

Translate this page into:
Conventional and microwave techniques for the synthesis and antimicrobial studies of novel 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)-(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones
⁎Corresponding author. dnisheeth@rediffmail.com (N.C. Desai)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In this paper, we have described the conventional and microwave methods for the synthesis of 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l). Through this method, we have observed a decrease in reaction time and a better yield than the previously described conventional method. The application of microwave irradiation (MWI) is used for carrying out chemical transformations which were pollution free and eco-friendly. The structures of the compounds were characterized by IR, NMR and mass spectral data. These compounds (4a–l) were evaluated for their in vitro antimicrobial screening on different strains of bacteria and fungi. Among the compounds tested, compound 4h showed the highest activity.
Keywords
Quinoline–oxadiazole based chalcones
2-Chloro-6-methylquinoline-3-carbaldehyde
Microwave method for quinoline–oxadiazole derivatives

1 Introduction
The quinoline nucleus is an important class of heterocyclic compounds found in many synthetic and natural products with a wide range of pharmacological activities, such as antimalarial (Larsen et al., 1996), anti-inflammatory (Chen et al., 2001), antiasthmatic (Roma et al., 2000), antibacterial (Dube et al., 1998) and tyrosine kinase inhibiting agents (Billker et al., 1998). The quinoline ring systems are important structural units in naturally occurring alkaloids and synthetic analogues with interesting biological activities. Therefore, the development of new and efficient synthetic route for the preparation of their analogues is an important aspect both in synthetic chemistry and medicinal chemistry (Balasubramanian and Keay, 1996; Ranu et al., 2003; Wada et al., 2003; Kobayashi et al., 2003).
The chemistry of 1,3,4-oxadiazoles has received considerable attention from synthetic organic chemists due to their diverse biological activities (Omar et al., 1996; Hui et al., 2002; Mohan et al., 2004). Several research groups have contributed for the development of methods for the synthesis of 1,3,4-oxadiazoles (Chiba and Mitsuhiro, 1992; Bacu et al., 2003; Bhat et al., 2004). However, these procedures are time consuming and proceed in low yields. Therefore, a convenient and eco-friendly method for the synthesis of 1,3,4-oxadiazoles is highly desirable.
Chalcones are natural biocides (Geiger and Conn, 1945) and are well known intermediates for synthesizing various heterocycles. Chalcones and their derivatives are also medicinally important. Many chalcone derivatives have been reported to possess antimalarial, antibacterial and antifungal properties (Katritzky, 1984). Anticancer properties of some simple chalcone derivatives have also been reported in literature (Rezig et al., 2000; Ducki et al., 1996). The synthesis of quinolinyl chalcones is scarcely reported in literature. Sayed et al. (1976) and Ibrahim et al. (1996) have synthesized a few quinoline–chalcone derivatives by Claisen–Schmidt condensation reaction. Dominguez et al. (2001) have reported the synthesis of some quinoline–chalcones and claimed their antimalarial activity. Moussaoui et al. (2002) have also described the synthesis of quinolinyl chalcones and claimed their cytotoxicity in K 562 human leukaemia cell lines.
The formation of heterocyclic rings by cyclocondensation reactions is typically a process well-suited for the microwave technology. Many of these condensation reactions require high temperatures and conventional reaction conditions very often involve heating the reactants in an oil, metal or sand bath for many hours or even days. An example is the formation of 4-hydroxy-1-H-quinoline-2-ones from anilines and malonic esters. The corresponding conventional, thermal protocol involves heating the two components in equimolar amounts in an oil bath at 220–300 °C for several hours (without solvent), whereas similar high yields can be obtained by microwave heating at 250 °C for 10 min (Kappe, 2004). This prompted us to adopt the MWI method for the synthesis of bio-active heterocyclic molecules.
The present study aims at preparing quinoline–oxadiazole–chalcone derivatives. The solvent-free organic reactions assisted by microwave irradiation in organic synthesis can increase the purity of the resulting products, enhance the chemical yield and shorten the reaction time. Solvent-free reaction leads to a clean, eco-friendly and economic technology. Reaction on solid support without using solvent usually with a closed vessel in Synthos-3000, Anton Paar microwave reaction system are currently popular among synthetic chemists to create eco-friendly atmosphere (Loupy, 2002; Varma et al., 1998; Rana et al., 2009).
Looking at the medicinal importance of quinolines, 1,3,4-oxadiazoles and chalcones, we have designed and synthesized a series of 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l) (Scheme 1) by conventional and microwave methods. The physical constants by conventional method are shown in Table 1 and by microwave method are shown in Table 2. The structures of the compounds synthesized were assigned on the basis of IR, 1H NMR, 13C NMR and mass spectral data.![Synthesis of 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l).](/content/184/2016/9/1_suppl/img/10.1016_j.arabjc.2011.05.004-fig1.png)
Synthesis of 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l).
Compd. No.
-R
M.F.
Yield (%)
m.p. (°C)
Reaction time (h)
Analysis (%)
C
H
N
4a
-2-C1
C27H18Cl2N4O4
76
278
6
60.80
60.85
3.40
3.47
10.50
10.55
4b
-3-C1
C27H18Cl2N4O4
64
243
5
60.80
60.84
3.40
3.46
10.50
10.56
4c
-4-C1
C27H18Cl2N4O4
73
223
7.5
60.80
60.86
3.40
3.46
10.50
10.55
4d
-2-NO2
C27H18ClN5O6
68
247
7
59.62
59.67
3.33
3.39
12.87
12.93
4e
-3-NO2
C27H18ClN5O6
62
231
6.5
59.62
59.68
3.33
3.37
12.87
12.93
4f
-4-NO2
C27H18ClN5O6
69
239
5.5
59.62
59.67
3.33
3.38
12.87
12.94
4g
-2-OH
C27H19ClN4O5
67
200
6
62.98
62.99
3.71
3.77
10.88
10.93
4h
-3-OH
C27H19ClN4O5
65
249
7
62.98
62.96
3.71
3.76
10.88
10.92
4i
-4-OH
C27H19ClN4O5
58
236
6.8
62.98
62.97
3.71
3.77
10.88
10.94
4j
-4-CH3
C28H21ClN4O4
73
220
6.5
65.56
65.62
4.12
4.18
10.92
10.97
4k
-4-OCH3
C28H21ClN4O5
73
198
8
63.58
63.64
4.00
4.06
10.59
10.65
4l
3,4,5-(OCH3)3
C30H25ClN4O7
64
221
7.5
61.17
61.22
4.27
4.32
9.51
9.56
Compd. No.
