

Computational screening of potential bromodomain-containing protein 2 inhibitors for blocking SARS-CoV-2 infection through pharmacophore modeling, molecular docking and molecular dynamics simulation
⁎Corresponding author at: Drug Clinical Trial Center, Gansu Wuwei Tumor Hospital, 16 Xuanwu Road, Wuwei, Gansu, PR China. shaohuaxu11@163.com (Shaohua Xu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Recently, human bromodomain-containing protein 2 (BRD2) has been reported as a potential drug target in host cells to combat SARS-CoV-2 as well as to treat COVID-19. In this study, we aimed to screen potential BRD2 inhibitors as anti- SARS-CoV-2 agents through computational approach. A pharmacophore model was generated with crystal structure of BRD2 in complex with RVX208 using ZINCPharmer server, and 10,842 compounds were screened from ZINC15 database against the pharmacophore model. Next, 102 compounds exhibited more affinity to BRD2 than control compound (RVX208) were screened using molecular docking. The top five hits (ZINC20417563, ZINC12322175, ZINC12391479, ZINC13098192, and ZINC20407881), which showed docking affinity to BRD2 with −10, −9.9, −9.8, −9.7 and −9.7 kcal/mol, respectively, were subjected to 100 ns molecular dynamics simulation to evaluate the stability of docked complexes and their interaction mechanisms. The appropriate stability of these complexes was displayed, and these compounds were found to be closely adhered to the binding site of the BRD2 protein based on the global dynamics analysis with RMSD, RMSF, Rg, SASA, DSSP and H-bond. The essential dynamics analysis with principal component analysis, dynamic cross-correlation matrix, and free energy landscape showed BRD2 binding of the screened compounds remained the stable conformation throughout molecular dynamics simulation. In addition, the pharmacokinetic and ADMET properties of the five compounds showed these compounds could be potential drug candidates. The findings of this study may contribute to further examinational validation and rational design of novel BRD2 inhibitors as anti-SARS-CoV-2 agents.
Keywords
Pharmacophore
Molecular docking
Molecular dynamics simulation
SARS-CoV-2
Bromodomain-containing protein 2

1 Introduction
Since the global pandemic of coronavirus disease 2019 (COVID-19), triggered by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), has been raging in December 2019, it has been a significant new challenge for healthcare (Zheng, 2020). As of March 2023, there have been 759, 408, 703 confirmed cases of COVID-19, including 6,866,434 deaths reported worldwide by World Health Organization (WHO) (https://covid19.who.int). SARS-CoV-2 can cause fever, upper respiratory tract infection, and severe pneumonia with respiratory failure (Lai et al., 2020). Owing to its high infectivity and transmissibility, SARS-CoV-2 is prevalent throughout the world and has had a long-lasting impact on human production and life. Effective therapeutic interventions are essential to the control of the epidemic. As the virus infection rages, the development of highly effective and low-toxic drugs to treat COVID-19 remains a top priority in the global.
Given that SARS-CoV-2 infection is inseparable from the two parts of the virus and the host, it is possible to develop methods to prevent virus infection from both the virus and the host cell perspectives (Robinson et al., 2022; Wagoner et al., 2022). Depending on the different targets of the existing antiviral drugs, they can be divided into drugs that target viruses (DAA, Direct Acting Antivirals Agent) as well as drugs that are targeted to host cells (HTA, Host-Targeting Antivirals Agent) (Bauer et al., 2017; Adamson et al., 2021). Currently, although global strategies for the prevention and control of SARS-CoV-2 infection, including vaccines, neutralizing antibodies, and small-molecule drugs, are for the most part mostly from the perspective of targeting viruses, there is still need to explore antiviral therapy against host factors (Arumugam et al., 2022; Johnson et al., 2021). Viruses are parasitic organisms that must rely on raw materials and machinery of host cells for the production and processing of nucleic acids and proteins in order to complete replication (de Chassey et al., 2014; Prasad et al., 2017; Dwek et al., 2022). Because HTA drugs can specifically target host proteins and interfere with the key process of SARS-CoV-2 replication in cells, in theory, it is more difficult for SARS-CoV-2 to generate resistance to this therapy through mutation (Liu et al., 2021; Gassen et al., 2021). Comparatively, drugs targeting host factors have obvious advantages in controlling the SARS-CoV-2 epidemic (Wagoner et al., 2022; Liu et al., 2021). Thus, the discovery of novel host-targeted compounds with antiviral activity has an essential role in SARS-CoV-2 blockade and the COVID-19 therapy.
Recently, several studies have reported that human bromodomain-containing protein 2 (BRD2) is a potential drug target in host cells for combating SARS-CoV-2 and treating COVID-19. Samelson et al identified bromodomain-containing protein 2 (BRD2) as a potential target for anti-SARS-CoV-2 in host cell using a large scale CRISPR screening (Samelson et al., 2022). They noted that knockdown of BRD2 gene or the use of small molecules to inhibit BRD2 can significantly block the infection and replication of the virus, indicating that BRD2 is an efficient target for inhibiting SARS-CoV-2 infection. Besides, the researchers found that BRD2 inhibition can reduce the expression of angiotensin-converting enzyme 2 (ACE2), the entry receptors for SARS-CoV-2, through reducing the mRNA transcription of ACE2 and regulating the expression of important factors in the interferon pathway such as STAT1, IRF9. It has also been in other research that the BRD2 inhibitor apabetalone (RVX-208), which targeting host cells, can block SARS-CoV-2 infection by inhibiting viral genome replication and down-regulating viral uptake receptors (ACE2) (Gilham et al., 2021). Moreover, Mills et al. disclosed that the BRD2 inhibitors, RXV-2157 and apabetalone, can decrease viral loading and viral titer in cardiomyocytes from SARS-CoV-2-infected K18-hACE2 mice to prevent SARS-CoV-2 caused cardiac injury and dysfunction through a reduction of ACE2 expression (Mills et al., 2021). These studies have demonstrated that BRD2 is an important therapeutic target for anti-SARS-CoV-2 and the treatment of COVID-19. Antiviral efficacy of targeting BRD2 is achieved through the specific regulation of host cell signaling pathways and interference with the intracellular environment upon which viral replication depends. Inhibition of BRD2 can make SARS-CoV-2 more difficult to produce drug resistance through mutation. However, due to the targeting of BRD2 to the host, more consideration should be given to safety and poor in vitro-in vivo efficacy conversion.
Bromodomain-containing protein 2 (BRD2) is a member of the bromodomain and extra-terminal family, which has two tandem bromodomains (bromodomain 1 and bromodomain 2) and one extra-terminal domain (Shang et al.,2011). BRD2 protein can specifically bind to histone acetylated lysine and participate in biological activities such as gene transcription regulation, chromatin remodeling, cell proliferation and apoptosis (Filippakopoulos and Knapp, 2014). Apabetalone (RVX-208) inhibits BRD2 with selectivity for binding to the acetyl-lysine binding pocket at second bromodomain (BD2) and the IC50 value is 510 nM (Picaud et al., 2013).
Computer-aided virtual screening involves the use of computers to the screen for the potential active compounds from virtual compound libraries on a computer (Yu and MacKerell, 2017). Compared to high throughput screening (HTS) in the laboratory, this method is rapid, efficient and low-cost (Baig et al., 2016). Such approaches are commonly used in modern medicinal chemistry, particularly for the discovery of novel lead compounds. The Computer-aided virtual screening allows researchers to rapidly find active compounds from large small-molecule libraries, and lock in the range of experimental determination from over a million molecules to hundreds of molecules in a short period of time, greatly improving the speed and efficiency of experimental screening (El-Gammal et al., 2021; Zhu et al.,2013). It is a powerful tool for the discovery and optimization of anti-SARS-CoV-2 lead compounds and shortens the development cycle for anti-SARS-CoV-2 drugs (El-Bindary and El-Bindary, 2022; El-Bindary et al., 2022).
Although several researches used computer-aided virtual screening approaches to discover anti-SARS-CoV-2 agents, those study focused on targeting the viral protein itself to achieve inhibition of SARS-CoV-2 entry into cells, replication and assembly (Lawal et al., 2023; Rizka et al., 2023; Tassakka et al., 2021). The researches of discovery of host-targeted compounds with anti-SARS-CoV-2 activity are merely. Thus, the main aim of this study was to discover host-targeted compounds against bromodomain-containing protein 2 via pharmacophore and docking based virtual screening. To demonstrate the stability of docked complexes, we performed a molecular dynamics simulation as well as the free binding energy using molecular mechanics Poisson-Boltzmann surface area (MM-PBSA). We propose that the compounds identified could be further BRD2 inhibitor candidates to be investigated both in vitro and in vivo.
2 Materials and methods
2.1 Pharmacophore modeling and database screening
A pharmacophore model was generated using the ZINCPharmer web service (https://zincpharmer.csb.pitt.edu/) (Koes and Camacho, 2012) on the basis of crystal structure of the second bromodomain of human BRD2 bound with an inhibitor of quinazolinone (RVX-208) (Picaud et al., 2013). The PDB file of the crystal structure was downloaded from RCSB PDB database (PDB ID: 4MR5, Resolution: 1.63 Å) (https://www.rcsb.org/structure/4mr6). The crystal structure of RVX-208 in complex with the second bromodomain of BRD2 was used to construct pharmacophore model by ZINCPharmer server. The searching against ZINC15 Purchasable database (Last Updataed 12/20/14) with the generated pharmacophore model was then performed in ZINCPharmer server. The hits were matched the all pharmacophore features and ranked with respect to the fitness scores based on root mean squared deviation (RMSD) between the query features and the hit compound features (Koes et al.,2015). And the compounds screening result was saved in SDF file format for further analysis.
2.2 Molecular docking
Compounds screened from ZINC database with pharmacophore model were docked to BRD2 protein. The screened compounds were added Gasteiger charges and hydrogen atoms and were converted into pdbqt format by Raccoon.py scripts using AutodockTools (Morris et al.,2009) (ADT; Scripps Research Institute, La Jolla,San Diego, USA). The crystal structure of the second bromodomain of human BRD2 bound with a quinazolinone inhibitor (RVX-208) was downloaded from RCSB PDB database (PDB ID: 4MR5, Resolution: 1.63 Å) (https://www.rcsb.org/structure/4mr6). After removing water molecular and co-crystallized ligand, the protein was prepared for docking by adding hydrogen atoms and Gasteiger partial charges using AutodockTools. The prepared protein structure was then converted into pdbqt format. The docking analysis between BRD2 protein and screened compounds was performed by AutoDock Vina (Trott and Olson, 2010) (The Scripps Research Institute, La Jolla, San Diego,USA). A grid box (X = 43.610000, Y = -7.375208, Z = 24.717417, 25*25*25) was generated around the binding sites of protein from co-crystallized ligand (RVX-208) using BIOVIA Discovery Studio Visualizer (Dassault Syst emes, San Diego, CA, USA). The parameters for docking procedures were set to default. The docking pose and docking score were generated from AutoDock Vina.
For each docking analysis, a total of 9 docking conformations are generated, and the conformation with the lowest docking energy is selected as the preferred docking conformation. The lower the binding affinity, indicating that the closer the binding with BRD2, the more stable the docked complex. Interaction between BRD2 and compound was analyzed and visualized using BIOVIA Discovery Studio Visualizer. Before docking procedures, co-crystallized ligand was re-docked to BRD2 protein to validate docking parameters and scheme.
2.3 Molecular dynamics simulation
To evaluate stabilization of the hits docked complexes, top five hits, control compound-BRD2 docked complexes and apo protein (unbound with ligand) were selected to carry out molecular dynamics simulation using Gromacs 2021.4 software (van der Spoel., 2013). The CHARMM36 force field in GROMACS format (charmm36-jul2021 version) (Huang and MacKerell, 2013) was employed for proteins to generate the topology of BRD2, whereas the CHARMM general force field version 4.6 (Vanommeslaeghe et al., 2010) was employed for ligands to generate the topology file using CGenFF server (Vanommeslaeghe and MacKerell, 2012; Vanommeslaeghe et al., 2012) (https://cgenff.umaryland.edu/). Each complex was placed in a dodecahedron box with 1 nm distance to the edges. The explicit water model in the water box is the TIP3P water model (Leontyev and Stuchebrukhov, 2011), and an appropriate amount of chloride ions or sodium ions was added to ensure that the entire simulation system is electrically neutral. The steepest descent energy minimization with 5000 steps was conducted for each system. Next, two steps of equilibration were performed. The first step was a NVT equilibration. In this step, the temperature of the system was gradually heated from 0 K to 310 K using V-rescale thermostat method and the simulation time was 200 ps under the NVT ensemble. The second step was a NPT equilibration. In this step, the pressure of 1.01325105 Pa was controlled using Parrinello-Rahman method and the simulation time was 200 ps under the NPT ensemble. The particle-mesh-Ewald method (Sagui and Darden, 1999) was used to deal with the long-range electrostatic interactions, and van der Waals interactions were evaluated by a cut-off of 10 Å. Finally, the molecular dynamics simulation of 100 ns was carried out under the NPT ensemble. The simulation time step is 2 fs, and the trajectories was saved every 10 ps for subsequent analysis.
