

Translate this page into:
Correlation analysis of reactivity in the oxidation of some organic diols by tripropylammonium fluorochromate in non-aqueous media
⁎Corresponding author. Tel.: +91 9944093020; fax: +91 4172 266487. smansoors2000@yahoo.co.in (S. Sheik Mansoor),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The kinetics of oxidation of some organic diols by tripropylammonium fluorochromate (TriPAFC) have been studied in dimethylsulfoxide (DMSO). The main product of oxidation is the corresponding hydroxy aldehydes. The reaction is first order with respect to TriPAFC and exhibited Michaelis-Menten type kinetics with respect to organic diols. The reaction is catalyzed by hydrogen ions. The hydrogen ion dependence has the form: kobs = a + b[H+]. Various thermodynamic parameters for the oxidation have been reported and discussed along with the validity of isokinetic relationship. Oxidation of diols was studied in 18 different organic solvents. The rate data are showing satisfactory correlation with Kamlet–Taft solvotochromic parameters (α, β and π∗). A suitable mechanism of oxidation has been proposed.
Keywords
Tripropylammonium fluorochromate
Diols
Solvent effect
Kinetics

1 Introduction
Specific and selective oxidation of organic compounds under non-aqueous conditions is an important reaction in synthetic organic chemistry. Chromium(VI) reagents are widely used in organic chemistry for oxidation of primary and secondary alcohols to carbonyl compounds. Cr(VI) as chromate or dichromate is highly soluble in water, and is reported to be highly toxic (Losi et al., 1994; Viamajala et al., 2004), there is continued interest in the development of new chromium(VI) reagents for the effective and selective oxidation of organic substrates, in particular alcohols, under mild conditions. Therefore, the search for new oxidizing agents is of interest to synthetic organic chemists.
In recent years, some new chromium(VI) based reagents like tetraethyl ammonium bromochromate (Mansoor and Shafi, 2011), tetraethylammonium chlorochromate (Swami et al., 2010), tetrabutylammonium bromochromate (Ghammamy et al., 2007), tetraheptylammonium bromochromate (Ghammamy et al., 2009a), tetrahexylammonium fluorochromate (Koohestani et al., 2008), tetramethylammonium fluorochromate (Sadeghy and Ghammami, 2005), benzyltrimethylammonium fluorochromate (Kassaee et al., 2004) and tributylammonium chlorochromate (Mansoor and Shafi, 2010) were proposed.
Tripropylammonium fluorochromate (Ghammamy and Hashemzadeh, 2004) is also one such oxidant developed recently. It is a more efficient and stronger oxidizing agent. This new compound is more efficient for quantitative oxidation of several organic substrates and has certain advantages over similar oxidizing agents in terms of the amount of oxidant and solvent required, short reaction times and high yields.
The kinetics of oxidation of organic diols has been studied by many reagents, such as 2,2′-bipyridiniumchlorochromate (Loonker et al., 1997), bromine in acid solution (Sharma et al., 1998), pyridinium bromochromate (Rao et al., 1998), hexamethylenetetramine-bromine (Gangwani et al., 1999), quinolinium fluorochromate (Choudhary et al., 1999), benzyltrimethyl ammonium dichloroiodate (Mehla et al., 2000), benzyltrimethyl ammonium tribromide (Goswami et al., 2001), and trialkyl fluorochromate (Ghammamy et al., 2009b). We have been interested in the kinetic and mechanistic studies of Cr(VI) species. Literature survey reveals that no report is available on the kinetics of the oxidation of organic diols by TriPAFC. It was considered important to investigate the oxidation by TriPAFC. Hence, we report herein the kinetics of the oxidation of some organic diols by TriPAFC in 18 different organic solvents.
2 Experimental
2.1 Materials
Tripropylamine and chromium trioxide were obtained from Fluka (Buchs, Switzerland). The organic diols used were ethane 1,2-diol, propane 1,2-diol, propane 1,3-diol, butane 1,2-diol, butane 1,4-diol, butane 2,3-diol, pentane 1,2-diol, pentane 1,3-diol and pentane 1,5-diol. The procedure used for the purification of alcohols has been described earlier (Banerji et al., 1993). [1,1,2,2-2H4] ethanediol (DED) was prepared by reducing diethyl oxalate with lithium aluminium deuteride (Kemp and Waters, 1963). Its isotopic purity, as ascertained by its NMR spectra, was 90 ± 4%.
