

Translate this page into:
Callistephin inhibits amyloid-β protein aggregation and determined cytotoxicity against cerebrovascular smooth muscle cells as an in vitro model of cerebral amyloid angiopathy
⁎Corresponding authors at: Hospital of Yan’an University, No.38, Wenlin Road, Xianyang 712000, China (W. Wang) No.193, Lianhe Road, Shahekou District, Dalian 116021, Liaoning Province, China (M. Qi). weiweiwang542@gmail.com (Weiwei Wang), ming_qi20@yahoo.com (Ming Qi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
It has been indicated that amyloid β (Aβ) plaques can be accumulated within the basement membranes of cerebrovascular smooth muscle cells (CVSMCs) and stimulate the induction of cerebral amyloid angiopathy (CAA). However, the exact mechanism(s) of which small molecules including callistephin mitigate the formation of Aβ aggregation and associated CAA is not well-understood. Therefore, in the present study, Aβ1–42 samples in the aggregation buffer were co-incubated for 36 h without or with of callistephin and the protein aggregation features along with the associated cytotoxicity against CVSMCs as the core components of cerebral arterial wall were explored by different biochemical and cellular methods. Fluorescence (ThT, Nile red) and CD techniques indicated the inhibition of Aβ1–42 fibrillization in the presence of callistephin. Cellular assays revealed that cytotoxicity of Aβ1–42 samples aged in the aggregation buffer with callistephin was much less against CVSMCs than Aβ1–42 amyloid alone through regulation of membrane leakage and downregulation of TNF-α and IL-6 at protein level. In conclusion, these data may provide useful information about the possible mechanisms by which callistephin can show its protective effect against CAA.
Keywords
Amyloid-β
Callistephin
Cerebral amyloid angiopathy

1 Introduction
The presence of extracellular amyloid plaques and intricate intracellular nerve fibers are known as the crucial morphological signs of neurodegenerative diseases (Ross and Poirier, 2005). One of the pathological factors of these kinds of diseases is the aggregation of amyloid β (Aβ) peptides around brain cells with associated neurofibrillary filaments within brain cells (Finder and Glockshuber, 2007; Walsh and Selkoe, 2020). Amyloid plaques, using a mechanism that is not yet known, cause loss of synaptic activity, inflammation, oxidative reactions, and neuronal depletion in various areas of the brain, and are among the most important factors involved (Aleksis et al., 2017 Sep). As a result of this neuronal damage, learning and memory are impaired (Aleksis et al., 2017 Sep).
The most common form of Aβ is Aβ (1–40), which is naturally secreted by cells. Another form is Aβ (1–42), the production and deposition of which play an important role in the development of Alzheimer's disease (Cerofolini et al., 2020; Ogawa et al., 2011; Hasegawa et al., 1999). Normally, the amount of these species in the cell is small and they are decomposed rapidly, but if their level in the neurons is imbalanced, overproduction of these species results in the development of Alzheimer's disease (Hasegawa et al., 1999; Schmidt et al., 2009).
In addition to the accumulation of Aβ plaques in the brain tissue, another potential location of extracellular Aβ deposition is within cerebral arteries which result in cerebral Aβ angiopathy (CAA) (Qi and Ma, 2017). Recent reports have revealed the cerebral microvascular protein accumulation in induction of neuroinflammation and CAA (Ghiso et al., 2014). For example, it has been shown that apolipoprotein E4 changes the amyloid-β 40: 42 ratio and increases the induction of CAA (Fryer et al., 2005). Also, it has been shown that immunotherapy mitigates CAA and amyloid accumulation while promoting cerebrovascular function (Xiong et al., 2021).
Epidemiological investigations have determined that phenolic and flavonoid compounds could provide preventive impacts on the progression of CAA or neurodegenerative diseases (Kim and Park, 2021). For example, it has been shown that some phenolic small molecules, including myricetin, rosmarinic acid, ferulic acid, curcumin, and nordihydroguaiaretic acid can show inhibitory effect on the aggregation of Aβ (Yamada et al., 2015).
The in vitro investigations have demonstrated that these phenolic small molecules potentially prevent amyloid fibrillization of Aβ by differential binding, whilst mitigating Aβ oligomer-triggered neurotoxicity (Doig and Derreumaux, 2015; Zaman et al., 2019). Moreover, phenolic small molecules revealed outstanding decrease of soluble Aβ oligomers and insoluble Aβ accumulation in the brain, in vivo (Saunders et al., 2016).
