

Translate this page into:
Current scenario on non-nucleoside reverse transcriptase inhibitors (2018-present)
⁎Corresponding author. yuminshi2021@163.com (Yu-min Shi)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
This review provides the recent developments (2018-present) in non-nucleoside reverse transcriptase inhibitors, and the structure–activity relationships are also discussed to facilitate further rational design of more effective candidates.
Abstract
Acquired immune deficiency syndrome (AIDS) mainly caused by human immunodeficiency virus (HIV) type 1 (HIV-1) is a deadliest infectious disease, in which the immune system becomes ineffective and opportunistic infections accompany the disease. Reverse transcriptase (RT) is the target for the majority of anti-HIV-1 agents, and HIV RT inhibitors can be categorized into nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs). In view of the structural diversity, unique mode of action, high efficacy and low toxicity of NNRTIs in comparison to NRTIs, NNRTIs represent one of the most significant antiretroviral drugs against HIV infections. The purpose of the present review article is to discuss the recent developments (2018-present) in NNRTIs, and the structure–activity relationships (SARs) are also discussed to facilitate further rational design of more effective candidates.
Keywords
Acquired immune deficiency syndrome
Human immunodeficiency virus
Non-nucleoside reverse transcriptase inhibitor
Structure–activity relationships

1 Introduction
Acquired immunodeficiency syndrome (AIDS), a chronic infectious disease characterized by multiple life-threatening illnesses and one of the most devastating pandemics in recorded history, is mainly caused by human immunodeficiency virus (HIV) type 1 (HIV-1) (Kabir et al., 2020; Voshavar, 2019). The currently available antiretroviral agents, inclusive of nucleoside reverse transcriptase inhibitors (NRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors (FIs), co-receptor inhibitors and integrase inhibitors (INIs), have improved the treatment efficacy of HIV infection significantly, and the mortality and morbidity decreased to those of a manageable chronic disease. However, the existing agents with some drawbacks such as the occurrence of viral mutants and the severe side effects are unable to make a complete cure (Motati et al., 2019; Chokkar et al., 2019). Nowadays, HIV infection remains one of the most important threats to global public health since around 38 million people worldwide live with HIV, and approximately 690,000 people died from AIDS-related complications in 2020 (Hokello et al., 2021; Ghosn et al., 2018). Hence, it is urgent to develop novel, more efficacious and less toxic anti-HIV therapeutics.
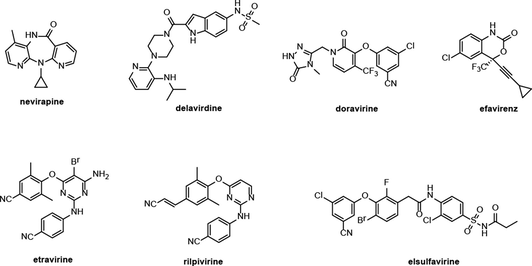
HIV-1 reverse transcriptase (HIV-1 RT) contains two drug binding sites. One is a substrate binding site, and another is an allosteric site. Both these two binding sites play a key role in the life cycle of HIV-1 and are deemed as one of the most successful targets for the development of novel anti-HIV agents (Namasivayam et al., 2019; Peddi et al., 2018). HIV RT inhibitors, categorized into NRTIs and NNRTIs, are taken as the ‘backbone’ of antiretroviral therapy (ART) (Battini and Bollini, 2019; Rai et al., 2018). Compared with NRTIs, NNRTIs demonstrated structural diversity, unique mode of action, high efficacy, and low toxicity. Several NNRTIs such as nevirapine (first-generation, Fig. 1), delavirdine (second-generation), doravirine (second-generation), efavirenz (second-generation), etravirine (second-generation), rilpivirine (second-generation), and elsulfavirine (second-generation) have already been approved for clinical application. Accordingly, NNRTIs represent a class of the most significant antiretroviral drugs for the treatment of HIV infections (Wang et al., 2020; Li et al., 2020; Gu et al., 2019; Gu et al., 2020). However, the lack of intrinsic exonucleolytic proofreading and rapid mutation of HIV-1 RT make the efficacy of the first generation of NNRTIs dramatically decreased (Shirvani et al., 2019; Ding et al., 2021). The second generation of NNRTIs can inhibit NNRTI-associated mutant species more effectively, but these NNRTIs suffer from poor pharmacokinetic properties and some side effects like Stevens-Johnson syndrome (Xavier Ruiz and Arnold, 2020; Song et al., 2014). Therefore, it is imperative to develop novel NNRTIs with high activity and low side effects.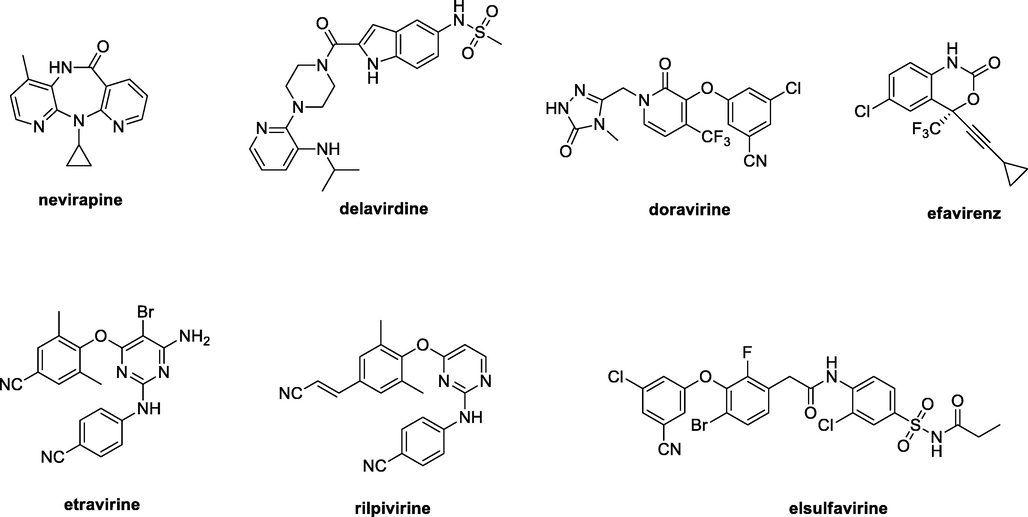
Chemical structures of nevirapine, delavirdine, doravirine, efavirenz, etravirine, rilpivirine, and elsulfavirine.
Azoles, coumarins and flavones, indoles, pyridines, pyrimidines, quinazolines and quinolines possess structural diversity, multiple anti-HIV mechanisms of action, favorable drug-like properties such as good oral bioavailability, efficient membrane penetration, excellent metabolic stability, desirable pharmacokinetic and pharmacodynamic profiles. Moreover, some of their derivatives have already applied in clinical practice or under different phases of clinical evaluations, demonstrating that coumarins and flavones, indoles, pyridines, pyrimidines, quinazolines and quinolines are useful scaffolds for the discovery of novel NNRTIs.
Recently, a great number of NNRTIs, inclusive of azoles, coumarins and flavones, indoles, pyridines, pyrimidines, quinazolines and quinolines, have been designed and synthesized, aiming to discover promising inhibitors with excellent anti-HIV activities and reduced side effects. This review summarizes the latest progresses on NNRTIs, covering articles published from 2018 to present. The structure–activity relationships (SARs) are also discussed to facilitate with the further rational design of more effective candidates.
2 Azoles
Azoles such as imidazole, pyrazole, 1,2,3-triazole and 1,2,4-triazole are the five-membered nitrogen-containing heteroaryl rings and possess unique physicochemical profiles. Azoles represent privileged scaffolds for the development of highly selective, physiological benevolent anti-HIV therapeutics (Feng et al., 2021; Prasher et al., 2021; Dixit et al., 2021; Lang et al., 2020).
A series of aryl substituted benzimidazolones 1 (Fig. 2, Table 1; half maximal inhibitory concentration/IC50: 0.026- > 44.1 μM) were designed, synthesized and evaluated for their potential as NNRTIs against wild-type HIV-1 strains by Pelly et al. (Pribut et al., 2019). The SAR revealed that the 3-chloro-5-cyanophenyl ring at C-7 position of benzimidazolone moiety was critical for the high activity, and replacement by pyridinyl or naphthyl group led to loss of activity. Substituent on the N-1 position also had great influence on the activity, and the relative contribution order was ethyl > methyl > propyl. Among them, compounds 1a,b (IC50: 0.026 and 0.029 μM) not only showed superior activity in comparison with nevirapine (IC50: 0.105 μM) against wild-type HIV-1 strains, but also possessed potent broad-spectrum activity (IC50: 0.007–0.415 μM and 0.055–0.494 μM, respectively) against a panel of clinically relevant single mutant HIV-1 strains (K103N, Y181C, V106M, G109A, Y188C and Y188H). Moreover, compound 1a (IC50: 0.052 μM) was also highly active against double mutant K103N/Y181C HIV-1 strains and displayed low toxic (half-cytotoxic concentration/CC50: 24.1 μM) towards MT-4 cells with selectivity index (SI: CC50/IC50) values of 58.0–3442.8. Docking studies demonstrated that compound 1a could be well accommodated in the non-nucleoside inhibitor binding pocket (NNIBP). Overall, compound 1a could serve as a useful candidate for anti-HIV therapy.
Chemical structures of azoles 1–10.
Azole
Structure
Mechanism of action/target
In vitro activity
Ref.
1a

accommodated in the non-nucleoside inhibitor binding pocket
IC50: 0.007–0.415 μM against WT HIV-1, single mutant HIV-1 (K103N, Y181C, V106M, G109A, Y188C and Y188H) and double mutant K103N/Y181C HIV-1 strains
(Costa et al., 2018)
2a
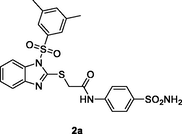
HIV‑1 RT
EC50: 0.073–1.58 μM against single mutants L100I, K103N, Y188L and E138K as well as double mutants F227L/V106A
(Cui et al., 2020)
3
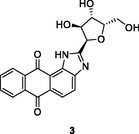
HIV-1 RT
IC50: 8.51 μM against HIV-1 RT
(Daelemans et al., 2021)
4a,b
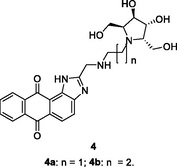
HIV-1 RT
IC50: 24.54 and 27.78 μM against HIV-1 RT
(Daelemans et al., 2021)
5
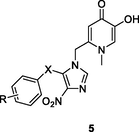
NRa
EC50: > 3.6 μM against HIV-1 strains
(De Clercq et al., 2021)
6a-c
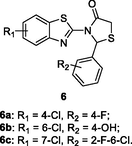
HIV-1 RT
IC50: 0.001–0.0047 μM against HIV-1 RT
(Desantis et al., 2020)
7

NR
IC50: 16.1 and 13.03 μM against HIV-1UG070 (X4) and HIV-1VB59 (R5) strains
(Ding et al., 2021)
8
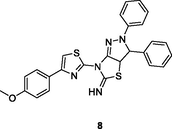
HIV-1 RT
IC50: 0.9 and 0.35 μM against HIV-1IIIB and HIV-1ADA5 strains
(Ding et al., 2021)
9
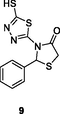
NR
EC50: 31.99 μM against HIV-1 strain
(Ding et al., 2021)
10

HIV-1 RT
IC50: 0.69 μM against HIV-1 RT
(Dixit et al., 2021)
The majority of 1-((3,5-dimethylphenyl)sulfonyl)-1H-benzo[d]imidazoles 2 (IC50: 0.047–0.222 μM; half effective concentration/EC50: 0.024–0.235 μM) demonstrated profound HIV-1 RT and HIV-1 inhibitory activity (Monforte et al., 2018). The SAR disclosed that substituent at para-position of phenyl ring was more favorable than ortho-position. Sulfonamide and sulfone on the phenyl ring could enhance the activity. Among them, compounds 2a,b (IC50: 0.05 and 0.047 μM; EC50: 0.024 and 0.028 μM) were more potent than nevirapine (IC50: 1.55 μM; EC50: 0.18 μM), and both of them (CC50: 30.94 and 10.59 μM) also displayed low toxicity towards MT-4 cells with SI values of 1289.2 and 378.2. In particular, compound 2a (EC50: 0.073–1.58 μM) exhibited promising activity against HIV-1 mutant strains, inclusive of the single mutants L100I, K103N, Y188L and E138K as well as the double mutants F227L/V106A.
The anthra[1,2–d]imidazole-6,11-dione 3 (IC50: 8.51 μM) was more active than zidovudine (IC50: 31.24 μM) in term of inhibition of HIV-1 RT (Wang et al., 2018). The SAR indicated that replacement of sugar moiety with azasugar was detrimental to the activity as evidenced by that compounds 4a,b (IC50: 24.54 and 27.78 μM) showed reduced activity when compared with compound 3. Extension of the carbon space between anthra[1,2–d]imidazole-6,11-dione and azasugar led to the loss of activity. For the 4-nitroimidazole-4-pyridinone hybrids 5 (EC50: >3.6 μM), none of them were active against HIV-1 strains regardless of electron-donating or electron-withdrawing groups on the phenyl ring (Shirvani et al., 2020).
The majority of benzothiazole-thiazolidin-4-ones 6 (IC50: 0.001–4.28 μM) showed promising inhibitory activity against HIV-1 RT (Petrou et al., 2019). The SAR revealed that chloro on the benzothiazole moiety was favorable to the activity, while methoxy and ethoxy groups decreased the activity. Additionally, electron-withdrawing group on the phenyl ring at C-2 position of thiazolidin-4-one motif enhanced the activity. The representative compounds 6a-c (IC50: 0.001–0.0047 μM) were 63.8–300 times superior to nevirapine (IC50: 0.3 μM) in inhibition of HIV-1 RT, suggesting the potential of these hybrids as novel NNRTIs.
The thiazolidin-4-one derivative 7 not only possessed considerable activity (IC50: 16.1 and 13.03 μM) against HIV-1UG070 (X4) and HIV-1VB59 (R5) strains, but also demonstrated promising activity (minimal inhibitory concentration/MIC: 7.88 μM) against Mycobacterium tuberculosis (MTB) H37Ra (Chitre et al., 2019). Accordingly, this compound could be used as a useful template for the discovery of novel candidates for the treatment of HIV and MTB co-infections.
The thiazole derivative 8 (IC50: 0.9 and 0.35 μM) exhibited excellent activity against HIV-1IIIB and HIV-1ADA5 strains, and it could inhibit 84.7 % HIV-1 RT at the concentration of 100 μM (Kasralikar et al., 2019). The thiadiazole derivative 9 (EC50: 31.99 μM) was active against HIV-1 strain, but non-toxic (EC50: >423.1 μM) towards MT-4 cells with SI value of 13.2, implying its potential as a novel anti-HIV template (Buemi et al., 2020). The 1,2,3-triazole-benzo[e] (Kabir et al., 2020; Motati et al., 2019)thiazin-4-one 10 (IC50: 0.69 μM) was 28.8 times more potent than zidovudine (IC50: 19.87 μM) against HIV-1 RT, revealing its potential as a novel NNRTI candidate (Yan et al., 2018).
3 Coumarins and flavones
Coumarins and flavones with fascinating structural diversity are widely distributed in natural resources (Ren et al., 2018). Some coumarins and flavones are potential NNRTIs, and the representative compound is (+)-calanolide A, which is currently under clinical evaluations for the treatment of HIV infections. (+)-calanolide A is active against HIV-1 on the two most common NNRTI-related mutations, K103N and Y181C (Xu et al., 2021; Saravanan et al., 2015). Accordingly, coumarin and flavone derivatives are of great importance for the development of novel anti-HIV candidates.
Three new prenylated coumarins, clauselenins A-C (11–13, Fig. 3, Table 2; EC50: 0.17–0.68 μM), isolated from the stems of Clausena lenis, possessed potent anti-HIV-1 activity and displayed non-toxicity (CC50: > 200 μM) towards C8166 cell line (Fu et al., 2020). Farnesiferol C (14, IC50: 30 μM), a well-known biologically active sesquiterpene coumarin derivative from genus Ferula, could inhibit HIV-1RT activity via mixed inhibition mechanism (Sistani et al., 2021). Herbacitrin (15; IC50: 21.5 μM), a favonoid 7-O-glucoside, isolated from cotton flowers (Gossypium herbaceum), showed considerable inhibitory activity against HIV-1 RT (Áy et al., 2019). Hence, these natural coumarin and flavone derivatives could serve as lead hits for the development of novel anti-HIV agents.
Chemical structure2 of coumarin and flavone derivatives 11–18.
Coumarin/ flavone
Structure
Mechanism of action/target
In vitro activity
Ref.
11

