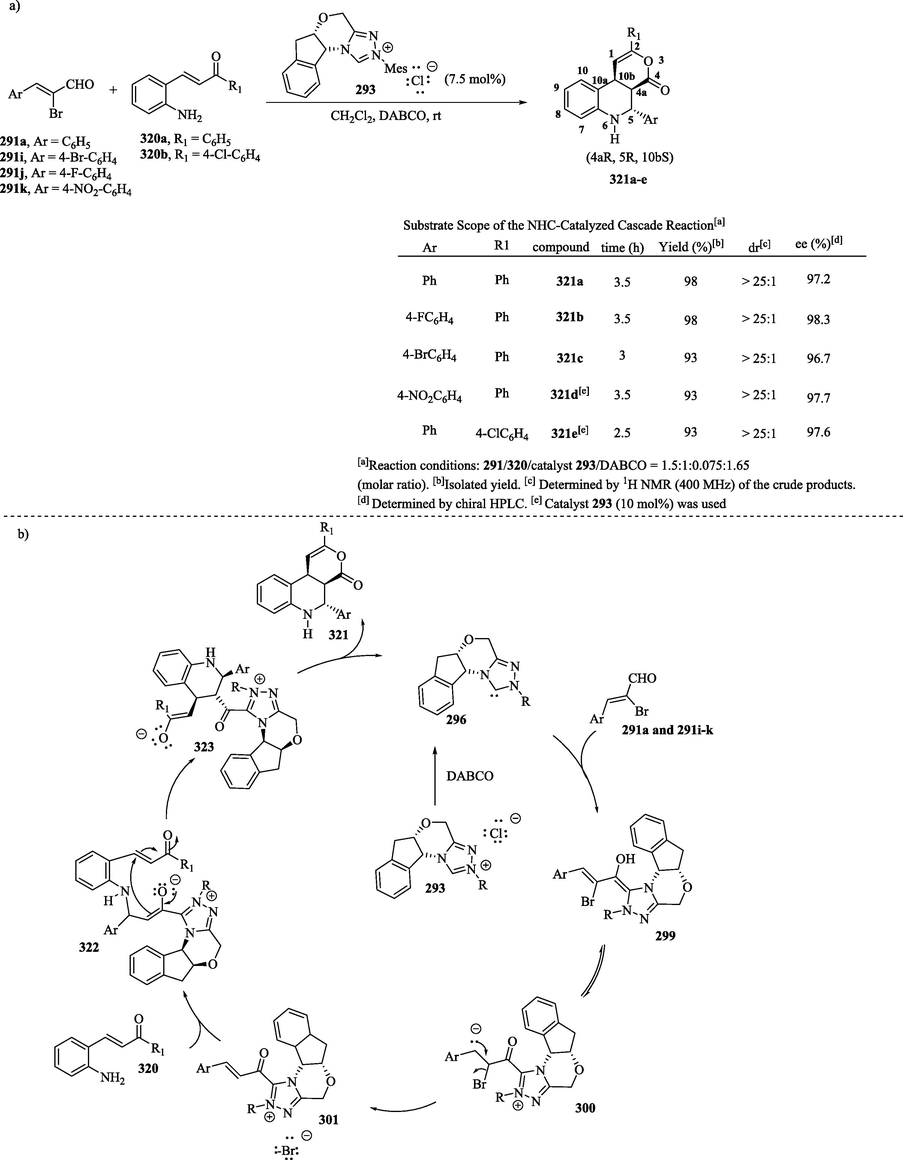
a) Synthesis of tetrahydroquinolines through the aza-Michael-Michael-lactonization cascade reaction of 2′-aminophenylenones and 2-bromoenals; b) Proposed mechanism.
1.8.8 Enantioselective synthesis of functionalized cyclopentenes through NHC-catalyzed reaction
The NHC-catalyzed synthesis of functionalized cyclopentenes 325 generated from 2-bromoenals 291a, 291j and 291 l–n and malonic ester 324a–d (Scheme 35a), which has a γ-aroyl group, proceeds via a Michael-intramolecular aldol-β-lactonization-decarboxylation sequence to provide the cyclopentene derivatives 325a–g in moderate to good yields (57–––85 % yield) and with high enantioselectivities (up to > 99 %) (Scheme 35a) (Mondal et al., 2019). The addition of NHC to 2-bromoenal derivatives 291a, 291j and 291 l–n, followed by proton transfer, results in the formation of the corresponding Breslow intermediates 299a, 299j and 299 l–n, shown also as the mesomeric structures 298a, 298j and 298 l–n, which then undergo debromination to generate the corresponding chiral α,β-unsaturated acyl azoliums 301a, 301j and 301 l–n (Flanigan et al., 2015; Mondal et al., 2019, 2014). The nucleophilic attack of 4-oxoenoates 326a–d to the species 301a, 301j and 301 l–n leads to the formation of the enolate intermediates 327a–g, which subsequently undergo a highly selective intramolecular aldol reaction, providing cyclopentenes 328a–g. The β-lactonization reaction of 328a–g, followed by the elimination of the catalyst, delivers the expected cyclopentane-fused β-lactones 329a–g. The decarboxylation reaction of these last compounds provides the desired cyclopentenes 325a–g (Scheme 35b). In the same scheme, when this reaction was performed with malonate derivatives containing an alkyl group at the γ-position, it led to the formation of highly substituted β-lactone 329 h (Flanigan et al., 2015; Mondal et al., 2019, 2014; Zhao et al., 2021).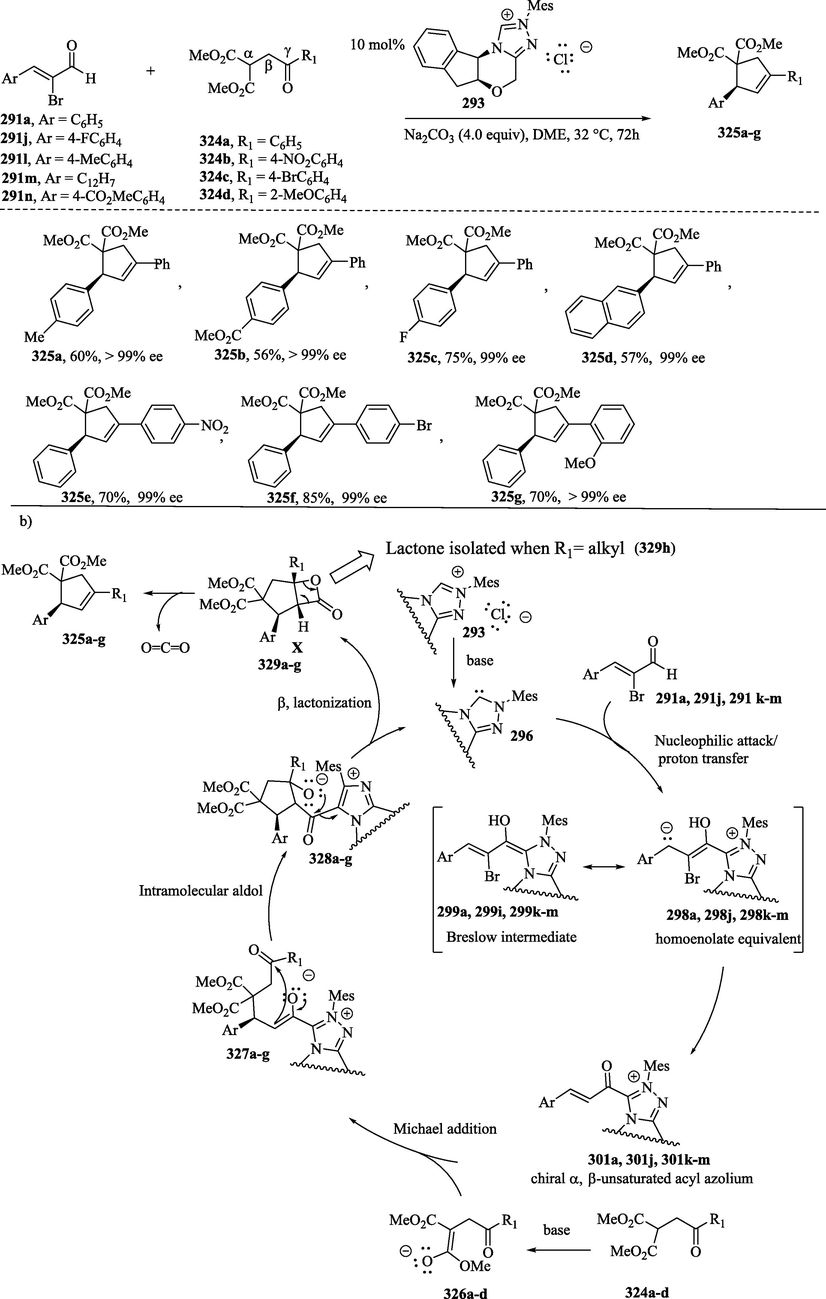
a) NHC-catalyzed enantioselective synthesis of cyclopentene derivatives; b) Proposed mechanism.