-R
Reaction time (min)
Yield (%)
m.p. (°C)
4a
-2-C1
1.7
77
278–280
4b
-3-C1
2.5
68
243–244
4c
-4-C1
1.5
78
223–225
4d
-2-NO2
3.5
71
247–249
4e
-3-NO2
4.2
65
231–233
4f
-4-NO2
3
72
239–240
4g
-2-OH
2.5
70
200–202
4h
-3-OH
3.5
74
249–250
4i
-4-OH
2
63
236–238
4j
-4-CH3
4
76
220–221
4k
-4-OCH3
2.5
77
198–200
4l
3,4,5-(OCH3)3
3
69
221–223
In the first step, acetic anhydride cleaves to give acetyl carbocation and acetate carbanion, which will be used in the reaction with aroylhydrazone intermediate. The lone pair present over nitrogen atom will attack on the cationic carbon of 2-chloro-6-methylquinoline-3-carbaldehyde moiety, which will undergo self-condensation reaction to form aroylhydrazone derivative. Due to the keto-imine tautomerization, the keto group of aroylhydrazone derivative converts to hydroxyl group. The negatively charged oxygen atom which is formed due to deprotonation of the hydroxyl group will attack on C⚌N bond to form oxadiazole ring. The nitrogen anion will attack on acetyl carbocation to form acetyl derivative of oxadiazole moiety. Here acetyl group undergoes Claisen–Schmidt condensation reaction which is a well known reaction. The reaction mechanism is described in Scheme 2 by taking the example of compound 4a–l.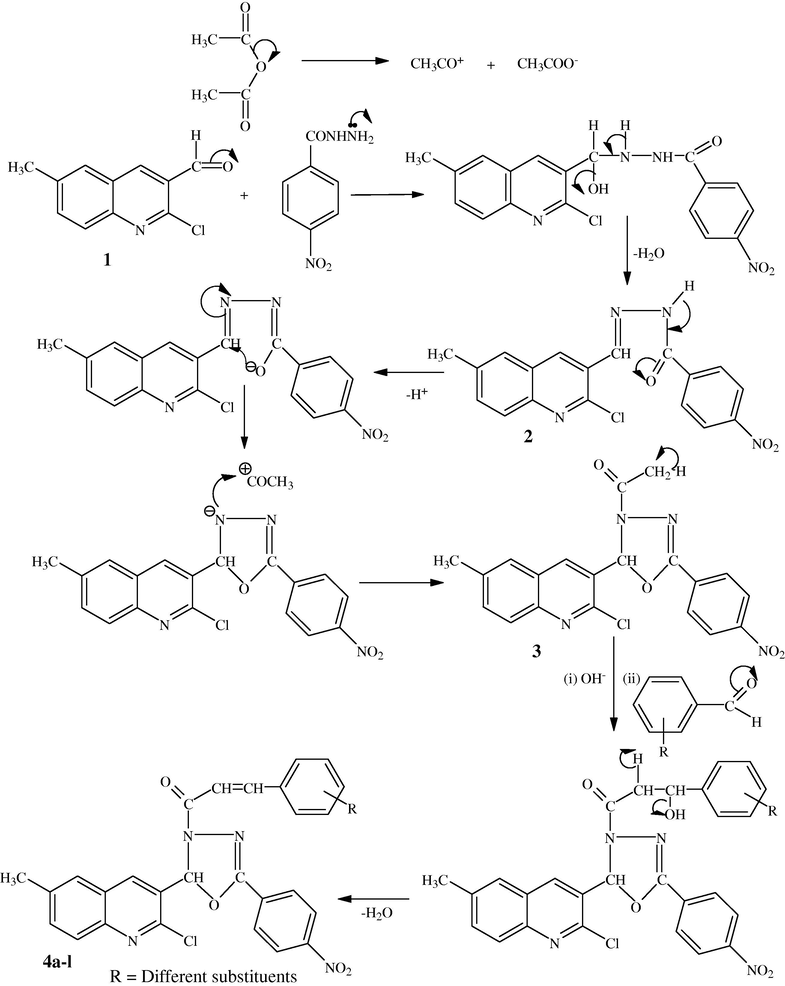
The mechanism of the synthesized compound 4a–l.
2 Experimental
2.1 Materials and methods
All the required chemicals were purchased from E. Merck. 2-Chloro-6-methylquinoline-3-carbaldehyde was synthesized by Meth-Cohn and Narine (1978) and modified (Kappe et al., 1994; Bawa and Kumar, 2009). IR spectra were recorded on a Perkin Elmer FT-IR spectrophotometer. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX-40C instrument at 400 MHz. Chemical shifts are reported in ppm referenced to the residual solvent signal. Mass spectra were recorded on JEOL SX-102. Elemental analysis was performed by a Perkin-Elmer 2400-CHN analyzer. Melting points were recorded on a Gallenkemp apparatus and were uncorrected. Aluminium coated TLC plates 60 F245 (E. Merck) were used for monitoring the reaction and purity of compounds. In the conventional method, compounds were synthesized by using a Random synthesizer. Bookie Rotavapour was used for the distillation, while microwave irradiation was carried out in Synthos-3000 Anton Paar, microwave reaction system.
