

Translate this page into:
Cr(II)-promoted internal cyclization of acyclic enediynes fused to benzo[b]thiophene core: Macrocycles versus 2-methylenecycloalkan-1-ols formation
⁎Corresponding authors. vpopik@uga.edu (V.V. Popik), i.balova@spbu.ru (I.A. Balova)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The utility of the intramolecular Nozaki-type coupling for the synthesis of macrocyclic benzo[b]thiophene-fused enediynes has been explored. The starting acyclic enediynes were prepared by the iodocyclization of 2-(buta-1,3-diynyl)thioanisoles followed by the Sonogashira cross-coupling of the resulting iodo-substituted benzo[b]thiophene with corresponding acetylenes. We found that Cr(II)-promoted intramolecular cyclization of 7-[2-(iodoethynyl)benzo[b]thiophen-3-yl]hept-6-ynal and 7-[3-(iodoethynyl)benzo-[b]thiophen-2-yl]hept-6-ynal resulted in the formation of 11-membered macrocyclic enediynes, while both expected 10-membered enediynes cannot be produced under the Nozaki-type reaction from corresponding 6-[3-(iodoethynyl)benzo[b]thiophen-2-yl]hex-5-ynal and 6-[2-(iodoethynyl)benzo[b]thiophen-3-yl]hex-5-ynal. In the case the reaction was catalyzed by Ni(II), the attack on a proximal triple bond led to the formation of 2-methylenecycloalkane-1-ol fragments, instead of macrocyclization. The DFT analysis of the ring strain in the benzo[b]thiophene-fused 10- and 11-membered enediyne-containing cycle provides the plausible explanation of the observed regioselectivity.
Keywords
Enediynes
Diacetylenes
Iodocyclization
Intramolecular Nozaki‐Hiyama‐Kishi reactions
Benzothiophene
Allylic alcohols

1 Introduction
Macrocyclic enediynes are the key part of powerful natural antitumor antibiotics (Galm et al., 2005; Hamann et al., 2011; Maretina and Trofimov, 2006; Minto and Blacklock, 2008; Nicolaou et al., 1992a; Siddiq and Dembitsky, 2008; Smith and Nicolaou, 1996). All members of this class of natural products have characteristic structural fragments containing two triple bonds conjugated to a double bond in a Z-configuration. Depending on the structural features, the enediynes undergo either Bergman (Bergman, 1973) (as in the case of the most of natural enediynes (Nicolaou et al., 1993a), for example: dynemicin, (Konishi et al., 1990; Nicolaou and Smith, 1992) calicheamicin (Lee et al., 1991, 1987; Thorson et al., 2000)) or Myers-Saito (Myers, 1987; Nagata et al., 1989) (for example, neocarzinostatin (Goldberg, 1991)) cyclization to yield a benzenoid biradical (Wang and Sondheimer, 1980). The biradical then abstracts two hydrogen atoms from the dDNA carbohydrate-phosphate backbone, resulting in the dDNA double strand cleavage (De Voss et al., 1990; Lee et al., 1987; Sugiura et al., 1991, 1990; Wolkenberg and Boger, 2002; Zein et al., 1988). It is known that the rate of enediyne cyclization correlates with the ring size. For biomedical applications, the intramolecular Bergman cyclization (BC) should proceed spontaneously around the human body temperature. This can be achieved by incorporating the enediyne scaffold into 9-membered or 10-membered ring structure, as in the case of naturally occurring enediynes (Advani et al., 2010; Nicolaou et al., 1988, 1993a; Nicolaou and Dai, 1991). It is known that about 70% of marketed antibiotics are derived from natural products, although in some times their mode of action remained unclear and is still under investigations (Keohane et al., 2018). Despite the mechanism of action of enediyne antibiotics has been well studied, their clinical use is limited due to the complex structure of enediyne natural products as well as low selectivity along with remarkable biological activity (Nicolaou et al., 1992a, 1992b, 1992c, 1993b; Siddiq and Dembitsky, 2008; Smith and Nicolaou, 1996; Zein et al., 1988). Thus, designing new enediyne structures with the goal of taming DNA-cleaving activity of this class of antibiotics remains the important goal of modern medicinal chemistry (Chari et al., 2014; Joshi and Rawat, 2012; Kraka et al., 2008; Mohamed et al., 2013; Nicolaou et al., 2015; Oku et al., 2003; Poloukhtine et al., 2010).
The fusion of the enediynes moiety to a heterocyclic core (Choy et al., 2000; Kim and Russel, 1999, 1998; Kim et al., 2000, 1999; Zhao et al., 2005, 2004) allows not only for the modulation of the cycloaromatization rate, but also permits to explore the additional DNA-binding affinity. Our group has previously reported a new efficient and facile strategy for the preparation of heterocyclic enediynes, which is based on the cyclization of ortho-functionalized butadiynylheteroarenes (Danilkina et al., 2014, 2011; Vinogradova et al., 2011). This strategy provides an efficient method for the introduction of various functional groups at the termini of the (Z)-3-en-1,5-diyne fragment fused to heteroindenes, allowing for the use of various macrocyclization techniques. Thus, we have recently reported the synthesis of macrocyclic enediynes fused to benzothiophene and indole using ring-closing metathesis (Danilkina et al., 2012, 2015, 2014) or the Nicholas-type macrocyclization (Lyapunova et al., 2016, 2018).
Nozaki-Hiyama-Kishi reaction (NHK) (Jin et al., 1986; Takai et al., 1986) is a useful tool for the intramolecular construction the new C—C bond between sp2-sp2 (Bolte et al., 2015; Iwamoto et al., 2004; LeClair et al., 2010; Lubineau and Billault, 1998; Mi and Maleczka, 2001; Mohapatra et al., 2010; Muller et al., 1998; Pilli et al., 2000; Pilli and Victor, 1998; Takao et al., 2009; Wang et al., 2016) and sp-sp2 carbon atoms (Boddenmann and Keese, 1993; Crévisy and Beau, 1991; Dai et al., 2001; Sandoval et al., 2002; Yamaguchi et al., 2012). This reaction is very selective and tolerant to various functional groups (ester, amide, alkene, etc.) (Furstner and Shi, 1996; Jin et al., 1986). A large number of 10- and 11-membered macrocyclic enediynes have been synthesized using NHK reaction as key step (Ban and Guanti, 2000; Banfi and Guanti, 2002a, 2002b; Brandstetter and Maier, 1994; Choy et al., 2000; Comanita et al., 2000; Crévisy and Beau, 1991; Dancy et al., 1995; Karpov et al., 2008; Karpov and Popik, 2007; Maier and Brandstetter, 1992; Nicolaou et al., 1992b; Nishikawa et al., 1994; Poloukhtine and Popik, 2005; Py et al., 1998; Semmelhack et al., 2002; Yamaguchi et al., 2012). We have also successfully applied Nozaki coupling in the synthesis of 10-membered macrocyclic cinnoline-fused endiyne (Vinogradova et al., 2011). It is important to note, that cinnolinemoiety quadruples the rate of cycloaromatization over the benzannulated analogue.
In this report we explore the scope and limitations of the intramolecular Nozaki reaction for the synthesis of 10- and 11- membered macrocyclic enediynes fused to benzothiophene.
2 Experimental section
2.1 General information and methods
Solvents and reagents were purchased from commercial suppliers and used without further purification, unless otherwise noticed. Solvents were dried and distilled using standard procedures. Starting compounds 6a-c, 7a-c, 9a,b, 10a,b 11a,b were prepared using previously reported procedures (Danilkina et al., 2014; Jones et al., 1987; Kulyashova et al., 2013). All reactions were carried out under argon atmosphere in flame-dried glassware. Evaporation of solvents and concentration of reaction mixtures were performed in vacuo at 30–40 °C on a rotary evaporator. Preparative chromatography was conducted using silica gel 60. Melting points (mp) are uncorrected. Differential scanning calorimetry (DSC) experiments were carried out with 0.11 mg of samples using crucibles with pierced caps under nitrogen atmosphere at a heating/cooling rate of 20 °C min−1 from a temperature of 20 °C up to 395 °C, followed by cooling to 20 °C and heating to 395 °C for the second time. 1H NMR and 13C NMR spectra were recorded using 300 MHz spec (for 9b) or 400 MHz instrument (for all other compounds) in CDCl3 with TMS as the internal standard or in CDCl3 without the internal standard (for 4b, 16a), or in DMSO‑d6. The 1H NMR data are reported as the chemical shift (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad), coupling constants (J, are given in Hz) and number of protons. The 13C NMR data are reported as the chemical shift (δ) and type of carbon, determined from DEPT 135 experiments). Chemical shifts are referenced to residual solvent (δ = 7.26 ppm for 1H in CDCl3, δ = 77.00 ppm for 13C in CDCl3, δ = 2.50 ppm for 1H in DMSO‑d6, δ = 39.50 ppm for 13C in DMSO‑d6). High-resolution mass spectra (HRMS) were measured using FAB or ESI. The single-crystal X-ray diffraction studies were carried out at 100.0 K using Cu Kα radiation (λ = 1.54184 Å).
2.2 General procedure for synthesis 9c, 10c
2.2.1 2-[4-(Trimethylsilyl)buta-1,3-diynyl]thioanisole (9c) (Danilkina et al., 2014)
PdCl2(PPh3)2 (0.440 mmol, 5 mol.%), PPh3 (0.890 mmol, 0.231 g, 10 mol.%), 4-(trimethylsilyl)buta-1,3-diyne 7c were added to a solution of 2-iodothioanisole 8 (8.80 mmol, 2.20 g,) in triethylamine (90 mL) at room temperature. In 5 min CuI (1.32 mmol, 0.251 g, 15 mol.%) was added, and reaction mixture was stirred at 40 °C for 2.5 h (TLC control). The reaction mixture was cooled, diluted with EtOAc (70.0 mL) and washed with a saturated aqueous solution of NH4Cl (2 × 70.0 mL), water (50.0 mL) and brine (50.0 mL). The combined aqueous layers were extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with a saturated aqueous solution of NH4Cl (50.0 mL), water (50.0 mL), brine (50.0 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, crude product was purified by Isolera™ prime flash-chromatography system eluting with acetone/hexane system (gradient from 1% to 5% of acetone) to give product as yellow oil (1.81 g, 84%). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.44 (dd, J = 7.7 Hz, J = 1.2 Hz, 1H), 7.31 (td, J = 8.0 Hz, J = 1.4 Hz, 1H), 7.16 (d, J = 7.9 Hz, 1H), 7.07 (td, J = 7.6 Hz, J = 0.9 Hz, 1H), 2.49 (s, 3H), 0.24 (s, 9H). 13C NMR (101 MHz, CDCl3), δ, ppm: 143.5, 133.8, 129.6, 124.5, 124.3, 119.6, 92.5, 87.6, 80.2, 74.0, 15.2, −0.42.
2.2.2 3-Iodo-2-(2-trimethylsilylethynyl)benzo[b]thiophene (10c) (Danilkina et al., 2014)
A solution of iodine (40.9 mmol, 1.04 g) in DCM (21.0 mL) was added dropwise to a degassed solution of 2-[4-(trimethylsilyl)buta-1,3-diynyl]thioanisole 9c (4.09 mmol, 1.00 g) in DCM (21.0 mL) under argon atmosphere at rt. Reaction was stirred at room temperature for 1.5 h (TLC control). The reaction mixture was diluted with DCM (30.0 mL) and washed with a saturated aqueous solution of Na2S2O3 (30.0 mL). The aqueous layer was extracted with DCM (2 × 15.0 mL). Combined organic layers were washed with brine (50.0 mL) and dried over Na2SO4. The solvent was removed under reduced pressure, crude product was purified by flash chromatography on silica gel eluting with pentane to give crystalline cream solid. Mp. = 61–62 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.73–7.69 (m, 2H), 7.46–7.39 (m, 2H), 0.32 (s, 9H). 13C NMR (101 MHz, CDCl3), δ, ppm: 140.4, 138.9, 126.6, 126.3, 125.7, 124.8, 122.1, 105.5, 98.3, 88.7, −0.24.
