

Translate this page into:
Chromenone-based GSK-3β inhibitors as potential therapeutic targets for cardiovascular diseases: In silico study, molecular dynamics, and ADMET profiles
⁎Corresponding authors. zhousann3@163.com (San Zhou), mirzaei22@yahoo.com (Hassan Mirzaei), salehia@goums.ac.ir (Aref Salehi), lotfor@ums.edu.my (Md. Lutfor Rahman)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Glycogen synthase kinase-3 beta (GSK-3β) regulates glycogen metabolism and many different cellulars, including apoptosis, signaling, and neural. It is a crucial therapeutic receptor in heart disease, type 2 diabetes, and Alzheimer's. In this study, using computational methods, flavonoid compounds were investigated for potential inhibitors against GSK-3β. Virtual screening was utilized to investigate flavonoid compounds obtained from the PubChem database. Structure of human heart mitochondria of GSK-3β receptor constructed by homology modeling. Best binding poses were discovered via in silico molecular docking simulation. We surveyed noncovalent interactions among amino acid residues involved in the active site of the modeled Protein and compounds via molecular docking and molecular dynamics (MD).
Moreover, ADMET characteristics of best docking conformers have been investigated. The obtained results revealed that compound 1 containing chromenone moiety with binding energy H-bond −11.4 kcal/mol inhibited effectively binding pocket of the GSK-3β receptor. Moreover, MD simulation analysis (RMSD and radius of gyration indicated complex of the compound and GSK-3β receptor remained stable throughout 100 ns MD simulation, and also analysis of ADMET profiles revealed that selected compounds had good drug-likeness and pharmacokinetic properties. Hence, it was suggested that compounds with chromenone scaffold could potentially inhibit GSK-3β. Structural modification of the chromenone derivatives may result in the discovery of promising candidates for identifying novel drugs as GSK-3β inhibitors.
Keywords
GSK-3β
Chromenone
Virtual screening
Molecular dynamics simulation
ADMET

1 Introduction
Glycogen synthase kinase-3 (GSK-3) is a cytoplasmic serine/threonine protein kinase identified as an required regulator in several signaling pathways of pathologies like diabetes, cancer, inflammation, neurodegenerative disorders, and cardiac (Takahashi-Yanaga, 2018; Khan et al., 2017; Lal et al., 2015). It shows a significant effect in regulatory switch in cell proliferation, apoptosis, and inflammation (Takahashi-Yanaga, 2018). The previous studies proposed that GSK-3 is needed to suppress cardiac hypertrophy, maintain normal cardiac development, and inhibit cardiac fibrosis (Takahashi-Yanaga, 2018; Tariq et al., 2021).
GSK-3α and GSK-3β are highly homologous mammalian isoforms of the GSK-3 family. Among two isoforms, current investigation has attended on GSK-3β inhibition in cardiac tissue, which could decrease hypertrophy and necrosis of cardiomyocytes in ischemic heart disease, cardiac regeneration, and heart failure (Tariq et al., 2021; Gao et al., 2008). Likewise, pharmacological GSK-3β inhibitors have been classified into three sections (Non-ATP-competitive, ATP-competitive, and substrate-competitive GSK-3β inhibitors as an attractive therapeutic target for human diseases (Khan et al., 2017). Despite these advances, there are some limitations in clinical application, including the complexity of the interplay between GSK-3β and disease therapy, determination of clinical dose/frequencies, and inefficiency for the prevention and treatment of diseases (Dou et al., 2018). Thus, there is a need to identify novel, safe, effective, and affordable scaffolds for GSK-3β inhibition.
Currently, available evidence reveals that natural products with their active moieties have a promising candidate for identifying newer drugs as a GSK-3β inhibitors (Saraswati et al., 2018; Kim et al., 2018). Because of this, in Alzheimer's disease, morin, a natural flavonoid, directly inhibits GSK-3β resulting in binding to the ATP and blocking tau phosphorylation in human neurons (Kim et al., 2018; Chen et al., 2021). Moreover, ursolic acid derivatives and catechin from Potentilla reptans L. root revealed a cardioprotective effect by inhibiting GSK-3β in competitive ATP in the in silico study (Enayati et al., 2022; Li et al., 2022; Yee, 2021; Alibak et al., 2022; Tang et al., 2020). Also, ethyl acetate fraction of Potentilla reptans root induces cardioprotective effect in ischemia/reperfusion injury by down-regulation of various signaling pathways (AKT, ERK1/2, STAT3) and up-regulation of Nrf2 expression, thereby inactivation of GSK-3β protein kinase led to cardiomyocytes protection of fraction during reperfusion injury (Enayati et al., 2021).
As far as most of the inhibitors act as effective competitors at the ATP-binding site, this study focuses on the assess specific and selective natural/synthetic inhibitors of GSK-3β to pave the way for the treatment of cardiac regeneration, inflammation, and their involved underlying mechanisms via molecular docking.
2 Materials and methods
2.1 Virtual screening
Molecular docking has been utilized to screen the PubChem compound database to identify molecules interacting to GSK-3β applying AutoDock software (Morris et al., 2009).
2.1.1 Target structure preparation
Homology modeling.
Inhibition of GSK-3β has a significant therapeutic for cardioprotection by ischemic preconditioning. To understand how the selected compounds interact binding site of GSK-3β, we searched in Protein Data Bank (PDB) (https://www.rcs.org) and did not find a similar crystal structure of GSK-3β in human heart mitochondria. Therefore, homology modeling has been applied to build an optimized 3D receptor of GSK-3β. The protein sequence of GSK-3β in human heart mitochondria (Acc: HGNC:4617) is retrieved from the ensemble database (https://www.ensembl.org) ) in FASTA format (Hollingsworth and Karplus, 2010) and considered as the GSK-3β sequence in this study. Accordingly, we used four similar templates (1J1B-1A, 1J1B-B, 6HOU, 3M1S). These templates with a high identity on the sequence of GSK-3β in human heart mitochondria were chosen. Moreover, the important configuration was determined through the Discrete relaxed Protein Energy (DOPE) score. Hence, the quality of the built configuration is determined by the Ramachandran plot and SAVE. Therefore, Φ/Ψ dihedral angles distribution of the molecule have been defined to demonstrate the Ramachandran graph (Sun and Ansari, 2021).
2.1.2 Ligand preparation
The 3D structure of natural and synthetic compounds was searched based on the similar structure of morin (Takahashi-Yanaga, 2018) (Fig. 1) as a chromenone scaffold and their medicinal characteristics; then, compounds were selected for further studies. 5640 compounds were obtained from two chemical libraries like the PubChem database (https://www.pubchem.ncbi.nlm. nih.gov). The structures were retrieved in SDF format converting into PDB format. Then, all collected compounds were optimized by density functional theory (DFT) and energy minimized at B3LYP functional (Cao et al., 2021; Soltani et al., 2018) and 6-31+G** basis set using the Gaussian 09 package (Frisch et al., 2009).
chemical structure of morin and chromenone.