2.2 Chemical synthesis
The conventional method involves the reaction of 2-chloro-6-methylquinoline-3-carbaldehyde (1) and 4-nitrophenyl hydrazide in the presence of a catalytic amount of glacial acetic acid in ethanol to form aroylhydrazone (2), the same reaction performed under the microwave irradiation, same starting material in 1,4-dioxane (5 drops) was subjected to microwave irradiation at 200 W intermittently at 30 s intervals for the specified time to obtain aroylhydrazone (2) as a product. In the third step, aroylhydrazone (2) was cyclized by refluxing with excessive amount of acetic anhydride for 6 h which resulted in product (3), while in microwave irradiation Silica gel (1 g) was added to the mixture of aroylhydrazone (2) and acetic anhydride (2 mL) and was irradiated in microwave oven at 400 W intermittently at 30 s intervals for 3–4 times to yield (3). In the final step, intermediate (3) was reacted with substituted aldehydes in the presence of KOH (0.01 mol) in 25 mL ethanol (95%) for 6 h to furnish the final product 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l), while the same product is obtained by microwave irradiation at 100 W intermittently at 30 s intervals for the specified time. Thus, the final product (4a–l) is obtained.
2.2.1 General procedure for the synthesis of N-[(1E)-1-aza-2-(2-chloro-6-methyl(3-quinolyl))vinyl]-(4-nitrophenyl) carboxamide (2)
2.2.1.1 Conventional method
A mixture of 2-chloro-6-methylquinoline-3-carbaldehyde (1) and 4-nitrophenyl hydrazide was mixed and dissolved in ethanol and a few drops of glacial acetic acid was added and the mixture was refluxed for 5–6 h by using a reflux condenser. After cooling, the crystals formed were filtered off and recrystallized from alcohol (99.5%) to give product (2). m.p. 128–130 °C, yield 71%; IR (cm−1): 3034 (aromatic–H), 1676 (C⚌O), 1608 (C⚌N), 1593, 1563, 1432 (C⚌C benzene and quinoline ring), 1340 (–NO2 symmetric stretching), 1522 (–NO2 asymmetric stretching), 761, 774 (C–Cl bond, mono substituted benzene). 1H NMR: δ 2.43 (s, 3H, –CH3), 7.50 (s, 1H, N⚌C–H), 7.51–8.90 (m, 8H, quinoline–H and Ar–H), 10.88 (s, 1H, O⚌C–N–H); 13C NMR: δ 24.7, 124.0, 124.1, 126.4, 126.6, 127.0, 128.4, 132.5, 136.6, 137.2, 140.3, 142.9, 147.8, 151.3, 151.6, 162.9; MS: m/z 368.78 (M+). Anal. calcd. for C18H13ClN4O3: C, 58.62; H, 3.55; N, 15.19. Found: C, 58.67; H. 3.61; N, 15.26%.
2.2.1.2 Microwave method
A mixture of 2-chloro-6-methylquinoline-3-carbaldehyde (0.01 mol), 4-nitrophenyl hydrazide (0.01 mol) and 1,4-dioxane (5 drops) was subjected to microwave irradiation at 200 W intermittently at 30 s intervals for the specified time (Table 2). On completion of the reaction, it was monitored by TLC, the reaction mixture was cooled and treated with chilled water. The precipitate thus obtained was filtered, washed with water and recrystallized from ethanol to afford 2.
The progress of the reaction and the purity of the compound were routinely checked on TLC aluminium sheet silica gel 60 F245 (E. Merck) using benzene–acetonitrile (4:1, v/v) as an irrigator and was developed in iodine chamber.
2.2.2 General procedure for the synthesis 3-acetyl-2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)-1,3,4-oxadiazoline (3)
2.2.2.1 Conventional method
Acetic anhydride (0.02 mol) was added to the aroylhydrazone (2) (0.01 mol) and refluxed for 2 h. After cooling, the reaction mixture was poured into ice-cold water. The precipitate was filtered off, washed with water, dried and recrystallized from DMF–ethanol to give the product (3). m.p. 185–187 °C, yield 67%; IR (cm−1): 3034 (aromatic–H), 1675 (C⚌O), 1607 (C⚌N), 1590, 1563, 1431 (C⚌C, benzene and quinoline ring), 1341 (–NO2 symmetric stretching), 1523 (–NO2 asymmetric stretching), 1204 (C–O–C linkage), 762, 774 (C–Cl bond, mono substituted benzene). 1H NMR: δ 2.02 (s, 3H, –CO–CH3), 2.43 (s, 3H, –CH3), 6.61 (s, 1H, N–CH–quinoline ring), 7.47–8.34 (m, 8H, quinoline–H and Ar–H); 13C NMR: δ 23.4, 24.7, 69.8, 124.0, 125.8, 126.4, 126.5, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 143.5, 150.2, 150.8, 155.0, 168.5; MS: m/z 410.81 (M+). Anal. calcd. for C20H15ClN4O4: C, 58.47; H, 3.68; N, 13.63. Found: C, 58.53; H, 3.74; N, 13.69%.
2.2.2.2 Microwave method
Silica gel (1 g) was added to the mixture of aroylhydrazone (0.01 mol) and acetic anhydride (2 mL) at room temperature. The reaction mixture was thoroughly mixed and the absorbed material was dried in air and irradiated in a microwave oven at 400 W intermittently at 30 s intervals for the specified time (Table 2). The reaction mixture was cooled and the product was extracted in methanol. Dilution of methanol solution with ice-cold water gave the crude product, which was filtered, washed with water and recrystallized from methanol to give product (3).
The progress of the reaction and the purity of the compound were routinely checked on TLC aluminium sheet silica gel 60 F245 (E. Merck) using benzene–acetonitrile (4:1, v/v) as an irrigator and was developed in iodine chamber.
2.2.3 General synthesis of 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(3-hydroxyphenyl)prop-2-en-1-one (4h)
2.2.3.1 Conventional method
A mixture of (3) (0.01 mol) and 3-hydroxybenzaldehyde (0.01 mol) was stirred in the presence of KOH (0.01 mol) in 25 mL ethanol (95%) for 6 h. The yellow crystals formed were filtered off, washed with water and crystallized from ethanol (99.5%). m.p. 249–251 °C, yield 74%. Anal. calcd. for C27H19ClN4O5: C, 62.98; H, 3.71; N, 10.88. Found: C, 62.95; H, 3.76; N, 10.92%.