2.3 General procedure for synthesis of enediynes 11c,d
A solution of 3-iodo-2-(2-trimethylsilylethynyl)benzo[b]thiophene 10c (1.00 mmol, 0.356 g) and corresponding alkyn-1-ol (2.5 mmol) in dry DCM (5.0 mL) were added to a degassed suspension of PdCl2(PPh3)2 (0.05 mmol, 0.035 g, 5 mol.%) and CuI (0.1 mmol, 0.019 g, 10 mol.%) in anhydrous triethylamine (5.0 mL). The reaction mixture was stirred at room temperature for 24 h (TLC control). The mixture was filtered through a shot pad of silica gel using EtOAc as an eluent. The solvent was removed under reduced pressure, crude product was purified by column chromatography on silica gel.
2.3.1 6-{2-[(Trimethylsilyl)ethynyl]benzo[b]thiophen-3-yl}hex-5-yn-1-ol (11c)
The compound 11c was synthesized following the standard procedure from 10c (1.60 mmol, 0.570 g) and hex-5-yn-1-ol (4.00 mmol, 0.393 g) in triethylamine (8.0 mL) and DCM (8.0 mL). Purification of crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 0.461 g (88%) of 11c as a reddish-yellow oil. Rf = 0.41 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.86–7.81 (m, 1H), 7.72–7.69 (m, 1H), 7.42–7.36 (m, 2H), 3.75 (t, J = 6.0 Hz, 2H), 2.62 (t, J = 6.6 Hz, 2H), 1.89–1.74 (m, 4H), 1.40 (br. s, 1H), 0.30 (s, 9H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.7, 138.4, 126.2, 125.0, 124.9, 124.2, 123.5, 122.0, 105.0, 97.3, 97.2, 74.1, 62.4, 31.8, 25.0, 19.6, −0.1. HRMS ESI: [M+Na]+ calculated for C19H22NaOSSi+: 349.1053; found 349.1043.
2.3.2 7-{2-[(Trimethylsilyl)ethynyl]benzo[b]thiophen-3-yl}hept-6-yn-1-ol (11d)
The compound 11d was synthesized following the standard procedure from 10c (2.25 mmol, 0.800 g) and hept-6-yn-1-ol (5.61 mmol, 0.630 g) in trimethylamine (11.0 mL) and DCM (11.0 mL). Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 0.688 g (90%) of 11d as a yellow oil. Rf = 0.51 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.86–7.82 (m, 1H), 7.72–7.68 (m, 1H), 7.42–7.36 (m, 2H), 3.69 (t, J = 6.3 Hz, 2H), 2.59 (t, J = 6.3 Hz, 2H), 1.77–1.69 (m, 2H), 1.68–1.59 (m, 4H), 1.41 (br. s, 1H), 0.30 (c, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.8, 138.4, 126.2, 124.93, 124.88, 124.3, 123.5, 122.0, 104.9, 97.5, 97.2, 73.9, 62.8, 32.3, 28.6, 25.0, 19.8, −0.1. HRMS ESI: [M+K]+ calculated for C20H24KOSSi+: 379.0949; found 379.0943.
2.4 General procedure for the synthesis of terminal triple bond of enediynes 12a-d
Procedure A. TBAF hydrate (1.10 mmol, 287 mg) was added to a degassed solution of corresponding TMS-compound (1.00 mmol) in anhydrous THF (0.05 M, 20.0 mL) at 0 °C. The color of the solution changed from yellow to dark green. Reaction mixture was stirred for 15 min at 0 °C. The progress of the reaction was followed by TLC. The reaction mixture was poured into water (40 mL) and extracted with EtOAc (5 × 30 mL). Combined organic layers were washed with brine (30 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to yield the crude product, which was purified by column chromatography on silica gel.
Procedure B. K2CO3 (8.00 mmol, 1.11 g) was added to a degassed solution of corresponding TMS-compound (1.00 mmol) in methanol (0.1 M, 10.0 mL) under argon atmosphere at rt. Reaction was stirred for 3 h (TLC control), then diluted with EtOAc (10 mL) and washed with brine (10 mL). The aqueous layer extracted EtOAc (3 × 5 mL). Combined organic layers were washed with brine (10 mL) and dried under Na2SO4. The solvent was removed under reduced pressure, crude product was purified by column chromatography on silica gel.
2.4.1 6-(3-Ethynylbenzo[b]thiophen-2-yl)hex-5-yn-1-ol (12a)
The enediyne alcohol 12a was synthesized in accordance with typical procedure A from the enediyne alcohol 11a (1.50 mmol, 490 mg) using a TBAF hydrate (1.65 mmol, 431 mg). Reaction time at room temperature – 15 min. Purification of the crude product by column chromatography using hexane/ethyl acetate (5:1) as the eluent gave 370 mg (96%) of 12a as a red oil. Rf = 0.26. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.88–7.83 (m, 1H), 7.73–7.69 (m, 1H), 7.45–7.36 (m, 2H), 3.73 (t, J = 6.0 Hz, 2H), 3.53 (s, 1H), 2.60 (t, J = 6.5 Hz, 2H), 1.85–1.71 (m, 4H), 1.45 (br. s, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.8, 137.9, 128.8, 126.0, 125.2, 123.1, 120.6, 101.3, 83.19, 83.15, 73.8, 62.4, 31.8, 24.7, 19.8, HRMS ESI: [M+H]+ calculated for C16H15OS+: 255.0838; found: 255.0838.
2.4.2 7-(3-Ethynylbenzo[b]thiophen-2-yl)hept-6-yn-1-ol (12b)
The enediyne alcohol 12b was synthesized in accordance with typical procedure from the enediyne alcohol 11b (1.99 mmol, 650 mg) using a TBAF hydrate (2.19 mmol, 572 mg). Reaction time at room temperature – 15 min. Purification of the crude product by column chromatography using hexane/ethyl acetate (5:1) as the eluent gave 520 mg (97%) of 12b as a dark red oil. Rf = 0.2. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.87–7.83 (m, 1H), 7.73–7.69 (m, 1H), 7.44–7.36 (m, 2H), 3.68 (t, J = 6.2 Hz, 2H), 3.54 (s, 1H), 2.57 (t, J = 6.9 Hz, 2H), 1.75–1.53 (m, 6H), 1.45 (br. s, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.8, 137.9, 128.9, 125.9, 125.1, 123.1, 122.0, 120.5, 101.5 83.15, 83.12, 73.6, 62.8, 32.3, 28.1, 25.0, 20.0. HRMS ESI: [M+H]+ calculated for C17H17OS+: 269.0995; found: 269.1005.
2.4.3 6-(2-Ethynylbenzo[b]thiophen-3-yl)hex-5-yn-1-ol (12c)
The enediyne alcohol 12c was synthesized in accordance with procedure B from the enediyne alcohol 11c (1.03 mmol, 350 mg) using a K2CO3 (8.22 mmol, 1.14 g). Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 251 mg (97%) of 12d as a dark red oil. Rf = 0.36 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.87–7.83 (m, 1H), 7.74–7.69 (m, 1H), 7.45–7.38 (m, 2H), 3.75 (t, J = 6.1 Hz, 2H), 3.68 (s, 1H), 2.62 (t, J = 6.6 Hz, 2H), 1.88–1.74 (m, 4H), 1.50 (br. s, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.7, 138.4, 126.4, 125.0, 124.8, 123.7, 123.6, 122.1, 97.5, 86.4, 76.8, 73.9, 62.4, 31.8, 25.0, 19.6 ppm. HRMS ESI: [M+Na]+ calculated for C16H14NaOS+: 277.0658; found 277.0650.
2.4.4 7-(2-Ethynylbenzo[b]thiophen-3-yl)hept-6-yn-1-ol (12d)
The enediyne alcohol 12d was synthesized in accordance with procedure B from the enediyne alcohol 11d (0.429 mmol, 140 mg) using a K2CO3 (3.43 mmol, 474 mg). Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 97 mg (89%) of 12d as a dark red oil. Rf = 0.36 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.88–7.84 (m, 1H), 7.74–7.70 (m, 1H), 7.45–7.38 (m, 2H), 3.71–3.68 (m, 3H), 2.59 (t, J = 6.9 Hz, 2H), 1.77–1.61 (m, 6H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.7, 138.4, 126.4, 125.1, 124.9, 123.61, 123.56, 122.1, 97.7, 86.4, 76.8, 73.7, 62.8, 32.3, 28.5, 25.0, 19.8. HRMS ESI: [M+Na]+ calculated for C17H16NaOS+: 291.0814; found 291.0807.
2.5 General procedure for the iodination of terminal triple bond of enediynes 13a-d
CuI (0.15 mmol, 28.6 mg) and N-iodomorpholine (Hein et al., 2009) (3.00 mmol, 1.02 g) were added to a degassed solution of starting material (1.00 mmol) in anhydrous THF (0.1 M, 10.0 mL) at rt. The progress of the reaction was checked by TLC. Upon completion the reaction mixture was washed with saturated aqueous solution of Na2S2O3 (20 mL) and extracted with EtOAc (10 mL). Organic layer was washed with a saturated aqueous solution NH4Cl (20 mL) and brine (20 mL). Combined water layers were extracted with EtOAc (3 × 10 mL). Combined organic layers were washed with a saturated aqueous solution of Na2S2O3 (20 mL), NH4Cl (20 mL), brine (20 mL) and dried over Na2SO4, concentrated under reduced pressure to yield the crude product, which was purified by column chromatography on silica gel.
2.5.1 6-[3-(Iodoethynyl)benzo[b]thiophen-2-yl]hex-5-yn-1-ol (13a)
The enediyne alcohol 13a was synthesized in accordance with typical procedure from the enediyne alcohol 12a (2.71 mmol, 690 mg) using a CuI (0.407 mmol, 77.0 mg) and N-iodomorpholine (8.14 mmol, 2.77 g). Reaction time at room temperature – 1 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (4:1) as the eluent gave 838 mg (82%) of 13a as a light yellow crystals. Rf = 0.12, mp = 63–65 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.83 (dd, J = 6.8 Hz, J = 1.6 Hz, 1H), 7.70 (dd, J = 6.8 Hz, J = 1.6 Hz, 1H), 7.44–7.36 (m, 2H), 3.77 (t, J = 6.0 Hz, 2H), 2.62 (t, J = 6.5 Hz, 2H), 1.88–1.73 (m, 5H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.9, 137.7, 129.3, 126.0, 125.2, 123.1, 122.1, 122.0, 101.5 (C .)87.8, 73.9, 62.5, 31.9, 24.7, 19.9, 12.0. HRMS ESI: [M+H]+ calculated for C16H14IOS+: 380.9805; found: 380.9813.
2.5.2 7-[3-(Iodoethynyl)benzo[b]thiophen-2-yl]hept-6-yn-1-ol (13b)
The enediyne alcohol 13b was synthesized following the standard procedure from the enediyne alcohol 12b (0.577 mmol, 155 mg) using a CuI (0.084 mmol, 22.0 mg) and N-iodomorpholine (1.67 mmol, 571 mg). Reaction time at room temperature – 1 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (5:1) as the eluent gave 205 mg (90%) of 13b as a light yellow crystals. Rf = 0.26, mp = 36–38 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.84–7.82 (m, 1H), 7.69 (d, J = 7.1 Hz, 1H), 7.43–7.36 (m, 2H), 3.71 (t, J = 7.1 Hz, 2H), 2.57 (t, J = 6.3 Hz, 2H), 1.74–1.55 (m, 7H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.9, 137.6, 129.3, 126.0, 125.1, 123.1, 122.0, 121.9, 101.7, 87.7, 73.7, 62.9, 32.3, 28.1, 25.0, 20.0, 12.0. HRMS ESI: [M+H]+ calculated for C17H16IOS+: 394.9961; found: 394.9965.
