

Translate this page into:
Comprehensive characterization of the in vitro and in vivo metabolites of xylocarpin H using UHPLC-Q-TOF-MS/MS
⁎Corresponding authors at: Hebei Key Laboratory of Innovative Drug Development and Evaluation, School of Pharmaceutical Sciences, Hebei Medical University, Shijiazhuang 050017, China. shixiaowei@hebmu.edu.cn (Xiaowei Shi), panzhenhua@hebmu.edu.cn (Zhenhua Pan)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Xylocarpin H, a limonoid from the Xylocarpin granatum, exhibits significant biological activities, including antioxidant, antiviral, analgesic, hypnotic, antimalarial, and antidepressant effects, demonstrating considerable potential for drug development. However, few studies have been reported to identify and classify active metabolites responsible for such excellent biological activities. In this study, we utilized ultra high-performance liquid chromatography/quadrupole time-of-flight mass spectrometry (UHPLC–Q–TOF–MS/MS) coupled with multiple data postprocessing techniques to rapidly identify in vivo and in vitro metabolites of xylocarpin H. Consequently, a total of 32 metabolites (23 in phase I and 9 in phase II) of xylocarpin H were detected both in rat feces, urine, bile, blood, intestinal flora, and rat and human liver microsomes. The metabolic pathways of xylocarpin H mainly involve hydrolysis, oxidation, reduction, addition, glucose conjugation, glucuronide conjugation, and sulfate conjugation. The metabolite M15 of xylocarpin H was synthesized by ester hydrolysis and its structure was determined by nuclear magnetic resonance spectroscopy. This study elucidates the metabolic pathways of xylocarpin H and provides insights into the origin of its pharmacological activity.
Keywords
Xylocarpin H
UHPLC-Q-TOF-MS/MS
Metabolite
Characterization
In vivo
In vitro

1 Introduction
The mangrove genus Xylocarpus granatum (X. granatum), commonly found in coastal regions, has a history of traditional medicinal use for treating fever, cholera, and diarrhea. Limonoids, classified as secondary plant metabolites, are the predominant constituents of X. granatum. (Cui et al., 2007). Our recent research has identified limonoid xylocarpin H as a significant component of X. granatum, with notable presence in both the fruits (0.04 %) and seeds (0.05 %) of the plant. (Shi et al., 2017). In our previous study, we established a superimposed multiple product ions liquid chromatography tandem triple-quadrupole mass spectrometry (SMPI-UHPLC-QTOF/MS) method for the determination of xylocarpin H in rat plasma(Zhang et al., 2022). The results show that xylocarpin H has better pharmacokinetic parameters than the limonoid nimbolide and limonin. Xylocarpin H has also exhibited significant biological activity in previous studies, including anticancer properties by inducing apoptosis, inhibiting cell proliferation, and suppressing angiogenesis in various cancer cell lines with selective toxicity towards cancer cells(Tan and Luo 2011). It also has broad-spectrum antimicrobial effects against bacteria and fungi, including drug-resistant strains, by disrupting microbial membranes and inhibiting critical enzymes necessary for pathogen survival. Additionally, xylocarpin H can suppress inflammatory mediators such as cytokines (e.g., TNF-α, IL-6) and enzymes (e.g., COX-2), and it exhibits antioxidant properties, reducing oxidative stress and inflammation. Furthermore, it protects neuronal cells from oxidative damage and reduces is chemia-reperfusion injury in heart cells, demonstrating potential benefits for neurodegenerative and cardiovascular diseases. (Yin et al., 2015). As a result, the wide range of biological activities of xylocarpin H makes it an ideal candidate for comprehensive metabolic studies. Investigating its metabolism can provide insights into its bioavailability, pharmacokinetics, and the identification of active metabolites, essential for developing effective therapeutic agents and dosing regimens. Their defined biological activity is closely connected with the furan ring and the multi-carbonyls in their structures. In vivo and in vitro biotransformation may influence their structures and further change their effects. Drug metabolism plays a pivotal role in the process of drug elimination(Stanley 2024). In vivo, the majority of chemical constituents present in natural products undergo extensive biotransformation, resulting in the generation of numerous metabolites. By investigating the metabolic pathways and metabolites associated with Chinese herbal medicine, we can elucidate its therapeutic efficacy, assess its safety and toxicity profiles, and optimize its medicinal administration regimen(Liu et al., 2024). These research findings hold significant guiding implications for the development and utilization of natural products. Therefore, to better understand the biological functions of limonoid, it is necessary to analyze the metabolic characteristics of limonoid more precisely. However, so far, the metabolic profile of xylocarpin H remains incompletely understood. Herein, we anticipate that our developed strategy will prove invaluable for elucidating the metabolism of various bioactive components in limonoid plants. Furthermore, these metabolic findings are expected to contribute significantly to future studies on the safety and efficacy of xylocarpin H.
In recent years, ultra high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC–Q–TOF–MS/MS) has been widely used in the rapid and accurate qualitative analysis of biological samples. Through liquid chromatography separation, taking advantage of the high sensitivity, high selectivity, and fast and accurate molecular weight qualitative advantages of high-resolution time-of-flight mass spectrometry, combined with characteristic secondary fragments, the known substances were analyzed and identified by liquid chromatography mass spectrometry (LC–MS) of biological samples. Comprehensive and rapid structural inference of major to secondary metabolites in vitro and in vivo. Even in the absence of corresponding standards, the drug metabolites can be qualitatively analyzed with high accuracy(Heiles 2021, Shao et al., 2021).
Therefore, the aim of this study was to develop a rapid and efficient UHPLC–Q–TOF–MS/MS based method for the identification and characterization of xylocarpin H metabolites in various biological matrices, including rat feces, urine, bile, blood, intestinal flora, and rat and human liver microsomes. Additionally, we present here the first proposed metabolic pathway for limonoids derived from X. granatum. The present findings are expected to establish a robust groundwork for future investigations on limonoids in X. granatum, thereby facilitating the elucidation of its pharmacological activity and offering novel insights into the underlying pharmacological mechanism.
2 Materials and methods
2.1 Instruments and conditions
An ultra high-performance liquid chromatography (UHPLC) system (Shimadzu Corp, Kyoto, Japan), equipped with an autosampler, a binary solvent delivery system and a column oven, was coupled to a Triple TOF™ 5600 + system (SCIEX, CA, USA) for analysis. Chromatographic separation was performed on a Kinetex-C18 column (100 mm × 2.1 mm, 3 μm) at 40 °C with a sample injection volume of 5 μL. The mobile phase consisted of water (A; containing 0.1 % formic acid-1 mM ammonium acetate) and acetonitrile (B), delivered at a flow rate of 400 μL/min using the following gradient elution program: 0–2 min, 5 % B; 2–8 min, 5–30 % B; 8–35 min, 30–45 % B; and 35–45 min, 45–95 % B.
Mass spectrometric analysis was operated in positive ion mode using a DuoSpray ESI source with a spray voltage of 5.5 kV and a temperature of 550 °C. The curtain gas, nebulizer gas and heater gas pressures were 35, 55 and 55 psi, respectively. The TOF-MS full scan mode acquisition range was 100–1000 Da, the declustering voltage (DP) was 80 V, the collision energy (CE) was 5 eV, accumulation time was 0.2 s. The TOF-MS/MS scan mode acquisition range was 50–1000 Da, with a declustering voltage (DP) of 80 V, a collision energy (CE) of 35 eV, an extended energy voltage (CES) of ± 15 eV, and an accumulation time of 0.1 s. MS data were acquired by information dependent acquisition (IDA) in combination with a mass defect filter (MDF) and dynamic background subtraction (DBS). A calibration delivery system was utilized for the automatic regulation of MS and MS/MS, ensuring that the error range for the acceptable mass was ≤ 5 ppm. The NMR spectra were recorded on Bruker Avance III 600 spectrometers (Bruker, USA) with tetramethylsilane (TMS) or deuterated solvent as the internal standard. Preparative HPLC was performed with OCTOPUS HS-2900P (Agela Technologies Ltd., Tianjin, China) equipped with an UV dual wavelength detector. Preparative HPLC was carried out using column (Luna 10 mm × 250 mm, 5 um C18 100 Å).
2.2 Chemicals and materials
Xylocarpin H (purity > 95 %, chemical structure was shown in Fig. 1) was isolated from X. granatum, which accumulated from Hainan Island in southern China and its structure was entirely characterized and affirmed by positive mode high resolution mass spectrometry (HRMS) and 1H and 13C nuclear magnetic resonance (NMR) spectroscopy as in our previous studies(Wu et al., 2014). Purified water was obtained from a Milli-Q water purification system (MA, USA), while acetonitrile, methanol, ammonium acetate, acetic acid, and formic acid were HPLC grade (Merck Life Science, Milano, Italy).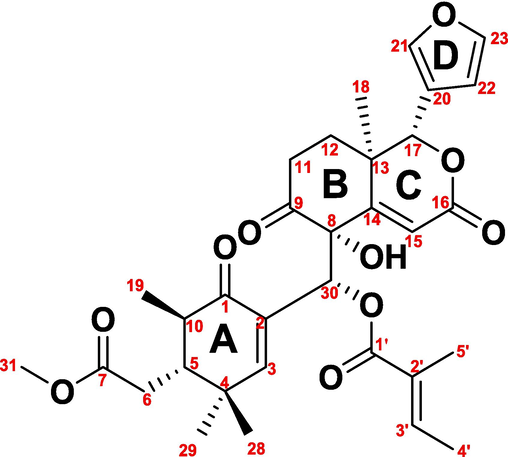
Chemical structure of xylocarpin H from Xylocarpus granatum.