2.2.3.2 Microwave method
A mixture of compound (3) (0.01 mol), 3-hydroxybenzaldehyde (0.01 mol), potassium hydroxide and ethanol (95%) (1 mL) was subjected to microwave irradiation at 100 W intermittently at 30 s intervals for the specified time (Table 2). On completion of the reaction as monitored by TLC, the reaction mixture was cooled. The precipitate thus obtained was filtered and recrystallized from ethanol (95%) to afford the final compound (4h).
The progress of the reaction and the purity of the compound were routinely checked on TLC aluminium sheet silica gel 60 F245 (E. Merck) using benzene–acetonitrile (4:1, v/v) as an irrigator and was developed in iodine chamber.
Other compounds of the series were prepared by using the same conventional and microwave method and their physical data are recorded (Tables 1 and 2).
2.3 Spectral characterization and elemental analysis of compounds 4a–l
2.3.1 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(2-chlorophenyl)prop-2-en-1-one (4a)
IR (KBr) (cm−1): 3038 (aromatic–H), 3058 (CH⚌CH group), 2911 (C–H stretching), 1672 (C⚌O), 1609 (C⚌N), 1595, 1561, 1433 (C⚌C, benzene and quinoline ring), 1342 (–NO2 symmetric stretching), 1543 (–NO2 asymmetric stretching), 1218 (C–O–C linkage), 760, 771 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.67 (d, 1H, O⚌C–C–H), 7.82 (d, 1H, ⚌C–H), 7.06–8.34 (m, 12H, quinoline–H and Ar–H); 13C NMR: δ 24.7, 70.1, 118.8, 124.0, 125.8, 126.4, 126.5, 126.8, 127.8, 128.7, 129.4, 130.1, 130.7, 131.1, 131.4, 133.0, 135.6, 135.9, 136.4, 143.5, 150.0, 150.8, 155.0, 167.0; MS: m/z 533.37 (M+). Anal. calcd. for C27H18Cl2N4O4: C, 60.80; H, 3.40; N, 10.50. Found: C, 60.85; H, 3.47; N, 10.55%.
2.3.2 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(3-chlorophenyl)prop-2-en-1-one (4b)
IR (KBr) (cm−1): 3052 (aromatic–H), 3063 (CH⚌CH group), 2911 (C–H stretching), 1682 (C⚌O), 1614 (C⚌N), 1583, 1541, 1433 (C⚌C, benzene and quinoline ring), 1458 (C–H bending), 1346 (–NO2 symmetric stretching), 1522 (–NO2 asymmetric stretching), 1209 (C–O–C linkage), 761, 775 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.84 (d, 1H, O⚌C–C–H), 7.55 (d, 1H, ⚌C–H), 6.61 (s, 1H, N–CH–quinoline ring), 7.15–8.34 (m, 12H, quinoline–H and Ar–H); 13C NMR: δ 24.5, 70.1, 118.8, 124.0, 125.8, 126.4, 126.5, 127.8, 128.7, 130.1, 130.7, 131.4, 133.3, 133.5, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.8, 155.1, 167.2; MS: m/z 533.37 (M+). Anal. calcd. for C27H18Cl2N4O4: C, 60.80; H, 3.40; N, 10.50. Found: C, 60.84; H, 3.46; N, 10.56%.
2.3.3 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(4-chlorophenyl)prop-2-en-1-one (4c)
IR (KBr) (cm−1): 3039 (aromatic–H), 3064 (CH⚌CH group), 2910 (C–H stretching), 1684 (C⚌O), 1609 (C⚌N), 1607, 1571, 1443 (C⚌C, benzene and quinoline ring), 1451 (C–H bending), 1343 (–NO2 symmetric stretching), 1521 (–NO2 asymmetric stretching), 1205 (C–O–C linkage), 757, 778 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.84 (d, 1H, O⚌C–C–H), 7.55 (d, 1H, ⚌C–H), 7.15–8.34 (m, 12H, quinoline–H and Ar–H); 13C NMR: δ 24.6, 70.1, 118.7, 124.0, 125.8, 126.4, 126.5, 127.8, 128.7, 130.7, 130.1, 131.4, 133.3, 133.5, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.8, 155.0, 167.3; MS: m/z 533.37 (M+). Anal. calcd. for C27H18Cl2N4O4: C, 60.80; H, 3.40; N, 10.50. Found: C, 60.86; H, 3.46; N, 10.55%.
2.3.4 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(2-nitrophenyl)prop-2-en-1-one (4d)
IR (KBr) (cm−1): 3063 (aromatic–H), 3058 (CH⚌CH group), 2917 (C–H stretching), 1687 (C⚌O), 1590 (C⚌N), 1585, 1545, 1427 (C⚌C, benzene and quinoline ring), 1461 (C–H bending), 1219 (C–O–C linkage), 1349 (–NO2 symmetric stretching), 1516 (–NO2 asymmetric stretching), 758, 775 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.91 (d, 1H, O⚌C–C–H), 7.56–8.34 (m, 12H, quinoline–H and Ar–H), 8.11 (d, 1H, ⚌C–H); 13C NMR: δ 24.5, 70.3, 118.8, 123.8, 124.0, 125.8, 126.4, 126.5, 127.2, 127.3, 128.9, 130.1, 130.7, 131.4, 134.8, 135.6, 135.9, 136.4, 143.5, 144.0, 145.0, 150.2, 150.8, 155.0, 167.2; MS: m/z 543.92 (M+). Anal. calcd. for C27H18ClN5O6: C, 59.62; H, 3.33; N, 12.87. Found: C, 59.67; H, 3.39; N, 12.93%.
