

Translate this page into:
Current perspectives on benzoflavone analogues with potent biological activities: A review
⁎Corresponding authors. shwang@cdutcm.edu.cn (Shaohui Wang), zhangyi@cdutcm.edu.cn (Yi Zhang)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Medicinal chemistry investigators have isolated and synthesized benzoflavone analogues with diverse bioactivities including enzyme inhibitory activity, central nervous system (CNS) activity, anti-inflammatory activity, anti-microorganism activity, hypoglycemic activity, and receptor modulating potential. SAR (Structure-Activity Relationship) studies have been conducted extensively and plenty of benzoflavone analogues have been prepared for potential targets. Herein, we review the pharmacology properties and SAR for benzoflavone analogues, and provide a brief summarization of synthetic strategies, wishing to give perspective on the research and development of benzoflavone derivatives.
Keywords
Benzoflavone
Cytochrome P450
Antitumor
Central nervous system
Anti-inflammatory
SAR
Derivatives

- ABCA1
-
ATP-binding cassette transporters A1
- AhR
-
Ah receptor
- ANF
-
α-naphthoflavone
- ASK1
-
apoptosis signal-regulating kinase 1
- ATP
-
adenosine triphosphate
- Bcl-2
-
B-cell lymphoma-2
- BCRP
-
Breast cancer resistance protein
- BNF
-
β-naphthoflavone
- B-V
-
Baker-Venkataraman
- CFTR
-
cystic fibrosis transmembrane conductance regulator
- CNS
-
central nervous system
- CYP
-
cytochrome P450
- DNA-PK
-
DNA-dependent protein kinase
- P-gp
-
P-glycoprotein
- GSH
-
glutathione
- HDL-C
-
high-density lipoprotein-cholesterol
- IL
-
interleukin
- LPS
-
lipopolysaccharide
- JNK
-
c-jun N-terminal kinase
- LXRs
-
liver X receptors
- MAO
-
monoamine oxidase
- MMP-2
-
metalloproteinase-2
- MRP1
-
multidrug resistance-associated protein 1
- MtbPtpB
-
Mycobacterium tuberculosis protein tyrosine phosphatases
- PDE
-
phosphodiesterase
- PK
-
pharmacokinetic
- PTP1B
-
protein tyrosine phosphatase 1b
- QSAR
-
quantitative structure-activity relationship
- ROS
-
reactive oxygen species
- SAR
-
Structure-Activity Relationship
- siRNA
-
small interfering RNA
- TIMP-2
-
Tissue inhibitor of metalloproteinase-2
Abbreviations
1 Introduction
Benzoflavones (Fig. 1) is a cluster of derivatives with a broad pharmacological spectrum. The most crucial application of benzoflavones is the interaction to cytochrome P450 (CYP), a cluster of heme-thiolate monooxygenases that participated in an NADPH-dependent electron transport pathway (Shimada et al., 2009), to detail, α-naphthoflavone (ANF, 1) and β-naphthoflavone (BNF, 41) are strong inhibitors of CYP1B1 and 1A2 (Shoda et al., 2000), and the potential of two compounds to modulate related receptors including Ah receptor (AhR) and estrogen receptor alpha were reported as well (Gasiewicz et al., 1996, Klinge et al., 2000), suggesting research value in diseases including breast cancer, liver cancer, cervical cancer. Besides, benzoflavones have been reported to show antitumor activity, central nervous system (CNS), anti-inflammatory activity, anti-microorganism activity, hypoglycemic activity, and receptor modulating potential, demonstrating a broad spectrum of pharmacological research.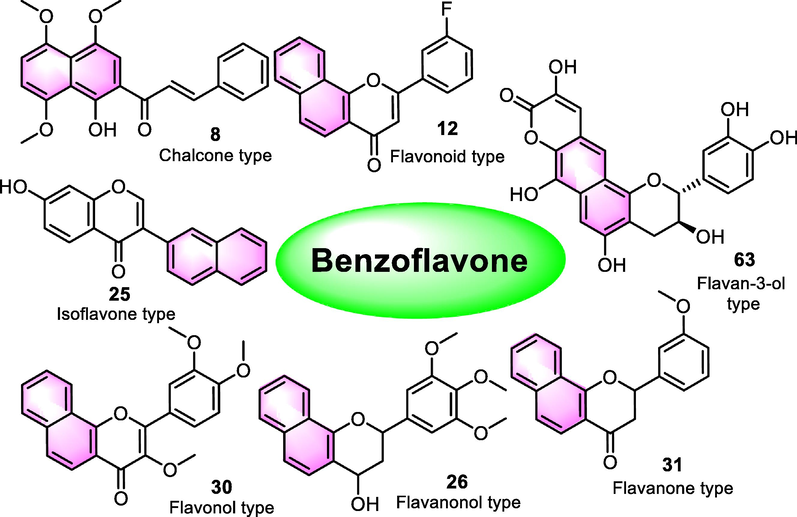
Nuclear family of benzoflavone.
As for benzoflavone derivatives, it is important to emphasize the variety of types of synthetic or natural benzoflavone derivatives (1–78) in existing literature. According to the types of the structure of different skeletons (Fig. 1), benzoflavone derivatives can be classified as chalcone type (8), flavonoid type (12), isoflavone type (25), flavanonol type (26), flavonol type (30), flavanone type (31) and flavan-3-ol type (63). The diversity of the structure type of benzoflavones gives multiple kinds of bioactivities (Fig. 2). Thus, it is necessary to summarize the bioactivities and the SAR analysis of benzoflavone analogues. Synthesis is an important method to develop benzoflavone derivatives as new drugs, thus it is necessary to summarize the investigations about the synthetic strategies of benzoflavones. This review focus on synthetic strategies and bioactive potential of natural isolated and synthesized benzoflavone derivatives.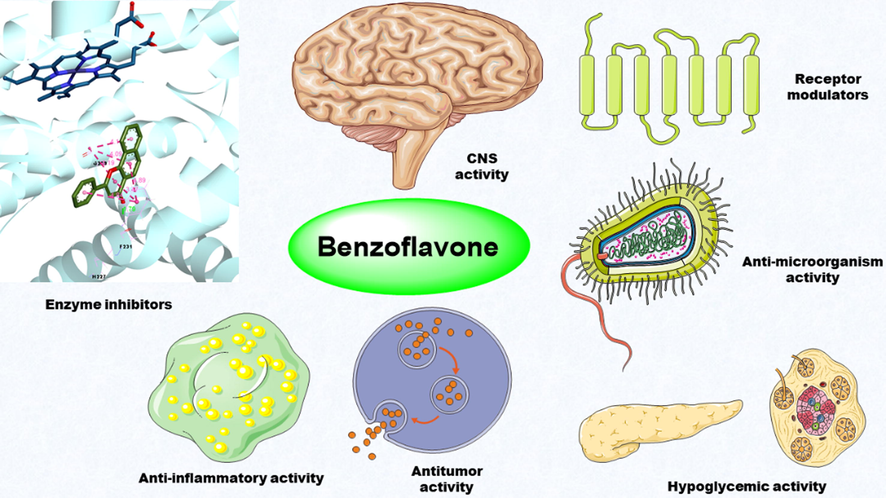
Broad-spectrum of pharmacological potencies for benzoflavone analogues.
Up to date, there is no published review article summarizing the bio Information and materials related to bioactivities of benzoflavones were obtained from scientific databases including Google Scholar, PubMed and SciFinder database, etc. activities of benzoflavone analogues. Thus, this review will fill a vacancy in this field. This review is to introduce the research progress in the synthetic strategies, biological activities and SAR of benzoflavone analogues which can be helpful for related investigators.
2 Synthetic strategies of benzoflavones
Same as the research situation of flavonoids, several kinds of novel benzoflavones are isolated from the medicinal plants, but the low level of the content limits further research. Hence, it is necessary to summarize the investigations about the synthetic strategies of benzoflavones. The synthetic strategies of benzoflavones are mainly based on the synthetic route of flavonoids. There are some differences between the different kinds of benzoflavones. Herein, we summarize some typical examples to explain the main constructive process of benzoflavone derivatives.
Take the synthesis of chalcone derivative 8 (Scheme 1) for instance (Dong et al., 2019), the author synthesized a cluster of analogues under a basic catalyst consisting of ethanol solution of potassium hydroxide. After 24 h of stirring of the mixture at ambient temperature, the disposition of different chalcone analogues was obtained with the yield ranging from 43% to 72%. The principle of these reactions follows the mechanism of aldol reaction, and the substrates are not limited to the methyl naphthyl ketone and substituted benzaldehydes, pyridine-carboxaldehydes are included as well. As for the types of catalyst used, with the development of the organic synthesis methodology, more and more types of catalysts have been exploited (Rammohan et al., 2020), such as metal oxide-catalyzed Claisen-Schmidt reaction under solvent-free condition, Claisen-Schmidt reaction, and biosynthesis method by tetraketide cyclization.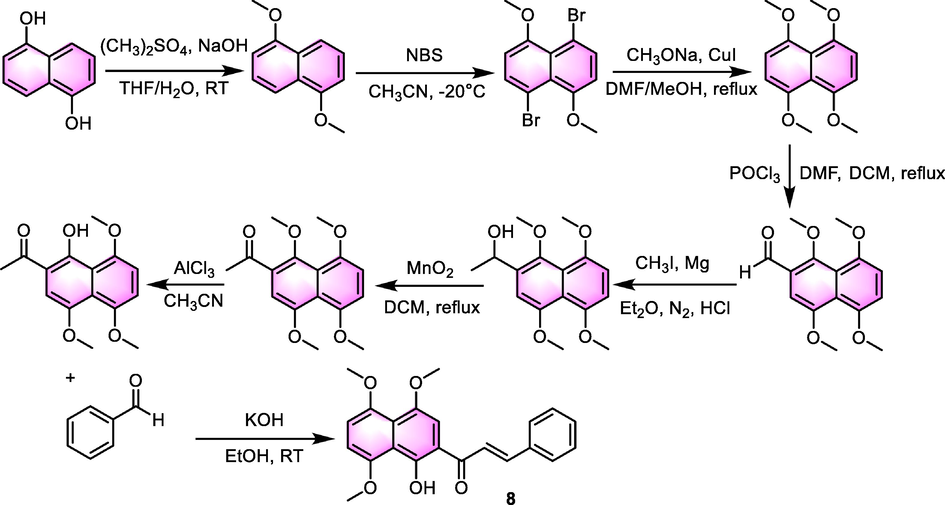
The synthesis of compound 8.
As for the reaction of synthesizing the flavonoid skeleton, different synthesis routes have been disclosed. The most commonly used method is Baker-Venkataraman (B-V) reaction, which is a reaction that mainly possesses nucleophilic processes. As we can see from the synthesis of compound 4 (Scheme 2) (Zhou et al., 2020), an esterification reaction was first processed to introduce benzoyl on the phenolic hydroxyl group of naphthalene ring. Then, backbone rearrangement was possessed to give the terminal product (St-Gelais et al., 2018, Xu et al., 2019). This reaction needs the presence of an enolate which is generally generated through deprotonation of acetophenone with a strong base, KOH or K2CO3 are the most classical catalysts with pyridine used as the solvent, then the ring-closing reaction happens under the catalysis of MnO2, H2SO4 or CH3COOH. The selection of catalysts of B-V reaction needs to be adjusted according to the different substrates. Phase transfer catalysts like tetrabutylammonium bromide are also included as the catalyst during the reaction sometimes. Apart from the B-V reaction, the Allan-Robinson reaction and Algar-Flynn-Oyamada reaction are often used in the synthesis of benzoflavones as well. We believe that more reactions will be explored in the future with the development of organic synthesis methodology.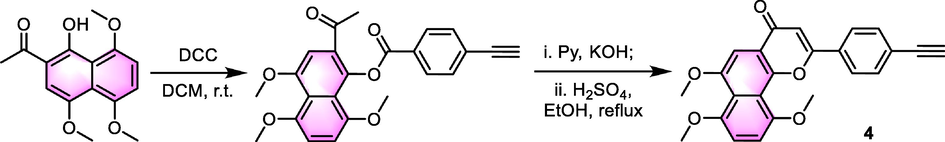
The synthesis of compound 4.