HIV‑1 RT
EC50: 0.29 μM against HIV-1 strain
(Famiglini and Silvestri, 2018)
12
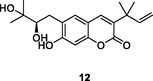
HIV‑1 RT
EC50: 0.68 μM against HIV-1 strain
(Famiglini and Silvestri, 2018)
13
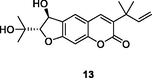
HIV-1 RT
EC50: 0.17 μM against HIV-1 strain
(Famiglini and Silvestri, 2018)
14
HIV-1 RT
IC50: 30 μM against HIV-1 RT
(Feng et al., 2021)
15
HIV-1 RT
IC50: 21.5 μM against HIV-1 RT
(Feng et al., 2021)
16
NR
EC50: 3.94–44.3 μM against HIV-1IIIB and single mutant L1001, K103N, Y181C, Y188L and E138K HIV-1 strains
(Feng et al., 2021)
17
HIV-1 PR and RT
IC50: 62.1 nM and 188.7 μM against HIV-1 PR and RT
(Fu et al., 2020)
18

HIV-1 PR and RT
IC50: 55.8 nM and 372.8 μM against HIV-1 PR and RT
(Fu et al., 2020)
The coumarin derivative 16 (EC50: 3.94–44.3 μM) exhibited considerable broad-spectrum activity against HIV-1IIIB and a panel of single mutant L1001, K103N, Y181C, Y188L and E138K HIV-1 strains (De Clercq et al., 2021). The coumarin-sulfamide hybrids 17 (IC50: 62.1 nM and 188.7 μM) and 18 (IC50: 55.8 nM and 372.8 μM) were active against HIV-1 PR and RT, so both of them could serve as dual HIV-1 PR/RT inhibitors (Zhu et al., 2020).
4 Indoles
Indoles are privileged scaffolds and possess favorable drug-like properties such as good oral bioavailability, efficient membrane penetration, excellent metabolic stability, desirable pharmacokinetic and pharmacodynamic profiles (Thanikachalam et al., 2019; Singh and Singh, 2018). Indoles could inhibit diverse HIV enzymes including RT. Several indole-based agents which are exemplified by delavirdine have already been used in clinics or under clinical evaluations as NNRTIs, demonstrating that indoles represent attractive scaffolds for the design and development of novel anti-HIV drugs (Chen et al., 2021; Famiglini and Silvestri, 2018).
The indolylarylsulfone 19 (Fig. 4, Table 3; IC50: 18.2 μM) could selectively inhibit Y181C mutant HIV-1 RT and it showed higher activity (EC50: 2.36 μM vs 9.74 μM) against HIV-1 Y181C strains than HIV-1 wide type (Gao et al., 2021). The SAR revealed that movement of pyrrolidinyl group from C-3 position to C-2 position in indole moiety was favorable to the activity (Zhao et al., 2019). In particular, indolylarylsulfone 20 (EC50: 0.0098–0.31 μM) demonstrated profound activity against HIV-1IIIB, single mutant (L1001, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A) HIV-1 strains. The activity was > 12.8 folds higher than that of nevirapine (EC50: 0.18- > 15.03 μM) (Zhao et al., 2019). Moreover, compound 20 (CC50: 28.07 μM) displayed low cytotoxicity towards MT-4 cells, and the SI values were in a range of 90.5 to 2864.2. Further study indicated that the pyrrolidinyl group was not essential for the high activity, and replacement by substituted benzyl (21) was also permitted (Famiglini et al., 2017; Nalli et al., 2020; Xu et al., 2022). The representative compounds 21a-d (EC50: 0.0007–0.086 μM) were > 160 times superior to nevirapine (EC50: 0.11->3.75 μM) against HIV-1IIIB, single mutant (K103N, Y181C, and Y188L) and double mutant (K103N/Y181C) HIV-1 strains. Moreover, compounds 21a-d were also non-toxic (CC50: > 26.9 μM) towards MT-4 cells (Famiglini et al., 2017; Nalli et al., 2020; Xu et al., 2022). Accordingly, indolylarylsulfones 20 and 21a-d could act as valuable candidates for further preclinical investigations.
Chemical structure of indole derivatives 19–24.
Indole
Structure
Mechanism of action/target
In vitro activity
Ref.
19

HIV‑1 RT
EC50: 9.74 and 2.36 μM vs μM) against RT HIV-1 and HIV-1 Y181C strains
(Gasparyan et al., 2021)
20
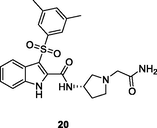
NR
EC50: 0.0098–0.31 μM against HIV-1IIIB, single mutant (L1001, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A) HIV-1 strains
(George and Reddy, 2018)
21a-d
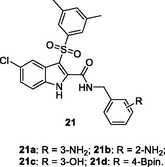
NR
EC50: 0.0007–0.086 μM against HIV-1IIIB, single mutant (K103N, Y181C, and Y188L) and double mutant (K103N/Y181C) HIV-1 strains
(Ghosn et al., 2018; Gu et al., 2019; Gu et al., 2020)
22a-d
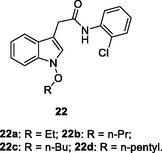
NR
EC50: 0.29–0.86 μM against HIV-1 strain
(Hokello et al., 2021)
23

NR
EC50: 0.04–1.89 μM against WT HIV-1, NNRTI-resistant HIV-1 strains, inclusive of single mutant (K103N, Y181C, and Y188L) and double mutant (K103N/Y101C, L100I/K103N, K103N/G190A and K103N/V108I) strains
(Hokello et al., 2021)
24a
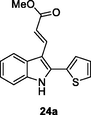
HIV-1 RT
IC50: 2.93 nM against HIV-1 RT
(Huang et al., 2019)
The anti-HIV-1 SAR of indole derivatives 22 proved that chloro at ortho-position of phenyl ring was crucial for the high activity (Xu et al., 2020). Linear alkyl group at R position was beneficial for the activity. In addition, replacement of the phenyl ring by pyridinyl ring could enhance the activity. Amongst them, compounds 22a-d (EC50: 0.29–0.86 μM) and 23a-c (EC50: 0.06–0.13 μM) exhibited excellent activity against HIV-1 strains. Moreover, compounds 23a,b (EC50: 0.18–1.89 μM and 0.04–1.42 μM, respectively) also possessed promising activity against a panel of NNRTI-resistant HIV-1 strains, inclusive of single mutant (K103N, Y181C, and Y188L) and double mutant (K103N/Y101C, L100I/K103N, K103N/G190A and K103N/V108I) strains.
The indole-thiophene hybrids 24a-c (IC50: 2.93–7.97 nM) were highly active against HIV-1 RT, and the representative hybrid 24a (IC50: 2.93 nM) was around 3 times more potent than efavirenz (IC50: 6.03 nM) (El-Hussieny et al., 2020). Thus, hybrid 24a could serve as a useful template for the development of novel NNRTIs.
5 Pyridines
Pyridines, one of the most widely used heterocycles in the drug discovery, could inhibit HIV with different action mechanisms such as RT inhibition (Alizadeh and Ebrahimzadeh, 2021; Ling et al., 2021). Several pyridine-containing compounds including nevirapine and delavirdine have been developed and used in clinics for the treatment of HIV infections. Accordingly, pyridines are attractive architectures for the development of novel NNRTIs.
The anti-HIV-1IIIB SAR of diarylpyridine derivatives 25 (Fig. 5, Table 4) illustrated that 4-methyl-2,6-dibromo group on the phenyl ring (R2 position) and ether linker (X = O) were favorable to the activity (Liu et al., 2017). Among them, derivatives 25a-d (EC50: 0.04–0.07 μM) were more potent than nevirapine (EC50: 0.25 μM) against HIV-1IIIB strains. The four derivatives (IC50: 0.05–0.65 μM) also showed higher inhibitory activity than nevirapine (IC50: 2.32 μM) towards HIV-1 RT polymerization. Therefore, these derivatives can potentially act as lead molecules in the development of novel anti-HIV chemotherapeutic agents.
Chemical structure of pyridine derivatives 25–29.
Pyridine
Structure
Mechanism of action/target
In vitro activity
Ref.
25a-d
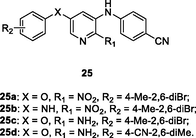
HIV‑1 RT
EC50: 0.04–0.07 μM against HIV-1IIIB strain
(Ippolito et al., 2021)
26a
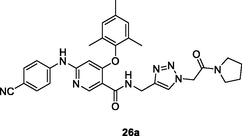
NR
EC50: 0.014–0.27 μM against HIV-1IIIB, HIV-1NL4-3, single mutant (K103N, Y181C, and Y138K) and double mutant (F227L/V106A) HIV-1 strains
(Jiang et al., 2021)
26b

NR
EC50: 0.02–0.61 μM, respectively) against HIV-1IIIB, HIV-1NL4-3, single mutant (K103N, Y181C, and Y138K) and double mutant (F227L/V106A) HIV-1 strains
(Jiang et al., 2021)
27