Scheme 36 shows the NHC-catalyzed Michael/Michael/esterification domino reaction for the construction of cyclopentanes 331a–f, which have four contiguous stereocenters (Shu et al., 2016). The NHC-catalyzed reaction between a variety of α, β-unsaturated aldehydes 121a, 121c, 121 g–h and 2-nitroallylic acetates 330a–c produced cyclopentane derivatives 331a–f in low to moderate yields (18–53 %) and with enantiomeric ratios from 86:14–98:2 (Scheme 36a). In the NHC-catalyzed [3 + 2] Michael/Michael/esterification cascade (Scheme 36b), the addition of carbanions 332a–d to unsaturated compounds 329a–c, which contain an electron-withdrawing group, followed by the elimination of the acetyl group from the intermediates 333a–f, provided Michael acceptors 334a–f. The Michael addition-ring closure reaction of 334a–f, followed by ethanolysis through nucleophilic acyl substitution on 335a–f, converted these compounds into the corresponding cyclopentane derivatives 331a–f.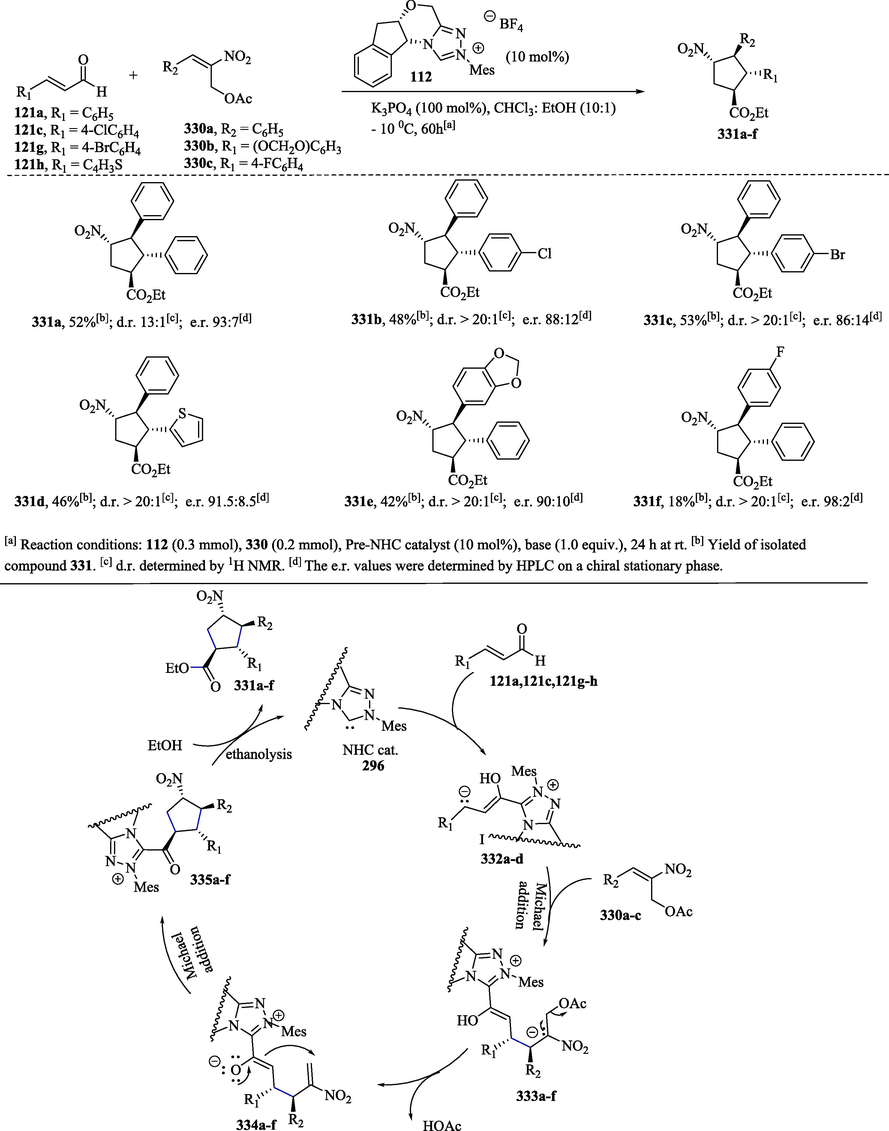
a) Asymmetric synthesis of cyclopentanes through nhc-catalyzed Michael/Michael/esterification domino reaction; b) Proposed mechanism.
Chiang et al. (2007) reported the synthesis of cyclopentene derivatives 337a–f with good enantioselectivity for the cis-1,3,4-trisubstituted diastereomers via an NHC-catalyzed benzoin-oxy-Cope reaction of aldehydes 121a, 121 g and 121i with ketones 336a–d (Scheme 37a) (Chen et al., 2018a; Chiang et al., 2007; Jones et al., 2014). In the mechanism, 4-oxoenoate 336a adopts preferably an s-cis conformation in the conjugate addition of homoenolate 336a to the enone providing the species 339a, which undergoes oxy-Cope rearrangement through a boat transition state conformation that leads to the formation of the cis-stereochemistry observed in the cyclopentene product 337a–f (Scheme 37b). The intermediate 341a undergoes tautomerization and subsequent intramolecular aldol addition to generate alkoxide derivative 343a. The intramolecular acylation of 343a, followed by the elimination of the NHC catalyst, provides β-lactone 344a. The [2 + 2] cycloreversion reaction of 344a to form the desired product 337a was carried out with the extrusion of CO2.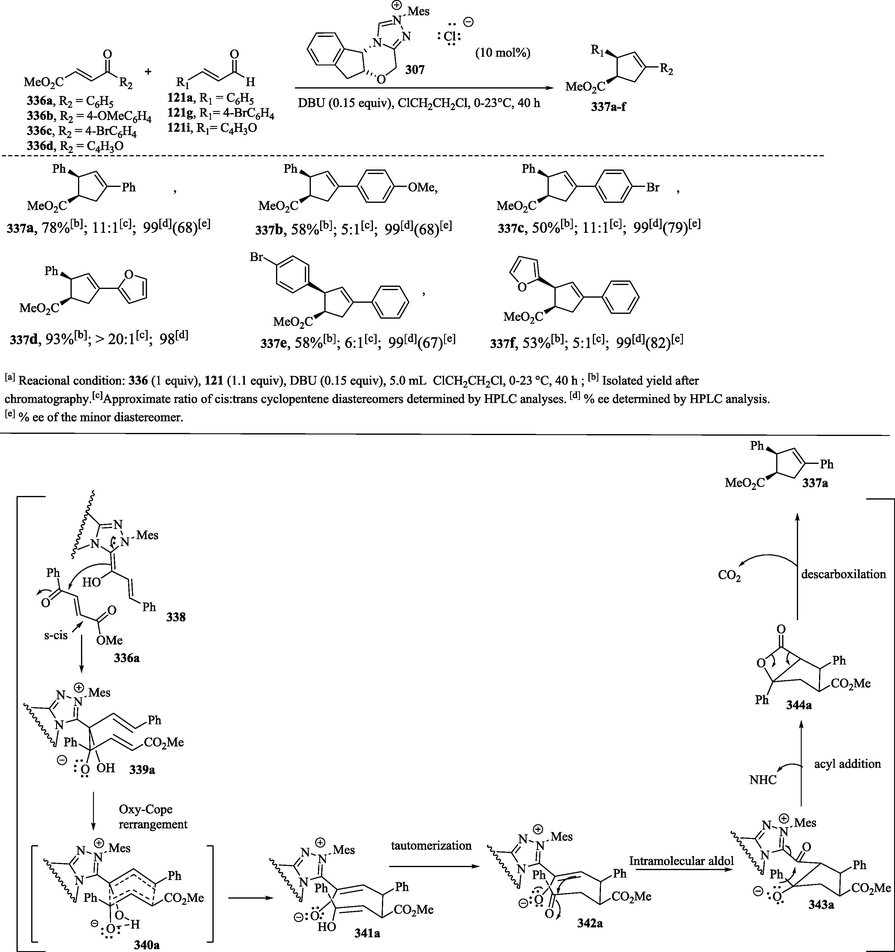
a) Synthesis of cyclopentene derivatives via NHC-catalyzed benzoin-oxy-Cope reactions; b) Proposed mechanism.