2.1.3 Molecular docking
To better understand the estimation of binding energy and binding mode between selected compounds and target structure in silico molecular docking investigation has been performed through the Auto Dock vina software (Howe et al., 2021). In order to find appropriate orientation and best poses of compounds inhibiting binding pocket of GSK-3β receptor. An optimized model of the structure of GSK-3β of the human heart mitochondria created by homology modeling was applied as the target for the docking simulation; then, the modeled Protein was prepared by the following several steps: adding Kollman atom partial charges and polar hydrogen atoms and elimination of water molecule and merged non-polar hydrogen atoms furthermore, a grid box of 60x60x60 with a point spacing of 0.375 Å was calculated around the active site in GSK-3β to provide the autogrid module (Mirzaei et al., 2020; Gong et al., 2011; Razzaghi-Asl et al., 2018; Mirzaei et al., 2017; Dou et al., 2018). 150 GA runs were implemented using the Lamarckian genetic algorithm. Maestro 11.0 Schrodinger suit has been utilized to visualize the 2D and 3D demonstration.
2.1.4 Molecular dynamic simulation study
Molecular dynamics simulation of the selected compound was accomplished using the GROMACS-2020 package allocating GROMOS9654A7 force field (Abraham et al., 2015). To generate topology selected ligand and prepare gromacs parameters, PRODRG webserver was applied. Minimizing the optimized recptor was implemented through two short runs at 100 ps using Amber 96 force fields to obtain the lowest energy conformations of the receptor. The system was solvated within a virtual cubic box containing about 28,900 water molecules (TIP3P model). To neutralize the system, seven chloride ions have been added. Equilibration of whole systems was performed employing both NVT and then NPT ensembles by position restrain on the optimized Protein at a temperature of 300 K and pressure of 1 bar during 100 ps MD runs. After equilibration of the system, production runs of 100 ns through a time step of 2 fs were carried out to examine the stability of the complex of Protein and selected compound. The Particle Mesh Ewald (PME) method has been utilized to measure the electrostatic interactions. QtGrace tools and VMD software were used for graph plotting and visualization of the complex.
2.1.5 Drug likeness and ADMET prediction
ADMET parameters (absorption, distribution, metabolism, excretion, and toxicity) and Pharmacokinetic charactreistics of the selected compounds have been estimated applying admetSAR database (Yang et al., 2019) then, swiss ADME (Daina et al., 2017), evaluating the drug-likeness of the selected compounds. Moreover, topological polar surface area (TPSA) has been estimated as a significant descriptor estimating oral bioavailability and absorption of the compounds then, the permeability of blood–brain barrier (BBB), inhibition profiles of cytochrome P450 including CYP3A4, CYP2C9and CYP2D6; AMES (Salmonella typhimurium reverse mutation assay) toxicity and carcinogenicity were evaluated. Hence, ADMET and pharmacological properties could help assess the selected compounds' drug-likeness properties.
3 Results and discussion
3.1 Frontier molecular orbital and molecular electrostatic potential
We have done the DFT calculations to explain the Frontier Molecular Orbital theory (FMO) and Molecular Electrostatic Potential (MEP) of the chromenone derivatives in the water phase applying the polarizable continuum model (PCM) method (Cao et al., 2021; Cao et al., 2021). The electronic distribution on the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) was characterized by FMO analysis. Fig. 2 demonstrates the HOMO and LUMO orbitals in order as electron donor and electron acceptor, which are more localized around carbonyl groups of chromenone moiety and carbon–carbon bonds (Soltani et al., 2022). The energies of HOMO (EHOMO) and LUMO (ELUMO) have been found to be −2.90 and −5.96 eV, respectively. The value of energy gap (EHLG) is 3.06 eV. MEP plot demonstrates the anionic fragment in the carbonyl (C⚌O) group with a negative charge (denoted by red color) around it, while the cationic fragment in around C—H and O—H groups have a positive (blue color) charge around them (Cao et al., 2021; Yang et al., 2022; Zhang et al., 2022; Antoine et al., 2022).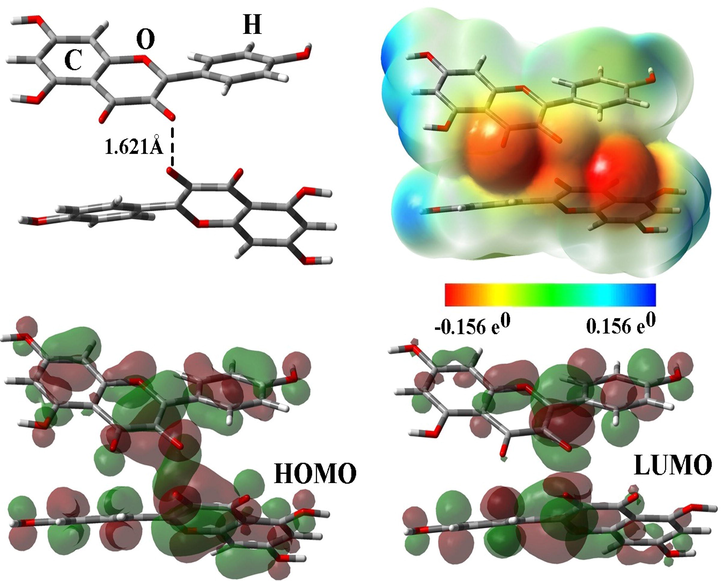
Optimized structure, FMO, and MEP plots of compound1.
3.2 Homology modeling
Based upon prior investigations, inhibition of GSK-3β acts as an underlying factor in cardiovascular disease (Juhaszova et al., 2009), but there is no similar structure of GSK-3β of the target in the PDB database. Hence, the 3D structure of GSK-3β of the target was created by homology modeling. First, we retrieved the amino acids sequence of GSK-3β (HGNC:4617) from The Ensembl (https://www.ensembl.org). Accordingly, searching BLAST and alignment of GSK-3β sequence were performed to find similar templates then 3D model protein of GSK-3β was built using the SWISS-MODEL server employing four templates (PDB IDs: 1J1B-1A, 1J1B-B, 6HOU, 3M1S) (Gao et al., 2018).