Therefore, active components of plants as bioactive small molecules have attracted a great deal of attention in human societies for the treatment of a wide range of neurodegenerative diseases due to their numerous therapeutic effects and negligible side effects. Callistephin as the bioactive components of pomegranate juice, strawberries and in purple corn show a wide range of healing characteristics, such as antioxidative (Yacout and Gaillard, 2017), neuroprotective (Winter et al., 2017), and anti-inflammatory responses (Zhao et al., 2019). Among the therapeutic effects of callistephin, its neuroprotective effects can be used as a potential feature for the treatment of a wide range of neurodegenerative diseases. Researches have shown that the use of callistephin promotes the protective effects of isoflurane on microglial damage (Zhao et al., 2019) and reduces Aβ-triggered neurotoxicity, in vivo (Ghasemi Moravej et al., 2017). With these potential properties, it is one of the small molecules that can be used as an effective compound in mitigating the protein amyloid-induced neurotoxicity and CAA. Therefore, based on our knowledge, it’s for the first time that the inhibitory effects of callistephin on Aβ fibrillization and associated cytotoxicity against cerebrovascular smooth muscle cells (CVSMCs) as the core components of cerebral arterial wall were aimed to be assessed.
2 Materials and methods
2.1 Materials
Aβ1–42 peptide was obtained from AnaSpec (CA, USA). Hexafluoroisopropanol (HFIP), callistephin chloride, and thioflavin T (ThT) were purchased from Sigma Co. (Shanghai, China).
2.2 Amyloid β preparation
1 mg of Aβ1–42 peptide was hydrated in 100 µL HFIP, where it was then removed with nitrogen to form a thin film. The samples were stored at −35 °C and for doing the assays, a sample was completely rehydrated in DMSO (5% final) followed by PBS (pH 7.4) to a final concentration of 30 µM, and sonicated (30 s, 25 °C). Stock of callistephin chloride (15 mM) was prepared in DMSO and 18 µL of 30 µM Aβ1–42 sample was then added to the callistephin samples (2 µL) over a broad final concentration range (0.3 µM–30 µM), followed by a gentle tapping. The samples were then covered and incubated in the dark at room temperature for 36 h without agitation.
2.3 ThT assay
At different time intervals, 480 µL of ThT solution [5 µM in glycine buffer (50 mM, pH 8.0] was added to protein samples and the fluorescence intensity was measured on a SpectraMax M5 (CA, USA) multi-mode plate reader with λex and λem at 444 nm and 488 nm, respectively. The results were then fit by nonlinear regression analysis with GRAPHPAD PRISM software. The ThT fluorescence intensity of callistephin and inner filter effect were subtracted from the final data. The fibrillization kinetic parameters [k (first-order rate constant), tlag (lag time), and relative fluorescence intensity) were calculated based on the equations reported in the previous studies (Cabaleiro-Lago et al., 2008; Pang et al., 2020).
2.4 Nile red fluorescence analysis
Nile red can be employed to assess the formation of folding intermediates the surface features of biomolecules (Fardanesh et al., 2019). This assay was done by mixing 15 μM Aβ1–42 samples that were already co-incubated with and without callistephin and 15 μM Nile red for 15 min. A Cary eclipse fluorescence spectrometer (Varian, Australia) was employed to characterize the fluorescence intensity with excitation at 560 nm.
2.5 Far-UV CD analysis
A Jasco-820 spectropolarimeter (Japan) was employed to characterize the CD spectra of Aβ1–42 samples that were co-incubated with or without callistephin at room temperature. The samples were assessed in a wavelength range of 190–260 nm at a scan rate of 60 nm/min at room temperature.
2.6 Cell culture and viability assay
Primary cultures of CVSMCs were well-characterized as described in previous reports (Van Nostrand et al., 1994). The cells were cultured in DMEM supplemented with 10% FBS (10%), 10 ng/ml growth factor, and 0.5 μg/ml antibiotics. The cells were used between passages 5–7 for all assays. Afterwards, 3 μM Aβ1–42 samples aged for 36 h with or without callistephin was added and incubated for 24 h at 37 °C. Subsequently, the cell viability was assessed by trypan blue exclusion assay (Hoskins et al., 2012).