1.8.9 Synthesis of cyclopentane-fused β-lactams through NHC-catalyzed annulations of enals and unsaturated N-sulfonyl ketimines
He and Bode (2008) expanded NHC catalysis for the preparation of cyclopentane-fused β-lactams 346a–d in good yields and with very good stereoselectivities from enals 121a-b and unsaturated N-sulfonyl ketimines 345a–c derived from chalcones (Scheme 38a) (He and Bode, 2008; Vora et al., 2012). These researchers suggest that the cis-relative configuration of the cyclopentane groups arises from the secondary orbital overlap considerations proceeding through a boat oxy-Cope transition state 347a–d (Scheme 38b), followed by the tautomerization reaction of 348a–d. The remaining stereocenter is established via a reversible intramolecular Mannich reaction of the enolate and the azomethenic carbon–nitrogen double bond of sulfonylimine, whose adjacent C–C bond is freely rotated. Finally, the lactamization reaction of 350a–d forms products 346a–d and catalyst release occurs.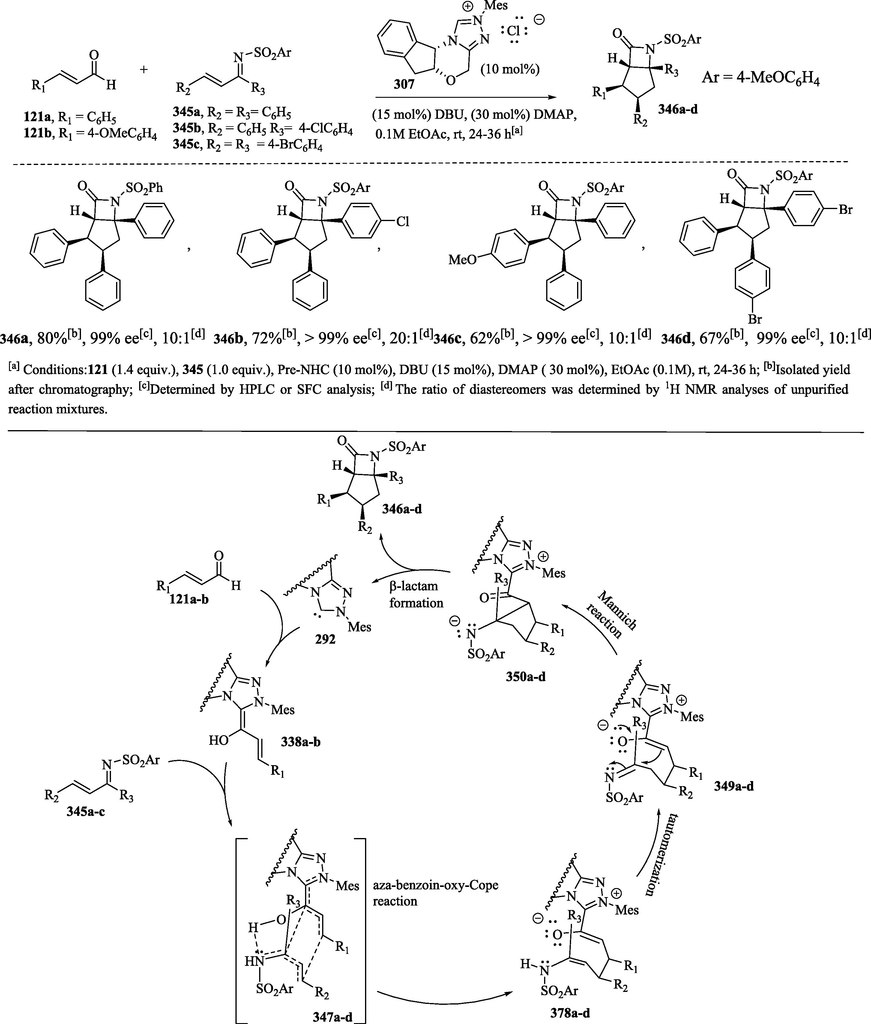
a) β-lactam synthesis through NHC catalysis; b) Proposed mechanism.
1.8.10 NHC-catalyzed Rauhut-Currier reaction for the construction of naphthyl lactones
An example of NHC-promoted esterification/Rauhut-Currier (RC) reactions was explored by Bae et al. (2019) for the construction of naphthyl lactones (Bae et al., 2019; Dzieszkowski et al., 2020). In Scheme 39a, the reaction of α,β-unsaturated acid fluoride 351a–c with NHC 355 produces anhydrous acyl azolium fluoride 356a–c, which subsequently reacts with naphthol derivatives 352a–b to provide bis(enoates) 357a–d. In the process known as RC reaction, the bis(enoates) 357a–d (Scheme 39b) participate as Michael donors after undergoing 1,2-addition with the NHC, providing the intermediate enolates 357a–d, which undergo intramolecular Michael addition to provide lactones 359a–d. In the RC method, also known as the vinylogous Morita–Baylis–Hillman reaction, the coupling of an active alkene / latent enolate (Michael donor) to a second Michael acceptor furnishes a new C–C bond between the α-position of the activated alkene and the β-position of a second alkene under the influence of a nucleophilic catalyst. From intermediates 359a–d, NHC diastereoselective elimination provides naphthyl lactones 252a–d and returns the catalyst.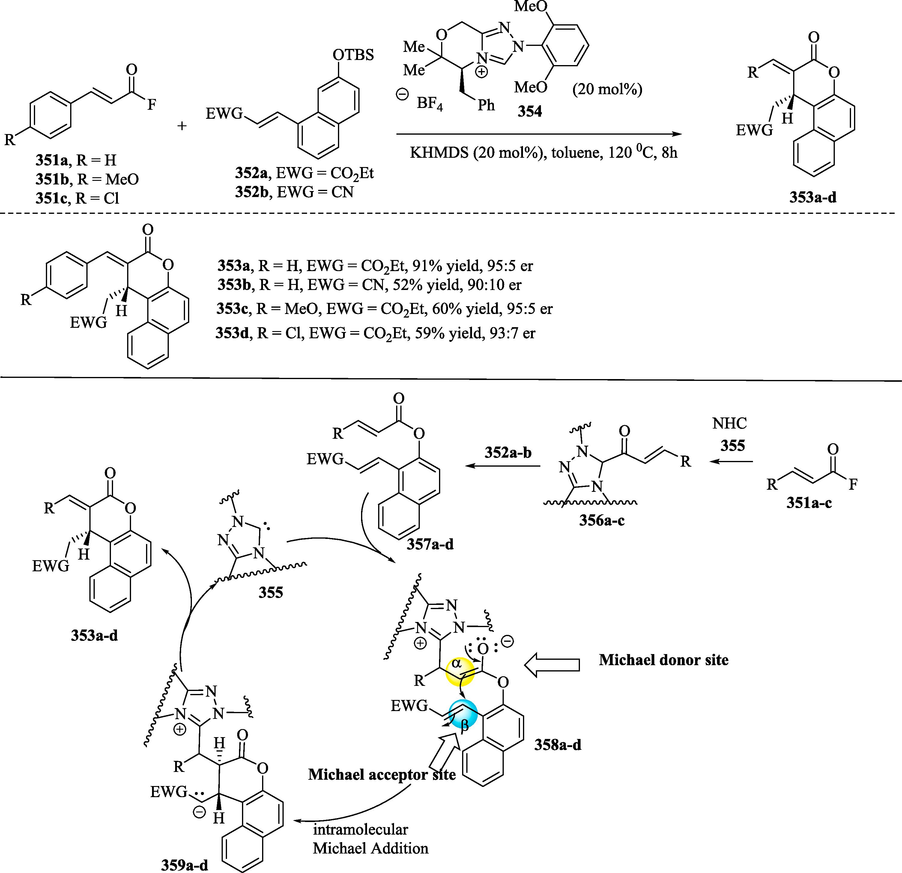
a) Enantioselective NHC-catalyzed bis(enoate) Rauhut‐Currier reaction; b) Plausible mechanism.
1.8.11 NHC-catalyzed [4 + 2] cycloaddition of enals with azodicarboxylates
Chen et al. (2013) reported that azolium dienolates 364a–l (Scheme 40b) could be synthesized by enals 360a–l, which contain a leaving group (-OCO2Me) at the γ-position of their backbones (X. Y. Chen et al., 2013). In Scheme 29a, the NHC-catalyzed reaction between a variety of enals 360a–l and azodicarboxylates 361a–c provided dihydropyridazinone compounds 362a–l with yields of 49–86 % and 94–99 % ee. In the proposed catalytic cycle, the addition of NHC catalyst 233 to enals 360a–l leads to the formation of vinyl Breslow intermediates 363a–l, which subsequently undergo the elimination of the leaving group to provide the corresponding azolium dienolates 364a–l. These latter compounds react with azodicarboxilates 361a–c to provide acyl azolium intermediates 365a–l, which undergo intramolecular cyclization to form the corresponding products 362a–l and release the NHC catalyst (Scheme 40b).![a) NHC-catalyzed [4 + 2] cycloaddition reaction of enals with azodicarboxylates; b) Plausible mechanism.](/content/184/2024/17/2/img/10.1016_j.arabjc.2023.105527-fig48.png)
a) NHC-catalyzed [4 + 2] cycloaddition reaction of enals with azodicarboxylates; b) Plausible mechanism.