Moreover, the 2-phenylchroman skeleton has been reported to be a promising scaffold as well. To establish this skeleton, a formyl group and an adjacent hydroxyl group need to be introduced into the naphthalene ring firstly, then ring-closing and reduction reactions are processed to give the terminal product. Taking the synthesis of compound 65 (Scheme 3) for example (Li et al., 2019), the ring-closing was triggered with L-(-)-piperidine-2-carboxylic acid in toluene as the solvent. Then, the NaBH4 restored the double bond to give enantiomer, and a chiral resolution was carried out to obtain the terminal compound. Take active natural products as to lead, more and more potential benzoflavone derivatives will be explored in the future, the progress of synthesis methodology in the process is helpful to cut down the period of research.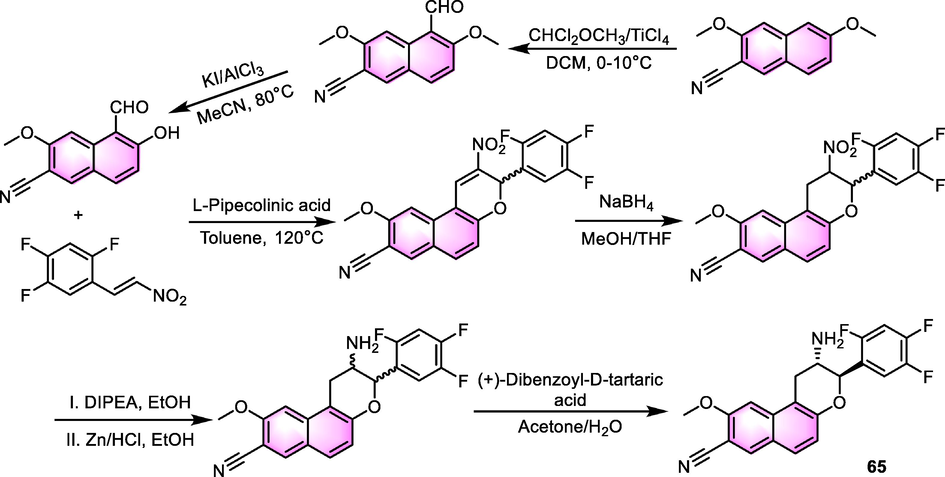
The synthesis of compound 65.
3 Biodiversity of benzoflavone analogues
Pharmacological potencies associated with benzoflavone analogues have been found to cover a wide range of applications, including enzyme inhibitory, antitumor, CNS, anti-inflammatory, anti-microorganism, hypoglycemic, and receptor modulating potential.
3.1 Enzyme inhibitors
3.1.1 CYP inhibitors
Cytochromes P450 consists of a family of metal-containing enzymes that performs a role in the metabolism of various endogenous as well as exogenous compounds (Zhao et al., 2019). The aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor that is encoded by the aryl hydrocarbon gene, has been found to regulate the transcription of the CYP1 family of enzymes. AhR is crucial for regulating the biological activity of CYP1-isoforms that metabolize estrogens (CYP1B1) and xenobiotics (CYP1A1). ANF exerted a substantial inhibitory effect on CYP1B1 (IC50: 1.3 nM). Additionally, ANF is a strong suppressor of human CYP1A1 (IC50: 11.9 nM) and 1A2 (IC50: 3.8 nM). The crystal structures of CYP1B1, CYP1A1, and CYP1A2 in combination with ANF have been determined and it may be used to develop highly selective CYP1B1 inhibitors. After analyzing the binding mechanism of CYP1B1, CYP1A1, and 1A2 to ANF, it was discovered that the relative location of a critical PHE residue (PHE 224 in the case of CYP1A1 and PHE 231 in the case of CYP1B1) with respect to ANF is varied by a small margin amongst the three proteins (Fig. 3). It has been demonstrated that the addition of electron-donating groups to ANF promotes the π-π stacking interactions and the residue of PHE 231 affects the selectivity of CYP1B1.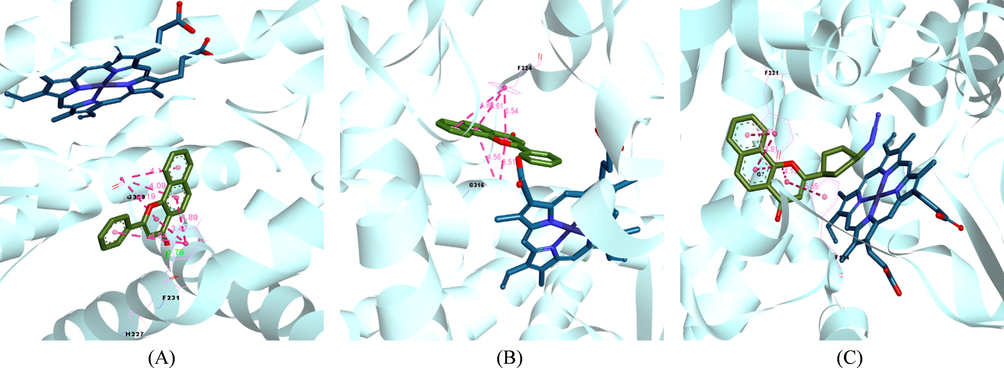
Crystal Structure of CYP1B1 (A, PDB code: 6OYU, blue molecular is heme) and CYP1A1 (B, PDB code: 4I8V, blue molecular is heme) in complex with ANF and CYP1B1(C, PDB code: 6IQ5, blue molecular is heme) in complex with compound 3.
In 2019, Kubo et al. (2019) designed and synthesized multiple selective CYP1B1 inhibitors through dearomatization of α-naphthoflavone. Compound 2 (Fig. 4) was identified as an appropriate cyclohexyl-core with enhanced solubility, based on a comparison of ring structures. The structural development of 3 resulted in the production of the azide-containing cis-4, which was proven to have excellent properties in three critical areas: (1) higher selectivity for CYP1B1 as opposed to CYP1A1 and CYP1A2 (120-times and 150-times, respectively); (2) higher inhibitory efficacy of > 2 times that of ANF; (3) enhanced solubility. The analysis of the X-ray crystal structure was carried out to elucidate the binding mechanism, which showed the interaction mechanism and described the subtype selectivity of cis-4.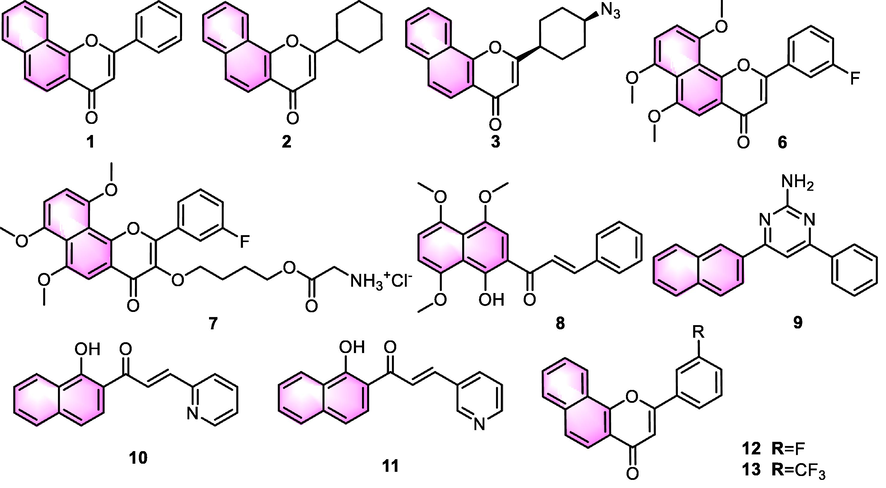
Structure of compounds benzoflavone derivatives as selective CYP inhibitors.
In 2020, Zhou et al. (2020) synthesized a sequence of ANF chimera derivatives eliminating CYP1B1-mediated drug resistance through targeting the degradation of CYP1B1. The results showed that ANF derivative 4 (Fig. 5) kept strong inhibition against CYP1B1 (IC50: 0.4 nM) and had a greater selectivity. The combination of 4 with thalidomide derivatives containing different chain lengths resulted in the formation of conjugates by the click reaction. The results of an in vitro cell-based assay demonstrated that 5 had higher efficacy than other analogues in reducing drug resistance of CYP1B1-overexpressed DU145 cells. The degradation of CYP1B1, as shown by Western blotting analyses, was a significant contributor to the reversal of drug resistance to docetaxel in a dose-dependent manner.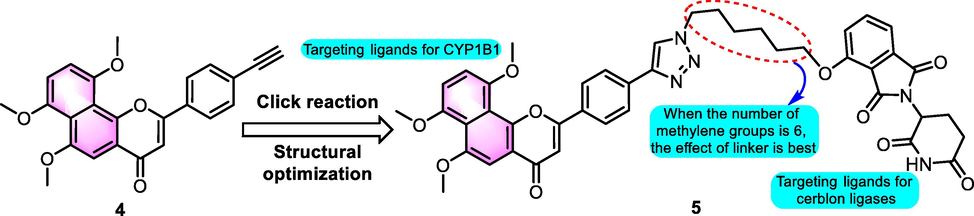
SAR of compounds 4 and 5 as selective CYP1B1 inhibitors.
Shaoshun Li task group made a great contribution to the advancement of benzoflavone derivatives as CYP inhibitors. Cui et al. (2015) designed and synthesized n 6,7,10-trimethoxy-ANF derivatives as CYP1B1 inhibitors for addressing docetaxel-resistance induced by the overexpression of CYP1B1. Compound 6 (Fig. 6) was found to be the most powerful and specific inhibitor of CYP1B1 ever described. Additional efforts were made to obtain water-soluble ANF derivatives for further cell-based studies to determine how to overcome anticancer medication resistance. Moreover, it has been shown that the water-soluble naphthoflavone 7 may completely eradicate docetaxel resistance induced by increased expression of CYP1B1 in MCF-7/1B1 cells. Using SAR analysis, it was discovered that adding methoxy groups at the positions of C-6, C-7, and C-10 on the naphthalene portion, as well as a fluoro atom at the position of C-3′ on the B-ring, might greatly improve the efficacy of CYP1B1 inhibition.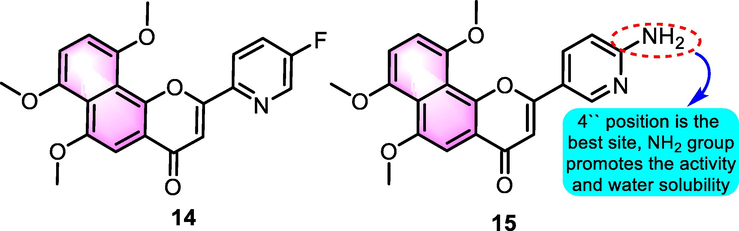
SAR of 6,7,10-trimethoxy-ANF derivatives as selective CYP1B1 inhibitors.
In 2019, the task group developed benzochalcone derivatives as CYP1B1 inhibitors with high selectivity and anticancer drugs (Dong et al., 2019). Obviously, compounds 8 and 9 (Fig. 4) were found to be the most effective inhibitors of CYP1B1 amongst all other benzochalcones, with IC50 values of 4.9 and 4.8 nM, correspondingly. Additionally, compounds 10 and 11 exhibited considerable growth-inhibiting properties both in the wild-type MCF-7 cell line and on drug-resistant MCF-7/1B1 and LCC6/P-gp cell lines, providing clues for the development of drugs to address drug resistance.