2.4 Simulation trajectories analysis
The obtained molecular dynamics simulation trajectories were analyzed to measure root mean square deviation (RMSD), root-mean-square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), hydrogen bond interaction and protein secondary structure using gmx rms, gmx rmsf, gmx gyrate, gmx sasa, gmx hbond and gmx do_dssp module in GROMACS. Principal component analysis and dynamics cross-correlation matrix analysis were performed using bio3d R package (Grant et al.,2021). The free energy landscape analysis was carried out using gmx covar, gmx anaeig and gmx sham module in GROMACS.
2.5 Binding free energy
The binding free energy (ΔGbind) of each system between ligand and protein was calculated by Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method (Wang et al.,2018) using g_mmpbsa module (Kumari et al.,2014) in GROMACS. The binding free energy is calculated according to the following formula:
Gcomplex, Gprotein and Gligand represent the free energy of complex, receptor and ligand, respectively. Each can be expressed separately as following:
2.6 ADME analysis
The physicochemical properties and ADME (absorption, distribution, metabolism and excretion) properties of the top five hits from docking were analyzed using swissADME web server (Daina et al.,2017) (https://www.swissadme.ch/).
3 Results and discussion
3.1 Construction of the pharmacophore model and pharmacophore-based virtual screening
Pharmacophore feature refers to a certain atom, atomic group, or some active energy characteristics, such as hydrophobic characteristics, in the drug molecules and target structure fusion and plays a key role in drug development (Seidel et al.,2019). A pharmacophore model based on interaction of RVX-208 with the second bromodomain of BRD2 was constructed using ZINCPharmer server and the pharmacophore features were shown in Fig. 1. The pharmacophore hypotheses were chosen for having two hydrogen bond acceptors, one hydrophobic characteristic and one aromatic characteristic. The radius of each hydrogen bond acceptor feature, the hydrophobic feature and aromatic feature were 0.5, 1 and 1.1 Å, respectively. The ZINC database was used to screen hits with the generated pharmacophore model. In total, 10,842 compounds out of 21,723,923 compounds of ZINC database were screened to match the all pharmacophore features using ZINCPharmer server.
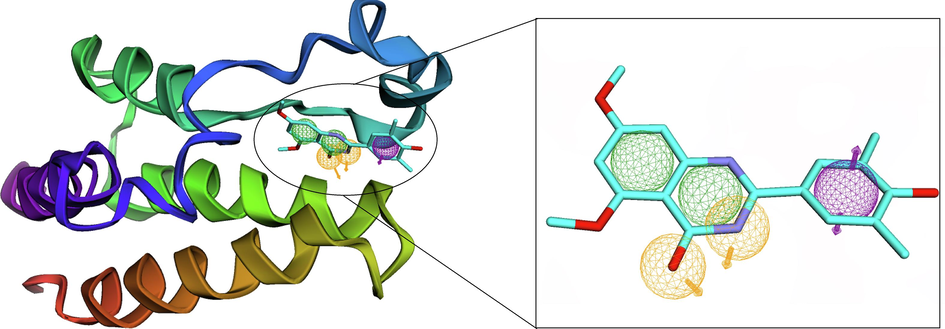
- Structure-based pharmacophore model derived from crystal structures of the second bromodomain of human BRD2 bound with a quinazolinone inhibitor (RVX-208) using ZINCPharmer. The hydrogen bond acceptors, the hydrophobic feature and aromatic feature were shown in yellow, green, and purple spheres, respectively. The arrows show the constraint direction.
3.2 Molecular docking
Before docking procedures, co-crystallized ligand was re-docked to BRD2 protein to validate reliability of docking parameters and scheme. The conformation of re-docked ligand almost overlapped with the original conformation, and root mean square deviation is 0.1 nm (Fig. S1), indicating that the docking parameters and scheme in this study could reliability predict the conformation of protein–ligand docked complex (Hevener et al., 2009). RVX208 (IC50 = 510 nM), co-crystallized ligand of BRD2 protein, was selected as a control compound. In docking analysis, 10,842 compounds screened from pharmacophore model were docked the BRD2 protein and docking affinity was generated.
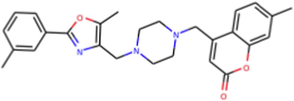
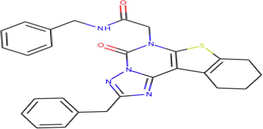
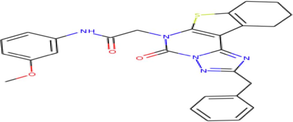
A total of 102 compounds were screened, which displayed docking affinity to BRD2 higher than the control compound (−8.1 kcal/mol). The top five hits, ZINC20417563, ZINC12322175, ZINC12391479, ZINC13098192, and ZINC20407881, which displayed high affinity against the BRD2 protein with docking score of −10, −9.9, −9.8, −9.7 and −9.7 kcal/mol in comparison of −8.1 kcal/mol for control compound were selected for subsequent analysis. The structure and docking scores of the top five hits were listed in Table 1. We then analyzed the interaction between the five compounds and residues of BRD2 protein using BIOVIA Discovery Studio Visualizer. Interaction details were listed in Table 2. ZINC20417563 molecule had hydrophobic interactions with Phe372, Lys374, Val376, Trp370, Cys425 and Val435 residues, formed two hydrogen bonds with Pro371 and Asn429 residues, and had a Pi-Sulfur interaction attributed to Cys425 residue of BRD2 protein (Fig. 2). In term of ZINC12322175, three hydrogen bonds were yielded with Val376, Asp377 and Asn429 residues, and hydrophobic interactions were formed with Trp370, Pro371, Val376, Leu381, Cys425 and Val435 residues, and a Pi-Sulfur interaction attributed to Trp370 residue of BRD2 protein (Fig. 3). In case of ZINC12391479, the formation of hydrogen bonds interacted with Lys374 and Val376 residues, hydrophobic interactions with Trp370, Pro371, Val376, Leu381, Cys425 and Val435 residues, and two Pi-Sulfur interactions with Trp370 and Cys425 residues were observed (Fig. 4). Fig. 5 showed the interaction diagram of ZINC13098192-BRD2 docked complex. Two residues of BRD2 protein, Trp370 and Pro371, formed two hydrogen bonds interacted with ZINC13098192. Resides Lys374, Val376, Leu381, Leu383, Cys425, His433 and Val435 of BRD2 protein yielded hydrophobic interactions with ZINC13098192. Moreover, a Pi-Sulfur interaction was formed between ZINC13098192 and TRP370 residue of BRD2 protein. As shown in Fig. 6, the BRD2 protein yielded hydrophobic interactions with ZINC20407881 through Lys374, Val376, Phe372, Cys425 and Val435 residues. Moreover, two hydrogen bonds for ZINC20407881 with Pro371 and Asn429 residues and a Pi-Sulfur interaction also formed with Trp370 residue of BRD2 