Human liver microsomes (lot no. 2010065) and rat liver microsomes (lot no. 1910100) were purchased from Shanghai Baiquan Biotechnology Co., Ltd. Phosphate-buffered salt solution (PBS, pH=7.4, lot no. 20200822), dimethyl sulfoxide (DMSO, lot No. 2018062), Tween-80(lot No. BCBT0817), PEG 300(lot No. HY-Y0873) and reduced coenzyme II tetrasodium (NADPH-4Na, lot no.1214L022) were purchased from Beijing Solaibao Technology Co., Ltd. L-ascorbic acid (lot no.108345), L-cysteine (lot no. 1013057), peptone (lot no. 163850), tryptone (lot no.184310) and nutrient agar (lot no.NAO01) were purchased from Beijing AoBoXing Bio-tech Co., Ltd. Potassium carbonate (K2CO3) (lot no.20180201), magnesium sulfate (MgSO₄) (lot no.20190401) sodium carbonate (Na2CO3) (lot no.20180421), hydrogen chloride (HCl) (lot no.20190621), sodium hydroxide (NaOH lot no.20190824), disodium hydrogen phosphate (Na2HPO4) (lot no.20190716), potassium dihydrogen phosphate (KH2PO4) (lot no.20190130), sodium chloride(NaCl) (lot no.20181014), sodium azide (NaN3) (lot no. DSZ-042251) and calcium chloride(CaCl2) (lot no.20191008) all of analytic grade, were purchased from Tianjin Yongda Chemical Reagent Co., Ltd.
2.3 Animals and drug administration
Thirty-six male Sprague–Dawley (SD) rats (220 ± 10 g) were purchased from the Animal Experimental Center of Hebei Medical University (Shijiazhuang, China) and were housed in an environmentally controlled animal room (temperature 22–25 °C, relative humidity 55–60 %, 12 h light/dark cycle) with freely available water and food for 5 days. All of the experimental protocols were approved by the Laboratory Animal Ethical and Welfare Committee of Hebei Medical University (IACUC-Hebmu-2022028). Thirty-six rats were randomly divided into six groups: three blank groups (blank plasma sample group, blank bile sample group, and blank urine and fecal sample group) and three experimental groups (experimental plasma sample group, experimental bile sample group, and experimental urine and fecal sample group). All rats were fasted overnight with free access to water prior to xylocarpin H administration. The experimental group was administered xylocarpin H powder dissolved in a 0.5 % sodium carboxymethylcellulose (CMC-Na) solution at a single dosage of 50 mg/kg, as per previous studies(Yin et al., 2015, Zhang et al., 2022). An equal 0.5 % CMC-Na solution without xylocarpin H was orally given to the blank groups.
2.4 Sample collection and pretreatment in vivo
Plasma samples were collected via retro-orbital puncture using a glass capillary from the fossa orbitalis vein at 0.083, 0.25, 0.5, 1, 2, 4, 8, 12 and 24 h post-dosing. The blood was collected into heparinized plastic tubes and centrifuged at 4500 g for ten minutes. All plasma samples were then combined into one sample and frozen at −80 °C. Blank plasma from unadministered rats was collected in the same manner. For bile sample collection, the rats were anesthetized with urethane physiological saline solution (1.5 − 2 g/kg) via intraperitoneal injection after oral administration, and bile was collected continually via a cannula inserted into the bile duct during the time intervals of 0–4, 4–8, and 8–24 h after dose. Unadministered rats underwent the same treatment, and the bile collected was used as a blank bile sample. The samples were tightly sealed with parafilm and properly stored at −80 °C until analysis. Prior to administration of the medication, rats were housed individually in clean metabolic cages lined with sterilized filter paper to collect urine and feces. Samples of urine and feces were collected at 0–4 h, 4–8 h, 8–12 h, and 12–24 h. Following defecation, fecal samples were immediately transferred using sterile forceps into cryovials. These samples were then rapidly frozen in liquid nitrogen and stored at −80 °C until analysis. Urine was collected using clean, dry, sterile collection tubes, which were kept on ice. A small amount of sodium azide (0.05 % wt/vol) was added to inhibit microbial growth. Subsequently, the urine samples were frozen in the same manner and stored at −80 °C until analysis. Urine and feces of unadministered rats were collected in the same way as blank controls. 2 mL of plasma, 2 mL of bile, and 2 mL of urine were used for protein precipitation with 3 times the volume of acetonitrile, vortexed for 5 min, and centrifuged at 15,000 g for 10 min. 2 g fecal samples were extracted by ultrasonic treatment with three volumes of acetonitrile for 30 min followed by centrifugation at 15,000 g for 10 min. Subsequently, the supernatant of each sample was concentrated via vacuum centrifugation, redissolved in 50 % acetonitrile, and analyzed using the UHPLC–Q–TOF–MS/MS system.
2.5 Sample preparation and treatment in vitro
2.5.1 Sample preparation
Xylocarpin H was prepared as a stock solution at a concentration of 1 mg/mL by dissolving xylocarpin H in a mixed clarified solution (10 % DMSO + 40 % PEG 300 + 5 % Tween 80 + 45 % PBS).
2.5.2 Preparation of rat intestinal flora culture solution and incubation reaction system
To prepare an anaerobic medium, the following ingredients were used: 1 g peptone, 1 g beef paste, 1 g nutrient agar, 0.5 g L-cysteine, 37.5 mL of a 0.78 % aqueous solution of disodium hydrogen phosphate, 37.5 mL of a 0.47 % potassium dihydrogen phosphate solution, 1.18 % sodium chloride, 1.2 % ammonium sulfate, 0.12 % calcium chloride, 0.25 % magnesium sulfate mixed solution, 50 mL of an 8 % aqueous solution of sodium carbonate, and 2 mL of a 25 % aqueous solution of L-ascorbic acid. These ingredients were dissolved in 1000 mL of purified water and the pH was adjusted to 7.1–7.2 using 1 mol/L HCl. The anaerobic medium was then sterilized by autoclaving. The experiments were classified into three groups: the blank group, control group, and experimental group. In the experimental group, the newly produced rat feces were gathered from Section 2.3 blank rat, and a suspension was created by combining 2 g of feces with normal saline at a ratio of 1 part mass to 4 parts volume. Following centrifugation for 10 min at a speed of 2,000 g, the resulting liquid above the sediment was extracted and combined with an anaerobic culture medium at a ratio of 1 part liquid to 9 parts anaerobic medium. The final mixed medium is the rat intestinal flora culture solution. In the experimental group, 1 mL of xylocarpin H stock solution at a concentration of 1 mg/mL in Section 2.5.1 was mixed with 1 mL of the aforementioned intestinal flora culture medium. The blank group did not receive any drug treatment, while in the control group, anaerobic medium was used instead of intestinal flora culture medium. All was incubated at room temperature (37 °C) for 24 h with collection of incubation solutions at intervals of 0–6 h, 6–12 h and 12–24 h. The collected samples were centrifuged at a speed of 15,000 g for 10 min followed by combination and drying under nitrogen flow. Subsequently, the dried supernatant was redissolved in a mixture containing acetonitrile (50 %) and analyzed after vortexing for five minutes(Yang et al., 2022).
2.5.3 Rat and human liver microsomal incubation reaction system
For phase I metabolism evaluation, a 200 μL incubation mixture containing 1.0 mg/mL rat liver microsomes, human liver microsomes, 100 μmol/L xylocarpin H, and NADPH-generating products was prepared in K2HPO4 buffer. After incubation for 60 min at 37 °C, the reaction was stopped with 600 μL of acetonitrile. For phase II metabolism analysis, a 200 μL K2HPO4 buffer incubation system composed of 1.0 mg/mL rat liver microsomes, human liver microsomes, 25 μg/mL alamethicin, 100 μmol/L xylocarpin H, and UDPGA-generating products was processed in a metabolic shaker and preincubated for 5 min at 37 °C. The reaction was incubated for 120 min and was terminated with 600 μL of acetonitrile. To evaluate cascade metabolism, a 200 μL K2HPO4 buffer incubation mixture composed of 1.0 mg/mL rat and human liver microsomes, 100 μmol/L xylocarpin H, and 25 μg/ mL alamethicin was preincubated for 20 min at 37 °C. Both phase I and II reactions were concurrently initiated after addition of β- NADPH and UDPGA. The incubation lasted for 120 min at 37 °C and was terminated with 600 μL of ice acetonitrile. After samples were vortexed for 5 min and centrifuged for 10 min at 12,000 g, all the organic layers were gathered and air-dried with nitrogen; all the driedsamples were redissolved in 100 μL of acetonitrile (50 %) for analysis. Additionally, blank samples (no xylocarpin H) and control samples (no NADPH or UDPGA) were prepared in parallel(Zhou et al., 2019).
2.6 Synthesis of crucial metabolites
The M15 metabolite of xylocarpin H was synthesized to elucidate its structural characteristics. Xylocarpin H (100 mg) was dissolved in methanol (10 mL), followed by the addition of 35 mg of potassium carbonate, and stirred at room temperature for 2 h. Subsequently, a solution containing 2 mL of acetic acid was added to adjust the pH to an acidic level. The resulting mixture was concentrated under vacuum, and the oily liquid obtained was dissolved in chromatographic methanol and filtered using a 0.45 μm oil film(Schuppe et al., 2019). The pure white solid of metabolite M15 (8 mg) was obtained by preparative liquid chromatography system. The separated mobile phase consisted of water (mobile phase A) and acetonitrile (mobile phase B), and the chromatographic column used was a (Luna 10 mm × 250 mm, 5 um C18 100 Å). The gradient elution procedure was as follows: 0–10 min, 35–55 % B, and 10–50 min, 55–70 % B. The flow rate was 3 mL/min, and the detection wavelength was 215 nm. The structures of the synthesized compounds were identified by HR-MS, 1H NMR, 13C NMR, 1H–1H correlation spectroscopy (COSY), heteronuclear single-quantum correlation (HSQC), and heteronuclear multiple-bond correlation (HMBC) with a Bruker Avance 600 (Bruker BioSpin, Rheinstetten, Germany). The chemical reaction equation is shown in Fig. 2.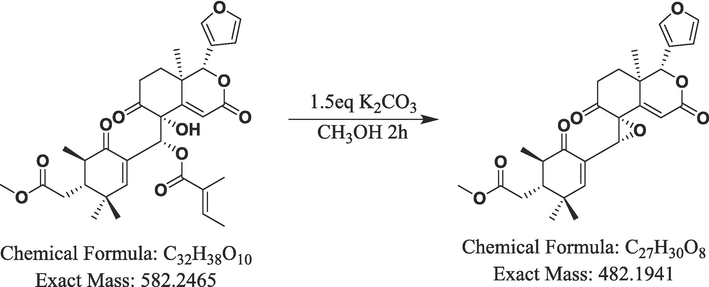
Synthesis route of metabolite M15.