NR
EC50: 7.61 μM against HIV-1 strain
(Jin et al., 2018)
28
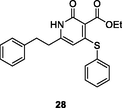
HIV-1 RT
IC50: 9.5 μM against HIV-1 RT
(Jin et al., 2019)
29
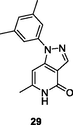
NR
EC50: 2.53–8.65 μM against HIV-1VB59, HIV-1UG070, HIV-1 N119, HIV-1VB51 (R5), HIV-192/BR/018 and HIV-1 N119 strains
(Jin et al., 2018)
The pyridine-1,2,3-triazole hybrids (EC50: 0.02–1.77 μM) showed promising activity against HIV-1IIIB strains, and the SAR disclosed that cyclic amide fragment at N-1 position of 1,2,3-triazole moiety could enhance the activity (Tian et al., 2018). In particular, hybrids 26a,b (EC50: 0.014–0.27 μM and 0.02–0.61 μM, respectively) were highly active against HIV-1IIIB, HIV-1NL4-3, single mutant (K103N, Y181C, and Y138K) and double mutant (F227L/V106A) HIV-1 strains. In addition, hybrids 26a,b (CC50: 58.09 and 180.9 μM) also displayed low cytotoxicity towards TZM-bl cells. Hence, hybrids 26a,b were worthy of further evaluations.
The pyridine derivative 27 (EC50: 7.61 μM) was also active against HIV-1 strains, but the activity was far inferior to that of nevirapine (EC50: 0.12 μM) (Changunda et al., 2021). Besides, this compound (CC50: 1.52 μM) was highly cytotoxic towards MT-4 cells. Thus, this compound still needs further structural modifications.
The inhibitory activity of pyridone 28 (IC50: 9.5 μM) towards HIV-1 RT was in the same level with that of nevirapine (IC50: 3.4 μM) (Yang et al., 2017), whereas pyrazolo (Áy et al., 2019; Al-Masoudi et al., 2020; Áy et al., 2019; Battini and Bollini, 2019; Buemi et al., 2020; Cai et al., 2018; Čechová et al., 2019; Cen et al., 2021; Chaivisuthangkura et al., 2020; Changunda et al., 2021; Chen et al., 2020; Chen et al., 2020; Chen et al., 2020; Chen et al., 2020; Chen et al., 2021; Chen et al., 2020; Chen et al., 2021; Cherukupalli et al., 2021; Chitre et al., 2019; Chokkar et al., 2019; Corbett and Rodgers, 2002; Costa et al., 2018; Cui et al., 2020; Daelemans et al., 2021; De Clercq et al., 2021; Desantis et al., 2020; Ding et al., 2021; Ding et al., 2021; Ding et al., 2021; Dixit et al., 2021; Duong et al., 2020; El-Hussieny et al., 2020; Famiglini et al., 2017; Famiglini and Silvestri, 2018; Feng et al., 2021; Feng et al., 2021; Feng et al., 2021; Fu et al., 2020; Gao et al., 2019; Gao et al., 2019; Gao et al., 2021; Gao et al., 2021; Gasparyan et al., 2021; George and Reddy, 2018; Ghosn et al., 2018; Gu et al., 2019; Gu et al., 2020; Hokello et al., 2021; Huang et al., 2019; Huang et al., 2021; Huo et al., 2018; Ippolito et al., 2021; Jiang et al., 2021; Jin et al., 2018; Jin et al., 2019; Jin et al., 2018; Jin et al., 2020; Kabir et al., 2020; Kang et al., 2019; Kang et al., 2019; Kang et al., 2019; Kang et al., 2020; Kang et al., 2020; Kang et al., 2020; Kang et al., 2020; Kang et al., 2020; Kasralikar et al., 2018; Kasralikar et al., 2019; Kaur and Kumar, 2021; Kumar et al., 2019; Lang et al., 2020; Li et al., 2020; Li et al., 2021; Ling et al., 2021; Liu et al., 2018; Liu et al., 2017; Lu et al., 2018; Makarasen et al., 2019; Monforte et al., 2018; Mostoufi et al., 2020; Motati et al., 2019; Nalli et al., 2020; Namasivayam et al., 2019; Overacker et al., 2019; Peddi et al., 2018; Petrou et al., 2019; Prasher et al., 2021; Pribut et al., 2019; Rai et al., 2018; Ren et al., 2018; Romeo et al., 2019; Sagma and Lakshmanan, 2020; Sang et al., 2019; Sang et al., 2019; Saravanan et al., 2015; Shirvani et al., 2019; Shirvani et al., 2020; Singh and Singh, 2018; Singh et al., 2021; Sistani et al., 2021; Smith et al., 2021; Song et al., 2014; Sun et al., 2018; Sun et al., 2021; T occo, G.; Esposito, F.; Caboni, P.; Laus, A.; Beutler, J. A.; Wilson, J. A.; Corona, A.; Le Grice, S. F. J.; Tramontano, E. Scaffold hopping and optimisation of 3’,4’-dihydroxyphenyl- containing thienopyrimidinones: Synthesis of quinazolinone derivatives as novel allosteric inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H. J Enzym Inhib Med Chem, 2020; Tang et al., 2019; Thanikachalam et al., 2019; Tian et al., 2018; Voshavar, 2019; Wang et al., 2018; Wang et al., 2020; Wang et al., 2018; Wang et al., 2018; Wang et al., 2018; Wang et al., 2020; Wang et al., 2021; Wu et al., 2020; Xavier Ruiz and Arnold, 2020; Xiao et al., 2020; Xu et al., 2021; Xu et al., 2022; Xu et al., 2020; Yan et al., 2018; Yang et al., 2018; Yang et al., 2017; Zhang et al., 2018; Zhao et al., 2019; Zhou et al., 2019; Zhu et al., 2020)pyridine-4-one 29 (EC50: 2.53–8.65 μM) possessed broad-spectrum anti-HIV-1 (HIV-1VB59, HIV-1UG070, HIV-1 N119, HIV-1VB51 (R5), HIV-192/BR/018 and HIV-1 N119 strains) activity and also displayed low cytotoxicity (CC50: 679 μM) towards MT-4 cells, demonstrating its potential to develop novel anti-HIV agents (Kumar et al., 2019).
6 Pyrimidines
Pyrimidines possess potent activity against various resistant HIV strains which are attributed to their potential to adopt multiple conformations of HIV RT (Sagma and Lakshmanan, 2020; Feng et al., 2021). Food and Drug Administration (FDA) has already approved pyrimidine-based etravirine and rilpivirine for the treatment of HIV infections, demonstrating that pyrimidines occupy an important position in the discovery of novel anti-HIV agents.
The diarylpyrimidine 30 (Fig. 6, Table 5; EC50: 7.19–77.9 nM) exhibited excellent broad-spectrum activity against HIV-1IIIB and a panel of NNRTIs-resistant single and double mutant strains, inclusive of L100I, K103N, Y181C, Y188L, E138K, F227L/V106A and K103N/Y181C HIV-1 strains (Jin et al., 2018). The activity was found not inferior to that of etravirine (EC50: 3.13–32.2 nM). The SAR elucidated that boronic acid fragment was not critical for the high activity, and compound 31 (EC50: 0.9–41.5 nM) demonstrated exceptionally potent against all tested HIV-1 strains (Kang et al., 2019; Daelemans et al., 2021). In addition, diarylpyrimidines 30 and 31 (IC50: 0.18 and 0.051 μM,CC50: 25.6 and > 250 μM) also displayed high inhibitory activity against HIV-1 RT and low cytotoxicity towards MT-4 cells. Furthermore, diarylpyrimidine 31 also showed favorable pharmacokinetic properties with an oral bioavailability (F) of 30.96 % and a half-life (t1/2) of 11.1 h. Overall, diarylpyrimidines 30 and 31 were worthy of further investigation as novel NNRTIs to circumvent drug resistance.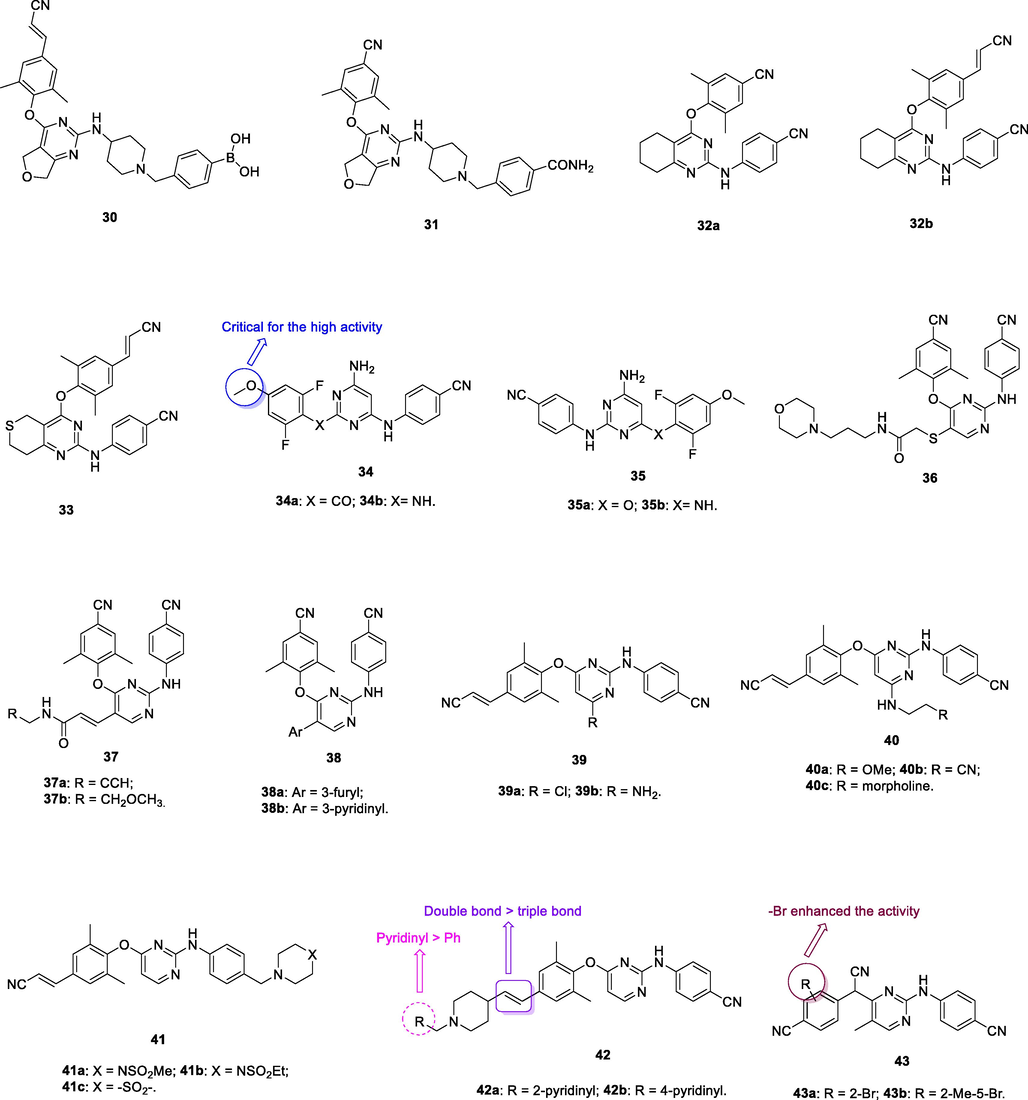
Chemical structure of diarylpyrimidine derivatives 30–43.
Pyrimidine
Structure
Mechanism of action/target
In vitro activity
Ref.
30
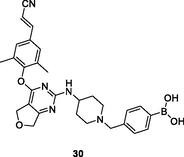
HIV-1 PR
EC50: 7.19–77.9 nM against HIV-1IIIB and NNRTIs-resistant single and double mutant strains, inclusive of L100I, K103N, Y181C, Y188L, E138K, F227L/V106A and K103N/Y181C HIV-1 strains
(Kang et al., 2019)
31
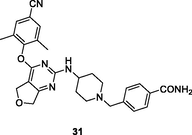
HIV-1 PR
EC50: 0.9–41.5 nM against HIV-1IIIB and NNRTIs-resistant single and double mutant strains, inclusive of L100I, K103N, Y181C, Y188L, E138K, F227L/V106A and K103N/Y181C HIV-1 strains
(Kang et al., 2019; Kang et al., 2019)
32a,b
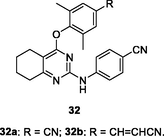
HIV-1 RT
EC50: 7.1–95.3 nM against HIV-1IIIB, L100I, K103N, Y181C, Y188L, E138K, and F227L/V106A HIV-1 strains
(Kang et al., 2020)
33
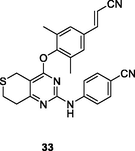
HIV-1 RT
EC50: 4.44–54.5 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Kang et al., 2020)
34a,b
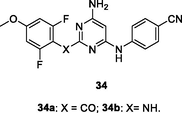
NR
EC50: 4.0–797 nM against HIV-1IIIB, K103N and Y181C HIV-1 strains
(Kang et al., 2020)
35a,b
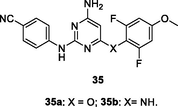
NR
EC50: 2.0–637 nM against HIV-1IIIB, K103N and Y181C HIV-1 strains
(Kang et al., 2020)
36
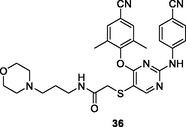
HIV-1 PR
EC50: 1.4–345.2 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant F227L/V106A and K103N/Y181C HIV-1 strains
(Kang et al., 2020)
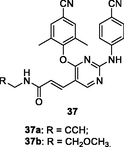
37a,b
NR
EC50: 5.2–1492 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant F227L/V106A and K103N/Y181C HIV-1 strains
(Kasralikar et al., 2018)
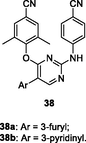
38a,b
NR
EC50: 2.5–394.9 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant F227L/V106A and K103N/Y181C HIV-1 strains
(Kasralikar et al., 2019)
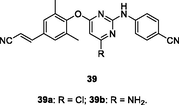
39a,b
HIV-1 PR
EC50: 0.8–160 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Kaur and Kumar, 2021)
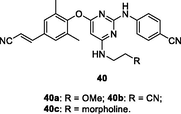
40a-c
HIV-1 PR
EC50: 1.2–220 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Kumar et al., 2019)
41a-c
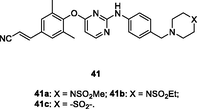
HIV-1 PR
EC50: 1.9–200 nM against HIV-1IIIB, L100I, K103N, Y181C, E138KF227L/V106A and K103N/Y181C HIV-1 strains
(Lang et al., 2020; Li et al., 2020)
42a