The solvents acetonitrile (MeCN), chloroform (CF), 1,2-dichloroethane (DCE), dichloromethane (DCM), dimethyl sulfoxide (DMSO), acetone (Me2CO), dimethylformamide (DMF), butanone (Bu), nitrobenzene(NB), benzene (Bz), cyclohexane (CH), toluene(TE), acetophenone (Ph2CO), tetrahydrofuran (THF), tert-butanol (t-BuOH), 1,4-dioxane (DO), 1,2-dimethoxyethane (DME) and ethyl acetate (EA), are of analytical grade and purified by conventional methods (Perrin et al., 1966). Due to non-aqueous nature of the solvent, p-toluene sulfonic acid (TsOH) was used as a source of hydrogen ions. TsOH is a strong acid and in a polar medium like DMSO; it is likely to be completely ionized.
2.2 Preparation of tripropylammonium fluorochromate
Tripropylammonium fluorochromate is easily prepared as follows: chromium(VI) oxide (15.0 g, 0.150 mol) was dissolved in water in a polyethylene beaker and 40% hydrofluoric acid (11.3 ml, 0.225 mol) was added with stirring at 0 °C. To the resultant orange solution, tripropylamine (28.3 ml, 0.150 mol) was added drop wise with stirring to this solution over a period of 0.5 h and stirring was continued for 0.5 h at 0 °C. The precipitated orange solid was isolated by filtration, washed with petroleum ether (3 × 60 ml) and dried in vacuum for 2 h at room temperature (Ghammamy and Hashemzadeh, 2004). Yield 37.5 g (95%); mp 142 °C:
The bright orange crystalline reagent can be stored in polyethylene containers for long periods without decomposition. The chromium(VI) content may be easily determined iodometrically.
2.3 Product analysis
Product analysis was carried out under kinetic conditions. In a typical experiment, diol (0.1 mol) and TriPAFC (0.01 mol) were made up to 100 ml in DMSO and kept in the dark for 24 h to ensure completion of the reaction. The solution was then treated with an excess (200 cm3) of a saturated solution of 2,4-dinitrophenyl hydrazine in 2 mol dm−3 HCl and kept over night in a refrigerator. The solvent was removed and the precipitated 2,4-dinitrophenylhydrazone (DNP) was filtered off, dried and recrystallized from ethanol. In the oxidation of ethanediol the identity of the product was confirmed by determining the mp with authentic samples of hydroxyethanal. Similar experiments were performed with other diols also.
3 Experimental procedure
3.1 Kinetic measurements
The pseudo-first-order conditions were attained by maintaining a large excess (×15 or more) of diol over TriPAFC. The solvent was DMSO, unless specified otherwise. The reactions were followed, at constant temperatures (±0.01 K), by monitoring the decrease in [TriPAFC] spectrophotometrically at 361 nm. No other reactant or product has any significant absorption at this wavelength. The pseudo-first-order rate constant, kobs was evaluated from the linear (r = 0.990–0.999) plots of log[TriPAFC] against time for up to 80% reaction. Duplicate kinetic runs showed that the rate constants were reproducible to within ±3%. The second order rate constant k2 was obtained from the relation k2 = kobs/[Diol]. All experiments, other than those for studying the effect of hydrogen ions, were carried out in the absence of TsOH.
3.2 Data analysis
Correlation analyses were carried out using Microcal origin (version 6) computer software. The goodness of the fit was discussed using the correlation coefficient (r in the case of simple linear regression and R in the case of multiple linear regressions) and standard deviation (SD).
4 Results and discussion
The rate data and other experimental data were obtained for all the diols investigated. As the results were similar, only representative data are given in Table 1.