Subsequently, they thoroughly investigated the SAR of ANF derivatives as CYP1B1 inhibitors with the help of the 3D-quantitative structure-activity relationship (QSAR) model (Dong et al., 2020). Previous studies demonstrated that modifying the C ring of ANF resulted in a reduction of the potency for CYP1B1 inhibition, whereas incorporating substituents into the various positions of the B ring resulted in the formation of analogues with varied capability for CYP1B1 inhibition. Based on this idea, more potential compounds were developed. Compounds 12 and 13 (Fig. 4) were proved to have the best potential for selective CYP1B1 inhibition with IC50 values of 0.49 and 0.52 nM, respectively, which were 10-times greater than that of the precursor ANF. Furthermore, 3D-QSAR study was carried out to provide a clear comprehension of the critical structural elements that influence the inhibitory action of CYP1B1. Their contour maps highlighted the significance of oxygen atom at 1 position, carbonyl oxygen atom at 4 position, and hydrophobic and electron-withdrawing groups (EWGs) at meta-position of the phenyl ring in inhibiting CYP1B1 action. Given the discovery that phenyl ring modification in ANF has a major influence on CYP1 inhibition activity, aromatic heterocycles are also well tolerated for this portion.
Recently, Dong et al. (2021) investigated heterocycle-containing ANF derivatives as selective and water-soluble inhibitors of CYP1B1. A series of 6,7,10-trimethoxy-ANF derivatives with different B rings were produced and selected against CYP1 enzymes, resulting in the discovery of fluorine-containing compound 14 (Fig. 6) as the most effective and selective inhibitor of CYP1B1 (IC50: 0.07 nM), with a potency that is 84-times greater than that of ANF. In addition, the amino-substituted derivative 15 was found to inhibit the activity of CYP1B1 (IC50: 0.98 nM) and was more water-soluble as opposed to the precursor ANF (311 mg/mL for 15 and < 5 mg/mL for ANF).
3.1.2 Other enzyme inhibitors
In addition to the studies mentioned above, benzoflavone derivatives were reported to show potential to be other types of enzyme inhibitors as well. Chiaradia et al. (2012) synthesized multiple chalcone derivatives that were shown to be effective inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (MtbPtpB). Derivative 16 of benzoflavone (Fig. 7) was the most promising MtbPtpB selective inhibitor (MtbPtpA IC50: >100 μM, MtbPtpB IC50: 12 μM, PTB1B IC50: 3090 μM). According to the suggested binding mechanism, this compound inhibited the narrow channel near the catalytic CYS160, forming favorable polar and hydrophobic interactions with the active site residues or MtbPtpB.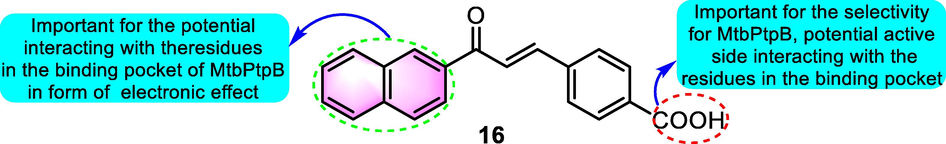
Structure of benzoflavone derivatives as MtbPtpB selective inhibitors.
In a 2013 study that used phosphodiesterase inhibition, Rodríguez-Ramos et al. (2013) examined the relaxant impact of 2-alkyl and 2-naphthylchromones on guinea-pig trachea and rat aorta. Though the benzoflavone derivative 17 (Fig. 8) showed significant inhibitory effects on both phosphodiesterase (PDE)-3, −4, and −5 in vitro with Ki values for 0.159, 9.27 and 10.24 μM, respectively, in vitro, compound 17 moderate relaxation in smooth muscle with ratios of 43.62% and 18.65% on rat aorta and guinea-pig trachea, respectively. The derivative 18 was proved to be the most potent compound as antihypertensive agent for maximal relaxation in smooth muscle with a ratio of 108.31%, which was better than the clinical drugs sildenafil and enoximone.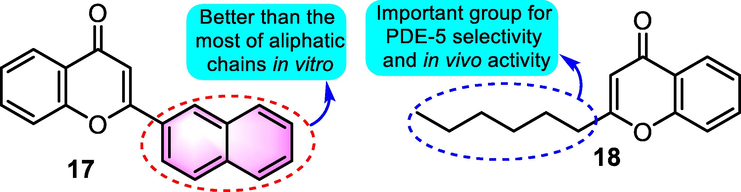
SAR of compounds 17 and 18 as PDE inhibitors.
Recently, Liu et al. (2020) generated a cluster of flavonoid-based derivatives as urease inhibitors, in which benzoflavone 19 (Fig. 9) showed moderate urease inhibitory activity with an IC50 value for 2.205 μM, which was better than the reference drug thiourea (IC50: 21.902 μM). Compound 20 was demonstrated to be the most potential urease inhibitor with an IC50 value of 1.207 μM, providing inspiration for further structural optimization for flavonoid derivatives as urease inhibitors.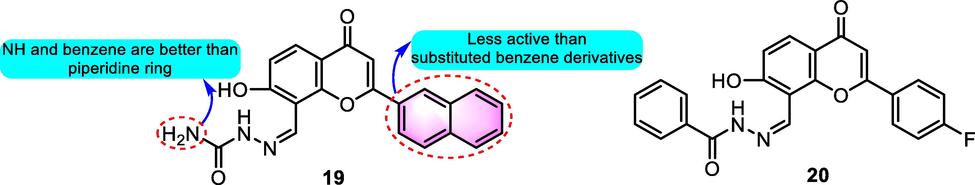
SAR of compounds 19 and 20 as urease inhibitors.
3.2 Antitumor agents
It has also been observed that benzoflavone derivatives perform an important function in the design of antitumor agents. Navarini et al. (2009) synthesized 13 kinds of hydroxychalcones to test antitumor potential on B16-F10 melanoma cells. Among the derivatives, benzoflavone analogues 21 and 22 (Fig. 10) exhibited remarked cytotoxicity with the reduction of cell viability for 97.7 % and 75.2 % at the dosage of 100 μM, respectively. Furthermore, compounds 21 and 22 were shown to trigger cell apoptosis, suggesting that this phenomenon occurred via depletion of mitochondrial glutathione (GSH) and adenosine triphosphate (ATP). SAR analysis indicated that the most striking commonality between chalcones 21 and 22 was the existence of a 1-naphtyl unit at the B-ring. The presence of a free hydroxyl group at the A-ring was shown to be responsible for the increased activity of compound 21.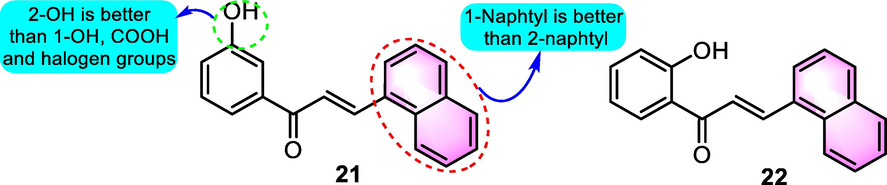
SAR of hydroxychalcones derivatives as antitumor agents.
The capacity of cancer cells to survive radiotherapy or chemotherapy is frequently attributed to their ability to repair the DNA damage elicited by these therapies. The serine/threonine protein kinase, DNA-dependent protein kinase (DNA-PK), is a critical enzyme that performs a vital function in repairing DNA double-strand breaks. In 2005, inspired by the structural fragments of existing inhibitors wortmannin and LY29400217, Griffin et al. (2005) prepared a number of pyrimido[2,1-α]isoquinolin-4-one and benzopyranone derivatives as DNA-PK inhibitors. Among the productions, benzoflavone-based compound 23 (Fig. 11) was the most promising candidate with IC50 value of 0.19 μM and a Ki value of 24 nM, besides, benzoflavone analogue 24 exhibited significant DNA-PK inhibitory activities as well with IC50 values for 0.23 μM. Furthermore, compound 23 was found to be selective for DNA-PK in contrast with the related enzymes including ATM, ATR, mTOR, and PI3K (IC50: >100, >100, 5.3 and 2.4 μM, respectively). Moreover, compound 23 was found to increase the sensitivity of the HeLa human tumor cell line to the cytotoxicity impacts of ionizing radiation in vitro, with a dosage adjustment coefficient of 2.3 at 10% survival being identified when used in conjunction with a 5 µM inhibitor concentration. The SAR analysis revealed that adding a 2-methyl substituent to the morpholine ring did not have any negative effects, and the racemic chromone 23 demonstrated strong DNA-PK inhibitory action. The 6-position was shown to be intolerant to an extra methyl group substitute. These findings revealed that the morpholino group was situated inside a sterically limited area of the ATP-binding domain and that the extra methyl group substitute places the morpholine oxygen into a suboptimal configuration for hydrogen bonding.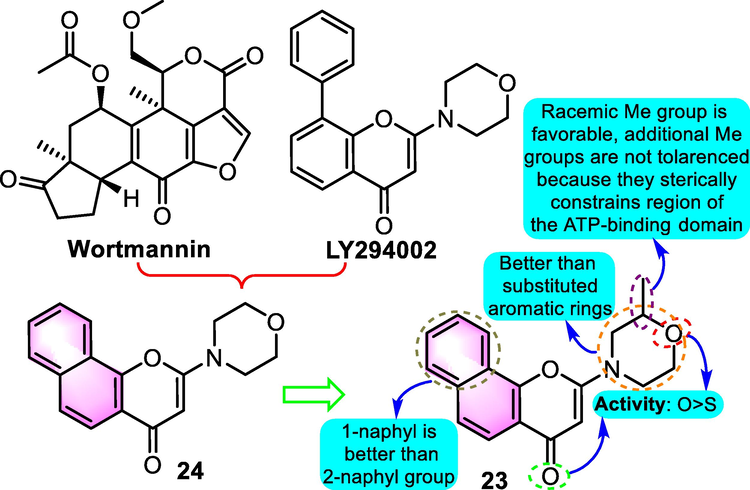
SAR of compounds 23 and 24 as antitumor agents.
Breast cancer resistance protein (BCRP) is an ATP-binding cassette transporter that performs a critical function in drug disposition and in the generation of cancer multidrug resistance. Zhang et al. (2005) reported that the possibility for in vivo pharmacokinetic (PK) interactions through a comparison of the pharmacokinetics of topotecan when 2 mg/kg topotecan was administered orally with and without varied ANF dosages in mdr1a/1b(-/-) mice and rats. ANF (5 µM) remarkedly suppressed the BCRP-mediated transport of topotecan in BCRP-overexpressing MCF-7 MX100 cells to a level that is equivalent to that identified with 10 µM of BCRP inhibitor fumitremorgin C. However, following coadministration of dosages up to 50 mg/kg of ANF orally, the pharmacokinetics of topotecan was not substantially changed in mdr1a/1b (-/-) mice or rats. The cause for this lack of in vivo-in vitro correlation was hypothesized to be the inability of flavonoids to potently inhibit the action of the rat or mouse BCRP.