2.5.3 6-[2-(Iodoethynyl)benzo[b]thiophen-3-yl]hex-5-yn-1-ol (13c)
The enediyne alcohol 13c was synthesized following the standard procedure from the enediyne alcohol 12c (0.944 mmol, 240 mg) using a CuI (0.142 mmol, 27.0 mg) and N-iodomorpholine (2.83 mmol, 965 mg). Reaction time at room temperature – 1.5 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 325 mg (91%) of 13c as a dark brown oil. Rf = 0.3 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.86–7.82 (m, 1H), 7.73–7.68 (m, 1H), 7.44–7.37 (m, 2H), 3.77 (t, J = 6.1 Hz, 2H), 2.63 (t, J = 6.6 Hz, 2H), 1.89–1.76 (m, 4H), 1.49 (br. s, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.1, 126.5, 125.1, 125.0, 123.6, 122.0, 97.6, 87.0, 73.9, 62.5, 31.8, 25.0, 19.6, 17.3. HRMS ESI: [M+H]+ calculated for C16H14IOS+: 380.9805; found 380.9813.
2.5.4 7-[2-(Iodoethynyl)benzo[b]thiophen-3-yl]hept-6-yn-1-ol (13d)
The enediyne alcohol 13d was synthesized following the standard procedure from the enediyne alcohol 12d (1.64 mmol, 440 mg) using a CuI (0.246 mmol, 47.0 mg) and N-iodomorpholine (4.92 mmol, 1.68 g). Reaction time at room temperature – 1.5 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 608 mg (94%) of 13d as a dark brown oil. Rf = 0.27 (hexane/ethyl acetate (1:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 7.86–7.82 (m, 1H), 7.72–7.69 (m, 1H), 7.44–7.38 (m, 2H), 3.72 (t, J = 6.2 Hz, 2H), 2.60 (t, J = 6.8 Hz, 2H), 1.77–1.59 (m, 6H) 1.43 (br. s, 1H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.4, 138.3, 126.5, 125.6, 123.6, 122.0, 97.8, 87.0, 73.7, 62.9, 32.2, 28.5, 25.0, 19.8, 17.2. HRMS ESI: [M+Na]+ calculated for C17H15NaIOS+: 416.9780; found 416.9796.
2.6 General procedure for the oxidation of enediyne alcohols 14a-d
DMP (3.00 mmol, 1.27 g) was added to a degassed solution of starting material (1.00 mmol) in freshly distilled DCM (0.1 M, 10.0 mL). The progress of the reaction was checked by TLC. Upon completion the reaction mixture was washed with saturated aqueous solution of Na2CO3 (20 mL) and extracted with EtOAc (10 mL). Organic layer was washed with saturated aqueous solution of NH4Cl (20 mL) and brine (20 mL). Combined water layers were extracted with EtOAc (3 × 10 mL). Combined organic layers were washed with saturated aqueous solutions of Na2S2O3 (20 mL), NH4Cl (20 mL), brine (20 mL) and dried over Na2SO4, concentrated under reduced pressure to yield the crude product, which was purified by column chromatography on silica gel.
2.6.1 6-[3-(Iodoethynyl)benzo[b]thiophen-2-yl]hex-5-ynal ( 14a)
The aldehyde 14a was synthesized following the standard l procedure from the enediyne alcohol 13a (2.15 mmol, 820 mg) using a Dess-Martin periodinane (6.45 mmol, 2.73 g). Reaction time at room temperature – 3 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (5:1) as the eluent gave 670 mg (82%) of 13a as a light yellow crystals. Rf = 0.56, mp = 76–79 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 9.91 (s, 1H), 7.87–7.81 (m, 1H), 7.70 (dd, J = 6.4 Hz, J = 1.5 Hz, 1H), 7.46–7.35 (m, 2H), 2.77 (t, J = 7.2 Hz, 2H), 2.65 (t, J = 6.7 Hz, 2H), 2.05–1.95 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 201.5, 138.8, 137.7, 128.9, 126.1, 125.3, 123.1, 122.3, 122.2, 100.2, 87.8, 74.6, 42.7, 20.8, 19.4, 12.3. HRMS ESI: [M+H]+ calculated for C16H12IOS+: 378.9648 found: 378.9654.
2.6.2 7-[3-(Iodoethynyl)benzo[b]thiophen-2-yl]hept-6-ynal (14b)
The aldehyde 14b was synthesized following the standard procedure from the enediyne alcohol 13b (1.32 mmol, 520 mg) using a Dess-Martin periodinane (3.96 mmol, 1.68 g). Reaction time at room temperature – 3 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (5:1) as the eluent gave 360 mg (70%) of 14b as a red oil. Rf = 0.41. 1H NMR (400 MHz, CDCl3), δ, ppm: 9.83 (s, 1H), 7.83 (d, J = 7.3 Hz, 1H), 7.70 (d, J = 7.3 Hz,1H), 7.43–7.37 (m, 2H), 2.60 (t, J = 6.8 Hz, 2H, 2.57–2.53 (m, 2H), 1.93–1.86 (m, 2H), 1.75–1.67 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 202.1, 138.9, 137.7, 129.2, 129.1, 126.0, 125.2, 123.1, 122.0, 100.9, 87.7, 74.0, 43.4, 27.7, 21.3, 19.9, 12.1. HRMS ESI: [M+H]+ calculated for C17H14IOS: 392.9805; found: 392.9817.
2.6.3 6-[2-(Iodoethynyl)benzo[b]thiophen-3-yl]hex-5-ynal (14c)
The aldehyde 14c was synthesized following the standard procedure from the enediyne alcohol 13c (0.842 mmol, 320 mg) using a Dess-Martin periodinane (1.68 mmol, 714 mg). Reaction time at room temperature – 0.5 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 219 mg (69%) of 14c as a brown crystals. Rf = 0.36 (hexane/ethyl acetate (2:1)), mp = 61–62 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 9.91 (s, 1H) 7.84–7.80 (m, 1H), 7.74–7.68 (m, 1H), 7.44–7.39 (m, 2H), 2.79 (td, J = 6.1 Hz, J = 1.0 Hz, 2H), 2.67 (t, J = 6.8 Hz, 2H), 2.05–1.98 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 201.5, 138.3, 138.2, 126.5, 125.5, 125.2, 124.8, 123.5, 122.1, 96.3, 87.0, 74.7, 42.7, 21.1, 19.6, 17.4. HRMS ESI: [M+H]+ calculated for C16H12IOS+: 378.9648; found 378.9654.
2.6.4 7-[2-(Iodoethynyl)benzo[b]thiophen-3-yl]hex-6-ynal (14d)
The aldehyde 14d was synthesized following the standard procedure from the enediyne alcohol 13d (0.254 mmol, 100 mg) using a Dess-Martin periodinane (0.507 mmol, 215 mg). Reaction time at room temperature – 0.5 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 70 mg (70%) of 14d as a dark brown oil. Rf = 0.34 (hexane/ethyl acetate (2:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 9.83 (t, J = 1.7 Hz, 1H), 7.85–7.81 (m, 1H), 7.72–7.68 (m, 1H), 7.45–7.38 (m, 2H), 2.62 (t, J = 6.8 Hz, 2H), 2.55 (td, J = 7.2 Hz, J = 1.7 Hz, 2H), 1.97–1.89 (m, 2H), 1.78–1.71 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 202.3, 138.3, 126.5, 125.2, 125.1, 124.9, 123.5, 122.0, 97.0, 87.0, 74.1, 43.4, 28.0, 21.3, 19.6, 17.3. HRMS ESI: [M+H]+ calculated for C17H14IOS+: 392.9805; found 392.9817.
2.7 General procedure for the synthesis of 2-methylenecycloalkan-1-ols 16a,b
Anhydrous NiCl2 (0.01 mmol, 1.27 mg) and anhydrous CrCl2 (1.00 mmol, 0.121 g) were added to degassed mix of anhydrous solvents DMF (2.00 mL) and THF (4.00 mL). Argon was bubbled through the solution for 1 h. Solution of aldehyde (0.10 mmol) in THF (4.00 mL) was slowly added to the suspension via syringe pump under flue of argon within 80 min. Complete conversion of starting material to the product was observed in 1.5 h (TLC control). Reaction mixture was diluted with Et2O (10 mL) and washed with brine (3 × 10 mL). Combined organic layers were dried over Na2SO4, concentrated under reduced pressure.
2.7.1 (E)-2-[(3-Ethynylbenzo[b]thiophen-2-yl)methylene]cyclopentanol (16a)
The alchohol 16a was synthesized following the standard procedure from the aldehyde 14a (0.634 mmol, 240 mg) using NiCl2 (0.063 mmol, 8.22 mg) and anhydrous CrCl2 (6.34 mmol, 0.780 g). Purification of the crude product by column chromatography on silica gel using hexane/ethyl acetate (3:1) as the eluent gave 140 mg (87%) of 16a as a light yellow crystals. Rf = 0.30, mp = 100–103 °C. 1H NMR (400 MHz, DMSO‑d6), δ, ppm: 7.99 (d, J = 7.9 Hz, 1H), 7.80 (d, J = 7.9 Hz, 1H), 7.48 (t, J = 7.9 Hz, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.14–7.11 (m, 1H), 5.35 (d, J = 6.2 Hz, 1H), 4.52–4.44 (m, 1H), 3.36 (s, 1H), 2.71–2.55 (m, 2H), 1.99–1.81 (m, 2H), 1.75–1.59 (m, 1H), 1.52–1.44 (m, 1H). 13C NMR (101 MHz, DMSO‑d6), δ, ppm: 153.2, 146.2, 138.6, 137.3, 125.5, 125.4, 122.5, 122.0, 114.4, 113.4, 87.6, 76.9, 75.1, 34.5, 29.5, 21.2. HRMS FAB: [M]+ calculated for C16H14OS+: 254.0765; found: 254.0767. IR (KBr) (ν, cm−1): 3292, 3058, 2960, 2860, 2097 (C≡C 1640, 1458, 1430, 1354, 1319, 1284, 1215, 1174, 1151, 1087, 1034, 1012, 946, 870, 831, 760, 730, 651, 632, 593.
Crystal of 16a was fixed on a micro mount and placed on an Agilent Technologies Supernova Atlas diffractometer and measured at a temperature of 100 K using micro focused monochromated Cu Kα radiation. The unit cell parameters were refined by least square techniques using 27,670 reflections in the 2θ range of 7.6–152.74°. The structure have been solved by the direct methods and refined R1 = 0.032 (wR2 = 0.084) for 4646 unique reflections with |Fo| ≥ 4σF by means of the SHELXL–97 program3 incorporated in the OLEX2 program package4. The carbon-bound H atoms were placed in calculated positions and were included in the refinement in the ‘riding’ model approximation, with Uiso(H) set to 1.2Ueq(C) and C—H 0.97 Å for the CH2 groups and Uiso(H) set to 1.5Ueq(N) and C—H 0.96 Å for the CH3 groups. Empirical absorption correction was applied in CrysAlisPr5 program complex using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm.