2.3.5 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(3-nitrophenyl)prop-2-en-1-one (4e)
IR (KBr) (cm−1): 3057 (aromatic–H), 3081 (CH⚌CH group), 2918 (C–H stretching), 1678 (C⚌O), 1608 (C⚌N), 1589, 1558, 1430 (C⚌C, benzene and quinoline ring), 1219 (C–O–C linkage), 1353 (–NO2 symmetric stretching), 1462 (C–H bending), 1524 (–NO2 asymmetric stretching), 763, 774 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 7.57–8.34 (m, 12H, quinoline–H and Ar–H), 7.09 (d, 1H, O⚌C–C–H), 7.66 (d, 1H, ⚌C–H); 13C NMR: δ 24.7, 70.1, 118.8, 120.0, 123.1, 124.0, 125.8, 126.4, 126.5, 129.6, 130.1, 130.7, 131.4, 132.5, 135.6, 135.9, 136.1, 136.4, 143.5, 144.0, 147.8, 150.2, 150.8, 155.0, 167.0; MS: m/z 543.92 (M+). Anal. calcd. for C27H18ClN5O6: C, 59.62; H, 3.33; N, 12.87. Found: C, 59.68; H, 3.37; N, 12.93%.
2.3.6 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(4-nitrophenyl)prop-2-en-1-one (4f)
IR (KBr) (cm−1): 3039 (aromatic–H), 3065 (CH⚌CH group), 2921 (C–H stretching), 1672 (C⚌O), 1616 (C⚌N), 1597, 1561, 1434 (C⚌C, benzene and quinoline ring), 1460 (C–H bending), 1216 (C–O–C linkage), 1342 (–NO2 symmetric stretching), 1523 (–NO2 asymmetric stretching), 756, 768 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 7.13 (d, 1H, O⚌C–C–H), 7.69 (d, 1H, ⚌C–H), 7.57–8.34 (m, 12H, quinoline–H and Ar–H); 13C NMR: δ 24.6, 70.2, 118.8, 123.8, 124.0, 125.8, 126.4, 126.5, 127.3, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 141.3, 143.5, 144.0, 147.1, 150.2, 150.7, 155.1, 167.2; MS: m/z 543.92 (M+). Anal. calcd. for C27H18ClN5O6: C, 59.62; H, 3.33; N, 12.87. Found: C, 59.67; H, 3.38; N, 12.94%.
2.3.7 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(2-hydroxyphenyl)prop-2-en-1-one (4g)
IR (KBr) (cm−1): 3060 (aromatic–H), 3420 (aromatic–OH group), 3087 (CH⚌CH group), 2906 (C–H stretching), 1681 (C⚌O), 1612 (C⚌N), 1608, 1565, 1405 (C⚌C, benzene and quinoline ring), 1212 (C–O–C linkage), 1353 (–NO2 symmetric stretching), 1468 (C–H bending), 1523 (–NO2 asymmetric stretching), 756, 768 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.67 (d, 1H, O⚌C–C–H), 6.68–8.34 (m, 12H, quinoline–H and Ar–H), 7.82 (d, 1H, ⚌C–H), 9.82 (s, 1H, aromatic–OH group); 13C NMR: δ 24.7, 70.1, 115.5, 116.5, 118.8, 121.3, 124.0, 125.8, 126.4, 126.5, 127.8, 129.4, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.8, 155.0, 158.3, 167.0; MS: m/z 514.92 (M+). Anal. calcd. for C27H19ClN4O5: C, 62.98; H, 3.71; N, 10.88. Found: C, 62.99; H, 3.77; N, 10.93%.
2.3.8 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(3-hydroxyphenyl)prop-2-en-1-one (4h)
IR (KBr) (cm−1): 3048 (aromatic–H), 3429 (aromatic–OH group), 3093 (CH⚌CH group), 2915 (C–H stretching), 1675 (C⚌O), 1611 (C⚌N), 1593, 1561, 1425 (C⚌C, benzene and quinoline ring), 1354 (–NO2 symmetric stretching), 1458 (C–H bending), 1533 (–NO2 asymmetric stretching), 1207 (C–O–C linkage), 758, 764 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.84 (d, 1H, O⚌C–C–H), 7.55 (d, 1H, ⚌C–H), 7.57–8.34 (m, 12H, quinoline–H and Ar–H), 9.82 (s, 1H, aromatic–OH group); 13C NMR: δ 24.5, 70.3, 112.1, 115.1, 188.8, 119.0, 124.0, 125.8, 126.4, 126.5, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 136.6, 143.5, 144.0, 150.2, 150.8, 155.0, 158.4, 167.2; MS: m/z 514.92 (M+). Anal. calcd. for C27H19ClN4O5: C, 62.98; H, 3.71; N, 10.88. Found: C, 62.96; H, 3.76; N, 10.92%.
2.3.9 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(4-hydroxyphenyl)prop-2-en-1-one (4i)
IR (KBr) (cm−1): 3064 (aromatic–H), 3429 (aromatic–OH group), 3075 (CH⚌CH group), 2911 (C–H stretching), 1673 (C⚌O), 1602 (C⚌N), 1608, 1564, 1423 (C⚌C, benzene and quinoline ring), 1460 (C–H bending), 1341 (–NO2 symmetric stretching), 1522 (–NO2 asymmetric stretching), 1219 (C–O–C linkage), 761, 769 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.67 (d, 1H, O⚌C–C–H), 7.57–8.83 (m, 12H, quinoline–H and Ar–H), 7.82 (d, 1H, ⚌C–H), 9.82 (s, 1H, aromatic–OH group); 13C NMR: δ 24.5, 70.1, 115.8, 118.8, 124.0, 125.8, 126.4, 126.5, 127.8, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.8, 155.0, 157.7, 167.0; MS: m/z 514.92 (M+). Anal. calcd. for C27H19ClN4O5: C, 62.98; H, 3.71; N, 10.88. Found: C, 62.97; H, 3.77; N, 10.94%.
2.3.10 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(4-methylphenyl)prop-2-en-1-one (4j)
IR (KBr) (cm−1): 3032 (aromatic–H), 3072 (CH⚌CH group), 2892, 2911 (C–H stretching), 1681 (C⚌O), 1605 (C⚌N), 1579, 1554, 1432 (C⚌C, benzene and quinoline ring), 1440, 1461 (C–H bending), 1345 (–NO2 symmetric stretching), 1535 (–NO2 asymmetric stretching), 1209 (C–O–C linkage), 761, 775 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 2.40 (s, 3H, –CH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.84 (d, 1H, O⚌C–C–H), 7.55 (d, 1H, ⚌C–H), 7.57–8.34 (m, 12H, quinoline–H and Ar–H); 13C NMR: δ 24.3, 24.7, 70.1, 118.8, 124.0, 125.8, 126.3, 126.4, 126.5, 128.9, 130.1, 130.7, 131.4, 132.2, 135.6, 135.9, 136.4, 137.6, 143.5, 144.0, 150.2, 150.8, 155.0, 167.3; MS: m/z 512.95 (M+). Anal. calcd. for C28H21ClN4O4: C, 65.56; H, 4.12; N, 10.92. Found: C, 65.62; H, 4.18; N, 10.97%.