Osteosarcoma has been recognized as the most common primal malignant tumor of the skeletal system, with an extremely aggressive clinical history and no effective therapy. An isoflavone-based analogue 25 (Fig. 12) was disclosed the in antitumor potential in human osteosarcoma cells by Hou et al. (2008). Human osteosarcoma cell lines exposed to compound 25 were shown to be susceptible to apoptosis in a dosage-dependent way. It was shown that the accumulation of reactive oxygen species (ROS) was a significant mediator in the cell death induced by compound 25. Compound 25 also caused the dephosphorylation of apoptotic signal-regulating kinase 1 (ASK1) as well as the phosphorylation of p38, JNK, and p53. The apoptosis induced by compound 25 was antagonized by the transfection with p38, ASK1, and c-jun N-terminal kinase (JNK) small interfering RNA (siRNA). Moreover, as shown by the alteration in the Bax/B-cell lymphoma-2 (Bcl-2) ratio, compound 25 also activated the mitochondrial apoptotic pathway. The knockdown of Bax was achieved by a Bax siRNA approach, which lowered the expression of Bax and caused cell death. Additionally, Bax expression and HBI-induced p53 phosphorylation were decreased by ASK1, p38, and JNK siRNAs.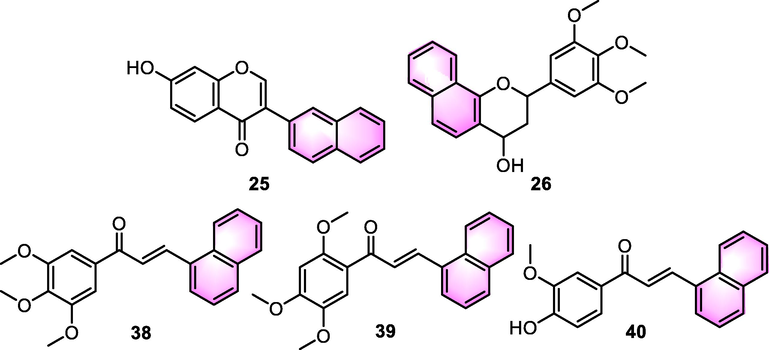
Structure of benzoflavone derivatives as antitumor agent.
It has been suggested that tissue inhibitor of metalloproteinases (TIMP-2), an endogenous inhibitor of matrix metalloproteinase-2 (MMP-2), is responsible for tumor invasion via regulating the activity of MMP-2. Additionally, TIMP-2 performs a critical function in the direct modulation of cell growth and angiogenesis, regardless of whether or not MMP is inhibited. Waleh et al. (2010) reported that synthetic benzoflavone-derived compound 26 (Fig. 12) suppressed the invasion of a highly metastatic human breast cancer cell line MCF-10CA1a via Matrigel, while simultaneously increasing the mRNA and protein levels of TIMP-2. Compound 26 considerably suppressed the MMP-2 gelatinolytic activity by>50% without impact on the expression of MMP-2 protein. Furthermore, compound 26 exhibited substantial anti-tumor action in a variety of tumor cell lines, independent of whether or not the cells expressed hormone receptors, p53, or were resistant to several drugs. In consideration of the multifunctional inhibitory impact of TIMP-2on tumor progression and invasion, the identified elevation in TIMP-2 expression by compound 26 may perform a significant function in the anti-invasive and anti-tumor activity of this new small molecule.
In the same year, Pedrini et al. (2010) synthesized a series of benzoflavone-based chalcones generated from 2-naphtaldehyde and performed preliminary screening for their ability to induce cell cycle arrest and apoptosis in L-1210 murine lymphoblastic leukaemia cells. Compared to other analogues, compound 27 (Fig. 13) exhibited the greatest cytotoxicity impact (IC50: 54 µM), but not in normal human lymphocytes. Furthermore, it has been proven that compound 27 causes cell cycle arrest in the G2/M phase as well as a substantial increase in the percentage of cells in the sub G0/G1 phase. Additionally, it was discovered that compound 27 caused a change in Bcl-2 ratio as well as elevated the expression of p53 and activation of caspase-3, providing initial evidence for the mechanism of the anti-leukaemia potency of compound 27.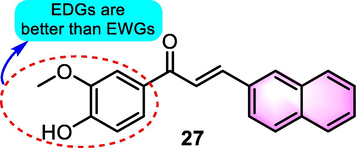
SAR of compound 27 as an antitumor agent.
Wang et al. (2012) designed and synthesized several millepachine derivatives as potent antiproliferative agents. Among the productions, benzoflavone derivative 28 (Fig. 14) displayed the strongest activity in inhibiting HepG2 cancer cell line (IC50: 0.21 µM), while weak inhibitory effects on other cell lines were detected. The most hopeful compound 29 showed better activity on multiple cancer cell lines including SW620, HT29, HCT116, SK-OV-3, K562, and HepG2 with IC50 values for 0.67, 0.55, 0.62, 0.61, 0.79, and 0.59 µM, respectively. Furthermore, compound 29 was found to inhibit tubulin polymerization in HepG2 cells in a dose-dependent manner and stimulated the HepG2 cell cycle arrest at the G2/M phase. Further studies proved that compared to the precursor millepachine and the anticancer medicine cisplatin, compound 29 suppressed the expansion of tumor volume and presented more anticancer activity in an A549 lung xenograft tumor model.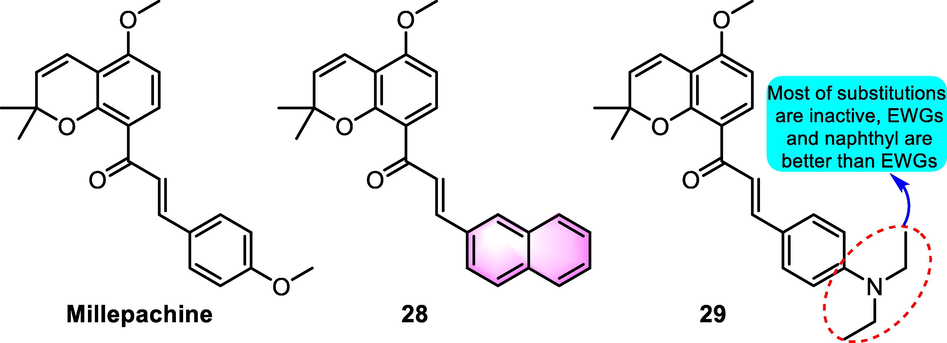
SAR of millepachine derivatives as antitumor agents.
Juvale et al. (2013) synthesized multiple flavones and benzoflavones as BCRP/ABCG2 inhibitors. All of the produced compounds were evaluated for BCRP inhibition and assessed for their inhibition effect against multidrug resistance-associated protein 1 (MRP1) and P-glycoprotein (P-gp) using the calcein-AM accumulation assay to verify that they were selective for BCRP. Compound 30 (Fig. 15), an ANF derivative with the most powerful and selective for BCRP, with a 50-fold increase in selectivity and extremely minimal cytotoxicity at increasing doses (IC50 values for Hoechst 33342, pheophorbide A, A2780 adr, and 2008 MRP1: 0.426, 0.468, 21.9 and 21.8 µM, respectively). SAR analysis revealed that in most cases, 7,8-benzoflavones had a higher potency when compared with the 5,6-benzoflavones. Moreover, the existence of a 3-OMe substituent elevated the activity as opposed to the existence of OH or no substitution at the C-3 position.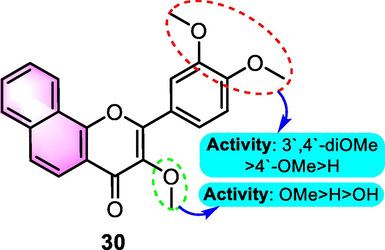
SAR of compound 30 as antitumor agents.
Shin et al. (2013) investigated SAR of polyphenols that cause cell cycle arrest at the G1 phase in HCT116 human colorectal cancer cell lines through the QSAR model. Typical benzoflavone derivative 31 (Fig. 16) displayed significant antitumor potential through arresting the cell cycle at the G1 phase. SAR analysis revealed that the bulky substituents at C-2 of the chromene group exhibited a positive contribution to the activity. Moreover, the 7,8-naphtho group demonstrated better performance as opposed to the 5,6-naphtho group, while the chromenyl group at the propenone showed a reduction in activity.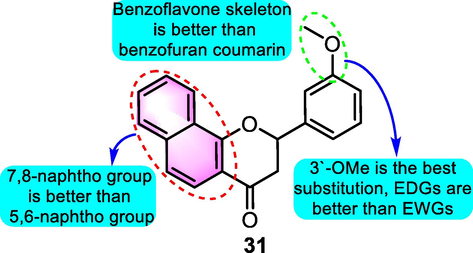
SAR of compound 31 as antitumor agents.
As medicinal chemistry continues to advance, an increasing number of naphthochalcones were proved to show antitumor effects. Inspired from the precursors like colchicine and combretastatin A-4, a sequence of cytotoxic 3,4,5-trimethoxychalcones were synthesized as inhibitors of cell migration and mitotic arresters (Salum et al., 2013). Benzoflavones analogue 32 (Fig. 17) was one of the most promising derivatives among the productions. Besides, compound 32 was found to have selective cytotoxicity and inhibit tubulin polymerization in L-1210, REH, JURKAT, NIH3T3, HUVEC, PBMC cell lines, and tubulin with IC50 values for 26, 0.73, 0.50, 34, 37, 18 and 2.2 µM, respectively. Furthermore, in the high-content analysis of mitotic arrest in HeLa cells, compound 32 showed promise in mitotic index, tubulin intensity, and nuclear condensation with minimal detectable effective doses of 397, 283 and 317 nM, respectively. Docking analysis suggested that trans configuration could be the favored orientation because it overlapped well within the colchicine binding domain, the carbonyl oxygen of 32 stabilized by 2 hydrogen bonds with the backbone NH of key residues like LEU b255 and ASP b251.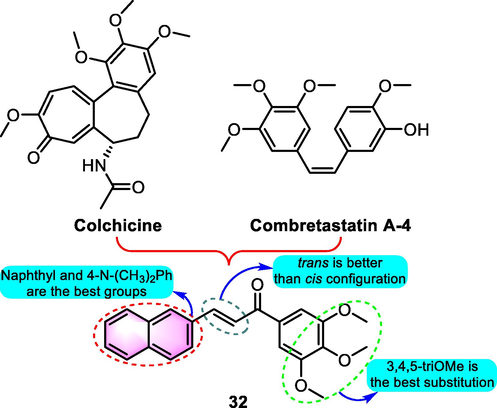
SAR of compound 32 as antitumor agents.
As for the antimitotic activity of benzoflavones, Shin et al. (2013) provided more evidence. Multiple benzochalcones were prepared and tested the inhibition of tubulin polymerization, in which compound 33 (Fig. 18) was proved to be the most effective inhibitor of the clonogenicity of Capan-1 human pancreatic cancer cells. Compound 33 was shown to suppress cell proliferation in a number of human solid tumor cell lines and inhibit the growth of xenografted tumors in nude mice. Compound 33 elicited a cell cycle arrest in the G2/M phase, accompanied by an increase in apoptotic cell death as a result of its mechanism of action. These processes were linked to the suppression of tubulin polymerization when compound 33 bided to tubulin, resulting in the production of aberrant mono- or multipolar mitotic microtubule structures, followed by the spherical organization of multinucleated chromosomes. Additionally, compound 34 induced the activation of caspase-2, caspase-3, caspase-7, and caspase-9. The phosphorylation of Erk1/2, JNK1/2, and p38 kinase was elevated by compound 33. SB600125, U0126, or SP600125 pretreatment was found to abrogate compound 33-induced caspase-3 and caspase-7 activation as well as PARP cleavage, indicating that the MAPK signaling pathway is involved in compound 33-induced apoptosis.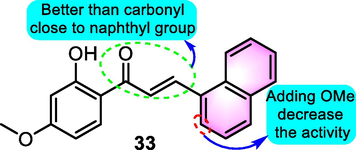
SAR of compound 33 as antitumor agents.