1.8.12 Synthesis of chiral cyclic ketones via NHC-catalyzed annulation of cis-homoenolate equivalents with unsaturated imines
Although the cis-enals are attractive raw materials for the development of catalytic reactions, their easy isomerization to the trans isomers under catalytic conditions often limits their use. Fortunately, the asymmetric reaction of α, β-unsaturated imines with cis-enals catalyzed by chiral NHCs in the presence of N,N’-diisopropylethylamine (DIEA) and benzene-1,2,3-triol acting as additives (Dirocco and Rovis, 2011) has been developed by Chen et al. (2013) (X. Chen et al., 2013a). The use of an additive and an NHC catalyst makes it possible to enhance the reaction performance in terms of both yield and stereoselectivity (Curti et al., 2020; Wang and Scheidt, 2016). Using this methodology, a variety of chiral cyclic ketones were obtained in good yields and with excellent enantioselectivities (Schemes 41 and 42). It should be emphasized that trans-enals (Schemes 40 and 41) and cis-enals (Schemes 41 and 42) show different stereoselectivities and reactivity patterns. The NHC-catalyzed reaction of cis-homoenolate intermediates from cis-enals with β-unsaturated imines preferentially leads to the formation of cyclic ketones 369, whereas trans-homoenolate intermediates provide bicyclic β-lactam compounds 346.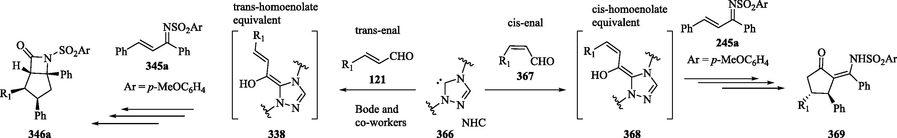
NHC-catalyzed reaction of a trans-enal and cis-enal with an
,β-unsaturated imine.
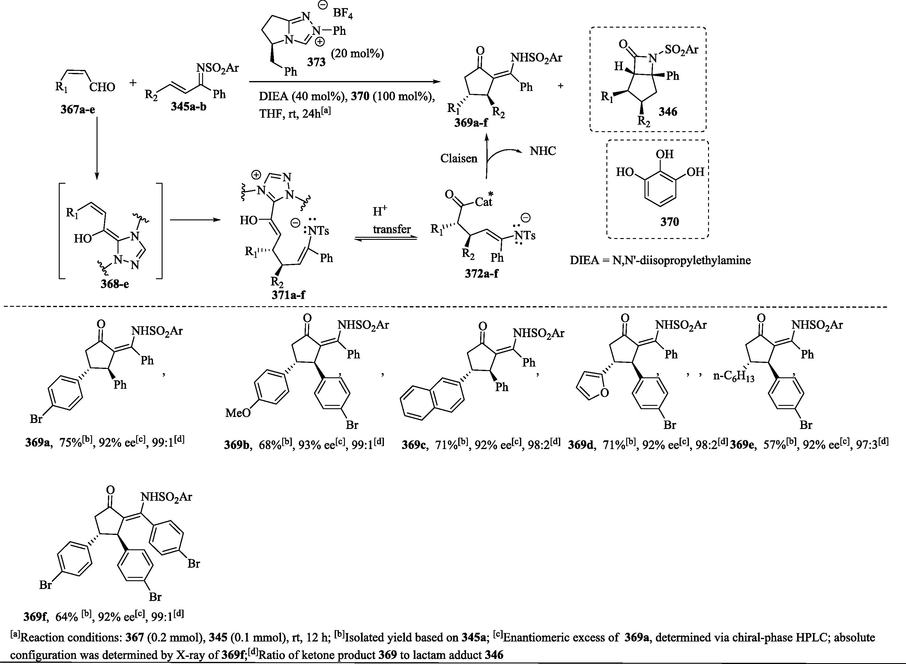
Synthesis of cyclic ketones via the NHC-catalyzed reaction of cis-enals with α,β-unsaturated imines.
According to Chen et al. (2013) the reaction between cis-α, β-unsaturated aldehydes 367a–e (Scheme 42) and unsaturated imines 345a–b occurs via a concerted benzoin/Cope rearrangement process or a direct Michael addition pathway, resulting in the formation of intermediates 371a–f, which subsequently undergo reversible proton transfer to produce the intermediate 372a–f. The cyclic ketones 369a–f were formed due to the intramolecular nucleophilic attack of the enamide carbon on the NHC-bound ester equivalent. Chen et al. (2013) report that the lactam derivative 346 can be detected under most conditions using different Pre-NHCs. They also suggest that this product can be formed by the reaction of the imine 345 with trans-enal 121 generated from the isomerization of the corresponding cis-enal 367 (X. Chen et al., 2013a).
1.8.13 NHC-catalyzed diastereoselective annulations of isatin with β,β-disubstituted enals
The stereoselective annulation of isatins 374a–e with β,β-disubstituted enals 375a–c through NHC/Brønsted acid dual catalysis leads to the formation of spirocyclic oxindoles 377a–g, which have two highly congested contiguous quaternary carbon centers (Scheme 43a). The reaction is highly dependent on the Brønsted acid cocatalyst, which is considered crucial to ensuring excellent reactivity and high stereoselectivity (Flanigan et al., 2015; Li et al., 2014). The absolute configuration of the products has been determined through the X-ray crystallographic analysis of 377a. In the mechanism proposed by Li et al. (2014) (Scheme 43b), the azolium enolate intermediates 378a–d generated from the NHC, such as 1,2,4-triazolidin-3-ylidene 86 and enals 375a–d, in the presence of an acid cocatalyst, preferentially react with isatin derivatives 374a–e via a pre-transition state 379 favored by the presence of two stabilizing hydrogen bonds; one of these bonds decreases the electron density on the carbonyl carbon of isatin, making it more electron-deficient and facilitating the attack by the nucleophile. From the diastereoselective synthesis of intermediates 381a–g, the intramolecular attack of the alkoxide on the carbonyl group occurs, leading to the formation of spirooxindoles 377a–g and the regeneration of the catalyst 86 (Li et al., 2014).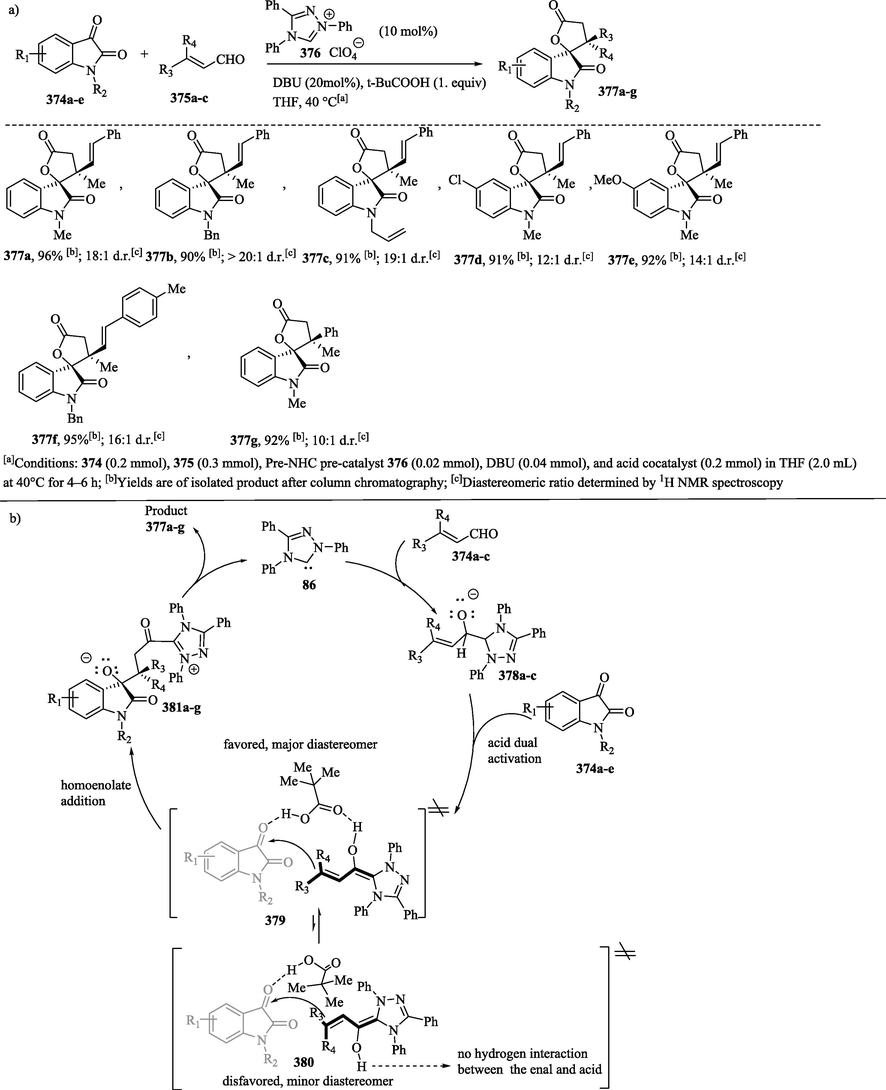
a) NHC-catalyzed diastereoselective annulations of isatin 374a–e with β,β-disubstituted enal 375a–c for the synthesis of oxindoles 377a–g; b) Proposed mechanism.