3 Results
3.1 Analytical strategy on metabolite analysis.
The analysis strategy(Kwak et al., 2022) of this experiment is divided into three steps. Initially, we collected UHPLC–Q–TOF–MS/MS data and used multiple mass defect filtering (MMDF) combined with dynamic background subtraction (DBS) to trigger IDA so that MS can quickly and effectively distinguish interfering ions from metabolite ions and obtain accurate MS and MS/MS information. In the second step, using xylocarpin H as a template, a variety of data postprocessing techniques were used to screen the metabolites, including multiple mass defect filtering (MMDF), neutral filtration (NLF), extracted ion chromatogram (XIC), and product ion filtration (PIF), to obtain the secondary mass spectrometry information of potential metabolites. Finally, based on the exact mass number of metabolites, combined with the cleavage law of xylocarpin H and the metabolic mode predicted by software, the structure of the possible metabolites was inferred, and the isomers in the metabolites were distinguished with the help of the Clog P value in the software. The metabolites with higher Clog P values appeared later in the reversed-phase chromatography system.
3.2 Mass fragmentation pathways of xylocarpin H (M0)
In positive ion mode, the parent drug (xylocarpin H, M0) with a molecular formula of C32H8O10 eluted at 35.29 min and exhibited a quasimolecular ion peak at m/z 583.2534. The corresponding ESI-MS2 spectrum featured product ions with m/z 483.1989 [M+H-H2O-C5H6O]+, 465.1888 [M+H-H2O-C5H6O-H2O]+, 369.1673, 345.1336, 321.1494, 263.0909, 245.0802, 239.0698, 217.0850, and 83.0512.
It has been suggested that there are two possible cleavage pathways. The first is that hydrogen protons in the solvent attack the hydroxyl groups in the structure to form hydroxyl cations [M+H]+ m/z 583.2534. From this position, a molecule of water is removed to obtain fragment ions of m/z 565.2513[M+H-H2O]+. Then the proton is transferred to the ester group of the side chain, and the ester bond is broken to remove the acyl group, resulting in the fragment ion of m/z 483.1989[M+H-H2O-C5H6O]+ and the tigloyl fragment ion m/z 83.0512. After proton progressive transfer, a molecule of water is removed from the structure to obtain a more stable sheet ion of m/z 465.1888[M+H-H2O-C5H6O-H2O]+, and then the fragment ion of m/z 369.1673 is obtained by RDA cleavage.
As a second approach, hydrogen protons are first attacked by the structure hydroxyl groups, forming [M+H]+ m/z 583.2534, which are then removed by a cyclohexenone ring and a CO molecule to obtain fragments m/z 345.1336 from the structure. Furthermore, [M+H]+ m/z 583.2534 can also be used to remove cyclohexanone fused rings to obtain fragment ions with m/z 321.1494 and m/z 263.0909.
When the ester bond of the fragment ion of m/z 321.1494 was broken, the fragment ion m/z 239.0698 and the tigloyl fragment ion m/z 83.0512 were obtained. The fragment ion of m/z 263.0909 removes a molecule of water to obtain the fragment ion of m/z 245.0802, and then the proton continues to transfer to the carbonyl group on the ring, so that the system removes a molecule of CO to obtain a more stable fragment ion of m/z 217.0850(Kumar et al., 2018). The MS/MS spectra and fragmentation pathways of M0 are presented in Fig. 3.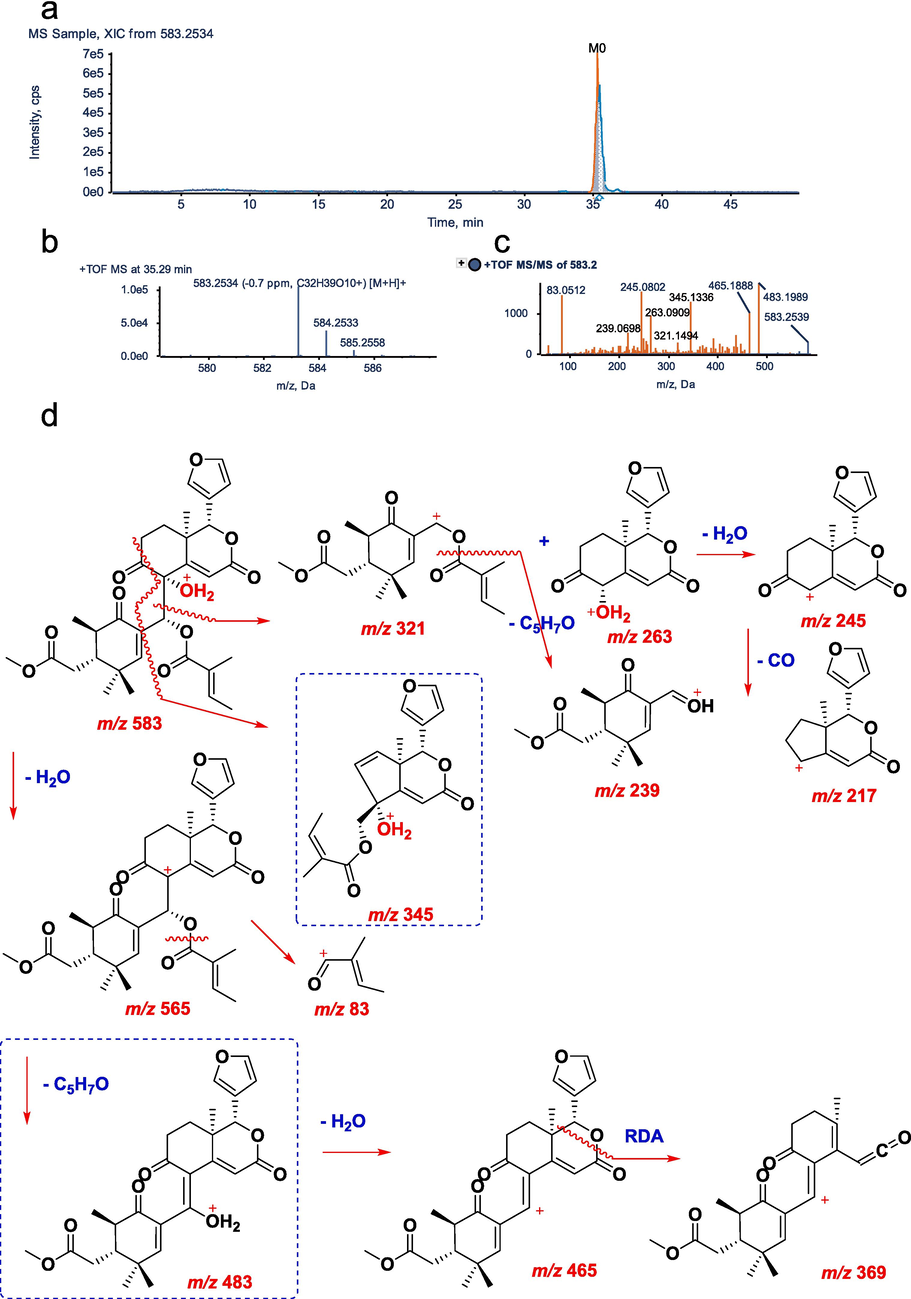
UHPLC–Q–TOF–MS/MS identification of parent compound M0. a, XIC; b, MS spectrum; c, MS/MS spectrum; d, fragmentation pattern.
3.3 Identification of metabolites in vitro and in vivo
According to the metabolite identification procedure, 32 metabolites of xylocarpin H were identified (23 metabolites in phase I and 9 metabolites in phase II), including 19 in feces, 5 in urine, 13 in bile, 5 in plasma, 7 in human liver microsomes, 8 in rat liver microsomes, and 15 in rat intestinal flora. The related information for xylocarpin H and its metabolites is presented in Table 1. The total ion chromatograms of 32 metabolites and the parent compound are displayed in Figure S1 and Figure S2, respectively. The metabolic pathways in vivo and in vitro are depicted in Fig. 4 and Figure S3, respectively. In the main text, representative metabolites of M6, M15 in vivo and V1 in vitro were selected for identification (Figs. 5-9). More detailed descriptions of the metabolite (M1-M5, M7-M14, M16-M22, V2-V10) identification are provided in the Supporting Information (Figure S4-S29). +: Detected −: Undetected P: Plasma U: Urine B: Bile F: Feces HL: Human liver microsome RL: Rat liver microsome RF: Rat gut flora.
Compound
IDFormula
Theoretical
Mass
[M+H]+
Measured
Mass
[M+H]+
Error
(ppm)tR
(min)MS/MS
fragmentsComposition shift
ClogP
F
U
B
P
HL
RL
RF
M0
C32H38O10
583.2547
583.2534
−0.7
35.29
483.1989,465.1888,345.1336,
263.0909,245.0802,239.0698–
2.2394
+
+
+
+
+
+
+
M1
C32H40O10
585.2694
585.2671
−4.0
36.31
483.1995,465.1903,347.1469,
263.0903,85.0655,57.0736Hydrogenation
2.4241
+
−
+
+
−
+
+
M2
C32H37O11
599.2480
599.2479
−1.2
28.83
581.2380,465.1906,263.0895,
245.0816,99.1013Oxidation
0.7564
+
−
−
+
−
−
−
M3
C32H36O11
597.2322
597.2301
−4.2
20.80
497.1807,423.1426,369.1547,
341.1541,267.1376Ketone Formation
0.6834
+
−
+
+
−
−
+
M4
C32H36O11
597.2322
597.2301
−4.2
20.80
497.1807,423.1426,369.1547,
341.1541,267.1376Ketone Formation
0.6834
+
−
+
+
−
−
+
M5
C32H40O11
601.2647
601.2648
0.7
16.73
519.2225,503.2176,485.2170,
265.2071Internal Hydrolysis
2.0094
−
+
−
−
−
+
−
M6
C27H32O9
501.2119
501.2095
−4.9
19.35
483.1997,465.1908,439.1959,
391.1523,257.0808,177.0905Loss of C5H6O
0.2014
+
+
+
−
−
+
−
M7
C32H37O11
487.1962
487.1946
−3.4
12.73
469.1855,451.1929,391.1926,
237.1112,177.0916,149.0952Loss of C5H6O and CH2
0.2108
+
+
−
−
+
+
−
M8
C27H33O10
519.2227
519.2206
−3.6
13.67
501.2137,483.2008,267.1227 217.0862,149.0958
Loss of C5H6O+Internal Hydrolysis
−0.0285
+
−
+
−
−
−
−
M9
C27H33O9
503.2274
503.2265
−2.1
20.31
485.2171,307.1526,251.1276, 177.0910,149.0960
Loss of C5H6O2 + Internal Hydrolysis
0.6295
+
−
+
−
−
−
+
M10
C26H31O10
505.2068
505.2078
1.9
10.61
487.1962,469.1857,453.1908, 149.0955,95.0853
Loss of C5H6O and CH2 + Internal Hydrolysis
−0.0191
+
−
−
−
+
−
−
M11
C27H32O10
517.2078
517.2058
−4.0
18.45
499.1949,483.1810,453.1893, 425.1959,261.0806,189.0920
Loss of C5H6O+Oxidation
−1.2741
+
−
−
−
−
−
−
M12
C27H32O10
517.2078
517.2058
−4.0
21.12
499.1949,483.1810,453.1893, 425.1959,261.0806,189.0920
Loss of C5H6O+Oxidation
−0.1919
+
−
−
−
−
−
−
M13
C27H32O10
517.2078
517.2058
−4.0
21.45
499.1949,483.1810,453.1893, 425.1959,261.0806,189.0920
Loss of C5H6O+Oxidation
−0.1919
+
−
−
−
−
−
−
M14
C28H33O9
515.2273
515.2251
−4.8
12.16
501.2026,497.2140,339.1939, 199.0213
Loss of C5H6O+Methylation
0.2014
−
−
−
+
−
−
+
M15
C27H30O8
483.1964
483.1944
−4.2
16.27
465.1990,245.0804,223.1159, 217.0850,189.0898,175.0749
Loss of C5H6O+Loss of Water
1.5798
+
+
+
−
−
−
−
M16
C27H32O11S
565.1740
565.1725
−3.8
11.56
483.1995,465.1892,387.1780
95.0503Loss of C5H6O2 + Sulfate Conjugation
0.8218
+
−
+
−
−
−
+
M17
C33H40O14
661.2494
661.2472
−4.5
11.16
485.2188,467.2166,437.2049
331.1614Loss of C5H6O2 + Glucuronidation
−0.6976
−
+
−
−
−
−
−
M18
C28H32NO9
528.2227
528.2207
−4.0
17.81
510.2216,492.2225,464.2234
266.1376Loss of C5H6O2 and CH2 + Glycine
0.4017
+
−
−
−
−
−
−
M19
C28H32NO9
528.2227
528.2215
−2.5
18.13
511.1963,471.2013,453.1908
320.2055,154.1235Loss of C5H6O2 and CH2 + Glycine
−1.6199
+
−
−
−
−
−
−
M20
C29H35NO9
542.2385
542.2384
−0.2
16.29
525.2119,485.2170,482.2173
304.1175,274.1074,156.0654Loss of C5H6O2 + Glycine
0.6406
+
−
+
−
−
−
−
M21
C30H36NO10S
604.2211
604.2182
−4.8
18.84
587.1945,558.2334,446.2863
342.1006,218.0478,188.0375Loss of C5H6O+Cysteine
0.3294
+
−
−
−
−
−
−
M22
C30H36NO9S
588.2304
588.2276
−4.8
15.56
571.1986,542.2385,485.2070
366.0855Loss of C5H6O2 + Cysteine
0.8431
+
−
+
−
−
−
−
V1
C32H36O11
613.2280
613.2288
1.3
5.71
483.2014, 457.1855,193.0693
Oxidation
1.1934
−
−
−
−
+
+
−
V2
C37H32O7
469.3195
469.2212
−2.5
28.89
297.1125, 141.0550
Loss of C5H6O3
2.354
−
−
−
−
−
−
+
V3
C26H30O7
455.2064
455.2047
−3.8
34.93
437.1963, 363.1595, 237.1480, 217.0855
Loss of C5H6O3 and CH2
2.3635
−
−
−
−
−
−
+
V4
C32H36O10
581.2381
581.2368
−2.2
36.22
481.1851, 339.1809, 83.0489
Hydrogenation
2.2394
−
−
−
−
+
+
+
V5
C31H34O11
581.2174
583.2175
−0.2
0.6
519.2021, 327.1599, 181.0862, 129.0913
Hydrogenation Internal Hydrolysis and loss of CH2
1.8634
−
−
−
−
+
+
−
V6
C27H30O9
499.1963
499.1942
−4.1
27.95
375.1794, 329.1735, 269.0810,159.0655
Hydrogenation and loss of C5H6O0.2014
−
−
−
−
−
−
+
V7
C28H34O10
531.2225
531.1822
−4.2
21.85
453.1910,375.1807,329.1381
Hydrogenation Internal Hydrolysis and Methylation
0.7729
−
−
−
−
−
−
+
V8
C27H30O10
515.1912
515.1896
−3.0
17.02
497.1806, 373.1646, 179.1071, 149.0965
Hydrogenation Internal Hydrolysis and Ketone Formation
0.7600
−
−
−
−
+
−
+
V9
C26H30O12S
567.1531
567.1524
−1.2
15.64
539.1583, 469.1862, 424.1882, 367.1901
Loss of C5H6O+Sulfation
0.1799
−
−
−
−
−
−
+
V10
C33H40O10
597.2694
597.2652
−4.1
8.46
579.2586, 505.2218,
Methylation
2.8624
−
−
−
−
+
+
+
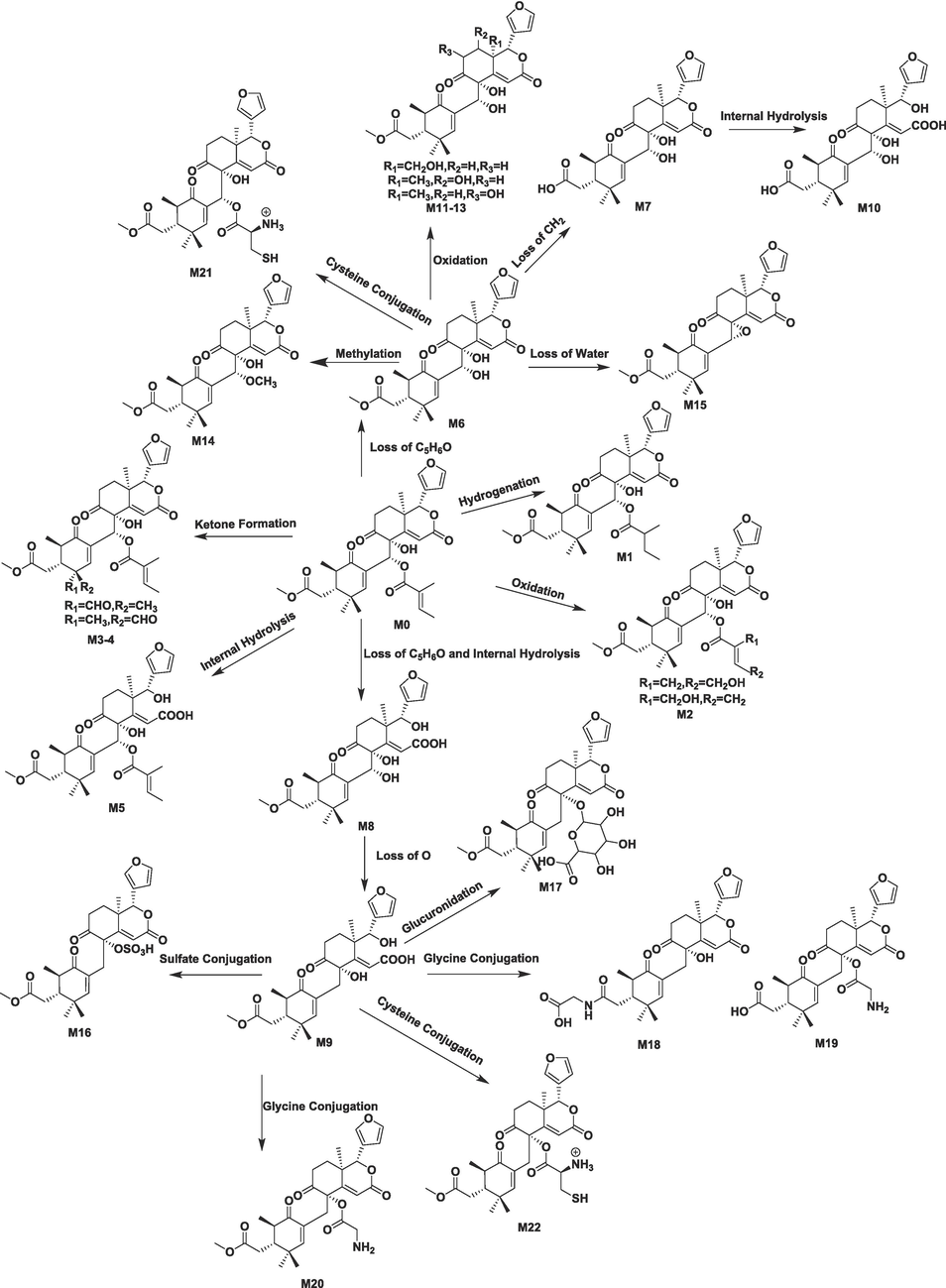
The proposed xylocarpin H metabolic pathways in vivo.
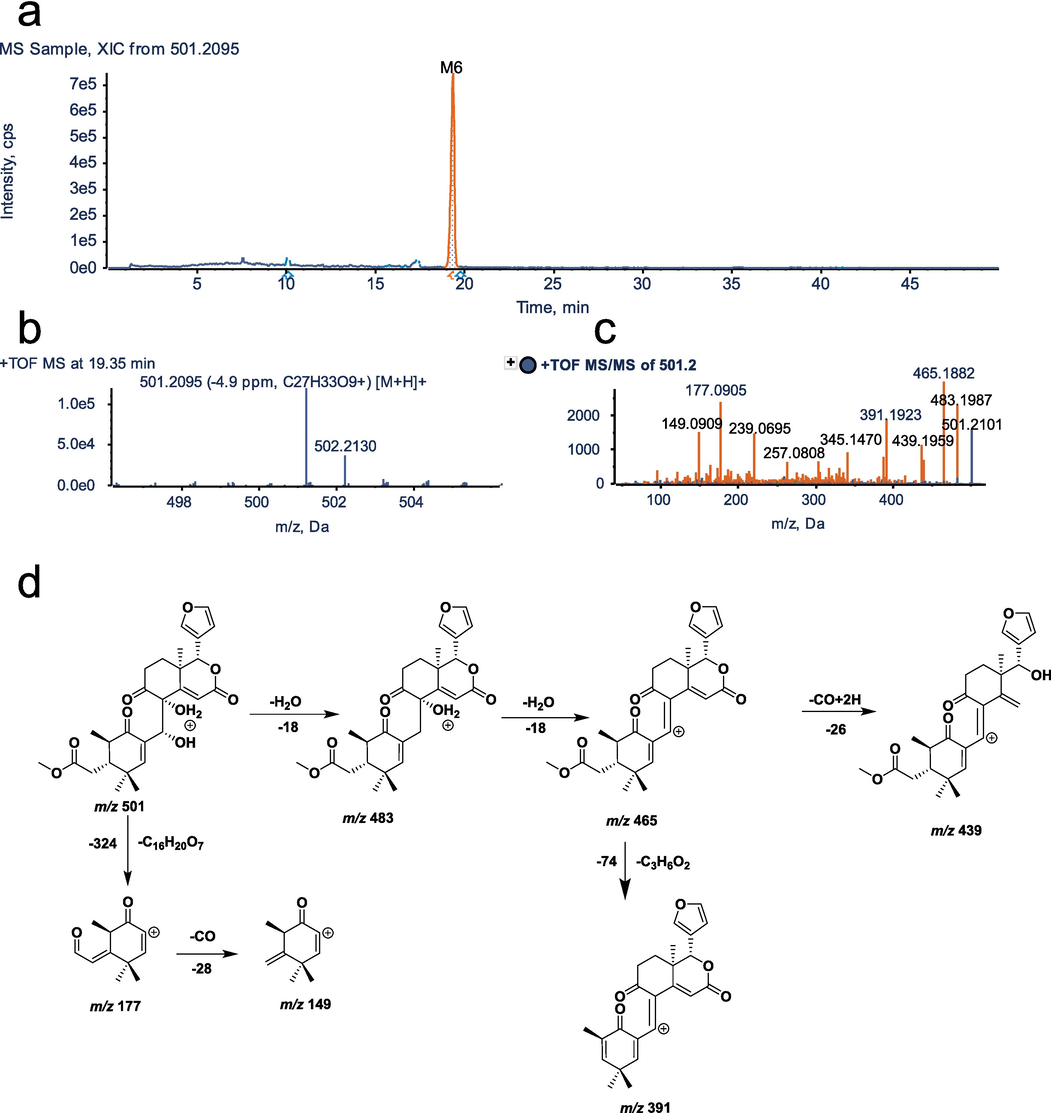
UHPLC–Q–TOF–MS/MS identification of metabolite M6. a, XIC; b, MS spectrum; c, MS/MS spectrum; d, fragmentation pattern.
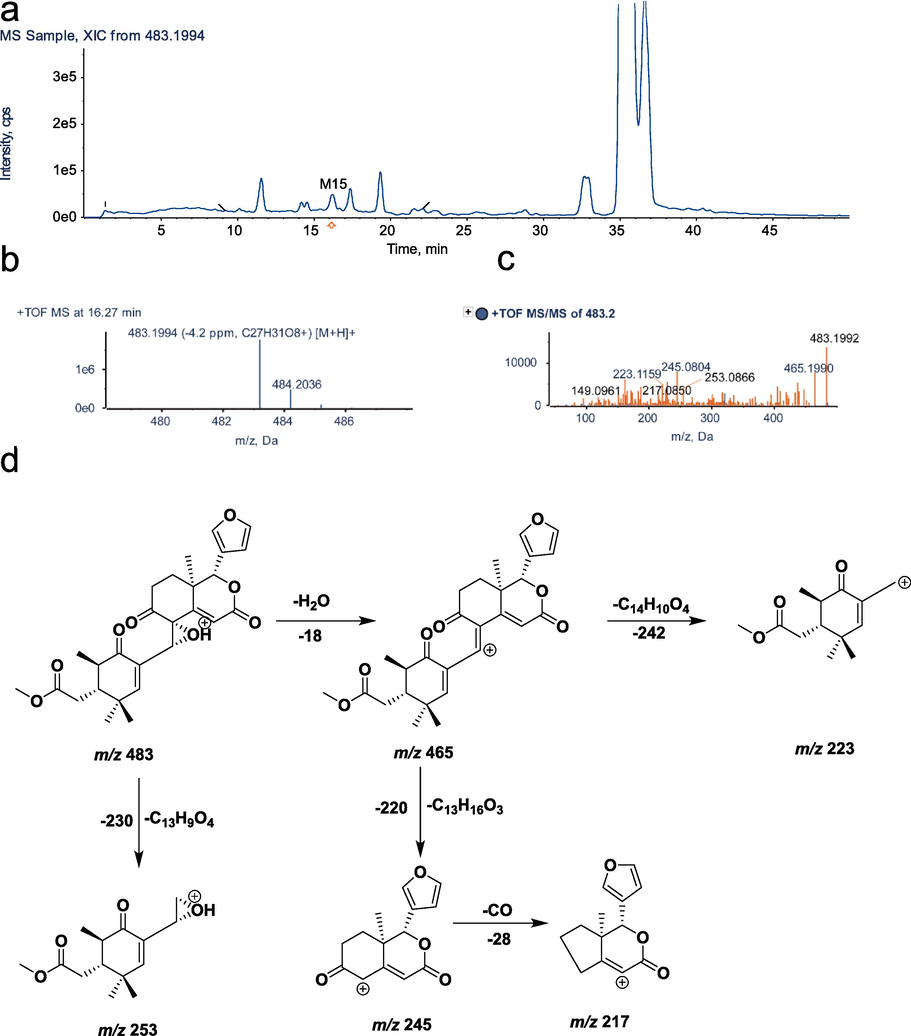
UHPLC–Q–TOF–MS identification of metabolite M15. a, XIC; b, MS spectrum; c, MS/MS spectrum; d, fragmentation pattern.
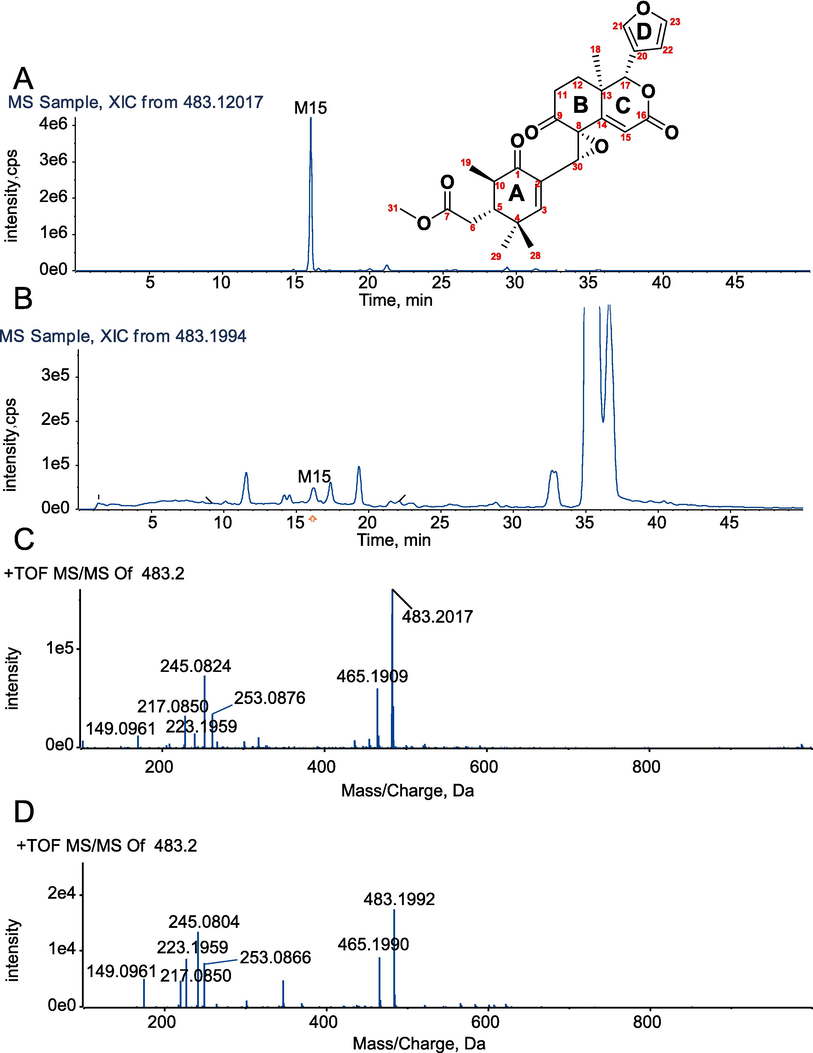
XIC (A) and MS/MS spectrum (C) of chemically synthesized metabolite M15. XIC (B) and MS/MS spectrum (D) of the identified metabolite M15.
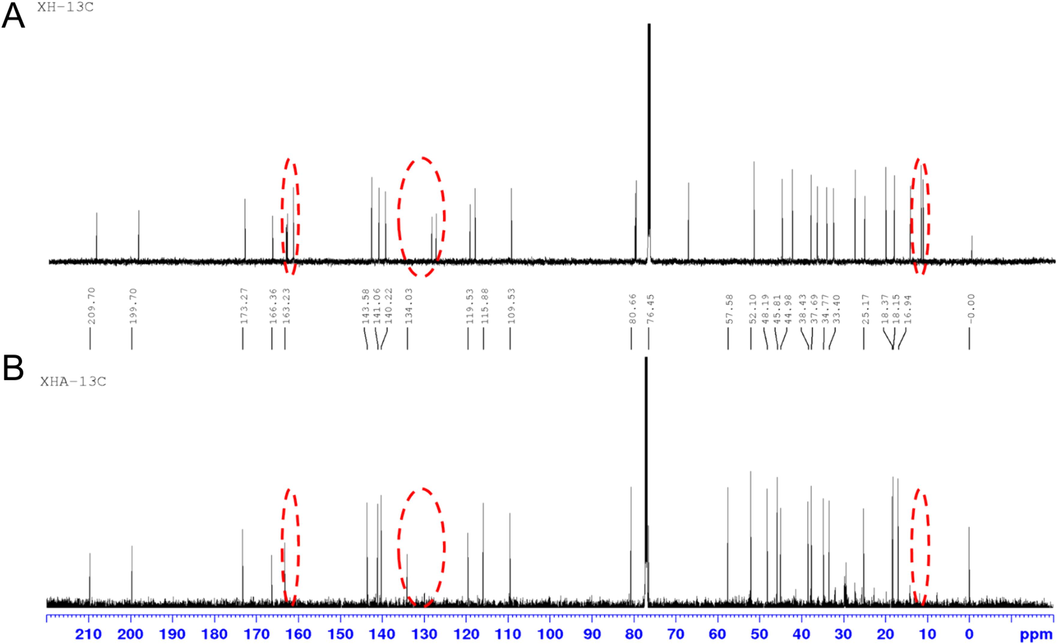
13C NMR spectra of xylocarpin H and metabolites. (A) xylocarpin H, (B) xylocarpin H-A(M15).
UHPLC–Q–TOF–MS/MS identification of metabolite V1. a, XIC; b, MS spectrum; c, MS/MS spectrum; d, fragmentation pattern.
3.4 Identification of metabolite M6
Molecular formula of M6 is C27H32O9 with an excimer ion of [M+H]+ m/z 501.2095 and a retention time of 19.35 min. The molecular formula and molecular weight of M6 are reduced by C5H6O and 82 Da, respectively, compared to M0.The major secondary characteristic fragments are m/z 439.1997 [M+H-2H2O-CO]+, 391.1523 [M+H-2H2O-CO-C3H6O2]+, 257.0808 [M+H-2H2O-CO-C12H16O3]+, 177.0905 [M+H-C16H20O7]+, and their characteristic ions m/z 483.1997, 465.1908, 345.1470, 239.0695 fragment structures were similar to the original drug. In addition, the tigloyl fragment 83.0512 could not be observed, so it is presumed that M6 is the metabolite of tigloyl C5H6O, which has lost its side chain from the original drug. The MS/MS spectra and fragmentation pathways of M6 are presented in Fig. 5.
3.5 Identification of the metabolite M15
As a result of an excimer ion of [M+H]+ m/z 483.1994 and a retention time of 16.27 min, M15 (C27H30O8) has a lower molecular formula of H2O and a lower molecular weight by 18 Da when compared with M6 (molecular formula: C27H32O9, [M+H]+ m/z 501.2095), which is believed to undergo one molecular water reaction. The fragment ions m/z 465.1990, m/z 245.0804 and m/z 217.0850 detected by secondary mass spectrometry had structurally similar backbones to the prodrug. The characteristic fragment ion m/z 223.1159 is 16 Da less than the excimer ion m/z 239.1434, presumably the C-30 carbonylation product or the loss of one molecule of two hydroxyl groups of C-8 and C-30 into an epoxy product. The metabolite M15 of xylocarpin H was prepared by ester hydrolysis. Finally, M15 was proven to be a C-8 and C-30 epoxide by chemical synthesis(Wu et al., 2017). The secondary mass spectra and cleavage pattern of M15 are shown in Fig. 6.
3.6 Structural identification of xylocarpin H chemical modification product M15 (Xylocarpin H-A)
Xylocarpin H-A is a white solid powder with an excimer ion peak detected by high-resolution mass spectrometry at m/z 483.2017 [M+H]+, giving a molecular formula of C27H30O8. The reaction produced a product with the same chromatographic and mass spectral characteristics as the M15 identified in the sample (Fig. 7). The1H-NMR, 13C NMR, 1H–1H COSY, HSQC, and HMBC spectra are shown in Figures S30-S34. The peak attribution table is given in Table S1. From the molecular formula, it is known that the compound is unsaturated to 13 and its 13C NMR has only 27 carbon atom signals and compared to the 1H and 13C NMR data of xylocarpin H(Yin et al., 2007) (Fig. 8), the compound lacks 2 methyl signals [δC 12.1 (C-4′); δC 14.7 (C-5′)], one pair of alkenyl carbon signals [δC 127.7 (C-2′); δC 139.9 (C-3′)] and one ester group signal [δC 166.8 (C-1′)], these missing C signals make up the side chain at position C-30 in the structure, i.e., the tigloyl side chain. The other neighboring positions are somewhat shifted by the carbon spectrum shift due to the loss of the side chain and the dehydration cyclization of the hydroxyl group at position 8 and the oxygen linked side chain at position 30.
In 2D NMR, H-22 [6.35 (dd, 1.9, 0.9)] is correlated with H-23 [7.45 (m)] from 1H–1H COSY, and in combination with HSQC and HMBC spectra, the tetrahydrofuran ring fragment can be inferred from H-12 [2.01 (dd, 13.6, 6.5)] and H-11 in HMBC [3.32 (ddd, 9.3, 6.4, 2.5)] are correlated with carbonyl C-9 (δC 209.7), and combined with H-18 and C-17 correlated with C-14, and H-15 correlated with C-13, C-8 and carbonyl C-16 (δC 163.2). It can be concluded that this endo-lactone ring fragment containing two six-membered rings is in agreement with that in xylocarpin H. From the HMBC, H-31 [3.70 (s)] is associated with carbonyl C-7 (δC 173.3), H-19 [1.20 (d, 7.0)] with C-1 (δC 199.7), C-10 (δC 45.8), and C-5 (δC 48.2), combined with H-29 [0.86 (s)] with C-4 (δC 37.7), C-5 (δC 48.2) and C-28 (δC 25.2) and the remote coupling of H-28 [1.02 (s)] to C-2 (δC 134.0) confirms that the hexameric ring fragment of this α,β-unsaturated ketone and the methyl acetate side chain at the C-5 position remain consistent with the fragment in xylocarpin H described above. Additionally, the unsaturation of the three structural fragments is 12, thus inferring the existence of a ring between the C-2 and C-8 positions. Furthermore, from HMBC, H-30 [3.70 (s)] is associated with C-2 and C-3, and combined with the molecular formula and the chemical shifts of C-30 and C-8, it can be inferred that C-30 is linked to C-2 and C-8 and forms a ternary ether ring with C-8, confirming the existence of dehydration cyclization in the above chemical reaction. Additionally, the unsaturation of the three structural fragments is 12, thus inferring the existence of a ring between the C-2 and C-8 positions. Furthermore, from HMBC, H-30 [3.70 (s)] is associated with C-2 and C-3, and combined with the molecular formula and the chemical shifts of C-30 and C-8, it can be inferred that C-30 is linked to C-2 and C-8 and forms a ternary ether ring with C-8, confirming the existence of dehydration cyclization in the above chemical reaction. Since NOESY was not performed, the stereo configuration of C-8 and C-30 can only be tentatively determined from the raw source; in summary, the compound was identified as M15, noted as xylocarpin H-A. Currently, this compound has not been reported in the literature.