2.3.11 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(4-methoxyphenyl)prop-2-en-1-one (4k)
IR (KBr) (cm−1): 3041 (aromatic–H), 3068 (CH⚌CH group), 2920 (C–H stretching), 2833 (Ar–OCH3), 1673 (C⚌O), 1609 (C⚌N), 1611, 1562, 1436 (C⚌C, benzene and quinoline ring), 1342 (–NO2 symmetric stretching), 1520 (–NO2 asymmetric stretching), 1277 (Ar–O–Me), 1462 (C–H bending), 1209 (C–O–C linkage), 758, 780 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 3.85 (s, 3H, –OCH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.68–8.34 (m, 12H, quinoline–H and Ar–H), 6.84 (d, 1H, O⚌C–C–H), 7.55 (d, 1H, ⚌C–H); 13C NMR: δ 24.4, 55.8, 70.1, 114.2, 118.8, 124.0, 125.8, 126.4, 126.5, 127.4, 127.5, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.8, 155.0, 159.8, 167.2; MS: m/z 528.95 (M+). Anal. calcd. for C28H21ClN4O5: C, 63.58; H, 4.00; N, 10.59. Found: C, 63.64; H, 4.06; N, 10.65%.
2.3.12 1-[2-(2-Chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(3,4,5–trimethoxyphenyl)prop-2-en-1-one (4l)
IR (KBr) (cm−1): 3070 (aromatic–H), 3064 (CH⚌CH group), 2914 (C–H stretching), 2841 (Ar–OCH3), 1671 (C⚌O), 1610 (C⚌N), 1608, 1570, 1442 (C⚌C, benzene and quinoline ring), 1343 (–NO2 symmetric stretching), 1522 (–NO2 asymmetric stretching), 1284 (Ar–O–Me), 1359 (–NO2 symmetric stretching), 1466 (C–H bending), 1525 (–NO2 asymmetric stretching), 1209 (C–O–C linkage), 756, 774 (C–Cl bond, mono substituted benzene). 1H NMR (DMSO-d6): δ 2.43 (s, 3H, –CH3 group), 3.71–3.83 (s, 9H, –OCH3 group), 6.61 (s, 1H, N–CH–quinoline ring), 6.84 (d, 1H, O⚌C–C–H), 6.78–8.34 (m, 9H, quinoline–H and Ar–H), 7.55 (d, 1H, ⚌C–H); 13C NMR: δ 24.5, 70.3, 56.1, 103.8, 118.8, 124.0, 125.8, 126.4, 126.5, 129.5, 130.1, 130.7, 131.4, 135.6, 135.9, 136.4, 143.5, 144.0, 150.2, 150.7, 150.8, 155.1, 167.1; MS: m/z 589.00 (M+). Anal. calcd. for C30H25ClN4O7: C, 61.17; H, 4.27; N, 9.51. Found: C, 61.22; H, 4.32; N, 9.56%.
3 Results and discussion
All the reactions (steps 2–4) were completed in 6–8 h by the conventional method, while under MWI, the same reactions were completed in 3–5 min, and also observed in good yield as compared to the conventional method. A comparative study in terms of yield and reaction period is shown in Tables 1 and 2.
3.1 IR-data
The IR spectrum of the title compound 4h (molecular formula C27H19ClN4O5, M.W. 514.92) has given vibration at 3048 cm−1 over the range which shows multiple weak absorption peaks corresponding to Qu–H and Ar–H stretching vibration absorption peaks. The absorption at 2915 cm−1 is due to the aromatic –CH3 group stretching vibration, while the absorption at 1458 cm−1 is due to the aromatic–CH3 group bending vibration. The strong absorption at 1675 cm−1 is due to the C⚌O stretching vibration and the moderate intensity absorption at 1611 cm−1 corresponds to a C⚌N stretching vibration and C⚌C linkage appeared stretching vibration at 1595 cm−1. The absorption at 1354 cm−1 is due to the symmetric stretching of –NO2 group while the absorption at 1533 cm−1 is due to the asymmetric stretching of –NO2 group. The high frequency region of the IR spectra of these compounds contains –OH stretching vibration bands at 3429 cm−1. While in oxadiazole nucleus C–O–C linkage appeared in the range of 1207 cm−1. Methylene (CH⚌CH) linkage appeared in the stretching vibration range of 3093 cm−1, while methylene linkage appeared in the bending vibration range of 825 cm−1, the absorption peak at 758 cm−1 arises due to the C–Cl group and the absorption peak at 764 cm−1 indicates that a mono substituted benzene ring is present.
3.2 1H NMR-data
It can be seen from the chemical structure of compound 4h that different pairs of carbons, e.g., C-14 and C-18, C-15 and C-17 are attached to the chemically equivalent protons, which appeared at δ = 8.09 and 8.34 ppm, respectively. The protons attached to C-10 position appeared as a singlet at δ = 2.43 ppm due to the –CH3 group. The proton attached to the C-7 position appeared as a doublet at 7.47 ppm, while the proton attached to the C-8 position also appeared as a doublet at δ = 7.83 ppm. Chemical shift in the aromatic region with a multiplet centered at δ = 8.05 ppm corresponds to C-3, while the C-11 (–CH group) present in the oxadiazole nucleus appeared at δ = 6.61 ppm. The protons, which are present in the alkene linkage, C-20 and C-21 give doublet, C-20 appeared at δ = 6.84 ppm, because of the vicinity of carbonyl group at C-19, while the proton of C-21 appeared at δ = 7.55 ppm, because it is directly attached to the phenyl ring. The proton of the –OH group appeared at δ = 9.83 ppm, –OH functional group is attached to C-24, while four other protons attached to carbons C-23, C-25, C-26, and C-27 in the phenyl ring appeared as a multiplet at δ = 6.61–7.04 ppm.