16a: colorless crystals, (C16H14OS), M = 254.33, crystal size 0.14 × 0.11 × 0.09, monoclinic, space group P21/a, a = 8.4743(2) Å, b = 22.3793(3) Å, c = 13.9195(3) Å, V = 2570.91(8) Å3, Z = 8, ρ = 1.314 g cm–3, μ = 2.092 mm–1. 27,670 reflections, 5098 unique (Rint = 0.0320), 327 parameters, R1 (|Fo| ≥ 4σF) 0.032, wR2 (all data) = 0.084, Gof = 0.937. Supplementary crystallographic data for this paper have been deposited at Cambridge Crystallographic Data Centre (CCDC 1479612) and can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
2.7.2 (E)-2-[(3-Ethynylbenzo[b]thiophen-2-yl)methylene]cyclohexanol (16b)
The alchohol 16b was synthesized in accordance with typical procedure from the enediyne 14b (0.250 mmol, 100 mg) using anhydrous NiCl2 (0.063 mmol, 8.22 mg) and anhydrous CrCl2 (6.34 mmol, 0.780 g). Purification of the crude product by column chromatography using hexane/ethyl acetate (3:1) as the eluent gave 40 mg (58%) of 16b as a yellowish crystals. Rf = 0.29, mp = 109–110 °C.
1H NMR (400 MHz, DMSO‑d6), δ, ppm: 7.87 (d, J = 7.9 Hz, 1H), 7.73 (d, J = 7.9 Hz, 1H), 7.40 (t, J = 7.9 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 7.11–7.01 (m, 1H), 4.30 (d, J = 6.2 Hz, 1H), 3.56 (s, 1H), 3.07–3.04 (m, 1H), 2.35–2.32 (m, 1H), 1.83–1.56 (m, 7H, water in solvent). 13C NMR (101 MHz, DMSO‑d6), δ, ppm: 148.6, 145.1, 139.4, 138.0, 125.3, 125.0, 122.8, 122.0, 116.3, 113.4, 84.2, 77.6, 74.0, 37.0, 28.9, 27.4, 23.3. HRMS FAB: [M]+ calculated for C17H16OS+: 268.0922; found: 268.0925.
2.8 Macrocyclic diol 4e
Anhydrous NiCl2 (0.005 mmol, 0.7 mg) and anhydrous CrCl2 (0.211 mmol, 0.026 g) were added to degassed anhydrous THF (4 mL) at 0 °C. Argon was bubbled through the solution for 1 h. Solution of aldehyde (0.053 mmol, 20.0 mg) in THF (2 mL) was slowly added to the suspension via syringe pump under flue of argon within 80 min at 0 °C. Reaction was left to stir overnight. Complete conversion of starting material to the product was observed in 14 h (TLC control). Reaction mixture was diluted with Et2O (10 mL) and washed with brine (3 × 10 mL). Combined organic layers were dried over Na2SO4, concentrated under reduced pressure. Purification of the crude product by column chromatography on silica gel using hexane/ethyl acetate (3:1) as the eluent gave 6 mg (46%) dimerization product as a light yellow crystals. Rf = 0.30. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.80–7.76 (m, 2H), 7.70–7.68 (m, 2H), 7.42–7.35 (m, 4H), 5.14–5.10 (m, 1H), 4.88–4.84 (m, 1H), 2.71–2.64 (m, 3H), 2.17–1.85 (m, 9H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.6, 138.0, 127.5, 127.4, 125.97, 125.96, 125.86, 125.2, 125.1, 125.0, 123.2, 123.0, 122.3, 122.1, 121.7, 121.6, 100.7, 100.4, 96.4, 96.3, 78.6, 78.5, 75.0, 74.7, 32.0, 29.73, 29.69, 22.7. HRMS FAB: [M]+ calculated for C32H2NaO2S2+: 527.1110; found: 527.1123.
2.9 General procedure for the oxidation of enediyne alcohols 14a-d
Anhydrous DMF (5.00 mL) was added in flask through the septum under argon atmosphere. Argon was bubbled through the solution for 15 min. The CrCl2 (1.00 mmol, 0.123 g) was added to the solvent under argon. Solution of corresponding compound 14 (0.10 mmol) in anhydrous DMF (5.00 mL) had been simultaneously added. The reaction was stirred for 24–60 h. The reaction mixture was diluted with water (30 mL) and extracted with EtOAc (30 mL). The organic layer was washed with water (3 × 30 mL) and brine (30 mL). Combined water layers were extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with water (30 mL), brine (30 mL) and dried over Na2SO4, and concentrated under reduced pressure to yield the crude product, which was purified by column chromatography on silica gel.
2.9.1 6,7,13,14-Tetradehydro-9,10,11,12-tetrahydro-8H-benzo[b]cycloundeca[d]thiophene-12-ol (4b)
The cyclic enediyne 4b was synthesized in accordance with typical procedure from the enediyne aldehyde 14b (0.076 mmol, 0.026 g) using a CrCl2 (0.094 g, 0.765 mmol). Reaction time at room temperature – 24 h. Purification of the crude product by column chromatography on silica gel using hexane/ethyl acetate (5:1) as the eluent gave 12 mg (60%) of 4b as a light yellow crystals. Rf = 0.30 (hexane/ethyl acetate (5:1)), mp = 100–101 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.81–7.79 (m, 1H), 7.73–7.71 (m, 1H), 7.42–7.34 (m, 2H), 4.82 (dd, J = 8.1 Hz, J = 4.1 Hz, 1H), 2.64–2.60 (m, 2H), 2.19–2.02 (m, 3H), 2.00–1.85 (m, 2H), 1.81–1.64 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 137.7, 136.8, 130.4, 125.8, 125.2, 125.1, 122.7, 122.4, 102.7, 99.3, 80.8, 77.5, 64.1, 35.9, 24.7, 22.0, 18.7. HRMS ESI: [M+Na]+ calculated for C17H14NaOS+: 289.0658; found: 289.0652.
2.9.2 6,7,13,14-Tetradehydro-9,10,11,12-tetrahydro-8H-benzo[b]cycloundeca[d]thiophen-8-ol (4d)
The cyclic enediyne 4d was synthesized in accordance with typical procedure from the enediyne aldehyde 14d (0.204 mmol, 80.0 mg) using a CrCl2 (2.04 mmol, 251 mg). Reaction time at room temperature – 60 h. Purification of the crude product by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 36 mg (66%) of 4d as a reddish-white crystals. Rf = 0.29 (hexane/ethyl acetate (2:1)), mp = 121–122 °C. 1H NMR (400 MHz, CDCl3), δ, ppm: 7.84–7.79 (m, 1H), 7.76–7.71 (m, 1H), 7.43–7.36 (m, 2H), 4.82 (t, J = 8.2 Hz, J = 4.2 Hz, 1H), 2.62 (t, J = 6.5 Hz, 2H), 2.22–2.03 (m, 2H), 2.01–1.85 (m, 3H), 1.82–1.67 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 138.4, 136.8, 128.1, 126.9, 126.1, 125.0, 123.1, 122.5, 102.0, 99.5, 80.8, 77.7, 64.1, 35.7, 25.0, 22.0, 18.6. HRMS ESI: [M+H]+ calculated for C17H15OS+: 267.0838; found 267.0832.
2.9.3 7-(3-Ethynylbenzo[b]thiophen-2-yl)hex-5-ynal (17a)
The aldehyde 17a was recognized in the mix with 14a according to NMR and HRMS as product of the deiodination of the latter. 1H NMR (400 MHz, CDCl3), δ, ppm: 9.86 (s, 1H), 7.86 (dd, J = 6.8 Hz, J = 1.8 Hz, 1H), 7.72 (dd, J = 6.9 Hz, J = 1.7 Hz, 1H), 7.41 (qd, J = 7.2 Hz, J = 3.6 Hz, 2H), 3.53 (s, 1H), 2.73 (td, J = 7.2 Hz, J = 0.9 Hz, 2H), 2.64 (t, J = 6.8 Hz, 2H), 1.99 (p, J = 7.0 Hz, 2H). HRMS ESI: [M+NH4]+ calculated for C16H16NOS+: 270.0947; found: 270.1764.
2.9.4 7-(3-ethynylbenzo[b]thiophen-2-yl)hept-6-ynal (17b)
The aldehyde 17b was isolated by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 14 mg (27%) of 17b as a yellow oil. Rf = 0.33. 1H NMR (400 MHz, CDCl3), δ, ppm: 9.80 (s, 1H), 7.85 (d, J = 7.3 Hz, 1H), 7.71 (d, J = 7.3 Hz, 1H), 7.44–7.37 (m, 2H), 3.54 (s, 1H), 2.59 (t, J = 6.8 Hz, 2H), 2.53 (t, J = 6.8 Hz, 2H), 1.91–1.83 (m, 2H), 1.74–1.62 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 202.2, 138.7, 137.8, 128.6, 126.0, 125.1, 123.1, 122.0, 120.6, 100.7, 83.3, 76.9, 73.9, 43.3, 27.6, 21.2, 19.8. HRMS FAB: [M]+ calculated for C17H14OS+: 266.0760; found: 266.0764.
2.9.5 6-(2-ethynylbenzo[b]thiophen-3-yl)hex-5-ynal (17c)
The aldehyde 17c was isolated by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 48 mg (60%) of 17c as a brown oil. Rf = 0.36 (hexane/ethyl acetate (2:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 9.87 (s, 1H), 7.86–7.81 (m, 1H), 7.74–7.70 (m, 1H), 7.45–7.39 (m, 2H), 3.69 (s, 1H), 2.76 (td, J = 7.2 Hz, J = 1.1 Hz, 2H), 2.66 (t, J = 6.8 Hz, 2H), 2.02 (p, J = 7.0 Hz, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 201.7, 138.5, 138.4, 126.4, 125.1, 124.4, 124.1, 123.5, 122.1, 96.2, 86.6, 76.7, 74.6, 42.7, 21.1, 19.2. HRMS ESI: [M+Na]+ calculated for C16H12NaOS+: 275.0501; found 275.0502.
2.9.6 7-(2-ethynylbenzo[b]thiophen-3-yl]hex-6-ynal (17d)
The aldehyde 17d was isolated by column chromatography using hexane/ethyl acetate (10:1) as the eluent gave 14 mg (26%) of 17d as a brown oil. Rf = 0.38 (hexane/ethyl acetate (2:1)). 1H NMR (400 MHz, CDCl3), δ, ppm: 9.82 (t, J = 1.6 Hz, 1H), 7.87–7.84 (m, 1H), 7.74–7.70 (m, 1H), 7.45–7.39 (m, 2H), 3.68 (s, 1H), 2.62 (t, J = 6.9 Hz, 2H), 2.54 (td, J = 7.3 Hz, J = 1.6 Hz, 2H), 1.96–1.88 (m, 2H), 1.78–1.71 (m, 2H). 13C NMR (101 MHz, CDCl3), δ, ppm: 202.2, 138.6, 138.4, 126.4, 125.1, 124.6, 123.8, 123.5, 122.1, 96.9, 86.5, 76.7, 74.0, 43.3, 28.0, 21.2, 19.5. HRMS ESI: [M+Na]+ calculated for C17H14NaOS+: 289.0658; found 289.0662.