NR
EC50: 0.019–1.61 μM against HIV-1IIIB, L100I, K103N, and E138K HIV-1 strains
(Li et al., 2021)
43a,b
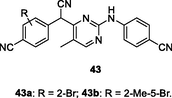
HIV-1 PR
EC50: 0.06–2.35 μM against HIV-1IIIB, L100I, K103N, Y181C, E138K and F227L/V106A HIV-1 strains
(Ling et al., 2021)
The diarylpyrimidines 32a,b (EC50: 7.1–95.3 nM) showed extremely high activity against HIV-1IIIB, L100I, K103N, Y181C, Y188L, E138K, and F227L/V106A HIV-1 strains and acceptable cytotoxicity (CC50: 3.1 and 7.2 μM) towards MT-4 cells (Wang et al., 2021). These two compounds (IC50: 52 and 119 nM) also possessed excellent inhibitory activity towards wide-type HIV-1 RT, favorable metabolic stability profiles and other drug-like properties. Compared with compound 32b, dihydrothiopyrano[4,3–d]pyrimidine 33 (EC50: 4.44–54.5 nM) exhibited enhanced activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains, revealing that introduction of sulfur atom improved the activity (Čechová et al., 2019). In addition, compound 33 (CC50: 284 μM) was found with reduced cytotoxicity towards MT-4 cells and increased inhibitory activity (IC50: 81 nM) towards wide-type HIV-1 RT. Moreover, compound 33 displayed good solubility (12.8 μg/mL) and no significant inhibition towards the main CYP enzymes happened. The in vivo acute toxicity evaluation revealed that no mortality and no poisoning symptoms occurred when oral administration of compound 33 at a single dose of 1000 mg/kg. After a single dose of 2 mg/kg intravenously, compound 33 showed a favorable mean CL (1380 mL/h/kg), volume of distribution (V: 702 mL/kg), and a long terminal half-life (T1/2: 1.94 h). Furthermore, after oral administration at a dose of 20 mg/kg, the absorption of compound 33 reached maximum (Tmax) at 0.25 h with a plasma concentration value (Cmax) of 16.6 ng/mL, and the mean residence time (MRT) was 2.90 h. Overall, dihydrothiopyrano[4,3–d]pyrimidine 33 endowed with huge potential to serve as a promising drug candidate due to its excellent potency, low toxicity, and favorable drug-like properties.
The anti-HIV-1 SAR of diarylpyrimidines 34 and 35 against HIV-1IIIB, K103N and Y181C HIV-1 strains illustrated that methoxy group at C-4 position of phenyl ring was critical for the high activity, and carboxyl (–CO-), amino (–NH-) as well as ether (-O-) were favorable linkers between pyrimidine and phenyl ring (Singh et al., 2021). However, replacement of two phenyl rings by pyridinyl group could not enhance the activity (Huo et al., 2018). In particular, diarylpyrimidines 34a,b (EC50: 4.0–797 nM) and 35a,b (EC50: 2.0–637 nM) were found to be most active against HIV-1IIIB, K103N and Y181C HIV-1 strains, and both of them both (CC50: 16.0–21.7 μM) also displayed low cytotoxicity towards MT-4 cells.
The diarylpyrimidine 36 (EC50: 1.4–16.2 nM) was > 2 folds more potent than etravirine (EC50: 3.3–34.1 nM) against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains (Feng et al., 2021). The activity was comparable to that of efavirenz (EC50: 105.9 and 345.2 nM vs 218.5 and 225.4 nM) against double mutant F227L/V106A and K103N/Y181C HIV-1 strains. In addition, diarylpyrimidine 36 (CC50: 27.2 μM) displayed lower cytotoxicity than etravirine (CC50: 2.2 μM) towards MT-4 cells and excellent inhibitory activity (IC50: 84 nM) against HIV-1 RT. Further study implied that thioether side chain was not essential for the high activity, and replacement of thioether side chain by acrylamide or aromatic ring was also permitted as evidenced by that compounds 37a,b (EC50: 5.2–1492 nM) and 38a,b (EC50: 2.5–394.9 nM) demonstrated great potency against wide-type, single mutant and double mutant HIV-1 strains (Gao et al., 2021; Lu et al., 2018).
The diarylpyrimidine 39a (EC50: 1.69–160 nM) was highly potent against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains (Smith et al., 2021). Further studies elucidated that replacement of chloro at C-6 position of pyrimidine moiety by amino or alkylamino side chain was also permitted as evidenced by that derivatives 39b (EC50: 0.8–44.0 nM) and 40a-c (EC50: 1.2–220 nM) exhibited profound activity against all tested HIV-1 strains (Smith et al., 2021; Jiang et al., 2021). Introduction of heterocycles into phenyl ring was also tolerated, and derivatives 41a-c (EC50: 1.9–200 nM) possessed remarkably activity against HIV-1IIIB, L100I, K103N, Y181C, E138KF227L/V106A and K103N/Y181C HIV-1 strains (Huang et al., 2019; Cherukupalli et al., 2021). In addition, the diarylpyrimidines (EC50: 0.02–0.34 μM; CC50: 1.0->173 μM) also displayed high inhibitory activity against wide-type and double-mutated (K103N/Y181C) HIV-1 RT enzymes as well as acceptable cytotoxicity towards MT-4 cells. Derivatives 40a-c (EC50: 0.32–1.56 nM) were found to possess profound activity against HIV-2ROD strains. In the in vivo toxicity study, diarylpyrimidine 41a did not display significant inhibition on main cytochrome P450 enzymes and no acute/subacute toxicity at doses of 2000 mg/kg (acute) and 50 mg/kg (subacute) in mice was observed, demonstrating its in vivo safety profiles (Huang et al., 2019). Collectively, these diarylpyrimidines could serve as useful prototypes for the development of novel anti-HIV-1 and anti-HIV-2 agents.
The anti-HIVIIIB SAR of diarylpyrimidines 42 (EC50: 19–110 nM) disclosed that the double bond between phenyl ring and piperidinyl group was more favorable than triple bond, and pyridinyl was better than phenyl group at R position (Chen et al., 2020). The molecular dynamics simulation studies revealed that the presence of pyridine moiety played a significant role in binding with HIV-1 RT. Among them, compound 42a (EC50: 0.019–1.61 μM) was found to be most active against HIV-1IIIB, L100I, K103N, and E138K HIV-1 strains, which merited further investigations.
The diarylpyrimidines 43 (EC50: 0.06–8.03 μM) showed considerable activity against HIV-1IIIB, L100I, K103N, Y181C, and E138K HIV-1 strains, and the SAR evinced that bromo on the phenyl ring could increase the activity (Jin et al., 2018). Among them, the most active compounds 43a,b (EC50: 0.06–0.86 μM) were not only more potent than nevirapine (EC50: 0.20–6.07 μM) against HIV-1IIIB, L100I, K103N, Y181C, and E138K HIV-1 strains, but also active (EC50: 2.35 and 1.52 μM) against F227L/V106A strains. Besides, these two compounds (IC50: 0.07 and 0.45 μM) also showed effective inhibition towards wide-type HIV-1 RT and acceptable cytotoxicity (CC50: 6.6 and 9.8 μM) against MT-4 cells.
The anti-HIV-1IIIB SAR of biphenyl-substituted diarylpyrimidines 44 (Fig. 7, Table 6; EC50: 1.0–34 nM) illustrated that electron-withdrawing group on the phenyl ring was favorable to the activity (Ding et al., 2021). Among them, the representative compounds 44a-c (EC50: 1.0–1.4 nM) were more potent than etravirine (EC50: 1.5 nM) and azidothymidine (EC50: 2.1 nM) against HIV-1IIIB strains. In particular, the most active compound 44a (IC50: 14 nM; CC50: 2.08 μM) displayed high inhibitory activity against wide-type HIV-1 RT enzymes and acceptable cytotoxicity towards MT-4 cells. Moreover, compound 44a (EC50: 0.84–10 nM) also exhibited profound activity against single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (K103N/Y181C) HIV-1 strains.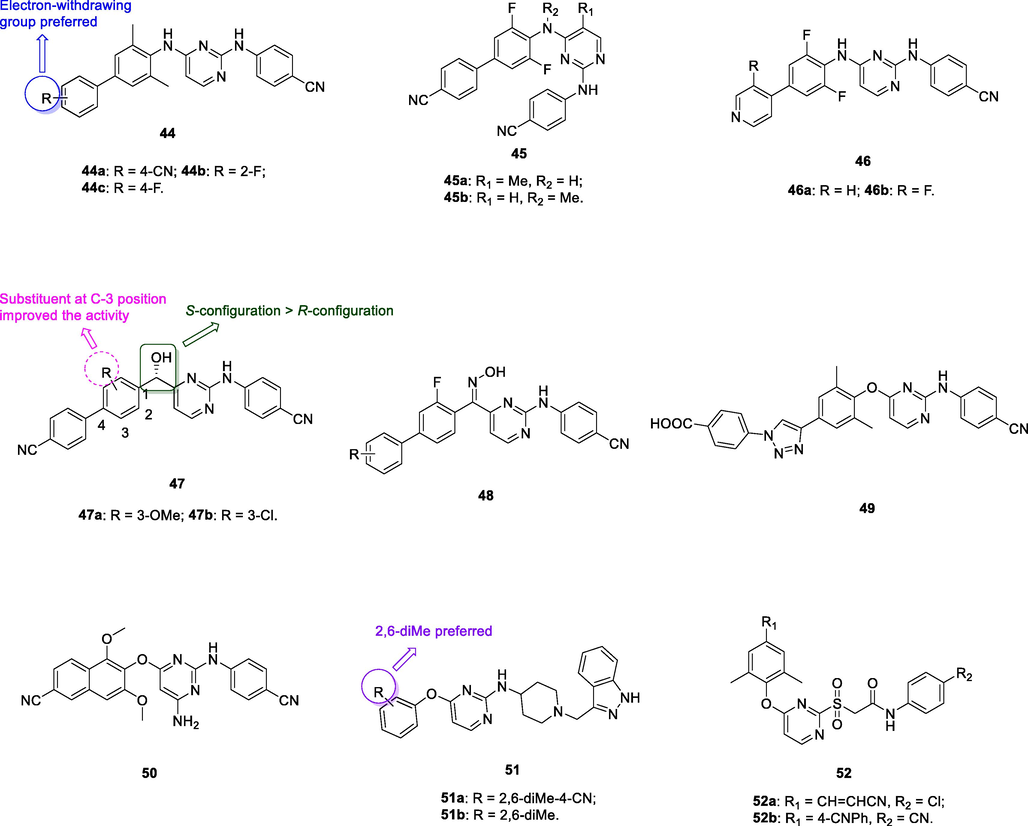
Chemical structure of diarylpyrimidine derivatives 44–52.
Pyrimidine
Structure
Mechanism of action/target
In vitro activity
Ref.
44a
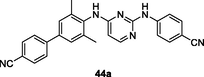
HIV-1 PR
EC50: 0.84–10 nM against WT, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (K103N/Y181C) HIV-1 strains
(Liu et al., 2018)
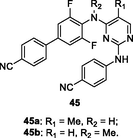
45a,b
HIV-1 PR
EC50: 1.37–16.3 nM and 6.2–524 nM against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains
(Liu et al., 2017)
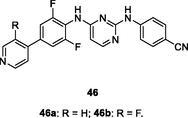
46a,b
HIV-1 RT
EC50: 1.0–417 nM and 1.3–92 nM against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains
(Lu et al., 2018)
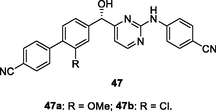
47
HIV-1 RT
EC50: 1.6–87.7 nM and 1.3–127.9 nM against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains
(Makarasen et al., 2019)
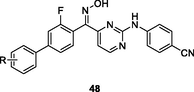
48
NR
EC50: 12.1–369 nM against HIV-1IIIB strain
(Monforte et al., 2018)
49
HIV-1 PR
EC50: 2.73–840 nM against HIV-1IIIB, L100I, K103N, Y181C, E138K, F227L/V106A and K103N/Y181C HIV-1 strains
(Mostoufi et al., 2020)
50
HIV-1 PR
EC50: 5.3–176 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Motati et al., 2019)
51a,b
NR
EC50: 6.4–110 nM and 8.6–400 nM against HIV-1IIIB and single mutant HIV-1 strains
(Nalli et al., 2020)
52a,b