3.3 13C NMR-data
The final compound 4h contains three moieties, such as quinoline, oxadiazole and chalcone. The chemical shifts of the carbons of the final compound 4h carbons vary from δ = 167.0 to 70.1 ppm. The carbon nuclei under the influence of a strong electronegative environment appeared downfield, e.g., the C-19 carbonyl, which is directly linked to the ring nitrogen that has a chemical shift at δ = 167.0 ppm, whereas the C-1 linked to one chlorine and other nitrogen atom, appeared at δ = 150.8 ppm. The chemical shift of the ring carbon at C-9 is affected by the presence of a directly attached ring nitrogen atom and appeared at δ = 143.5 ppm. While the alkene carbon at C-20 is directly attached to a carbonyl group and then appeared at δ = 118.8 ppm. The alkene carbon at C-21 directly conjugated to the phenyl ring, indicates a downfield chemical shift at δ = 144.0 ppm. The carbons of the phenyl ring (C-22, C-23, C-25, C-26 and C-27) conjugated to the alkene functionality have chemical shifts in the range δ = 115.1–136.6 ppm. While the C-24, which is attached to a hydroxyl group appeared at δ = 158.4 ppm, under the strong electron withdrawing influence exerted by the hydroxyl group. The carbon of the methyl group C-10 appeared at δ = 24.7 ppm. While, the carbons which are present in the oxadiazole nucleus C-11 and C-12 are directly attached to oxygen atom on one side and on the other side C-11 is attached to the nitrogen with a single bond. So, it gives a chemical shift at δ = 70.1 ppm, while on the other side C-12 is attached to the nitrogen atom by a double bond, so, it gives a chemical shift at δ = 155.0 ppm. The carbons of the quinoline ring (C-2, C-3, C-4, C-5, C-6, C-7 and C-8) appeared upfield between δ = 125.8 and 136.4 ppm compared to those quinoline ring carbons at C-1 and C-9. The carbon C-13 which is present in the phenyl ring and which is attached to the oxadiazole nucleus carbon C-12, appeared at 135.9 ppm. The equivalent carbons C-14 and C-18 appeared at δ = 130.1 ppm, while other equivalent carbons C-15 and C-17 appeared at 124.0 ppm. While C-16 which is directly attached to the nitro group appeared at 150.2 ppm, respectively. Carbon numbering is described in Fig. 1.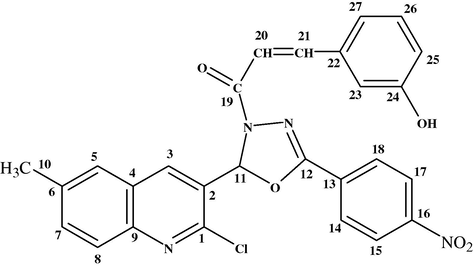
Carbon numbering of the compound 4h.
3.4 Antimicrobial activity
Minimum inhibitory concentration for bacteria (MICb) of all the synthesized compounds was determined against four different strains, viz. two gram positive bacteria (Staphylococcus aureus and Staphylococcus pyogenes and two gram negative bacteria (Escherichia coli and Pseudomonas aeruginosa) compared to the standard drug ampicillin by broth dilution method (Rattan, 2000). For antifungal activities, minimum inhibitory concentration for fungi (MICf) of all the synthesized compounds was determined against Candida albicans, Aspergillus niger and Aspergillus clavatus organisms. The results were compared with standard drug griseofulvin by the same method, which showed 100 μg/mL MICf against all fungi used for antifungal activity. We have synthesized 1-[2-(2-chloro-6-methyl(3-quinolyl))-5-(4-nitrophenyl)(1,3,4-oxadiazolin-3-yl)]-3-(aryl)prop-2-en-1-ones (4a–l) derivatives (Table 3). ±SD = Standard deviation. *p ⩽ 0.0001.
Sr. No.
Compd.
-R
Minimum inhibitory concentration for bacteria (μg/mL) ± SD
Minimum inhibitory concentration for fungi (μg/mL) ± SD
Gram positive
Gram negative
C. albicans
A. niger
A. clavatus
E. coli
P. aeruginosa
S. aureus
S. pyogenes
MTCC-443
MTCC-1688
MTCC-96
MTCC-442
MTCC-227
MTCC-282
MTCC-1323
1
4a
-2-C1
250 ± 2.34∗
250 ± 3.35∗
500 ± 3.6∗
500 ± 3.34∗
250 ± 4.36∗
500 ± 3.50∗
500 ± 3.11∗
2
4b
-3-C1
500 ± 3.65∗
500 ± 3.55∗
100 ± 4.04∗
100 ± 3.43∗
100 ± 3.55∗
100 ± 2.44∗
500 ± 3.20∗
3
4c
-4-C1
100 ± 3.30∗
100 ± 2.13∗
500 ± 3.34∗
500 ± 4.13∗
500 ± 4.34∗
500 ± 3.02∗
1000 ± 3.23∗
4
4d
-2-NO2
50 ± 3.23∗
50 ± 4.57∗
500 ± 3.23∗
200 ± 2.53∗
500 ± 2.48∗
1000 ± 3.5∗
100 ± 3.36∗
5
4e
-3-NO2
500 ± 3.56∗
1000 ± 2.18∗
1000 ± 3.04∗
100 ± 4.55∗
100 ± 4.24∗
100 ± 3.4∗
1000 ± 3.41∗
6
4f
-4-NO2
100 ± 4.45∗
500 ± 2.58∗
100 ± 2.56∗
500 ± 2.53∗
500 ± 4.25∗
1000 ± 3.5∗
100 ± 3.31∗
7
4g
-2-OH
500 ± 3.32∗
250 ± 2.69∗
100 ± 4.13∗
250 ± 3.05∗
1000 ± 3.2∗
500 ± 2.02∗
500 ± 3.25∗
8
4h
-3-OH
25 ± 3.56∗
50 ± 4.57∗
200 ± 3.08∗
100 ± 4.93∗
500 ± 3.4∗
200 ± 3.12∗
100 ± 3.35∗
9
4i
-4-OH
250 ± 3.12∗
500 ± 2.66∗
100 ± 3.45∗
500 ± 4.73∗
500 ± 3.53∗
100 ± 3.55∗
500 ± 3.3∗
10
4j
-4-CH3
250 ± 4.35∗
100 ± 2.42∗
200 ± 3.36∗
250 ± 3.6∗
250 ± 3.75∗
100 ± 3.4∗
1000 ± 3.40∗
11
4k
-4-OCH3
100 ± 4.24∗
500 ± 3.45∗
100 ± 3.53∗