3 Results and discussion
3.1 Preliminary calculations of enediynes activity in the Bergman cyclization
Before the synthesis of macrocyclic enediynes, the DFT calculations were performed to predict their activity in the Bergman cyclization. As was mentioned above, BC proceeds via the formation of p-benzyne biradicals. Therefore, we have calculated the relative activation energy for the formation of these intermediates for known 10-membered macrocyclic enediynes 1–3 and 9-membered macrocyclic enediynes 5 and compared these values with 10-,11-membered benzothiophene fused enediynes 4a,c,d (Fig. 1). Geometry optimizations of reactants and transition stated were performed using B3LYP functional with the 6-31G++(d,p) basis set. Because of the open shell nature of the transition-state and product, calculations on these structures were performed using BS-UB3LYP (broken-spin-symmetry, unrestricted) calculations. Values calculated are the same for enediyne 4a and isomeric macrocycle 4c.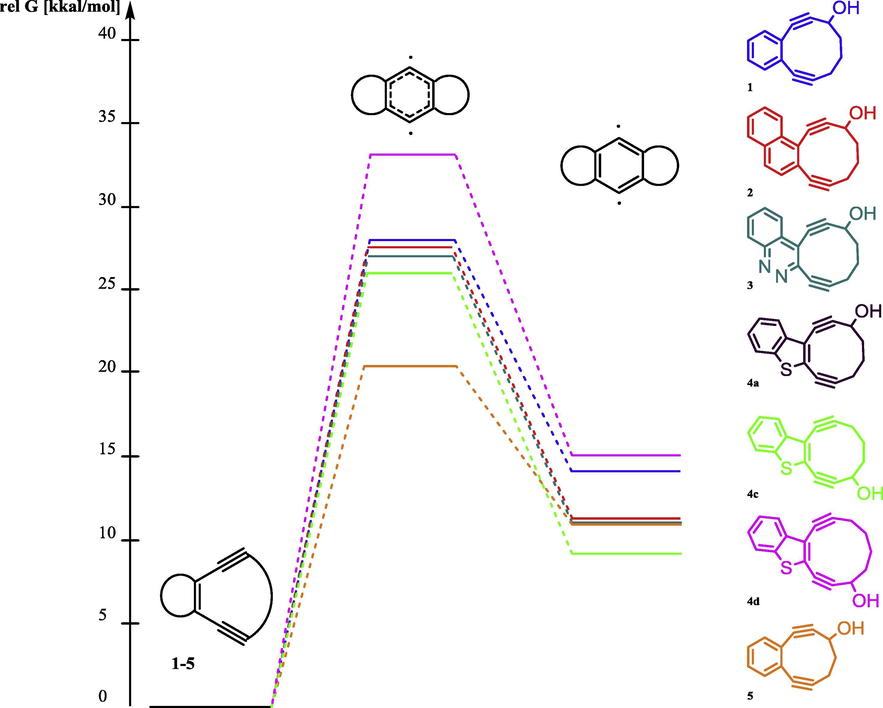
Relative activation energy and relative stability of biradical intermediates of Bergman cyclization.
We found out that calculation data related to the 10-membered enediynes fused with benzo- and cinnoline core correlate with the experimental data obtained before (Vinogradova et al., 2011) and predicted macrocyclic enediynes annulated with benzothiophene to be more reactive in this series. It should be noted, that only the precursor of the most reactive nine-membered enediyne 5 with one triple bond masked as a cyclopropenone was obtained under Nozaki-Hiyama-Kishi conditions, and after photochemical decarbonylation 4,5-benzocyclonona-2,6-diyn-1-ol (5) underwent spontaneous Bergman cyclization (Pandithavidana et al., 2009). Thus 10-membered enediyne 4a and isomeric macrocycle 4c which could be obtained by NHK cyclization became synthetic targets of the research whereas 11-membered enediynes 4b,d were selected for test NHK reaction (Fig. 2).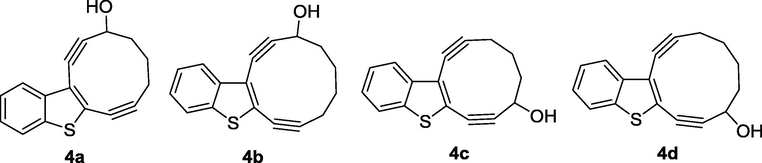
Targets of research.
3.2 Synthesis of starting materials
For the synthesis of 10- and 11-membered macrocycles enediynes fused to benzo[b]thiophene 4a-d by the NHK-cyclization, two regioisomers of the starting acyclic enediynes 11a-d were produced by varying initial diacetylene derivatives for iodocyclization (Scheme 1).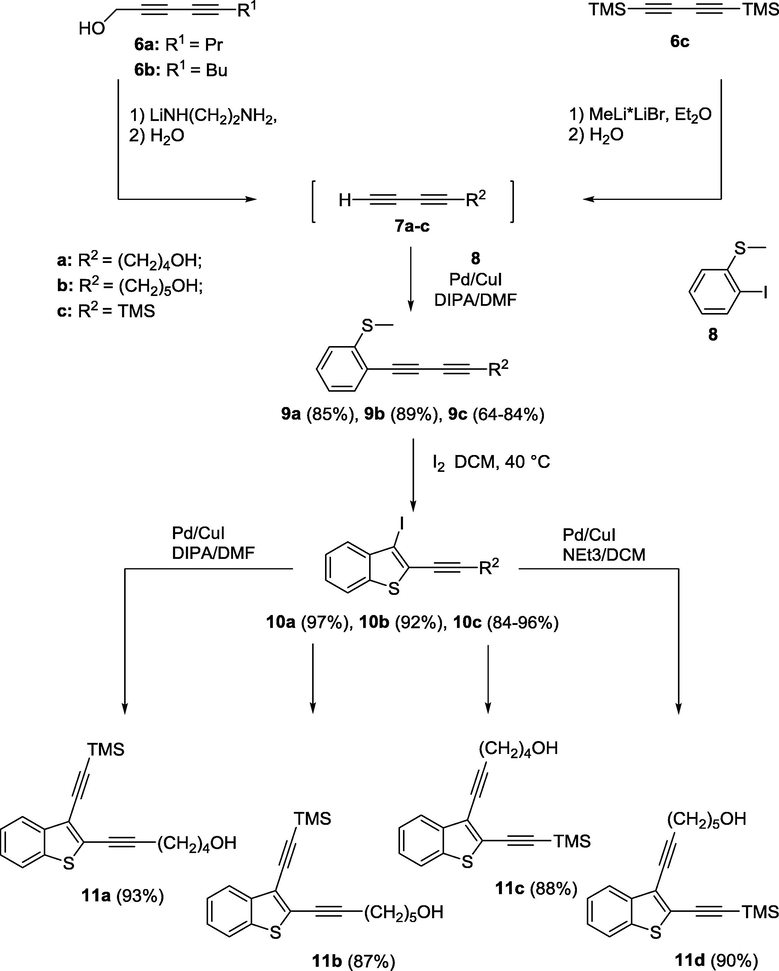
Synthesis of two regioisomers of starting acyclic enediynes 11a-d.
Terminal alkadiynols 7a,b were readily obtained by the treatment of the corresponding internal diacetylenes 6a,b with lithium 2-aminoethylamide (LAETA) («diacetylene zipper reaction») (Kulyashova et al., 2013). TMS-buta-1,3-diyne 7c was synthesized according to the reported procedure (Danilkina et al., 2014). Both isomers of terminal diacetylenes were used in the next step without purification. The Sonogashira coupling of ortho-iodothioanisole 8 with terminal diacetylenes 7a-c opened access to the compounds 9a-c. Iodocyclization of thioanisoles 9a-c followed by the Sonogashira cross-coupling (Scheme 1) with corresponding acetylenes led to acyclic enediynes fused to S-heteroindene 11a-d as key intermediates.
While Pd/Cu-promoted coupling of compounds 10a,b with TMS-acetylene using diisopropanolamine (DIPA) as a base in DMF gave products 11a,b in high yields, the synthesis of 11c,d under the same conditions led to the formation of products in moderate yields (∼40%) due to a visible decomposition of reaction mixtures. Meanwhile, the coupling of 2-(2-trimethylsilyl)ethynyl-3-iodobenzo[b]thiophene (10c) with hex-5-yn-1-ol or hept-6-yn-1-ol was carried out successfully in the presence of less active base (NEt3) in DCM.
Precursors for NHK macrocyclization have to contain iodo- and aldehyde moieties at opposite termini of (Z)-3-ene-1,5-diyne system. These compounds were obtained in several steps from enediynes 11a-d (Scheme 2). Compounds 12a-d were synthesized from TMS derivatives 11a-d upon treatment with TBAF hydrate. Next, the reaction with iodine-morpholine complex in presence of CuI (Hein et al., 2009) gave iodides 13a-d, which were then oxidized using Dess-Martin periodinane to give starting materials for Nozaki reaction 14a-d.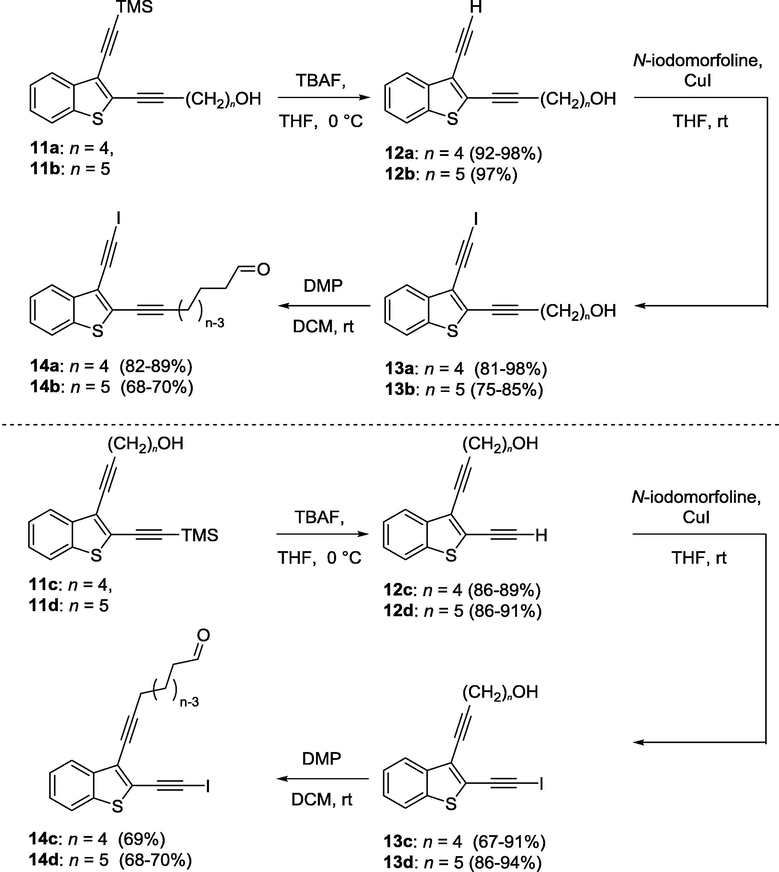
Synthesis of precursors for Nozaki-reaction.
3.3 Investigation of Cr(II)-promoted cyclizations
Intramolecular macrocyclization under NHK conditions has several features: (1) diluted solutions in polar solvents like DMF (Takai et al., 1986), DMSO (Jin et al., 1986), THF and their mixtures (Panek et al., 2000) are used to avoid intermolecular processes; (2) a mixture of anhydrous CrCl2 as a reagent and NiCl2 as a catalyst, has been found as the more effective combination for NHK reaction (Takai et al., 1986); (3) excess of CrCl2 are required due to the formation of chromium (III) alcoholates as the by-products; (4) inert atmosphere and absence of moisture are needed (Jin et al., 1986; Takai et al., 1986) to avoid oxidation and decomposition of chromium salts and intermediates (Takai et al., 1983). Therefore, all experiments were carried out at 10−2–10−3 M concentration of the starting material using 4–20-fold excess of CrCl2 under argon.
According to Vinogradova et al. (2011), a facile intramolecular NHK reaction of acyclic enediyne fused with cinnoline gave desired 10-membered macrocycle using CrCl2–NiCl2 mixture in DMF (0.01 M concentration of starting material) at 0 °C. Therefore, these conditions were initially tested for the cyclization of benzo[b]thiophenes 14a. However, instead of cyclization, compound 14a underwent partial convertiion into the product of deiodination 17a (Entry 1, Table 1). Exploring of DMSO and THF as solvents with different concentrations has revealed the fast formation of complicated mixture of oligomers (Entries 2–4, Table 1). When the concentration of starting aldehyde 14a was increased from 0.001 M to 0.01 M under the action of 4-fold excess of CrCl2 in the presence of catalytic amount of NiCl2 (Entry 5, Table 1, Scheme 3), only a dimeric product was isolated from the reaction mixture in low yield.
Entry
Solvent
t °C
Time, h
CM, mol/L
CrCl2/NiCl2
Product (Yield, %)a
1 (14a)
DMF
r.t.
14
0.01
4/0.1
17ab
2 (14a)
DMSO
r.t.
14
0.01
4/0.1
Mixture of oligomers
3 (14a)
THF
0 °C → r.t.
14
0.005
4/0.1
Mixture of oligomers
4 (14a)
THF
0 °C → r.t.
14
0.001
4/0.1
Mixture of oligomers
5 (14a)
THF
0 °C → r.t.
14
0.01
4/0.1
Dimer 4e (46)
6 (14a)
THF/DMF (3 : 1)
0 °C → r.t.
14
0.01
10/0.1
–
7 (14a)
THF/DMF (3 : 1)
r.t.
1
0.01
20/0.1
16a (56–67)
8 (14b)
THF/DMF (3 : 1)
r.t.
1
0.01
20/0.1
16b (58)
9 (14a)
THF/DMF (4 : 1)
r.t.
1.5
0.007
10/0.1
16a (87)
10c (14b)
THF/DMF (1 : 4)
r.t.
18
0.002
10/0
4b (33), 17b (27)
11c (14b)
THF/DMF (1 : 4)
r.t.
24
0.005
10/0
4b (47), 17b (29)
12c (14b)
THF/DMF (1 : 4)
r.t.
24
0.01
10/0
4b (47), 17b (47)
13c (14b)
DMF
r.t.
48
0.01
10/0
4b (60)
14c (14a)
DMF
r.t.
48
0.01
10/0
17ab
15c (14c)
DMF
r.t.
48
0.01
10/0
17c (60)
16c (14d)
DMF
r.t.
48
0.01
10/0
4d (51–66) 17d (13–26)
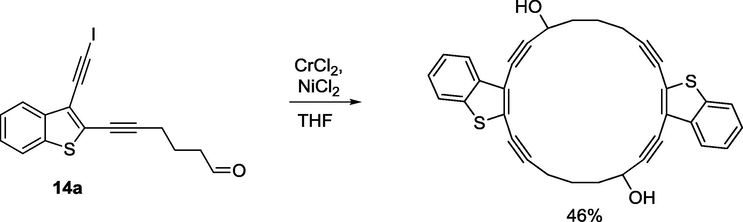
Synthesis of the macrocyclic diol.
NMR 1H spectra of dimer showed signals of two diastereomers namely two individual signals (multiplicity-triplet) of stereocentres at hydroxyl group in spectra. In addition, double set of signals had been detected in NMR 13C (see SI).
Then intramolecular cyclization of substrates 14a,b has been investigated in the THF/DMF mixture. No reaction was observed for the compound 14a when tenfold access of CrCl2 was used in THF/DMF (3:1) (Panek et al., 2000) at 0 °C (Entry 6, Table 1). When 20 equivalents of CrCl2 were employed at room temperature, a single product has been formed from both starting aldehydes 14a and 14b (Entry 7, 8, Table 1). NMR 1H spectra of new product did not show the signal of an aldehyde group, but rather presented characteristic signals CH-OH group.
HRMS ESI spectra contain signals of molecular ions at 254.0763 and 268.0925 m/z. This masses corresponds to the chemical formulae C16H14OS+ and C17H16OS+ respectively, which agrees with the structure of the Bergman cyclization products 15a and 15b (Scheme 4, route A).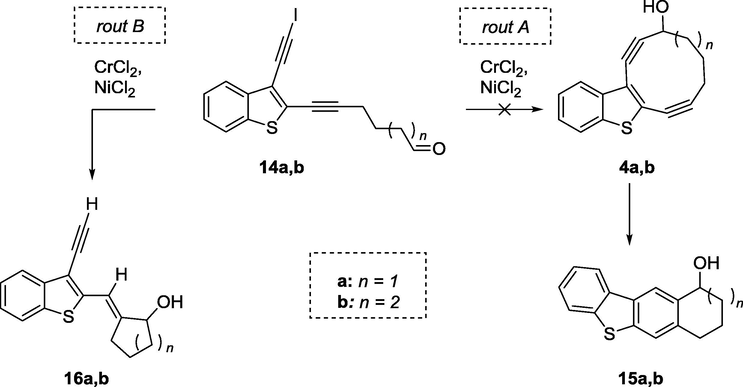
Synthesis of 2-methylenecycloalkan-1-ols as alternative way of Nozaki reaction.
Therefore, we have initially suggested a reaction sequence involving intramolecular NHK reaction followed by the in situ cycloaromatization. However, the analysis of spectral data, including IR, NMR 1H, 13C (in CDCl3 and DMSO‑d6) revealed the presence of only one triple bond and a new double bond in the products.
The analysis of the spectral data, including NOESY, allowed us to establish the structure of the unexpected products of the intramolecular cyclization as vinyl cyclopentanol 16a,b with E-configuration of double bond (Scheme 4, route B). In addition, the structure of 16a has been established by single-crystal X-ray analysis (Fig. 3). Compounds 16a,b are the apparent products of the electrophilic addition of acetylene to Cr (II) – activated aldehyde. The dilution of a reaction mixture by THF (ratio of THF:DMF was changed from 3:1 to 4:1) and reduction of excess of CrCl2 to10 equivalents have led to the formation of cyclic allylic alcohol 16a in a good yield (Entry 9, Table 1).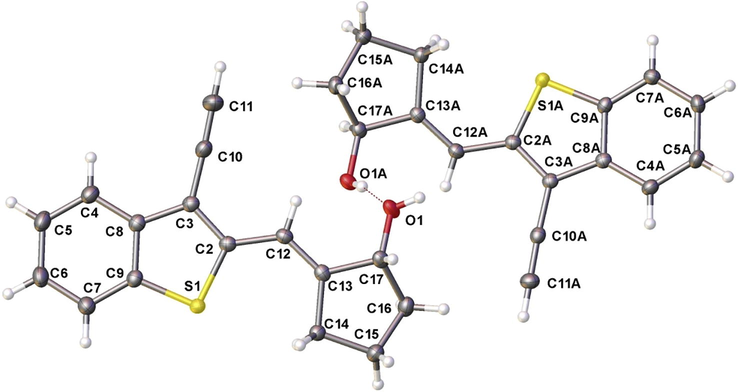
Molecular structure of compound 16a according to X-ray data.
The authors know only one other example of the similar intramolecular formation of allylic alcohol under NHK conditions (Boddenmann and Keese, 1993). At the same time, the nickel-catalyzed intermolecular formation of 2-methylenecyclopentane-1-ol and 2-methylenecyclohexane-1-ol from derivatives of hex-5yn-1-al and hept-6-yn-1al has been reported previously (Malik et al., 2010; McCarren et al., 2009; Montgomery, 2004; Oblinger and Montgomery, 1997; Tang and Montgomery, 1999). Apparently nickel coordination in THF activates both the triple bond and the carbonyl group (Hodgson and Wells, 1994). Therefore, we decided to elucidate the influence of NiCl2 on the cyclization.
In the absence of NiCl2 aldehyde 14b under the action of 10-fold excess of CrCl2 in the mixture THF/DMF in ratio 1:4 produces the target 11-membered enediyne 4b (Scheme 5, entry 10–12, Table 1) along with the product of reduction of iodoacetylene 14b to terminal alkyne 17b.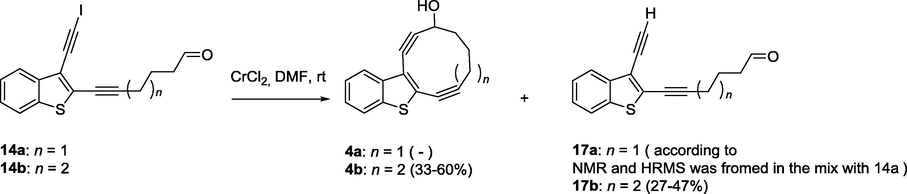
Cr(II)-promoted reaction of compounds 14a,b.
The best yield of 4b (60%) without the traces of byproduct 17b has been obtained under the action of 10-fold access of CrCl2 in DMF (0.01 M) at ambient temperature (Entry 13, Table 1). These results suggest that the formation of by-product 17 can be minimized by avoiding NiCl2 and THF. The cyclization of aldehyde 14a to 10-membered macrocycle 4a under the same conditions failed (Entry 14, Table 1). Purification and separation of a crude reaction mixture allowed to yield only product of deiodination in mixture with starting material (80% of starting material was recovered).
The cycloaromatization of enediyne 4b has been studied by DSC, which is commonly used to characterize the efficiency of the enediyne cycloaromatization (Basak et al., 2004; Danilkina et al., 2016, 2015, 2014; Hickenboth et al., 2008; König and Rütters, 1994). The onset of the cycloaromatization was recorded at 172 °C, which agrees well with the calculated activation energy of 33.1 kcal/mol and previous experimental data (Danilkina et al., 2016).
The treatment of isomeric compounds 14c,d under NHK conditions showed very similar results. While the macrocyclization of acyclic enediyne 14d in the prence of Cr(II) in DMF (no NiCl2) produced good yield of 11-membered 4d (Entry 16, Table 1, Scheme 6), all attempts of cyclization of 14c resulted in the formation of the deiodination product 17c (Entry 15, Table 1) or complex mixture of oligomeric products.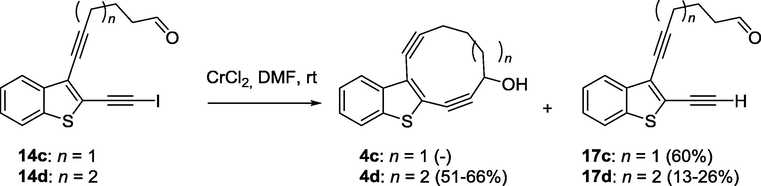
Cr(II)-promoted reaction of compounds 14c,d.
In order to explain the difference in reactivity between 14a,c and 14b,d, the DFT analysis of the ring strain in the target cyclic systems has been carried out. The relative strain energies of structures 1–5 were calculated at B3LYP/6-31G++(d,p) level using isodesmic reactions and benzannulated 10-membered ring enediyne as a reference (Fig. 4).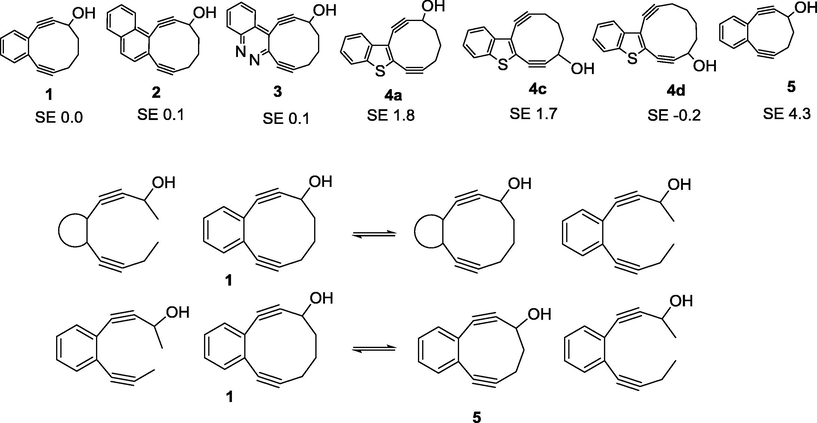
Scheme of calculation of relative SE (kcal) for enediyne macrocycles, the strain energy of molecule 1 was admitted as zero.