We implement the Ramachandran plot to evaluate the quality of modeled Protein and structural integrity (Lovell et al., 2003). As illustrated in Fig. 3, 90.5 % of the amino acid residues were in favored region, 8.8 % of total residues in allowed region, and only 0.6 % of the amino acid residues in disallowd region. Based on the results mentioned, the quality of modeled Protein would be expected to employ in silico molecular docking simulation.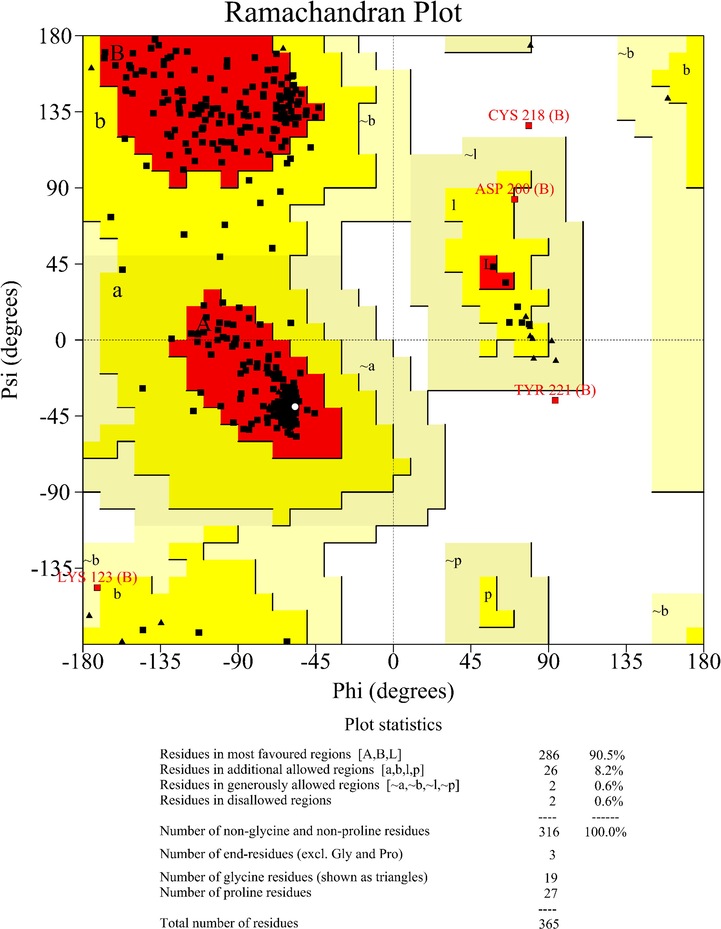
Ramachandran plot for the homology model protein. 90.5 % of amino acid residues are in favored regions, 8.8 % of amino acid residues are in the allowed regions, and 0.6 % are in the disallowed regions.
Additionally, this 3D structure was analyzed using the PROCHECK server to estimate the receptor's sterochemical quality, indicating that 99.4 % of the total amino acid residues had accurate geometry. Therefore, the modeled protein structure generated based on Homology modeling method is reliable and can be used for drug design and finding promising drug candidates.
3.3 Virtual screening
To investigate new potential natural and synthetic compounds for inhibiting GSK-3β, in silico molecular docking simulation was performed. To validate the molecular docking precise of modeled protein structure and compare it with experimental binding affinity, we executed the redocking procedure. ANP (phosphor aminophosphonic acid-adenylate ester) as a cognate ligand was used to evaluate molecular docking protocol; redocking results indicated an RMSD value of 1.46 Å. Based on similarity structure with morin as an inhibitor of GSK-3β, 4650 compounds were extracted from the PubChem database. All screened compounds follow Lipinski's rule of five (Table 1). To investigate the potential inhibition of the compounds to interact to the binding pocket of GSK-3β, a molecular docking simulation was performed. Therefore, based on the lowest binding free energy, the best 12 docked molecules were selected for more analysis (Fig. 4). Abbreviation: BE: Binding Energy.
Compound
PDB ID: GSK3B
NO
Pubchem-CID
BE
(kcal/mol)Ki (µM)
1
127039278
−11.4
3.1
2
44378184
−11.1
3.9
3
1608168
−10.9
4.3
4
15478904
−10.2
5.1
5
12019530
−10.1
5.4
6
14309760
−9.9
6.2
7
44258296
−9.8
6.3
8
52949348
−9.7
6.7
9
10617404
−9.5
7.2
10
4020240
−9.4
7.8
11
14583584
−9.3
8.0
12
53305954
−9.1
8.4
13
5281670
−9.8
6.1
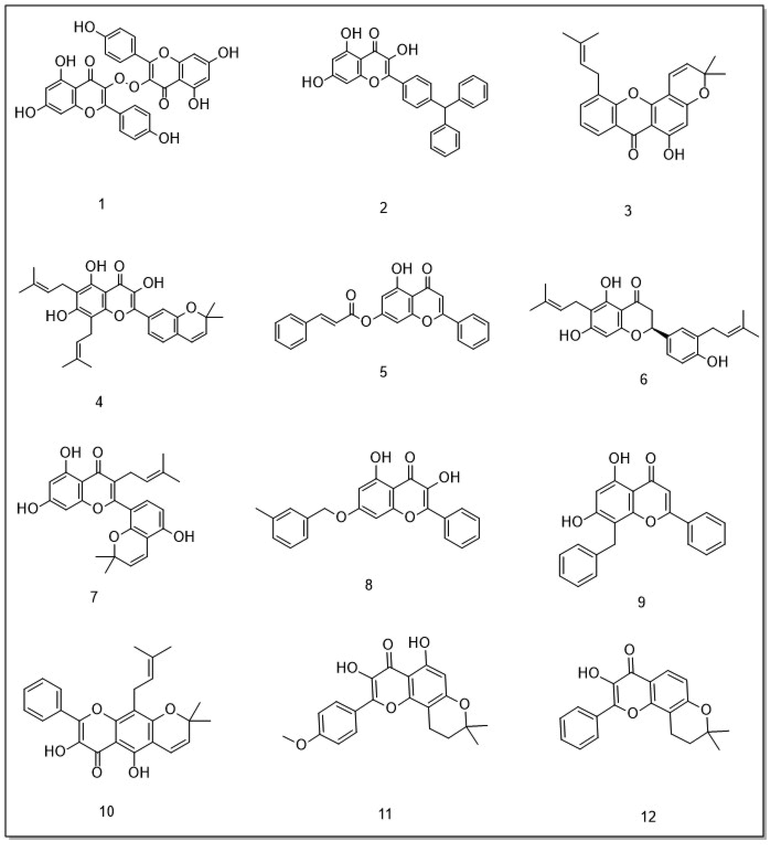
chemical structure of 12 top chromenone derivatives.