2.7 LDH, TNF-α, and IL-6 assays
After treatment for 24 h, the cell culture media were collected. The samples were also centrifuged at 15,000 × g for 15 min to discard any cellular pellets. The level of LDH and inflammatory mediators in the supernatant was then determined employing the LDH, TNF-α, and IL-6 ELISA kits as instructed by the manufacturer (BioSource International Inc., Camarillo, CA).
2.8 Statistical analysis
Data were reported as the mean ± SD of five experiments and were analyzed by student's t test at P < 0.05 significance level using SPSP software. The final spectrum analysis and cell assays were reported after correction against control groups.
3 Results
3.1 Kinetic analysis
The aggregation kinetics of Aβ1–42 was assessed employing ThT assay with the purpose of exploring the effect of callistephin on the fibrillogenesis phases. It was indicated that the amyloid formation process was followed by a typical sigmoidal curve (Fig. 1). The fibrillization kinetic parameters were then calculated and summarized in Table 1. It was observed that callistephin showed potential inhibitory effects on the aggregation of Aβ1–42 by slowing down the rate of fibrillization and reducing the fluorescence intensity at the steady-state step. Also, callistephin demonstrated a concentration-dependent manner to prevent the aggregation of Aβ1–42. Fig. 2. Nile red fluorescence assay of Aβ1–42 without and with different concentrations of callistephin as described in Section 2.4.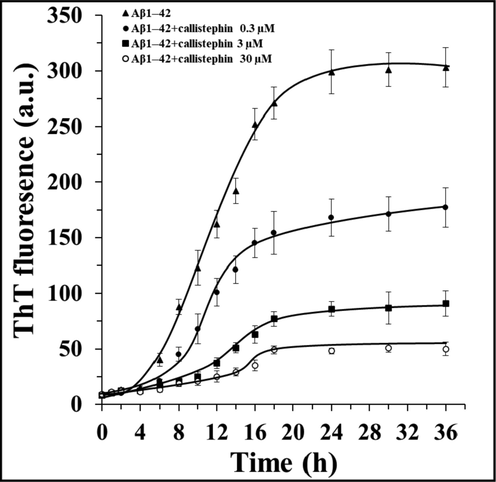
The inhibitory influence of different concentrations of callistephin on the kinetics of Aβ1–42 aggregation as described in Sections 2.2 and 2.3.
k (10-2h−1)
Lag time (h)
Relative fluorescence intensity (%)
Aβ1–42
4.14 ± 0.39
3.89 ± 0.04
100.00 ± 5.88
Aβ1–42: callistephin (1:0.01)
3.98 ± 0.26
5.83 ± 0.07
58.41 ± 10
Aβ1–42: callistephin (1:0.1)
1.98 ± 0.17
6.91 ± 0.11
30.03 ± 4.35
Aβ1–42: callistephin (1:1)
1.88 ± 0.15
9.35 ± 0.14
16.50 ± 2.37
3.2 Nile red fluorescence assay
As a hydrophobic probe, Nile red can be used as a potential dye to reflect the alterations of surface hydrophobicity of biomolecules by interaction with the formation of hydrophobic patches during fibrillization (Gilan et al., 2019; Zhang et al., 2021). The control sample, Aβ1–42 (0h), emitted at 632 nm and the fluorescence intensity of Nile red enhanced with a significant blue shift at 605 nm when it interacted with the formed hydrophobic moieties of Aβ1–42 (36h) (Fig. 2). For samples incubated with different concentrations of callistephin, the Nile red fluorescence intensity and corresponding blue shift were reduced continuously. Based on this data, we speculated that callistephin can mitigate the formation of hydrophobic patches by binding to the partially misfolded Aβ1–42 peptides to stabilize their structure.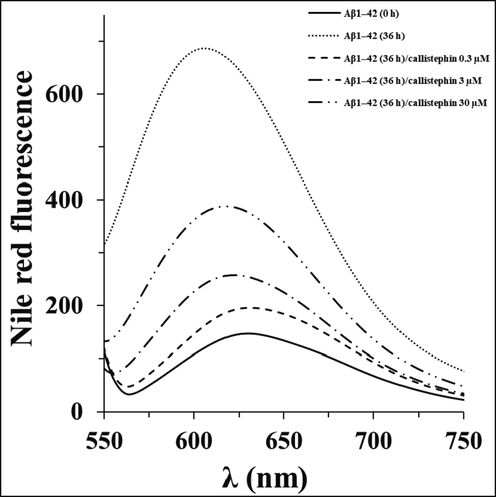
Nile red fluorescence assay of Aβ1–42 without and with different concentrations of callistephin as described in Section 2.4.