HIV-1 PR
EC50: 7–760 nM and 6–642 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Namasivayam et al., 2019)
The biphenyl-substituted diarylpyrimidines 45a,b (EC50: 1.37–16.3 nM and 6.2–524 nM, respectively) exhibited remarkable activity against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains as well as low cytotoxicity (CC50: >285 and 148 μM) towards MT-4 cells (Ding et al., 2021). In addition, diarylpyrimidines 45a,b (IC50: 13 and 15 nM) were comparable to efavirenz (IC50: 10 nM) and etravirine (IC50: 11 nM) in terms of inhibition of wide-type HIV-1 RT. Moreover, no apparent in vivo acute toxicity was observed in 45a-treated female pregnant mice, at a dose of 2 g/kg through intragastric administration. Notably, diarylpyrimidine 45a showed excellent stability in human liver microsomes (Clint: 5.0 μL/min/mg proteins, T1/2: 138 min), with a suitable clearance and a long half-life. After a single 1 mg/kg dose of compound 45a (intravenous injection), the mean clearance rate and half-lives were 15.8 mL/min/kg and 1.18 h, respectively. At a dose of 5.0 mg/kg, diarylpyrimidine 45a reached maximum concentration (Tmax) at 0.36 h with a maximum concentration (Cmax) of 38 ng/mL, and t1/2 was up to 2.13 h. However, the oral bioavailability (F) was only 2.39 %.
The biphenyl-substituted diarylpyrimidine 46a (EC50: 1.0–417 nM) with a 4-pyridine group displayed excellent inhibitory activity against HIV-1IIIB and single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains, and this compound (CC50: > 313 μM) was non-toxic towards MT-4 cells (Chen et al., 2020). Further study indicated that introduction of fluoro into C-3 position of pyridine moiety could enhance the activity, but increase the cytotoxicity as evidenced by that compound 46b (EC50: 1.3–92 nM; CC50: 3.3 μM) showed improved activity but higher cytotoxicity. In addition, diarylpyrimidine 46a (IC50: 7.4 nM) was superior to efavirenz (IC50: 10 nM) and etravirine (IC50: 11 nM) in terms of inhibiting wide-type HIV-1 RT. Compound 46a also exhibited a decent improvement in druggability than etravirine and rilpivirine. The hydrochloric acid salt of diarylpyrimidine 46a showed significantly improved water solubility in different pH conditions. It did not show apparent CYP enzymatic inhibitory activity or acute toxicity at a dose of 2 g/kg via intragastric administration. Besides, hydrochloric acid salt of diarylpyrimidine 46a possessed excellent oral bioavailability (F: 126 %). Collectively, hydrochloric acid salt of diarylpyrimidine 46a represented a promising drug candidate for HIV clinical therapy.
The anti-HIV-1IIIB SAR of hydroxyl-containing biphenyl-substituted diarylpyrimidines 47 (EC50: 1.4–21.0 nM) elucidated that the configuration of hydroxyl group was crucial for the activity, and S-configuration > R-configuration (Chen et al., 2020). Substituent at C-3 position of phenyl ring (R) was favorable to the activity, while replacement of hydroxyl group by hydroxime resulted in loss of activity as evidenced by that diarylpyrimidines 48 (EC50: 12.1–369 nM) showed decreased activity (Zhou et al., 2019). Among them, diarylpyrimidines 47a,b (EC50: 1.6 and 1.4 nM against HIV-1IIIB strains; EC50: 2.3–87.7 nM and 1.3–127.9 nM against single mutant strains) not only were superior to efavirenz (EC50: 2.9 nM) and etravirine (EC50: 3.7 nM) against HIV-1IIIB strains, but also were not inferior to efavirenz (IC50: 4.8–171.1 nM) against single mutant (L100I, K103N, Y181C, Y188L and E138K) HIV-1 strains. Diarylpyrimidines 47a,b (IC50: 9.5 and 6.9 nM) also showed higher inhibitory effect than efavirenz (IC50: 9.8 nM) and etravirine (IC50: 11 nM) against wide-type HIV-1 RT and acceptable cytotoxicity (CC50: 9.07 and 4.25 μM) towards MT-4 cells. Furthermore, compound 47a did not show any acute toxicity at a dose of 2 g/kg via intragastric administration, demonstrating its excellent safety profile. Overall, compound 47a could serve as a promising candidate for further preclinical evaluations.
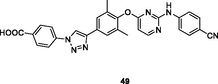
The 1,2,3-triazole-containing diarylpyrimidine 49 (EC50: 65–840 nM) with EC50 values in nanomolar level against HIV-1IIIB, L100I, K103N, Y181C and E138K HIV-1 strains was also active (EC50: 2.73 and 3.27 μM) against double mutant F227L/V106A and K103N/Y181C HIV-1 strains (Chen et al., 2021). This compound also displayed low cytotoxicity (CC50: 33.17 μM) towards MT-4 cells and high inhibitory activity (IC50: 20 nM) against wide-type HIV-1 RT.
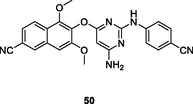
The naphthyl-diarylpyrimidine 50 (EC50: 5.3–176 nM) possessed profound activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains, and the activity was comparable to that of etravirine (EC50: 4.3–220 nM) (Xiao et al., 2020). Besides, compound 50 (EC50: 7.28 μM) was also active against HIV-2ROD strains, but it displayed low cytotoxicity (CC50: 19.0 μM) towards MT-4 cells. The SAR revealed that introduction of linear or branched alkyl group into amino group at C-6 position of pyrimidine moiety led to loss of the activity. Further study disclosed that the inhibitory effect of compound 50 (IC50: 20 nM) against wide-type HIV-1 RT was 24 times superior to that of efavirenz (IC50: 480 nM). The molecular docking results indicated that the interactions between the compound 50 and the NNIBP not only maintained the hydrogen-bonding interaction network with the backbone of Lys101 and π-π stacking effect, but also formed a new hydrogen bond with Glu138 in the entrance channel.
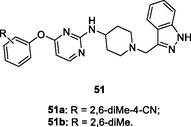
The anti-HIV-1 SAR of indazole-containing diarylpyrimidines 51 (EC50: 0.0064–10.0 μM) against HIV-1IIIB, K103N, Y181C and E138K strains disclosed that 2,6-dimethyl group on the phenyl ring was favorable to the activity (Chen et al., 2020). Amongst them, diarylpyrimidines 51a,b (EC50: 6.4–110 nM and 8.6–400 nM) were comparable to efavirenz (IC50: 2.6–76 nM) against HIV-1IIIB and single mutant HIV-1 strains, whereas compound 51a (EC50: 8.7 μM) was also active against double mutant (K103N/Y181C) HIV-1 strains. Moreover, compound 51a (CC50: 16 μM) displayed low cytotoxicity towards MT-4 cells, so this derivative represented a promising lead compound for future discovery.
The sulfone-containing diarylpyrimidines 52a,b (EC50: 7–760 nM and 6–642 nM, respectively) exhibited excellent activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains (Kang et al., 2020). The activity was in the same level with that of efavirenz (IC50: 7–292 nM). In addition, these two derivatives also displayed low cytotoxicity (CC50: 4.29 and 24.44 μM) towards MT-4 cells and potential inhibitory activity (IC50: 186 and 142 nM) against wide-type HIV-1 RT.
The thiophene[2,3–d]pyrimidine 53 (Fig. 8, Table 7; EC50: 2.05–7.59 nM) showed single-digit nanomolar level activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains, and the activity was superior to that of efavirenz (IC50: 2.35–29.4 nM) (Wang et al., 2020). The SAR illustrated that sulfonamide on the phenyl ring was critical for the high activity and replacement by amide or ester resulted in loss of activity. Compound 53 (CC50: 10.1 μM) also displayed low cytotoxicity towards MT-4 cells, and the SI values were 1330–4926. In addition, thiophene[2,3–d]pyrimidine 53 (IC50: 114–452 nM) possessed great inhibitory activity against wide-type and a panel of mutant HIV-1 RT. The pharmacokinetics study showed that the plasma concentration reached maximum (Tmax) at 3.2 h with Cmax value of 614 ng/mL, and the t1/2 was 2.8 h at a dose of 20 mg/kg (oral administration). Notably, the oral bioavailability (F) was 37.06 %, and no acute toxicity (2000 mg/kg) was observed, highlighting that thiophene[2,3–d]pyrimidine 53 could serve as a promising anti-HIV-1 drug candidate.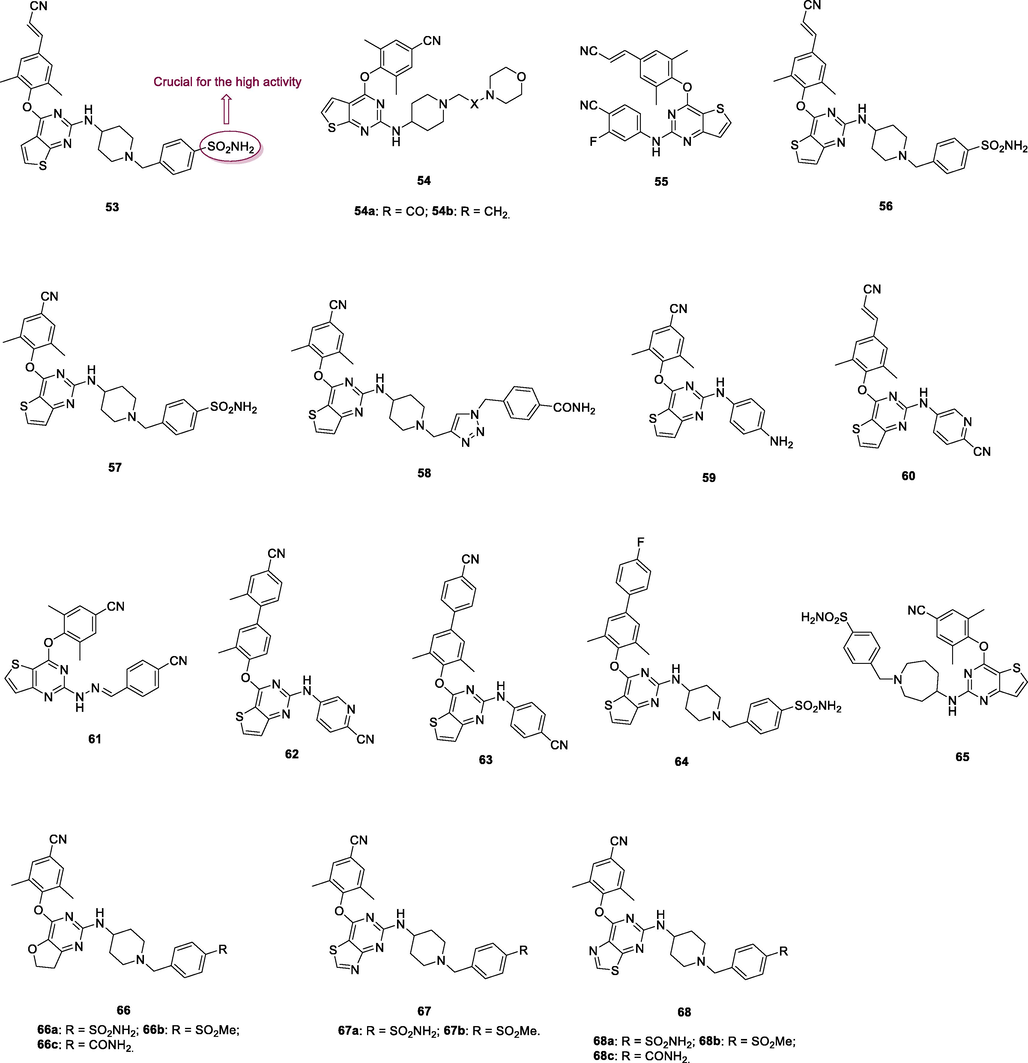
Chemical structure of pyrimidine derivatives 53–68.
Pyrimidine
Structure
Mechanism of action/target
In vitro activity
Ref.
53
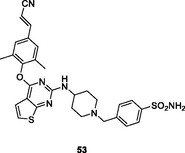
HIV-1 PR
EC50: 2.05–7.59 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K), and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Overacker et al., 2019)
54a,b
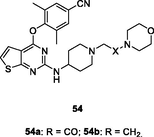
NR
EC50: 65 and 82 nM against HIV-1IIIB strain; 0.334–45.2 μM against single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Peddi et al., 2018)
55
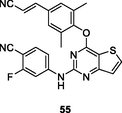
HIV-1 RT
EC50: <3.62–21.5 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Liu et al., 2017)
56
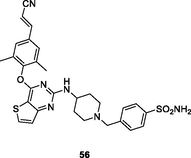
HIV-1 RT
EC50: 7.0–175 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A) HIV-1 strains
(Prasher et al., 2021)
57

HIV-1 RT
EC50: 1.4–31 nM against HIV-1IIIB, L100I, K103N, Y181C, E138K and K103/Y181C HIV-1 strains
(Pribut et al., 2019)
58
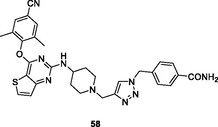
NR
EC50: 3.28 and 480 nM against HIV-1IIIB and K103N/Y181C strains
(Rai et al., 2018)
59
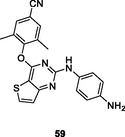
NR
EC50: 14–790 nM against HIV-1IIIB, single mutant and double mutant HIV-1 strains
(Ren et al., 2018)
60
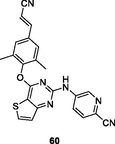
NR
EC50: 6.4–110 nM and 8.6–400 nM against HIV-1IIIB and single mutant HIV-1 strains
(Romeo et al., 2019)
61

NR
EC50: 21.2 nM against HIV-1IIIB strain
(Sagma and Lakshmanan, 2020)
62
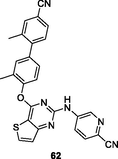
HIV-1 PR
EC50: 14–130 nM against HIV-1IIIB, L100I, K103N, Y181C and E138K strains
(Sang et al., 2019)
63
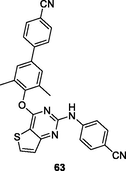
HIV-1 PR
EC50: 11.3–388 nM against HIV-1IIIB, single mutant (Y188L, K101P, Y181l, P225H, P236L) and double mutant (K103N/Y181C, V106A/F227L and K103N/181 l) HIV-1 strains
(Sang et al., 2019)
64
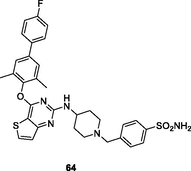
HIV-1 PR
EC50: 23.2–253.6 nM against HIV-1IIIB, single mutant (Y188L, K101P, Y181l, P225H, P236L) and double mutant (K103N/Y181C, V106A/F227L and K103N/181 l) HIV-1 strains
(Saravanan et al., 2015)
65
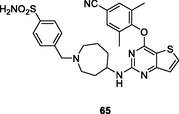
NR
EC50: 2.2 nM against HIV-1IIIB strain
(Shirvani et al., 2019)
66
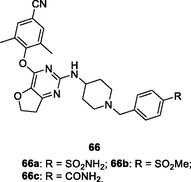
NR
EC50: 1.6–7.8 nM against HIV-1IIIB strain
(Shirvani et al., 2019)
67a,b

NR
EC50: 2.2–27.4 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Shirvani et al., 2020)
68a-c
NR
EC50: 1.6–28.1 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Shirvani et al., 2020)
The thiophene[2,3–d]pyrimidines 54a,b (EC50: 65 and 82 nM) showed remarkable activity against HIV-1IIIB strains and low cytotoxicity (CC50: 37.9 and 36.3 μM) towards MT-4 cells (Kang et al., 2020). However, both of them (EC50: 0.334–45.2 μM) only possessed moderate activity against most of the tested single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains, suggesting that these compounds had cross resistance with the currently available NNRTIs.
The thiophene[3,2–d]pyrimidine 55 (EC50: <3.62–21.5 nM; IC50: 36.2 nM) was not inferior to etravirine (EC50: 2.55–37.9 nM; IC50: 11.0 nM) against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains as well as wide-type HIV-1 RT (Sun et al., 2021). In addition, significantly low cytotoxicity (CC50: 155 μM) against MT-4 cells and no hERG inhibition (IC50: >30 μM) were also observed. The acute toxicity results showed that no mortality, poisoning symptoms, abnormal behaviors or significant changes of body weight were observed after a single oral dose of 2000 mg/kg. However, the oral bioavailability (F) was only 5.38 % in mice. Hence, further structural modification could focus on improving the oral bioavailability.
The thiophene[3,2–d]pyrimidine 56 (EC50: 7.0–175 nM; IC50: 961 nM; CC50: 145.3 μM) not only showed comparable activity to efavirenz (IC50: 5.0–380 nM) against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A) HIV-1 strains, but also displayed potential inhibitory activity against HIV-1 RT and low cytotoxicity towards MT-4 cells (Yang et al., 2018). The thiophene[3,2–d]pyrimidine 57 (EC50: 1.4–31 nM; CC50: >227 μM) was highly potent against HIV-1IIIB, L100I, K103N, Y181C, E138K and K103/Y181C HIV-1 strains and non-toxic against MT-4 cells (Kang et al., 2020). Compound 57 (IC50: 2.5–412 nM) also exhibited great inhibitory activity against wide-type, single mutant (Y188L, K101P, Y181l, P225H, P236L) and double mutant (K103N/Y181C, V106A/F227L and K103N/181 l) HIV-1 RT. The 1,2,3-triazole-containing thiophene[3,2–d]pyrimidine 58 (EC50: 3.28 and 480 nM; CC50: >210 μM) showed profound activity against HIV-1IIIB and K103N/Y181C and non-toxicity against MT-4 cells, implying that sulfamide fragment was not the indispensable functional group for the high activity (Kang et al., 2019).
The thiophene[3,2–d]pyrimidines 59 (EC50: 14–790 nM against HIV-1IIIB, single mutant and double mutant HIV-1 strains; CC50: 19.39 μM) and 60 (EC50: 3.3 and 22.6 nM against HIV-1IIIB and K103N/Y181C; CC50: >256 μM) demonstrated excellent anti-HIV-1 activity and low cytotoxicity against MT-4 cells (Kang et al., 2020; Wang et al., 2018). Furthermore, compound 60 (IC50: >50 and > 30 μM) did not inhibit CYP and hERG, and had favorable metabolic stability (T1/2: 77.5 min; Clint: 17.3 μL/min/mg) (Wang et al., 2018). Collectively, compound 60 was potential as a therapeutic drug and held great promise in the treatment of HIV-1 infections. The hydrazone-containing thiophene[3,2–d]pyrimidine 61 (EC50: 21.2 nM) possessed great potency against HIV-1IIIB strains, but it (EC50: >58.9 μM) was devoid of activity against K103N/Y181C strains, suggesting that this compound had cross resistance with the currently available anti-HIV agents (Sang et al., 2019).
The diaryl thiophene[3,2–d]pyrimidine 62 (EC50: 14–130 nM; IC50: 27 nM; CC50: 65.96 μM) exhibited strong inhibitory activity against HIV-1IIIB, L100I, K103N, Y181C and E138K strains as well as HIV-1 RT (Kang et al., 2020). Low cytotoxicity against MT-4 cells was also observed for compound 62. The diaryl thiophene[3,2–d]pyrimidines 63 (EC50: 11.3–388 nM against HIV-1 strains and 3.28 μM against HIV-2ROD strains; IC50: 211 nM; CC50: 16.9 μM) and 64 (EC50: 23.2–253.6 nM against HIV-1 strains and 1.40 μM against HIV-2ROD strains; IC50: 44 nM; CC50: 223.8 μM) were highly active against HIV-1IIIB, single mutant (Y188L, K101P, Y181l, P225H, P236L) and double mutant (K103N/Y181C, V106A/F227L and K103N/181 l) HIV-1 strains as well as HIV-1 RT (Sang et al., 2019; Chen et al., 2020). They also displayed low cytotoxicity against MT-4 cells and considerable activity against HIV-2ROD strains. Accordingly, diaryl thiophene[3,2–d]pyrimidines can be considered as promising therapeutic candidates for treatment of various HIV infections.
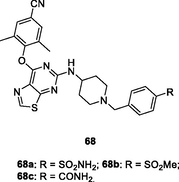
Further studies revealed that introduction of azepane or replacement of thiophene moiety by furan or thiazole was tolerated, and pyrimidines 65–68 (EC50: 1.1–8.2 nM) demonstrated profound anti-HIV-1IIIB activity (Kang et al., 2019; Desantis et al., 2020); while 1,2,4-triazole reduced the activity (Huang et al., 2021; Ippolito et al., 2021). Among them, pyrimidines 67a,b (EC50: 2.2–27.4 nM) and 68a-c (EC50: 1.6–28.1 nM) possessed potent broad-spectrum activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains. In particular, pyrimidines 68a-c (EC50: 3.11–6.06 μM) were also active against HIV-2ROD strains. The pharmacokinetics study of compound 68a suggested that the plasma concentration reached maximum (Tmax) at 2.8 h with Cmax value of 2982 ng/mL, and the t1/2 was 2.0 h at a dose of 20 mg/kg (oral administration). Notably, the oral bioavailability (F) was 83.8 %, highlighting that pyrimidine 68a could serve as a promising candidate for further evaluations.
The pyrimidine-2,4-dione 69 (Fig. 9, Table 8; EC50: 0.055 nM; IC50: 3.0 nM) was 12.1–2000 folds more potent than rilpivirine (EC50: 0.67 nM; IC50: 38 nM) and nevirapine (EC50: 110 nM; IC50: 1060 nM) against HIV-1IIIB strains and HIV-1 RT (Duong et al., 2020). This compound (CC50: 10 μM) also displayed low cytotoxicity towards MT-2 cells, proving its potential as a novel NNRTI candidate. Further study disclosed that the naphthalene-containing pyrimidine-2,4-dione 70 (EC50: 1.9–410 nM) possessed promising activity against HIBV-1IIIB, Y181C and K103N/Y181C HIV-1 strains (Romeo et al., 2019), while introduction of isoxazolidine (Gasparyan et al., 2021), methyl (Tang et al., 2019) or hydroxyl group (Gao et al., 2019; Mostoufi et al., 2020; Wang et al., 2018; Wang et al., 2018; Cui et al., 2020) into N-3 position of pyrimidine-2,4-dione moiety decreased the activity.
Chemical structure of pyrimidine derivatives 69–73.
Pyrimidine
Structure
Mechanism of action/target
In vitro activity
Ref.
69