103[TriPAFC] (mol dm−3)
102[Diol] (mol dm−3)
[TsOH] (mol dm−3)
105 kobs (s−1)
0.5
2.0
0.0
25.36 ± 0.08
1.0
2.0
0.0
25.00 ± 0.06
1.5
2.0
0.0
25.66 ± 0.10
2.0
2.0
0.0
25.04 ± 0.04
2.5
2.0
0.0
25.44 ± 0.11
1.0
4.0
0.0
43.56 ± 0.05
1.0
6.0
0.0
64.60 ± 0.20
1.0
8.0
0.0
84.20 ± 0.26
1.0
10.0
0.0
103.20 ± 0.18
1.0
2.0
0.2
27.43 ± 0.24
1.0
2.0
0.4
33.20 ± 0.16
1.0
2.0
0.6
38.34 ± 0.12
1.0
2.0
0.8
45.20 ± 0.26
1.0
2.0
1.0
52.60 ± 0.28
1.0
4.0
0.0
24.40a ± 0.08
4.1 Stoichiometric studies
The stoichiometry of the reaction was determined by carrying out several sets of experiments with varying amounts of TriPAFC largely in excess over ethanediol. The estimation of unreacted TriPAFC showed the following reaction:
4.2 Order of reaction
The reaction is first order with respect to TriPAFC. Michaelis–Menten type kinetics were observed with respect to ethanediol (Table 1). A plot of log kobs vs log[Diol] is linear with a slope value of 0.887 (Fig. 1). This indicates the following Eqs. (3) and (4) to represent for the mechanism. Eq. (5) represents the rate law:
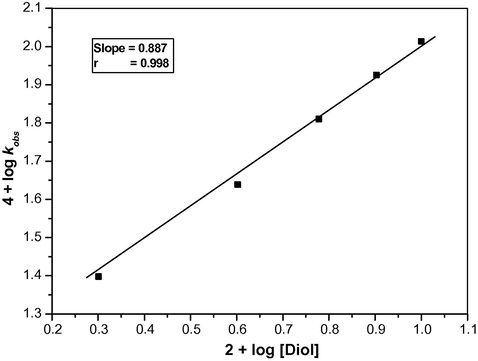
Order plot for the oxidation of ethanediol by tripropylammonium fluorochromate at 303 K.
4.3 Effect of acidity
The reaction is catalyzed by hydrogen ions (Table 1). The hydrogen ion dependence has the following form kobs = a + b[H+]. The values of a and b, for ethanediol at 303 K are 25.09 ± 0.5 × 10−5 s−1 and 34.08 ± 0.9 × 10−5 mol−1 dm3 s−1, respectively (r2 = 0.996).
4.4 Induced polymerization of acrylonitrile
The oxidation of diol by TriPAFC, in an atmosphere of nitrogen failed to induce the polymerization of acrylonitrile. Further, an addition of a radical scavenger, acrylonitrile, has no effect on the rate (Table 1).
4.5 Kinetic isotope effect
To ascertain the importance of the cleavage of the α-C–H bond in the rate-determining step, oxidation of [1,1,2,2-2H4]ethanediol (DED) was studied. Results showed the presence of a substantial primary kinetic isotope effect (Table 2). [Diol] = 2.0 × 10–2 M; [TriPAFC] = 1.0 × 10−3 M.
Substrate
105 × k1 (s−1)
298 K
303 K
308 K
313 K
Ethane-1,2-diol
18.2
25.0
34.4
46.0
[1,1,2,2-2H4]Ethane-1,2-diol
2.9
4.1
6.2
8.6
kH/kD
6.27
6.09
5.55
5.35
4.6 Thermodynamic parameters
The variation in all the diols concentration was studied at four temperatures and the values of k2 were evaluated. The activation parameters have been evaluated from the slope and intercept of Eyring’s plot of log(k2/T) against 1/T, from k2 at 298, 303, 308 and 313 K by the method of least square and presented in Table 3. The least square method gives the values and standard errors of enthalpy and entropy of activation, respectively. Statistical analysis of the Eyring equation clearly confirms that the standard errors of ΔH# and ΔS# correlate. 102[Substrate] = 2.0 mol dm−3; 103[TriPAFC] = 1.0 mol dm−3.