There is virtually no difference in the ring strain between 1 and 2, 3, 4d and all of these compounds have been successfully prepared using Nozki cyclization (e.g., Vinogradova et al., 2011). On the other hand, ring strain in macrocycles 4a,c is about 2 kcal/mol higher than in 1. Apparently, this additional ring strain prevents macrocyclization of acyclic enediynes 14a,c. The cycle 5 with the highest ring strain calculated was obtained under Nozaki-Hiyama-Kishi only with one triple bond masked as a cyclopropenone (Pandithavidana et al., 2009).
Another possible cause of the inefficient cyclization 14a,c is an increased distance between reacting groups due to the lager angles between substituents attached to the 5-membered rings in comparison to the 6-membered analogs. Compounds 18–20 containing chlorine as halogen were used in DFT calculations as models of precursors for NHK reaction. A noticeable increase in the angles 1 and 2 in compound 20 over 18, 19 is observed, supporting this suggestion (Fig. 5). Consequently, NHK intramolecular cyclization producing 10-membered enediyne cycle conjugated with benzothiophene is forbidden.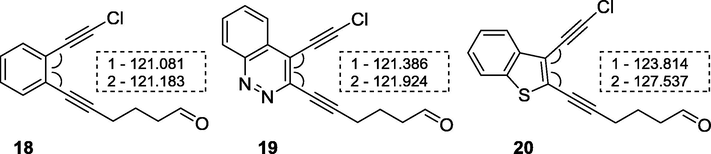
Values of angles 1 and 2 calculated of model substrates for NHK-cyclization.
4 Conclusion
The investigation of intramolecular Nozaki-type coupling of acyclic enediynes fused to benzo[b]thiophene elucidated that geometry and structure of starting material as well as nature of catalytic system influence dramatically on the reaction results. We found, that two different metal (chromium and nickel) commonly employed in this reaction, expand the number of coordination sites and might lead to the formation of different types of cycles. Thus, in the presence of Ni(II) NHK coupling of the compounds discussed in the present manuscript instead of a macrocycle formation gives the product of nucleophilic addition of the triple bond to the carbonyl group within the same fragment. Cyclic allylic alcohols were obtained as the result of exo-cyclization under the action of Cr2+/Ni2+ system in THF/DMF in the case of 6-[3-(iodoethynyl)benzo[b]thiophen-2-yl]hex-5-ynal and 7-[3-(iodoethynyl)benzo[b]thiophen-2-yl]hept-6-ynal. The use of CrCl2 without NiCl2 co-catalyst in DMF (0.01 M) led to the formation of 11-membered enediyne macrocycles. The formation of more strained 10-membered benzotiophene-fussed enediynes by Nozaki cyclization is restricted probably due to the high values of activation energy barrier which correlates with the ring strain. Thus, our findings demonstrated both limitations of NHK reaction in the synthesis of 10-membered macrocyclic enediynes and intriguing challenge for study of cheap NiCl2–catalyzed intramolecular cyclization of easily formed hex-5-ynal and hept-6-ynal fragments as facile and universal approach to the synthesis of important cyclic allylic alchohols.
Acknowledgment
This study was supported by Saint Petersburg University - Russia (SPbU) (grant numbers 12.40.515.2017). A.E.K. is grateful to RFBR (grant numbers 16-33-00817, mol-a). A.E.K. and V. V. P. are thankful to NIH (Award #R01-CA175480). Scientific research was performed at the Center for Magnetic Resonance, the Center for Chemical Analysis and Materials Research, the Thermogravimetric and Calorimetric Research Centre and the Center for X-ray Diffraction Methods of Research park of St. Petersburg State University.
References
- Safety, pharmacokinetics, and preliminary clinical activity of inotuzumab ozogamicin, a novel immunoconjugate for the treatment of B-cell non-Hodgkin’s lymphoma: results of a phase I study. J. Clin. Oncol.. 2010;28:2085-2093.
- [CrossRef] [Google Scholar]
- Synthesis of a methoxy-substituted lactenediyne. Tetrahedron Lett.. 2000;41:6523-6526.
- [CrossRef] [Google Scholar]
- Synthesis of a new lactenediyne scaffold equipped with three handles. Tetrahedron Lett.. 2002;43:7427-7429.
- [CrossRef] [Google Scholar]
- Synthesis of intramolecularly activated lactenediynes and evaluation of their activity against plasmid DNA. Eur. J. Org. Chem.. 2002;2002:3745-3755.
- [CrossRef] [Google Scholar]
- Effect of remote trigonal carbons on the kinetics of bergman cyclization: synthesis and chemical reactivity of pyridazinedione-based enediynes. J. Org. Chem.. 2004;69:6927-6930.
- [CrossRef] [Google Scholar]
- Synthesis of cyclododeca-2,8-diyne-1,7-dione. Tetrahedron Lett.. 1993;34:1467-1470.
- [CrossRef] [Google Scholar]
- Modular total syntheses of the marine-derived resorcylic acid lactones cochliomycins A and B Using a late-stage Nozaki-Hiyama-Kishi macrocyclization reaction. J. Org. Chem.. 2015;80:460-470.
- [CrossRef] [Google Scholar]
- Synthesis and oxidative activation of an oxabicyclo[7.2.1]enediyne. Tetrahedron. 1994;50:1435-1448.
- [CrossRef] [Google Scholar]
- Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chemie - Int. Ed.. 2014;53:3796-3827.
- [CrossRef] [Google Scholar]
- Photochemical and thermal bergman cyclization of a pyrimidine enediynol and enediynone. Org. Lett.. 2000;2:3761-3764.
- [CrossRef] [Google Scholar]
- A mild route to α -alkoxyacetylenes mediated by Lewis Acids and synthetic routes to 10–11- and 12-membered ring enediyne carbocycles. Isr. J. Chem.. 2000;40:241-253. https://onlinelibrary.wiley.com/doi/abs/10.1560/7J4F-D8BG-3FQR-E3TD
- [Google Scholar]
- The esperamicin- calicheamicin aglycones: ring closure of a simple strained system mediated by chromium(II)-nickel(II) salts. Tetrahedron Lett.. 1991;32:3171-3174.
- [CrossRef] [Google Scholar]
- Intramolecular Nozaki-Hiyama-Kishi reactions and Ln(III)-catalyzed allylic rearrangement as the key steps towards 10-membered ring enediynes. Tetrahedron Lett.. 2001;42:4211-4214.
- [CrossRef] [Google Scholar]
- Synthetic studies related to the esperamicin/calicheamicin aglycone: efficient construction of a homochiral oxabicyclo [7:3:1] analogue from D-xylose. J. Chem. Soc. Chem. Commun.. 1995;3:799-800.
- [CrossRef] [Google Scholar]
- Electrophilic cyclization of buta-1,3-diynylarenes: synthesis of precursors of (z)-3-ene-1,5-diyne systems fused to heterocycles. Synlett. 2011;517–520
- [CrossRef] [Google Scholar]
- Towards isocoumarin-fused enediyne systems through the electrophilic cyclization of methyl o -(Buta-1,3-diynyl)benzoates. Eur. J. Org. Chem.. 2016;2016:739-747.
- [CrossRef] [Google Scholar]
- Electrophilic cyclization of aryldiacetylenes in the synthesis of functionalized enediynes fused to a heterocyclic core. J. Org. Chem.. 2014;79:9018-9045.
- [CrossRef] [Google Scholar]
- Ring-closing metathesis of Co2(CO)6–alkyne complexes for the synthesis of 11-membered dienediynes: overcoming thermodynamic barriers. J. Org. Chem.. 2015;80:5546-5555.
- [CrossRef] [Google Scholar]
- Electrophilic cyclization and ring-closing metathesis as key steps in the synthesis of a 12-membered cyclic enediyne. Eur. J. Org. Chem.. 2012;5660–5664
- [CrossRef] [Google Scholar]
- Characterization of the in vitro cyclization chemistry of calicheamicin and its relation to DNA cleavage. J. Am. Chem. Soc.. 1990;112:4554-4556.
- [CrossRef] [Google Scholar]
- Nozaki - Hiyama - Kishi Reactions catalytic in chromium. J. Am. Chem. Soc.. 1996;118:12349-12357.
- [CrossRef] [Google Scholar]
- Antitumor antibiotics: Bleomycin, enediynes, and mitomycin. Chem. Rev.. 2005;105:739-758.
- [CrossRef] [Google Scholar]
- Mechanism of neocarzinostatin action: role of DNA microstructure in determination of chemistry of bistranded oxidative damage. Acc. Chem. Res.. 1991;24:191-198.
- [CrossRef] [Google Scholar]
- Hamann, P.R., Upeslacis, J., Borders, D.B., 2011. Enediynes. In: Cragg, G.M., Kingston, D.G.I., Newman, D.J. (Eds.), Anticancer Agents from Natural Products, Boca Raton, pp. 575−621.
- Copper(I)-catalyzed cycloaddition of organic azides and 1-iodoalkynes. Angew. Chemie - Int. Ed.. 2009;48:8018-8021.
- [CrossRef] [Google Scholar]
- Preparation of enediyne-crosslinked networks and their reactivity under thermal and mechanical conditions. Tetrahedron. 2008;64:8435-8448.
- [CrossRef] [Google Scholar]
- Chromium(II)-mediated nickel(II)-catalysed cyclisations of (iodoaryl)-substituted alkynes and alkynals. Tetrahedron Lett.. 1994;35:1601-1604.
- [CrossRef] [Google Scholar]
- Synthetic studies on the seven- and eight-membered rings by the intramolecular Nozaki-Hiyama reaction of the allylic phosphates. Tetrahedron Lett.. 2004;45:8653-8657.
- [CrossRef] [Google Scholar]
- Catalytic effect of nickel(II) chloride and palladium(II) acetate on chromium(II)-mediated coupling reaction of iodo olefins with aldehydes. J. Am. Chem. Soc.. 1986;108:5644-5646.
- [CrossRef] [Google Scholar]
- Recent developments in enediyne chemistry. Chem. Biodivers.. 2012;9:459-498.
- [CrossRef] [Google Scholar]
- Enhancement of the reactivity of photochemically generated enediynes via keto-enol tautomerization. J. Am. Chem. Soc.. 2008;130:11771-11777.
- [CrossRef] [Google Scholar]
- Triggering of the bergman cyclization by photochemical ring contraction. Facile cycloaromatization of benzannulated cyclodeca-3,7-diene-1,5-diynes. J. Am. Chem. Soc.. 2007;129:3792-3793.
- [CrossRef] [Google Scholar]
- Promysalin elicits species-selective inhibition of pseudomonas aeruginosa by targeting succinate dehydrogenase. J. Am. Chem. Soc.. 2018;140:1774-1782.
- [CrossRef] [Google Scholar]
- Solvent dependent Bergman cyclization of 2,3-diethynylquinoxaline. Tetrahedron Lett.. 1999;40:3835-3838.
- [CrossRef] [Google Scholar]
- Tautomer-dependent Bergman cyclization of novel uracil-enediyne chimeras. Chem. - A Eur. J.. 2000;6:1555-1558. https://doi.org/10.1002/(SICI)1521-3765(20000502)6:9<1555::AID-CHEM1555>3.0.CO;2-M
- [Google Scholar]
- Rapid Bergman cyclization of 1,2-diethynylheteroarenes. J. Org. Chem.. 1998;63:8229-8234.