protein were observed. Compared to the RVX208 (exited BRD2 inhibitor) (Picaud et al.,2013) and NSC127133 (a new BRD2 inhibitor identified by Tripathi) (Tripathi et al., 2016), the interactions between the screened compounds in this study and BRD2 include not only hydrogen bonds and hydrophobic interactions but also Pi-Sulfur interactions. Above docking results show that the five compounds can potentially interfere with some of the important residues interactions such as Trp370, Pro371, Leu381 and Cys425 (Picaud et al.,2013). These specific residues are located at the binding interface between substrate and BD2 domain of BRD2. According to docking analysis, the top compounds were occupied in substrate binding pocket of BRD2, and formed hydrogen bond, hydrophobic interaction, and Pi-Sulfur interaction with specific residues located at substrate binding interface. It indicated that the screened compounds can prevent the acetyl-lysine substrate from entering the binding sites to consistent BRD2 activity.
| Molecular Formula | IUPAC Name | Structure | Affinity (kcal/mol) |
|
|---|---|---|---|---|
| ZINC20417563 | C27H29N3O3 | 7-methyl-4-[[4-[[5-methyl-2-(3-methylphenyl)-1,3-oxazol-4-yl]methyl]piperazin-1-yl]methyl]chromen-2-one |
|
−10 |
| ZINC12322175 | C27H25N5O2S | N-benzyl-2-(2-benzyl-5-oxo-8,9,10,11-tetrahydro[1]benzothieno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidin-6(5H)-yl)acetamide |
|
−9.9 |
| ZINC12391479 | C27H25N5O3S | 2-(2-benzyl-5-oxo-8,9,10,11-tetrahydro[1]benzothieno[3,2-e][1,2,4]triazolo[1,5-c]pyrimidin-6(5H)-yl)-N-(3-methoxyphenyl)acetamide |
|
−9.8 |
| ZINC13098192 | C27H26FN5OS | [4-[[4-Benzyl-3-(2-fluorophenyl)-5-sulfanylidene-1,2,4-triazol-1-yl]methyl]piperazin-1-yl]-phenylmethanone |
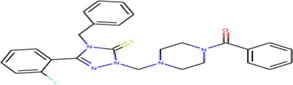
|
−9.7 |
| ZINC20407881 | C26H27N3O3 | 7-methyl-4-({4-[(5-methyl-2-phenyl-1,3-oxazol-4-yl)methyl]piperazin-1-yl}methyl)–2H-chromen-2-one |
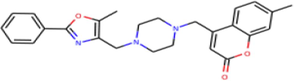
|
−9.7 |
| Residues | Category | Type | |
|---|---|---|---|
| ZINC20417563 | ASN429 | Hydrogen Bond | Conventional Hydrogen Bond |
| PRO371 | Hydrogen Bond | Carbon Hydrogen Bond | |
| VAL376 | Hydrophobic | Pi-Sigma | |
| LYS374 | Hydrophobic | Alkyl | |
| CYS425 | Hydrophobic | Alkyl | |
| TRP370 | Hydrophobic | Pi-Alkyl | |
| PHE372 | Hydrophobic | Pi-Alkyl | |
| VAL376 | Hydrophobic | Pi-Alkyl | |
| VAL435 | Hydrophobic | Pi-Alkyl | |
| CYS425 | Other | Pi-Sulfur | |
| ZINC12322175 | ASP377 | Hydrogen Bond | Conventional Hydrogen Bond |
| VAL376 | Hydrogen Bond | Carbon Hydrogen Bond | |
| ASN429 | Hydrogen Bond | Pi-Donor Hydrogen Bond | |
| VAL376 | Hydrophobic | Pi-Sigma | |
| LEU381 | Hydrophobic | Pi-Sigma | |
| TRP370 | Hydrophobic | Pi-Pi T-shaped | |
| TRP370 | Hydrophobic | Pi-Alkyl | |
| PRO371 | Hydrophobic | Pi-Alkyl | |
| VAL376 | Hydrophobic | Pi-Alkyl | |
| LEU381 | Hydrophobic | Pi-Alkyl | |
| CYS425 | Hydrophobic | Pi-Alkyl | |
| VAL435 | Hydrophobic | Pi-Alkyl | |
| TRP370 | Other | Pi-Sulfur | |
| ZINC12391479 | LYS374 | Hydrogen Bond | Conventional Hydrogen Bond |
| VAL376 | Hydrogen Bond | Conventional Hydrogen Bond | |
| TRP370 | Hydrophobic | Pi-Pi T-shaped | |
| TRP370 | Hydrophobic | Pi-Alkyl | |
| VAL376 | Hydrophobic | Pi-Alkyl | |
| LEU381 | Hydrophobic | Pi-Alkyl | |
| PRO371 | Hydrophobic | Pi-Alkyl | |
| VAL435 | Hydrophobic | Pi-Alkyl | |
| CYS425 | Other | Pi-Sulfur | |
| TRP370 | Other | Pi-Sulfur | |
| ZINC13098192 | TRP370 | Hydrogen Bond | Carbon Hydrogen Bond |
| PRO371 | Hydrogen Bond | Carbon Hydrogen Bond | |
| HIS433 | Hydrophobic | Pi-Pi Stacked | |
| VAL376 | Hydrophobic | Pi-Alkyl | |
| LYS374 | Hydrophobic | Pi-Alkyl | |
| LEU381 | Hydrophobic | Pi-Alkyl | |
| LEU383 | Hydrophobic | Pi-Alkyl | |
| CYS425 | Hydrophobic | Pi-Alkyl | |
| VAL435 | Hydrophobic | Pi-Alkyl | |
| TRP370 | Other | Pi-Sulfur | |
| ZINC20407881 | ASN429 | Hydrogen Bond | Conventional Hydrogen Bond |
| PRO371 | Hydrogen Bond | Carbon Hydrogen Bond | |
| LYS374 | Hydrophobic | Alkyl | |
| CYS425 | Hydrophobic | Alkyl | |
| PHE372 | Hydrophobic | Pi-Alkyl | |
| LYS374 | Hydrophobic | Pi-Alkyl | |
| VAL376 | Hydrophobic | Pi-Alkyl | |
| VAL435 | Hydrophobic | Pi-Alkyl | |
| CYS425 | Other | Pi-Sulfur |
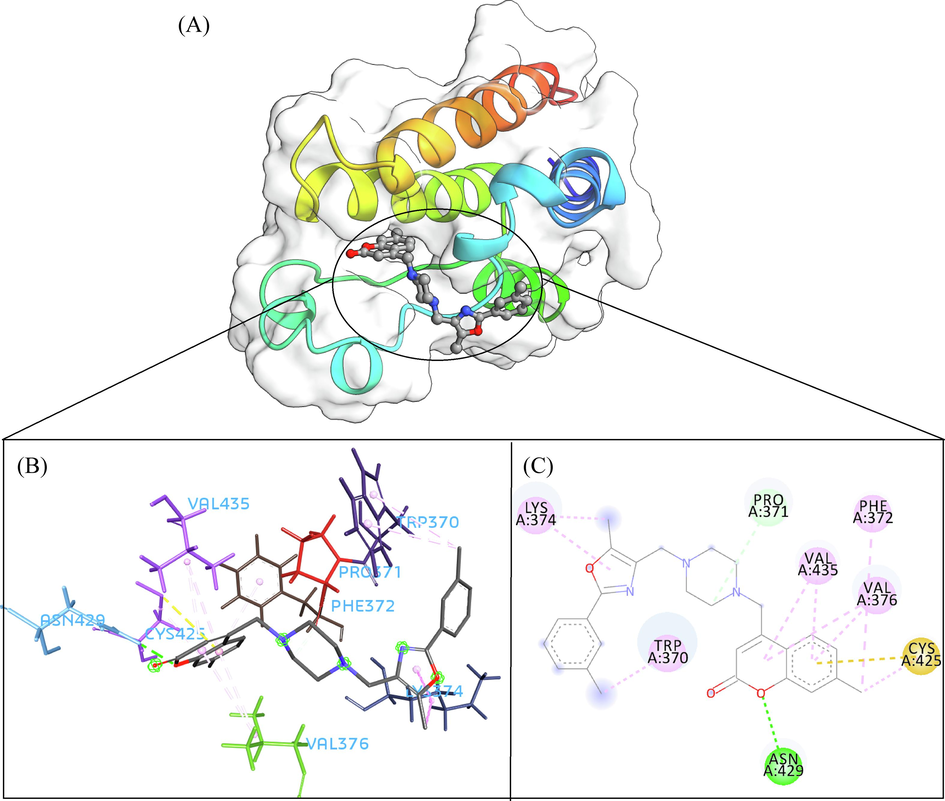
- The 3D interaction (a), docked pose view (b) and 2D interaction (c) profile of ZINC20417563 docked with BRD2 protein. Hydrogen bonds are displayed as green ball and stick, Pi-Sulfur are displayed as gold ball and stick, hydrophobic (pi-pi/pi-alkyl stacking) are displayed as pink ball and stick, and carbon-hydrogen bond are displayed as white ball and stick.
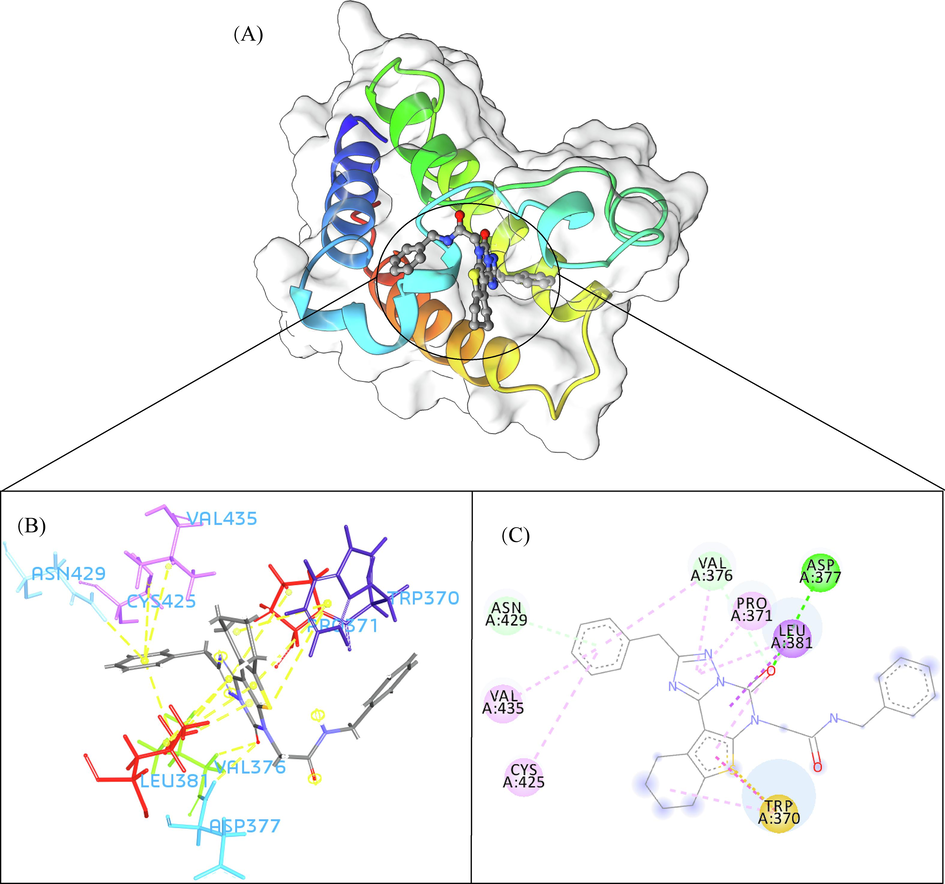
- The 3D interaction (a), docked pose view (b) and 2D interaction (c) profile of ZINC12322175 docked with BRD2 protein. Hydrogen bonds are displayed as green ball and stick, Pi-Sulfur are displayed as gold ball and stick, hydrophobic (pi-pi/pi-alkyl stacking) are displayed as pink ball and stick, and carbon-hydrogen bond are displayed as white ball and stick.
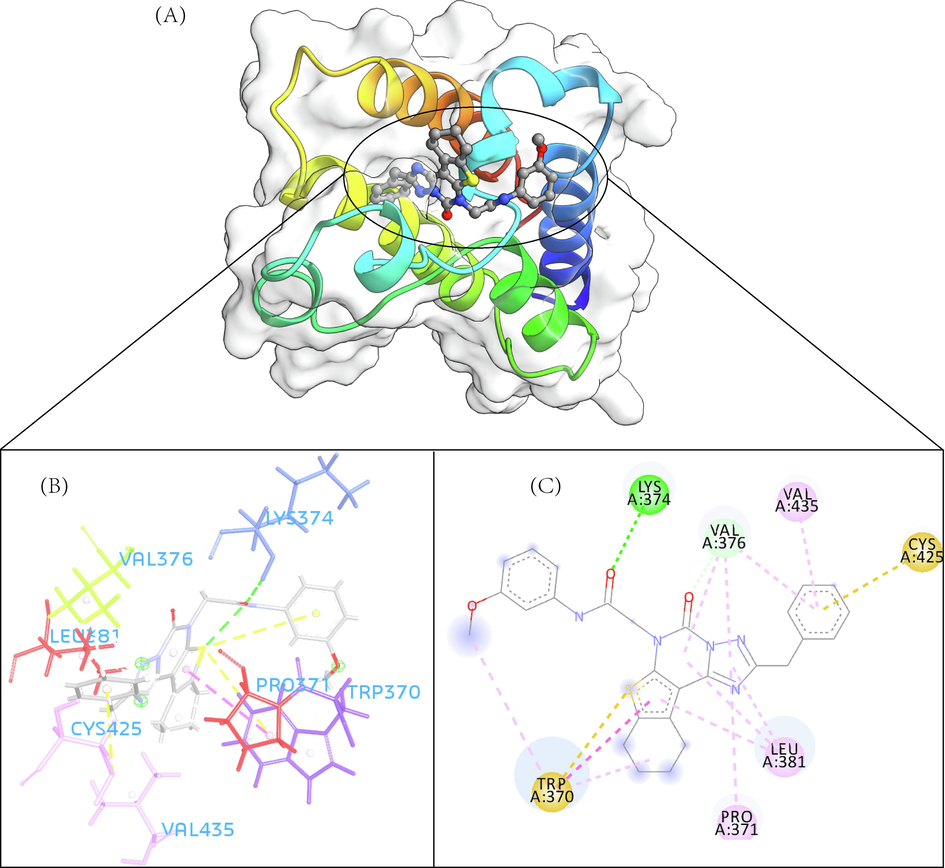
- The 3D interaction (a), docked pose view (b) and 2D interaction (c) profile of ZINC12391479 docked with BRD2 protein. Hydrogen bonds are displayed as green ball and stick, Pi-Sulfur are displayed as gold ball and stick, hydrophobic (pi-pi/pi-alkyl stacking) are displayed as pink ball and stick, and carbon-hydrogen bond are displayed as white ball and stick.
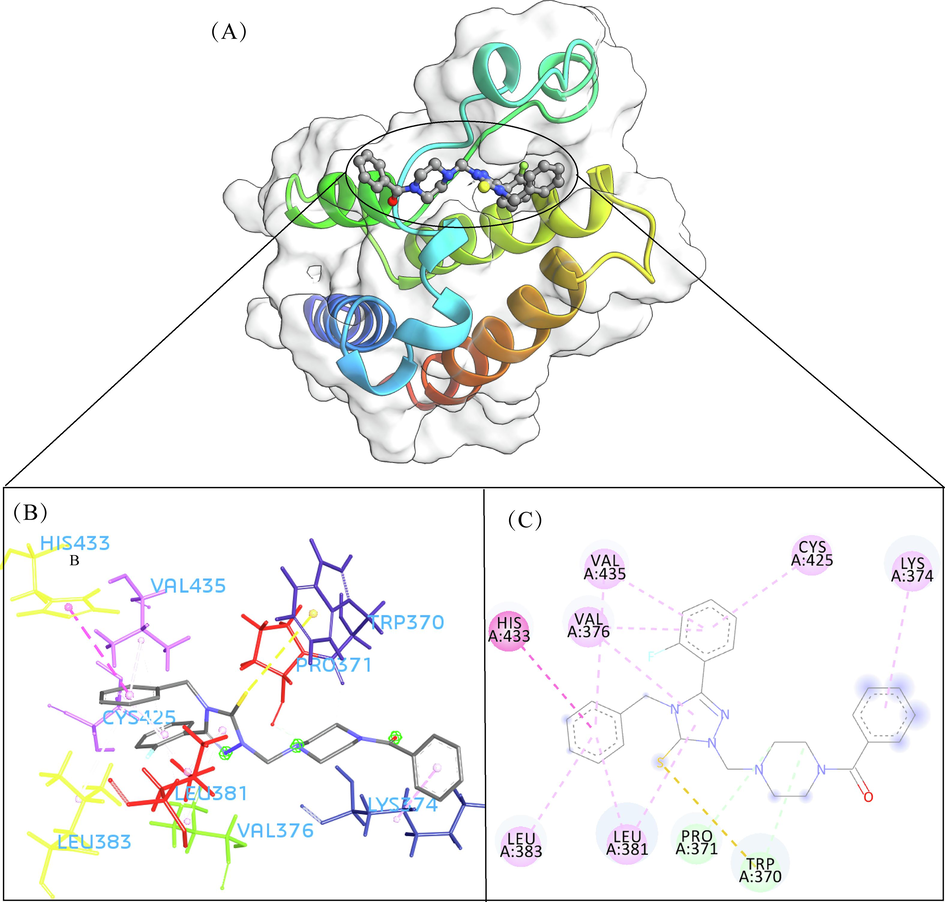
- The 3D interaction (a), docked pose view (b) and 2D interaction (c) profile of ZINC13098192 docked with BRD2 protein. Hydrogen bonds are displayed as green ball and stick, Pi-Sulfur are displayed as gold ball and stick, hydrophobic (pi-pi/pi-alkyl stacking) are displayed as pink ball and stick, and carbon-hydrogen bond are displayed as white ball and stick.