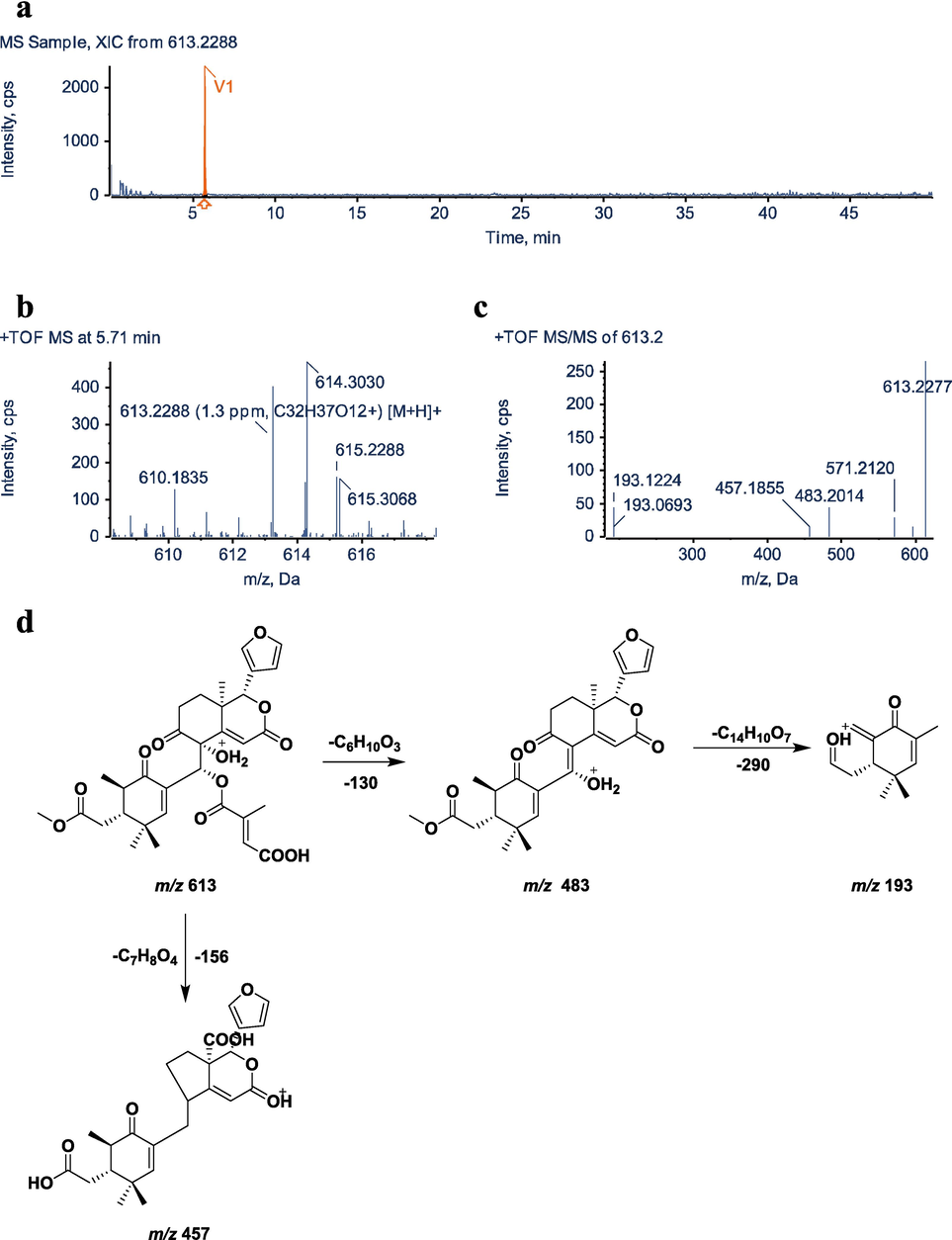
3.7 Identification of metabolite V1
The molecular formula of metabolite V1 is C32H36O12, its excimer ion is [M+H]+m/z 613.2288,and its retention time is 5.71 min. Compared with the parent drug M0 (molecular formula is C32H38O10, [M+H]+m/z 583.2534), The molecular formula was reduced by two hydrogen atoms, two oxygen atoms were added, and the molecular weight was increased by 30 Da. The characteristic fragment ions m/z 483.1650, m/z 457.1857 and m/z 193.1223 were obtained by secondary mass spectrometry scanning, which had a structurally similar skeleton to the parent drug. The V1 structure was presumed to be the product of the C-4′ site of the parent drug being oxidized to carboxylic acid. The MS/MS spectra and fragmentation pathways of V1 are presented in Fig. 9.
3.8 Relative quantitative estimations of metabolites in vivo and in vitro
The relative contents of xylocarpin H and its metabolites in all samples were calculated using area-normalization, and the components were classified according to different compound types and different samples. The relevant data are presented in Table S2, and the relative contents of metabolites for each sample type are shown in Figure S35. The ratio (W%) for each compound was determined using the formula: W% = (peak area value of each compound/sum of peak area values of parent drug and all metabolites) × 100 %. Due to the prevalence of isomeric metabolites and the inability to ascertain their specific structural formulas, the peak areas of metabolites with identical reaction types were collectively calculated(Huang et al., 2022). Xylocarpin H was detected in all in vitro and in vivo samples, as well as in relatively higher proportions in each biological sampleThe most metabolites detected in the gut microbiota, followed by feces, and the least in blood and urine. It is speculated that xylocarpin H has low bioavailability and depends mainly on the intestine for absorption.
4 Discussion
4.1 Mass spectrometric condition selection
In the mass spectrometry analysis, both positive and negative ion modes were evaluated to obtain abundant product ions for the identification of xylocarpin H metabolites in biological samples. The results indicated that the positive ion mode yielded higher mass spectrometric responses for the metabolites. In contrast, the negative ion mode provided fewer metabolites and less secondary mass spectrometry information for xylocarpin H. Therefore, the positive ion mode was selected for metabolite analysis. Limonoids typically possess multiple oxygen-containing functional groups, such as hydroxyl and carbonyl groups, which readily accept protons (H+) to form positive ions. High proton affinity implies easier ionization and stronger signals in the positive ion mode. Additionally, parameters for declustering potential (DP), collision energy (CE), and collision energy spread (CES) were optimized. In MS/MS mode, the optimal DP, CE, and CES in the positive ion mode were 80 V, 35 eV, and 15 eV, respectively.
4.2 Chromatographic condition selection
Compared to the Shim-pack XRODS (2.2 μm, 100 × 2.0 mm, Shimadzu, Japan), the Kinetex XB-C18 column (3 μm, 100 × 2.1 mm, Phenomenex, USA) is preferred due to its core–shell technology, which generates lower backpressure and improved peak shapes. Comparative studies between acetonitrile–water and methanol–water systems demonstrated that the acetonitrile–water system more effectively separated xylocarpin H. Considering the structural properties of limonoids, optimization experiments were conducted with gradient elution using aqueous solutions of formic acid (0.01 %, 0.05 %, 0.1 %, and 0.2 %) and 1 mM ammonium acetate to achieve better peak shapes and higher responses for most metabolites. The results indicated that the mobile phase consisting of water with 0.1 % formic acid and acetonitrile yielded the best peak shape. The addition of a small amount of ammonium acetate increased the peak response, significantly enhancing ionization efficiency and reducing compound dissociation in the mobile phase. The final optimized mobile phase conditions were water with 0.1 % formic acid and 1 mM ammonium acetate (solvent A) and acetonitrile (solvent B).
4.3 Selection of conditions for the synthesis of the metabolite M15
The chemical synthesis of the crucial metabolite M15 was conducted in this experiment, and the reaction conditions of 1, 1.5, 3 and 7 equivalents of K2CO3 and 1, 2, 4 and 6 h were studied. The reaction was found to be essentially complete with 1.5 equivalents of K2CO3 at a reaction time of 2 h. The best results were obtained using 1.5 equivalents of K2CO3 at a reaction time of 2 h. The reaction condition for structural modification was 1.5 equivalents of K2CO3 with a reaction time of 2 h.
4.4 Metabolic pathways of xylocarpin H
Drug metabolism studies in vivo targeting the limonoid were reported in previous studies. Liu et al established a UHPLC-Q-TOF-MS/MS method for the determination of the biotransformation profiles of limonin and its derivatives. The formation and metabolic profiles of limonin in liver microsomes, bile, faeces and urine of rats were investigated. The results demonstrated that reduction and hydrolysis are the two main pathways in the in vivo and in vitro metabolism of limonin.(Liu et al., 2018) Based on the experimentally detected metabolites, we hypothesize that the sites susceptible to biotransformation of xylocarpin H are the double bond at the C-2′ of the tigloyl fragment ion, the carbon atom at the C-4′ or C-5′ allyl position, and the C-1′ ester bond breakage, resulting in the removal of the hydroxyl group at the C-8 and C-30 positions formed by the tigloyl group, hydrolysis of the side chain methyl ester at the C-7 position of the A ring, and hydrolysis of the lactone at the C-16 position in the C ring. The main metabolic pathways are ester hydrolysis reactions, dehydration reactions, addition reactions, reduction reactions and oxidation reactions, while phase II metabolic reactions are mainly sulfate esterification reactions, glucuronidation reactions, and binding reactions with glycine and cysteine, which gives us a richer metabolic pathway compared to that of limonin. In addition, the relatively high content of the metabolite M6 suggests that this pathway is one of the major metabolic pathways of xylocarpin H in vivo and in vivo. It is speculated that xylocarpin H may first lose the tigloyl fragment before further exerting its pharmacological effects.
4.5 Comparison of metabolites from different biological samples
By comparing the number and type of metabolites detected in different biological samples, it was found that the most metabolites were detected in the intestinal flora, followed by feces, and the least were detected in urine and blood. The primary route of excretion for limonoids is through the feces of both rats and humans. Certain drugs undergo initial absorption into the bloodstream and subsequent hepatic metabolism, while a small portion of drugs can also be eliminated via urine (Liu et al., 2017). When oral drugs traverse the gastrointestinal tract, they undergo various metabolic reactions influenced by gastric acid, digestive enzymes, and intestinal flora, leading to the inactivation of certain drugs during intestinal metabolism and a corresponding reduction in the absorption of the parent drug into the systemic circulation. The biotransformation of 80 % of all the clinical drugs is catalyzed by CYP families. In previous studies of individual recombinant enzymes, it was demonstrated that P450 2C9, 2C19 and 3A4 were the enzymes responsible for limonoid bioactivation, with P450 3A4 being the most potent enzyme(Deng et al., 2018). Following oral administration, the bioavailability of limonoid is extremely low. This phenomenon is primarily attributed to the actions of P-gp efflux and CYP3A4 metabolism (Zhang et al., 2012). Therefore, it is speculated that possible mechanism of drug action is that it exerts its effect directly on the target tissue of the rat intestine (Zhang et al., 2016). The study of metabolite activity is very important for the discovery of new pharmacologically active species. Fig. 6a shows that 483 is present in many metabolites and is the crucial ion. M15 may have become a crucial metabolite following the hypothesis that the parent ion is 483. Therefore, it is possible that this is a very important material basis for the pharmacological and toxic effects of xylocarpin H in vivo. The ester hydrolysis of the metabolite synthesized by our method provides further insight into the pharmacological mechanism of action of xylocarpin H and the potential for clinical studies.
4.6 Comparison of in vivo and in vitro metabolism
Considering the importance of drug metabolism for the study of drug safety and toxicity, it plays a crucial role in the discovery and development of drugs(He and Wan 2018).The use of rats for in vivo metabolism investigations has been proven to be feasible and efficient, while liver microsomes and intestinal flora(Xie et al., 2020, Wang et al., 2024) are well suited for metabolite identification, metabolite preparation, and toxicity research in vitro. The results revealed a significant disparity in the metabolism of xylocarpin H between in vivo and in vitro, with the metabolites exhibiting higher abundance in vitro compared to in vivo. It is hypothesized that certain metabolites have widespread distribution throughout the organism, resulting in relatively lower concentrations within the body. Metabolite M15 was exclusively detected in vivo, suggesting that variations in metabolite profiles may be attributed to differential expression levels of metabolic enzymes across various tissues and organs. The differences between in vivo and in vitro drug metabolites can be attributed to several key factors. Firstly, the in vivo system involves various interacting cell types, tissues, and organs, which significantly influence drug metabolism pathways, whereas the in vitro system usually lacks this complexity. Secondly, the concentration and availability of enzymes and cofactors are dynamically regulated in vivo, while these factors are typically fixed in vitro. Thirdly, in vivo microenvironment conditions such as pH, oxygen levels, and nutrient availability are tightly controlled, but these conditions are difficult to replicate fully in vitro. Additionally, in vivo metabolism is influenced by the immune system, which regulates metabolic pathways through cytokines and other signaling molecules, whereas these regulatory factors are generally absent in vitro. Finally, cells in vivo are modulated by various signaling molecules and hormones, which may be incomplete or missing in vitro, leading to differences in metabolic pathways and final metabolites.
4.7 Discussion of metabolite differences in phase I, phase II, and cascade reactions
Phase I reactions primarily involve oxidation, reduction, and hydrolysis, often mediated by cytochrome P450 enzymes. According to the results, hydrolysis, dehydration, oxidation, reduction, and addition reactions were the main metabolic transformation pathways of xylocarpin H during phase I. In contrast, phase II reactions involve conjugation processes, where the metabolites from phase I are further modified by the addition of endogenous molecules like glucuronic acid, sulfate, or glutathione, enhancing the solubility and excretion of the compounds. In phase II metabolic reactions, glucuronidation, sulfate esterification, and glycine and cysteine binding reactions were predominant. The cascade reaction combines both phase I and phase II processes, resulting in a sequential transformation where phase I metabolites are immediately subjected to phase II modifications, often leading to a more complex metabolite profile. The metabolites in this phase include: Loss of C5H6O2 + Sulfate Conjugation; Loss of C5H6O2 + Glucuronidation; Loss of C5H6O2 and CH2 + Glycine; Loss of C5H6O2 and CH2 + Glycine; Loss of C5H6O2 + Glycine. The metabolites from the cascade reactions generally exhibit higher structural complexity compared to those from individual phases due to the sequential enzymatic modifications. Additionally, phase II metabolites and cascade reaction products are typically more polar and water-soluble than phase I metabolites, facilitating easier excretion. The biological activity of metabolites can greatly differ; phase I metabolites may retain or enhance the activity of the parent compound, while phase II metabolites are usually less active due to their increased polarity and reduced interaction with biological targets.
5 Conclusions
This study comprehensively investigated xylocarpin H related metabolites from in vivo and in vitro samples by LC-MS/MS and NMR. The results show that 32 metabolites (23 in phase I and 9 in phase II) were identified in urine, feces, bile, blood, rat liver microsomes, human liver microsomes and rat intestinal flora using UHPLC–Q–TOF–MS. Among them, 19 were in feces, 5 in urine, 13 in bile, 5 in plasma, 7 in human liver microsomes, 8 in rat liver microsomes, and 15 in rat intestinal flora. According to the results, hydrolysis, dehydration, oxidation, reduction, and addition reactions were the main metabolic transformation pathways of xylocarpin H during phase I, whereas in phase II metabolic reactions, glucuronidation, sulfate esterification, and glycine and cysteine binding reactions were the predominant reactions. The majority of metabolites were detected in the intestinal flora, followed by feces, which may be attributed to their limited bioavailability. The metabolite M15 of xylocarpin H was synthesised by ester hydrolysis and its structure was subsequently determined by nuclear magnetic resonance spectroscopy. The approach and outcomes presented herein will contribute to future investigations on the efficacy and safety of xylocarpin H while providing a reference for exploring related mechanisms and facilitating further advancements in drug development.
CRediT authorship contribution statement
Xin Li: Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Binliang Tong: Writing – original draft, Visualization, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xiaoliang Zhu: Validation, Methodology, Investigation, Formal analysis, Data curation. Yuqian Chi: Validation, Methodology, Investigation, Formal analysis, Data curation. Ziyi Jiang: Methodology, Investigation, Formal analysis, Data curation. Xiaoyang Jian: Visualization, Validation, Investigation, Formal analysis, Data curation. Yibing Wu: Supervision, Resources. Tao Lv: Resources, Project administration. Lei Wang: Supervision, Funding acquisition. Xiaowei Shi: Writing – review & editing, Supervision, Resources, Project administration, Funding acquisition, Conceptualization. Zhenhua Pan: Writing – review & editing, Supervision, Funding acquisition.
Acknowledgments
This work was supported by National Natural Science Foundation of China (82073681) and Hebei Natural Science Foundation (H2023206028 and H2022206327).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Xylocarpins A− I, Limonoids from the Chinese mangrove plant Xylocarpus granatum. J. Nat. Prod.. 2007;70:772-778.
- [CrossRef] [Google Scholar]
- Detection and structural characterization of nucleophiles trapped reactive metabolites of limonin using Liquid Chromatography-Mass Spectrometry. J. Anal. Methods Chem.. 2018;2018:3797389.
- [CrossRef] [Google Scholar]
- Drug metabolism and metabolite safety assessment in drug discovery and development. Expert Opin. Drug Metab. Toxicol.. 2018;14:1071-1085.
- [CrossRef] [Google Scholar]
- Advanced tandem mass spectrometry in metabolomics and lipidomics—methods and applications. Anal. Bioanal. Chem.. 2021;413:5927-5948.
- [CrossRef] [Google Scholar]
- A Systematic strategy for the characterization of 2, 3, 5, 4′-tetrahydroxystilbene-2-O-β-d-glucoside metabolites in vivo by Ultrahigh Performance Liquid Chromatography Coupled with a Q Exactive-Orbitrap Mass System. J. Agric. Food Chem.. 2022;70:7773-7785.
- [CrossRef] [Google Scholar]
- Rapid identification of limonoids from Cipadessa baccifera and Xylocarpus granatum using ESI-Q-TOF-MS/MS and their structure-fragmentation study. J. Pharm. Biomed. Anal.. 2018;152:224-233.
- [CrossRef] [Google Scholar]
- Methods of metabolite identification using MS/MS data. J. Comput. Inform. Syst.. 2022;62:12-18.
- [CrossRef] [Google Scholar]
- Biotransformation and metabolic profile of limonin in rat liver microsomes, bile, and urine by high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J. Agric. Food Chem.. 2018;66:10388-10393.
- [CrossRef] [Google Scholar]
- Excretion pathway analysis of limonin in rats. J. Exp. Med. Formul.. 2017;23:91-94.
- [CrossRef] [Google Scholar]
- Metabolic profiles of Fufang Xiling Jiedu capsule in rats by ultra-performance liquid chromatography coupled with quadrupole time-of-flight tandem mass spectrometry. J. Sep. Sci.. 2024;47:2300788.
- [CrossRef] [Google Scholar]
- Total synthesis of (+)-Granatumine A and related bislactone limonoid alkaloids via a pyran to pyridine interconversion. J. Am. Chem. Soc.. 2019;141:9191-9196.
- [CrossRef] [Google Scholar]
- Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol. Neurodegener.. 2021;16:1-15.
- [CrossRef] [Google Scholar]
- A chemometric-assisted LC–MS/MS method for the simultaneous determination of 17 limonoids from different parts of Xylocarpus granatum fruit. Anal. Bioanal. Chem.. 2017;409:4669-4679.
- [CrossRef] [Google Scholar]
- Meliaceous limonoids: chemistry and biological activities. Chem. Rev.. 2011;111:7437-7522.
- [CrossRef] [Google Scholar]
- Effects of in vitro fecal fermentation on the metabolism and antioxidant properties of cyanidin-3-O-glucoside. Food Chem.. 2024;431:137132
- [CrossRef] [Google Scholar]
- Chemical constituents of Xylocarpus granatum. Chem. Nat. Compd.. 2014;50:549-551.
- [CrossRef] [Google Scholar]
- Xylomexicanins I and J: limonoids with unusual B/C rings from Xylocarpus granatum. J. Nat. Prod.. 2017;80:2547-2550.
- [CrossRef] [Google Scholar]
- The metabolic effect of gut microbiota on drugs. Drug Metab. Rev.. 2020;52:139-156.
- [CrossRef] [Google Scholar]
- Targeting intestinal flora and its metabolism to explore the laxative effects of rhubarb. Appl. Microbiol. Biotechnol.. 2022;106:1615-1631.
- [CrossRef] [Google Scholar]
- Xylocarpin H, a limonoid of Xylocarpus granatum, produces antidepressant-like activities in mice. J. Behav. Brain. Sci.. 2015;05:524-532.
- [CrossRef] [Google Scholar]
- Limonoids from the seeds of the marine mangrove Xylocarpus granatum. J. Nat. Prod.. 2007;70:682-685.
- [CrossRef] [Google Scholar]
- Study on limonin distribution in rat intestine by LC-MS/MS method. J. Nanjing Univ. Tradit. Chin. Med.. 2016;32:379-382.
- [CrossRef] [Google Scholar]
- Transport of limonin in rat intestine in situ and Caco-2 cells in vitro. Acta Pharm. Sin.. 2012;2:229-232.
- [CrossRef] [Google Scholar]
- A UHPLC-QTOF-MS/MS method with a superimposed multiple product ion strategy and esterase inhibitor improved sensitivity for the determination of xylocarpin H in rat plasma. J. Pharm. Biomed. Anal.. 2022;216:114803
- [CrossRef] [Google Scholar]
- Elaboration of the comprehensive metabolic profile of Salvianolic acid a in vivo and in vitro using UFLC-Q/TOF-MS. J. Agric. Food Chem.. 2019;67:12199-12207.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105920.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