Substrate
104 k2 (dm3 mol−1 s−1)
ΔH# (kJ mol−1)
−ΔS# (J K−1 mol)
ΔG# (kJ mol−1) (at 303 K)
298 K
303 K
308 K
313 K
Ethane 1,2-diol
91
125
172
230
45.76 ± 0.2
130.55 ± 0.5
85.32 ± 0.4
Propane 1,2-diol
370
480
618
820
38.30 ± 1.2
143.95 ± 3.8
81.92 ± 2.1
Propane 1,3-diol
230
320
450
640
49.78 ± 1.2
107.19 ± 4.2
82.25 ± 2.4
Butane 1,2-diol
560
760
998
1360
44.04 ± 1.0
124.81 ± 3.0
81.61 ± 2.0
Butane 1,4-diol
420
600
860
1180
51.12 ± 0.6
99.73 ± 1.8
81.33 ± 1.2
Butane 2,3-diol
1450
1904
2448
3200
38.10 ± 0.6
134.19 ± 1.9
78.75 ± 1.4
Pentane 1,2-diol
630
900
1310
1890
54.38 ± 1.0
85.56 ± 3.0
80.30 ± 2.0
Pentane 1,3-diol
686
980
1388
1950
51.50 ± 0.4
94.56 ± 1.2
80.15 ± 0.8
Pentane 1,5-diol
402
572
890
1320
59.54 ± 2.3
71.96 ± 7.4
81.34 ± 4.5
(ΔS#) = 1/Tav(ΔH#), where Tav is the center of the temperature range used. It follows that in most solution phase studies (ΔS#) ≈ (ΔH#) × 0.003 K−1 (Lente et al., 2005). The negative entropy of activation suggests a definite orientation in the transition state and this may be in part due to the solvation of the activated complex.
The entropy of activation is negative for all the diols. The negative entropy of activation in conjunction with other experimental data supports the mechanism outlined in (Scheme 1).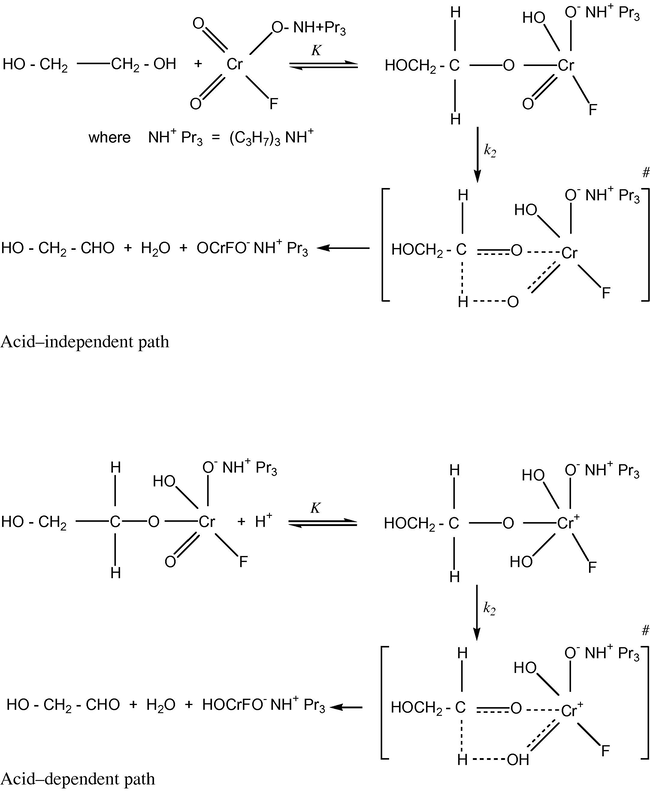
Mechanism of oxidation of diols by TriPAFC.
4.7 Isokinetic relationship
The reaction is neither isoenthalpic nor isoentropic but complies with the compensation law also known as the isokinetic relationship:
The isokinetic temperature β is the temperature at which all the compounds of the series react equally fast. Also, at the isokinetic temperature, the variation of the substituent has no influence on the free energy of activation. In an isoentropic reaction, the isokinetic temperature lies at infinite and only enthalpy of activation determines the reactivity. The isokinetic temperature is zero for an isoenthalpic series, and the reactivity is determined by the entropy of activation (Bhuvaseshwari and Elango, 2007). The isokinetic relationship is tested by plotting the logarithms of rate constants at two different temperatures (T2 > T1) against each other according to the following equation:
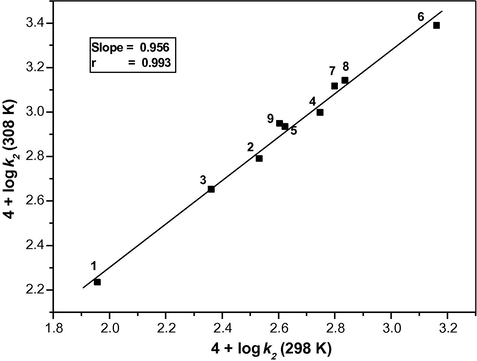
Exner’s plot for the oxidation of diols by tripropylammonium fluorochromate between 4 + log k2 (at 308 K) and 4 + log k2 (at 298 K). (1) Ethane-1,2-diol; (2) propane-1,2-diol; (3) propane-1,3-diol; (4) butane-1,2-diol; (5) butane-1,4-diol; (6) butane-2,3-diol; (7) pentane-1,2-diol; (8) pentane-1,3-diol; (9) pentane-1,5-diol.
4.8 Solvent–reactivity correlation
The rate of oxidation of ethanediol was determined in 18 different organic solvents viz., MeCN, CF, DCE, DCM, DMSO, Me2CO, DMF, Bu, NB, Bz, CH, TE, Ph2CO, THF, t-BuOH, DO, DME and EA. The choice of the solvents was limited by the solubility of TriPAFC and its reactivity with primary and secondary alcohols. There was no noticeable reaction with the solvents chosen. The kinetics were similar in all the solvents. The values of k2 at four different temperatures were determined. The thermodynamic parameters were also calculated for the oxidation of ethanediol in 18 different organic solvents and the values are recorded in Table 4. Negative entropy of activation indicates a greater degree of ordering in the transition state than in the initial state, due to an increase in solvation during the activation process. The isokinetic relationship of ethanediol in 18 different organic solvent is shown in Fig. 3. The existence of a linear relationship (slope = 0.933, r = 0.995, isokinetic temperature = 578 K) between 4 + log k2 (298 K) and 4 + log k2 (308 K) indicates that a common mechanism is operating in all studied solvent systems. 102[Substrate] = 2.0 mol dm−3; 103[TriPAFC] = 1.0 mol dm−3.
Solvents
103 k2 (dm3 mol−1 s−1)
ΔH# (kJ mol−1)
−ΔS# (J K−1 mol)
ΔG# (kJ mol−1) (at 303 K)
298 K
303 K
308 K
313 K
MeCN
99
140
188
250
45.18 ± 1.0
131.70 ± 3.2
85.09 ± 1.9
CF
9.06
14.3
22.0
34.0
66.25 ± 1.9
80.78 ± 6.0
90.72 ± 3.4
DCE
14.2
22.5
35.0
56.6
68.54 ± 1.5
69.48 ± 4.6
89.59 ± 2.8
DCM
13.6
20.6
28.8
43.0
56.29 ± 1.9
111.22 ± 6.0
89.98 ± 3.7
DMSO
86.0
125
180
252
53.03 ± 0.2
106.13 ± 0.6
85.18 ± 0.4
Me2CO
11.6
18.4
28.0
45.2
67.20 ± 1.7
75.66 ± 5.4
90.12 ± 3.3
DMF
30.0
42.6
58.8
82.0
48.06 ± 0.6
128.07 ± 1.9
86.86 ± 1.2
Bu
7.8
12.4
19.2
31.4
68.92 ± 1.9
72.92 ± 5.6
91.01 ± 3.6
NB
19.6
28.0
40.4
60.2
55.33 ± 1.7
111.95 ± 5.0
89.08 ± 3.2
Bz
2.8
4.6
7.6
12.8
76.20 ± 1.4
57.03 ± 4.2
93.48 ± 2.6
CH
0.24
0.42
0.60
0.98
68.35 ± 2.8
103.56 ± 8.6
99.72 ± 5.3
TE
3.6
5.08
7.2
10.8
53.99 ± 2.3
130.17 ± 6.5
93.43 ± 4.2
Ph2CO
23.6
33.2
46.2
64.7
49.40 ± 1.6
129.02 ± 4.8
88.55 ± 3.0
THF
4.8
7.9
13.2
20.8
73.71 ± 0.6
61.44 ± 1.5
92.32 ± 1.0
t-BuOH
2.56
4.0
6.4
10.4