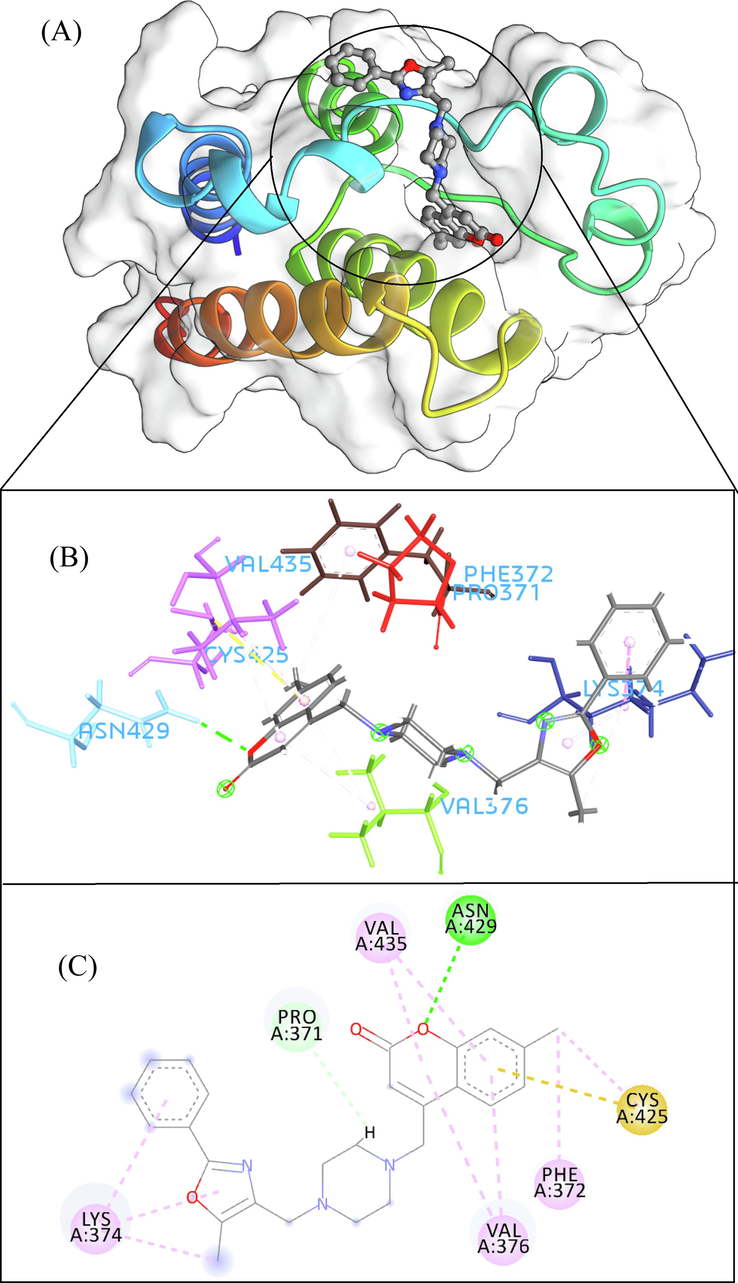
- The 3D interaction (a), docked pose view (b) and 2D interaction (c) profile of ZINC20407881 docked with BRD2 protein. Hydrogen bonds are displayed as green ball and stick, Pi-Sulfur are displayed as gold ball and stick, hydrophobic (pi-pi/pi-alkyl stacking) are displayed as pink ball and stick, and carbon-hydrogen bond are displayed as white ball and stick.
3.3 Molecular dynamics simulation
3.3.1 Root mean standard deviation (RMSD)
The root mean standard deviation (RMSD) is a quantitative parameter that can evaluate the stability of protein–ligand system (Stark et al., 2003; Brüschweiler et al., 2003). In order to study the conformational stability of the protein–ligand complex system, the conformation of each frame in the trajectory is compared with the initial conformation, and RMSD of the Cα atom is calculated. As shown in Fig. 7A, the fluctuations in the curves of all complex systems tend to be balanced after 10 ns and remain at ∼ 0.25 nm, indicating that the structures of all systems are in a stable state after a dynamic simulation. The averaged RMSD values of the ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, control-BRD2 docked complexes and apo BRD2 protein were 0.252 nm, 0.251 nm, 0.247 nm, 0.253 nm, 0.248 nm, 0.253 nm, and 0.246 nm, respectively. The RMSD value of apo BRD2 protein was similar as BRD2 bound with compounds over the simulation, indicating that binding of the compounds had no effect on protein stability.
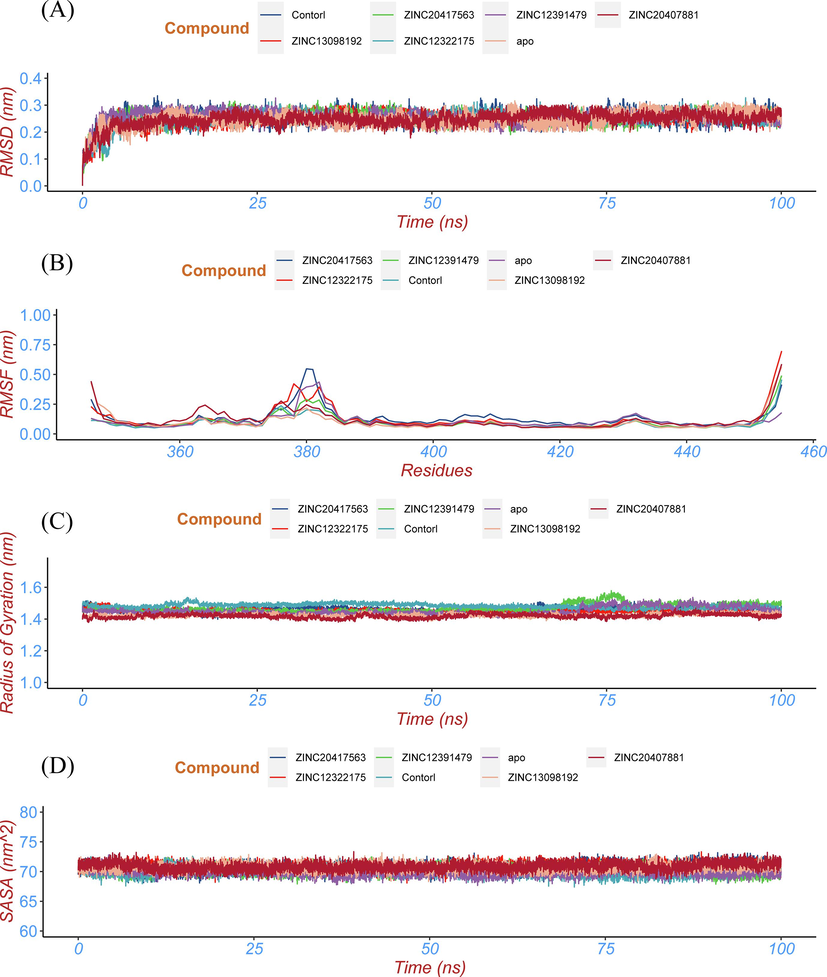
- Conformational stability analysis of the top five hits, control compound-BRD2 docked complexes and apo protein. (a) the RMSD of the Cα atom, (b) the RMSF profile, (c) the Rg profile, and (d) the SASA profile of the protein of top five hits, control compound-BRD2 docked complexes and apo protein.
3.3.2 Root mean square fluctuation (RMSF)
Root mean square fluctuation (RMSF) is a physical parameter that can depict the fluctuation of each amino acid residue in a protein over an overall simulation (Omoboyowa et al.,2021). Higher RMSF value indicates that the residues has greater volatility, conversely, a smaller RMSF value indicates that the residues is less volatile (Shafique et al., 2021). In order to evaluate the effect of top five hits on the fluctuation of each amino acid residue in BRD2 protein through MD simulation, we calculated the RMSF of each amino acid residue in the BRD2 bound with the five top hits and control compound and apo BRD2 (Fig. 7B). The average value of RMSF was 0.133 nm, 0.122 nm, 0.100 nm, 0.099 nm, 0.118 nm, 0.099 nm and 0.109 nm for ZINC20417563, ZINC12322175, ZINC12391479, ZINC13098192, ZINC20407881, control-BRD2 docked complexes and BRD2 unbound protein, respectively. Fig. 7B showed that the six complex systems have RMSF values similar to the dynamic characteristics and the fluctuation of amino acid residues (375 ∼ 383) in all systems is relatively consistent, indicating the binding modes between these compounds and BRD2 protein are relatively similarity.
3.3.3 Radius of gyration (Rg)
The radius of gyration is a physical quantity used to describe the compactness of the protein structure (Yanao et al.,2007). The more stable the structure of the protein, the better the compactness of the protein, and the smaller the corresponding Rg value (Yamamoto et al., 2021). The averaged Rg values of the ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, control-BRD2 docked complexes and BRD2 unbound with ligand were 1.423 nm, 1.458 nm, 1.451 nm, 1.469 nm, 1.419 nm, 1.491 nm and 1.450 nm, respectively. This six docked complexes and apo BRD2 had approximately similar Rg value during the entire simulation period (Fig. 7C). These results proposed that binding of the compounds had no affect on compactness in BRD2 protein structure, indicting BRD2 protein and selected hits formed stable and compact docked complexes.
3.3.4 Solvent accessible surface area (SASA)
Solvent accessible surface area (SASA) is an important parameter to describe the hydrophobicity of proteins (Chen et al.,2019). The smaller the SASA value, the smaller the exposed area of macromolecules in the solution, the tighter the protein folding and the more stable the conformation (Ali et al.,2014). The averaged SASA values of the ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, control-BRD2 docked complexes and BRD2 unbonded protein were 70.73 nm2, 70.86 nm2, 70.65 nm2, 70.22 nm2, 70.86 nm2, 70.09 nm2 and 69.99 nm2, respectively. Six docked complexes and apo protein represented similar SASA value over the 100 ns simulation (Fig. 7D), revealing all systems remained relatively the stable conformational dynamics and protein–ligand interactions had no effect on protein swelling.
Overall, the molecular dynamics analysis showed these compound-BRD2 docked complexes were properly stable and these compounds were tightly adhered to active site of BRD2.
3.4 Hydrogen bond analysis
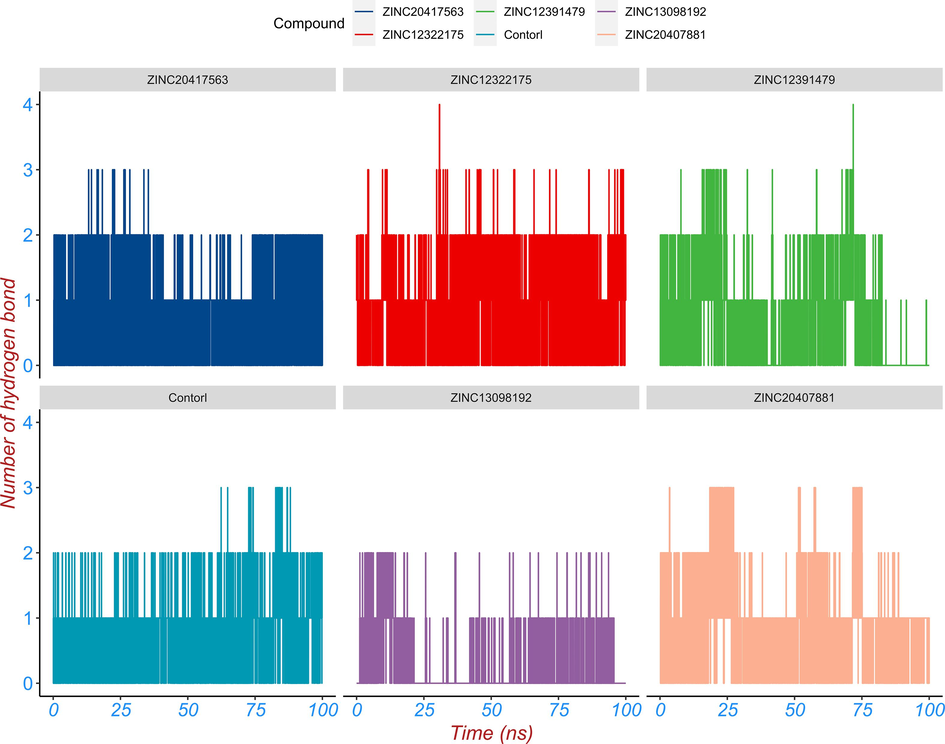
Hydrogen bonding between proteins and their ligands provides the basic directional and specific interactions for molecular recognition (Korth, 2011). The number of hydrogen bonds between top five hits and control compound and BRD2 protein over simulation time were analyzed using gmx_hbond module in GROMACS. The number of the hydrogen bond formed during protein–ligand complex interaction throughout the 200 ns MD simulation was presented in Fig. 8. ZINC20417563 compound formed up to three hydrogen bonds, ZINC12322175 compound formed up to four hydrogen bonds, ZINC12391479 compound formed up to four hydrogen bonds, ZINC12391479 compound formed up to four hydrogen bonds, ZINC20407881 compound formed up to three hydrogen bonds and control compound formed up to four three bonds, whereas ZINC13098192 compound rarely formed two hydrogen bonds over the 100 ns MD simulation (Fig. 8). These results revealed that the hydrogen bond interaction between all compounds and BRD2 was relatively stable and continuous over simulation time.