In 2015, there was new progress in synthetic benzoflavone derivatives as cell migration inhibitors against acute lymphoblastic leukemia (Salum et al., 2015). Starting from the benzochalcone 34 (Fig. 19, B-cell precursor ALL REH cell line IC50: 0.79 µM), a synthetic benzoflavone derivative derived from the precursors like colchicine and combretastatin A-4, a cluster of analogues were prepared. Among the derivatives, compound 35 (B-cell precursor ALL REH cell line IC50: 0.015 µM) was the most effective agent, exhibiting relatively mild toxic side effects toward normal T lymphocytes as well as an in vivo model of acute toxicity. Notably, in a mouse model of human acute lymphoblastic leukemia, compound 35 was as effective as vincristine. The postulated underlying mechanism in which the derivatives function is to interfere with microtubule polymerization, as evidenced by their ability to block microtubule assembly in vitro. An attractive feature of the derivatives was that the colchicine binding site was the binding area, which differs from that of the taxanes and vinca alkaloids, although there is a binding site similar to that of combretastatin A4 for solid tumors.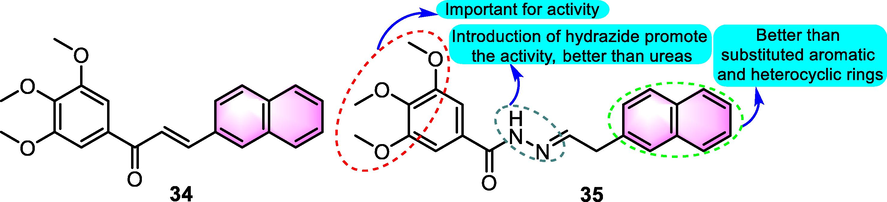
SAR of compounds 34 and 35 as antitumor agents.
Based on the structure of benzoflavone precursor 36 (Fig. 20) as a potential antitumor agent (Brunhofer-Bolzer et al., 2015), after the abundant structural optimizations, compound 37 was selected as the most promising antitumor agent on diverse types of human malignant cells of the myeloid and lymphoid lineage. As for the specific cancer cell lines, both compounds 36 and 37 showed remarkable antitumor potency on three kinds of malignant hematological cell lines. A further experiment of compound 37 on peripheral blood mononuclear cells (PBMCs) of chronic lymphocytic leukemia (CLL) patients, it was shown that the cytotoxicity was greater in PBMC from CLL patients as opposed that from healthy donors (HD).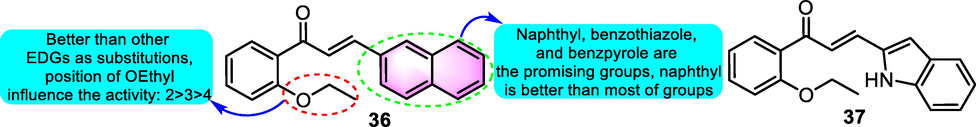
SAR of compounds 36 and 37 as antitumor agents.
Bittencourt et al. (2016) assessed the activity of three types of benzoflavone (38, 39, and 40, Fig. 12) inducing apoptosis on surgery obtained-glioma cells and A172. In all the tests, chalcones demonstrated a reduction in cell viability, in which compound 39 exhibited the strongest activity. There was an elevation in apoptosis levels, but no additional rise in necrosis levels was observed. This enhancement was considered to associated with the strong oxidative effects observed, which were induced by the overwhelming presence of ROS and nitric oxide generation. An analysis of cell cycle distribution indicated that the G0/G1 and S stages of the cell cycle were arrested, implying that 39 interfered with cell cycle regulation.
Recently, Saavedra et al. (2020) synthesized multiple 6-methoxy-2-(naphthalen-1-yl)chroman-4-one derivatives and assessed their antiproliferative activity against the U-937 human tumor cell line. Among the benzoflavone derivatives, compound 40 (Fig. 21) with flavanone skeleton showed the strongest antitumor activity (IC50: 1.3 μM) and was selected for more experiments. When compared with other human leukaemia cells, compound 40 exerted substantial cytotoxicity capabilities against three human leukaemia cell lines, including the acute lymphoblastic leukaemia MOLT-3, human B cell precursor leukaemia NALM-6 and human acute myeloid leukaemia HL-60. When the U-937 cells were treated with compound 40, a G2-M cell cycle arrest was induced, followed by the increase in sub-G1 ratio and annexin-V positive cells, poly (ADP-ribose)polymerase processing, caspase activation, and mitochondrial cytochrome c release. Overexpressed anti-apoptotic protein Bcl-2 inhibited the induction of apoptosis induced by compound 40. Following different kinetics, compound 40 promoted the phosphorylation of JNK/stress-activated protein kinases (SAPK), extracellular signal-regulated kinases, and p38 mitogen-activated protein kinases. Furthermore, cell death activity was weakened by the inhibition of mitogen-activated extracellular kinases and JNK/SAPK, and this effect occurred independently without the involvement of ROS. The SAR of the derivatives was investigated by the changing different substituents (chlorine, benzyloxy, methoxy) or altering the location of the benzyloxy or methoxy groups on the A ring. The findings revealed that both the chalcone bearing the methoxy group at the 5′ position of the A ring and the flavanones derived from it were potentially active.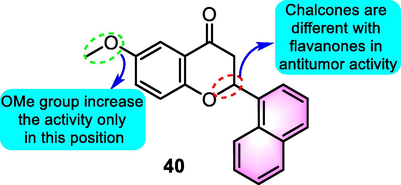
SAR of 6-methoxy-2-(naphthalen-1-yl)chroman-4-one derivatives as antitumor agents.
3.3 CNS agents
Benzoflavones derivatives have been found to perform a function in the development of CNS agents. Goutman et al. (2003) reported that multiple flavonoids can modulate the ionic currents facilitated by GABAC and GABAA receptors, while ANF and flavone showed almost non-activity compared to active derivatives including quercetin, apigenin, morine, and chrysin. SAR analysis showed that hydroxyl groups were crucial for the regulation of ionotropic GABA receptors. Additionally, anxiolytic flavonoids, including apigenin or chrysin, did not act as a potentiator but served as an antagonist on GABAA receptors. The impacts of quercetin and apigenin on GABAA receptors were found to be unresponsive to the benzodiazepine antagonist flumazenil. In 2008, a liner QSAR modeling of the interaction of flavonoids with GABAA receptor was set up by Duchowicz et al. (2008). The results indicated that compared to other types of scaffolds, BNF (41, Fig. 22) could be the promising skeleton, which can highly improve the binding affinities towards the benzodiazepine binding site of the GABAA receptor complex considering to the factors including 2D autocorrelations, topological, radial distribution and function Leverage-weighted autocorrelation, which provided inspiration for the application of benzoflavone in this field.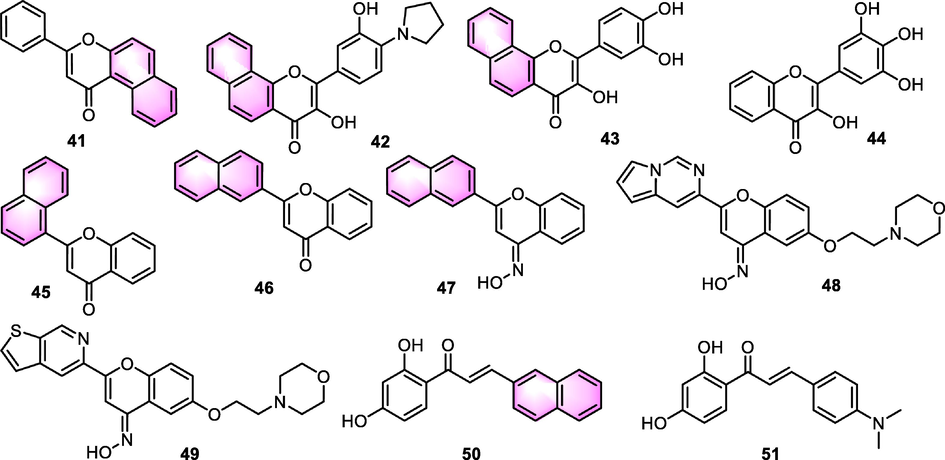
Structure of benzoflavone derivatives as CNS agents.
In 2012, Chiruta and co-workers (Chiruta et al., 2012) performed a chemical modification of the multitarget neuroprotective compound fisetin. Among the derivatives, benzoflavones presented favorable activities in ischemia assay in vitro. The most promising compound 42 (Fig. 22) exhibited potency with EC50 for 5 nM, which was 600 folds lower than the lead compound fisetin. While the potency of anti-inflammatory and GSH-maintaining activities of 42 was lower than fisetin, indicating there was room for medicinal chemical improvement.
In 2013, several flavonoids were selected and evaluated the neuroprotective activities by Lapchak (2013). The findings illustrated that all the test compounds exhibited better activity as opposed to the reference fisetin utilizing the cortical neuron glutamate assay and HT22 cell IAA assay. Benzoflavones analogue 43 (Fig. 22) showed cortical protection with a ratio of 86 at the dose of 100 nM. In further investigation, 43 showed toxicity in normal cell line and CYP9 inhibition tests. In this regard, non-naphthol flavone 44 was identified to be the ideal candidate.
Nayak et al. (2015) synthesized a series of 2-aryl-4H-chromen-4-ones and assessed their monoamine oxidase (MAO) inhibitory effects. Benzoflavones derivatives 45 and 46 (Fig. 22) exhibited moderate MAO inhibitory effects with Ki values for MAO-A: 0.8 and 2.55 μM, Ki values for MAO-B: 0.39 and 1.30 μM, respectively. The most potential MAO-B inhibitor was the 3,4-dimethoxy substituted derivative, which displayed MAO-A and MAO-B inhibitory effects for Ki values of 4.8 and 0.16 μM, respectively. SAR analysis illustrated that 2 position of O on the C ring of the flavonoid was important to the potential binding affinity with both the pockets of MAO-B and MAO-A, while the presence of naphthol group was unfavorable for the binding to some degree.
Charvin et al. (2017) carried out chemical modifications and SAR analysis based on the flavonoid derivatives for the purpose of developing antiparkinsonian agents. Though the benzoflavone derivative 47 (Fig. 22) was inactive, a similar aromatic group substituted derivative 48 exerted excellent activity on mGluR4 positive allosteric modulator (EC50: 46 nM) with enhanced water solubility, showing similar activity within substantiated preclinical rodent models of Parkinson’s disease motor symptoms following intraperitoneal administration: haloperidol-induced catalepsy in mouse and the rat 6- hydroxydopamine lesion model. Furthermore, after changing the substitution again, compound 49 emerged with a better pharmacokinetic profile but weaker activity (EC50: 79 nM), which has been nominated as a feasible candidate for clinical development.
Selvaraj et al. (2020) synthesized isoliquiritigenin derivatives as a neuroprotective agent against glutamate-mediated neurotoxicity in HT22 cells for the development of neurodegenerative curing agents. Compared to the lead compound isoliquiritigenin, most of the synthetic derivatives including benzoflavone 50 (Fig. 22) showed weaker activity. Only the N,N-dimethyl derivative 51 exhibited stronger activity than isoliquiritigenin. Moreover, compound 51 decreased the ROS level and concentration of cellular Ca2+ once exposed to glutamate and decreased the MAPK activation, and the translocation of apoptosis stimulating factor to the nucleus was inhibited by compound 51 as well.
3.4 Anti-inflammatory agents
Benzoflavone derivatives have extensively been studied for their anti-inflammatory properties. Batt et al. (1993) generated a series of 2′-substituted chalcone derivatives as inhibitors of the biosynthesis of inflammatory-related biomarker interleukin (IL)-1. In the production of IL-10 from human peripheral blood monocytes triggered with lipopolysaccharide (LPS) model, synthetic productions showed inhibitory potential with IC50 values that span between 0.2 and 5.0 μM, in which benzoflavone analogues 52 and 53 (Fig. 23) exhibited moderate activities (IC50: 0.22 and 1.35 μM, respectively). Further investigations revealed that the in the septic shock model in vivo, the survival ratio was related to the activity in vitro, indicating good potential for further development.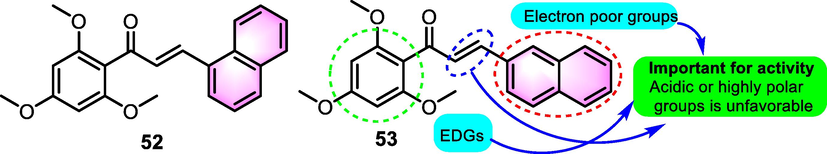
SAR of compounds 52 and 53 as anti-inflammatory agents.