3.3 CD analysis
To further investigate the changes of the secondary structure during the Aβ1–42 aggregation, CD spectroscopy analysis was employed. It was shown that the native Aβ1–42 had a typical random coil conformation (Fig. 3). After incubation in the aggregation buffer for 36 h, an obvious minimum appeared around 217 nm in the CD band of Aβ1–42 without callistephin, indicating the transition from native structures to β-sheet conformers (Fig. 3). However, in the presence of callistephin, the structural transition was inhibited to some extent in a concentration-dependent manner.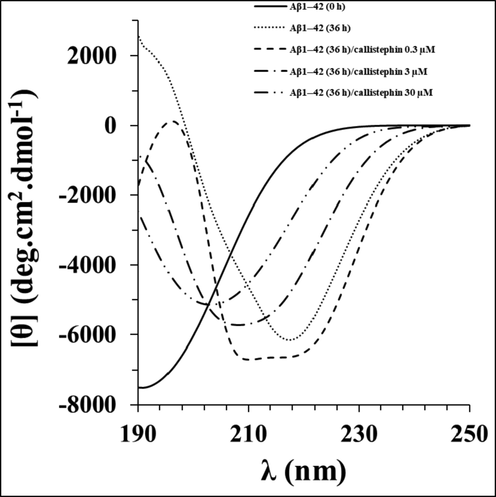
Far -UV CD spectra of Aβ1–42 without and with different concentrations of callistephin as described in Section 2.6.
3.4 Callistephin mitigates the cytotoxicity of Aβ1–42 aggregates
To explore the promising protective impacts of callistephin against Aβ1–42-stimulated cytotoxicity, membrane leakage, and inflammation, trypan blue, LDH release, and protein levels of TNF-α/IL-6 assays, respectively were performed in the primary cell culture of CVSMCs after 24 h. Tryptophan blue exclusion assay revealed that the cell viability was significantly reduced after incubation of cells with Aβ1–42 fibrils with a concentration of 3 µM relative to control sample (Fig. 4a). As a control, CVSMC viability was assessed in the presence of callistephin with a concentration of 3 µM (corresponding to highest concentration of Aβ1–42: callistephin molar ratio: 1.1) and no remarkable cytotoxicity was detected. It was seen that the incubation of Aβ1–42 with callistephin mitigated the cell mortality induced by Aβ1–42 fibrils alone in a concentration-dependent manner (Fig. 4a). Fig. 4b also indicated that LDH release was increased followed by 24 h cells incubation with 3 μM Aβ1–42 fibrils, and the incubation of Aβ1–42 by callistephin attenuated the LDH release induced by Aβ1–42 fibrils in a dose-dependent mode.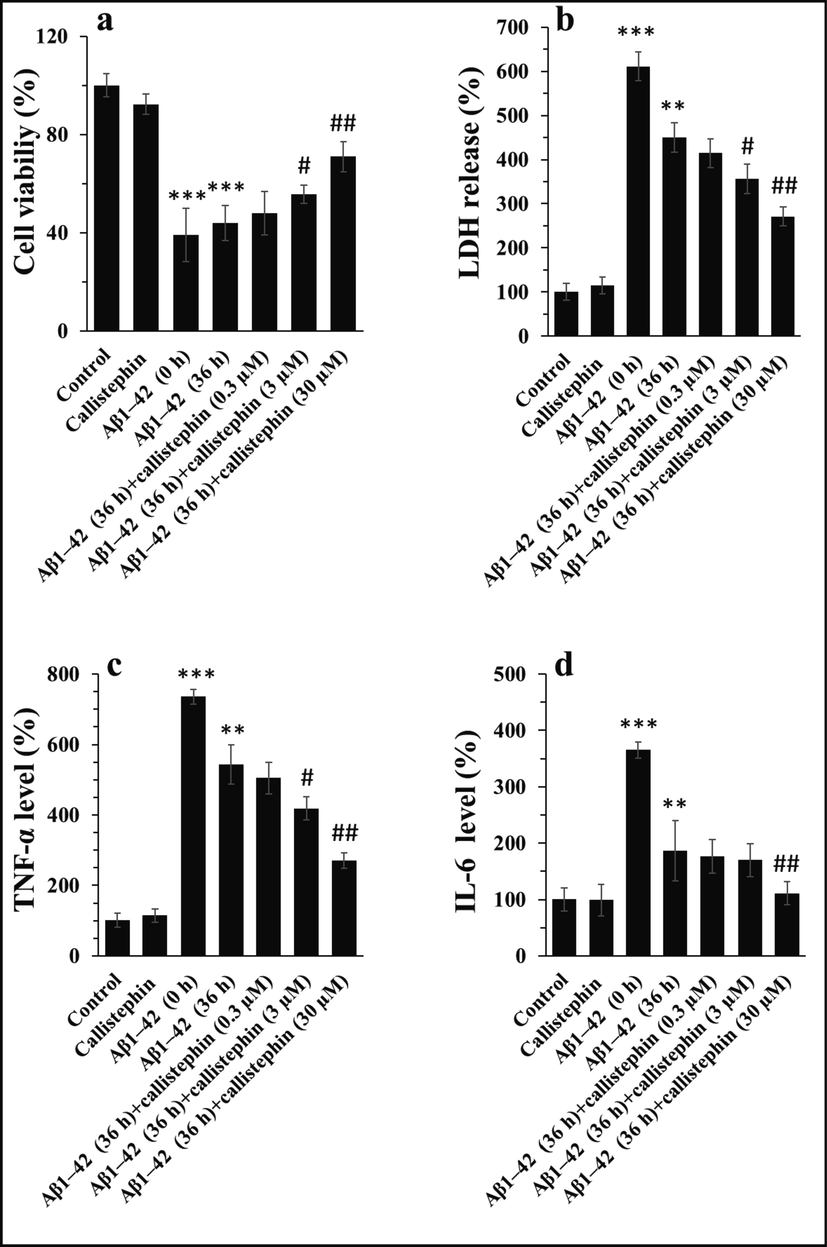