As presented in Table 1, 12 top compounds with the lowest binding affinity interacted with the binding pocket of GSK-3β by noncovalent interactions. Table 1 shows Compound 1 as a derivative of chromenon had the lowest binding free energy. As depicted in Fig. 5. Compound 1 interacted with amino acid residues like Leu132, Cys199, Ala83, Val70, Val87, Val69, Phe67, and Leu188 through hydrophobic interaction. Furthermore, compound 1 was established in the target's active site by seven H-bond interactions. Carbonyl group of chromenone of compound 1 interacted with Lys85 at a distance 2.88 Å, and also the compound formed H-bond with residues Ser66, Lys183, Gln185, Val69, and Lys86 with the hydroxyl group of the receptor at a distance 2.76, 2.52, 3.14, 2.77, 2.99, and 2.81 Å, respectively. The compound has also interacted with residues of Asp133, Asp200, Lys85, Lys86, and Lys183 using charged interactions. The chromenone moiety of compound 1 is covered by different noncovalent interactions indicating These interactions play an important role in the inhibition of GSK-3β (Elangovan et al., 2020; Emami et al., 2018).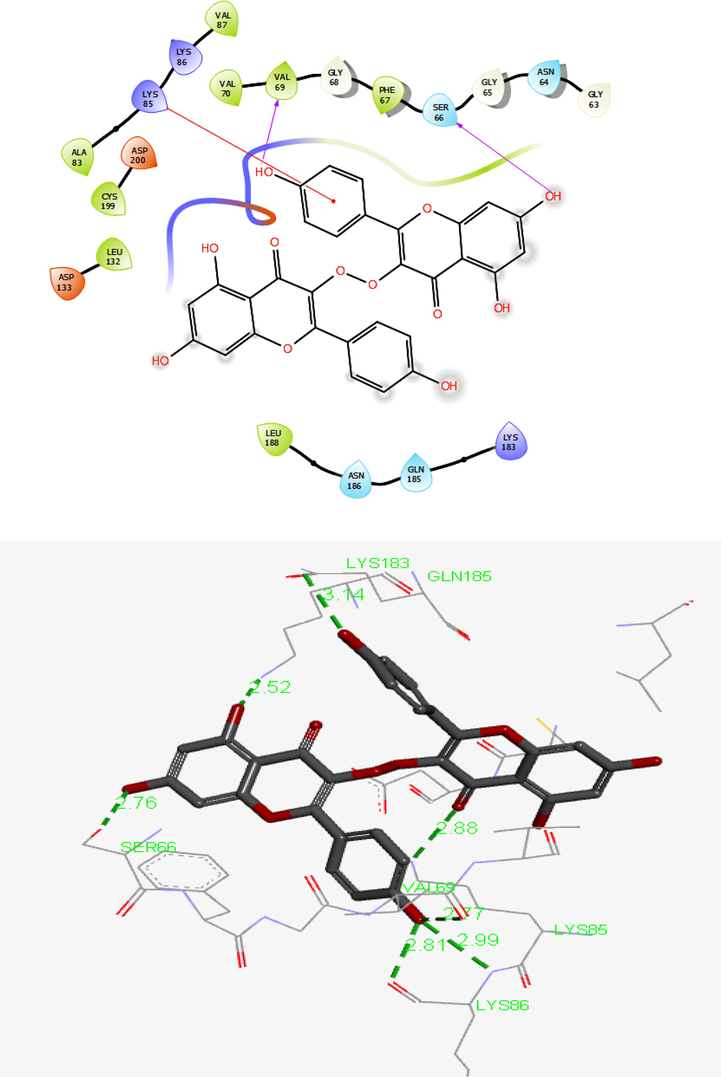
Presentation of 2D and 3D models of interactions between compound1 and modeled Protein (GSK-3β).
As depicted in Fig. 6, molecular docking analysis revealed that Compound 2, consisting of chromenone moiety, showed an excellent dock score of −11.1 kcal/mol. Compound 2 exhibited hydrophobic interaction with Amino acid residues such as Val70, Phe67, Ile62, Cys199, Ala83, Val110, Leu132, Tyr134, Val135, Pro136, and Leu188 in the active site of the target. Moreover, three hydrogens bound surrounded chromenone moiety of the compound in the receptor's binding site. Amino acid residues such as Lys183, Asn186, and Gly68 interact with the compound at distances of 3.29, 2.85, and 2.79 Å, respectively. Additionally, this compound has a notable affinity for the structure of GSK-3β with residues Asp200, Asp133, Glu137, Arg141, and Lys85 through electrostatic interactions. Consequently, it could be proposed as a potential GSK-3β inhibitor.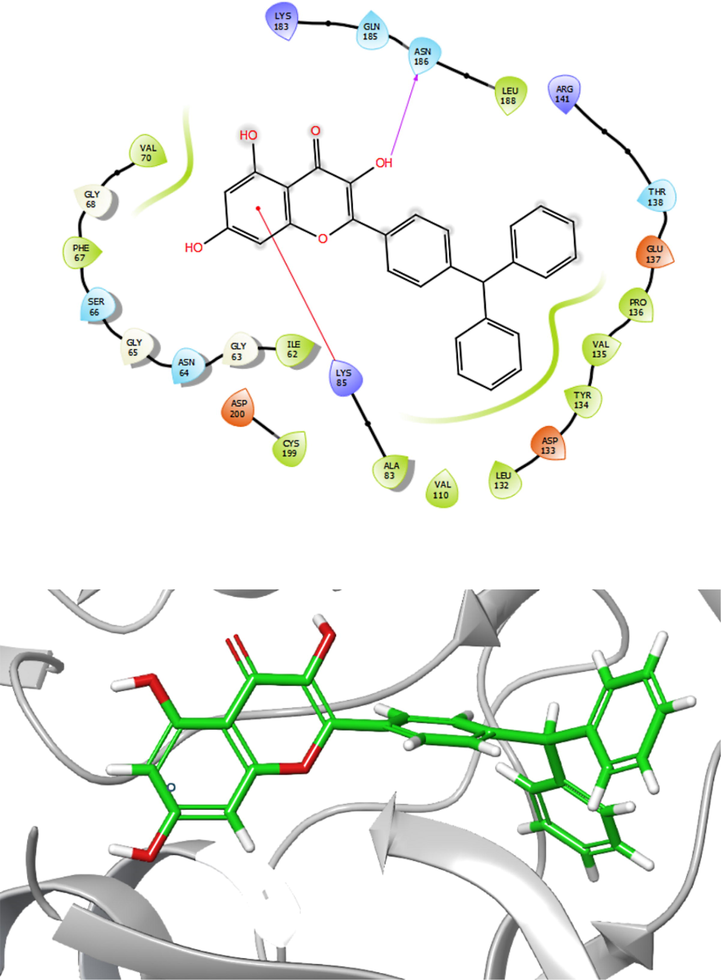
Presentation of 2D and 3D models of interactions between compound 2 and modeled Protein (GSK-3β).
2D and 3D presentation of the best pose of Laurentixanthone A (Compound 3) as a derivative of chromenone is presented in Fig. 7. The obtained results demonstrated that the compound was established in the active site. Residues like Pro136, Val135, Tyr134, Leu132, Ala83, Val110, Leu188, Cys199, and Ile62 formed hydrophobic interaction with the ligand. Moreover, binding free energy of −10.9 kcal/mol is presented in Table1 interacting hydrogen bond with Asp133 and Val135 residues with the hydroxyl group of chromenone of the compound at a distance of 2.92 and 2.98 Å, respectively. In addition, the compound has interacted with residues such as Glu137, Lys85, Asp200, Arg141, and Lys85 using electrostatic interactions.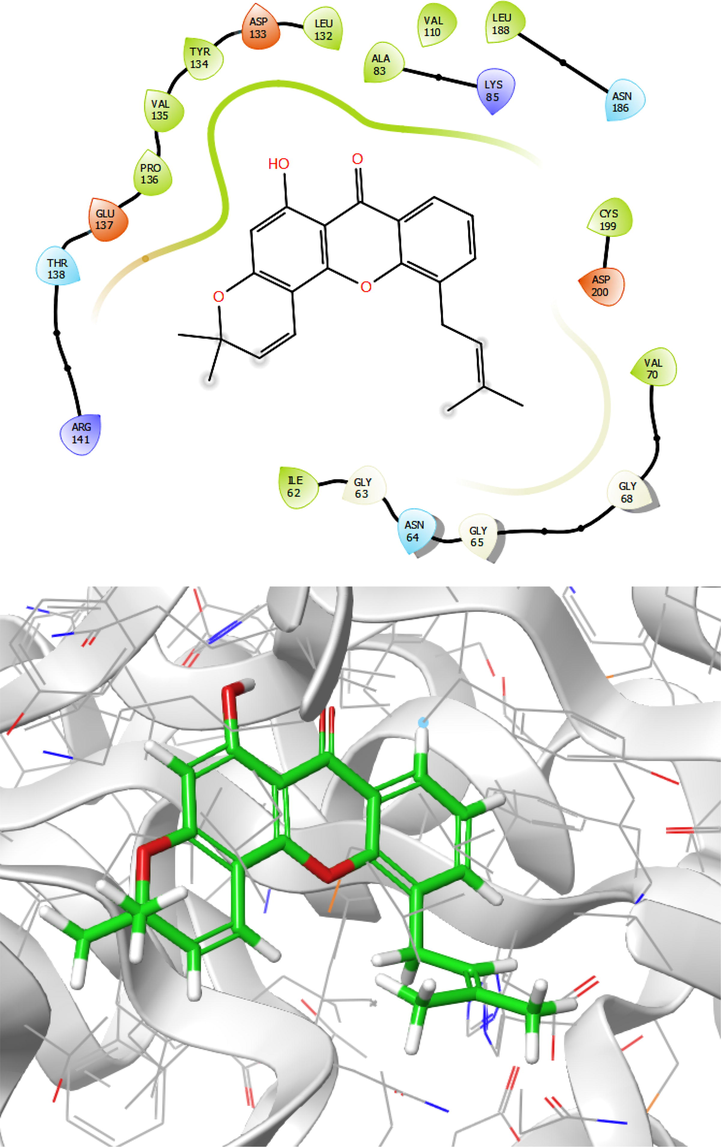
Presentation of 2D and 3D models of interactions between compound 3 and modeled Protein (GSK-3β).
Consequently, chemical structure analysis indicated compounds consisting of chromenon moiety had high binding affinity to the modeled Protein's binding pocket and resulted in occupy the active site of the target. Furthermore, these compounds inhibit key amino acid residues in the binding mode of the Protein through the different types of interactions (electrostatic, polar, hydrogen bond, and hydrophobic) (Daggupati et al., 2018; Daddam et al., 2020). Interestingly, most compounds shown in Table 1 are more persuasive than morin as a reference ligand (Pubchem-CID: 5281670 ). It seems that this effect of the mentioned compounds is associated with existing of electron donor substitutes or electron resonance extenders in the chromone moiety, especially in the pyrone ring. Moreover, in comparing the structure of compounds, some points are important in their GSK-3β inhibition potential: the presence of hydroxyl group or oxygenated substitution in carbon 5 and 7 of chromone, to be free hydroxyl groups, electronic resonance should be easily accessible by suitable stereochemistry of substituents. Therefore, compounds consisting of chromenone moiety could be considered promising potential inhibitors of GSK-3β.
3.4 Assessment of pharmacological properties, ADME and toxicity prediction
Pharmacokinetic properties and ADMET parameters of the top 12 compounds are depicted in Tables 2 and 3. These compounds examined for further research were chosen to understand the top compounds' complying with Lipinski's Rule of Five. Remarkably, the majority of the compounds followed the Lipinski'sRO5 and did not violate the standardized Lipinski criteria (Cao et al., 2021; Cao et al., 2021). This study contributed to the identification of the top flavonoids related to pharmacokinetic properties. Table 2 summarizes the anticipated ADMET features of chosen compounds, including number of H-bond acceptors (HBA), number of H-bond donors (HBD), topological polar surface area (TPSA), solubility, Molecular refractivity (MR), drug likeness (DL), and toxicity profiles. Abbreviations:LogS: Logarithm of water solubility; MW: molecular weight; logP: Logarithm of compound partition coefficient between n-octanol and water; HBA: Number of hydrogen bonds acceptors; HBD: Number of hydrogen bond donors; TPSA: Topological polar surface area; NRB: Number of rotatable bonds, MR: Molecular refractivity, DL: Drug likeness. Abbreviations: BBB: Blood Brain Barrier; HIA: Human Intestinal Absorption, P-GI: P-glycoprotein inhibitor, CIG: Carcinogens, HPT: hepatotoxicity, AOC: Acute oral Toxicity, Caco-2: a model of the intestinal epithelial barrier, AMES: (Salmonella typhimurium reverse mutation assay).
Compound
MW
Log S
Clog P
HBA
HBD
NRB
MR
TPSA
DL
ref
–
>-4
<=5
<=10
<=5
<=10
40–130
less than140
–
1
570
−3.7
4.84
12
6
5
147
192
0.72
2
436
−3.14
5.75
5
3
4
126
90.9
0.70
3
362
−4.1
5.34
4
1
2
108
56
0.48
4
488
−3.65
6.7
6
3
5
142
96
0.42
5
382
−3.96
4.7
5
1
4
110
73
0.44
6
408
−3.9
5.5
5
3
5
116
87
0.56
7
420
−3.78
5.3
6
3
3
119
96
0.51
8
374
−3.31
4.7
5
2
4
106
76
0.41
9
344
−2.88
4.4
4
2
3
100
67
0.57
10
404
−4.0
5.5
5
2
3
118
76
0.53
11
368
−4.16
3.9
6
2
2
100
85
0.38
12
322
−4.05
4.2
4
1
1
92
56
0.51
Compound
BBB
HIA
Caco-2
P-GI
CYP450-2C9
CYP450-2D6
CYP450-3A4
AMES
CIG
HPT
AOC
ref
–
–
No
No
No
No
No
No
No
–
1
Yes
Yes
No
No
No
No
No
No
No
No
2.36
2
Yes
Yes
No
No
inhibitor
No
inhibitor
No
No
No
3.08
3
Yes
Yes
Yes
No
inhibitor
inhibitor
No
No
No
No
2.80
4
Yes
Yes
No
inhibitor
inhibitor
No
No
No
No
No
2.60
5
No
Yes
No
No
No
No
No
No
No
No
2.67
6
No
Yes
No
No
inhibitor
No
inhibitor
No
No
No
2.63
7
No
Yes
No
No
inhibitor
No
No
No
No
No
3.2
8
No
Yes
No
No
inhibitor
No
No
No
No
No
2.36
9
Yes
Yes
No
No
inhibitor
No
No
No
No
No
2.68
10
Yes
Yes
Yes
No
inhibitor
No
No
No
No
No
1.53
12
Yes
Yes
Yes
No
No
No
No
No
No
No
3.1
Molecular weights of most compounds are less than 500 Da; their predicted solubility values are −3.3 to −5.64, suggesting slightly soluble for most compounds. Furthermore, their calculated Log P ranges between 3.9 and 6.7. Moreover, the number of hydrogen bond acceptors and donors in all selected compounds except compound PubChem CID-127039278 were acceptable and obeyed Lipinski's rule of five. Additionally, the molar refractivity value of most compounds was less than 130, while two compounds, 1 and 4, were higher than 140.
Molar refractivity is related to volume and polarizability of the molecule, indicating volume occupied by functional amino acid residues (Prasanna and Doerksen, 2009). Another important descriptor in ADMET prediction is topological polar surface area (TPSA), indicating intestinal permeability of compounds (Fagerholm et al., 2021). So TPSA values higher than 140 results in decreasing permeability of compounds. Based on obtaining results, TPSA values of all compounds were agreeable range except compound 1.
One important molecular descriptor in predicting human oral bioavailbilty and structural stability is the number of Hydrogen bond donors and acceptors (Pourbavarsad et al., 2021; Mishra et al., 2021; Hachem et al., 2022). On the other hand, increasing H-bond donors and acceptors increase selected compounds' solubility. It also leads to essential interactions with key amino acid residues of the receptor. Likewise, too many H-bond donors and acceptors could negatively affect the permeability of compounds (Wang et al., 2022). According to Table 2 number of Hydrogen bond acceptors and donors of all compounds except compound 1 has no violation of the Lipinski rule of five.
Table 3 presents the findings of ADMET properties. Human intestinal absorption of all selected compounds is in the acceptable range, indicating that the compounds could be absorbed appropriately by the intestine. The blood brain barrier (BBB) of most compounds showed moderate absorption.
Therefore, most compounds comply with drug-likeness rules illustrating good absorption and permeability of the compounds. Prediction inhibitors of CYP450 enzymes (3A4, 2D6, and 2C9) exhibited most compounds as non-inhibitors of CYP450 enzymes. They are essential for drug metabolism. Biotransformation of drugs is an important function of Cytochrome P450 isoforms containing heme prosthetic group found abundant in different types of tissues (Zhou et al., 2022). Based upon the data obtained from Table 3 that all selected compounds except compound 4 were revealed as non-inhibitors of the P-glycoprotein inhibitor (P-GI). Inhibition of P-glycoprotein by drugs may decrease drug bioavailability (Lu et al., 2022; Yu et al., 2022; Bai et al., 2021; De Vivo et al., 2016; Lobanov et al., 2008). Moreover, prediction of carcinogenicity, hepatotoxicity and AMES toxicity of selected compounds was performed, indicating all compounds were to be as non-toxic. As a result, it was concluded that most compounds followed the Lipinski rule of five and had good drug-likeness properties and ADMET profiles. Therefore, the selected compounds were determined as appropriate inhibitors of GSK-3β considering for the molecular dynamics (MD) simulation study (Mirzaei et al., 2020).
3.5 Molecular dynamics simulation
To determine molecular dynamics simulation and structural stability of the receptor and geometry changes of the complexes of the modeled Protein and best binding pose, we performed molecular dynamics simulation for 100 ns (Wang et al., 2019). The best pose docking (compound 1) was evaluated to assess modeled Protein's residual flexibility by molecular dynamic study. To analyze the stability of complex of protein structure and compound 1 the RMSD value was calculated. as depicted in Fig. 8A, the RMSD graph provides information about stability state of the structural backbone of the modeled Protein (black) and complex Protein – compound 1 (red) throughout 100 ns MD simulation (Mirzaei et al., 2020).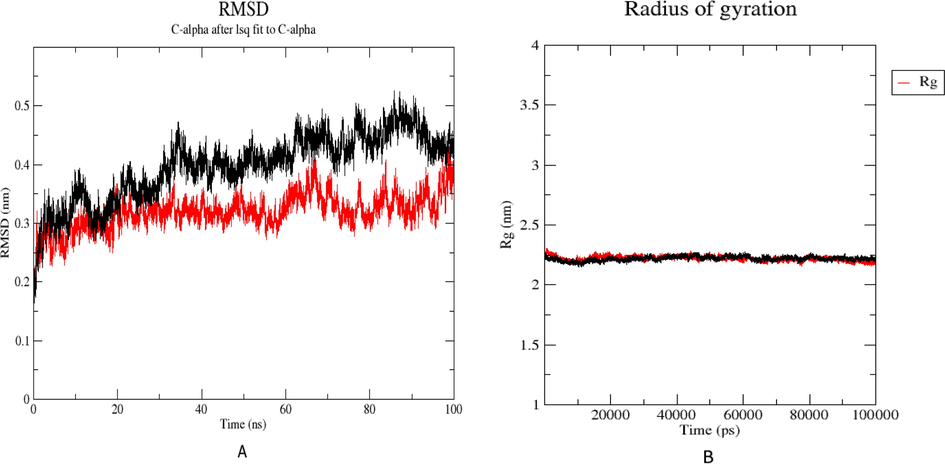
A). RMSD plots of the modeled Protein alone (black) and in complex with compound1 (red), in water at 300 K, during 100 ns MD simulation. B) Radius of gyration (Rg) of the modeled Protein alone (black) and in complex with compound1 (red) in water during 100 ns MD simulation.
The calculated RMSD value of the Protein without inhibitor was 0.46 during 100 ns MD simulation. Following an initial fluctuation, the RMSD graph reached equilibrium at 40 ns. It remained stable throughout the MD simulation, indicating that the system changed to a more perfect equilibrium status than the initial structure. In contrast, the calculated RMSD value of the Protein-compound complex was 0.67, reached stable status at 40 ns, and remained stable during the rest of the MD simulation.
To assess the compactness changes of protein–ligand complex, the radius of gyration is usually applied to indicate geometrical and conformational changes of the protein–ligand complex; furthermore, it displayed the stability status of the complex during 100 ns MD simulation (Mirzaei et al., 2020; Wang et al., 2019).
Fig. 8B demonstrated that the obtained outputs illustrates the radius of gyration value of compound 1 remained stable between 2 and 2.5 nm during the MD simulation. The comparison results of Protein alone and complex of protein–ligand revealed that after binding the compound, folding of complex protein–ligand remained stable from 20 ns throughout MD simulation.
4 Conclusions
Glycogen synthase kinase-3 (GSK-3) is a cytoplasmic serine/threonine protein kinase identified as an essential regulator in several different signaling pathways of pathogenesis like type 2 diabetes, Alzheimer and cardiac, particularly glycogen metabolism. GSK-3β act as a competitive inhibitor that effectively inhibits the ATP binding pocket of the Protein. Inhibition of GSK-3β in cardiac tissue leads to decreased hypertrophy and cardiomyocyte necrosis in ischemic heart disease, cardiac regeneration, and heart failure. In this study, we proposed morin as a natural flavonoid consisting of chromenone moiety inhibited GSK-3β directly, resulting in binding to the ATP and blocking tau phosphorylation in human neurons hence based on similarity with morin as chromenone structure and inhibitor of GSK-3β, 4650 compounds were retrieved from PubChem database.
Molecular docking has been performed to determine hydrophobic, H-bond, and electrostatic interactions between compounds and target structures on the active site. Furthermore, analysis of docking results indicated that compound 1with lowest binding free energy (-11.4 kcal/mol), interacted with key amino acid residues of binding pocket of GSK-3β and effectively inhibited the target. Key amino acid residues including Leu132, Cys199, Ala83, Val70, Val87, Val69, Phe67, Leu188, Lys85, Ser66, Lys183, Gln185, Val69, Lys86, Asp133, and Asp200 play a significant role in the binding pose of GSK-3β. Moreover, in silico ADMET parameters evluatrion revealed that most compounds follow the Lipinski rule of five and had good pharmacokinetic properties and ADMET profiles. Finally, compound 1, as the top-ranked pose subjected to molecular dynamics (MD) to evaluate the stability binding of the complex of Protein-compound. Analysis of RMSD and radius gyration demonstrated complex of the compound 1 and GSK-3β receptor remained stable during 100 ns MD simulation. It can be concluded that flavonoid compounds consisting of chromenone moiety could act as promising molecules for drug design and development of GSK-3β inhibitors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX.. 2015;1(1):19-25.
- [Google Scholar]
- Simulation the adsorption capacity of polyvinyl alcohol/carboxymethyl cellulose based hydrogels towards methylene blue in aqueous solutions using cascade correlation neural network (CCNN) technique. J. Cleaner Prod.. 2022;130509
- [Google Scholar]
- Rapid, Point-of-Care scFv-SERS Assay for Femtogram Level Detection of SARS-CoV-2. ACS Sens.. 2022;7(3):866-873.
- [Google Scholar]
- The attachment-detachment mechanism of ionic/nanoscale/microscale substances on quartz sand in water. Powder Technol.. 2021;394:1158-1168.
- [Google Scholar]
- Penicillamine functionalized B12N12 and B12CaN12 nanocages act as potential inhibitors of proinflammatory cytokines: A combined DFT analysis, ADMET and molecular docking study. Arab. J. Chem.. 2021;14
- [Google Scholar]
- Predicting adsorption behavior and anti-inflammatory activity of naproxen interacting with pure boron nitride and boron phosphide fullerene-like cages. J. Mol. Liq.. 2021;339:116678
- [Google Scholar]
- Spectroscopic, density functional theory, cytotoxicity and antioxidant activities of sulfasalazine and naproxen drugs combination, Arabian. J. Chem.. 2021;14(6):103190
- [Google Scholar]
- Ng Kay Lup A, Norouzi A, Tazikeh Lemeski E, Pishnamazi M, Soltani A, Albadarin AB, Investigations of Adsorption Behavior and Anti-cancer Activity of Curcumin on Pure and Platinum-Functionalized B12N12 Nanocages. J. Mol. Liq.. 2021;334:116516
- [Google Scholar]
- Corynoxine Protects Dopaminergic Neurons Through Inducing Autophagy and Diminishing Neuroinflammation in Rotenone-Induced Animal Models of Parkinson's Disease. Front. Pharmacol.. 2021;12
- [Google Scholar]
- Designing, docking and molecular dynamics simulation studies of novel cloperastine analogues as anti-allergic agents: homology modeling and active site prediction for the human histamine H1 receptor. RSC Adv.. 2020;10(8):4745-4754.
- [Google Scholar]
- In silico screening and identification of potential GSK3β inhibitors. J. Recept. Signal Transduction. 2018;38(4):279-289.
- [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7(1):1-3.
- [Google Scholar]
- Role of molecular dynamics and related methods in drug discovery. J. Med. Chem.. 2016;59(9):4035-4061.
- [Google Scholar]
- Discovery of new GSK-3β inhibitors through structure-based virtual screening. Bioorg. Med. Chem. Lett.. 2018;28(2):160-166.
- [Google Scholar]
- Discovery of new GSK-3β inhibitors through structure-based virtual screening. Bioorg. Med. Chem. Lett.. 2018;28(2):160-166.
- [Google Scholar]
- Screening of potential drug for Alzheimer's disease: a computational study with GSK-3 β inhibition through virtual screening, docking, and molecular dynamics simulation. J. Biomol. Struct. Dyn.. 2020;10:1-5.
- [Google Scholar]
- Acetophenone benzoylhydrazones as antioxidant agents: synthesis, in vitro evaluation and structure-activity relationship studies. Food Chem.. 2018;268:292-299.
- [Google Scholar]
- Potentilla reptans L. postconditioning protects reperfusion injury via the RISK/SAFE pathways in an isolated rat heart. BMC Complement Med Ther.. 2021;21:288.
- [Google Scholar]
- Six new triterpenoids from the root of Potentilla reptans and their cardioprotective effects in silico. Nat. Prod. Res.. 2022;36:2504-2512.
- [Google Scholar]
- Advances in Predictions of Oral Bioavailability of Candidate Drugs in Man with New Machine Learning Methodology. Molecules. 2021;26(9):2572.
- [Google Scholar]
- Gaussian 09, Revision A01. Wallingford, CT: Gaussian Inc; 2009.
- Glycogen synthase kinase 3 inhibition protects the heart from acute ischemia-reperfusion injury via inhibition of inflammation and apoptosis. J. Cardiovasc. Pharmacol.. 2008;52(3):286-292.
- [Google Scholar]
- Discovery and anti-inflammatory evaluation of benzothiazepinones (BTZs) as novel non-ATP competitive inhibitors of glycogen synthase kinase-3β (GSK-3β) Bioorg. Med. Chem.. 2018;26(20):5479-5493.
- [Google Scholar]
- Morin attenuates tau hyperphosphorylation by inhibiting GSK3β. Neurobiology of disease.. 2011;44(2):223-230.
- [Google Scholar]
- Ng Kay Lup A, Arian Nia A, Anti-inflammatory effect of functionalized sulfasalazine boron nitride nanocages on cardiovascular disease and breast cancer: An in-silico simulation. J. Mol. Liq.. 2022;356:119030
- [Google Scholar]
- A fresh look at the Ramachandran plot and the occurrence of standard structures in proteins. Biomol Concepts. 2010;1(3–4):271-283.
- [Google Scholar]
- Role of glycogen synthase kinase-3β in cardioprotection. Circ. Res.. 2009;104(11):1240-1252.
- [Google Scholar]
- Natural and synthetic bioactive inhibitors of glycogen synthase kinase. Eur. J. Med. Chem.. 2017;5(125):464-477.
- [Google Scholar]
- Crystal structure of GSK3β in complex with the flavonoid, morin. Biochem. Biophys. Res. Commun.. 2018;504(2):519-524.
- [Google Scholar]
- The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res.. 2015;116(1):138-149.
- [Google Scholar]
- Two-Dimensional Material-Based Electrochemical Sensors/Biosensors for Food Safety and Biomolecular Detection. Biosensors-Basel. 2022;12(5)
- [Google Scholar]
- Radius of gyration is indicator of compactness of protein structure] Mol Biol (Mosk).. 2008;42(4):701-706.
- [Google Scholar]
- Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinf.. 2003;50(3):437-450.
- [Google Scholar]
- Met1-specific motifs conserved in OTUB subfamily of green plants enable rice OTUB1 to hydrolyse Met1 ubiquitin chains. Nat. Commun.. 2022;13(1):4672.
- [Google Scholar]
- Synthesis, cytotoxic activity and docking study of two indole-chalcone derivatives. J. Mazandaran Univ. Med. Sci.. 2017;27(154):12-25.
- [Google Scholar]
- Indole-derived chalcones as anti-dermatophyte agents: In vitro evaluation and in silico study. Comput. Biol. Chem.. 2020;84:107189
- [Google Scholar]
- Synthesis, Pharmacological Evaluation, and In-Silico Studies of Thiophene Derivatives. Oncologie. 2021;23(4):493-514.
- [Google Scholar]
- AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30(16):2785-2791.
- [Google Scholar]
- Nitrogen oxidation and carbon removal from high strength nitrogen habitation wastewater with nitrification in membrane aerated biological reactors. J. Environ. Chem. Eng.. 2021;9(5):106271
- [Google Scholar]
- Topological polar surface area: a useful descriptor in 2D-QSAR. Curr. Med. Chem.. 2009;16(1):21-41.
- [Google Scholar]
- Identification of COX-2 inhibitors via structure-based virtual screening and molecular dynamics simulation. J. Mol. Graph. Model.. 2018;83:138-152.
- [Google Scholar]
- Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions. Eur. J. Med. Chem.. 2018;20(144):843-858.
- [Google Scholar]
- Serine adsorption through different functionalities on the B12N12 and Pt-B12N12 nanocages. Mater. Sci. Eng., C. 2018;92:216-227.
- [Google Scholar]
- Improvement of anti-inflammatory and anticancer activities of poly(lactic-co-glycolic acid)-sulfasalazine microparticle via density functional theory, molecular docking and ADMET analysis. Arabian J. Chem.. 2022;15:103464
- [Google Scholar]
- Ng Kay Lup A, Javan M, Soltani A, Khandoozi SR, Arian Nia A, Tavassoli S, Rahman ML, Sani Sarjadi M, Sarkar SM, Su C-H, Chinh Nguyen H, Molecular docking and Density functional theory simulation: Improved anti-inflammatory and anticancer properties of celecoxib using Zinc oxide and magnesium oxide nanoclusters. Arabian J. Chem.. 2021;15:103568
- [Google Scholar]
- Roles of glycogen synthase kinase-3 (GSK-3) in cardiac development and heart disease. Journal of UOEH.. 2018;40(2):147-156.
- [Google Scholar]
- Competitive-Type Pressure-Dependent Immunosensor for Highly Sensitive Detection of Diacetoxyscirpenol in Wheat via Monoclonal Antibody. Anal. Chem. (Washington). 2020;92(5):3563-3571.
- [Google Scholar]
- Role of GSK-3 in Cardiac Health: Focusing on Cardiac Remodeling and Heart Failure. Curr. Drug Targets 2021
- [Google Scholar]
- Porous Organic Polymer-Derived Nanopalladium Catalysts for Chemoselective Synthesis of Antitumor Benzofuro[2,3-b]pyrazine from 2-Bromophenol and Isonitriles. Org. Lett.. 2019;21(13):4929-4932.
- [Google Scholar]
- Ng Kay Lup A, Enayati A, Mirzaei H, Soltani A, Su C-H, Nguyen HC, Electrostatic interaction assisted Ca-decorated C20 fullerene loaded to anti-inflammatory drugs to manage cardiovascular disease risk in rheumatoid arthritis patients. J. Mol. Liq.. 2022;350:118564
- [Google Scholar]
- Rh(iii)-catalyzed synthesis of dibenzo[b, d]pyran-6-ones from aryl ketone O-acetyl oximes and quinones via C-H activation and C–C bond cleavage. RSC Adv.. 2022;12(23):14435-14438.
- [Google Scholar]
- admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019;35(6):1067-1069.
- [Google Scholar]
- Medicinal Properties Of Bioactive Compounds And Antioxidant Activity In Durio Zibethinus. Malaysian Journal Of Sustainable Agriculture. 2021;5(2):82-89.
- [Google Scholar]
- A redox-active perylene-anthraquinone donor-acceptor conjugated microporous polymer with an unusual electron delocalization channel for photocatalytic reduction of uranium (VI) in strongly acidic solution. Appl. Catal. B Environ.. 2022;314
- [Google Scholar]
- The crystal structure of (E)-1-(4-aminophenyl)-3-(p-tolyl)prop-2-en-1-one, C16H15NO. Zeitschrift für Kristallographie. New Cryst. Struct.. 2022;237(3):385-387.
- [Google Scholar]
- Usefulness of enzyme-free and enzyme-resistant detection of complement component 5 to evaluate acute myocardial infarction. Sensors Actuators B: Chem.. 2022;369:132315.
- [Google Scholar]