Chiaradia et al. (2008) produced a cluster of compounds derived from 2,4,6-trimethoxyacetophenone to evaluate the anti-inflammatory activity in RAW 264.7 cells triggered by LPS. Compared to other productions, benzoflavone derived compound 54 (Fig. 24) exhibited a significant iNOS inhibitory effect with an IC50 value for 4.62 μM (no obvious cytotoxicity on this dosage), which was better than the highly selective iNOS inhibitor as positive control drug. The results indicated that compound 54 was probably active in inflammatory responses, as NO synthesis was a critical stage of the inflammation.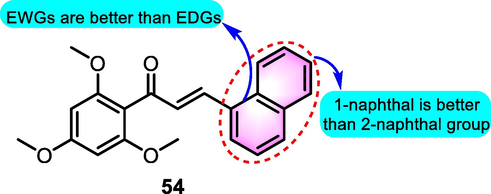
SAR of compound 54 as anti-inflammatory agents.
3.5 Anti-microorganism agents
Additionally, it was reported that benzoflavone derivatives perform a function in the production of antimicrobial agents. Liu et al. (2001) produced a cluster of substituted chalcones and assessed in vitro activity against Plasmodium falciparum. The results demonstrated the potential of benzoflavone derivatives developing as antimalarial agents. Benzoflavones 55, 56, 57, 58 and 59 (Fig. 25) inhibited [3H] hypoxanthine uptake into P. falciparum with IC50 values for 24.8, 320, 20, 39.9 and 27.5 μM, respectively, in which compound 59 showed the strongest activity among the benzoflavones thus was selected for further research. In the survival of P. berghei infected mice model in vivo, the significant activity exhibited by compound 59 enhanced the survival of the mice compared to control infected mice that received DMSO treatment.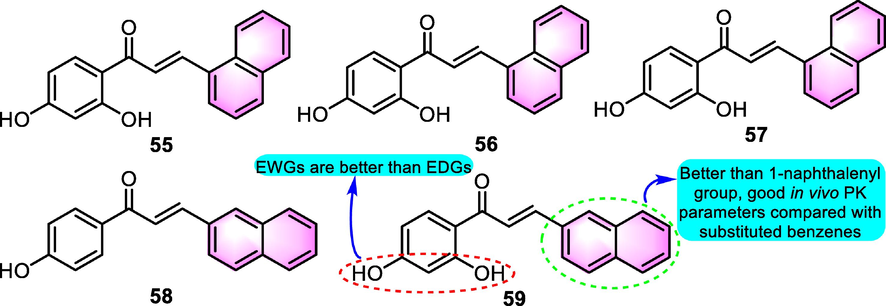
SAR of benzochalcone derivatives as anti-microorganism agents.
Chiaradia et al. (2008) produced a cluster of chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Among the synthetic derivatives, 2-naphthyl analogues 60 and 61 (Fig. 26) exerted the strongest actives against Mycobacterium tuberculosis PtpA with IC50 values for 8.4 and 23.1 μM, respectively. The SAR can be initially summarized and listed in Fig. 26.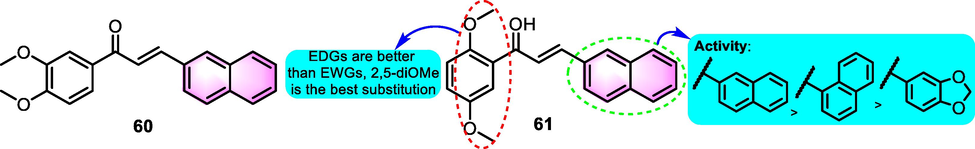
SAR of compounds 60 and 61 as anti-microorganism agents.
Janeczko et al. (2018) evaluated the potential that α- and β-naphthoflavones as monooxygenase inhibitors of Chaetomium sp. KCh 6651, Syncephalastrum racemosum KCh 105, and Absidia coerulea KCh 93. The findings illustrated that compound 62 (Fig. 27) was the most effective derivative (recovery of the untransformed substrate on Chaetomium, A. coerulea and S. racemosum: 46%, 42%, and 36%) with a similar impact of ANFs suppressing P450 1A1 binding to benzo[a]pyrene through a classical competitive process.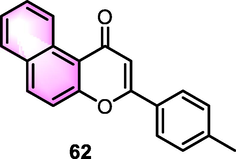
Structure of compound 62 as anti-microorganism agents.
3.6 Hypoglycemic agents
Jeong et al. (2015) isolated and identified multiple monomers from Euonymus alatus as hypoglycemic agents. Among the isolated compounds, benzoflavone analogue euonymalatus (63, Fig. 28) showed significant activity with IC50 values for 13.1 and 9.1 µM targeting the enzymes tyrosine phosphatases 1B (PTP1B) and α-glucosidase, respectively, which were better than most of triterpenoids and flavonoids, indicating a valuable potential for further development.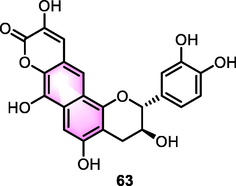
Structure of compound 63 as hypoglycemic agents.
Li et al. (2016) designed 2‑phenyl-3,4-dihydro‑2H‑benzo[f ]chromen-3-amine compounds derived from natural products as new and effective dipeptidyl peptidase 4 (DPP-4) inhibitors for treating type 2 diabetes. Illuminated by the promising hit isodaphnetin isolated from Daphne odora and the active moiety of clinical drug sitagliptin, a cluster of chiral benzoflavones was prepared. Among the synthesized derivatives, benzoflavone compound 64 (Fig. 29) showed the most effective DPP-4 inhibitory function (IC50: 2.06 nM) in vitro with good PK profiles in vivo in ob/ob mice. Compared to the precursor, the modified design approaches provided activity with about 7400-fold enhancement in potency. In-depth preclinical investigation, compound 64 showed potential as a long-acting antidiabetic drug that targets DPP-4 that could be administered orally once daily, which was predominantly closely related to the potential of benzoflavone scaffold. Subsequently, the structure of the inhibitor was optimized again by incorporating quantum chemistry and molecular dynamics simulation technologies (Li et al., 2019). Compound 65 was screened and selected as the preclinical candidate for the once-weekly treatment of type 2 diabetes. In the pharmacological screen, compound 65 exhibited excellent DPP-4 inhibitory potential for KD value for 1.77 × 10−10 M (66 KD: 2.38 × 10−9 M). As for the PK tests in diabetic mice, the administration of a single dose of 65 (3 mg/kg) orally impeded > 80% of DPP-4 action for over 7 days. The long-term antidiabetic efficacy of 5 was superior to that of once-weekly omarigliptin and trelagliptin, particularly in lowering the level of hemoglobin A1c.(See Fig. 30).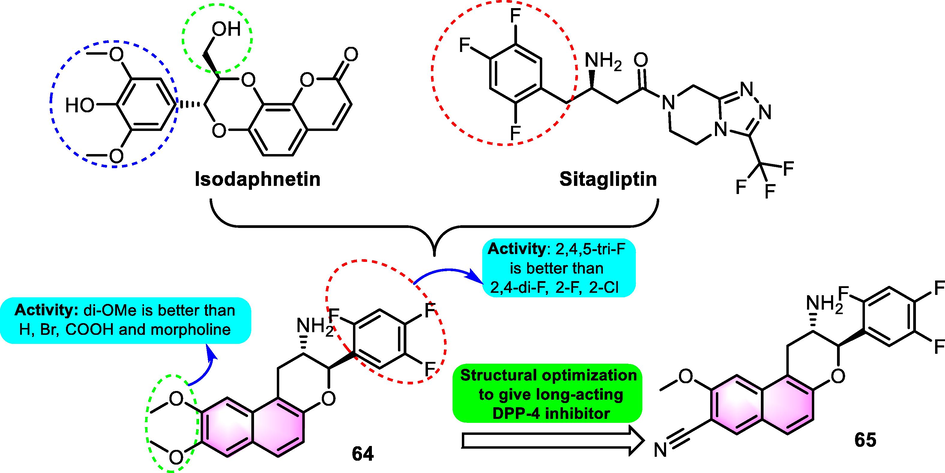
SAR of compounds 64 and 65 as hypoglycemic agents.
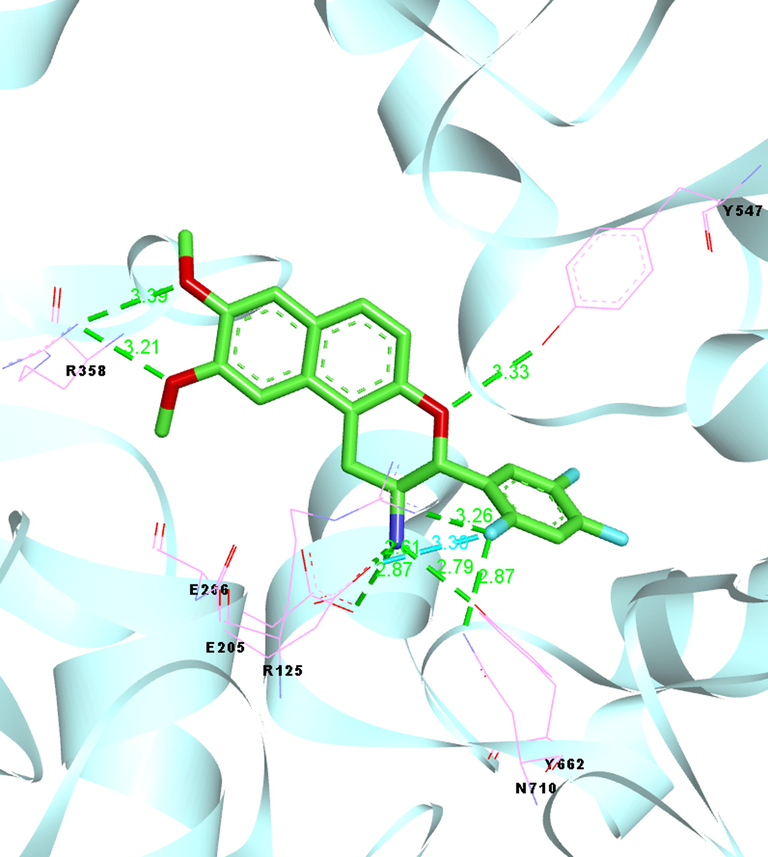
Crystal Structure of human DPP-IV in complex with compounds 64 (PDB code: 5J3J).
Inspired from the lead compound catechin, Spasov et al. (2019) synthesized multiple 3-aryl-1H-benzo[f]chromene and 2-aryl-4H-chromene derivatives as new inhibitors for α-glucosidase. The results showed that normal flavonoid-based derivatives were better than benzoflavone derivatives. The most potent benzoflavone was compound 66 (Fig. 31, IC50: 648.8 µM), which was even weaker than the reference drug acarbose (IC50: 543.6 µM), while normal flavonoid-based derivative 67 exerted the strongest inhibitory potency with IC50 value for 62.26 µM, which was nearly 10-folds better than acarbose.(See Fig. 33).
SAR of compounds 66 and 67 as hypoglycemic agents.
3.7 Receptor modulators
3.7.1 Ah receptor (AhR) modulators
Benzoflavones were reported to show activity on modulating AhR, which was a transcription factor as well as a member of the bHLH family of DNA-binding proteins. Gasiewicz et al. (1996) evaluated the SAR of ellipticines and flavones affecting AhR. The results showed that benzoflavone analogues exhibited significant competitive AhR binding effects. Firstly, ANF was tested and showed moderate activity (competitive AhR binding IC50: 226 nM) among the test candidates. Then more types of flavonoids were determined, in which compound 68 (Fig. 32) demonstrated the greatest activity with IC50 value for 10 nM in the competitive AhR binding test.