69.88 ± 1.9
79.25 ± 5.6
93.88 ± 3.6
DO
5.2
7.70
11.6
17.2
59.54 ± 1.0
108.35 ± 3.0
92.37 ± 1.9
DME
1.60
2.40
3.72
5.78
63.94 ± 1.9
103.18 ± 6.0
95.20 ± 3.7
EA
2.72
4.40
7.04
11.40
71.41 ± 1.2
73.69 ± 3.4
93.73 ± 2.2
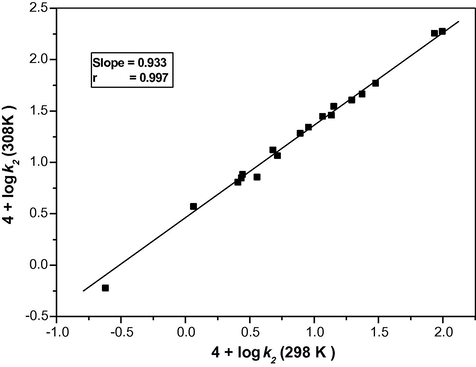
Exner’s plot for the oxidation of ethanediol by tripropylammonium fluorochromate between 4 + log k2 (at 308 K) and 4 + log k2 (at 298 K) at 18 different organic solvents.
The influence of solvent on the rate of any reaction can be described in terms of solvation which is a stabilization process. Two view points have been established on the solvation phenomenon. According to the first, a solvent is considered as a homogeneous continuum which surrounds the solute molecules and exerts long range interactions. The strength of these interactions with the solute molecules is described in terms of macroscopic physical properties of the solvent like dielectric constant (ε) and refractive index (η). According to the second view point, a solvent is considered to be anisotropic and inhomogeneous which exerts short range forces on the solute molecules. These forces are chemical in nature, and result in the formation of solvation complexes through donor–acceptor bonds which are localized and directed in space. The strength of these interactions is described in terms of solvation parameters namely hydrogen bond donor acidity (α), hydrogen bond acceptor basicity (β), etc. Thus the solvent can solvate the solute by exhibiting any of these interactions with the specific sites in the solute. Hence, the effect of solvent on the rate of the reaction can be described in terms of multiparameteric equation which involves different solvation parameters given by Kamlet–Taft (Kamlet et al., 1983).
4.9 The Kamlet–Taft method for the examination of solvent effect
In order to get a deeper understanding about the various solvent–solvent–solute interactions which influence reactivity, we applied the following Kamlet–Taft equation:
In this equation, π∗ represents an index of solvent dipolarity/polarizability, which measures the ability of the solvent to stabilize a charge or a dipole by virtue of its dielectric effect, α is the solvent hydrogen bond donor (HBD) acidity, β is the solvent hydrogen bond acceptor (HBA) basicity of the solvent in a solute to solvent hydrogen bond. Ao is the intercept term, it is the regression value of the solute property in the reference solvent cyclohexane.
Kamlet et al. (1981) established that the effect of a solvent on the reaction rate should be given in terms of the following properties: (i) the behavior of the solvent as a dielectric, facilitating the separation of opposite charges in the transition state, (ii) the ability of the solvent to donate a proton in a solvent-to-solute hydrogen bond and thus stabilize the anion in transition state and (iii) the ability of the solvent to donate an electron pair and, therefore, stabilize the initial alcohol, by way of a hydrogen bond between the alcoholic proton and the solvent electron pair. The parameter π∗ is an appropriate measure of the first property, while the second and third properties are governed by the parameters α and β, respectively. The solvent parameters (π∗, α and β) are taken from the literature (Kamlet et al., 1983) and are given in Table 5. The linear dependence (LSER) on the solvent properties was used to correlate and predict a wide variety of solvent effect.