- The hydrogen bond analysis between the top five hits, control compound and SGLT2 protein during MD simulation. The hydrogen bond number varies with simulation time.
Next, hydrogen bond existence maps for six complexes were plotted to explore the consistency of key residues involved in hydrogen bonding (Fig. 9). Fig. 9A showed TRP370, LYS374, SER379, TYR386, ASN429, and HIS433 residues participated in formation of hydrogen bonds interacted with ZINC20417563. Among them, the hydrogen bond between residue ASN433 and atom O3 of ZINC20417563 was seen constantly during the 200 nm simulation. PRO371 and ASN429 residues of BRD2 protein were found to be participated in forming hydrogen bonds with ZINC12322175. And these were observed incessantly throughout the simulation (Fig. 9B). For ZINC12391479, it has been observed to form hydrogen bonds with these residues LYS363, ALA366, TRP370, PRO371, LYS374 and ASP377 of BRD2 protein. Among them, TRP370, LYS374 and ASP377 residues yielded hydrogen bonds with different atoms of ZINC12391479 in which only one hydrogen bond between ASP377 residue and atom N1 of ligand was seen insistently during 0 ∼ 25 ns and 40 ∼ 80 ns simulation time (Fig. 9C). In case of ZINC13098192, BRD2 protein residues TRP370, TYR373, LYS374, ASP377 and HIS433 were found to yield several hydrogen bonds. However, these hydrogen bonds were fleeting and less consistent over simulation time (Fig. 9D). ZINC20407881 formed eight hydrogen bonds with LYS374, TYR386, ASN429 and HIS433 residues in which only one hydrogen bond established between ASN429 residue and atom O3 of ligand was observed constantly throughout the simulation time (Fig. 9E). Similarly, only one hydrogen bond of ASN429 residue of BRD2 protein was found for atom O5 of control compound among several hydrogen bonds formed between BRD2 protein and ligand which was seen consistently over the simulation. (Fig. 9F). These results revealed that the top five hits interacted with the BRD2 with higher stability than control compound. Combining the activity of each system with the hydrogen bond, we believe that the hydrogen bond had a positive effect on the inhibitory activity of the selected hits and BRD2 protein.
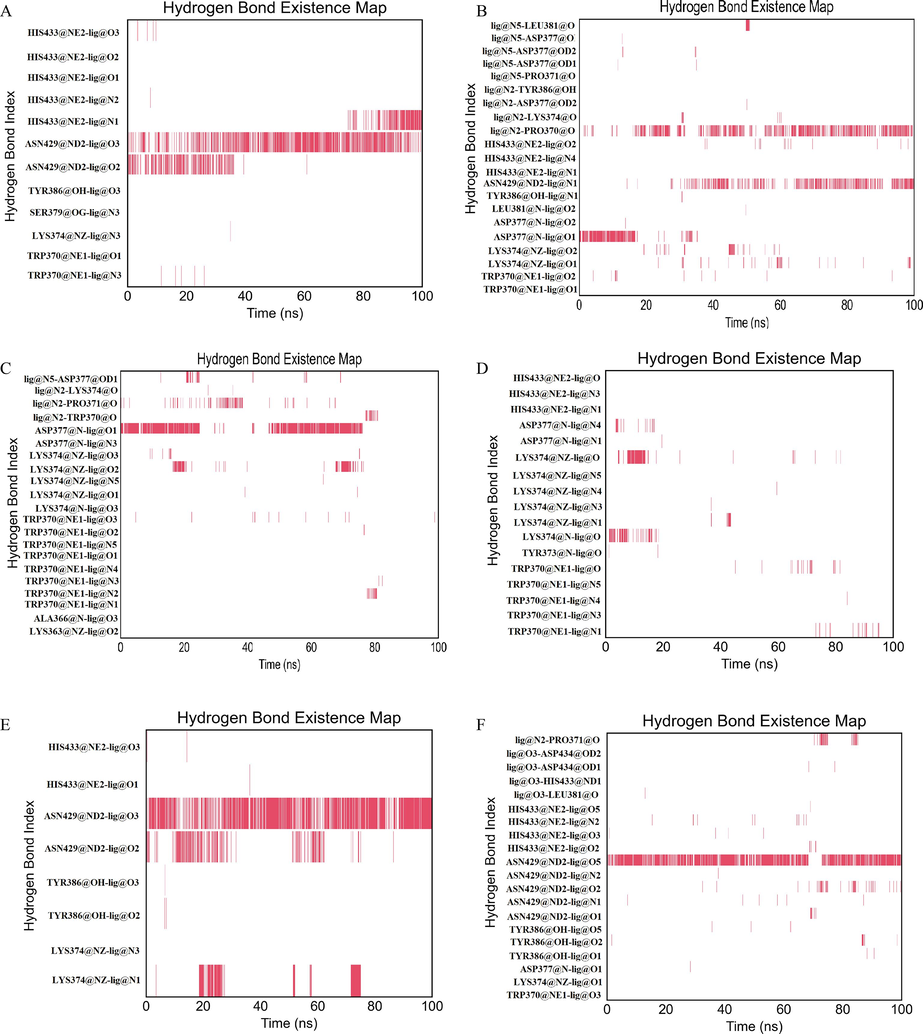
- The hydrogen bond existence map for ligands (a) ZINC20417563 (b) ZINC12322175 (c) ZINC12391479 (d) ZINC13098192 (e) ZINC20407881 (f) control compound over simulation.
3.5 Secondary structure analysis
Protein secondary structure analysis can be utilized to monitor the folding process of protein, the change of protein structure caused by mutation, and the effect of ligand binding on protein structure. The secondary structural elements (a-helix, b-sheet, b-brige, trun, coil, bend, and 5-helix) changes of BRD2 protein bound with hits and control compound during MD simulation were analyzed by dictionary of secondary structure for proteins (DSSP) module (Taghvaei et al.,2022) in GROMACS. DSSP uses hydrogen bonds and special geometric features to identify different secondary structures. The output of DSSP can be displayed as a color map to visually observe the secondary structure changes in each region of the protein during the simulation. The percentage of various secondary structures in the whole simulation time can also be obtained by processing the output data of DSSP. The distribution of the secondary structural elements was seen in Fig. 10, the percentage of various secondary structures was detailed in Table 3, and the secondary structure changes of each region of BRD2 protein were observed in Fig. 11. The secondary structure profile of BRD2 bound with hits and apo BRD2 had similar distribution and changes of the secondary structural elements in most regions. The conserved α-helices and coils with a little of turn and bend elements were mainly formed over majority simulation time in secondary structure profile of BRD2-hit complexes and apo protein. When binding to hits, the percentage of the structure (α-helix + β-sheet + β-bridge + turn) and α-helices were decreased as shown in Fig. 10 and Table 3. The binding of ligands contributed to transforming α-helix to turn and bend in residues 378 ∼ 382 region during the period of 25 ∼ 100 ns simulation as seen in Fig. 11. The binding of the identified compounds caused fluctuation of some residues in BRD2 during simulation between different secondary structural elements. These transformed residues region was located at acetyl-lysine binding pocket at second bromodomain of BRD2. These changes in the secondary structure will affect the conformation of the binding pocket, resulting in the inability of BRD2 to accurately recognize and bind the substrate (acetyl-lysine), thereby reducing the activity of BRD2.
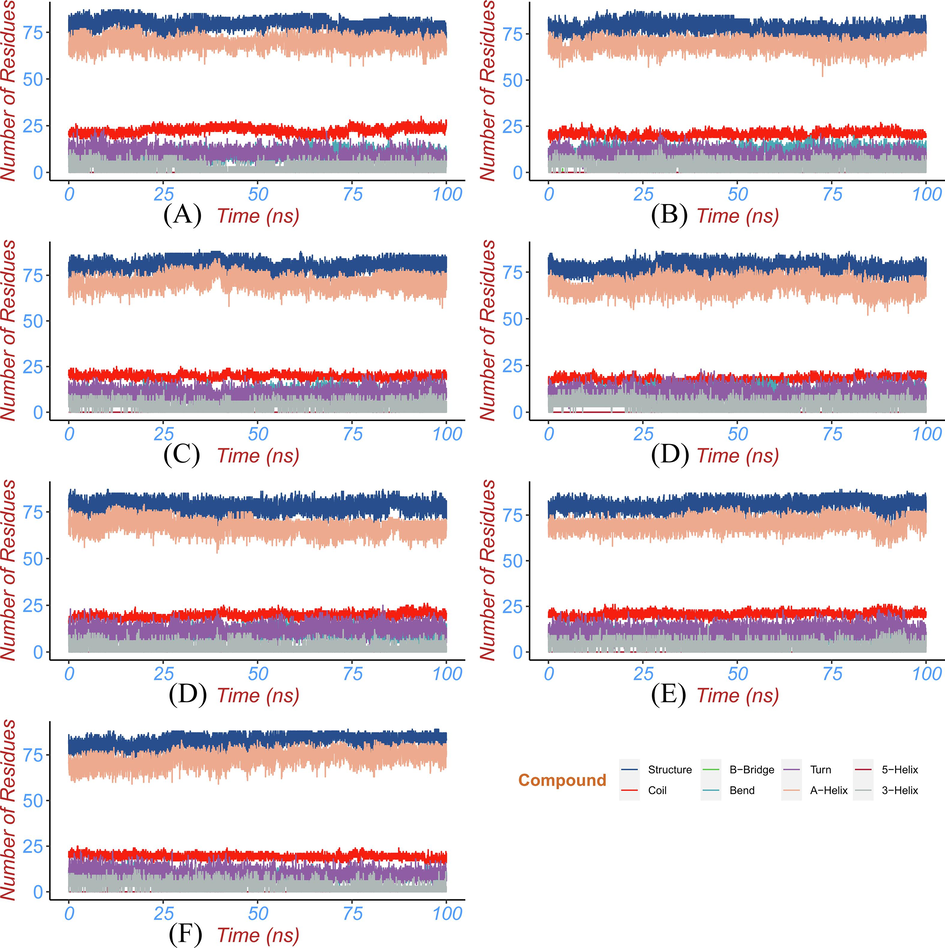
- The percentage of the secondary structure of the protein during the simulation time for (a) ZINC20417563 (b) ZINC12322175 (c) ZINC12391479 (d) ZINC13098192 (e) ZINC20407881 (f) control compound and (g) apo protein.