SAR of compound 68 as AhR modulators.
Murray et al. (2011) disclosed the activity of compound 69 (Fig. 32) as a selective AhR modulator. Competitive ligand binding validated compound 69 as the most promising AhR ligand among the test compounds. DRE-dependent reporter assays with mRNA quantification of AhR target genes showed minimal agonist activity associated with AhR binding. Importantly, mRNA analysis indicated that compound 69 retained the inhibitory ability of αNF with regard to cytokine-mediated SAA1 expression in Huh7 cells. The docking modeling suggested that compound 69 adopted a specific configuration inside the AhR ligand-binding pocket in comparison to αNF, which might expedite the rational design of more selective AhR modulators, providing potential perspective to the treatment of inflammatory reactions.
3.7.2 Liver X receptors modulators
The ATP-binding cassette transporters A1 (ABCA1) performs a crucial function in reverse cholesterol transport, which facilitates the rate-controlling step in the formation of high-density lipoprotein-cholesterol (HDL-C) particles. ABCA1 mutations or dysfunction result in enhanced atherogenesis. The expression of ABCA1 is transcriptionally modulated by liver X receptors (LXRs). In addition, LXRβ binds directly to the C-terminal domain of ABCA1 to regulate its post-translational modulation. Searching for highly effective ABCA1 up-modulators that target LXRβ is important for the balance of HDL-C and the reduction of lipid accumulation. Hu et al. (2013) synthesized several new flavonoids targeting LXRs pathway involved in LXRs as prospective mediators in the clearance of Aβ that reduced lipid accumulation. Among the productions, benzoflavone derivatives 70, 71, 72, and 73 (Fig. 34) showed activities to different degrees with ABCA1 promoter activation effect for 1.57, 2.64, 1.35, and 0.88 at the dosage of 1 µM, in which compound 71 was selected to be the greatest ABCA1 promoter activator and LXRβ agonist in the work for further investigations. As a selective LXRβ agonist, compound 71 significantly reduced lipid and triglyceride accumulation in liver cells compared to the precursor. Furthermore, compound 71 was proved to increase 1.7 folds of the expression of apoE protein in BV2 microglia, and the degradation of intracellular Aβ was improved by 71 dose-dependently as well, providing evidence for benzoflavones as a hopeful skeleton for further development as pleiotropic molecule for treating AD with an enhanced therapeutic index.
SAR of compound 69 as AhR modulators.
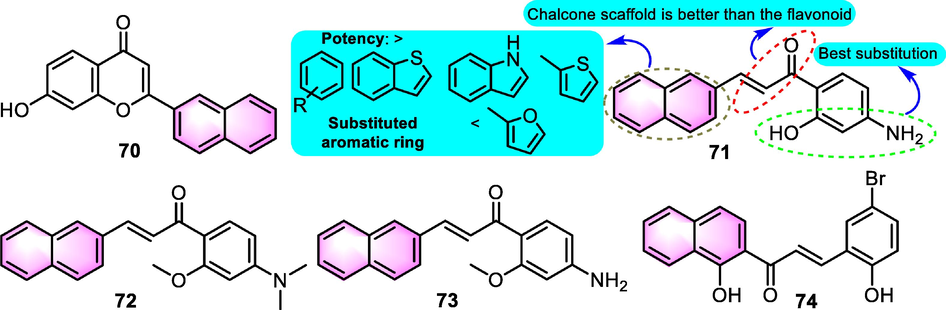
SAR of benzoflavones as LXRβ modulators.
Subsequently, based on the QSAR modeling, more potential benzoflavone derivatives were excavated (Chen et al., 2016). After a thorough investigation of the binding domain of LXRβ, some residues of the active site of LXRβ including THR 316, HIS 435, YYR 335, and PHE 271 were regarded to be key residues for the binding affinity of the interaction between ligands and LXRβ. The mentioned active benzoflavone analogue 71 was set as one of template molecular and tested for the binding affinity with a score of −10.997, and the new benzoflavone derivative 74 (Fig. 34) excavated from ZINC database showed more potential than 71 (score: −11.060), which was a relatively high score among the test ligands, providing perspective for further structural optimization.
3.8 Other bioactivities
3.8.1 Cystic fibrosis transmembrane conductance regulator (CFTR) activity
Benzoflavones derivatives were associated with the activity of human cystic fibrosis transmembrane conductance regulator (CFTR). Springsteel et al. (2003) developed a pharmacophore model for the nucleotide-binding domain of CFTR, starting a naturally occurring flavonoid apigenin (Kd: 4 µM), a cluster of derivatives were excavated and investigated the CFTR activation. The representative derivatives were 75 and 76 (Fig. 35), which were proved to be more potent than the precursor with CFTR Kd values for 4.8 and 1.7 µM, respectively. The SAR analysis revealed that benzannulation at the 7,8-positions of the flavone scaffold considerably improved the CFTR activation, and the existence of a rigid π-system in this region is superior to benzannulation at other A-ring positions. The pyridyl B-ring appeared to have less influence than 7,8-phenylglycine in activating CFTR. In addition, modification of the flavonoid B ring with pyridyl nitrogen or fluorine substituents at the 3 or 4 position was equally well tolerated. Ferrera et al. (2007) disclosed the double effect of compound 75 on CFTR Cl- channel activity. They investigated the interaction between the protein and compound 75 utilizing the patch-clamp method in the resected inside-out orientation to investigate the underlying molecular mode of action for this potentiator on totally phosphorylated channels with fairly reduced ATP levels. At low doses, compound 75 increased the open probability, promoting the channel transition to an activated state, whereas high concentrations of compound 75 (<50 nM) levels inhibit the CFTR by reducing the total open time. The findings indicated that compound 75 may enhance CFTR activity by binding to a particular location on the nucleotide-binding domain and boosting dimer formation. The response of CFTR to different concentrations of ATP was not altered by the application of low (activating) or high (inhibiting) concentrations of potentiator compound 75. They therefore concluded that the potentiator may not interfere with ATP binding, but may act at a separate site of the protein, interacting directly with CFTR to regulate channel activity.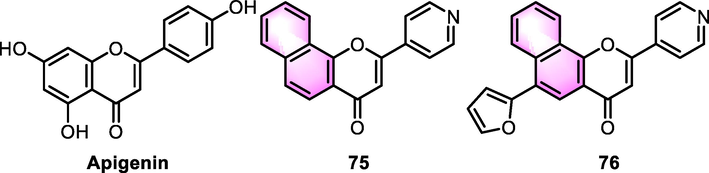
Structure of benzoflavone derivatives as CFTR modulators.
3.8.2 Anti-hepatitis C virus (HCV) activity
Fukazawa et al. (2012) screened multiple flavonoids as the hepatitis C virus (HCV) inhibitors. The results showed that ANF showed strong anti-HCV activity while BNF was inactive. The authors reported that ANF was more active than quercetin or naringenin in suppressing HCV. Furthermore, BNF displayed genotype specificity but was not demonstrated as active against the genotype 1b replicon, indicating potency to block later phases of the HCV life cycle.
3.8.3 Xanthine oxidase (XO) inhibitory activity
Singh et al. (2019) evaluated the potential of synthetic benzoflavone derivatives as anti-hyperuricemic agents. After the initial screen, compounds 77 and 78 (Fig. 36) were selected to be representative derivatives with xanthine oxidase (XO) IC50 values for 0.62 and 5.21 μM, respectively. Enzyme kinetic studies showed that compounds 77 and 78 are hybrid inhibitors of the XO enzyme. Molecular modelling studies were also carried out to investigate the binding of compounds 77 and 78 to amino acid residues present in the active site of the enzyme. The docking results confirmed that their favorable binding conformation at the XO active site could completely block the catalytic activity of the enzyme. Compounds 77 and 78 with good XO enzyme inhibition also exhibited promising activities on the serum uric acid levels in hyperuricemic mice model in vivo.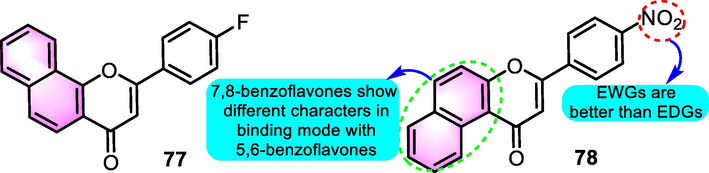
SAR of compounds 77 and78 as anti-hyperuricemic agents.
4 Conclusion
Natural products have been known to have a long history of serving as vital sources of insight into medicine, biology, and chemistry. Numerous studies have established strategies for obtaining various natural products and their derivatives for the development of drugs (Li and Lou 2018). Benzoflavones, which are derived from natural products and have a structural motif, have therapeutic promise in a variety of domains. The benzoflavone-based active compounds have been identified, indicating that they have a wide application potential based on careful drug design considerations and extensive structural optimization experiments. This review discusses the latest developments in benzoflavone analogues, including but not restricted to their synthetic characteristics and pharmacological qualities (Zhao et al., 2020, Zhao et al., 2021, Zhao et al., 2021). A discussion is also delivered on the SAR of synthesized benzoflavone analogues, with the enhanced SAR potentially opening the path to more rational creation of benzoflavone derivatives. The most widely reported usages of benzoflavone derivatives were CYP inhibitors and antitumor agents, herein, we not only summarized these well-known contents in the article, but also cover other aspects in the bioactivities of benzoflavones, especially in the hypoglycemic agent section, the perspective was not restricted in the traditional flavonoid skeleton, the anthocyanin scaffolds were incorporated as well, which help us find out more promising clinical candidates. In bioorganic chemistry research, the emergence of privileged scaffolds is usually the hotspot. However, with the advancement of chemoinformatic technologies and computational chemistry, it seems that an efficacious systematic growth of the chemical space revolving around the benzoflavone scaffold to reach the peak of biological binding sites is now feasible. Exploratory studies of new synthetic methods have led to a rapid expansion of the diversity of substituents around the core of benzoflavones, opening up new avenues for future inquiry into the mechanism of action, selectivity, and specificity of benzoflavone-based medicines. Without a doubt, these thorough examinations will provide additional favorable outcomes.
Acknowledgments
This work was supported by the National Key R&D Program of China (grant number 2017YFC1703904) and the Regional Innovation and Cooperation Project of the Science & Technology Department of Sichuan Province (2020YFQ0032).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 2'-substituted chalcone derivatives as inhibitors of interleukin-1 biosynthesis. J. Med. Chem.. 1993;36:1434-1442.
- [CrossRef] [Google Scholar]
- Novel synthetic chalcones induces apoptosis in human glioblastoma cells. Chem. Biol. Interact.. 2016;252:74-81.
- [CrossRef] [Google Scholar]
- SAR-guided development and characterization of a potent antitumor compound toward B-cell neoplasms with no detectable cytotoxicity toward healthy cells. J. Med. Chem.. 2015;58:1244-1253.
- [CrossRef] [Google Scholar]
- Discovery, structure-activity relationship, and antiparkinsonian effect of a potent and brain-penetrant chemical series of positive allosteric modulators of metabotropic glutamate receptor 4. J. Med. Chem.. 2017;60:8515-8537.
- [CrossRef] [Google Scholar]
- Multi-layer identification of highly-potent ABCA1 up-regulators targeting LXRβ using multiple QSAR modeling, structural similarity analysis, and molecular docking. Molecules. 2016;21:1639.
- [CrossRef] [Google Scholar]
- Synthesis and pharmacological activity of chalcones derived from 2,4,6-trimethoxyacetophenone in RAW 264.7 cells stimulated by LPS: quantitative structure-activity relationships. Bioorg. Med. Chem.. 2008;16:658-667.