Solvent
π∗
α
β
Acetonitrile
0.75
0.19
0.31
Chloroform
0.58
0.44
0.00
1,2-Dichloroethane
0.81
0.00
0.00
Dichloromethane
0.82
0.30
0.00
DMSO
1.00
0.00
0.76
Acetone
0.71
0.08
0.48
DMF
0.88
0.00
0.69
Butanone
0.67
0.06
0.48
Nitrobenzene
1.01
0.00
0.39
Benzene
0.59
0.00
0.10
Cyclohexane
0.00
0.00
0.00
Toluene
0.54
0.00
0.11
Acetophenone
0.90
–
0.49
THF
0.58
0.00
0.55
tert-Butyl alcohol
0.41
0.68
1.01
1,4-Dioxane
0.55
0.00
0.37
1,2-Dimethoxyethane
0.53
0.00
0.41
Ethyl acetate
0.55
0.00
0.45
In order to explain the kinetic results through the solvent polarity and basicity or acidity, the rate constants were correlated with the solvatochromic parameters π∗, α and β using total solvatochromic equation, Eq. (8). The correlation of kinetic data was realized by means of multiple linear regression analysis. The regression coefficients s, a, and b measure the relative susceptibilities of the solvent-dependent solute property log k to the indicated solvent parameter. The rates of oxidation for all the compounds studied showed good correlations with solvent via the above LSER. The correlation results obtained are as follows:
It was found that the rate constants in 18 solvents showed satisfactory correlation with the π∗, α and β solvent parameters. The results of correlation analysis in terms of Eq. (8), a biparametric equation involving π∗ and β are given in the following equations:
Kamlet’s (Kamlet et al., 1983) triparametric equation explains ca. 93% of the effect of solvent on the oxidation. However, by Exner’s criterion (Exner, 1966) the correlation is not even satisfactory (cf. Eq. (12)). The major contribution is of solvent polarity. It alone accounted for ca. 90% of the data. Both α and β play relatively minor roles.
4.10 The Swain’s method
Swain et al. (1983) believed that the specific solvation is determined principally by the acidity and the basicity of the solvent. The data are analyzed using a two-parameter equation involving anion-solvating tendency (A) and cation-solvating tendency (B):
The rates of oxidation of ethanediol in different solvents showed an excellent correlation in Swain’s equation with the cation-solvating power playing the major role. In fact, the cation solvation alone accounts for ca. 99% of the data. The correlation with anion-solvating power was very poor. The solvent polarity, represented by (A + B), also accounted for ca. 80% of the data.
4.11 Mechanism of oxidation
The absence of any effect of a radical scavenger, acrylonitrile (Table 1), indicated that a hydrogen abstraction mechanism, giving rise to free radicals, is unlikely. The presence of a substantial kinetic isotope effect (Table 2) confirms the cleavage of an α-C–H bond in the rate determining step (Scheme 1). The hydride ion transfer may take place either by a cyclic process via an ester intermediate or by an acyclic one-step bimolecular process. Negative reaction constants are traditionally associated with an electron deficient center in transition states: a convention originally developed from the analysis of substituent effects in nucleophilic displacement reactions. Negative reaction constants have been used by Banerji (1978a,b, 1988) as supporting evidence for oxidation mechanisms involving a hydride-ion transfer in the rate determining step.
Kwart and Nickel (1973) have shown that dependence of kinetic isotope effect on the temperature can be gainfully employed to determine whether the loss of hydrogen proceeds through a concerted cyclic process or by an acylic one. The data for protio- and deuterio-benzhydrols, fitted to the expression: kH/kD = AH/AD(−ΔH∗/RT) (Kwart and Latimer, 1971; Kwart and Slutsky, 1972) show direct correspondence with the properties of a symmetrical transition state in which activation energy difference for protio and deuterio compounds is equal to the difference in the zero-point energy for the respective C–H and C–D bonds (≈4.5 kJ mol−1) and the entropies of activation of the respective reactions are almost equal.