HIV-1 PR
EC50: 0.055 nM against HIV-1IIIB strain
(Sistani et al., 2021)
70
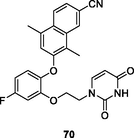
NR
EC50: 1.9–410 nM against HIBV-1IIIB, Y181C and K103N/Y181C HIV-1 strains
(Smith et al., 2021)
71
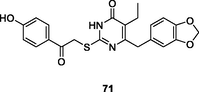
NR
EC50: 60–645 nM) possessed potent broad-spectrum activity against HIV-1IIIB, HIV-1 resistant strains (HIV-1RF/V82F/184V, pNL4-3gp41(36G)V38A, and HIV-14755-5) and clinical isolates (HIV-1TC-1, HIV-1WAN, HIV-1KIZ001and HIV-1KM018)
(Voshavar, 2019)
72
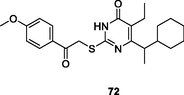
NR
EC50: 18 nM against HIV-1IIIB strain
(Wang et al., 2018)
73
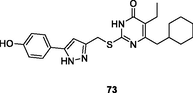
NR
EC50: 3.8–56.6 nM against wild-type strains (HIV-1IIIB and HIV-1Ba-L), resistant strains (HIV-1A17, HIV-174V and HIV-1RF/V82F/184V) and clinical isolated strains (HIV-1TC-1 and HIV-1WAN)
(Wang et al., 2018)
The 2-mercaptopyrimidin-4(3H)-one 71 (EC50: 60–645 nM) possessed potent broad-spectrum activity against HIV-1IIIB, HIV-1 resistant strains (HIV-1RF/V82F/184V, pNL4-3gp41(36G)V38A, and HIV-14755-5) and clinical isolates (HIV-1TC-1, HIV-1WAN, HIV-1KIZ001and HIV-1KM018), and this compound (CC50: 96.23 μM) also displayed low cytotoxicity towards normal C8166 cells (Li et al., 2021). The SAR indicated that replacement of benzo[d] (Kabir et al., 2020; Motati et al., 2019)dioxole by cyclohexane (compound 72, EC50: 18 nM against HIV-1IIIB) was tolerated (Kasralikar et al., 2018). Introduction of 1,2,3-triazole resulted in reduced activity (Wu et al., 2020),while incorporation of pyrazole was beneficial for the activity. Compound 73 (EC50: 3.8–56.6 nM) was found highly active against wild-type strains (HIV-1IIIB and HIV-1Ba-L), resistant strains (HIV-1A17, HIV-174V and HIV-1RF/V82F/184V) and clinical isolated strains (HIV-1TC-1 and HIV-1WAN) (Ajani et al., 2017). Additionally, compound 73 (CC50: 96.78 μM) also displayed low cytotoxicity towards normal C8166 cells. The in vivo pharmacokinetic studies revealed that compound 73 (5 mg/kg, oral administration) was rapidly absorbed with a Tmax of 0.5 h, T1/2 of 2.5 h and Cmax of 654 ng/mL. However, only 10.4 % oral bioavailability was observed.
7 Miscellaneous derivatives
Quinazoline and quinoline frameworks have attracted considerable attention due to their diverse spectrum of therapeutic potential (Kaur and Kumar, 2021; Corbett and Rodgers, 2002). Quinazoline/quinoline derivatives showed potential inhibitory activity against HIV-1 RT, demonstrating that rational design of quinazoline/quinoline derivatives may be used for the treatment of HIV infections (Jin et al., 2019; T occo, G.; Esposito, F.; Caboni, P.; Laus, A.; Beutler, J. A.; Wilson, J. A.; Corona, A.; Le Grice, S. F. J.; Tramontano, E. Scaffold hopping and optimisation of 3’,4’-dihydroxyphenyl- containing thienopyrimidinones: Synthesis of quinazolinone derivatives as novel allosteric inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H. J Enzym Inhib Med Chem, 2020). The dihydroquinazoline 74 (Fig. 10;Table 9 ; EC50: 0.84, 3.5 and 66 nM) exhibited profound activity against HIV-1IIIB, E138K and K103N/Y181C HIV-1 strains, and the activity was not inferior to that of efavirenz (IC50: 1.6, 2.0, and 55 nM) and etravirine (IC50: 2.2, 6.3 and 15 nM) (Gao et al., 2019). The SAR elucidated that cyano group on the phenyl ring was essential for the high activity, and replacement by halogen atoms, nitro, amide, methyl and methoxy led to significant loss of activity. Substitution of dihydroquinazoline with quinazolinone resulted in reduced activity (Gao et al., 2019; Overacker et al., 2019; Cai et al., 2018). Most of quinolone derivatives showed micromolar anti-HIV activity (Costa et al., 2018; Chaivisuthangkura et al., 2020; Makarasen et al., 2019; George and Reddy, 2018; Jin et al., 2020; Zhang et al., 2018), and among them, quinoline 75 (EC50: 4.7–8.9 μM) showed considerable activity against HIV-1Ba-L, HIV-1LAI, HIV-1HXB2, HIV-1YU2 and HIV-189.6 strains. Additionally, compound 75 (EC50: 34.2 μM) was also active against multidrug-resistant HIV-1v13-03413B isolate, demonstrating its potential to fight against both drug-sensitive and drug-resistant HIV-1 infections (Costa et al., 2018).
Chemical structure of compounds 74–77.
Compounds
Structure
Mechanism of action/target
In vitro activity
Ref.
74
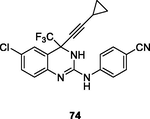
NR
EC50: 0.84, 3.5 and 66 nM against HIV-1IIIB, E138K and K103N/Y181C HIV-1 strains
(Wu et al., 2020)
75
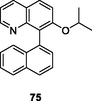
NR
EC50: 4.7–8.9 μM against HIV-1Ba-L, HIV-1LAI, HIV-1HXB2, HIV-1YU2 and HIV-189.6 strains
(Xu et al., 2021)
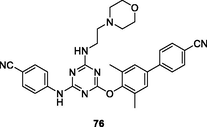
76
NR
EC50: 3.0–34 nM against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains
(Zhang et al., 2018)
77
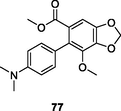
NR
EC50: 44–740 nM against wide-type HIV-1NL4-3, resistant HIV-174V and HIV-1A018 strains
(Zhao et al., 2019)
The triazine 76 (EC50: 3.0–34 nM and 50 nM against HIV-1 and HIV-2 strains, respectively) showed excellent activity against HIV-1IIIB, single mutant (L100I, K103N, Y181C, Y188L and E138K) and double mutant (F227L/V106A and K103N/Y181C) HIV-1 strains as well as HIV-2ROD strains, and the activity was not inferior to that of efavirenz (IC50: 1.3–84 nM) and etravirine (IC50: 1.2–15 nM) against HIV-1 strains (Al-Masoudi et al., 2020). In addition, this compound (CC50: 1.08 μM) also displayed acceptable cytotoxicity towards MT-4 cells, and SI values were 21.6–360. The pharmacokinetic studies indicated that triazine 76 reached Tmax at 13.3 h with Cmax of 43.23 ng/mL, but the oral bioavailability (F) was only 0.485 %.
Besides the derivatives mentioned above, some other compounds also displayed certain inhibitory activity against HIV-1 RT and HIV-1, but most of them were far inferior to the reference drugs [128-132]. In particular, SJP-L-5 (77, EC50: 44–740 nM) showed potent activity against wide-type HIV-1NL4-3 and resistant HIV-174V as well as HIV-1A018 strains. Further studies disclosed that SJP-L-5 as a new NNRTI could inhibit HIV-1 polypurine tract primed plus-strand DNA synthesis, initiating HIV-1 down-stream plus-strand DNA synthesis at multiple sites under drug pressure (Cen et al., 2021).
8 Conclusions
The advent of highly active antiretroviral treatment (HAART) has already caused the HIV disease trajectory and made the course from a terminal disease to a chronic disease model. However, fast-growing drug-resistant mutations are still unpredictably in clinics, resulting in deactivation or reduction of the existing drugs. Accordingly, it is imperative to develop novel anti-HIV drug candidates with more efficacy against HIV viruses and less toxicity. HIV-1 RT occupies an important position in the life cycle of HIV-1 and has been considered as one of the most successful targets for the development of novel anti-HIV agents. Among the RT inhibitors, NNRTIs represent one of the most significant antiretroviral drugs for the treatment of HIV infection due to their structural diversity and unique mode of action. However, the currently available NNRTIs possess some defects such as unsatisfactory efficacy against single/double resistant mutant strains, poor pharmacokinetic profiles, and adverse effects.
To address the above issues, a variety of novel potential NNRTIs including azoles, coumarins, flavones, indoles, pyridines and pyrimidines have been developed in recent years, and some of them have the potential to reduce toxicity, enhance efficacy, improve specificity, and overcome drug resistance. In particular, pyrimidines usually exhibited nanomolar level activity against both drug-sensitive and drug-resistant HIV-1 strains, and some of them also possessed low in vitro and in vivo toxicity as well as favorable pharmacokinetic profiles. For example, pyrimidine-based etravirine and rilpivirine have already been approved for the treatment of HIV infections. Accordingly, pyrimidines constitute versatile and interesting scaffolds in the discovery of novel NNRTIs, and the future research can be focused on this field. The natural products such as coumarins and flavones have also shown fascinating structural diversity, which could provide novel lead hits for further modification. Hybrid molecules have the potential to act on multiple targets and reduce side effects, so hybridization of two or more anti-HIV pharmacophores is a promising strategy to provide valuable therapeutic intervention for the treatment of various HIV infections.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Undeniable pharmacological potentials of quinazoline motifs in therapeutic medicine. Am. J. Drug Discov. Dev.. 2017;7(1):1-24.
- [Google Scholar]
- Antiviral activities of pyridine fused and pyridine containing heterocycles, a review (From 2000 to 2020) Mini-Rev. Med. Chem.. 2021;21(17):2584-2611.
- [Google Scholar]
- Synthesis, in vitro anti-HIV activity, cytotoxicity, and computational studies of some new steroids and their pyrazoline and oxime analogues. Russ. J. Bioorg. Chem.. 2020;46(5):822-836.
- [Google Scholar]
- Flavonol 7-O-glucoside herbacitrin inhibits HIV-1 replication through simultaneous integrase and reverse transcriptase inhibition. Evid. Based Complement. Altern. Med.. 2019;2019:e1064793.
- [Google Scholar]
- Challenges and approaches in the discovery of human immunodeficiency virus type-1 non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev.. 2019;39(4):1235-1273.
- [Google Scholar]
- Inhibition of HIV-1 RT activity by a new series of 3-(1,3,4-thiadiazol-2-yl)thiazolidin-4-one derivatives. Bioorg. Med. Chem.. 2020;28(8):e115431.
- [Google Scholar]
- Allosteric mechanism of quinoline inhibitors for HIV RT-associated RNase with MD simulation and dynamics fluctuation network. Chem. Biol. Drug Des.. 2018;91(3):805-816.
- [Google Scholar]
- Synthesis and anti-human immunodeficiency virus activity of substituted (o, o-difluorophenyl)-linked-pyrimidines as potent non-nucleoside reverse transcriptase inhibitors. Antiviral Chem. Chemother.. 2019;27:1-18.
- [Google Scholar]
- Design and biological evaluation of cinnamic and phenylpropionic amide derivatives as novel dual inhibitors of HIV-1 protease and reverse transcriptase. Eur. J. Med. Chem.. 2021;220:e113498.
- [Google Scholar]
- Binding interaction of potent HIV-1 NNRTIs, amino-oxy-diarylquinoline with the transport protein using spectroscopic and molecular docking. Spectrochim. Acta Part A: Mol. Biomol. Spectr.. 2020;233:e118159.
- [Google Scholar]
- Synthesis of novel pyridine and pyrimidine derivatives as potential inhibitors of HIV-1 reverse transcriptase using palladium-catalysed C-N cross-coupling and nucleophilic aromatic substitution reactions. Arkivoc. 2021;2020(3):152-170.
- [Google Scholar]
- Bioisosterism-based design and enantiomeric profiling of chiral hydroxyl-substituted biphenyl-diarylpyrimidine nonnucleoside HIV-1 reverse transcriptase inhibitors. Eur. J. Med. Chem.. 2020;202:e112549.
- [Google Scholar]
- Scaffold hopping in discovery of HIV-1 non-nucleoside reverse transcriptase inhibitors: from CH(CN)-DABOs to CH(CN)-dapys. Molecules. 2020;25(7):e1581.
- [Google Scholar]
- Privileged scaffold inspired design of novel oxime-biphenyl-DAPYs in treatment of HIV-1. Bioorg. Chem.. 2020;99:e103825.
- [Google Scholar]
- Pharmacophore-fusing design of pyrimidine sulfonylacetanilides as potent non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Chem.. 2020;96:e103595.
- [Google Scholar]
- Design of the naphthyl-diarylpyrimidines as potent non-nucleoside reverse transcriptase inhibitors (NNRTIs) via structure-based extension into the entrance channel. Eur. J. Med. Chem.. 2021;226:e113868.
- [Google Scholar]
- In silico design of novel HIV-1 NNRTIs based on combined modeling studies of dihydrofuro[3,4-d]pyrimidines. Front. Chem.. 2020;8:e164.
- [Google Scholar]
- Therapeutic potential of indole derivatives as anti-HIV agents. Curr. Top. Med. Chem.. 2021;22(12):993-1008.
- [Google Scholar]
- Design, synthesis, and antiviral evaluation of novel piperidine-substituted arylpyrimidines as HIV-1 NNRTIs by exploring the hydrophobic channel of NNIBP. Bioorg. Chem.. 2021;116:e105353.
- [Google Scholar]
- Non-nucleoside reverse transcriptase inhibitors, molecular docking studies and antitubercular activity of thiazolidin-4-one derivatives. Curr. Comput.-Aided Drug Des.. 2019;15(5):433-444.
- [Google Scholar]
- A review on quinoline derived scaffolds as anti-HIV agents. Mini-Rev. Med. Chem.. 2019;19(6):510-526.
- [Google Scholar]
- Discovery of second generation quinazolinone non-nucleoside reverse transcriptase inhibitors of HIV-1. Progress Med. Chem.. 2002;40:63-105.
- [Google Scholar]
- Costa, C. C. P.; Boechat, N.; Bastos, M. M.; Da Silva, F. de C.; Marttorelli, A.; Souza, T. M. L.; Baptista, M. S.; Hoelz, L. V. B.; Cafffarena, E. R. New efavirenz derivatives and 1,2,3-triazolyl-phosphonates as inhibitors of reverse transcriptase of HIV-1. Curr Top Med Chem 2018, 18(17), 1494-1505
- Design, synthesis and anti-HIV evaluation of 5-alkyl-6-(benzo[d][1,3]dioxol-5-alkyl)-2-mercaptopyrimidin-4(3H)-ones as potent HIV-1 NNRTIs. Bioorg. Chem.. 2020;102:e104041.
- [Google Scholar]
- Structure-based design and discovery of pyridyl-bearing fused bicyclic HIV-1 inhibitors: synthesis, biological characterization, and molecular modeling studies. J. Med. Chem.. 2021;64(18):13604-13621.
- [Google Scholar]
- Discovery, optimization, and target identification of novel coumarin derivatives as HIV-1 reverse transcriptase-associated ribonuclease H inhibitors. Eur. J. Med. Chem.. 2021;225:e113769.
- [Google Scholar]
- 1,2,4-Triazolo[1,5-a]pyrimidines as a novel class of inhibitors of the HIV-1 reverse transcriptase-associated ribonuclease H activity. Molecules. 2020;25:e1183.
- [Google Scholar]
- Hydrophobic pocket occupation design of difluoro-biphenyl-diarylpyrimidines as non-nucleoside HIV-1 reverse transcriptase inhibitors: from N-alkylation to methyl hopping on the pyrimidine ring. J. Med. Chem.. 2021;64(8):5067-5081.
- [Google Scholar]
- Improving druggability of novel diarylpyrimidine NNRTIs by a gragment-based replacement strategy: from biphenyl-DAPYs to heteroaromatic-biphenyl-DAPYs. J. Med. Chem.. 2021;64(14):10297-10311.
- [Google Scholar]
- Druggability modification strategies of the diarylpyrimidine-type non-nucleoside reverse transcriptase inhibitors. Med. Res. Rev.. 2021;41(3):1255-1290.
- [Google Scholar]
- A review on ‘triazoles’: their chemistry, synthesis and pharmacological potentials. J. Iran. Chem. Soc.. 2021;18(10):2535-2565.
- [Google Scholar]
- Structural investigation of 2-naphthyl phenyl ether inhibitors bound to WT and Y181C reverse transcriptase highlights key features of the NNRTI binding site. Protein Sci.. 2020;29(9):1902-1910.
- [Google Scholar]
- Synthesis, molecular docking and biological evaluation of 2-(thiophen-2-yl)-1H-indoles as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Chem.. 2020;95:e103521.
- [Google Scholar]
- Chiral indolylarylsulfone non-nucleoside reverse transcriptase inhibitors as new potent and broad-spectrum anti-HIV-1 agents. J. Med. Chem.. 2017;60(15):6528-6547.
- [Google Scholar]
- Indolylarylsulfones, a fascinating story of highly potent human immunodeficiency virus type 1 non-nucleoside reverse transcriptase inhibitors. Antiviral Chem. Chemother.. 2018;26:1-19.
- [Google Scholar]
- Boronic acid-containing diarylpyrimidine derivatives as novel HIV-1 NNRTIs: design, synthesis and biological evaluation. Chin. Chem. Lett.. 2021;32(12):4053-4057.
- [Google Scholar]
- 1,2,3-Triazole hybrids with anti-HIV-1 activity. Arch. Pharm.. 2021;354(1):e2000163.
- [Google Scholar]
- Design, synthesis, and evaluation of “dual-site”-binding diarylpyrimidines targeting both NNIBP and the NNRTI adjacent site of the HIV-1 reverse transcriptase. Eur. J. Med. Chem.. 2021;211:e113063.
- [Google Scholar]
- Bioactive prenylated coumarins as potential anti-inflammatory and anti-HIV agents from Clausena lenis. Bioorg. Chem.. 2020;97:e103699.
- [Google Scholar]
- In silico study of 3-hydroxypyrimidine-2,4-diones as inhibitors of HIV RT-associated RNase H using molecular docking, molecular dynamics, 3D-QSAR, and pharmacophore models. New J. Chem.. 2019;43(43):17004-17017.
- [Google Scholar]
- Design, synthesis and biological evaluation of 3-hydroxyquinazoline-2,4(1H,3H)-diones as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and integrase. Bioorg. Med. Chem.. 2019;27(17):3836-3845.
- [Google Scholar]