Trypan blue exclusion (a), LDH release (b), the level of TNF-α (c), and the level of IL-6 (d) in CVSMCs after incubation with callistephin (3 µM), Aβ1–42 monomer (3 µM), Aβ1–42 (36h) (3 µM) and Aβ1–42 (36h) with callistephin for 24 h. as described in sections 2.8 and 2.9. **P < 0.01, ***P < 0.001 relative to control, #P < 0.05, ##P < 0.01, ###P < 0.001, relative to Aβ1–42 (36 h).
The protein levels of inflammatory mediators were also assessed by ELISA assay. It was shown that the expression patterns of TNF-α (Fig. 4c) and IL-6 (Fig. 4d) in the protein levels were similar to preceding cytotoxicity and LDH release assays. Indeed, it was detected that the callistephin reduced the upregulation of TNF-α (Fig. 4c) and IL-6 (Fig. 4d) triggered by Aβ1–42 fibrils in a dose-dependent manner.
4 Discussion
The present study focused on the protective effect of callistephin against aggregation of Aβ1–42 and relevant cytotoxicity against CVSMCs as an in vitro model of CAA. The aggregation of Aβ1–42 in the human brain suffered from neurodegenerative diseases have been linked with CVSMCs toxicity and CAA (Urmoneit et al., 1997). We realized that incubation of Aβ1–42 with callistephin prevented the aggregation of Aβ1–42 through the control of kinetic parameters of aggregation and subsequently modulated the cytotoxicity, LDH release and overexpression of inflammatory mediators. It has been reported that Aβ1–42 accumulation stimulates CAA (Attems et al., 2004) and some small molecules/drugs including neprilysin (Miners et al., 2011), apolipoprotein E (Endo et al., 2019), and clusterin (Endo et al., 2019) can show promising protective influence against CAA and Aβ‐triggered degeneration of CVSMCs. Indeed, prevention of amyloid accumulation can be a potential strategy in delaying the onset of CAA (Wilhelmus et al., 2006). It is known that Aβ is crucial for activating the pathogenic signaling cascades, thereby resulting in neural death, CVSMC toxicity, and induction of CAA (Burwinkel et al., 2018).
Several reports have shown that membrane leakage (Zhao et al., 2011) and inflammation (Previti et al., 2006) in CVSMC lead to onset of CAA. For example, it has been displayed that apigenin inhibits Aβ-stimulated toxicity in CVSMCs (Zhao et al., 2011) or dexamethasone prevents the overexpression of pro-inflammatory mediators induced by Aβ in CVSMCs (Previti et al., 2006). Here, we also determined a significant increase in overexpression of inflammatory markers (TNF-α and IL-6) in Aβ1–42-incubated CVSMCs as well as an enhancement in the amount of LDH release, both of which were reversed by callistephin incubation. It has been also determined that callistephin improves the protective influence of isoflurane on microglial damage through downregulation of inflammation (Zhao et al., 2019). Also, it has been indicated that callistephin shows neuroprotective effects through modulation of oxidative stress (Winter et al., 2017). Also, in agreement with our data, it has been indicated that soluble Aβ peptide show significant pathologic effects in cultured CVSMCs which is may due to their aggregation in the presence of cells (Davis-Salinas and Van Nostrand, 1995).
In addition to reported protective and anti-inflammatory influences of callistephin, hence introducing its potential as a therapeutic and/or inhibitory small molecule for cerebral Aβ amyloidosis like CAA can be considered as the novelty of this work. However, well-established in vivo assays and clinical trials are necessary to report their potency as disorder-modifying compounds.
5 Conclusion
In the present study, several analytical approaches were employed to examine the effect of callistephin on the aggregation of Aβ1–42. ThT binding analysis showed the potential preventive effects of callistephin on the aggregation of Aβ1–42 in a concentration-dependent manner. Due to the presence of callistephin, the formation of hydrophobic patches and the transition of the secondary conformation were inhibited as evidenced by Nile red fluorescence and CD studies, respectively. Hydrophobic and π- π interactions between aromatic rings of callistephin and hydrophobic patches of Aβ1–42 may be the major reason for potential stabilization of Aβ1–42–callistephin complex and prevention of subsequent Aβ1–42 aggregation. Moreover, callistephin showed promising protective influences against Aβ1–42-stimulated cytotoxicity against CVSMCs.
In conclusion, the outcomes presented may provide useful details for elucidating the interaction of callistephin with Aβ1–42 and developing potential amyloid inhibitors in future.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol.. 2005;6(11):891-898.
- [Google Scholar]
- Amyloid β-protein and beyond: The path forward in Alzheimer’s disease. Curr. Opin. Neurobiol.. 2020;1(61):116-124.
- [Google Scholar]
- Structural studies of amyloid-β peptides: Unlocking the mechanism of aggregation and the associated toxicity. Biochimie. 2017 Sep;1(140):176-192.
- [Google Scholar]
- Mixing Aβ (1–40) and Aβ (1–42) peptides generates unique amyloid fibrils. Chem. Commun.. 2020;56(62):8830-8833.
- [Google Scholar]
- Ganglioside-mediated aggregation of amyloid β-proteins (Aβ): comparison between Aβ-(1–42) and Aβ-(1–40) J. Neurochem.. 2011;116(5):851-857.
- [Google Scholar]
- Interaction between Aβ (1–42) and Aβ (1–40) in Alzheimer's β-amyloid fibril formation in vitro. Biochemistry. 1999;38(47):15514-15521.
- [Google Scholar]
- Comparison of Alzheimer Aβ (1–40) and Aβ (1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci.. 2009;106(47):19813-19818.
- [Google Scholar]
- The role of amyloid beta clearance in cerebral amyloid angiopathy: more potential therapeutic targets. Trans. Neurodegeneration. 2017;6(1):1-2.
- [Google Scholar]
- Amyloidosis associated with cerebral amyloid angiopathy: cell signaling pathways elicited in cerebral endothelial cells. J. Alzheimers Dis.. 2014;42(s3):S167-S176.
- [Google Scholar]
- Human apolipoprotein E4 alters the amyloid-β 40: 42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci.. 2005;25(11):2803-2810.
- [Google Scholar]
- APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Trans. Med.. 2021;13(581):1-8.
- [Google Scholar]
- Flavonoids for treatment of Alzheimer's disease: An up to date review. EXCLI J.. 2021;26(20):495-502.
- [Google Scholar]
- Yamada M, Ono K, Hamaguchi T, Noguchi-Shinohara M. Natural phenolic compounds as therapeutic and preventive agents for cerebral amyloidosis. InNatural Compounds as Therapeutic Agents for Amyloidogenic Diseases 2015 (pp. 79-94). Springer, Cham.