It is well-established that intrinsically concerted sigmotropic reactions, characterized by transfer of hydrogen in a cyclic transition state, are the only truly symmetrical processes involving a linear hydrogen transfer (Woodward and Hoffmann, 1969). Littler (1971) has shown that a cyclic hydride transfer, in which the oxidation of alcohols by Cr(VI), involving six electrons and, being a Huckel-type system, is an allowed process. Thus, a transition state having a planar, cyclic and symmetrical structure can be envisaged for the decomposition of the ester intermediate. Hence, the overall mechanism is proposed to involve the formation of a chromate ester in a fast pre-equilibrium step and then a decomposition of the ester in a subsequent slow step via a cyclic concerted symmetrical transition state leading to the product.
5 Conclusion
The oxidation of nine organic diols by tripropylammonium fluorochromate (TriPAFC) in dimethyl sulfoxide was studied. The oxidation leads to the formation of corresponding hydroxy aldehyde compounds. The reaction is first order with respect to TriPAFC. The reaction exhibited Michaelis–Menten type kinetics with respect to the organic diols. The reaction is catalyzed by hydrogen ions. The hydrogen-ion dependence has the form: kobs = a + b[H+]. Oxidation of ethanediol was studied in 18 different organic solvents. The solvent effect has been analyzed using Kamlet’s and Swain’s multi parametric equation. A hydrogen abstraction mechanism has been proposed.
References
- J. Chem. Soc., Perkin Trans.. 1978;2:639-641.
- Bull. Chem. Soc. Jpn.. 1978;51:2732-2734.
- J. Org. Chem.. 1988;53:2154-2159.
- J. Chem. Soc., Perkin Trans.. 1993;2:205.
- Int. J. Chem. Kinet.. 2007;39:657-663.
- Indian J. Chem.. 1999;38A:325.
- Nature 1964:488.
- Collect. Czech. Chem. Commun.. 1966;31:3222-3251.
- Progress in Physical Organic Chemistry. New York: John Wiley; 1973. p. 41
- J. Chem. Res.. 1999;180:0854-0871.
- Bull. Korean Chem. Soc.. 2004;25(8):1277-1279.
- Afr. J. Pure Appl. Chem.. 2007;1(1):008-010.
- J. Chil. Chem. Soc.. 2009;54:491-493.
- J. Mex. Chem. Soc.. 2009;53(2):41-43.
- Proc. Indian Acad. Sci., Chem. Sci.. 2001;113:43.
- Prog. Phys. Org. Chem.. 1981;13:485-633.
- J. Org. Chem.. 1983;48:2877-2887.
- Acta Chim. Slov.. 2004;51:743-750.
- Proc. Roy. Soc. Ser. A. 1963;274:480-499.
- J. Mex. Chem. Soc.. 2008;52:116-119.
- J. Am. Chem. Soc.. 1971;93:3770.
- J. Am. Chem. Soc.. 1973;95:3394-3396.
- J. Chem. Soc. Chem. Commun. 1972:1182-1183.
- Rates and Equilibrium of Organic Reactions. New York: Wiley; 1963.
- New J. Chem.. 2005;29:759.
- Tetrahedron. 1971;27:81-91.
- J. Chem. Res.. 1997;242:1663.
- Rev. Environ. Contam. Toxicol.. 1994;136:91-121.
- React. Kinet. Mech. Catal.. 2010;100:21-30.
- Z. Phys. Chem.. 2011;225:249-263.
- Oxid. Commun.. 2000;23:229.
- Purification of Organic Compounds. Pergamon Press; 1966.
- Int. J. Chem. Kinet.. 1998;30:285.
- Russ. J. Gen. Chem.. 2005;75:1886-1888.
- Proc. Indian Acad. Sci., Chem. Sci.. 1998;110:65.
- J. Am. Chem. Soc.. 1983;105:502-513.
- Int. J. Chem. Kinet.. 2010;42:50-55.
- Biotechnol. Prog.. 2004;20:87-95.
- Angew. Chem., Int. Ed. Engl.. 1969;8:781-853.