- Novel indolylarylsulfone derivatives as covalent HIV-1 reverse transcriptase inhibitors specifically targeting the drug-resistant mutant Y181C. Bioorg. Med. Chem.. 2021;30:e115927.
- [Google Scholar]
- Design, synthesis and anti-HIV evaluation of novel 5-substituted diarylpyrimidine derivatives as potent HIV-1 NNRTIs. Bioorg. Med. Chem.. 2021;40:e116195.
- [Google Scholar]
- Design, antihuman immunodefciency activity and molecular docking studies of synthesized 2-aryl and 2-pyrimidinyl pyrrolidines. Mol. Divers.. 2021;25:2045-2052.
- [Google Scholar]
- George, A.; Gopi Krishna Reddy, A.; Satyanarayana, G.; Raghavendra, N. K. 1,2,3,4-Tetrahydroisoquinolines as inhibitors of HIV-1 integrase and human LEDGF/p75 interaction. Chem Biol Drug Des 2018, 91(6), 1133-1140
- Lancet. 2018;392(10148):685-697.
- Recent progress in HIV-1 inhibitors targeting the entrance channel of HIV-1 non-nucleoside reverse transcriptase inhibitor binding pocket. Eur. J. Med. Chem.. 2019;174:277-291.
- [Google Scholar]
- Recent discoveries in HIV-1 reverse transcriptase inhibitors. Curr. Opin. Pharmacol.. 2020;54:166-172.
- [Google Scholar]
- Exploiting the tolerant region i of the non-nucleoside reverse transcriptase inhibitor (NNRTI) binding pocket: Discovery of potent diarylpyrimidine-typed HIV-1 NNRTIs against wild-type and E138K mutant virus with significantly improved water solubility and favorable safety profiles. J. Med. Chem.. 2019;62(4):2083-2098.
- [Google Scholar]
- Design, synthesis, and biological evaluation of piperidinyl-substituted [1,2,4]triazolo[1,5-a]pyrimidine derivatives as potential anti-HIV-1 agents with reduced cytotoxicity. Chem. Biol. Drug Des.. 2021;97(1):67-76.
- [Google Scholar]
- Discovery of novel diarylpyrimidine derivatives as potent HIV-1 NNRTIs targeting the “nNRTI adjacent” binding site. ACS Med. Chem. Lett.. 2018;9(4):334-338.
- [Google Scholar]
- Covalent inhibition of wild-type HIV-1 reverse transcriptase using a fluorosulfate warhead. ACS Med. Chem. Lett.. 2021;12(2):249-255.
- [Google Scholar]
- Exploiting the tolerant region I of the non-nucleoside reverse transcriptase inhibitor (NNRTI) binding pocket. Part 2: Discovery of diarylpyrimidine derivatives as potent HIV-1 NNRTIs with high Fsp3 values and favorable drug-like properties. Eur. J. Med. Chem.. 2021;213:e113051.
- [Google Scholar]
- Design and synthesis of a novel series of non-nucleoside HIV-1 inhibitors bearing pyrimidine and N-substituted aromatic piperazine. Bioorg. Med. Chem. Lett.. 2018;28(22):3491-3495.
- [Google Scholar]
- Synthesis and biological evaluation of dihydroquinazoline-2-amines as potent non-nucleoside reverse transcriptase inhibitors of wild-type and mutant HIV-1 strains. Eur. J. Med. Chem.. 2019;176:11-20.
- [Google Scholar]
- Discovery of biphenyl-substituted diarylpyrimidines as non-nucleoside reverse transcriptase inhibitors with high potency against wild-type and mutant HIV-1. Eur. J. Med. Chem.. 2018;145:726-734.
- [Google Scholar]
- Improving the positional adaptability: structure-based design of biphenyl-substituted diaryltriazines as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Acta Pharm. Sin. B. 2020;10(2):344-357.
- [Google Scholar]
- Advances in HIV diagnosis and monitoring. Crit. Rev. Biotechnol.. 2020;40(5):623-638.
- [Google Scholar]
- Discovery of potent HIV-1 non-nucleoside reverse transcriptase inhibitors by exploring the structure-activity relationship of solvent-exposed regions I. Chem. Biol. Drug Des.. 2019;93(4):430-437.
- [Google Scholar]
- Discovery of piperidine-substituted thiazolo[5,4-d]pyrimidine derivatives as potent and orally bioavailable HIV-1 non-nucleoside reverse transcriptase inhibitors. Commun. Chem.. 2019;2(1):e74.
- [Google Scholar]
- Identification of dihydrofuro[3,4-d]pyrimidine derivatives as novel HIV-1 non-nucleoside reverse transcriptase inhibitors with promising antiviral activities and desirable physicochemical properties. J. Med. Chem.. 2019;62(3):1484-1501.
- [Google Scholar]
- In situ click chemistry-based rapid discovery of novel HIV-1 NNRTIs by exploiting the hydrophobic channel and tolerant regions of NNIBP. Eur. J. Med. Chem.. 2020;193:e112237.
- [Google Scholar]
- Structure-activity relationship exploration of NNIBP tolerant region I leads to potent HIV-1 NNRTIs. ACS Infect. Dis.. 2020;6(8):2225-2234.
- [Google Scholar]
- Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm. Sin. B. 2020;10(5):878-894.
- [Google Scholar]
- Discovery and characterization of fluorine-substituted diarylpyrimidine derivatives as novel HIV-1 NNRTIs with highly improved resistance profiles and low activity for the hERG ion channel. J. Med. Chem.. 2020;63(3):1298-1312.
- [Google Scholar]
- Structure-based bioisosterism yields HIV-1 NNRTIs with improved drug-resistance profiles and favorable pharmacokinetic properties. J. Med. Chem.. 2020;63(9):4837-4848.
- [Google Scholar]
- Design and synthesis of novel 1,2,3-triazolyl-pyrimidinone hybrids as potential anti-HIV-1 NNRT inhibitors. J. Heterocyclic Chem.. 2018;55(4):821-829.
- [Google Scholar]
- Design, synthesis and molecular docking of pyrazolo[3,4d]thiazole hybrids as potential anti-HIV-1 NNRT inhibitors. Bioorg. Chem.. 2019;86:437-444.
- [Google Scholar]
- Synthetic and medicinal perspective of quinolines as antiviral agents. Eur. J. Med. Chem.. 2021;215:e113220.
- [Google Scholar]
- Synthesis and in vitro anti-HIV-1 evaluation of novel pyrazolo[4,3-c]pyridine-4-one derivatives. Eur. J. Med. Chem.. 2019;183:e111714.
- [Google Scholar]
- Nitrogen-containing heterocycles as anticancer agents: an overview. Anti-Cancer Agents Med. Chem.. 2020;20(18):2150-2168.
- [Google Scholar]
- Pyridin-2(1H)-ones as HIV-1 NNRTIs: a combinatorial optimization strategy. J. Chin. Pharm. Sci.. 2020;29(2):79-89.
- [Google Scholar]
- Structure-based linker optimization of 6-(2-cyclohexyl-1-alkyl)-2-(2-oxo-2-phenylethylsulfanyl)pyrimidin-4(3H)-ones as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Chin. Chem. Lett.. 2021;32(3):1020-1024.
- [Google Scholar]
- The expanding role of pyridine and dihydropyridine scaffolds in drug design. Drug Des. Dev. Ther.. 2021;15:4289-4338.
- [Google Scholar]
- Design, synthesis and biological evaluation of substituted (+)-SG-1 derivatives as novel anti-HIV agents. Bioorg. Med. Chem. Lett.. 2018;28(10):1699-1703.
- [Google Scholar]
- Design, synthesis and anti-HIV evaluation of novel diarylpyridine derivatives as potent HIV-1 NNRTIs. Eur. J. Med. Chem.. 2017;140:383-391.
- [Google Scholar]
- The discovery of novel diarylpyri(mi)dine derivatives with high level activity against a wide variety of HIV-1 strains as well as against HIV-2. Bioorg. Med. Chem.. 2018;26(8):2051-2060.
- [Google Scholar]
- Molecular docking studies and synthesis of amino-oxy-diarylquinoline derivatives as potent non-nucleoside HIV-1 reverse transcriptase inhibitors. Drug Res.. 2019;69(12):671-682.
- [Google Scholar]
- Structural optimization of N1-aryl-benzimidazoles for the discovery of new non-nucleoside reverse transcriptase inhibitors active against wild-type and mutant HIV-1 strains. Bioorg. Med. Chem.. 2018;26(3):661-674.
- [Google Scholar]
- 3-Hydroxypyrimidine-2,4-dione derivatives as HIV reverse transcriptase-associated RNASE h inhibitors: QSAR analysis and molecular docking studies. Iran. J. Pharm. Res.. 2020;19(1):84-97.
- [Google Scholar]
- The discovery and development of oxalamide and pyrrole small molecule inhibitors of gp120 and HIV entry-a review. Curr. Top. Med. Chem.. 2019;19(18):1650-1675.
- [Google Scholar]
- New indolylarylsulfone non-nucleoside reverse transcriptase inhibitors show low nanomolar inhibition of single and double HIV-1 mutant strains. Eur. J. Med. Chem.. 2020;208:e112696.
- [Google Scholar]
- The Journey of HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) from lab to clinic. J. Med. Chem.. 2019;62(10):4851-4883.
- [Google Scholar]
- Biological evaluation of molecules of the azaBINOL class as antiviral agents: Inhibition of HIV-1 RNase H activity by 7-isopropoxy-8-(naphth-1-yl)quinolone. Bioorg. Med. Chem.. 2019;27(16):3595-3604.
- [Google Scholar]
- Multiple-receptor conformation docking, dock pose clustering, and 3D QSAR-driven approaches exploring new HIV-1 RT inhibitors. Struct. Chem.. 2018;29(4):999-1012.
- [Google Scholar]
- Novel thiazolidin-4-ones as potential non-nucleoside inhibitors of HIV-1 reverse transcriptase. Molecules. 2019;24(21):e3821.
- [Google Scholar]
- Synthesis and anticancer properties of ‘Azole’ based chemotherapeutics as emerging chemical moieties: a comprehensive review. Curr. Org. Chem.. 2021;25(6):654-668.
- [Google Scholar]
- Aryl substituted benzimidazolones as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. ACS Med. Chem. Lett.. 2019;10(2):196-202.
- [Google Scholar]
- Emerging reverse transcriptase inhibitors for HIV-1 infection. Expert Opin. Emerg. Drugs. 2018;23(2):149-157.
- [Google Scholar]
- Bis-coumarin derivatives and their biological activities. Curr. Top Med. Chem.. 2018;18(2):101-113.
- [Google Scholar]
- Pyrimidine 2,4-diones in the design of new HIV RT inhibitors. Molecules. 2019;24(9):e1718.
- [Google Scholar]
- A review on therapeutic potential of heterocyclic pyrimidine derivatives as potent antiviral agents. Asian J. Pharm. Clin. Res.. 2020;13(7):30-34.
- [Google Scholar]
- Conformational restriction design of thiophene-biphenyl-DAPY HIV-1 non-nucleoside reverse transcriptase inhibitors. Eur. J. Med. Chem.. 2019;182:e111603.
- [Google Scholar]
- Follow on-based optimization of the biphenyl-DAPYs as HIV-1 nonnucleoside reverse transcriptase inhibitors against the wild-type and mutant strains. Bioorg. Chem.. 2019;89:e102974.
- [Google Scholar]
- Anti-HIV flavonoids from natural products: a systematic review. Int. J. Res. Pharm. Sci.. 2015;6(3):248-255.
- [Google Scholar]
- Recent advances in the design and development of non-nucleoside reverse transcriptase inhibitor Scaffolds. ChemMedChem.. 2019;14(1):52-77.
- [Google Scholar]
- Synthesis, anti-HIV-1 and antiproliferative evaluation of novel 4-nitroimidazole derivatives combined with 5-hydroxy-4-pyridinone moiety. J. Mol. Struct.. 2020;1202:e127344.
- [Google Scholar]
- Recent progress in biological activities of indole and indole alkaloids. Mini-Rev. Med. Chem.. 2018;18(1):9-25.
- [Google Scholar]
- Anti-HIV potential of diarylpyrimidine derivatives as non-nucleoside reverse transcriptase inhibitors: design, synthesis, docking, TOPKAT analysis and molecular dynamics simulations. J. Biomol. Struct. Dyn.. 2021;39(7):2430-2446.
- [Google Scholar]
- Structural and kinetic insights into HIV-1 reverse transcriptase inhibition by farnesiferol C. Int. J. Biol. Macromol.. 2021;174:309-318.
- [Google Scholar]
- Structure-based non-nucleoside inhibitor design: Developing inhibitors that are effective against resistant mutants. Chem. Biol. Drug Des.. 2021;97(1):4-17.
- [Google Scholar]
- Recent advances in the discovery and development of novel HIV-1 NNRTI platforms (Part II): 2009–2013 update. Curr. Med. Chem.. 2014;21(3):329-355.
- [Google Scholar]
- 5-Hydroxypyrido[2,3-b]pyrazin-6(5H)-one derivatives as novel dual inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H and integrase. Eur. J. Med. Chem.. 2018;155:714-724.
- [Google Scholar]
- Identification of novel potent HIV-1 inhibitors by exploiting the tolerant regions of the NNRTIs binding pocket. Eur. J. Med. Chem.. 2021;214:e113204.
- [Google Scholar]
- T occo, G.; Esposito, F.; Caboni, P.; Laus, A.; Beutler, J. A.; Wilson, J. A.; Corona, A.; Le Grice, S. F. J.; Tramontano, E. Scaffold hopping and optimisation of 3’,4’-dihydroxyphenyl- containing thienopyrimidinones: Synthesis of quinazolinone derivatives as novel allosteric inhibitors of HIV-1 reverse transcriptase-associated ribonuclease H. J Enzym Inhib Med Chem 2020, 35(1), 1953-1963
- Pharmacophore-based design of novel 3-hydroxypyrimidine-2,4-dione subtypes as inhibitors of HIV reverse transcriptase-associated RNase H: Tolerance of a nonflexible linker. Eur. J. Med. Chem.. 2019;166:390-399.
- [Google Scholar]
- An insight into the medicinal perspective of synthetic analogs of indole: a review. Eur. J. Med. Chem.. 2019;180:562-612.
- [Google Scholar]
- Targeting the entrance channel of NNIBP: Discovery of diarylnicotinamide 1,4-disubstituted 1,2,3-triazoles as novel HIV-1 NNRTIs with high potency against wild-type and E138K mutant virus. Eur. J. Med. Chem.. 2018;151:339-350.
- [Google Scholar]
- Protease inhibitors for the treatment of HIV/AIDS: recent advances and future challenges. Curr. Top. Med. Chem.. 2019;19(18):1571-1598.
- [Google Scholar]
- Design, synthesis, and antiviral evaluation of novel hydrazone-substituted thiophene[3,2-d]pyrimidine derivatives as potent human immunodeficiency virus-1 inhibitors. Chem. Biol. Drug Des.. 2018;92(6):2009-2021.
- [Google Scholar]
- Targeting dual tolerant regions of binding pocket: Discovery of novel morpholine-substituted diarylpyrimidines as potent HIV-1 NNRTIs with significantly improved water solubility. Eur. J. Med. Chem.. 2020;206:e112811.
- [Google Scholar]
- Synthesis of novel sugar or azasugar modified anthra[1,2-d] imidazole-6,11-dione derivatives and biological evaluation. Carbohydr. Res.. 2018;460:29-33.
- [Google Scholar]
- 6-Biphenylmethyl-3-hydroxypyrimidine-2,4-diones potently and selectively inhibited HIV reverse transcriptase-associated RNase H. Eur. J. Med. Chem.. 2018;156:680-691.
- [Google Scholar]
- 6-Arylthio-3-hydroxypyrimidine-2,4-diones potently inhibited HIV reverse transcriptase-associated RNase H with antiviral activity. Eur. J. Med. Chem.. 2018;156:652-665.
- [Google Scholar]
- New strategy for identifying potential natural HIV-1 non-nucleoside reverse transcriptase inhibitors against drug-resistance: an in silico study. J. Biomol. Struct. Dyn.. 2020;38(11):3327-3341.
- [Google Scholar]
- Discovery of novel dihydrothiopyrano[4,3-d]pyrimidine derivatives as potent HIV-1 NNRTIs with significantly reduced hERG inhibitory activity and improved resistance profiles. J. Med. Chem.. 2021;64(18):13658-13675.
- [Google Scholar]
- Synthesis and biological evaluation of a series of 2-(((5-akly/aryl-1H-pyrazol-3-yl)methyl)thio)-5-alkyl-6-(cyclohexylmethyl)-pyrimidin-4(3H)-ones as potential HIV-1 inhibitors. Acta Pharm. Sin. B. 2020;10(3):512-528.
- [Google Scholar]
- Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr. Opin. Struct. Biol.. 2020;61:113-123.
- [Google Scholar]
- Indazolyl-substituted piperidin-4-yl-aminopyrimidines as HIV-1 NNRTIs: Design, synthesis and biological activities. Eur. J. Med. Chem.. 2020;186:e111864.
- [Google Scholar]
- Coumarin-based derivatives with potential anti-HIV activity. Fitoterapia. 2021;150:e104863.
- [Google Scholar]
- Indolylarylsulfones bearing phenylboronic acid and phenylboronate ester functionalities as potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem.. 2022;53:e116531.
- [Google Scholar]
- Discovery, synthesis, and optimization of an N-alkoxy indolylacetamide against HIV-1 carrying NNRTI-resistant mutations from the Isatis indigotica root. Eur. J. Med. Chem.. 2020;189:e112071.
- [Google Scholar]
- Synthesis of pentacyclic iminosugars with constrained butterfly-like conformation and their HIV-RT inhibitory activity. Bioorg. Med. Chem. Lett.. 2018;28(3):425-428.
- [Google Scholar]
- Yang, Y.; Kang, D.; Nguyen, L. A.; Smithline, Z. B.; Pannecouque, C.; Zhan, P.; Liu, X.; Steitz, T. A. Structural basis for potent and broad inhibition of HIV-1 RT by thiophene[3,2-d] pyrimidine non-nucleoside inhibitors. eLife 2018, 7, E36340.
- Design, synthesis and activity evaluation of novel pyridinone derivatives as anti-HIV-1 dual (RT/IN) inhibitors. J. Chin. Pharm. Sci.. 2017;26(1):31-44.
- [Google Scholar]
- SJP-L-5 inhibits HIV-1 polypurine tract primed plus-strand DNA elongation, indicating viral DNA synthesis initiation at multiple sites under drug pressure. Sci. Rep.. 2018;8(1):e2574.
- [Google Scholar]
- Discovery of novel indolylarylsulfones as potent HIV-1 NNRTIs via structure-guided scaffold morphing. Eur. J. Med. Chem.. 2019;182:e111619.
- [Google Scholar]
- Targeting the hydrophobic channel of NNIBP: Discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus. Org. Biomol. Chem.. 2019;17(12):3202-3217.
- [Google Scholar]
- Rational design and structure-activity relationship of coumarin derivatives effective on HIV-1 protease and partially on HIV-1 reverse transcriptase. Eur. J. Med. Chem.. 2020;186:e111900.
- [Google Scholar]