- [CrossRef] [Google Scholar]
- Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg. Med. Chem. Lett.. 2008;18:6227-6230.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation, and molecular modeling of chalcone derivatives as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatases (PtpA and PtpB) J. Med. Chem.. 2012;55:390-402.
- [CrossRef] [Google Scholar]
- Chemical modification of the multitarget neuroprotective compound fisetin. J. Med. Chem.. 2012;55:378-389.
- [CrossRef] [Google Scholar]
- Design and synthesis of new alpha-naphthoflavones as cytochrome P450 (CYP) 1B1 inhibitors to overcome docetaxel-resistance associated with CYP1B1 overexpression. J. Med. Chem.. 2015;58:3534-3547.
- [CrossRef] [Google Scholar]
- Development of benzochalcone derivatives as selective CYP1B1 inhibitors and anticancer agents. Medchemcomm.. 2019;10:1606-1614.
- [CrossRef] [Google Scholar]
- Synthesis and structure-activity relationship studies of α-naphthoflavone derivatives as CYP1B1 inhibitors. Eur. J. Med. Chem.. 2020;187:111938
- [CrossRef] [Google Scholar]
- Discovery of heterocycle-containing α-naphthoflavone derivatives as water-soluble, highly potent and selective CYP1B1 inhibitors. Eur. J. Med. Chem.. 2021;209:112895
- [CrossRef] [Google Scholar]
- QSAR modeling of the interaction of flavonoids with GABA(A) receptor. Eur. J. Med. Chem.. 2008;43:1593-1602.
- [CrossRef] [Google Scholar]
- Characterization of a 7,8-benzoflavone double effect on CFTR Cl- channel activity. J. Membr. Biol.. 2007;220:1-9.
- [CrossRef] [Google Scholar]
- A cell-based, microplate colorimetric screen identifies 7,8-benzoflavone and green tea gallate catechins as inhibitors of the hepatitis C virus. Biol. Pharm. Bull.. 2012;35:1320-1327.
- [CrossRef] [Google Scholar]
- Analysis of structural requirements for Ah receptor antagonist activity: ellipticines, flavones, and related compounds. Biochem. Pharmacol.. 1996;52:1787-1803.
- [CrossRef] [Google Scholar]
- Flavonoid modulation of ionic currents mediated by GABA(A) and GABA(C) receptors. Eur. J. Pharmacol.. 2003;461:79-87.
- [CrossRef] [Google Scholar]
- Selective benzopyranone and pyrimido[2,1-a]isoquinolin-4-one inhibitors of DNA-dependent protein kinase: synthesis, structure-activity studies, and radiosensitization of a human tumor cell line in vitro. J. Med. Chem.. 2005;48:569-585.
- [CrossRef] [Google Scholar]
- The novel isoflavone 7-hydroxy-3',4'-benzoisoflavone induces cell apoptosis in human osteosarcoma cells. Cancer Lett.. 2008;271:117-128.
- [CrossRef] [Google Scholar]
- Synthesis and identification of new flavonoids targeting liver X receptor β involved pathway as potential facilitators of Aβ clearance with reduced lipid accumulation. J. Med. Chem.. 2013;56:6033-6053.
- [CrossRef] [Google Scholar]
- Application of α- and β-naphthoflavones as monooxygenase inhibitors of Absidia coerulea KCh 93, Syncephalastrum racemosum KCh 105 and Chaetomium sp. KCh 6651 in transformation of 17α-methyltestosterone. Bioorg. Chem.. 2018;78:178-184.
- [CrossRef] [Google Scholar]
- Chemical constituents of Euonymus alatus (Thunb.) Sieb. and their PTP1B and α-glucosidase inhibitory activities. Phytother. Res.. 2015;29:1540-1548.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of flavones and benzoflavones as inhibitors of BCRP/ABCG2. Eur. J. Med. Chem.. 2013;67:115-126.
- [CrossRef] [Google Scholar]
- The aryl hydrocarbon receptor interacts with estrogen receptor alpha and orphan receptors COUP-TFI and ERRalpha1. Arch. Biochem. Biophys.. 2000;373:163-174.
- [CrossRef] [Google Scholar]
- Design and synthesis of selective CYP1B1 inhibitor via dearomatization of α-naphthoflavone. Bioorg. Med. Chem.. 2019;27:285-304.
- [CrossRef] [Google Scholar]
- Drug-like property profiling of novel neuroprotective compounds to treat acute ischemic stroke: guidelines to develop pleiotropic molecules. Transl. Stroke Res.. 2013;4:328-342.
- [CrossRef] [Google Scholar]
- Strategies to diversify natural products for drug discovery. Med. Res. Rev.. 2018;38:1255-1294.
- [CrossRef] [Google Scholar]
- Discovery and rational design of natural-product-derived 2-phenyl-3,4-dihydro-2H-benzo[f]chromen-3-amine analogs as novel and potent dipeptidyl peptidase 4 (DPP-4) inhibitors for the treatment of type 2 diabetes. J. Med. Chem.. 2016;59:6772-6790.
- [CrossRef] [Google Scholar]
- Discovery of a natural-product-derived preclinical candidate for once-weekly treatment of type 2 diabetes. J. Med. Chem.. 2019;62:2348-2361.
- [CrossRef] [Google Scholar]
- Flavonoid analogues as urease inhibitors: synthesis, biological evaluation, molecular docking studies and in-silico ADME evaluation. Bioorg. Chem.. 2020;105:104370
- [CrossRef] [Google Scholar]
- Antimalarial alkoxylated and hydroxylated chalcones [corrected]: structure-activity relationship analysis. J. Med. Chem.. 2001;44:4443-4452.
- [CrossRef] [Google Scholar]
- Suppression of cytokine-mediated complement factor gene expression through selective activation of the ah receptor with 3 ',4 '-dimethoxy-alpha-naphthoflavone. Mol. Pharmacol.. 2011;79:508-519.
- [CrossRef] [Google Scholar]
- Hydroxychalcones induce apoptosis in B16–F10 melanoma cells via GSH and ATP depletion. Eur. J. Med. Chem.. 2009;44:1630-1637.
- [CrossRef] [Google Scholar]
- Monoamine oxidase inhibitory activity of 2-aryl-4H-chromen-4-ones. Bioorg. Chem.. 2015;58:72-80.
- [CrossRef] [Google Scholar]
- Induction of apoptosis and cell cycle arrest in L-1210 murine lymphoblastic leukaemia cells by (2E)-3-(2-naphthyl)-1-(3'-methoxy-4'-hydroxy-phenyl)-2-propen-1-one. J. Pharm. Pharmacol.. 2010;62:1128-1136.
- [CrossRef] [Google Scholar]
- Chalcone synthesis, properties and medicinal applications: a review. Environ. Chem. Lett.. 2020;18:433-458.
- [CrossRef] [Google Scholar]
- Synthesis, docking study and relaxant effect of 2-alkyl and 2-naphthylchromones on rat aorta and guinea-pig trachea through phosphodiesterase inhibition. Bioorg. Chem.. 2013;50:17-25.
- [CrossRef] [Google Scholar]
- The synthetic flavanone 6-methoxy-2-(naphthalen-1-yl)chroman-4-one induces apoptosis and activation of the MAPK pathway in human U-937 leukaemia cells. Bioorg. Chem.. 2020;94:103450
- [CrossRef] [Google Scholar]
- Cytotoxic 3,4,5-trimethoxychalcones as mitotic arresters and cell migration inhibitors. Eur. J. Med. Chem.. 2013;63:501-510.
- [CrossRef] [Google Scholar]
- N-(1'-naphthyl)-3,4,5-trimethoxybenzohydrazide as microtubule destabilizer: synthesis, cytotoxicity, inhibition of cell migration and in vivo activity against acute lymphoblastic leukemia. Eur. J. Med. Chem.. 2015;96:504-518.
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of isoliquiritigenin derivatives as a neuroprotective agent against glutamate mediated neurotoxicity in HT22 cells. Bioorg. Med. Chem. Lett.. 2020;30:127058
- [CrossRef] [Google Scholar]
- Reverse type I binding spectra of human cytochrome P450 1B1 induced by flavonoid, stilbene, pyrene, naphthalene, phenanthrene, and biphenyl derivatives that inhibit catalytic activity: a structure-function relationship study. Chem. Res. Toxicol.. 2009;22:1325-1333.
- [CrossRef] [Google Scholar]
- Novel antimitotic activity of 2-hydroxy-4-methoxy-2',3'-benzochalcone (HymnPro) through the inhibition of tubulin polymerization. J. Agric. Food Chem.. 2013;61:12588-12597.
- [CrossRef] [Google Scholar]
- Structural properties of polyphenols causing cell cycle arrest at G1 phase in HCT116 human colorectal cancer cell lines. Int. J. Mol. Sci.. 2013;14:16970-16985.
- [Google Scholar]
- Liver tumor-promoting effect of beta-naphthoflavone, a strong CYP 1A1/2 inducer, and the relationship between CYP 1A1/2 induction and Cx32 decrease in its hepatocarcinogenesis in the rat. Toxicol. Pathol.. 2000;28:540-547.
- [CrossRef] [Google Scholar]
- Benzoflavone derivatives as potent antihyperuricemic agents. Medchemcomm.. 2019;10:128-147.
- [CrossRef] [Google Scholar]
- Synthesis, in vitro and in vivo evaluation of 2-aryl-4H-chromene and 3-aryl-1H-benzo[f]chromene derivatives as novel α-glucosidase inhibitors. Bioorg. Med. Chem. Lett.. 2019;29:119-123.
- [CrossRef] [Google Scholar]
- Benzoflavone activators of the cystic fibrosis transmembrane conductance regulator: towards a pharmacophore model for the nucleotide-binding domain. Bioorg. Med. Chem.. 2003;11:4113-4120.
- [CrossRef] [Google Scholar]
- Soft-enolization baker-venkataraman rearrangement enabled total synthesis of dirchromones and related 2-substituted chromones. Org. Lett.. 2018;20:7424-7428.
- [CrossRef] [Google Scholar]
- Increase in tissue inhibitor of metalloproteinase-2 (TIMP-2) levels and inhibition of MMP-2 activity in a metastatic breast cancer cell line by an anti-invasive small molecule SR13179. Cancer Lett.. 2010;289:111-118.
- [CrossRef] [Google Scholar]
- Advances on synthesis of flavonoid glycosides. Chinese J. Org. Chem.. 2019;39:1875-1890.
- [CrossRef] [Google Scholar]
- Flavonoids chrysin and benzoflavone, potent breast cancer resistance protein inhibitors, have no significant effect on topotecan pharmacokinetics in rats or mdr1a/1b (-/-) mice. Drug Metab. Dispos.. 2005;33:341-348.
- [CrossRef] [Google Scholar]
- Pyrazolone structural motif in medicinal chemistry: retrospect and prospect. Eur. J. Med. Chem.. 2020;186:111893
- [CrossRef] [Google Scholar]
- Research progress in biological activities of isochroman derivatives. Eur. J. Med. Chem.. 2021;210:113073
- [CrossRef] [Google Scholar]
- Strategies for the development of highly selective cytochrome P450 inhibitors: several CYP targets in current research. Bioorg. Med. Chem. Lett.. 2019;29:2016-2024.
- [CrossRef] [Google Scholar]
- Research progress in biological activities of succinimide derivatives. Bioorg. Chem.. 2021;108:104557
- [CrossRef] [Google Scholar]
- Design and synthesis of α-naphthoflavone chimera derivatives able to eliminate cytochrome P450 (CYP)1B1-mediated drug resistance via targeted CYP1B1 degradation. Eur. J. Med. Chem.. 2020;189:112028
- [CrossRef] [Google Scholar]