The central objective of this review is to document the NHC-catalyzed reactions, anchoring the discussion within the scope of experimental data. However, NHC-catalyzed transformation reactions have also been studied using theoretical methods based on density functional theory. Examples of theoretical studies involving reactions catalyzed by NHC have been very well explored in the literature by Lan and Wei and other groups of researchers. (Alam, 2022; S. Li et al., 2019; Li et al., 2018b, 2018a; X. Li et al., 2019; Li et al., 2020; C. Liu et al., 2022; Liu et al., 2020; Yang Wang et al., 2016; Wang et al., 2017, 2019a, 2019c, 2019b, 2020a, 2020b, 2021, 2018a, 2018b; Yanyan Wang et al., 2016; Wei et al., 2011; Wu et al., 2020; Zhang et al., 2018, 2014; W. Zhang et al., 2016; X. Zhang et al., 2016).
2 Conclusion
N-Heterocyclic carbenes have multiple uses in many areas of organic and inorganic chemistry. Most NHC applications require coordination to transition metals, including applications in the areas of organometallic materials, coordination to surfaces, metallopharmaceuticals and homogenous catalysis. This review summarized the properties of NHC and metal-free triazolium carbene-catalyzed reactions involving the formation of C–C and C-heteroatom bonds with regioselectivity and stereoselectivity through umpolung and non-umpolung processes for unconventional access to various target molecules. The polarity inversion allows exploring new synthetic possibilities that cannot be realized from the normal reactivity of functional groups. NHC-catalyzed polarity reversal (umpolung) strategies have been explored in the generation of homoenolate, acyl anion and enolate equivalents, which are reactive species for a multitude of reactions, including annulation, benzoin, Stetter, Claisen rearrangement, cycloaddition and C–C bond functionalization reactions, among others. This review has provided examples of aerobic oxidative/oxygenative carbene catalysis, specifically highlighting NHC-catalyzed reactions involving oxygen and strategies employed for activation of oxygen in NHC-catalytic processes.
The future of NHCs looks highly promising, given the significant advances achieved thus far. In this sense, NHC chemistry can be used for the construction of molecules with great economic potential.
Acknowledgments
This work was supported by the Brazilian agency FAPERJ. Fellowships granted to UFF, by CAPES, CNPq, CNPq-PIBIC and FAPERJ are gratefully acknowledged.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. (Weinheim). 2022;355:2100158.
- [CrossRef] [Google Scholar]
- Perfluorinated 1,2,3- and 1,2,4-Triazolium Ionic Liquids. Eur. J. Org. Chem.. 2018;2018:4331-4337.
- [CrossRef] [Google Scholar]
- Analyses of the Structural and Electronic Properties of NHCs with Bicyclic Architectures. Organometallics. 2020;39:3839-3848.
- [CrossRef] [Google Scholar]
- A. Axelsson, E. Hammarvid, L.T., H. Sundén, 2016. Asymmetric aerobic oxidative NHC-catalysed synthesis of dihydropyranones utilising a system of electron transfer mediators. Chem. Commun. 52, 11571–11574. https://doi.org/10.1039/c6cc06060a.
- Enantioselective N-heterocyclic carbene catalyzed bis (enoate) Rauhut- Currier reaction. Angew. Chem. Int. Ed. 2019;58:13370-13374.
- [CrossRef] [Google Scholar]
- Synthetic routes to N-heterocyclic carbene precursors. Chem. Rev.. 2011;111:2705-2733.
- [CrossRef] [Google Scholar]
- Synthetic strategies for free & stable N-heterocyclic carbenes and their precursors. Mini. Rev. Org. Chem.. 2013;10:180-197.
- [CrossRef] [Google Scholar]
- Extending NHC-catalysis: Coupling aldehydes with unconventional reaction partners. Acc. Chem. Res.. 2011;44:1182-1195.
- [CrossRef] [Google Scholar]
- Acyl donor intermediates in N-heterocyclic carbene catalysis: Acyl azolium or azolium enolate? Angew. Chem.. 2021;133:4557-4561.
- [CrossRef] [Google Scholar]
- An insight into the mechanism of the aerobic oxidation of aldehydes catalyzed by N-heterocyclic carbenes. Chem. Commun.. 2014;50:2008-2011.
- [CrossRef] [Google Scholar]
- Formation, oxidation, and fate of the Breslow intermediate in the N-heterocyclic carbene-catalyzed aerobic oxidation of aldehydes. J. Org. Chem.. 2017;82:302-312.
- [CrossRef] [Google Scholar]
- Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev.. 2012;41:3511-3522.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbenes: An introductory overview. In: Cazin C.S.J., ed. N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis. Dordrecht, Dordrecht: Springer; 2010. p. :1-22.
- [CrossRef] [Google Scholar]
- J. Chen, Y. Huang, 2019. N-heterocyclic carbenes as brønsted base catalysts, in: Biju, A.T. (Ed.), N-Heterocyclic Carbenes in Organocatalysis. Wiley-VCH, Weinheim, pp. 261–286. https://doi.org/10.1002/9783527809042.ch9.
- cis-Enals in N-heterocyclic carbene-catalyzed reactions: distinct stereoselectivity and reactivity. Chem. Sci.. 2013;4:2613-2618.
- [CrossRef] [Google Scholar]
- N-heterocyclic-carbene-catalyzed domino reactions via two or more activation modes. iScience. 2018;2:1-26.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene catalysis via azolium dienolates: An efficient strategy for remote enantioselective functionalizations. Angew. Chemie - Int. Ed.. 2018;57:3862-3873.
- [CrossRef] [Google Scholar]
- Functionalization of benzylic C(sp3) - H bonds of heteroaryl aldehydes through N-heterocyclic carbene organocatalysis. Angew. Chem. Int. Ed.. 2013;52:11134-11137.
- [CrossRef] [Google Scholar]
- N-Heterocyclic Carbene Organocatalysis: Activation Modes and Typical Reactive Intermediates. Chin. J. Chem. 2020;38:1167-1202.
- [CrossRef] [Google Scholar]
- Highly enantioselective γ-amination by N-heterocyclic carbene catalyzed [4+2] annulation of oxidized enals and azodicarboxylates. Angew. Chem. Int. Ed.. 2013;52:10644-10647.
- [CrossRef] [Google Scholar]
- Enantioselective, cyclopentene-forming annulations via NHC-catalyzed benzoin - oxy-cope reactions. J. Am. Chem. Soc.. 2007;129:3520-3521.
- [CrossRef] [Google Scholar]
- New developments of the principle of vinylogy as applied to π - extended enolate-type donor systems. Chem. Rev.. 2020;120:2448-2612.
- [CrossRef] [Google Scholar]
- Recent advances in N-heterocyclic carbene-catalyzed radical reactions. Chinese Chem. Lett.. 2021;32:660-667.
- [CrossRef] [Google Scholar]
- NHC-catalysed aerobic aldehyde-esterifications with alcohols: No additives or cocatalysts required. Chem. Commun.. 2013;49:6510-6512.
- [CrossRef] [Google Scholar]
- Oxidative coupling of aldehydes with alcohol for the synthesis of esters promoted by polystyrene-supported N-heterocyclic carbene: Unraveling the solvent effect on the catalyst behavior using NMR relaxation. Org. Lett. 2020;22:4927-4931.
- [CrossRef] [Google Scholar]
- Catalytic asymmetric intermolecular stetter reaction of enals bifunctional additives. J. Am. Chem. Soc. 2011;133:10402-10405.
- [CrossRef] [Google Scholar]
- SET processes in Lewis acid-base reactions: The tritylation of N-heterocyclic carbenes. Chem. Sci.. 2020;11:7615-7618.
- [CrossRef] [Google Scholar]
- A review on generation and reactivity of the N-heterocyclic carbene-bound alkynyl acyl azolium intermediates. Molecules. 2022;27:7990.
- [CrossRef] [Google Scholar]
- The measure of all rings — N-heterocyclic carbenes. Angew. Chem. Int. Ed.. 2010;49:6940-6952.
- [CrossRef] [Google Scholar]
- Computational predictions of stereochemistry in asymmetric thiazolium- and triazolium-catalyzed benzoin condensations. Proc. Natl. Acad. Sci. U. S. A.. 2004;101:5770-5775.
- [CrossRef] [Google Scholar]
- Organocatalytic name reactions enabled by NHCs. Materials (Basel). 2020;13:3574.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene catalysed asymmetric cross-benzoin reactions of heteroaromatic aldehydes with trifluoromethyl ketones. Chem. Commun.. 2010;46:6282-6284.
- [CrossRef] [Google Scholar]
- An efficient nucleophilic carbene catalyst for the asymmetric benzoin condensation. Angew. Chem. Int. Ed.. 2002;41:1743-1745.
- [CrossRef] [Google Scholar]
- Theoretical NMR spectroscopy of N-heterocyclic carbenes and their metal complexes. Coord. Chem. Rev.. 2017;344:101-114.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbenes and parent cations: Acidity, nucleophilicity, stability, and hydrogen bonding— Electrochemical study and ab initio calculations. ChemElectroChem. 2016;3:1133.
- [CrossRef] [Google Scholar]
- Advances in the knowledge of N-heterocyclic carbenes properties. The backing of the electrochemical investigation. Catalysts. 2016;6:178.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbenes (NHCs) as organocatalysts and structural components in metal-free polymer synthesis. Chem. Soc. Rev.. 2013;42:2142-2172.
- [CrossRef] [Google Scholar]
- Organocatalyzed anodic oxidation of aldehydes. J. Am. Chem. Soc.. 2012;134:12374-12377.
- [CrossRef] [Google Scholar]
- Organocatalytic reactions enabled by N - heterocyclic carbenes. Chem. Rev.. 2015;115:9307-9387.
- [CrossRef] [Google Scholar]
- Synthesis of 2-aryl naphthoquinones by the cross-dehydrogenative coupling involving an NHC-catalyzed endo-Stetter reaction. J. Org. Chem.. 2019;84:1103-1110.
- [CrossRef] [Google Scholar]
- Cyclizations of alkynes: Revisiting Baldwins rules for ring closure. Chem. Rev.. 2011;111:6513-6556.
- [CrossRef] [Google Scholar]
- Quantifying and understanding the steric properties of N-heterocyclic carbenes. Chem. Commun.. 2017;53:2650-2660.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-mediated oxidative electrosynthesis of esters in a microflow cell. Org. Lett.. 2015;17:3290-3293.
- [CrossRef] [Google Scholar]
- A facile synthetic route to benzimidazolium salts bearing bulky aromatic N-substituents. Beilstein J. Org. Chem.. 2015;11:1656-1666.
- [CrossRef] [Google Scholar]
- V.B. Gudimetla, B.P. Joy, S. Paul, 2022. Imidazolium-Based N-Heterocyclic Carbenes (NHCs) and Metal-Mediated Catalysis, in: Satyen Saha, Arunava Manna (Eds.), Carbene. IntechOpen. https://doi.org/10.5772/INTECHOPEN.102561.
- Biomimetic carbene-catalyzed oxidations of aldehydes using TEMPO. Angew. Chem. - Int. Ed.. 2008;47:8727-8730.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene/carboxylic acid co-catalysis enables oxidative esterification of demanding aldehydes/enals, at low catalyst loading. Angew. Chem. - Int. Ed.. 2021;60:19631-19636.
- [CrossRef] [Google Scholar]
- The mechanism of the Stetter reaction - A DFT study. Eur. J. Org. Chem. 2008:5563-5570.
- [CrossRef] [Google Scholar]
- Enantioselective, NHC-catalyzed bicyclo-lactam formation via direct annulations of enals and unsaturated N-sulfonyl ketimines. J. Am. Chem. Soc.. 2008;130:418-419.
- [CrossRef] [Google Scholar]
- Theoretical investigations on the mechanism of benzoin condensation catalyzed by pyrido[1,2- a ]-2-ethyl[1,2,4]triazol-3-ylidene. J. Phys. Chem. A. 2011;115:1408-1417.
- [CrossRef] [Google Scholar]
- Recent advances in Stetter reaction and related chemistry: An update. Asian J. Org. Chem.. 2020;9:1999-2034.
- [CrossRef] [Google Scholar]
- pKas of the conjugate acids of N -heterocyclic carbenes in water w. Chem. Commun.. 2011;47:1559-1561.
- [CrossRef] [Google Scholar]
- Formation of Breslow intermediates under aprotic conditions: A computational study. J. Org. Chem.. 2022;87:2501-2507.
- [CrossRef] [Google Scholar]
- Catalytic preparation of 1-aryl-substituted 1,2,4-triazolium salts. ACS Omega. 2019;18:17923.
- [CrossRef] [Google Scholar]
- Electronic properties of N-heterocyclic carbenes and their experimental determination. Chem. Rev.. 2018;118:9457.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-catalyzed decarboxylative alkylation of aldehydes. J. Am. Chem. Soc.. 2019;141:3854-3858.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-catalyzed radical relay enabling vicinal alkylacylation of alkenes. J. Am. Chem. Soc.. 2019;141:14073-14077.
- [CrossRef] [Google Scholar]
- Recent advances in N-heterocyclic carbene-based radical catalysis. Chem. Sci.. 2020;11:5630-5636.
- [CrossRef] [Google Scholar]
- M.C. Jahnkea, F.E. Hahn, 2017. Introduction to N-Heterocyclic Carbenes: Synthesis and Stereoelectronic Parameters, in: Laboratory Curiosities to Efficient Synthetic Tools. The Royal Society of Chemistry, Münster, pp. 1–45. https://doi.org/10.1039/9781782626817-00001.
- Annulation of o - quinodimethanes through N - heterocyclic carbene catalysis for the synthesis of 1 - isochromanones. Org. Lett.. 2016;18:4444-4447.
- [CrossRef] [Google Scholar]
- Toward a symphony of reactivity : Cascades involving catalysis and sigmatropic rearrangements angewandte. Angew. Chem. Int. Ed.. 2014;53:2556-2591.
- [CrossRef] [Google Scholar]
- Highly enantioselective synthesis of α-amino acid derivatives by an NHC-catalyzed intermolecular stetter reaction. Angew. Chem. - Int. Ed.. 2011;50:1410-1414.
- [CrossRef] [Google Scholar]
- An enantioselective claisen rearrangement catalyzed by N-heterocyclic carbenes. J. Am. Chem. Soc.. 2010;132:8810-8812.
- [CrossRef] [Google Scholar]
- Synthesis of polyarylates and aliphatic polyesters by divalent acyl-1,2,4-triazole: a route to metal-free synthesis at low temperature. Polym. J.. 2021;53:887-893.
- [CrossRef] [Google Scholar]
- The impact of cation structure upon the acidity of triazolium salts in dimethyl sulfoxide. Org. Biomol. Chem.. 2020;18:66-75.
- [CrossRef] [Google Scholar]
- Theoretical study on the mechanism of N-heterocyclic carbene catalyzed transesterification reactions. Tetrahedron Lett.. 2005;46:6265-6270.
- [CrossRef] [Google Scholar]
- In fl uence of the N - substituents on the nucleophilicity and Lewis basicity of N - heterocyclic carbenes. Org. Lett.. 2016;18:3566-3569.
- [CrossRef] [Google Scholar]
- An acidity scale of triazolium-based NHC precursors in DMSO. J. Org. Chem. 2017;82:9675-9681.
- [CrossRef] [Google Scholar]
- Insights into NHC-catalyzed oxidative α-C(sp3)-H activation of aliphatic aldehydes and cascade [2 + 3] cycloaddition with azomethine imines. Catal. Sci. Technol.. 2019;9:2514-2522.
- [CrossRef] [Google Scholar]
- Oxidative radical NHC catalysis: Divergent difunctionalization of olefins through intermolecular hydrogen atom transfer. Angew. Chem. - Int. Ed.. 2022;61
- [CrossRef] [Google Scholar]
- Conjugate umpolung of β, β -disubstituted enals by dual catalysis with an N-heterocyclic carbene and a Brønsted acid : Facile construction of contiguous quaternary stereocenters. Angew. Chem. Int. Ed.. 2014;53:10515-10519.
- [CrossRef] [Google Scholar]
- Insights into the N-Heterocyclic Carbene (NHC)-catalyzed intramolecular cyclization of aldimines: General mechanism and role of catalyst. Chem. - an Asian J.. 2018;13:1710-1718.
- [CrossRef] [Google Scholar]
- NHC-catalyzed aldol-like reactions of allenoates with isatins: Regiospecific syntheses of γ-functionalized allenoates. Org. Lett.. 2019;21:1306-1310.
- [CrossRef] [Google Scholar]
- Insights into the N-Heterocyclic Carbene (NHC)-catalyzed oxidative γ-C(sp3)-H deprotonation of alkylenals and cascade [4 + 2] cycloaddition with alkenylisoxazoles. J. Org. Chem.. 2018;83:8543-8555.
- [CrossRef] [Google Scholar]
- Prediction of NHC-catalyzed chemoselective functionalizations of carbonyl compounds: A general mechanistic map. Chem. Sci.. 2020;11:7214-7225.
- [CrossRef] [Google Scholar]
- Single-electron transfer reactions enabled by N-heterocyclic carbene organocatalysis. Chem. - Eur. J.. 2021;27:3238-3250.
- [CrossRef] [Google Scholar]
- Dual N-heterocyclic carbene/photocatalysis: A new strategy for radical processes. Org. Chem. Front.. 2020;7:2082-2087.
- [CrossRef] [Google Scholar]
- Efficient aerobic oxidation of organic molecules by multistep electron transfer. Angew. Chem. Int.ed.. 2021;60:15686-15704.
- [CrossRef] [Google Scholar]
- NHC-catalyzed transformation reactions of imines: Electrophilic versus nucleophilic attack. J. Org. Chem.. 2022;87:7989-7994.
- [CrossRef] [Google Scholar]
- Computational study on N-heterocyclic carbene (NHC)-catalyzed intramolecular hydroacylation-Stetter reaction cascade. Mol. Catal.. 2020;484:110723
- [CrossRef] [Google Scholar]
- A computational study of the origin of stereoinduction in NHC-catalyzed annulation reactions of α, β-unsaturated acyl azoliums. Chem. Sci.. 2012;3:2346-2350.
- [CrossRef] [Google Scholar]
- On the mechanism of N-heterocyclic carbene-catalyzed reactions involving acyl azoliums. Acc. Chem. Res.. 2014;47:696-707.
- [CrossRef] [Google Scholar]
- Chiral N-heterocyclic carbene catalyzed annulations of enals and ynals with stable enols: A highly enantioselective coates– Claisen rearrangement. ACS Catal.. 2012;2:494-503.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbenes : Organocatalysts with moderate nucleophilicity but extraordinarily high Lewis basicity. Angew. Chem. Int. Ed. 2011;50:6915-6919.
- [CrossRef] [Google Scholar]
- Nucleophilic reactivities of deoxy breslow intermediates : How does aromaticity affect the catalytic activities of N-heterocyclic carbenes ? Angew. Chem. Int. Ed. 2012;51:6231-6235.
- [CrossRef] [Google Scholar]
- Synthesis of thioesters from aldehydes via N-Heterocyclic Carbene (NHC) catalyzed radical relay. Chem. - Eur. J.. 2023;29
- [CrossRef] [Google Scholar]
- Proton transfer reactions of triazol-3-ylidenes: Kinetic acidities and carbon acid p K. J. Am. Chem. Soc.. 2012;134:20421-20432.
- [CrossRef] [Google Scholar]
- Aryl radical-mediated N-heterocyclic carbene catalysis. Nat. Commun.. 2021;12:1-8.
- [CrossRef] [Google Scholar]
- A quantitative approach to nucleophilic organocatalysis. Beilstein J. Org. Chem.. 2012;8:1458-1478.
- [CrossRef] [Google Scholar]
- Recent advances in N-heterocyclic carbene (NHC)-catalysed benzoin reactions. Beilstein J. Org. Chem.. 2016;12:444-461.
- [CrossRef] [Google Scholar]
- Oxidative γ -addition of enals to trifluoromethyl ketones: enantioselectivity control via lewis acid/N-heterocyclic carbene cooperative catalysis. J. Am. Chem. Soc.. 2012;134:8810-8813.
- [CrossRef] [Google Scholar]
- N-Heterocyclic carbene-catalyzed enantioselective synthesis of functionalized cyclopentenes via α, β-unsaturated acyl azoliums. Chem. Commun.. 2014;50:14539-14542.
- [CrossRef] [Google Scholar]
- NHC-catalyzed generation of α, β-unsaturated acylazoliums for the enantioselective synthesis of heterocycles and carbocycles. Acc. Chem. Res.. 2019;52:425-436.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-catalyzed amidation of unactivated esters with amino alcohols. Org. Lett.. 2005;7:2453-2456.
- [CrossRef] [Google Scholar]
- How to enhance the efficiency of breslow intermediates for SET catalysis. J. Org. Chem.. 2023;88:2535-2542.
- [CrossRef] [Google Scholar]
- Polymer Chemistry Organo-catalyzed synthesis of aliphatic polycarbonates in solvent-free conditions. Polym. Chem.. 2012;3:1475-1480.
- [CrossRef] [Google Scholar]
- Quantum tunneling on carbene organocatalysis: Breslow intermediate formation via water-bridges. ACS Catal.. 2021;11:14836-14841.
- [CrossRef] [Google Scholar]
- Liberation of N-heterocyclic carbenes (NHCs) from thermally labile progenitors: Protected NHCs as versatile tools in organo- and polymerization catalysis. Catal. Sci. Technol.. 2014;4:2466-2479.
- [CrossRef] [Google Scholar]
- N-Heterocyclic carbenes for metal-free polymerization catalysis: An update. Polym. Int.. 2016;65:16-27.
- [CrossRef] [Google Scholar]
- Quantifying and understanding the electronic properties of N -heterocyclic carbenes. Chem. Soc. Rev.. 2013;42:6723-6753.
- [CrossRef] [Google Scholar]
- NHC-catalyzed activation of α, β-unsaturated N-acyltriazoles: An easy access to dihydropyranones. Chem. Commun.. 2015;51:14628-14631.
- [CrossRef] [Google Scholar]
- Experimental and computational gas phase acidities of conjugate acids of triazolylidene carbenes: Rationalizing subtle electronic effects. J. Am. Chem. Soc.. 2017;139:14917-14930.
- [CrossRef] [Google Scholar]
- Electrochemical characterization of azolium salts. Chem. Lett.. 2014;43:907-909.
- [CrossRef] [Google Scholar]
- Heterocyclic synthesis via catalysis of N-heterocyclic carbenes: Very classical and very modern chemical species. Heterocycl. Commun.. 2013;19:311-326.
- [CrossRef] [Google Scholar]
- Electrochemically promoted N-heterocyclic carbene polymer- catalyzed cycloaddition of aldehyde with isocyanide acetate. Sci. China Chem.. 2022;65:1873-1878.
- [CrossRef] [Google Scholar]
- Tale of the Breslow intermediate, a central player in N-heterocyclic carbene organocatalysis: then and now. Chem. Sci.. 2021;12:7973-7992.
- [CrossRef] [Google Scholar]
- Predictive energetic tuning of C-Nucleophiles for the electrochemical capture of carbon dioxide. ISCIENCE. 2022;25:103997
- [CrossRef] [Google Scholar]
- Advances in N-heterocyclic carbene catalysis for natural product synthesis. Eur. J. Org. Chem. 2020:5917-5925.
- [CrossRef] [Google Scholar]
- P. Quinn, M.S. Smith, J. Zhu, D.R.W. Hodgson, A.C. O’donoghue, 2021. Triazolium salt organocatalysis: Mechanistic evaluation of unusual ortho-substituent effects on deprotonation. Catalysts 11, 1055. https://doi.org/10.3390/catal11091055.
- Exploring oxidative NHC-catalysis as organocatalytic polymerization strategy towards polyamide oligomers. Chem. Eur. J.. 2021;27:1839-1848.
- [CrossRef] [Google Scholar]
- Origin of stereoselectivity in cooperative asymmetric catalysis involving N-heterocyclic carbenes and Lewis acids toward the synthesis of spirooxindole lactone. ACS Catal.. 2017;7:530-537.
- [CrossRef] [Google Scholar]
- Organocatalyzed cross-nucleophile couplings: Umpolung of catalytic enamines. Acc. Chem. Res.. 2022;55:1703-1717.
- [CrossRef] [Google Scholar]
- Chiral N-heterocyclic biscarbenes based on 1,2,4-triazole as ligands for metal-catalyzed asymmetric synthesis. Dalt. Trans.. 2011;40:41-43.
- [CrossRef] [Google Scholar]
- First example of organocatalysis by cathodic N-heterocyclic carbene generation and accumulation using a divided electrochemical flow cell. J. Org. Chem.. 2022;18:979-990.
- [CrossRef] [Google Scholar]
- Acyl anion free N-heterocyclic carbene organocatalysis. Chem. Soc. Rev.. 2013;42:4906-4917.
- [CrossRef] [Google Scholar]
- Nucleophilic addition of enols and enamines to α, β-unsaturated acyl azoliums: Mechanistic studies. Angew. Chem. Int. Ed.. 2012;51:5234-5238.
- [CrossRef] [Google Scholar]
- N-Heterocyclic carbenes as chiral Brønsted base catalysts: a highly diastereo- and enantioselective 1,6-addition reaction. Chem. Sci.. 2018;9:6446-6450.
- [CrossRef] [Google Scholar]
- Electrochemical generation of N-heterocyclic carbenes for use in synthesis and catalysis. Adv. Synth. Catal.. 2021;363:3189-3200.
- [CrossRef] [Google Scholar]
- Alternating polarity for enhanced electrochemical synthesis. React. Chem. Eng.. 2021;6:147-151.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-catalyzed cyclization of unsaturated acyl chlorides and ketones. Adv. Synth. Catal.. 2011;353:1943-1948.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene coordinated metal nanoparticles and nanoclusters. Coord. Chem. Rev.. 2022;458:214425
- [CrossRef] [Google Scholar]
- Asymmetric synthesis of cyclopentanes bearing four contiguous stereocenters via an NHC-catalyzed Michael/Michael/esterification domino reaction. Chem. Commun.. 2016;52:2609-2611.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene catalyzed radical coupling of aldehydes with redox-active esters. Angew. Chem. - Int. Ed.. 2019;58:8628-8630.
- [CrossRef] [Google Scholar]
- Aerobic oxidative N-heterocyclic carbene catalysis. Chem. Rec.. 2023;23:1-24.
- [CrossRef] [Google Scholar]
- Attractive aerobic access to the α, β-unsaturated acyl azolium intermediate: oxidative NHC catalysis via multistep electron transfer. Green Chem.. 2016;18:686-690.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene catalyzed ester synthesis from organic halides through incorporation of oxygen atoms from air. Angew. Chem.. 2021;60:2140-2144.