100 ± 4.02∗
500 ± 4.8∗
1000 ± 3.2∗
100 ± 2.30∗
12
4l
3,4,5-(OCH3)3
200 ± 4.36∗
200 ± 3.55∗
500 ± 3.36∗
500 ± 3.53∗
100 ± 3.43∗
100 ± 3.54∗
100 ± 4.15∗
Ampicillin
100 ± 4.57∗
100 ± 4.12∗
250 ± 4.15∗
100 ± 3.55∗
–
–
–
Griseofulvin
–
–
–
–
500 ± 2.64∗
100 ± 3∗
100 ± 3.46∗
3.4.1 Antibacterial activity
From screening results, it has been observed that final compounds 4c, 4f and 4k possess good activity against E. coli while compound 4d has better activity against E. coli and compound 4h has excellent activity against E. coli as compared to standard drug ampicillin. Final compounds 4c and 4j possess good activity against P. aeruginosa and compounds 4d and 4h possess very good activity against P. aeruginosa as compared to the standard drug ampicillin. Final compounds 4b, 4f, 4g, 4i and 4k possess very good activity against S. aureus, while compounds 4c and 4j possess excellent activity against S. aureus as compared to standard drug ampicillin. Final compounds 4b, 4e, 4h and 4k were considered as good active against S. pyogenes and compound 4f possesses very good activity as compared to the standard drug ampicillin. The remaining compounds of the series possess moderate to poor antibacterial activity.
3.4.2 Antifungal activity
Antifungal screening data showed that final compounds 4c, 4d, 4f, 4h, 4i and 4k possess good activity against C. albicans, while compound 4j possesses very good activity against C. albicans and compounds 4b, 4e and 4l possess excellent activity as compared to the standard drug griseofulvin. Compounds 4b, 4e, 4i, 4j and 4l possess good activity against A. niger as compared to the standard drug griseofulvin. Compounds 4d, 4f, 4h, 4k and 4l possess good activity against A. clavatus as compared to the standard drug griseofulvin. The remaining compounds of the entire series possess only moderate to poor antifungal activity.
3.4.3 Statistical analysis
The standard deviation value is expressed in terms of ±SD. On the basis of the calculated value by using ANOVA method, it has been observed that the differences below 0.0001 level (p ⩽ 0.0001) were considered as statistically significant.
4 Conclusion
We have synthesized a variety of quinoline–oxadiazole containing chalcone derivatives under classical and microwave conditions. In general, improvements in the rate and yield of reactions were observed in MWI. When organic reactions were carried out under microwave as compared to classical conditions, it may be observed that activation occurs at different temperatures with these techniques and, therefore, strict comparisons will require a balance between effectiveness and energy costs. In general, compounds with electron withdrawing groups showed good antibacterial and antifungal activities. These results proved that novel quinoline, oxadiazole and chalcone based heterocyclic compounds are found to be interesting lead molecules for further synthetic and biological evaluation.
Acknowledgements
The authors are thankful to Department of Chemistry, Bhavnagar University, Bhavnagar for providing research facilities. One of authors A.M.D. is thankful to the University Grants Commission, New Delhi for providing the UGC-meritorious fellowship.
References
- Synth. Commun.. 2003;33:143.
- Balasubramanian M., Keay J.G., eds. Pyridine and their Benzo Derivatives: Application in Comprehensive Heterocyclic Chemistry. Vol vol. 5. Oxford: Pergamon; 1996. p. :245-300.
- Indian J. Chem.. 2009;48B:142.
- Indian J. Chem.. 2004;43B:1765.
- Nature. 1998;392:289.
- J. Med. Chem.. 2001;44:2374.
- J. Org. Chem.. 1992;57:1375.
- Eur. J. Med. Chem.. 2001;36:555.
- Bioorg. Med. Chem. Lett.. 1998;8:1255.
- Planta Med.. 1996;62:185.
- J. Am. Chem. Soc.. 1945;67:112.
- Indian J. Chem.. 2002;41B:2176.
- Chem. Papers. 1996;51:33.
- Angew. Chem., Int. Ed.. 2004;43:6250.
- J. Parkat. Chem.. 1994;336:596.
- Comprehensive Heterocyclic Chemistry. Oxford: Pergamon Press; 1984. pp. 25–85
- Tetrahedron Lett.. 2003;44:4733.
- J. Org. Chem.. 1996;61:3398.
- Loupy A., ed. Microwaves in Organic Synthesis. Weinhein: Wiley–VCH Verlag Gmbh & Co. KgaA; 2002.
- Tetrahedron Lett.. 1978;19:2045.
- Indian J. Chem.. 2004;43B:1798.
- J. Soc. Alger. Chim.. 2002;12:71.
- Eur. J. Med. Chem.. 1996;31:819.
- Indian J. Chem.. 2009;48B:1601.
- Tetrahedron. 2003;59:813.
- Antimicrobials in Laboratory Medicine (first ed.). B.I. Churchill Livingstone Pvt. Ltd.; 2000.
- J. Soc. Alger. Chim.. 2000;10:111.
- Eur. J. Med. Chem.. 2000;35:1021.
- Egypt. J. Chem.. 1976;19:811.
- J. Chem. Soc., Perkin Trans.. 1998;I:4093.
- Chem. Lett.. 2003;32:1000.