- [CrossRef] [Google Scholar]
- A Pd-catalyzed double coupling reaction to 4,5-disubstituted imidazole alkynes. Synth. Commun.. 1999;29:507-512.
- [CrossRef] [Google Scholar]
- Synthesis and reactivity of the first bis(crown ether) enediyne. Tetrahedron Lett.. 1994;35:3501-3504.
- [CrossRef] [Google Scholar]
- Crystal and molecular structure of dynemicin A: a novel 1,5-diyn-3-ene antitumor antibiotic. J. Am. Chem. Soc.. 1990;112:3715-3716.
- [CrossRef] [Google Scholar]
- Design of a new warhead for the natural enediyne dynemicin A. An increase of biological activity. J. Phys. Chem. B. 2008;112:2661-2670.
- [CrossRef] [Google Scholar]
- An acetylene zipper-Sonogashira reaction sequence for the efficient synthesis of conjugated arylalkadiynols. Tetrahedron Lett.. 2013;54:2235-2238.
- [CrossRef] [Google Scholar]
- Total synthesis of LL-Z1640-2 utilizing a late-stage intramolecular Nozaki–Hiyama–Kishi reaction. Tetrahedron Lett.. 2010;51:6852-6855.
- [CrossRef] [Google Scholar]
- Calichemicins, a novel family of antitumor antibiotics. 2. Chemistry and structure of calichemicin.gamma. 1I. J. Am. Chem. Soc.. 1987;109:3466-3468.
- [CrossRef] [Google Scholar]
- Calicheamicins: discovery, structure, chemistry, and interaction with DNA. Acc. Chem. Res.. 1991;24:235-243.
- [CrossRef] [Google Scholar]
- New access to unsaturated keto carba sugars (gabosines) using an intramolecular nozaki-kishi reaction as the key step. J. Org. Chem.. 1998;63:5668-5671.
- [CrossRef] [Google Scholar]
- Oxaenediynes through the Nicholas-Type Macrocyclization Approach. European J. Org. Chem.. 2016;28:4842-4851.
- [CrossRef] [Google Scholar]
- Relative reactivity of benzothiophene-fused enediynes in the bergman cyclization. J Org Chem.. 2018;83:2788-2801.
- [CrossRef] [Google Scholar]
- Synthesis of an oxabicyclo[7.2.1] enediyne from a furanoside derivative. Tetrahedron Lett.. 1992;33:7511-7514.
- [CrossRef] [Google Scholar]
- A general strategy for regiocontrol in nickel-catalyzed reductive couplings of aldehydes and alkynes. J. Am. Chem. Soc.. 2010;132:6304-6305.
- [CrossRef] [Google Scholar]
- Enediyne antibiotics and their models: new potential of acetylene chemistry. Russ. Chem. Rev.. 2006;75:825-845.
- [CrossRef] [Google Scholar]
- Mechanism and transition-state structures for nickel-catalyzed reductive alkyne-aldehyde coupling reactions. J. Am. Chem. Soc.. 2009;131:6654-6655.
- [CrossRef] [Google Scholar]
- A Nozaki−Hiyama−Kishi Ni(II)/Cr(II) coupling approach to the phomactins. Org. Lett.. 2001;3:1491-1494.
- [CrossRef] [Google Scholar]
- Biosynthesis and function of polyacetylenes and allied natural products. Prog. Lipid Res.. 2008;47:233-306.
- [CrossRef] [Google Scholar]
- Concerted reactions that produce diradicals and zwitterions: electronic, steric, conformational, and kinetic control of cycloaromatization processes. Chem. Rev.. 2013;113:7089-7129.
- [CrossRef] [Google Scholar]
- Protecting-group directed stereoselective intramolecular Nozaki-Hiyama-Kishi reaction: a concise and efficient total synthesis of amphidinolactone A. Eur. J. Org. Chem.. 2010;2010:4775-4784.
- [CrossRef] [Google Scholar]
- Nickel-catalyzed reductive cyclizations and couplings. Angew. Chemie - Int. Ed.. 2004;43:3890-3908.
- [CrossRef] [Google Scholar]
- “Ab normal” Eight - Membered Ring Formation through SN2’ intramolecular Nozaki/Kishi reaction in a synthetic approach to a taxane precursor Benoit Muller, Jean-Pierre F6r6zou*, Jean-Yves Lallemand, Ange Panerazi, Jo∼lle Pru. Tetrahedron Lett.. 1998;39:279-282.
- [CrossRef] [Google Scholar]
- Proposed structure of the neocarzinostatin chromophore-methyl thioglycolate adduct; A mechanism for the nucleophilic activation of neocarzinostatin. Tetrahedron Lett.. 1987;28:4493-4496.
- [CrossRef] [Google Scholar]
- Biradical formation from acyclic conjugated eneyne-allene system related to neocarzinostatin and esperamicin-calichemicin. Tetrahedron Lett.. 1989;30:4995-4998.
- [CrossRef] [Google Scholar]
- J. Am. Chem. Soc.. 1988;110:4868-4869.
- [CrossRef]
- Chemistry and biology of the enediyne anticancer antibiotics. Angew. Chemie - Int. Ed.. 1991;30:1387-1416.
- [CrossRef] [Google Scholar]
- Designed enediynes: a new class of DNA-cleaving molecules with potent and selective anticancer activity. Science. 1992;80(256):1172-1178.
- [CrossRef] [Google Scholar]
- On the mechanism of activation of designed enediynes with selective cytotoxicity. Bioorg. Med. Chem. Lett.. 1992;2:1155-1160.
- [CrossRef] [Google Scholar]
- Redox-controlled Bergman cycloaromatizations. Designed enediynes with DNA-cleaving properties and antitumor activity. J. Am. Chem. Soc.. 1992;114:9279-9282.
- [CrossRef] [Google Scholar]
- Total synthesis of Shishijimicin A. J. Am. Chem. Soc.. 2015;137:8716-8719.
- [CrossRef] [Google Scholar]
- Molecular design, chemical synthesis, and biological action of enediynes. Acc. Chem. Res.. 1992;25:497-503.
- [CrossRef] [Google Scholar]
- Chemistry and biology of natural and designed enediynes. Proc. Natl. Acad. Sci.. 1993;90:5881-5888.
- [CrossRef] [Google Scholar]
- Cell-specific regulation of apoptosis by designed enediynes. Proc. Natl. Acad. Sci.. 1993;90:3142-3146.
- [CrossRef] [Google Scholar]
- One pot synthesis of haloacetylenes from trimethylsilylacetylenes. Synlett. 1994;1994:485-486.
- [CrossRef] [Google Scholar]
- A new stereoselective method for the preparation of allylic alcohols. J. Am. Chem. Soc.. 1997;119:9065-9066.
- [CrossRef] [Google Scholar]
- Shishijimicins A-C, novel enediyne antitumor antibiotics from the ascidian Didemnum proliferum. J. Am. Chem. Soc.. 2003;125:2044-2045.
- [CrossRef] [Google Scholar]
- Photochemical generation and reversible cycloaromatization of a nine-membered ring cyclic enediyne. J. Am. Chem. Soc.. 2009;131:351-356.
- [CrossRef] [Google Scholar]
- Total synthesis of the actin-depolymerizing agent (-) -Mycalolide A: application of chiral silane-based bond construction methodology. J. Am. Chem. Soc.. 2000;122:11090-11097.
- [Google Scholar]
- Total synthesis of (−)-decarestrictine D through a stereoselective intramolecular Nozaki-Hiyama-Kishi reaction. Tetrahedron Lett.. 1998;39:4421-4424.
- [CrossRef] [Google Scholar]
- First Total Synthesis of Aspinolide B, a New Pentaketide Produced by Aspergillus o chraceus. J. Org. Chem.. 2000;65:5910-5916.
- [CrossRef] [Google Scholar]
- Application of photochemical decarbonylation of cyclopropenones for the in situ generation of reactive enediynes. construction of a cyclopropenone-containing enediyne precursor by using a cyclopropenone acetal building block. J. Org. Chem.. 2005;70:1297-1305.
- [CrossRef] [Google Scholar]
- Nucleophilic cycloaromatization of ynamide-terminated enediynes. J. Org. Chem.. 2010;75:5953-5962.
- [CrossRef] [Google Scholar]
- Taxamycin studies: Synthesis of taxoid-calicheamicin hybrids. Tetrahedron Lett.. 1998;39:6139-6142.
- [CrossRef] [Google Scholar]
- Suitable entry to a 10-membered ring with eleutheside functionality through Nozaki-Hiyama condensation. Tetrahedron Lett.. 2002;43:6521-6524.
- [CrossRef] [Google Scholar]
- A simple synthesis and evaluation of the bicyclo[8.3.0] enediyne framework. Tetrahedron Lett.. 2002;43:4947-4950.
- [CrossRef] [Google Scholar]
- Reductive and nucleophilic activation products of dynemicin A with methyl thioglycolate. A rational mechanism for DNA cleavage of the thiol-activated dynemicin A. Biochemistry. 1991;30:2989-2992.
- [CrossRef] [Google Scholar]
- DNA intercalation and cleavage of an antitumor antibiotic dynemicin that contains anthracycline and enediyne cores. Proc. Natl. Acad. Sci.. 1990;87:3831-3835.
- [CrossRef] [Google Scholar]
- Selective grignard-type carbonyl addition of alkenyl halides mediated by chromium(II) chloride. Tetrahedron Lett.. 1983;24:5281-5284.
- [CrossRef] [Google Scholar]
- Reactions of alkenylchromium reagents prepared from alkenyl trifluoromethanesulfonates (triflates) with chromium(II) chloride under nickel catalysis. J. Am. Chem. Soc.. 1986;108:6048-6050.
- [CrossRef] [Google Scholar]
- Total syntheses of (+)- and (-)-pestalotiopsin A. J. Org. Chem.. 2009;74:6452-6461.
- [CrossRef] [Google Scholar]
- Nickel catalysis in the stereoselective preparation of quinolizidine, pyrrolizidine, and indolizidine alkaloids: total synthesis of (+)- allopumiliotoxin 267A. J. Am. Chem. Soc.. 1999;121:6098-6099.
- [CrossRef] [Google Scholar]
- Understanding and exploiting natures chemical arsenal: the past, present and future of calicheamicin research. Curr. Pharm. Des.. 2000;6:1841-1879.
- [CrossRef] [Google Scholar]
- Synthesis and reactivity of cinnoline-fused cyclic enediyne. J. Org. Chem.. 2011;76:6937-6941.
- [CrossRef] [Google Scholar]
- Concise synthesis of calystegines B 2 and B 3 via intramolecular Nozaki–Hiyama–Kishi reaction. Org. Biomol. Chem.. 2016;14:4885-4896.
- [CrossRef] [Google Scholar]
- Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem. Rev.. 2002;102:2477-2496.
- [CrossRef] [Google Scholar]
- Synthetic study of the angular tetracyclic core skeleton of landmycine a via Masamune-Bergman cyclization. Synlett. 2012;23:1327-1330.
- [CrossRef] [Google Scholar]
- Calicheamicin gamma 1I: an antitumor antibiotic that cleaves double-stranded DNA site specifically. Science. 1988;80(240):1198-1201.
- [CrossRef] [Google Scholar]
- Photoinduced Bergman cycloaromatization of imidazole-fused enediynes. Tetrahedron Lett.. 2005;46:1373-1375.
- [CrossRef] [Google Scholar]
- Bergman cycloaromatization of imidazole-fused enediynes: the remarkable effect of N-aryl substitution. Tetrahedron Lett.. 2004;45:3621-3624.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.arabjc.2018.05.005.
Appendix A
Supplementary material
Supplementary data 1
Supplementary data 1