- Inhibition of protein aggregation and amyloid formation by small molecules. Curr. Opin. Struct. Biol.. 2015;1(30):50-56.
- [Google Scholar]
- Protein misfolding, aggregation and mechanism of amyloid cytotoxicity: an overview and therapeutic strategies to inhibit aggregation. Int. J. Biol. Macromol.. 2019;1(134):1022-1037.
- [Google Scholar]
- An in vivo platform for identifying inhibitors of protein aggregation. Nat. Chem. Biol.. 2016;12(2):94-101.
- [Google Scholar]
- The anthocyanins, oenin and callistephin, protect RPE cells against oxidative stress. Photochem. Photobiol.. 2017;93(2):590-599.
- [Google Scholar]
- Chemical basis for the disparate neuroprotective effects of the anthocyanins, callistephin and kuromanin, against nitrosative stress. Free Radical Biol. Med.. 2017;1(103):23-34.
- [Google Scholar]
- Callistephin enhances the protective effects of isoflurane on microglial injury through downregulation of inflammation and apoptosis. Mol. Med. Rep.. 2019;20(1):802-812.
- [Google Scholar]
- The Effect of Callistephin on Amyloid Beta-Induced Neurotoxicity in the Hippocampus of Male Rats. Anatomical Sci. J.. 2017;14(2):73-78.
- [Google Scholar]
- Inhibition of amyloid β protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc.. 2008;130(46):15437-15443.
- [Google Scholar]
- Effects of lithospermic acid on hIAPP aggregation and amyloid-induced cytotoxicity by multiple analytical methods. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics.. 2020;1868(1):140283-140289.
- [Google Scholar]
- Amorphous aggregation of tau in the presence of titanium dioxide nanoparticles: biophysical, computational, and cellular studies. Int. J. Nanomed.. 2019;14:901.
- [Google Scholar]
- Amyloid β-protein precursor in cultured leptomeningeal smooth muscle cells. Amyloid. 1994;1(1):1-7.
- [Google Scholar]
- Dilemmas in the reliable estimation of the in-vitro cell viability in magnetic nanoparticle engineering: which tests and what protocols? Nanoscale Res. Lett.. 2012;7(1):1-2.
- [Google Scholar]
- α-synuclein interaction with zero-valent iron nanoparticles accelerates structural rearrangement into amyloid-susceptible structure with increased cytotoxic tendency. Int. J. Nanomed.. 2019;14:4637.
- [Google Scholar]
- Evaluation of heptelidic acid as a potential inhibitor for tau aggregation-induced Alzheimer's disease and associated neurotoxicity. Int. J. Biol. Macromol.. 2021;31(183):1155-1161.
- [Google Scholar]
- Cerebrovascular smooth muscle cells internalize Alzheimer amyloid beta protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Laboratory investigation; a J. Technical Methods Pathol.. 1997;77(2):157-166.
- [Google Scholar]
- Amyloid β peptide 1–42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol.. 2004;107(4):283-291.
- [Google Scholar]
- Neprilysin Protects against Cerebral Amyloid Angiopathy and Aβ-Induced Degeneration of Cerebrovascular Smooth Muscle Cells. Brain Pathol.. 2011;21(5):594-605.
- [Google Scholar]
- Apolipoprotein E and clusterin inhibit the early phase of amyloid-β aggregation in an in vitro model of cerebral amyloid angiopathy. Acta Neuropathologica Commun.. 2019;7(1):1.
- [Google Scholar]
- Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res.. 2006;1089(1):67-78.
- [Google Scholar]
- Intravenous injection of beta-amyloid seeds promotes cerebral amyloid angiopathy (CAA) Acta Neuropathologica Commun.. 2018;6(1):1-6.
- [Google Scholar]
- Apigenin isolated from the medicinal plant Elsholtzia rugulosa prevents β-amyloid 25–35-induces toxicity in rat cerebral microvascular endothelial cells. Molecules. 2011;16(5):4005-4019.
- [Google Scholar]
- Dexamethasone diminishes the pro-inflammatory and cytotoxic effects of amyloid β-protein in cerebrovascular smooth muscle cells. J. Neuroinflammation. 2006;3(1):1-8.
- [Google Scholar]
- Amyloid β-protein aggregation nullifies its pathologic properties in cultured cerebrovascular smooth muscle cells. J. Biol. Chem.. 1995;270(36):20887-20890.
- [Google Scholar]