| system | Structure | Coil | Bend | Turn | A-Helix | 3-Helix |
|---|---|---|---|---|---|---|
| apo | 83 | 19 | 8 | 9 | 73 | 3 |
| control | 81 | 21 | 9 | 11 | 70 | 3 |
| ZINC20417563 | 79 | 22 | 9 | 10 | 70 | 3 |
| ZINC12322175 | 78 | 21 | 11 | 10 | 69 | 4 |
| ZINC12391479 | 81 | 20 | 10 | 9 | 71 | 3 |
| ZINC13098192 | 78 | 18 | 11 | 10 | 68 | 4 |
| ZINC20407881 | 78 | 19 | 10 | 11 | 67 | 2 |
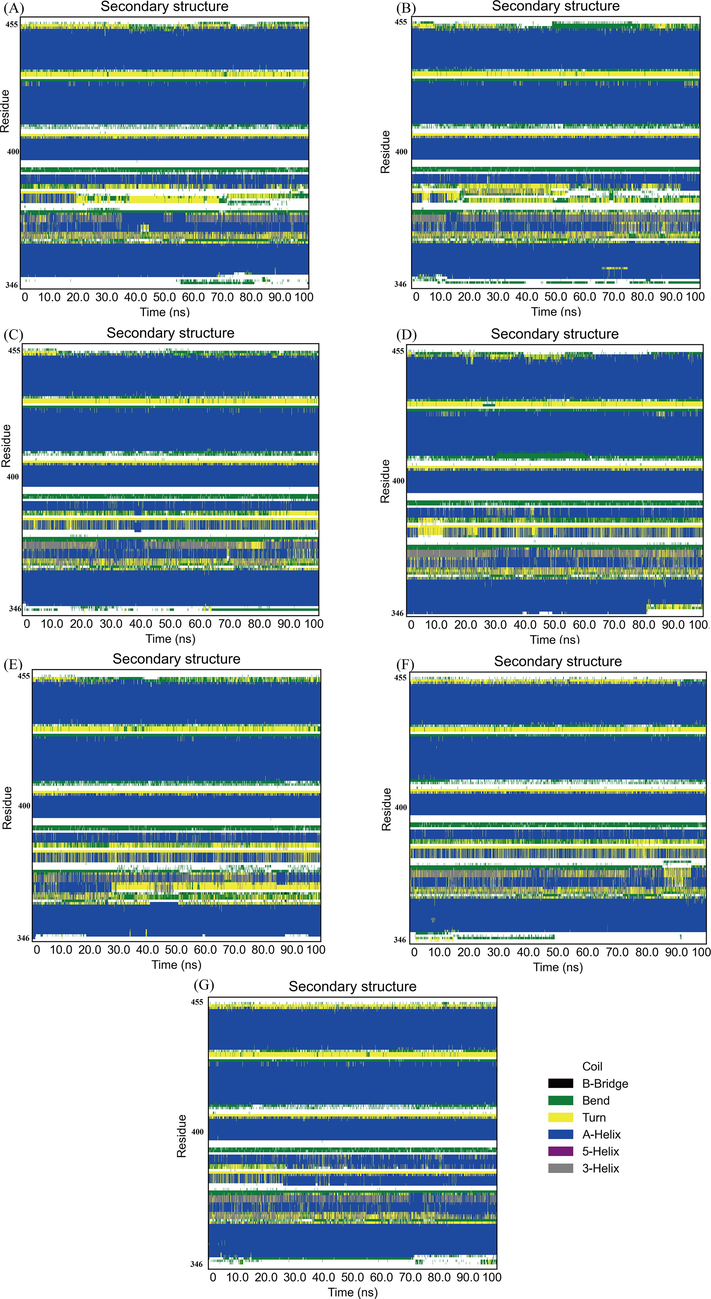
- Color maps to for the secondary structure changes of each region of the protein during the simulation time for (a) ZINC20417563 (b) ZINC12322175 (c) ZINC12391479 (d) ZINC13098192 (e) ZINC20407881 (f) control compound and (g) apo protein.
3.6 Principal component analysis
Studies have shown that although the degree of freedom of the protein is very large, most of its motion is concentrated on a very small number of degrees of freedom. These degrees of freedom form a basic subspace, in which the motion is closely related to the function of the protein. Principal component analysis (PCA) can separate this subspace of proteins, making it possible to understand the functional movement patterns of proteins more clearly (Amadei et al.,1993). Based on the MD simulation trajectory of the protein–ligand complex and apo protein system, the covariance matrix is constructed for Cα atom, and the covariance matrix is then diagonalized into eigenvalues and corresponding eigenvectors. Two eigenvectors with the largest eigenvalue are selected as the projection basis vectors, that is, the direction of functional motion. The simulated trajectory is projected onto the basis vectors, and the two projection coordinates with the largest deviation are stacked to visually see the trend and amplitude of functional motion. The PCA results were visualized by the percentage of variance of eigenvectors graph and essential subspace projection of PC1 vs PC2 graph as shown in Fig. 12. We found that the first three PCs is primarily responsible for the variance seen in the collective motion of BRD2 protein bound with or without ligand. It has been shown that the first three PCs of ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, control-BRD2 docked complexes account for 51.4 %, 71.6 %, 54.1 %, 51.7 %, 55.8 %, and 56.8 % of the variance, whereas the apo BRD2 protein account for 75.8 % of the variance. It should be observed that the first PC was adequate to account for around 50 % of the overall variance in apo BRD2 protein, whereas contribution of PC2 and PC3 was necessary to account for the identical variance in BRD2-ligand complexes. When analyzing the dominating motions in PCA, the first PC is a vital factor. As seen in the graph (Fig. 12), the first PC of apo BRD2 protein accounts for 53.1 % of the variance, while the ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, and control-BRD2 complexes account for 23.6 %, 52.2 %, 33.7 %, 26.9 %, 26 %, and 27.4 % of the overall variance, respectively. The apo BRD2 protein had a considerable drop in variance regarding the initial three PCs, which indicated that the unbound protein exhibited significant conformational motions. However, when binding ligand, BRD2 exhibited a reduction in such conformational motions.
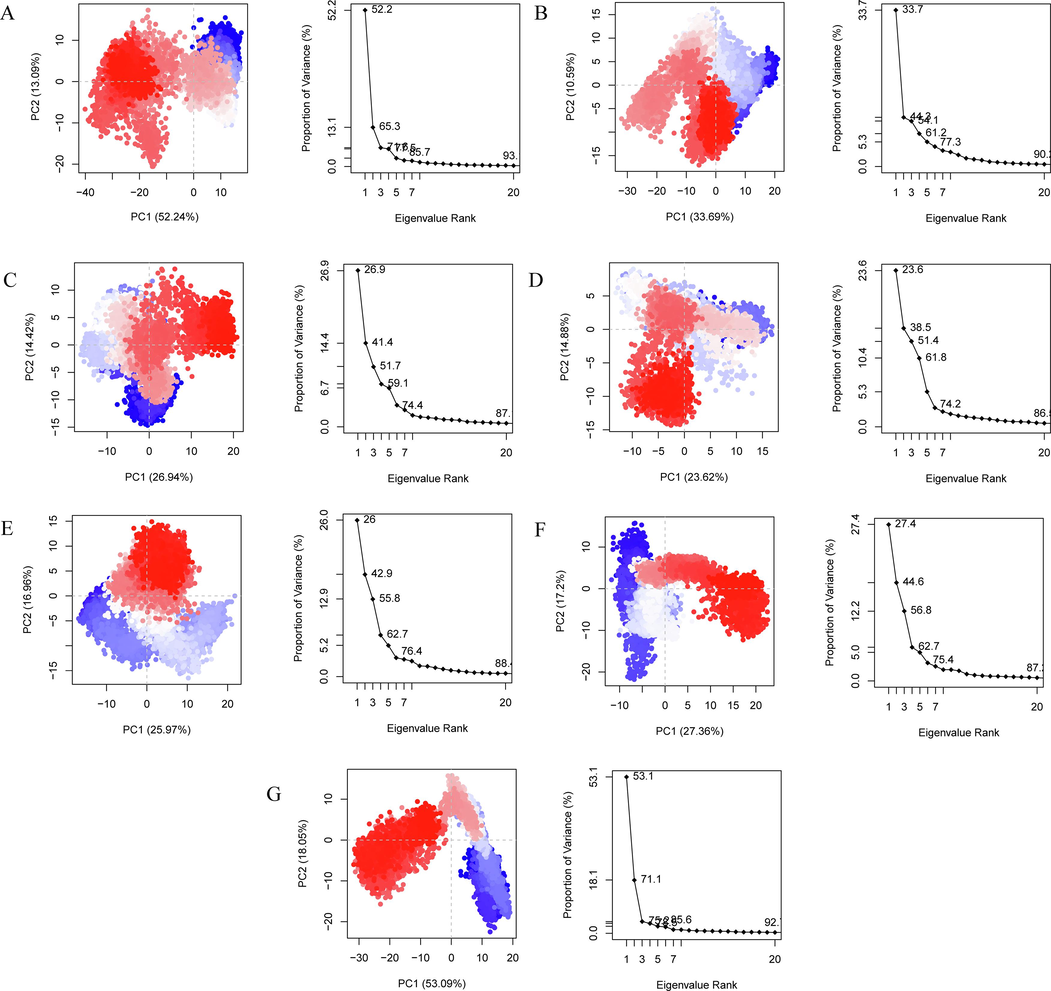
- PCA analysis for (a) ZINC20417563-BRD2 complex (b) ZINC12322175-BRD2 complex (c) ZINC12391479-BRD2 complex (d) ZINC13098192-BRD2 complex (e) ZINC20407881-BRD2 complex (f) control compound-BRD2 complex and (g) apo protein. The percentage of variance of eigenvectors graphs (on the right), and essential subspace projection of PC1 vs PC2 graphs (on the left).
The influence of ligand molecules on the essential dynamics of BRD2 protein was seen in the graphs of essential subspace projection of PC1 vs PC2 (Fig. 12). Protein-ligand complexes had bigger covered area in comparison with the apo BRD2 protein, indicating that a greater portion of the conformational space is sampled, particularly along the first PC. As a result, proteins bound with ligand exhibited more dynamic tertiary structural conformations. Such dynamic tertiary structural conformations are considered to contribute to the formation of stable complexes by ligands in the active pocket of the protein. Furthermore, the region covered by the BRD2 protein bound with hits was also bigger than the protein bound with control compound, showed that this scenario is more flexible than the control case.
3.7 Free energy landscape generated
To depict the energy minimum landscape of control and hit compounds docked BRD2 protein, free energy landscape (FEL) analysis was produced. The Gibbs free energy map of the resulting PC1 and PC2 of eigenvectors was used to investigate the conformational stabilities of the docked complexes as shown in Fig. 13. The values of the Gibbs free energy for ZINC13098192, ZINC20417563, ZINC12322175, ZINC12391479, ZINC20407881, control-BRD2 docked complexes and apo protein ranged from 0 kJ/mol to 12.29 kJ/mol, 14.91 kJ/mol, 12.83 kJ/mol, 11.89 kJ/mol, 11.89 kJ/mol, 15.19 kJ/mol, and 15.44 kJ/mol, respectively. The findings of the global free energy minima demonstrated that the docked complexes displayed more stable states on the folding behavior of BRD2 than apo protein. According to the FEL contour plot (Fig. 13), the binding of selected hits in the binding pocket was more significant and stable than the binding of control compounds. It found that the docked complexes were persistent with significant energy minima, suggesting that the residues involved the docking poses are critical for BRD2 stability and function.