- [CrossRef] [Google Scholar]
- T. Ukai, R. Tanaka, T. Dokawa, 1943. A new catalyst for acyloin codensation. J. Pharm. Soc. Jpn 63, 296–300. https://doi.org/DOI: 10.1248/yakushi1881.63.6_296.
- From imidazole toward imidazolium salts and N-heterocyclic carbene ligands: Electronic and geometrical redistribution. ACS Omega. 2017;2:1392-1399.
- [CrossRef] [Google Scholar]
- Electrogenerated NHCs in organic synthesis: Ionic liquids vs organic solvents effects. Chem. Rec.. 2021;21:2130-2147.
- [CrossRef] [Google Scholar]
- Exploiting acyl and enol azolium intermediates via N-heterocyclic carbene catalyzed reactions of alpha-reducible aldehydes. Adv. Synth. Catal.. 2012;354:1617-1639.
- [CrossRef] [Google Scholar]
- Carbene-catalyzed enantioselective addition of benzylic carbon to unsaturated acyl azolium for rapid synthesis of pyrrolo[3,2- c]quinolines. ACS Catal.. 2018;8:9859-9864.
- [CrossRef] [Google Scholar]
- DFT perspective toward [3 + 2] annulation reaction of enals with α-ketoamides through NHC and Brønsted acid cooperative catalysis: Mechanism, stereoselectivity, and role of NHC. Org. Chem. Front.. 2016;3:190-203.
- [CrossRef] [Google Scholar]
- N-Heterocyclic Carbene (NHC)-catalyzed sp3 β-C-H activation of saturated carbonyl compounds: Mechanism, role of NHC, and origin of stereoselectivity. ACS Catal.. 2016;6:279-289.
- [CrossRef] [Google Scholar]
- Computational study on NHC-catalyzed enantioselective and chemoselective fluorination of aliphatic aldehydes. Org. Chem. Front.. 2017;4:1987-1998.
- [CrossRef] [Google Scholar]
- Unravelling the mechanism and selectivity of the NHC-catalyzed three-membered ring-opening/fluorination of epoxy enals: A DFT study. ChemCatChem. 2019;11:2919-2925.
- [CrossRef] [Google Scholar]
- Origin of regio- and stereoselectivity in the NHC-catalyzed reaction of alkyl pyridinium with aliphatic enal. ChemCatChem. 2020;12:1068-1074.
- [CrossRef] [Google Scholar]
- Unveiling the chemo- and stereoselectivities of NHC-catalyzed reactions of an aliphatic ester with aminochalcone. J. Org. Chem.. 2020;85:8437-8446.
- [CrossRef] [Google Scholar]
- Predicting the origin of selectivity in NHC-catalyzed ring opening of formylcyclopropane: A theoretical investigation. Catal. Sci. Technol.. 2021;11:332-337.
- [CrossRef] [Google Scholar]
- Cooperative catalysis and activation with N-heterocyclic carbenes. Angew. Chem. Int. Ed.. 2016;55:14912-14922.
- [CrossRef] [Google Scholar]
- Competing mechanisms and origins of chemo- and stereo-selectivities of NHC-catalyzed reactions of enals with 2-aminoacrylates. Catal. Sci. Technol.. 2018;8:4229-4240.
- [CrossRef] [Google Scholar]
- Prediction on the origin of selectivities in base-controlled switchable NHC-catalyzed transformations. Chem. - Asian J.. 2019;14:293-300.
- [CrossRef] [Google Scholar]
- Prediction on the origin of selectivities of NHC-catalyzed asymmetric dearomatization (CADA) reactions. Catal. Sci. Technol.. 2019;9:465-476.
- [CrossRef] [Google Scholar]
- The importance of N-heterocyclic carbene basicity in organocatalysis. Org. Biomol. Chem.. 2018;16:6852-6866.
- [CrossRef] [Google Scholar]
- Insights into the NHC-catalyzed cascade Michael/aldol/lactamization reaction: Mechanism and origin of stereoselectivity. Org. Chem. Front.. 2018;5:2065-2072.
- [CrossRef] [Google Scholar]
- Polymer N - Heterocyclic Carbene (NHC) ligands for silver nanoparticles. ACS Appl. Mater. Interfaces. 2022;14:55227-55237.
- [CrossRef] [Google Scholar]
- Methyl cation affinities of commonly used organocatalysts. J. Am. Chem. Soc.. 2008;130:3473-3477.
- [CrossRef] [Google Scholar]
- A DFT study on enantioselective synthesis of aza-β-lactams via NHC-catalyzed [2 + 2] cycloaddition of ketenes with diazenedicarboxylates. J. Mol. Catal. A Chem.. 2011;334:108-115.
- [CrossRef] [Google Scholar]
- Formation of Breslow intermediates from N-heterocyclic carbenes and aldehydes involves autocatalysis by the breslow intermediate, and a hemiacetal. Angew. Chem. - Int. Ed.. 2022;61
- [CrossRef] [Google Scholar]
- Carbene-catalyzed oxidative acylation promoted by an unprecedented oxidant CCl3CN. Org. Chem. Front.. 2019;6:688-693.
- [CrossRef] [Google Scholar]
- NHC-catalyzed β-specific addition of N-based nucleophiles to allenoates. Org. Chem. Front.. 2020;7:1593-1599.
- [CrossRef] [Google Scholar]
- Polyhalides as efficient and mild oxidants for oxidative carbene organocatalysis by radical processes. Angew. Chem. - Int. Ed.. 2017;56:2942-2946.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene catalyzed reactions of α-bromo-α, β- unsaturated aldehydes/α, β-dibromoaldehydes with 1,3-dinucleophilic reagents. Chem. Eur. J.. 2012;18:1914-1917.
- [CrossRef] [Google Scholar]
- Enantioselective N-heterocyclic carbene-catalyzed annulations of 2-bromoenals with 1,3-dicarbonyl compounds and enamines via chiral α, β-unsaturated acylazoliums. Adv. Synth. Catal.. 2013;355:1089-1097.
- [CrossRef] [Google Scholar]
- Stereoselective synthesis of (E) - α, β -unsaturated esters via carbene-catalyzed redox esterification. Org. Lett.. 2006;8:637-640.
- [CrossRef] [Google Scholar]
- N - heterocyclic carbene-catalyzed stereoselective cascade reaction : Synthesis of functionalized tetrahydroquinolines. Org. Lett.. 2013;15:4750-4753.
- [CrossRef] [Google Scholar]
- DFT study on the mechanism and stereoselectivity of NHC-catalyzed synthesis of substituted trifluoromethyl dihydropyranones with contiguous stereocenters. J. Org. Chem.. 2016;81:868-877.
- [CrossRef] [Google Scholar]
- A DFT study on NHC-catalyzed intramolecular aldehyde-ketone crossed-benzoin reaction: Mechanism, regioselectivity, stereoselectivity, and role of NHC. Org. Biomol. Chem.. 2016;14:6577-6590.
- [CrossRef] [Google Scholar]
- DFT study on the reaction mechanisms and stereoselectivities of NHC-catalyzed [2 + 2] cycloaddition between arylalkylketenes and electron-deficient benzaldehydes. Org. Biomol. Chem.. 2014;12:6374-6383.
- [CrossRef] [Google Scholar]
- Theoretical study on the mechanism and enantioselectivity of NHC-catalyzed intramolecular SN2′ nucleophilic substitution: What are the roles of NHC and DBU? Org. Chem. Front.. 2018;5:1493-1501.
- [CrossRef] [Google Scholar]
- Asymmetric reactions of N-heterocyclic carbene (NHC)-based chiral acyl azoliums and azolium enolates. Green Synth. Catal.. 2021;2:198-215.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene complexes in C-H activation reactions. Chem. Rev.. 2020;120:1981-2048.
- [CrossRef] [Google Scholar]
- Cooperative N-Heterocyclic Carbene (NHC) and ruthenium redox catalysis: Oxidative esterification of aldehydes with air as the terminal oxidant. Adv. Synth. Catal.. 2013;355:1098-1106.
- [CrossRef] [Google Scholar]
- Electroredox carbene organocatalysis with iodide as promoter. Nat. Commun.. 2022;13:3827.
- [CrossRef] [Google Scholar]
- The role of the fused ring in bicyclic triazolium organocatalysts: Kinetic, X-ray, and DFT insights. J. Org. Chem.. 2022;87:4241-4253.
- [CrossRef] [Google Scholar]
- N-heterocyclic carbene-catalyzed reaction of alkynyl aldehydes with 1,3-keto esters or 1,3-diketones. Adv. Synth. Catal.. 2010;352:2455-2458.
- [CrossRef] [Google Scholar]
- Chiral N-heterocyclic carbene catalyzed annulation of α, β- unsaturated aldehydes with 1,3-dicarbonyls. Chem. Commun.. 2011;47:8670-8672.
- [CrossRef] [Google Scholar]