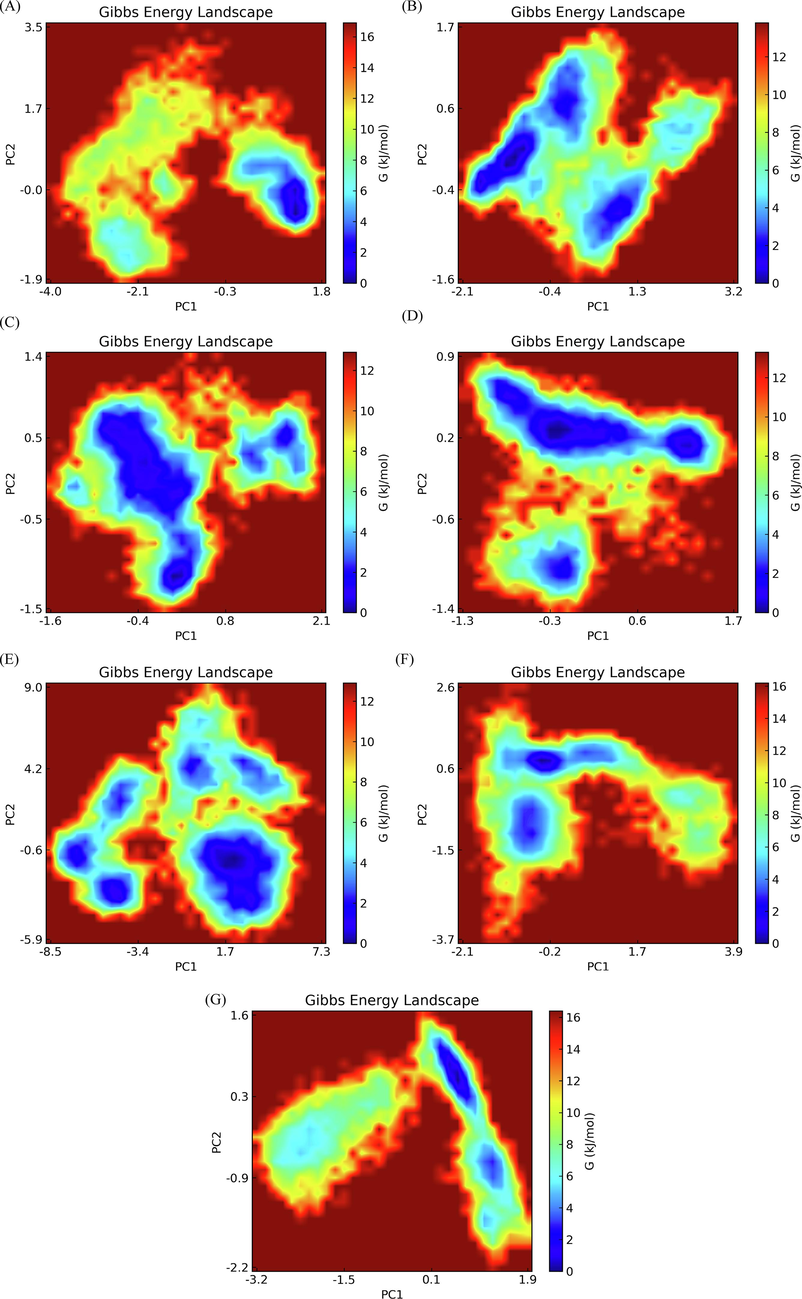
- Free energy landscape for (a) ZINC20417563-BRD2 complex (b) ZINC12322175-BRD2 complex (c) ZINC12391479-BRD2 complex (d) ZINC13098192-BRD2 complex (e) ZINC20407881-BRD2 complex (f) control compound-BRD2 complex and (g) apo protein.
3.8 Dynamics cross-correlation matrix analysis
The concerted movement of protein domains plays an important role in the binding of proteins to their ligands and other specific biological functions. The atomic motion synergy of domains which can play a specific function in protein system is higher. At present, MD simulation sampling is generally used, the dynamic characteristics of the trajectory are analyzed by statistical methods, and finally the correlation of the system motion is obtained using bio3d R package (Yu and Dalby, 2020). The dynamic cross-correlation matrix (DCCM) was calculated by using the coordinates of Cα atoms from the 100 ns MD trajectories. The DCCM plots showed the difference in correlated motions between BRD2 protein bound with and without compound (Fig. 14). The reduction of correlated motions were observed for BRD2 protein bound with selected five hits and control compound compared to apo protein. The BRD2-ZINCZINC20417563 docked complex displayed a slight decrease in correlated motions, while maximum decrease in correlated motions was seen for BRD2 protein bound with ZINC13098192 compound. The DCCM analysis revealed that the activity of BRD2 protein decreased when binding with compounds.
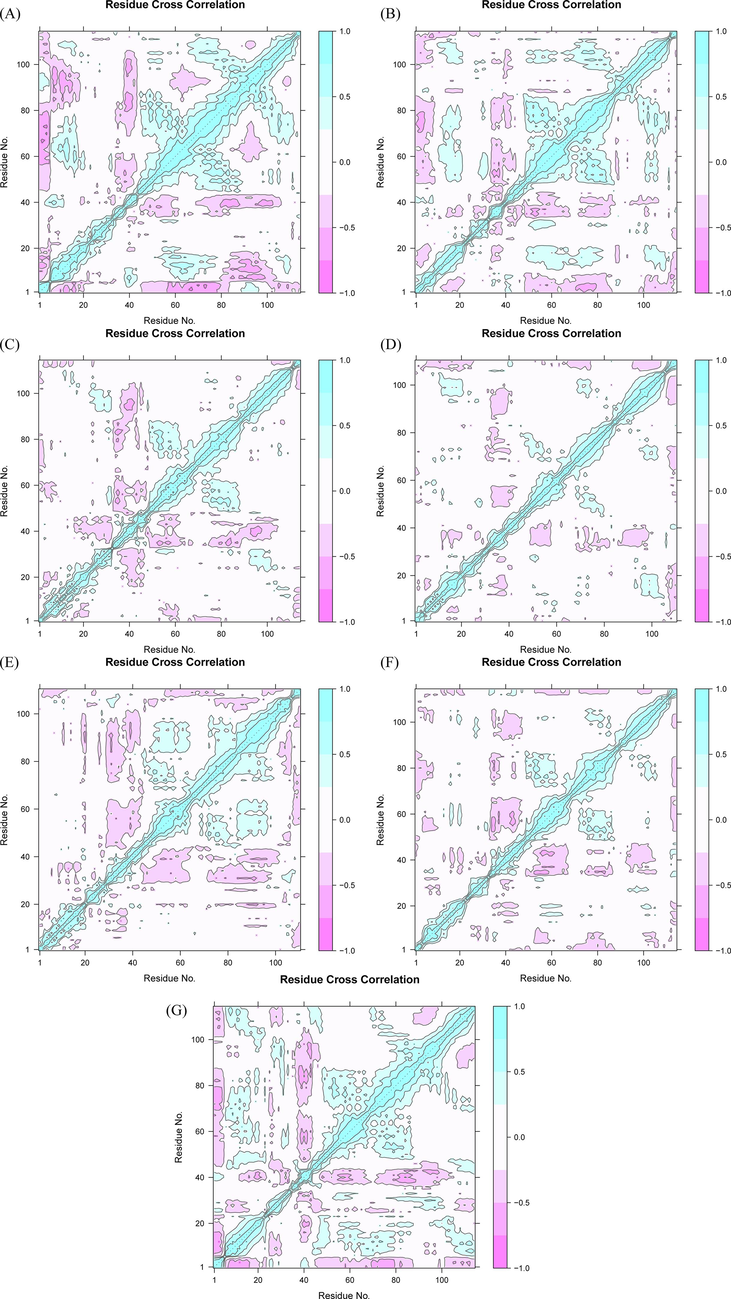
- The dynamics cross-correlation matrix (DCCM) plots for (a) ZINC20417563-BRD2 complex (b) ZINC12322175-BRD2 complex (c) ZINC12391479-BRD2 complex (d) ZINC13098192-BRD2 complex (e) ZINC20407881-BRD2 complex (f) control compound-BRD2 complex and (g) apo protein. The positive value represents correlated motions in cyan, while the negative values represent the anti-correlated motions in pink.
3.9 Binding free energy
In order to study the interaction between BRD2 protein and the hits in more detail, binding free energy of each system between hit and protein can be calculated by the Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) method using the MM-PBSA method in g-mmpbsa tools. We selected 1000 frames uniformly from the 100 ns trajectory of simulation to calculate its binding free energy and the listed the results in Table 4. The order of binding free energiey of six systems is ZINC20417563 (−12.988 kcal/mol) < ZINC20407881 (−9.176 kcal/mol) < ZINC12322175 (−8.47 kcal/mol) < ZINC12391479 (−7.172 kcal/mol) < ZINC13098192 (−7.02 kcal/mol) < control (−6.257 kcal/mol). It indicated that control interacted with the BRD2 with lower stability than selected hits. From the results of binding free energy calculation, it can be seen that the van der Waals force (ΔEvdw), electrostatic interaction (ΔEele) and non-polar solvation free energy (ΔEnonpolar) are favorable for the binding of protein receptors and ligands, while the polar solvation free energy (ΔEpolar) plays a major adverse effect on the binding. In short, van der Waals force, electrostatic interaction and nonpolar interaction provided the main driving forces for binding these compounds to BRD2.
| Energy (kcal/mol) | △Evdw | △Eelec | △Epolar | △Enonpolar | -T△S | △Gbind |
|---|---|---|---|---|---|---|
| ZINC13098192 | –32.638 | −5.779 | 22.772 | −4.346 | 12.971 | −7.020 |
| ZINC20407881 | −34.915 | −4.904 | 21.968 | −5.302 | 13.981 | −9.176 |
| ZINC20417563 | −35.353 | −1.812 | 19.932 | −5.471 | 9.722 | −12.988 |
| ZINC12391479 | −38.674 | −9.160 | 27.124 | −5.622 | 19.163 | −7.172 |
| ZINC12322175 | −43.749 | −7.834 | 32.586 | −6.094 | 16.624 | −8.470 |
| control | −30.877 | −6.027 | 22.547 | −4.757 | 12.860 | −6.257 |
△Evdw: van der Waals interaction energy; △Eelec:electrostatic interaction enery; -T△S:entropic energy; △Epolar: polar solvation energy; △Enonpolar: nonpolar solvation energy; △Gbind:binding free energy.
To further analyze the amino acid residues that play an important role in the binding process, we decomposed the binding free energy between the protein and ligand into each amino acid residue. Most of the key residues in six systems are similar, suggesting that the binding patterns of the six small compounds are not significantly different (Fig. 15). Among the six complexes, Pro371, Val376, Leu381, Leu383, Ile663 have important contributions.
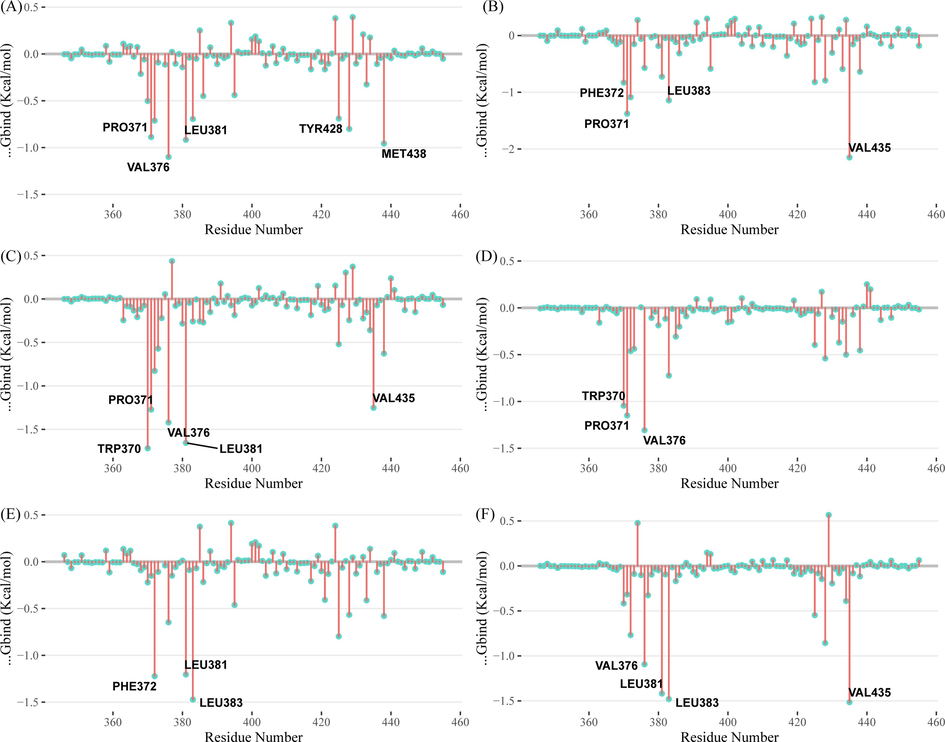
- Residue binding energy contribution of (a) ZINC20417563-BRD2 complex (b) ZINC12322175-BRD2 complex (c) ZINC12391479-BRD2 complex (d) ZINC13098192-BRD2 complex (e) ZINC20407881-BRD2 complex and (f) control compound-BRD2 complex.
3.10 The ADME properties
The ADME properties refer to the absorption, distribution, metabolism and excretion of drugs in the human body, involving the pharmacokinetic properties of drugs in the body, and are key factors in measuring whether a compound can be a drug (Daoud et al.,2021). The physicochemical properties and ADME properties of the top five hits from docking were assessed using swissADME web server as shown in Table 5. The results showed that pharmacokinetic and ADMET properties of the five selected compounds were in an acceptable range, suggesting these compounds may be potential drug candidates.
| Molecule | ZINC20417563 | ZINC12322175 | ZINC12391479 | ZINC13098192 | ZINC20407881 |
|---|---|---|---|---|---|
| MW | 443.54 | 483.58 | 499.58 | 487.59 | 429.51 |
| Heavy atoms | 33 | 35 | 36 | 35 | 32 |
| Rotatable bonds | 5 | 7 | 7 | 7 | 5 |
| H-bond acceptors | 6 | 4 | 5 | 4 | 6 |
| H-bond donors | 0 | 1 | 1 | 0 | 0 |
| Log Po/w (XLOGP3) | 3.84 | 5.23 | 5.27 | 4.48 | 3.48 |
| Log S (ESOL) | −5.15 | −6.18 | −6.29 | −5.71 | −4.85 |
| GI absorption | High | High | High | High | High |
| BBB permeant | Yes | No | No | No | Yes |
| Pgp substrate | Yes | Yes | Yes | Yes | Yes |
| CYP1A2 inhibitor | No | No | No | No | No |
| CYP2C19 inhibitor | No | Yes | Yes | Yes | No |
| CYP2C9 inhibitor | Yes | Yes | Yes | Yes | Yes |
| CYP2D6 inhibitor | Yes | No | Yes | No | Yes |
| CYP3A4 inhibitor | Yes | Yes | Yes | Yes | Yes |
| log Kp (cm/s) | −6.28 | −5.54 | −5.61 | −6.09 | −6.45 |
| Lipinski #violations | 0 | 1 | 0 | 1 | 0 |
| Bioavailability Score | 0.55 | 0.55 | 0.55 | 0.55 | 0.55 |
4 Conclusion
This study aimed to discover potent inhibitor from ZINC database against BRD2 to block SARS-CoV2 infection through pharmacophore-based virtual screening, docking-based virtual screening and molecular dynamics simulation. Through structure-based virtual screening, five compounds (ZINC20417563, ZINC12322175, ZINC12391479, ZINC13098192, and ZINC20407881) with high binding affinity compared to the existing BRD2 inhibitor (RVX-208) were screened from ZINC database. The docking analysis showed that the screened compounds tightly bound to the substrate binding pocket of BRD2 and hindering the binding of the acetyl-lysine substrate to active sites thereby inhibiting BRD2 activity and Compared with pp, the screened compounds bind to BRD2 more closely and the existing BRD2 inhibitor (RVX-208). The molecular dynamics analysis showed these compound-BRD2 docked complexes were properly stable and these compounds were tightly adhered to active site of BRD2. The principal component analysis, DCCM analysis and free energy landscape analysis also showed that the stable conformation observed in BRD2 bound of the screened compounds throughout molecular dynamics simulation. MM-PBSA analysis showed the screened compounds are more likely to form stable complexes with BRD2 in comparison to existing BRD2 inhibitor (RVX-208). The identified compounds could reduce the expression of ACE2 through inhibiting BRD2, thereby the blocking SARS-CoV-2 entry into host cells through ACE2 receptor. The screened compounds necessary appropriate experimental validation to determine their inhibitory efficacy against SARS-CoV-2 and subsequently the treatment efficacy of COVID-19.
Funding
This work was supported by Science and Technology Plan Project of Wuwei, Grant/Award Numbers:WW2202RPZ032 (to S.X.).
CRediT authorship contribution statement
Shaohua Xu: Writing – review & editing, Conceptualization, Writing – original draft, Writing – review & editing. Huicheng Yuan: Methodology, Writing – original draft. Ling Li: Methodology. Kai Yang: Software, Visualization. Liangcun Zhao: Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Antiviral drug discovery: preparing for the next pandemic. Chem. Soc. Rev.. 2021;50(6):3647-3655.
- [CrossRef] [Google Scholar]
- A review of methods available to estimate solvent-accessible surface areas of soluble proteins in the folded and unfolded states. Curr. Protein Pept. Sci.. 2014;15:456-476.
- [CrossRef] [Google Scholar]
- Essential dynamics of proteins. Proteins Struct. Funct. Bioinform.. 1993;17(4):412-425.
- [CrossRef] [Google Scholar]
- Significant perspectives onvarious viral infections targeted antiviral drugs and vaccines including COVID-19pandemicity. Mol. Biomed.. 2022;3(1):21.
- [CrossRef] [Google Scholar]
- Computer aided drug design: success and limitations. Curr. Pharm. Des.. 2016;22(5):572-581.
- [CrossRef] [Google Scholar]
- Direct-acting antivirals and host-targeting strategies to combat enterovirus infections. Curr. Opin. Virol.. 2017;24:1-8.
- [CrossRef] [Google Scholar]
- Efficient RMSD measures for the comparison of two molecular ensembles Root-mean-square deviation. Proteins. 2003;50:26-34.
- [CrossRef] [Google Scholar]
- Molecular modeling of surfactant micellization using solvent-accessible surface area. Langmuir. 2019;35:2443-2450.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- ADMET profiling in drug discovery and development:perspectives of in silico, in vitro and integrated approaches. Curr. Drug Metab.. 2021;22(7):503-522.
- [CrossRef] [Google Scholar]
- Virus-host interactomics: new insights and opportunities for antiviral drug discovery. Genome Med.. 2014;6(11):115.
- [CrossRef] [Google Scholar]
- Host-targeting oral antiviral drugs to prevent pandemics. Lancet. 2022;399(10333):1381-1382.
- [CrossRef] [Google Scholar]
- Synthesis, characterization, DNA binding, and biological action of dimedone arylhydrazone chelates. Appl. Organomet. Chem.. 2022;36(4):e6576.
- [Google Scholar]
- Metal–organic frameworks encapsulated with an anticancer compound as drug delivery system: synthesis, characterization, antioxidant, anticancer, antibacterial, and molecular docking investigation. Appl. Organomet. Chem.. 2022;36(5):e6660.
- [Google Scholar]
- Synthesis, characterization, catalytic, DNA binding and antibacterial activities of Co(II), Ni(II) and Cu(II) complexes with new Schiff base ligand. J. Mol. Liq.. 2021;326:115381
- [CrossRef] [Google Scholar]
- Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov.. 2014;13(5):337-356.
- [CrossRef] [Google Scholar]
- SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun.. 2021;12(1):3818.
- [CrossRef] [Google Scholar]
- Bromodomain and extraterminal protein inhibitor, Apabetalone (RVX-208), Reduces ACE2 expression and attenuates SARS-Cov-2 infection in vitro. Biomedicines. 2021;9(4):437.
- [CrossRef] [Google Scholar]
- The Bio3D packages for structural bioinformatics. Protein Sci.. 2021;30(1):20-30.
- [CrossRef] [Google Scholar]
- Validation of molecular docking Programs for virtual screening against dihydropteroate Synthase. J. Chem. Inf. Model.. 2009;49:444-460.
- [CrossRef] [Google Scholar]
- all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem.. 2013;34(CHARMM36):2135-2145.
- [CrossRef] [Google Scholar]
- Perspectives on the use and risk of adverse events associated with cytokine-storm targeting antibodies and challenges associated with development of novel monoclonal antibodies for the treatment of COVID-19 clinical cases. Hum. Vaccin. Immunother.. 2021;17(9):2824-2840.
- [CrossRef] [Google Scholar]
- ZINCPharmer: pharmacophore search of the ZINC database. Nucleic Acids Res.. 2012;40(Web Server issue):W409-W414.
- [CrossRef] [Google Scholar]
- A teach-discover-treat application of zincpharmer: an online interactive Pharmacophore modeling and virtual screening tool. PLoS One. 2015;10(8):e0134697.
- [Google Scholar]
- Empirical hydrogen-bond potential functions–an old hat reconditioned. ChemPhysChem. 2011;12:3131-3142.
- [CrossRef] [Google Scholar]
- g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model.. 2014;54:1951-1962.
- [CrossRef] [Google Scholar]
- Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Antimicrob. Agents. 2020;55(3):105924
- [CrossRef] [Google Scholar]
- In silico study of novel niclosamide derivatives, SARS-CoV-2 nonstructural proteins catalytic residue-targeting small molecules drug candidates. Arab. J. Chem.. 2023;16(5):104654
- [CrossRef] [Google Scholar]
- Accounting for electronic polarization in non- polarizable force fields. PCCP. 2011;13:2613-2626.
- [CrossRef] [Google Scholar]
- SARS-CoV-2-host proteome interactions for antiviral drug discovery. Mol. Syst. Biol.. 2021;17(11):e10396
- [CrossRef] [Google Scholar]
- BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. 2021;184(8):2167-2182.e22.
- [CrossRef] [Google Scholar]
- AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem.. 2009;30:2785-2791.
- [CrossRef] [Google Scholar]
- Identification of terpenoids from abrus precatorius against parkinson's disease proteins using in silico approach. Bioinf. Biol. Insights. 2021;15:11779322211050757
- [CrossRef] [Google Scholar]
- RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U.S.A.. 2013;110(49):19754-19759.
- [CrossRef] [Google Scholar]
- Virus-Host interactions: new insights and advances in drug development against viral pathogens. Curr. Drug Metab.. 2017;18(10):942-970.
- [CrossRef] [Google Scholar]
- Identification of potential Indonesian marine invertebrate bioactive compounds as TMPRSS2 and SARS-CoV-2 Omicron spike protein inhibitors through computational screening. Arab. J. Chem.. 2023;16(9):104984
- [CrossRef] [Google Scholar]
- COVID-19 therapeutics: challenges and directions for the future. PNAS. 2022;119(15)
- [CrossRef] [Google Scholar]
- Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct.. 1999;28:155-179.
- [CrossRef] [Google Scholar]
- BRD2 inhibition blocks SARS-CoV-2 infection by reducing transcription of the host cell receptor ACE2. Nat. Cell Biol.. 2022;24(1):24-34.
- [CrossRef] [Google Scholar]
- The pharmacophore concept and its applications in computer-aided drug design. Prog. Chem. Org. Nat. Prod.. 2019;110:99-141.
- [CrossRef] [Google Scholar]
- A computational approach to modeling an antagonistic angiogenic vegfr1-il2 fusion protein for cancer therapy. Bioinf. Biol. Insights. 2021;15:11779322211043297
- [CrossRef] [Google Scholar]
- The bromodomain-containing gene BRD2 is regulated at transcription, splicing, and translation levels. J. Cell. Biochem.. 2011;112(10):2784-2793.
- [CrossRef] [Google Scholar]
- A model for statistical significance of local similarities in structure. J. Mol. Biol.. 2003;326:1307-1316.
- [CrossRef] [Google Scholar]
- Identification of novel anti-cancer agents, applying in silico method for SENP1 protease inhibition. J. Biomol. Struct. Dyn.. 2022;40(14):6228-6242.
- [CrossRef] [Google Scholar]
- Potential bioactive compounds as SARS-CoV-2 inhibitors from extracts of the marine red alga Halymenia durvillei (Rhodophyta) - a computational study. Arab. J. Chem.. 2021;14(11):103393
- [CrossRef] [Google Scholar]
- A novel phenanthridionone based Scaffold as a potential inhibitor of the BRD2 bromodomain: crystal structure of the complex. PLoS One. 2016;11(5):e0156344.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [CrossRef] [Google Scholar]
- GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845-854.
- [CrossRef] [Google Scholar]
- CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem.. 2010;31:671-690.
- [CrossRef] [Google Scholar]
- Automation of the CHARMM General force field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model.. 2012;52:3144-3154.
- [CrossRef] [Google Scholar]
- Automation of The CHARMM General Force Field (CGenFF) II: assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model.. 2012;52:3155-3168.
- [CrossRef] [Google Scholar]
- Combinations of host- and virus-targeting antiviral drugs confer synergistic suppression of SARS-CoV-2. Microbiol. Spectr.. 2022;10(5):e0333122
- [CrossRef] [Google Scholar]
- Recent developments and applications of the MMPBSA method. Front. Mol. Biosci.. 2018;4(87)
- [Google Scholar]
- Universal relation between instantaneous diffusivity and radius of gyration of proteins in aqueous solution. Phys. Rev. Lett.. 2021;126:128101
- [CrossRef] [Google Scholar]
- Gyration-radius dynamics in structural transitions of atomic clusters. J. Chem. Phys.. 2007;126:124102
- [CrossRef] [Google Scholar]
- A beginner's guide to molecular dynamics simulations and the identification of cross-correlation networks for enzyme engineering. Methods Enzymol.. 2020;643:15-49.
- [CrossRef] [Google Scholar]
- Computer-aided drug design methods. Methods Mol. Biol.. 2017;1520:85-106.
- [CrossRef] [Google Scholar]
- SARS-CoV-2: an emerging coronavirus that causes a global threat. Int. J. Biol. Sci.. 2020;16(10):1678-1685.
- [CrossRef] [Google Scholar]
- Hit identification and optimization in virtual screening: practical recommendations based on a critical literature analysis. J. Med. Chem.. 2013;56:6560-6572.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105365.
Appendix A
Supplementary material
The following are the Supplementary data to this article:




