

Translate this page into:
Current status and future perspectives of efficient catalytic conversion of bioethanol to value-added chemicals and fuels
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University. Production and hosting by Elsevier.
Abstract
This review offers a detailed analysis of how catalytic bioethanol conversion has the potential to greatly contribute to the advancement of sustainable energy and resource utilisation. This resource fills a significant gap in current literature by bringing together and evaluating scattered information on this subject. It serves as a comprehensive reference for ongoing research, identifies emerging trends, and highlights potential areas for future research. The review discusses different aspects of converting bioethanol, including its natural characteristics and benefits. It also explores the complex reaction mechanisms involved in catalytic transformation and the wide variety of valuable chemicals that can be produced, such as olefins, aldehydes, ketones, alcohols, and lubricants. Furthermore, it explores the process of converting bioethanol into renewable fuels, such as hydrogen, as well as hydrocarbon fuels like petrol, diesel, and jet fuels. The review examines the techno-economic aspects of process modelling, cost estimation methods, and economic evaluations to determine the feasibility and commercial potential of these conversion processes. As we face the challenges and consider the future of this changing field, this review emphasizes the importance of catalytic bioethanol conversion in creating a sustainable and diverse energy and chemical landscape.
Keywords
Catalysts
Bioethanol, Value added fuel and Chemicals
Techno-economic Analysis
Waste Management
Feedstock

- SR
-
Steam reforming
- ESR
-
Ethanol steam reforming
- WGS
-
Water gas shift
- TPD
-
Temperature programmed desorption
- AC
-
Activated carbon
- CNT
-
Carbon nanotube
- CNF
-
Carbon nanofibre
- ETE
-
Ethanol-to-ethylene
- DEE
-
Diethyl ether
- ETP
-
Ethanol-to-propylene
- MPV
-
Meerwein-Ponndorf-Verley
- BD
-
1,3-Butadiene
- TOS
-
Time-on-stream
- ETG
-
Ethanol-to-gasoline
- MTG
-
Methanol-to-gasoline
- BTX
-
Benzene toluene xylene
- HAP
-
Hydroxyapatite
- CHAP
-
Carbonate hydroxyapatite
- ETO
-
Ethanol to light olefins
- MPV
-
Methylcyclopentadiene dimerization
- SRE
-
Steam Reforming of Ethanol
- ETH
-
Ethanol to higher hydrocarbons
- CZA
-
Cu/ZnO/Al2O3
Abbreviations
1 Introduction
Liquid fuel can be made in a variety of ways by applying basic and practical procedures that are now widely accessible in the commercial market. Designing commercialization strategies that are both financially viable and promising requires attempting to use currently available methodologies. This is true even though producing liquid fuel often costs more than extracting fossil fuels directly. Instead, than emphasizing the technology, the focus is more on finding low-cost solutions than on the mechanism. In order to achieve large-scale production in huge quantities, it is vital to use profitable and viable procedures rather than strategies that are only marginally profitable or technically feasible. The widespread usage of fossil fuels brought about by the world's growing energy demand resulted in environmental problems such as air pollution, global warming, and climate change (Li et al., 2019), (Wu et al., 2018). As a result, scientists are paying close attention to the development of sustainable and renewable energy sources. The environmental benefits and sustainability of biofuels, such biodiesel and bioethanol, made from biomass valorization, make them attractive substitutes for fossil fuels (Gouda et al., 2022), (Cirujano and Dhakshinamoorthy, 2021a), (Cirujano and Dhakshinamoorthy, 2021b), (Dhakshinamoorthy et al., 2023), Takei et al., 2011]. The goals of US and EU biofuel policies are to lower greenhouse gas emissions, increase the biofuels' life cycle energy efficiency, and support domestic biofuel production, which includes the manufacture of bioethanol, biobutanol, and biodiesel. The development and consumption of biofuels are also highly promoted in Asia. For instance, the Chinese government declared in 2017 that 10 % ethanol must be blended into all gasoline by 2020 (“Biofuel China,” 2020). Considerable progress has been made to increase the capacity for producing bioethanol in order to reach the goal. The strong biofuel policies of the Association of Southeast Asian Nations (ASEAN) emerge from the fact that domestic biofuel usage ensures energy security and fosters socio-economic development by guaranteeing the demand for strategically important agricultural commodities (Ying et al., 2020).
Numerous investigations have indicated the considerable potential of the catalytic process as a workable strategy for turning low-alcohol beverages into eco-friendly substitutes. Catalytic conversion of bioethanol, or its transformation into the aforementioned compounds and fuels, is often made possible by catalysts. Zeolites, metal oxides, and supported metal catalysts are often employed as catalysts for the conversions (Sun and Wang, 2014a), (Abdulrazzaq and Schwartz, 2019a). The kind of catalyst used depends on the intended product since different products made from bioethanol require different chemistry to be produced. In the last ten years, a lot of research has been done to create technology for converting ethanol into other valuable compounds. Reviews also particularly address the conversion of bioethanol. As an illustration, the catalytic conversion of ethanol to butanol and bioethanol to butanol has been reviewed (Wu et al., 2018), (Aitchison et al., 2016), (Galadima and Muraza, 2015). The effect of bioethanol impurities on steam reforming for hydrogen production was studied by Sanchez et al. (2020). Nevertheless, the majority of those evaluations on the catalytic conversion of bioethanol concentrate on a single product. Our goal in this article is to present a thorough analysis of the catalytic conversion of bioethanol to a range of compounds and fuels, such as gasoline, hydrogen, and C2-C4 olefins. We pay particular attention to the connections that exist between the catalyst, the reaction process, and the stability and catalytic activity. Nowadays, activated Al2O3-based catalysts are the most widely used ones in industrial facilities. Furthermore, certain by-products are no longer acceptable for use in the synthesis of other products once the reaction temperature reaches a high enough level. At lower temperatures, zeolites can be used as a catalyst for the production of bioethanol. Zeolite catalysts can completely utilize the commodities value of low-concentration ethanol, such as the leftover liquid recovered following bio-fermentation. Regarding ethylene selectivity and catalyst stability, zeolite acidity significantly affects catalytic performance (MOSER, 1989). Strong acidic sites at high concentration may facilitate ethylene secondary reactions, such as oligmerization, cracking, and coking, which result in the formation of long-chain hydrocarbons and even carbonaceous deposits on the catalyst (Chaudhuri et al., 1990). At temperatures below 300 °C, zeolites with a consistent pore shape and modifiable acidity prove to be effective catalysts for enzymatic thermolysis. To get high activity, zeolites' pore size (by steaming treatment) and acidity (by adjusting the Si/Al ratio and doping modifiers) may be readily adjusted. Subsequent investigations will mainly focus on gaining a comprehensive comprehension of the activated alumina catalyst, which is presently in widespread use. It will also entail the creation of a rational design and suggested adjustments to lower process temperature and enhance product dispersion. Moreover, it is essential to focus on improving the durability of the zeolite catalyst under varying process conditions. Mesoporous zeolites were designed to enhance mass transfer during reaction since large molecules can readily obstruct the zeolite's micropores, resulting in catalyst deactivation. This effort aims to expand its practical use in the commercial sphere, facilitate the development of an efficient low-alcohol conversion system, and boost the overall competitiveness of the process within the market. The finding of the starting effect may represent a positive development for the technique and a workable way to address the coking issue. There is an urgent need for more innovative studies and research that employ a range of catalytic systems. On the other hand, it is imperative to take into account and resolve issues pertaining to the impact of the catalyst generation process and the design of effective catalysts. Catalyst deactivation and a decline in catalytic effectiveness occur when various substances accumulate on the catalyst's surface during catalytic processes, as they block the active sites and pores of the catalyst. The sintering process is the most important stage in the catalyst deactivation process (Bartholomew, 2001). Catalyst recovery is one of the primary challenges in asymmetric homogeneous catalysis. Recovering the catalyst via filtration, recycling, or using it in conjunction with a continuous liquid flow reduces its activity throughout the leaching process (Cole-Hamilton, 2003).
2 Bioethanol characteristics and advantages
Bioethanol is a form of single-chain alcohol characterized by a two-carbon structure, typically obtained from renewable resources like sugarcane, cereal grains, and sugar beetroot. It is liquid in normal environmental conditions after two stages of yeast-fermentation of carbohydrates and dehydration (Araújo, 2016a; Puricelli et al., 2021). The classification is made according on the kind of feedstock that is used. First, starch and saccharine feedstock, such as sugarcane, maize, and beetroot, are generated. 2. The term “generation” refers to lignocellulosic biomass. 3. Generation is utilized to calculate the biomass of both macro- and microalgae. 4. Generation refers to cyanobacteria that have been genetically modified to directly make ethanol from light and CO2 (de Souza Abud and de Farias Silva, 2019). Bioethanol production is currently capped at 1 G, but new techniques using various feedstocks and technologies may allow for greater production of this sustainable fuel while reducing waste—all without further impacting land use, food prices, or availability (Cherubini, 2010). According to a study (Araújo, 2016b), this alcohol is primarily used as biofuel. It may be utilized at lower mix percentages in normal spark-ignition engines that have not had considerable changes to replace gasoline, or at higher blend percentages (up to 15 % in Brazil) in engines that have undergone minimal modifications (flex-fuel cars). Furthermore, because bioethanol is easily biodegradable, it has a minimal environmental impact. It is a fantastic fuel additive that increases the performance and economy of gasoline engines while reducing emissions due to its high octane rating. The main fuel attribute used to characterize a gasoline's resistance to “knocking” in a spark-ignited engine is its octane rating. Research engines are used to determine octane ratings, which measure a fuel's resistance to knocking relative to key reference fuels. When ethanol is blended into gasoline, the result is typically a higher octane rating rise than what the volume-weighted average suggests, particularly for low- and mid-level blends (up to E30) (Anderson et al., 2012). The idea of a volumetric “blending” octane number (bONvol,e) has been applied in order to explain the octane blending behavior of ethanol–gasoline mixtures. When ethanol–diesel blends and regular diesel are compared for engine performance in engines that have not been upgraded, power decreases are typically observed that roughly correspond to the blends' lower energy content when compared to diesel fuel. When Meiring et al. (1983) evaluated a 30 % ethanol–diesel blend in a tractor engine equipped with a rotary distributor pump, they found a 5 % decrease in the maximum fuel delivery. Different kinds of hydrocarbons or additives are frequently used in blended fuels to accomplish certain performance goals. The standard octane rating for regular unleaded gasoline is between 87 and 89. Most traditional engines that don't require great performance may use it. The octane range for mid-grade gasoline is 91 to 93. It works well with some higher-performance engines and offers more knock resistance than ordinary unleaded. The standard octane rating for premium unleaded gasoline is 91 or higher. It is intended for use with engines that are more powerful or have greater compression ratios. In certain vehicles, premium fuels can reduce engine knocking and increase overall engine efficiency (Graham et al., 2008). E85 is a mixture of 85 % ethanol and 15 % gasoline. Although it might vary, E85 typically has an octane value between 100 and 105. Despite having a higher octane rating than gasoline, E85 may have less fuel economy because of its lower energy content. To raise the octane rating of ordinary gasoline, there are a few additives and octane boosters available. To stop knocking, these materials are frequently used in high-performance or modified engines. It is noteworthy that a fuel's overall quality or efficiency cannot be determined only by its octane rating.
Nonetheless, it is also utilized by the petrochemical, nutraceutical, and cosmetic industries (Bórawski et al., 2019). Because of advancements in the previously indicated production capacities, bioethanol has the potential to displace even more fossil fuels in chemical and transportation processes. If ethanol was utilized as a platform molecule, this percentage might rise even further (Gallo et al., 2014). While other fuels are currently used, these systems must take into account in its corrosivity. Other advantages that haven't been discussed yet include safety, ease of handling, storage, and transportation, as well as the availability of the infrastructure needed (Sakthivel et al., 2018; Yusoff et al., 2015). As transportation electrification becomes more widespread, the chemical industry could potentially integrate its production by utilizing bio refineries. This shift would enhance environmental sustainability by reducing nonrenewable carbon emissions. In the realm of energy, bioethanol stands as the most extensively manufactured biofuel worldwide, comprising 61 % of the total production in 2020 (REN21 Renewables Report, 2018). Its production is highly specialized, and its number is 978–3-948393–03-8 (Paris: REN21 Secretariat). As a matter of fact, in 2019 the United States (54 %) and Brazil (30 %) produced over 84 % of the world's fuel ethanol (“Ethanol exports and trade (2020),” n.d.). Concerning the production and use of bioethanol, the European Union (5 %), China (3 %), India (2 %), Canada (2 %), and other producing nations have interests and policies in place.
Over the last 20 years, bioethanol has been considered the primary candidate in substituting for a fraction of gasoline (Megawati et al., 2011). The history of first-generation bioethanol production is very ancient. As of 2017, the United States has more than 200 first generation ethanol production facilities (Lennartsson et al., 2014). The plants have an average production capacity of approximately 260,000 m3 /year and produce ethanol mainly from corn or sorghum. Table 1 illustrates brief information on commercial-scale bioethanol plants currently in operation. With a 500,000-ton annual production capacity, the Henan Tianguan Alcohol Chemical Group is the world's largest producer of first-generation bioethanol, mostly from maize. A 227 million liters-per-year corn-based ethanol manufacturing plant was inaugurated in Mato Grosso, Brazil, in August 2017 (REN21 Renewables Report, 2018). A joint venture between the world's largest steel manufacturer, Shougang Group, and carbon recycling firm LanzaTech has just constructed the world's first commercial facility for the sustainable ethanol production from flue gas from the steel sector. The factory can produce 46,000 tons annually (see Table 2). ETE: ethanol to ethylene; ETP: ethanol to propylene; ETA; ethanol to acetaldehyde, ETnB; ethanol to n-butane, ETIB; ethanol to iso-butane. W/F: catalyst weight/flowrate; Mc: catalyst weight, X, S, Y; conversion, selectivity, and yield, respectively.
Company
Startup Year
Region
Capacity
(ton/year)Feed material
Technology
Gevo
2006
Luverne, United States
54,000
Forest residues
Fermentation
Longlive Biotechnology Co. Ltd.
2012
Yucheng, China
60,000
Corn cob
Fermentation
Beijing Shougang LanzaTech New Energy Technology Co., Ltd.
2018
Caofeidian, China
46,000
Waste gases
Fermentation
Cane Technology Center
2012
Piracicaba, Brazil
2,400
Bagasse
Fermentation
Enerkem Alberta Biofuels LP
2014
Edmonton, Canada
30,000
Postsorted municipal solid waste
Gasification
Henan Tianguan Group
2011
Nanyang, China
30,000
Lignocellulosics
Fermentation
GranBio
2014
Sao Miguel, Brazil
65,000
Sugarcane bagasse and straw
Fermentation
Quad-County Corn Processors
2014
Galva, United States
6,000
Corn kernel fiber
Fermentation
Henan Tianguan Group
2009
Zhenping, China
10,000
Wheat/corn stover
Fermentation
Raizen Energia
2015
Costa Pinto, Brazil
31,600
Bagasse
Fermentation
POET-DSM Advanced Biofuels
2014
Emmetsburg, United States
75,000
Agricultural residues
Fermentation
Sr no.
Catalyst
Process
Synthesis method
Process conditions
Results
product
Key findings
Ref
1
Zr/HZSM-5
ETE
Impregnation
T = 450 °C
P = 1 bar
Mc = 0.2 gX = 100
S = 61
Y = 61C3+ olefins, Propylene
The incorporation of Zr into H-ZSM-5 zeolite enhances in the initial production of C3+ olefins.
(Inaba et al., 2012)
2
Ni/ZSM-5
Ethylene oligomerization.
Hydrothermal
T = 250–450 °C,
P = 1.5–20 bar,
MC = 0.5 g catalystX = 92 %
C5+
Ethylene conversion rates are 9 % at 250 °C and 20 bar; and 92 % at 450 °C and 1.5 bar, respectively.
(Jin et al., 2020)
3
Ni-MCM-41/ZSM-5
Ethylene oligomerization.
Hydrothermal
T = 250–450 °C.
P = 1.5–20 bar,
MC = 0.5 g catalystX = 63 %
C5+
Ethylene conversion rates are 19 % at 250 °C and 20 bar; and 63 % at 450 °C and 1.5 bar, respectively.
(Jin et al., 2020)
4
HZSM-5
Ethanol to ethylene
Methanol to hydrocarbon.
W/F = 0.01–0.4 g.min ML-1
Y = 100 %
C5+
To generate C5+ hydrocarbons, a process temperature exceeding 300 °C is necessary.
(Gayubo et al., 2001b)
5
H2O2-ZSM-5
ETP
Impregnation method
T = 280 °C.
P = 40 bar,
WHSV = 4.0 h−1
X = 97 %
S = 93 %Properlyene
Compared with the parent ZSM-5, the Fenton-ZSM-5 catalyst exhibited the best activity and stability for propene oligomerization reaction because of acid sites.
(ZI et al., 2020)
6
Zr-ZSM-5
ETP
–
T = 260 °C.
P = 40 bar,
WHSV = 1 h−1
X = 80 %
S = 61.28 %
Y = 87.65 %Properlyene
Partial incorporation of Zr into the zeolite yielded a catalyst with moderate acidity and medium pore size having better catalytic properties of Zr-ZSM-5
(Li and Jiang, 2013)
7
Ni-ZSM-5/MCM-41
Ethylene oligomerization.
Hydrothermal
T = 250–450 °C.
P = 1.5–20 bar,
MC = 0.5 g catalystX = 29 %
C5+
30 & 29 % ethylene conversion at 250 °C & 20 bar, & 450 °C & 1.5 bar respectively
(Jin et al., 2020)
8
Rh/CeO2
ESR
Precipitation
T = 400 °C
Reactor = fixed bed
P = 1 bar
–
Hydrogen and C1 products (CO, CO2, CH4)
The strong contact between Rh and ceria support effectively suppressed Rh particle sintering (stable at about 2 nm) and coke production to ensure catalyst stability.
(Hou et al., 2015)
9
0.02AgCeO2 + t-ZrO2(2:1)
ETP
ETEPrecipitation
calcinationT = 400 °C.
P = 1 bar, TOS = 17 h
WHSV = 0.19X = 72 %
Spropene = 50 %
Sethene = 15 %
Y = 40 %Propene Ethene
High selectivity toward propene is obtained in a narrow temperature range
(R. V. Matheus et al., 2018b)
10
WO3-ZrO2
ETE
Impregnation
T = 500 °C.
P = 1 bar,
X = 100 %
S = 100 %
Y = 100 %Acetaldehyde
Incorporating WO3 into ZrO2 results in the formation of robust Brønsted acid sites and effectively inhibiting the formation of byproducts.
(Phung et al., 2015)
11
MoO3-ZrO2
ETE
Impregnation
T = 300 °C
X = 98 %
S = 69 %
Y = 100 %Ethene
Enhancing the percentage loading (up to 15 wt%) of MoO3 led to an improvement in surface area and its further addition will decrease the N2-SBET.
(El-Sharkawy et al., 2007)
12
Y(20)/CeO2
ETP
ETE–
T = 693–703 K.
P = 1 bar, TOS = 56 h
X = 100
Spropene = 30 %
Sethene = 37 %
Y = 40 %Propene Ethene
Introducing water into the reaction system boosted the propene yield to 30 % while reducing the ethene yield to 37 %.
(Hayashi and Iwamoto, 2013)
13
Sr-ZrO2
ETE
Coprecipitation method
T = 450 °C.
P = 11 bar, TOS = 28 h
Mc = 0.7 g
WHSV = 0.05 g min. M L -1
X = 99 %
S = 43 %
Y = 42 %
Ethane
Incorporating alkaline earth metals led to an increase in ethylene synthesis, attributed to the heightened presence of strong acid sites.
(Xia et al., 2018)
14
La-ZrO2
ETP
Coprecipitation method
T = 450 °C.
P = 11 bar, TOS = 28 h
Mc = 0.7 g
WHSV = 0.05 g min. M L -1
X = 94 %
S = 45 %
Y = 42 %
Propene
The significant enhancement in process efficiency from La/ZrO2 catalysts can be attributed to the coordination of redox and acid-base characteristics.
(Xia et al., 2020)
15
ZrO2
ETP
Precipitation method
T = 450 °C.
P = 11.1 bar,
Mc = 0.7 g
WHSV = 0.05 g min. M L -1
X = 98 %
S = 36 %
Y = 35 %
Propene
ZrO2 enhanced the efficiency of ETP conversion.
(Xia et al., 2017)
16
2Ag-4ZrO2/SiO2
ETB
Precipitation
T = 320 °C.
P = 1 bar,X = 55.2 %
S = 71 %
Butadiene
Tetrahedral Zr atom sites isolated within the crystalline structure of zeolite BEA were found to be the most efficient.
(Sushkevich et al., 2014)
17
0.02AgCeO2
–
Precipitation and co-precipitation methods
T = 400 °C.
P = 1 bar,
TOS = 14 hX = 30 %
S = 65 %
Acetone
The primary impact of doping CeO2 with Ag is an enhancement in the reducibility of the oxide, resulting in greater selectivity towards acetone and reduced selectivity towards ethylene.
(de Lima et al., 2017)
18
γ-Al2O3
ETE
Strong electrostatic adsorption (SEA) method
T = 300 °C.
P = 1 bar, TOS = 24 h
WHSV = 1.26X = 97 %
S = 94 %
Y = 70 %Ethene
–
(Srinivasan et al., 2019a)
19
0.3Co-Al2O3 a
ETE
Strong electrostatic adsorption (SEA) method
T = 300 °C.
P = 1 bar, TOS = 24 h
WHSV = 1.26X = 99 %
S = 95 %
Y = 70 %Ethene
Co-Al2O3 possessed higher surface area because of additional microporosity in the c-Al2O3 support.
(Srinivasan et al., 2019b)
20
50In2O3-BET a
Ethanol to propylene (ETP)
Deposition precipitation method
T = 460 °C.
P = 1 bar, TOS = 43 h
WHSV = 0.20X = 100 %
S = 50 %
Y = 60 %Propene
The role of beta in the In2O3-beta composite catalyst is to promote the conversion to propylene.
(Xue et al., 2017)
21
3 %Sc/In2O3 p
ETP
–
T = 500 °C.
P = 1 bar, TOS = 56 h
WHSV = 0.20 h−1
X = 100
Spropene = 30 %
Sethene = 10 %
Y = 40 %Propene
The inclusion of Sc prevented the reduction of In2O3 to In metal during the reaction, while the introduction of water reduced the formation of coke.
(Iwamoto et al., 2013)
22
γ-χ Al2O3/B (M−Al−B)
ETE
Solvothermal method
T = 400 °C.
P = 1 bar, Mc = 0.05 g
X = 100 %
S = 92 %
Y = 92 %
Ethene
Increasing temperatures, from 200 to 400 °C, led to a surge in ethanol transformation.
(Chaichana et al., 2019)
23
Cu10ZA
ETA
Impregnation
T = 500 °C.
P = 5 bar,
X = 80 %
S = 93 %
Y = 90 %Acetaldehyde
The high selectivity at low temperatures is due to the ability of copper to eliminate the dehydration activity of the zinc aluminate support to diethyl ether.
(Garbarino et al., 2020a)
24
Cu30ZA
ETA
Impregnation
T = 400 °C.
P = 5 bar,
X = 94 %
S = 94 %
Acetaldehyde
The high selectivity at low temperatures is in part due to the ability of copper to kill the dehydration activity of the zinc aluminate support to diethyl ether.
(Garbarino et al., 2020b)
25
SAPO-34
ETE
Hydrothermal method
T = 400 °C.
P = 1 bar, TOS = 600 min
Mc = 0.5 g
WHSV = 3.5 h−1
X = 92 %
S = 52 %
Ethylene
Lower conversion and selectivity than modified catalyst
(Kang, 2000)
26
Zn1Ce5O2 + HBETA (Si/Al = 43) (10:1)
ETP
–
T = 450 °C.
P = 1 bar, TOS = 28 h
WHSV = 0.15 h−1
X = 66 %
S = 55 %
Y = 30 %
Propene
Dealuminated HBeta zeolite was employed as an additive to the composite catalyst and exhibits excellent catalytic performance in the ETP process.
(Xu et al., 2020)
27
Cr1Zn1Zr8O n
ETIB
–
T = 500 °C.
P = 1 bar, TOS = 15 h
WHSV = 1.90 h−1
X = 100 %
S = 45 %
Y = 52 %
Isobutene
The introduction of chromium into the mesoporous Zn1Zr8On catalyst to give the Cr1Zn1Zr8On composite results in a 38 % increase in the selectivity to isobutene
(Liu et al., 2017)
28
Nano Zn1Zr10Oz
ETIB
–
T = 450 °C.
P = 1 bar,
WHSV = 0.80 h−1
X = 100 %
S = 55.3 %
Y = 27 %
Isobutane
Undesirable reactions such as bio-ethanol dehydration and acetone polymerization/coking are inhibited.
(Sun et al., 2011c)
29
CS2/AMA
ETE
–
T = 400 °C.
P = 1 bar,X = 94 %
S = 93 %
Y = 88 %
Ethylene
The catalyst boosts the catalytic performance, increasing the amount of ethylene produced.
(Banzaraktsaeva et al., 2019)
30
4Ag-4ZrO2/SBA-16
ETnB
Wetness impregnation
T = 375 °C.
P = 7 bar,
WHSV = 0.23 h−1
X = 93 %
S = 66 %
Y = 100 %
1,3- butadiene, ethylene
With sufficient hydrogen partial pressure, 1,3-butadiene is completely and selectively hydrogenated to form n-butene.
(Dagle et al., 2020)
3 Reaction mechanism of catalytic conversion of bioethanol
A complicated reaction process is involved in the catalytic conversion of bioethanol, and it is regulated by the catalyst of choice and the reaction circumstances. As seen in Fig. 1, Matheus et al.'s analysis (Matheus and Sousa-Aguiar, 2022) focused on the primary pathways involved in the conversion of ethanol to chemicals and fuels. When ethanol is dehydrated, acid catalysts such as alumina, zeolites (ZSM-5, Beta, Faujasite), and SAPO are typically used (Liu et al., 2016), which produces ethylene at higher temperatures and diethyl ether at lower ones (Tran et al., 2021a). On the other hand, acetaldehyde is produced by dehydrogenating or basic catalysts like supported copper or magnesium oxide (Tran et al., 2021a). Because it goes through oligomerization, metathesis, and aromatization, among other processes, ethylene typically occurs before Ethanol to higher hydrocarbons (ETH) reactions. The product distribution and stability of HZSM-5 make it the ideal prospect for increased hydrocarbon output (Madeira et al., 2009a). Zeolites' acidity and porosity are crucial in the conversion of ethanol into hydrocarbons with three or more carbon atoms (C3+). This paper (Zeng et al., 2022) examines the ethanol-to-hydrocarbons (ETH) process in response to the need for a sustainable manufacturing pathway for light olefins like propene and ethene. A homologous reaction system is involved in the methanol-to-hydrocarbons (MTH) process, which serves as the foundation for the majority of the mechanistic insights of the ETH process. The acid-catalyzed mechanism (Gayubo et al., 2001a), the radical-intermediate mechanism (Madeira et al., 2011a), and the dual cycle mechanism (Johansson et al., 2009) are the three processes that have been postulated thus far for the ETH process. There was a tendency to believe that ethanol dehydrates first to produce an ethene intermediate, which then undergoes secondary processes such oligomerization, cyclization, aromatization, and cracking, just as MTH, to turn into C3+ hydrocarbons and aromatics (Boronat et al., 2008), (Inaba et al., 2006a).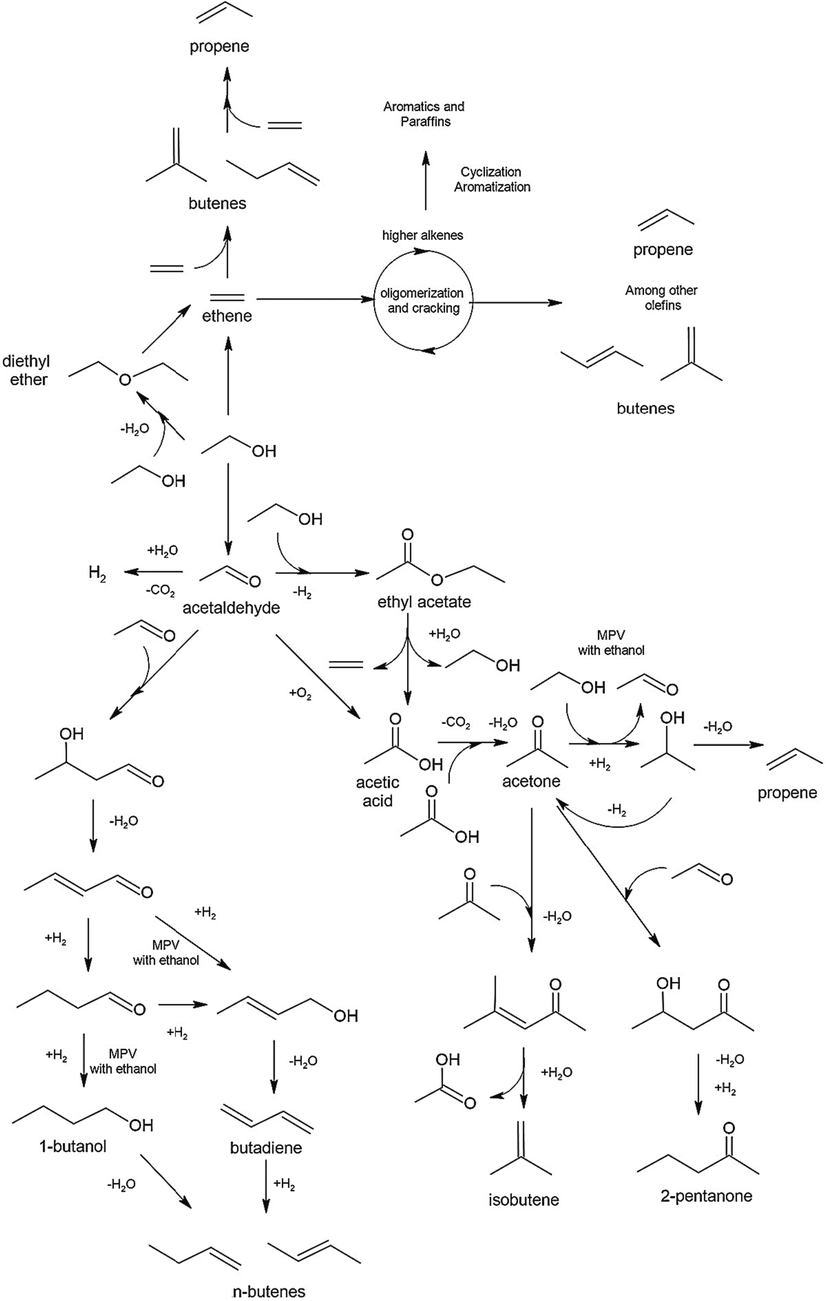
Principal pathways for converting ethanol into different compounds (Matheus and Sousa-Aguiar, 2022).
Certain catalysts possess both acidic and basic sites, however they generally exhibit a predominant characteristic. This enables individuals to execute a broader spectrum of reactions, hence resulting in a greater variety of goods. One example is the conversion of bioethanol into a blend of oxygenated and non-oxygenated hydrocarbons, resembling conventional fossil fuels, by the utilization of carbonate hydroxyapatite (Lovón-Quintana et al., 2017a). Another type of bifunctional catalyst is hydrotalcite, which can have its MgO/Al2O3 ratio changed to give it the desired characteristics (Ramasamy et al., 2016). As MPV catalysts, certain bifunctional catalysts also have an intriguing function. In this instance, a hydride can be transferred from an adsorbed alcohol to an acetaldehyde or ketone via a hydrogenation pathway. The aforementioned method exerts a significant influence on processes associated with the production of propylene, butadiene, and butanol through the utilization of ethanol. The reduction in MPV (methylcyclopentadiene dimerization) was shown to be influenced by the strength and density of Lewis acids as well as the presence of basic sites in ZrO2.
These factors exhibited a synergistic effect on the reaction, despite the potential capability of both acid and basic catalysts to facilitate this reaction (Komanoya et al., 2015). Additionally, the alcohol hydride donor affects this reaction. A secondary alcohol is chosen over a primary one because the intermediate is stabilized and the reduction is reversible. However, ethanol itself can be used as a donor of hydrides, particularly if the recipient responds rapidly and upsets the equilibrium (R. V. Matheus et al., 2018a). By adding certain elements, catalyst qualities can also be adjusted. For example, nickel is known to improve the stability of the catalyst in ethanol to higher hydrocarbon reactions, changing the acidity of the reaction and producing a different distribution of products (Van der Borght et al., 2015a). Another method of influencing the distribution of the product is to block or promote hydrogen transfer processes, which can be achieved by adding gallium or iron, respectively. Aromatic and paraffinic compounds are formed through hydrogen transfer, which is impeded by reaction suppression. The reactivity of alumina-based catalysts is altered when readily reducible elements such as Co or Cu replace Al, following the dehydrogenation of ethanol to acetaldehyde. The aforementioned phenomenon leads to a heightened inclination towards ketonization processes as opposed to aldol coupling reactions, resulting in the formation of compounds with an odd number of carbon atoms (Tran et al., 2021b).
Zanchet et al., (2015) in the course of conducting metal-catalyzed ethanol reforming, the study provides an overview of potential reaction pathways occurring on the metal surface during Steam Reforming of Ethanol (SRE). These pathways are derived from a combination of experimental findings and theoretical modeling. Ethanol activation follows three primary routes. In this route, ethanol's OH bond is cleaved, leading to a series of successive dehydrogenation reactions. This process leads to the generation of various intermediate compounds, including acetaldehyde (CH3CHO*), acetyl (CH3C*O), ketene (*CH2C*O), and ketenyl (*CHC*O), as shown in steps 1–5 of Fig. 2. In this pathway, the CH bond in ethanol is activated, followed by a series of dehydrogenation reactions. Importantly, the O–H bond remains intact during this process, resulting in the creation of intermediates like *CHyCHx*OH, as depicted in steps 1A to 2A. This pathway involves the breaking of both O–H and C–H bonds, leading to the formation of the intermediate *CH2CH2O* (referred to as an oxametallacycle), as indicated in step 1B of Fig. 2. It's worth noting that these pathways can be interconnected, and the specific reactions depend on the type of metal catalyst used. Additionally, the cleavage of C–C bonds within these intermediates is followed by hydrogenation/dehydrogenation of CHx* species, activation of water, and oxidation of C* species.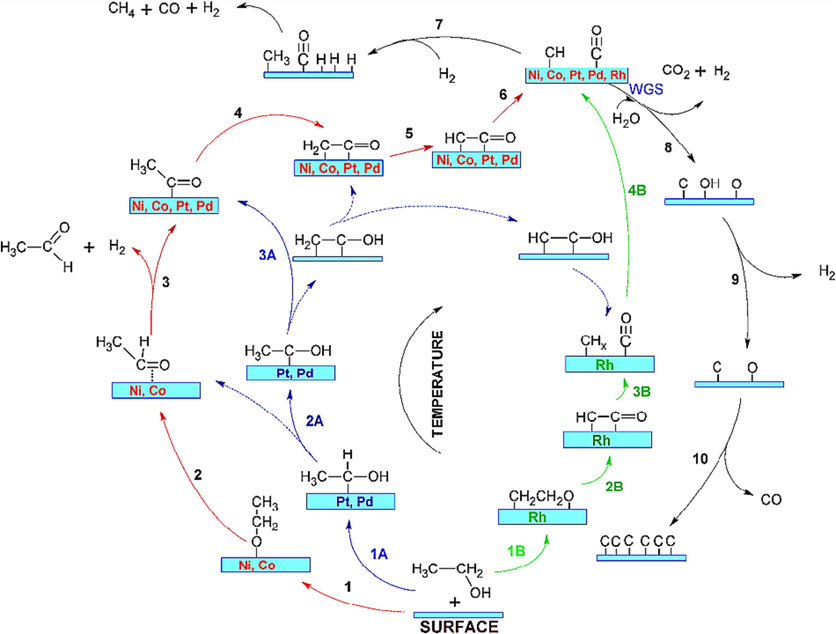
Diagram showing the SRE reaction paths for various metal surfaces as a function of temperature: dashed lines represent secondary routes; red, blue, and green colours denote the major routes on Ni or Co, Pt or Pd, and Rh, respectively (Zanchet et al., 2015).
Sun and Wang., (Sun and Wang, 2014b) examined ethanol's catalytic conversion to a range of compounds. The following reaction mechanisms are displayed: ethanol conversion to hydrocarbons ranging from light olefins to longer chain alkenes/alkanes and aromatics; ethanol conversion to other oxygenates such as 1-butanol, acetaldehyde, acetone, diethyl ether, and ethyl acetate; and ethanol steam reforming (ESR) to produce hydrogen as shown in Fig. 3 (Nguyen and Le Van Mao, 1990), (Mikkelsen and Kolboe, 1999), (Talukdar et al., 1997). For the effective conversion of many products, however, the creation of highly active and stable catalysts remains a serious issue. Using acid-base catalysts, ethanol was converted into useful chemicals such propylene using In2O3 and CeO2-based catalysts, isobutene using ZnxZryOz, and 1,3-butadiene using (mixed) metal oxides like ZnxZryOz. The basic-site-catalyzed ethanol dehydrogenation process toward acetaldehyde is the starting point for all other reactions. The ethanol-to-propylene and ethanol-to-isobutene processes have now been suggested to use acetaldehyde to acetone (Sun et al., 2011a). Nevertheless, for the 1,3-butadiene production, it was suggested to add acetaldehyde to acetaldol and then dehydrate acetaldol to crotonaldehyde (Jones et al., 2011a). The three reactions' reaction conditions imply that H2O and reaction temperature may be important factors in identifying the acetaldehyde reaction routes. Acetaldehyde-to-acetone conversion is promoted by cofeeding H2O at a substantially higher reaction temperature (>673 K) (Sun et al., 2011b), but aldol-condensation of acetaldehyde at a relatively lower temperature (<673 K) appears to be favored (Jones et al., 2011b). It was discovered that H2 is essential to the conversion of acetone into propylene and isobutene. The acetone-to-isobutene reaction predominates when hydrogen is absent or present in small amounts (Liu et al., 2013). However, propylene was greatly increased when hydrogen was present in high concentrations (e.g., 30 mol%) under the same reaction conditions (Mizuno et al., 2012). To convert ethanol to 1,3-butadiene, crotonaldehyde must be selectively hydrogenated toward the production of crotyl alcohol rather than hydrogenating the C–C link.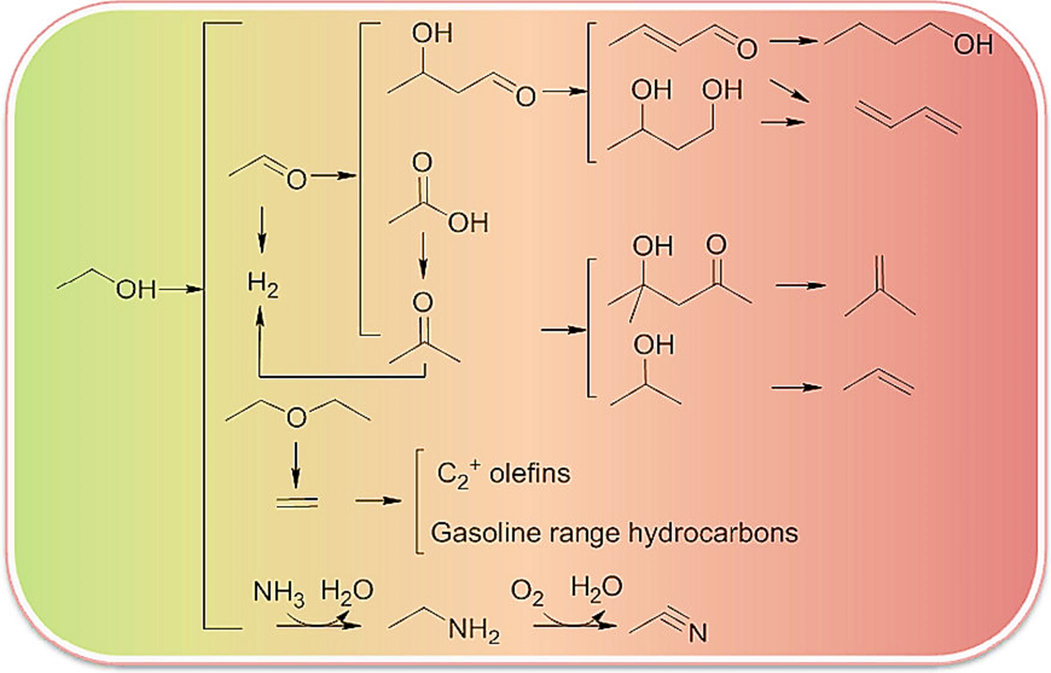
The widely recognized process for converting ethanol to hydrocarbons (Sun and Wang, 2014b).
4 Catalytic conversion of bioethanol to Value-Added chemicals
There is a wide range of processes involved in the catalytic conversion of bioethanol into valuable added compounds. The several kinds of catalysts used in the catalytic breakdown of bio-ethanol to create compounds with added value are displayed in Table 1. Zeolites present excellent conversion percentage to valuable hydrocarbons. They exhibit an average temperature range from 250 to 450 °C and a pressure range of 1–40 bar as shown in Table 1. They have the highest yield among the various catalysts. Moreover, acidic oxides also exhibit better performance and conversion rates. So, bioethanol can be converted into a variety of useful olefins such as ethylene, propene, isobutene, butadiene, and other butenes. Applications for these olefins include the production of plastics, polymers, and several chemical intermediates. Along with these additional benefits, the conversion of bioethanol yields aldehydes and carboxylic acids, specifically acetaldehyde and acetic acid, which have applications in the food, pharmaceutical, and chemical manufacturing industries. Acetone and 2-pentanone, two ketones that are crucial for the synthesis of chemicals and solvents, are also produced by the catalytic process. A valuable biofuel and chemical feedstock, butanol, is one of the alcohols that are also produced by the conversion. The final product of bioethanol conversion is lubricants, which are essential for use in equipment and automobiles (Sun and Wang, 2014b). All things considered, these catalytic routes provide a viable and appealing means of converting bioethanol into a variety of valuable compounds with a wide range of industrial applications.
4.1 Olefins
Given that ethanol is created by a straightforward dehydration process and that ethylene is the most widely manufactured petrochemical globally, ethylene is most likely the most obvious molecule to be obtained from ethanol. Nearly 80 % of the ethylene produced goes towards making polyethylene, ethylene oxide, ethylene dichloride, and ethylbenzene. Ethylene is utilised nearly exclusively as a chemical building block (Zimmermann and Walzl, 2009). Enzymatic solid acids catalyse the endothermic conversion of ethanol to ethylene. In the process, ethanol is either etherified to produce diethyl ether or directly dehydrated to produce ethylene. Since the ether can be later transformed into ethylene, it is regarded as a reactive intermediate. Diethyl ether is typically seen as a result of the reaction when it is conducted at temperatures between 150 and 300 °C, but at higher temperatures, the ether is easily changed to ethylene (Morschbacker, 2009). The principal secondary products generated from the reaction include acetic acid, ethyl acetate, acetaldehyde, acetone, methanol, methane, propane, propylene, butane, butenes, various hydrocarbons, carbon dioxide, carbon monoxide, and hydrogen. Ouyang et al., (2009) examined how bio-ethanol was catalytically converted to ethylene in a bioreactor using La-modified HZSM-5 catalysts. According to the stability test, selectivity and ethanol conversion over this catalyst could be sustained at levels over 98 % for over 950 h. High reactivity and stability lasting up to 830 h were also demonstrated by the regenerate catalyst. Shetsiri et al., (2019) examined ethylene synthesis from bioethanol in a sustainable manner using hierarchical ZSM-5 nanosheets. Two different models are employed to portray hierarchical ZSM-5 catalyst structures in order to compare reaction processes across various catalyst designs. These models include Brønsted acid sites situated on the external silanol surfaces, which contain Q3 species, and the interior surfaces of the bulk MFI framework, containing Q4 species, as depicted in Fig. 4A. A traditional model is also presnted in Fig. 4B. Mechanistic pathways that have been confirmed are illustrated in Fig. 4C. In the initial phase of the reaction, an ethanol molecule experiences dehydration over the Brønsted acid site of ZSM-5, leading to the creation of an ethoxide intermediate (I) through an ethyl carbenium transition state. The breakdown of this ethoxide intermediate into ethylene (I → II → III → IV) represents the first reaction pathway, while the conversion of a second ethanol molecule on the ethoxide surfaces to diethyl ether (DEE) (I → V → VI → VII → VIII) constitutes the second pathway (Chiang and Bhan, 2010), (Mortensen et al., 2011). Moreover, the Time on Stream (TOS) causes ethanol conversion to decrease from 89.3 % to 59.4 % even after several hours. However, as demonstrated in Fig. 4 (D, E), a significant conversion is still observed over the MFI-HieNS (37) catalyst (∼91.9). The MFI-HieNS (37) sample's activity has changed compared to the (MFI-CON(38)) catalyst, and there is a noticeable difference in the two samples' product selectivity. It is worth mentioning that ethylene demonstrates a significant level of selectivity, reaching nearly 90 %, in comparison to MFI-HieNS(37). Nevertheless, the dominant output of MFI-CON(38) is DEE, exhibiting a selectivity of 60.4 %, whereas the selectivity for ethylene stands at a mere 39.6 %.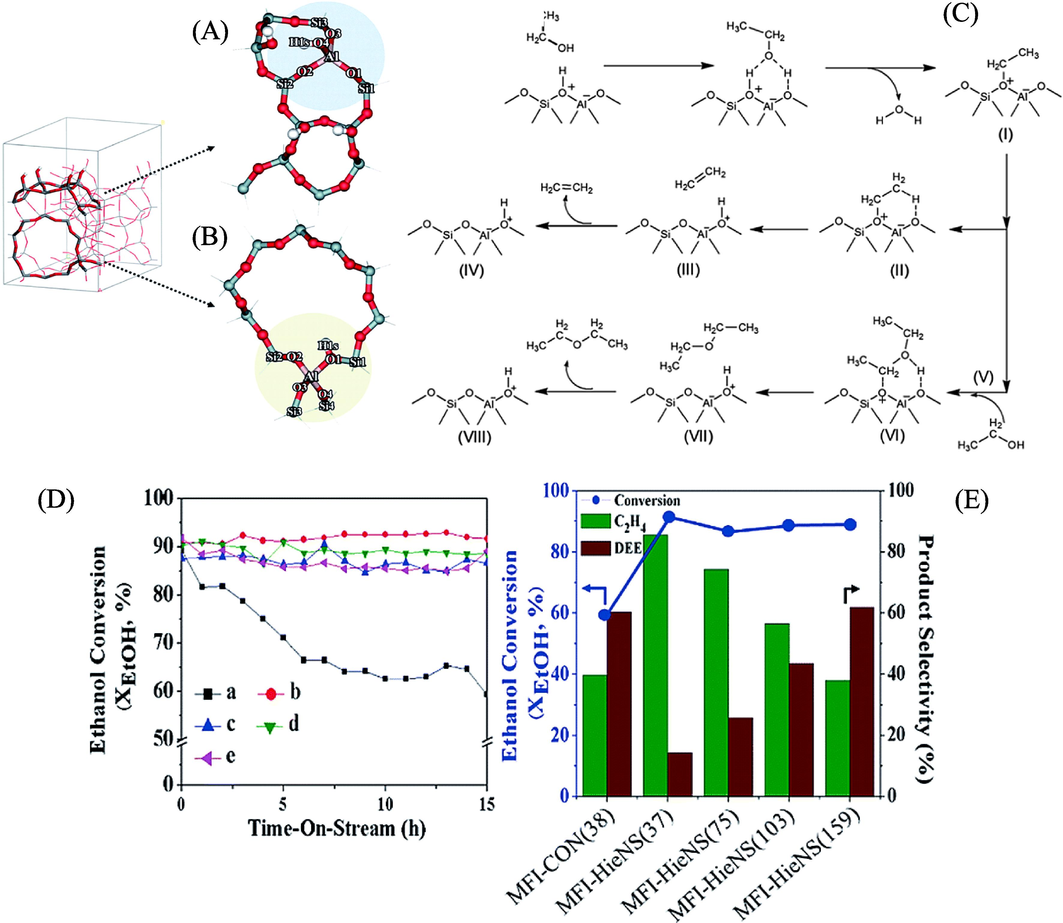
Optimized structures in (A) and (B) represent the Brønsted acid sites at the external silanol surfaces of hierarchical ZSM-5 and the Brønsted acid sites at the interior surfaces of a standard MFI framework, respectively. Pathways for the proposed ethanol dehydration to ethylene and diethyl ether (DEE) are illustrated in (C). The time-on-stream (TOS) dependence of ethanol conversion is shown in (D), while (E) displays the selectivity and conversion of the products obtained after 15 h of TOS at a reaction temperature of 300 °C, a weight hourly space velocity (WHSV) of 10 h−1, and a molar ratio of N2 to EtOH of 2.5 (Shetsiri et.al., 2019).
Using specialized catalysis, Iwamoto et al., (2015). investigated the conversion of propene from bio-ethanol. The study investigated the conversion of ethanol to propene using different catalysts: Ni ion-loaded silica MCM-41 (Ni-M41), Sc-modified In2O3 (Sc/In2O3), and a solid solution of Y2O3-CeO2. The activity for propene synthesis followed the sequence Sc/In2O3 > Y2O3-CeO2 > Ni-M41, but their stability during the process ranked as Y2O3-CeO2 ∼ Sc/In2O3 > Ni-M41. The addition of water and hydrogen to the reactant stream significantly improved both the propene output and endurance of Sc/In2O3. The choice of catalyst had a profound impact on the reaction process. On Ni-M41, the dimerization of ethene played a crucial role, leading to a metathesis reaction between ethene and butenes, which was an essential step in propene synthesis. Conversely, on the other two oxide catalysts, the main pathway for ethanol conversion to propene involved the following steps: ethanol → acetaldehyde → acetone → propene. A study conducted by Zhang et al. (2017) investigated the conversion of bioethanol into propene using nano-HZSM-5 zeolite. Their research revealed that fluorinated HZSM-5 had rougher surfaces with etched crystals and defects compared to pure zeolite, which had a smoother surface. These irregular surfaces, fragile particles, and gaps between them were found to facilitate the formation of new mesopores, increasing the external surface area of the particles, as confirmed by N2 adsorption–desorption experiments. XRD patterns (Fig. 5C) of HZ and HZ-F-20 before and after the reaction showed that the fresh catalysts had two peaks around 23°, which merged into a single peak after deactivation (Fig. 5D), indicating some damage to the zeolite HZSM-5 framework during the reaction. The active sites for converting ethanol to propene were identified as Brønsted acid sites (Bun et al., 1990). Various reaction pathways (Takamitsu et al., 2014), (Takahashi et al., 2013), (Inaba et al., 2006b) were proposed for the ethanol-to-propene (ETP) process, with ethene initially forming from ethanol and subsequently being converted in parallel to propene and butene. Due to the catalyst's large pore size, propene and butene had limited interaction time with the active acid sites, leading to their diffusion out of the channels. This effectively hindered the synthesis of aromatics, C5 + aliphatics, and paraffins (Xia et al., 2016). To assess the impact of time on stream (TOS) on the selectivity of C2–C4 olefins (propene, butylene, and ethylene) and aromatics during bioethanol-to-propene conversion, experiments were conducted and the results are shown in Fig. 5 (E, F, G, and H). The ethene selectivity increased gradually with TOS, while aromatics selectivity decreased as reaction time increased. Propene and butene selectivity exhibited similar fluctuations, initially rising, reaching a maximum, and then declining, as illustrated in Fig. 5 (F) and (G). This suggests that propene and butene are produced through both common intermediate and parallel pathways in HZSM-5 zeolites (Huangfu et al., 2016). Similar trends were observed in the fluorinated samples, but the initial propene selectivity was significantly higher, and catalytic stability improved compared to the parent HZ. Notably, the HZ-F-20 catalyst exhibited even greater advantages, reaching a maximum propene selectivity of 24.9 % within nine hours and extending the catalyst's lifetime (the duration it maintained a propene selectivity of at least 10 %) to 214 h, surpassing the original HZ catalyst's lifespan by more than five times. The long-term propene selectivity ranking was as follows: HZ-F-20 > HZ-F-15 > HZ-F-25 > HZ-F-10 > HZ-F-5 > HZ. In summary, the fluorine-modified HZSM-5 catalysts, particularly HZ-F-20, demonstrated significantly enhanced resistance to coke deposits, as evidenced by the catalytic test results (Fig. 5).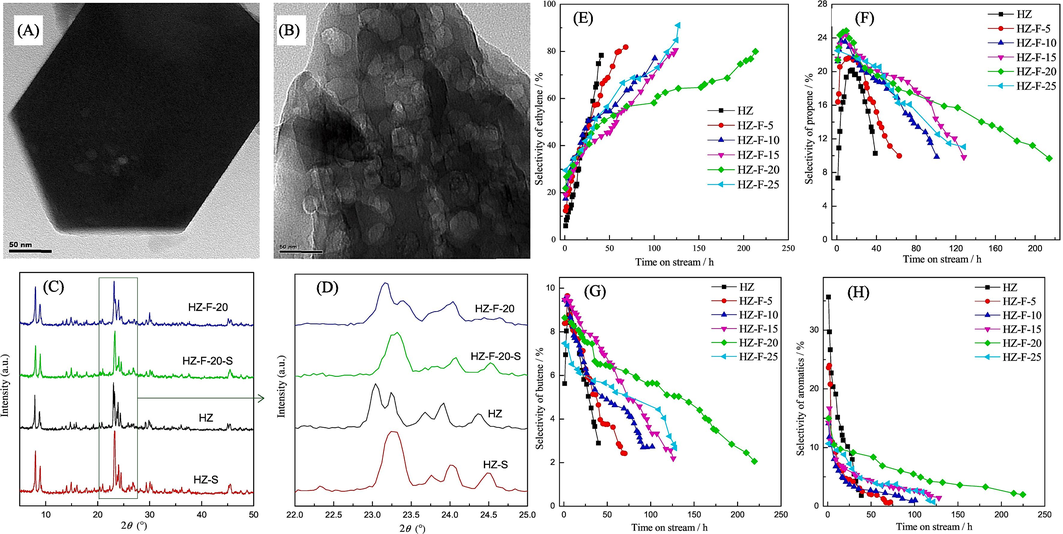
The HZ (A) and HZ-F-20 (B) TEM pictures. (D, C) The HZSM-5 samples' fresh and spent XRD patterns. E, F, G, and H HZ-S stands for 39 h, and HZ-F-20-S for 214 h. Time in the stream has an impact on the aromatic and C2–C4 olefin selectivity across various HZSM-5 zeolites. 500 °C, atmospheric pressure, ethanol/water = 9/1 vol/vol, and ethanol WHSV = 10 h− 1 are the reaction conditions (Iwamoto, 2015).
By using the HZSM-5 zeolite to perform the ethanol-to-hydrocarbon reaction, Madeira et al. (2011b) were able to identify radical species and investigate their evolution through catalyst deactivation. Every coked sample was subjected to an infrared analysis in order to compare the primary vibration bands of the fresh zeolite with those linked to the carbon deposit region (Fig. 6A) and the OH region (Fig. 6B). At first, the researchers noticed that all of the coked samples in the area (Fig. 6A) had uniform coke molecule vibration bands. The strength of these bands is the only change that can be seen. The research indicates that vibration bands between 1500 and 1640 cm−1 are frequently connected to aromatic rings. In contrast, vibration bands ranging from 1350 to 1470 cm−1 are generally linked to the aromatics' alkyl branches; that is, they are connected to CH stretching δs(CH2), CH stretching δs(CH3), and/or δs([CH3]2C < ). This suggests that the carbon deposit formed in the framework is the result of alkylated aromatic molecules, which make up the compounds occluded in the framework. It was anticipated that the coke molecules would reduce the intensity of the silanol band (about 3740 cm−1) and balance the acidic bands, which are bridged by OH groups, at about 3600 cm−1. External cocaine is frequently linked to this band (MAGNOUX, 1987). The OH-bridging band (Fig. 6B) vanished from the original sample after just one hour of Time-On-Stream (TOS), suggesting a quick deactivation process. As the sample's time on stream (TOS) rises, the silanol band's intensity progressively diminishes, with the degree of this drop becoming increasingly noticeable. The yields of the two main products—ethylene and diethyl ether—obtained from the dehydration of ethanol are shown in Fig. 6C. Additionally, it displays the yield of C3+ hydrocarbon production as well as the full conversion of ethanol. For all four runs, the ethanol conversion was finished. Diethyl ether was identified at the end of the thirty-hour trial, albeit in negligible amounts. All of the ethanol was transformed entirely into C3+ hydrocarbons during the one-hour run. The majority of the higher hydrocarbons (C3+) are paraffinic and aromatic compounds, with five to eleven carbon atoms on average (Madeira et al., 2009b). Initially, the four coked samples as well as the fresh samples were analyzed using the EPR-Continuous Wave (EPR-CW) method. The resulting spectra are shown in Fig. 6D. With respect to the samples that were subjected to coke treatment, it is clear that every sample shows some sort of signal, indicating that this specific species has been present and formed since the reaction began. Each of these signals in the case of an organic radical species is uniquely defined by a Lorentzian line form. Depending on the particular sample, the line width fluctuates between 10 and 14 G, with the g factor at the center of these signals being 2.007. When examining the evolution of signal form and intensity with time of sample (TOS), an increase in signal intensity is seen from the 1-hour to the 16-hour TOS. This observation points to an increase in the concentration of spin. We appear to have reached a limit when comparing the 16- and 30-hour signals. Still, there is a small change in the signal shapes, giving the appearance of being slimmer. This suggests a shift in the species' characteristics. Fig. 6 displays the chromatograms for the soluble coke in the runs with a 1-hour and 30-hour TOS. The compounds found are clearly the same in both samples, although the peaks' intensities are different. The peaks that are seen in the first fifteen minutes of the one-hour run (Fig. 6E) are more intense than those that are seen in the thirty-hour run (Fig. 6F). In the 30-hour chromatogram, the peaks in the 23–30-minute period are more prominent than in the 1-hour chromatogram of the identical sample, where these peaks were not displayed. This suggests that the properties of TOS have changed, and there has been a rise in the amount that has accumulated on the catalyst. It also provides insights about the features of the current carbon deposit. These compounds are responsible for most of the highly alkylated monoaromatic compounds seen in the first 20 min of the chromatograms. It was found that these compounds were also found by online GC analysis over the course of our catalytic testing. Over the course of the next twenty to thirty minutes, more condensed aromatics that are still highly alkylated are added. These large, bulky molecules are most likely the main cause of the pore blockage, which prevents reactants from entering the channel (also known as micropore blockage) or products from being released.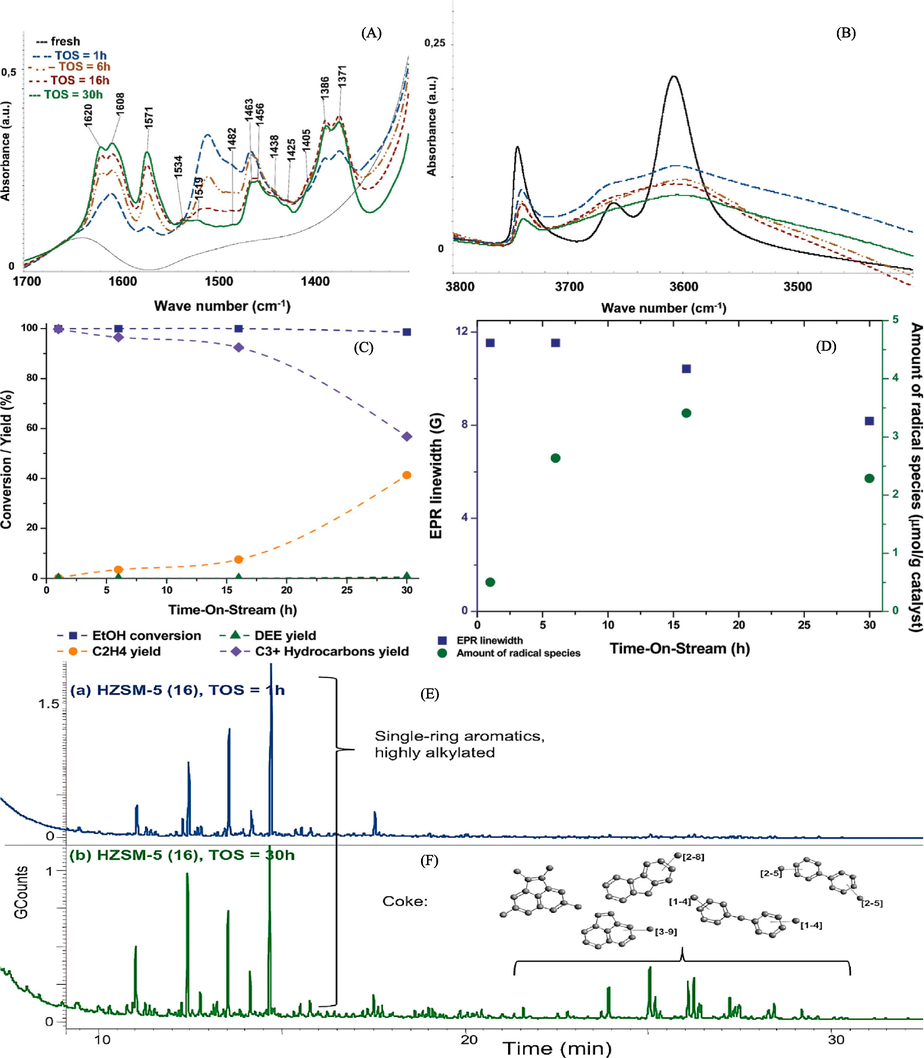
In (A) and (B), the infrared spectra are presented for both the fresh and coked samples in the 1300–1700 cm−1 and 3500–3800 cm−1 regions. The reaction conditions for these tests were as follows: Temperature (T) = 623 K, Pressure (P) = 30 bar, N2/EtOH = 4, and WHSV = 15 h−1.(C) displays data on ethanol conversion and the production of ethylene, diethyl ether, and C3+ hydrocarbons (wt%) for the four tests conducted at different times on stream (TOS) of 1, 6, 16, and 30 h.(D) illustrates the relative EPR line width and the content of radical species (derived from spin concentration) for both the fresh and coked HZSM-5 (16) samples at TOS intervals of 1 h, 6 h, 16 h, and 30 h. Finally, (E) and (F) provide results from a GC–MS study of coke compounds recovered by CH2Cl2 from the coked samples of HZSM-5 (16) at TOS intervals of 1 h and 30 h, respectively, following solubilization by HF solution (Madeira et al., 2011b).
4.2 Aldehydes and carboxylic acid
The production of either aldehydes or carboxylic acids can occur during the oxidation of ethanol, depending on the specific reaction conditions. Acetaldehyde is an essential industrial chemical that is used in the production of various chemicals. It can be selectively oxidized from ethanol under specific regulated conditions. Acetic acid is a widely utilized carboxylic acid in the production of plastics, textiles, and vinegar. It is produced through the additional oxidation of acetaldehyde. The adaptable dual-pathway process is crucial to the chemical industry due to its capability to generate significant intermediates for various applications (Jørgensen et al., 2007), (Redina et al., 2015). Tembe et al., (2009) conducted a study on the synthesis of acetic acid through the selective oxidation of ethanol using Au catalysts that were supported on various metal oxides. The capacity to oxidize ethanol was evaluated by employing catalysts containing 1 wt% of gold supported on TiO2, Al2O3, and ZnO. The results indicate that Al2O3 exhibited the highest level of activity as the initial support for gold in the ethanol oxidation process, followed by ZnO and TiO2.
In the study by Raynes and Taylor (2021), a catalyst consisting of zinc oxide-modified mordenite was employed for the dehydrogenation of (bio)ethanol to acetaldehyde. The research involved performing pXRD (powder X-ray diffraction) investigations on various ZnO/Na–MOR-(7) material loadings. All the samples successfully maintained the MOR framework type after the impregnation and calcination treatment, as indicated in Fig. 7A. Furthermore, pXRD reflections corresponding to ZnO were observed in the sample loaded with 10.0 wt% Zn, suggesting the presence of ZnO clusters in this material that were sufficiently large to produce a pXRD response (Ouyang et al., 2020). In Fig. 7B, solid-state Al NMR spectra of each ZnO/Na–MOR-(7) material confirmed that aluminum existed solely in tetrahedral framework positions (δAl ≈ 60 ppm), ruling out any impact of extra-framework alumina on catalysis (Nagy et al., 1984). SEM (scanning electron microscopy) imaging of the four catalyst variations did not reveal any changes in catalyst morphology or the presence of large ZnO clusters on the surface of the catalyst crystals (Fig. 7C). To further investigate the Zn distribution in the materials, a sample of each was embedded in resin, mechanically ground, polished with a diamond wheel, and then subjected to SEM-EDS (energy-dispersive X-ray spectroscopy) analysis. This preparation exposed the crystal interiors, allowing for the evaluation of element distribution within the zeolite crystals. Fig. 7E displays the selectivities to the main products for ZnO/Rb–MOR–(7), ZnO, and a physical mixture of ZnO and Rb–MOR–(7) at 0.2 h (A) and 4.0 h (B) TOS (total organic matter reaction time). After a 4-hour TOS, Fig. 7D shows that all catalysts achieved comparable acetaldehyde selectivities of over 80 %, with supported ZnO/Rb–MOR–(7) exhibiting the highest selectivity of 89 %.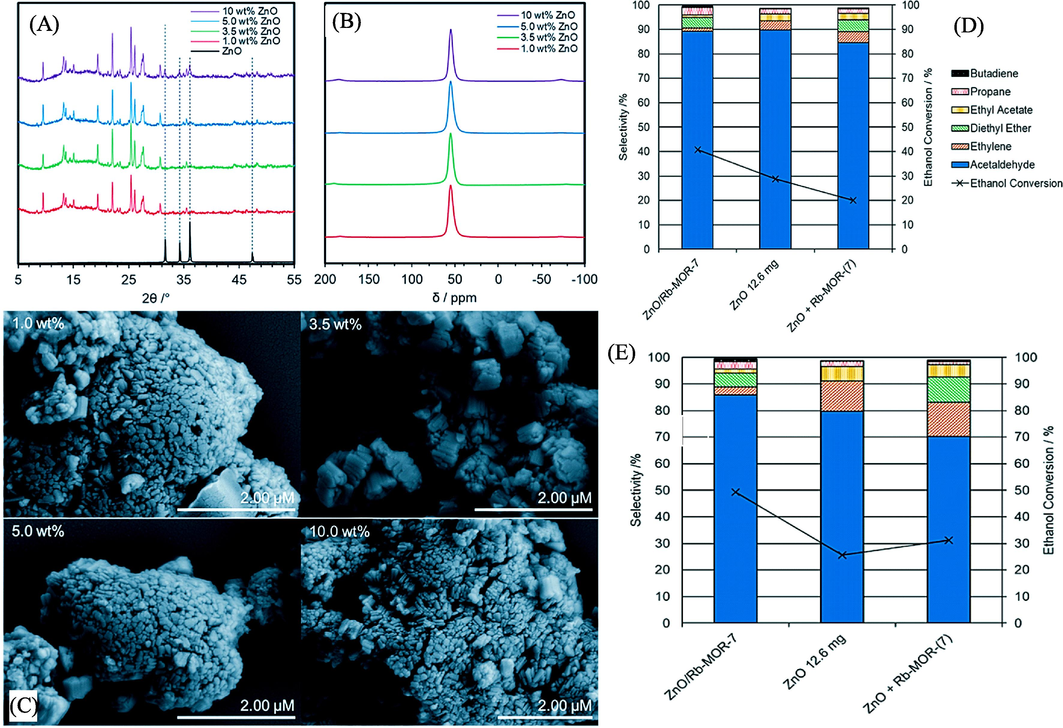
(A) shows the pXRD (powder X-ray diffraction) patterns in the 2θ range of 5–55° for ZnO/Na–MOR-(7) materials containing different percentages of ZnO by weight (1.0, 3.5, 5.0, and 10 wt% ZnO). These samples were mounted on a glass pXRD slide during data acquisition. A reference diffractogram of ZnO (99.99 %, Sigma-Aldrich) was also acquired using the same analysis conditions as those used for the zeolite materials.(B) displays solid-state Al NMR spectra of the ZnO/Na–MOR-(7) materials with varying ZnO content (1.0, 3.5, 5.0, and 10 wt% ZnO by Zn).(C) presents conventional SEM (scanning electron microscopy) images of the ZnO/Na–MOR-(7) materials loaded with nominal percentages of 1.0, 3.5, 5.0, and 10 wt% ZnO by Zn.(D) and (E) provide information on the selectivities for major products at 4.0 h and 0.2 h TOS (total organic matter reaction time), respectively, for ZnO/Rb–MOR-(7) (marked with “×,” 300 mg), ZnO (marked with “▲,” 12.6 mg), and a physical mixture of ZnO and Rb–MOR-(7) (marked with “■,” 12.6 mg + 300 mg) at a reaction temperature of 400 °C for 4 h TOS (Raynes and Taylor, 2021).
4.3 Ketones
Acetone finds extensive application as a solvent in medicine and polymer manufacturing, as well as serving as a feedstock for the synthesis of diacetone alcohol, methyl isobutyl ketone, isophorone, and cyanohydrins, which are precursors to methyl methacrylate (Chagas et al., 2018). Rodrigues et al., (2013) examined the one-pot acetone synthesis from ethanol, and Fig. 8 depicts the reaction process. The selectivity of various catalytic systems at isoconversion (∼80 %) is displayed in Fig. 8A. The primary byproducts of using ZrO2 or CZA (Cu/ZnO/Al2O3) are acetaldehyde and ethylene, respectively. According to extensive literature research, ZrO2 exhibits pairs of basic and acid Lewis sites (Zonetti et al., 2011a), which facilitate the conversion of ethanol to ethylene by dehydration. Conversely, ethanol is dehydrogenated on the Cu0 sites of CZA to produce acetaldehyde (Zonetti et al., 2011b). Almost reaching thermodynamic equilibrium, this reaction is at the experimental conditions used (Fig. 8A), known to create hydrogen. These two catalysts also produce trace amounts of carbon dioxide and acetone, as Fig. 8A illustrates. When the catalytic behaviour of the physical mixture of these oxides (CZA + ZrO2 (1:2)) is compared with that of ZrO2 and CZA, it is entirely different. The selectivity to acetone and carbon dioxide rises when ZrO2 is added to CZA, whereas the selectivity to acetaldehyde falls. This time, under the same experimental conditions, Fig. 8B displays the distribution of the products as well as the conversion of ethanol utilising various catalytic systems. Low conversion from the CZA catalyst deviates significantly from thermodynamic equilibrium. The ethanol conversion increases when ZrO2 is added to CZA (CZA + ZrO2 (1:1), and the selectivity towards acetone and carbon dioxide likewise rises while it falls towards acetaldehyde. When ZrO2 concentration in the physical mixture (CZA + ZrO2 (1:2) is doubled, selectivity to acetone and carbon dioxide as well as ethanol conversion rise even more, but selectivity to acetaldehyde falls. Thus, the ZrO2 surface may be the site of acetone production.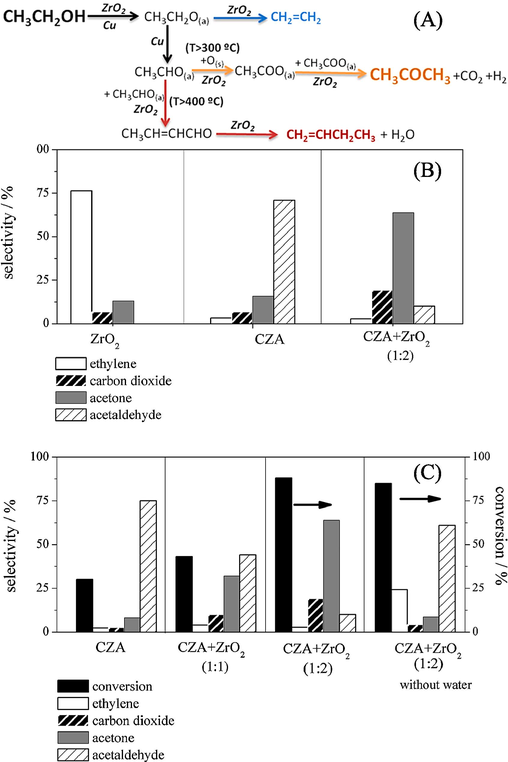
(A) The one-pot synthesis of acetone from ethanol and its reaction mechanism. (B) Distinct catalytic system selectivities during isoconversion. The temperature, weight of ZrO2, weight of CZA, and composition of the gas mixture are N2:H2O:C2H5OH = 91:8:1 mol%, 400 °C, 25 mg, and 50 mg, respectively. CZA + ZrO2 (1:2) is the physical mixture that uses these similar weights. To achieve the isoconversion, the flow rate was adjusted. (C) The different catalytic systems' selectivities and ethanol conversion. The gas mixture composition, temperature, weight, and flow rate of CZA are as follows: N2:H2O:C2H5OH = 91:8:1 mol%, 400 °C, and 25 mg, respectively; the flow rate is 70 mL min−1. ZrO2 weighs 25 mg and 50 mg, respectively, when it is used in the physical mixes CZA + ZrO2 (1:1) and CZA + ZrO2 (1:2) (Zonetti et al., 2011a and 2011b).
The main disadvantage of Pt, Pd, and Ru based catalysts is their high cost, even if they show great activity in SRE (Song and Ozkan, 2010). As a result, bimetallic catalysts with non-noble metal additions and trace amounts of noble metal are being studied for SRE. Sliwa et al. assessed SRE for CuO/ZrO2 catalysts doped with Mn, Ni, and Ga at 350 °C (Śliwa and Samson, 2021). By using the co-precipitation approach, the copper-based catalysts were created at a set (weight percentage) CuO/ZrO = 2.3 and a constant pH of 7. CuO and tetragonal ZrO2 phases (t-ZrO2) are detected for all catalysts. However, there are no discernible XRD peaks for MnO, Ga2O3, or NiO. The conversion of ethanol to all synthesized catalysts is high. Compared to Cu/Zr catalyst (XEtOH = 90 %), the inclusion of Ni and Ga results in an increase in ethanol conversion (XEtOH = 99 % and 98 %, respectively). Conversely, ethanol conversion shows a little reduction with Cu/Zr doping with Mn (XEtOH = 86 %). The observed variations in conversion are caused by variations in the synthesized catalysts' BET surface area. The BET surface area of the Cu/Zr catalyst increases with Ni and Ga doping, going from 35 m2/gcat for Cu/Zr to 50 m2/gcat and 51 m2/gcat for Cu/Zr/Ni and Cu/Zr/Ga, respectively. Furthermore, the inclusion of Ga causes a notable 16 % decrease in selectivity to acetic acid. Gallium addition improves both BET surface area and copper dispersion. While adding Ga reduces the crystallization of the t-ZrO2 phase and increases the thermal stability of a-ZrO2, adding Mn promotes the crystallization of tetragonal ZrO2.
2-Pentanone finds industrial applications primarily as a solvent, especially in the dewaxing process of high-boiling petroleum fractions like lubricating oils. It is also used in the manufacturing of pharmaceuticals, pesticides, nitrocellulose sprays, and synthetic resin coatings (He et al., 2005), (WEI et al., 2021). In their study, Subramaniam et al., (2020) explored the direct catalytic conversion of ethanol into C5+ ketones and investigated the role of a Pd–Zn alloy in catalytic activity and stability. They achieved highly selective ketone production (around 71 %) by directly impregnating 0.1 % Pd onto a ZnO-ZrO2 catalyst. These ketones had a higher average carbon number (approximately 6) and minimal acetone selectivity (less than 10 %). A notable portion of the ketones belonged to the C7+ category, resulting from the sequential cross-condensation of ketones with aldehydes. The detailed product distribution from this experiment can be found in Fig. 9A inset. In order to gain a more comprehensive understanding of the reaction mechanism, the research collected data for the 0.1 wt% Pd–ZnO-ZrO2 catalyst at lower conversion levels by varying the space velocity, as depicted in Fig. 9C. Fig. 9B illustrates the corresponding changes in product distribution, indicating that as the weight hourly space velocity (WHSV) increases, there are lower conversions and a significant shift in selectivity away from ketones towards esters and alcohols/aldehydes. Additionally, there is a reduction in the length of the product chains. These trends were also observed when the reaction temperature was lowered, suggesting that the in-situ formation of acetone was a slower secondary process. Fig. 9B provides information on the rates of formation of key compounds as a function of contact time. It's worth noting that lower contact times (<20 min) were not tested due to the formation of cyclic compounds resulting from unsaturated condensations. Importantly, the data shows that the total production of ketones is roughly equivalent in moles to the amount of CO2 produced. This observation aligns with two possible pathways for intermediate production: either through a decarboxylation pathway or through water gas shift from CO generated via a decarbonylation pathway (Dagle et al., 2008).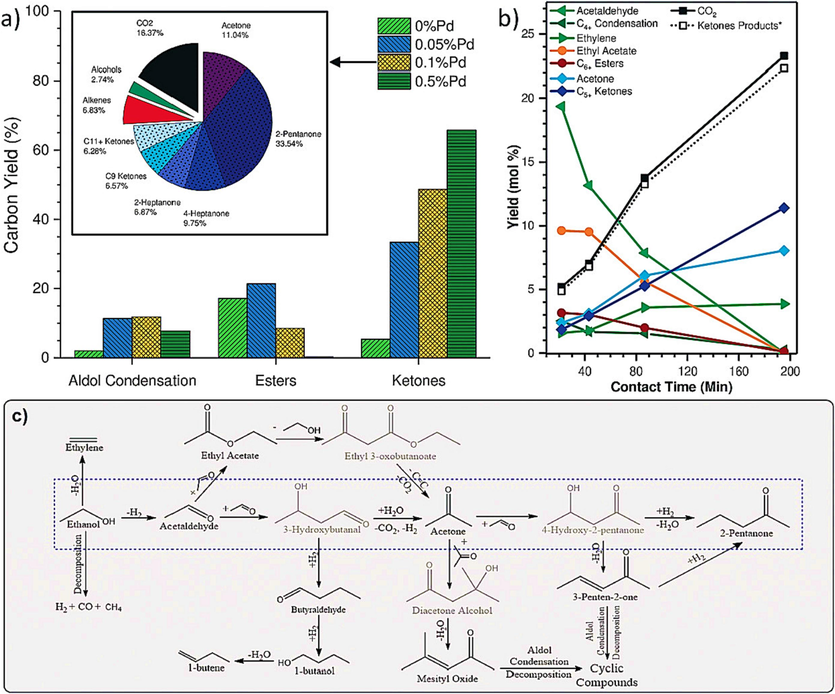
A) the carbon yield distribution of major product groups with varying pd loading on zno-zro2 at 340 °C, 0.21 MPa EtOH, 1.86 MPa N2, and 0.15 h−1 WHSV is depicted. The inset provides a closer look at the detailed product carbon selectivity derived from ethanol conversion over 0.1 % Pd-ZnO-ZrO2 at 370 °C, 0.21 MPa EtOH, 1.86 MPa N2, and 0.15 h−1 WHSV. b) The molar yield of key products is shown as a function of contact time, which was conducted by varying the catalyst loading for ethanol conversion over 0.1 % Pd-ZnO-ZrO2 at 370 °C, 0.21 MPa EtOH, 1.86 MPa N2, and 0.15 h−1 WHSV. The “*” denotes the sum of products, including all ketones such as acetone, 2-pentanone, and 3-penten-2-one, their respective alcohols like isopropanol and 2-pentanol, their respective dehydration products such as propene and trans-2-pentene, as well as isobutene from acetone self-condensation. c) A simplified reaction mechanism is provided for ethanol conversion to C5+ ketones, along with potential side reactions. Compounds in gray were not found in the product stream (Subramaniam et al., 2020), (Dagle et al., 2008).
4.4 Alcohols
n-Butanol is a valuable chemical that finds use in the paint and coating, cosmetics, and pharmaceutical sectors as well as an extractant and solvent. In addition, it serves as a fuel by being mixed with petrol and utilised as a feedstock for the synthesis of other compounds, including butyl acrylate, methacrylate, acrylic acid, and acrylic esters (Ndaba et al., 2015), (Earley et al., 2015). An analysis of the one-pot liquid-phase catalytic conversion of ethanol to 1-butanol over aluminium oxide was conducted by Riittonen et al., (2012a). Among all the catalysts examined, the commercial HTC-500 (20.7 % Ni on alumina) had the highest level of selectivity (62 %). Additionally, a 20 % Ni solution made on alumina by the user had a modest level of selectivity (37 %) towards 1-butanol; nonetheless, the conversion was nearly four times higher than that of the HTC-500, yielding a sizable amount of acetaldehyde. XRD and TEM were used to characterize both nickel catalysts (Fig. 10A and B) (Riittonen et al., 2012b). It's interesting to note that the self-prepared catalyst has a much narrower particle size range and is devoid of aggregates, which are thought to be inert components. Fig. 10C depicts an example kinetic curve that displays the conversion of ethanol and the 1-butanol selectivity among liquid carbon products as a function of time.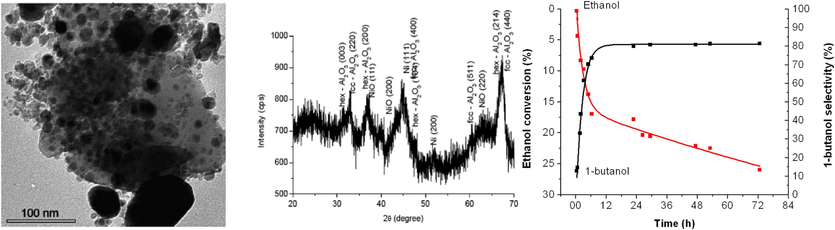
TEM and XRD images of the commercial model catalyst 20.7 wt% Ni/Al2O3 HTC-500. The average particle size was found to be about twice as much as for the self-made Ni-catalyst and showed considerably broad size dispersion (Riittonen et al., 2012b).
Sun et al., (2017) analyzed the efficient catalytic conversion of ethanol to 1-butanol via the Guerbet reaction over copper- and nickel-doped porous. The hydrogen neutral reaction sequence of the Guerbet coupling of ethanol to 1-butanol involves the following key steps: (a) dehydrogenation of ethanol to acetaldehyde, (b) aldol condensation of acetaldehyde including dehydration to afford the corresponding unsaturated C4 products, and (c) hydrogenation to form saturated longer chain alcohols (Fig. 11A) (Gabriëls et al., 2015), (Kozlowski and Davis, 2013). In Fig. 11B, the study investigated the impact of varying reaction temperature (ranging from 180 to 320 °C) and changes in catalyst loading (from 0.05 to 0.2 g) on both conversion and yield values. As expected, the conversion consistently increased, starting at 2.3 % at 180 °C and reaching 56.5 % at 320 °C. Simultaneously, the yield of 1-butanol increased, peaking at 22.2 %, and the space–time yield achieved an impressive value of 704.6 g kgcat−1h−1 at 320 °C. However, it's important to note that the carbon balance decreased from 98.5 % to 75.0 % as the temperature increased. To delve deeper into the product formation profiles, experiments were carried out at 310 °C using 0.1 g of Cu10Ni10-PMO catalyst over a 24-hour period. Fig. 11C illustrates the results, where the conversion of ethanol exhibited a linear increase up to approximately 10 h, after which no significant change was observed. In contrast, the yield of 1-butanol reached a constant value of 21 % after 6 h. The initial increase in ethanol conversion during the first 10 h can be attributed to the competitive conversion of 1-butanol into higher alcohols, particularly between the 6th and 10th hour of the reaction (Riittonen et al., 2012c).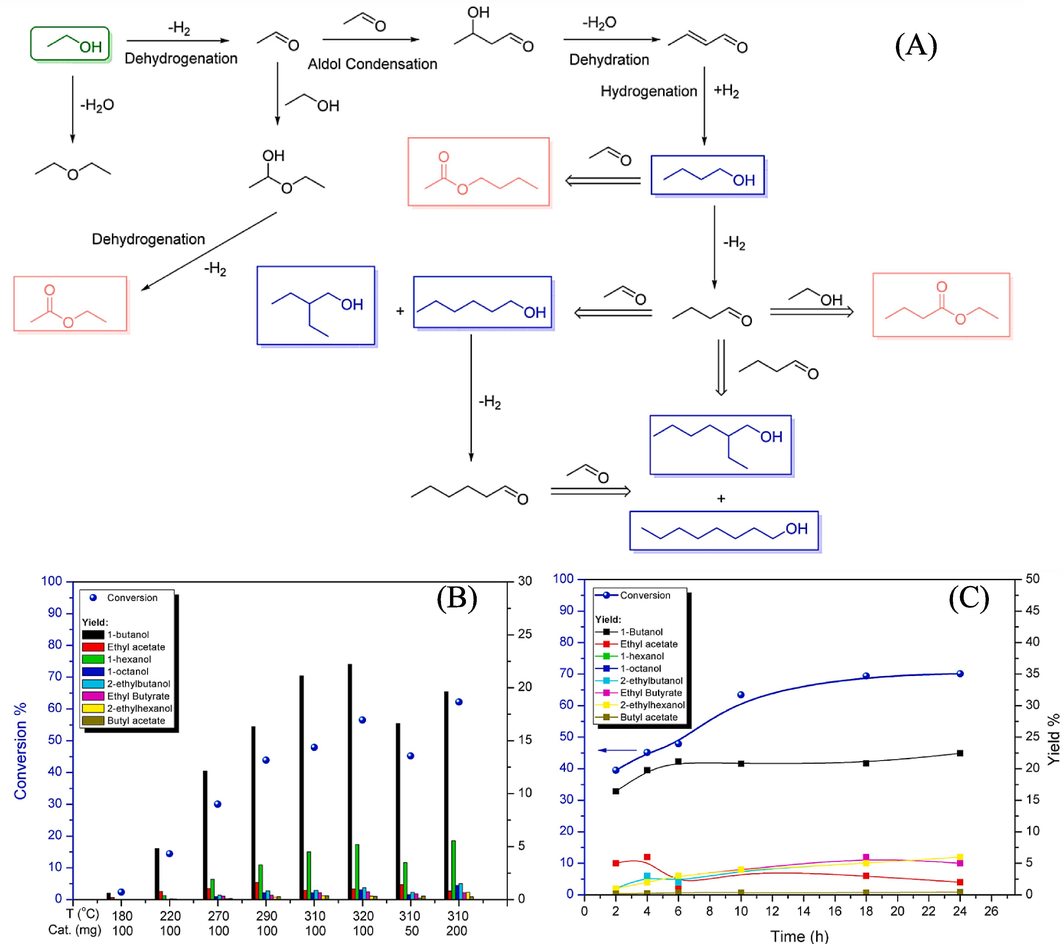
(A) Reaction network for Guerbet coupling of ethanol. (B) Influence of temperature and catalyst loading on ethanol conversion and distribution of products. Reaction conditions: Cu10Ni10-PMO (50–––200 mg), ethanol (3 mL), 6 h, decane (20 μL). (C) Product formation profile for the Guerbet reaction of ethanol to 1-butanol for 24 h. Reaction conditions: Cu10Ni10-PMO (100 mg), ethanol (3 mL), 310 °C, decane (20 μL) (Riittonen et al., 2012c).
4.5 Lubricants
Carbon-carbon bond formation has a history in ethanol processing (as discussed earlier with the Lebedev and Guerbet reactions), new processes have emerged that combine ethanol with other small molecules derived from biomass. Notably, the Toste and Bell research groups have made significant contributions in this area, developing pathways to create novel bio-based lubricants and long-chain hydrocarbon fuels. They utilize a mixture of acetone, butanol, and ethanol, which is produced during acetone, butanol, and ethanol fermentation (Goulas and Toste, 2016). By employing reduced transition metals and K3PO4 as a base, it becomes possible to perform alkylation reactions with acetone, butanol, and ethanol. This alkylation process leads to a mixture of methyl ketones, which are subsequently doubly alkylated, resulting in mixed C7-C11 ketones, as illustrated in Fig. 12 (Anbarasan et al., 2012), (Sreekumar et al., 2015). These products, particularly when reactions are conducted at moderate space velocities and result in linear molecules, can be further processed through hydrodeoxygenation to produce bio-based alkanes suitable for use as fuels. To enhance the processing efficiency, it is preferable to use a solid base rather than K3PO4. Studies have shown that hydrotalcite-supported Pd and Cu catalysts can achieve the same reaction. Additionally, replacing butanol with i-propanol leads to improved product yields (Sreekumar et al., 2014). The use of Pd-Cu bimetallic nanoparticles supported on hydrotalcite is particularly advantageous because it mitigates decarbonylation reactions, which can reduce yields compared to monometallic Pd catalysts (Goulas et al., 2016). Furthermore, life cycle analyses of this process indicate a significant reduction (approximately 50 %-80 %) in greenhouse gas emissions when it is optimized for diesel production. By modifying the feedstock to include additional methyl ketones obtained from furans, further coupling reactions can occur, yielding higher molecular weight products (up to C45) suitable for use as lubricants (Balakrishnan et al., 2015).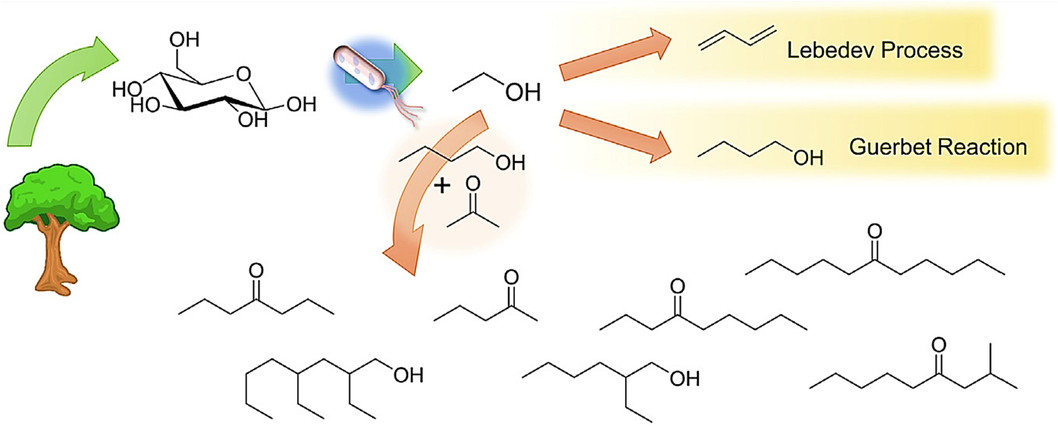
Routes for carbon–carbon coupling of ethanol, including the Lebedev process, the Guerbet reaction, and cross-coupling with the products of acetone-butanol-ethanol fermentation (Abdulrazzaq and Schwartz, 2019b).
5 Catalytic conversion of bioethanol to renewable fuels
The catalytic transformation of bioethanol into sustainable fuels is a pivotal pathway for advancing the cause of environmentally friendly energy production. In the quest for hydrogen generation, bioethanol can undergo steam reforming or autothermal reforming processes, facilitated by metal catalysts, to yield a hydrogen-rich gas. This hydrogen resource can serve as an immaculate energy carrier, finding applications in cutting-edge technologies such as fuel cells and other hydrogen-based systems (BISWAS and KUNZRU, 2007a). Concurrently, the catalytic conversion of bioethanol extends to the synthesis of hydrocarbon fuels such as gasoline, diesel, and aviation fuels. By judiciously employing suitable catalysts and precise reaction conditions, bioethanol can undergo a series of transformations, including dehydration, oligomerization, and hydrodeoxygenation processes, ultimately resulting in hydrocarbon molecules closely resembling conventional fossil fuels. This presents an extraordinary solution for curtailing the carbon footprint of the transportation and aviation sectors (Lovón-Quintana et al., 2017b). In summary, the multifaceted catalytic conversion of bioethanol into both hydrogen and hydrocarbon fuels underscores its potential to usher in a transformative era in the renewable energy landscape while concurrently addressing environmental concerns.
5.1 Hydrogen
Hydrogen has garnered substantial attention from both the scientific community and industries due to its potential as a clean-burning fuel. Presently, the primary method of hydrogen production involves methane steam reforming. However, alcohols, such as EtOH, have emerged as viable alternatives. They exhibit the advantage of facile decomposition in the availability of water at lessen temperatures (typically in the range of 250–600 ℃), offering the prospect of cleaner hydrogen generation (Jalowiecki-Duhamel et al., 2010), (BISWAS and KUNZRU, 2007b). Mondal et al., (2015a) conducted a study on the catalytic oxidative steam reforming of bio-ethanol for hydrogen production using a Rh-promoted Ni/CeO2–ZrO2 catalyst. TEM of the catalyst, both fresh and after 36 h of operation, are presented in Fig. 13 (A, B). It revealed that the size of NiO crystals fall within the range of 50–100 nm at the very early stage, consistent with XRD data. Notably, the TEM images of the catalyst after use show carbon deposits agglomerated on the catalyst surface (Bespalko et al., 2011). It's worth mentioning that there was no significant change in NiO crystallite size among both stages that represents a negligible sintering of the catalyst. However, the deactivation of catalysts mostly arises from the accumulation of carbon on the surface of the catalyst, hence diminishing the number of active sites available for the reaction. Fig. 13C presents the results of the investigation, showcasing the ethanol conversion, product selectivities, and hydrogen yield of two catalysts: a 30 %Ni/CeO2–ZrO2 catalyst and b 1 %Rh–30 %Ni/CeO2–ZrO2 catalyst. Notably, the incorporation of the noble metal Rh (1 wt%) to the 30 %Ni/CeO2–ZrO2 catalyst resulted in a significant increase in ethanol conversion and hydrogen yield (ranging from 3.5 to 4.6 mol/mol). The inclusion of this additional component resulted in an enhancement of the product, leading to an increase in the selectivity of hydrogen from 60 % to 71 %. Simultaneously, the selectivity towards carbon monoxide (CO) and methane (CH4) decreased to roughly 4 % and 3 % respectively. The improved production of hydrogen and its increased selectivity can be due to the catalytic properties of rhodium (Rh), which facilitate the water gas shift reaction and methane steam reforming processes which inhibit the creation of unwanted by-products. Furthermore, the study revealed that with increase in temperature the conversion capacity also enhanced upto 99 % at 600 °C (Fig. 13D). At 500 to 600 °C, the production of H2 also rises from 2.5 to 3.5 mol/mol in reactor. But beyond 600 °C, this decreased because of the decomposition reaction (Rxn-2). The highest hydrogen yield attained was roughly 3.5 mol of H2 per mole of ethanol supplied, which is lower than the thermodynamic value of 5. The rise in temperature from 500 to 600 °C resulted in an increase in hydrogen selectivity from 63 % to 65 % and CO2 selectivity from 18 % to 19 %. Conversely, a declining trend was noted in the selectivity of CH4 (Mondal et al., 2015b).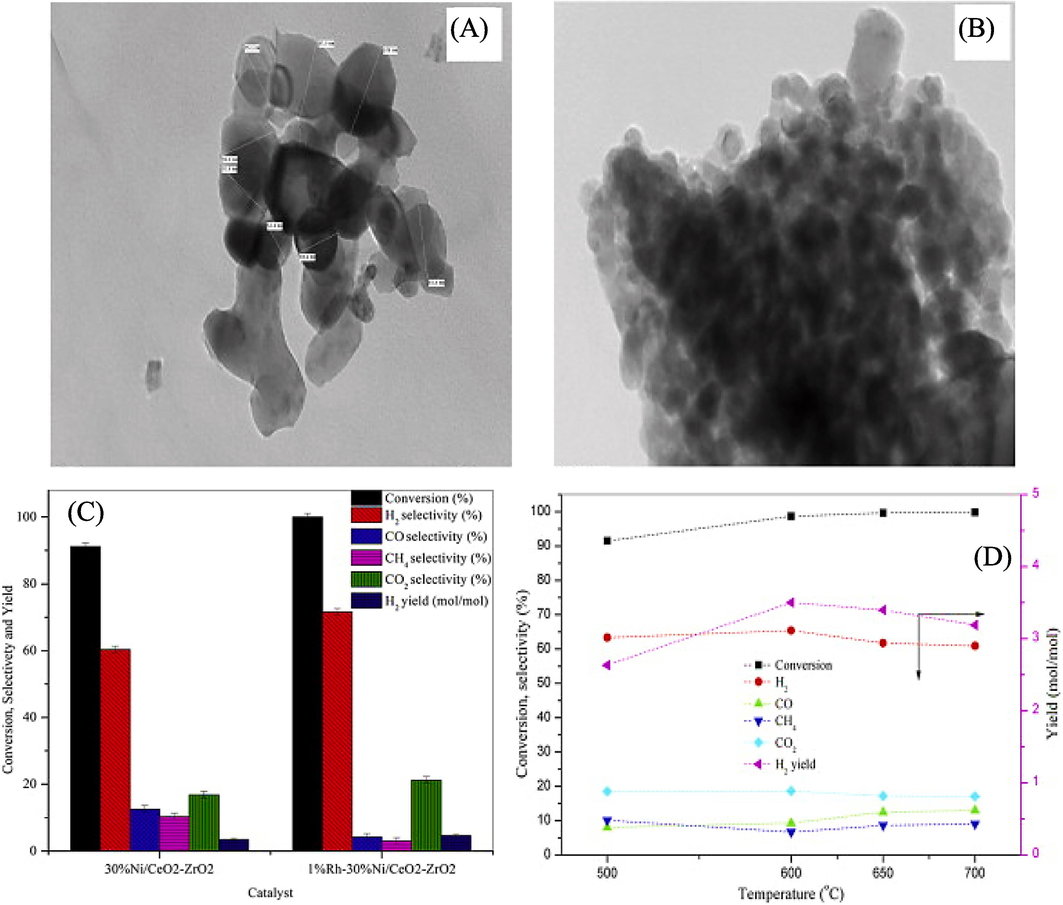
Transmission Electron Microscopy (TEM) images (A) Represents the fresh 1 %Rh–30 %Ni/CeO2–ZrO2 catalyst (B) Represents the used 30 %Ni/CeO2–ZrO2 catalyst after a certain duration of operation. (C) The effects of introducing a noble metal at an operating temperature of 600 °C, atmospheric pressure (P = 1 atm), and a molar ratio of EtOH/H2O/O2 = 1:9:0.35. (D) The impact of temperature on ethanol conversion, product selectivity, and hydrogen (H2) yield (Mondal et al., 2015b).
Fang et al. (2014) used NiXMg2AlOY ex-hydrotalcite catalysts to do a thorough investigation of hydrogen production from bioethanol. They looked at the catalysts' active sites, which are shown in Fig. 14 and comprise two cations in strong interaction—aluminum or nickel-magnesium. The standard expression for this active site is XNi–YM, where x and y are the degrees of unsaturation of each cation. Earlier studies on Ni-based catalysts (Pirez et al., 2011) and a suggested active site for CeNiXOY catalysts are the foundations of this approach. This provides an understanding of the possible process by which ethanol transforms on NiXMg2AlOY compounds. In the context of the XNi–YM ensemble, Fig. 14A illustrates the mechanism by which acetaldehyde and H2 can be produced by the heterolytic abstraction of hydrogen from ethanol when the site (1Ni–1M) has less anionic vacancies. Every individual XNi–YM ensemble is linked to a unique reaction, and the conversion of ethanol might result in a variety of products based on the active site's degree of unsaturation. For example, Fig. 14B demonstrates that H2, CO, and CH4 are formed at the 3Ni–1M site. This model fits the observed synergistic effect in mixed oxides well, when several strongly interacting cations are present. It is worth mentioning that the presence of metallic Ni0 can also contribute to the carbon generation process from CH4 (Ziebro et al., 2010) and/or CO (Yan and Liu, 2013). Nevertheless, blaming the activity on this species alone will not adequately explain the data obtained. Using in-situ X-ray diffraction, the characteristics of the NiXMg2AlOY in the presence of H2 at different temperatures were also investigated. The XRD profile of the Ni3Mg2AlOY catalyst, acquired during in-situ treatment in H2, is shown in Fig. 14C. When comparing the diffraction patterns of the fresh catalyst to those at temperatures below 450 °C, no discernible changes were found. On the other hand, diffraction peaks at 2θ = 44.5 and 51.8°, which correspond to the (1 1 1) and (2 0 0) planes of metallic nickel, show that the metallic Ni0 phase began to form at 450 °C for 10 h. As the temperature rose to 620 °C, these peaks grew somewhat stronger, and at 76.2 °C, a new peak connected to the Ni (2 2 0) plane appeared. To prevent volume variation problems resulting from gas production, Fig. 14D shows the time history of the Single-Step Reforming (SRE) process under diluted circumstances (EtOH/H2O/N2 = 3/9/88). The Ni3Mg2AlOY catalyst fully converted ethanol at 450 °C and generated the anticipated SRE products, such as H2 and CO2, along with some CH4. However, no CO production was seen. In these conditions, the H2 yield was measured at 3.0 mol molEtOH−1. For this reaction, a partial order to ethanol is indicated by the increase in conversion with decreasing ethanol partial pressure. The formation of CH4 dropped to almost nothing as the reaction temperature rose to 650 °C, whereas the formation of CO rose to approximately 11 %. On a dry basis, the product distribution at 650 °C was 13 % CO2, 76 % H2, 11 % CO, and 0.2 % CH4 (Mattos et al., 2012). One of the greatest findings published for inexpensive catalysts is the H2 productivity of 5.0 mol molEtOH−1on the Ni3Mg2AlOY catalyst at 650 °C. Importantly, at high temperatures, the water gas shift equilibrium limits reach the amount of 6.0 mol molEtOH−1, and the obtained result exceeds previous reports (Lucrédio et al., 2010), where 4.5 mol molEtOH−1 was reported using 150 mg of LaNiMgAl catalyst. and (with 200 mg of NiMg4ZnAl catalyst at 700 °C) 4.9 mol molEtOH−1 (Zeng et al., 2011).
Active site and mechanism modeling of NiXMg2AlOY catalysts for H2 production from transformation of bio-ethanol. Nin+: Ni2+ or Niδ+; Mm+: Mg2+ or Al3+. (A) 1Ni–1M site, (B) 3Ni–1M site. (C) In situ XRD patterns in H2 for (A) Ni1Mg2AlOY and (B) Ni3Mg2AlOY mixed oxides. MgO (○), Ni–Mg–O (■), NiO (▽), Ni (▾). (D) Efficiency of the Ni3Mg2AlOY catalyst pre-treated in H2 at 450 °C toward H2 production. D) Ethanol conversion (♦), H2 (♢), CO2 (○), CO (●), CH4 (▵) formation and H2 yield (♢). EtOH/H2O/N2 = 3/9/88 (Fang et al., 2014).
5.2 Hydrocarbon fuels
Fuels classified as hydrocarbons, such as petrol, diesel, and jet fuel, are essential energy sources for industry and transportation. They are produced by refining crude oil and are the main energy source used by automobiles and aeroplanes around the globe. Seeking greener and more sustainable substitutes for traditional hydrocarbon fuels is a major emphasis in the energy sector (Van der Borght et al., 2015b). The study conducted by Quintana et al. (2017) examined the potential of carbonate hydroxyapatite (CHAP) as a catalyst for the conversion of ethanol into hydrocarbon fuels (Lovón-Quintana et al., 2017c). Fig. 15 illustrates the ethanol reaction profiles over the CHAP catalyst with respect to time (h), reaction temperature (°C), and modified residence time W/FEthanol (g h mol−1). To facilitate investigation, the reaction products were categorized into two primary groups: namely, lighter hydrocarbons including carbon chain lengths ranging from C1 to C3, and hydrocarbons containing carbon chain lengths spanning from C4 to C18 +. Fig. 15A illustrates that after around two hours of continuous reaction, ethanol conversion reached a steady state. Ethanol conversion was roughly 15 % at lower reaction temperatures (300 °C), but it rose with temperature and peaked at 500 °C (nearly 100 %) before falling to 41 % at 600 °C. Fig. 15B shows that from 300 to 500 °C, the production of C1–C3 stayed below 8 %. At temperatures more than 500 °C, on the other hand, the yield abruptly rose to 40 %. Fig. 15C shows that as reaction temperature rose, the yield of hydrocarbons higher than C3 has been increased. This yield peaked at about 97 % at 500 °C and then dropped to 1 % at 600 °C. Significant quantities of dienes, aromatics (Fig. 15E), and ketones (Fig. 15F) were found between 400 and 500 °C. Reactions such as the Meerwein-Ponndorf-Verley (eq. (1) (Tsuchida et al., 2006), (Jones, 2014) and consecutive dehydration of crotyl alcohol to 1,3-butadiene (eq. (2) reaction and Lebedev's reaction (eq.3) can generate dienes, which can then produce butadiene (La-Salvia et al., 2015), (Makshina et al., 2012).
Ethanol reaction profiles over CHAP catalyst: (a) ethanol conversion as a function of time-on reaction; (b) product yield to lighter hydrocarbons with carbon chain lengths C1–C3 as a function of time-on reaction; (c) product yield to hydrocarbons with carbon chain lengths C4–C18+ as a function of time-on reaction; (d–f) evolution of ethanol reaction rate (-r'Ethanol) or product formation rate (r'Product) as a function of reaction temperature at constant W/FEthanol of 40 g h mol−1; and, (g–i) evolution of ethanol reaction rate or product formation rate as a function of modified residence time W/FEthanol at constant reaction temperature of 500 °C.
At reaction temperatures near 500 °C, the CHAP catalyst showed greater activity for generating hydrocarbons C4–C18+, despite diffusional restrictions (Fig. 15D). At a fixed temperature of 500 °C, the effects of changed residence time (W/FEthanol ) on the reaction were investigated. There was a small increase observed in the production rate of product molecules per unit mass of catalyst and per unit time when the modified residence time was decreased (Fig. 15G), suggesting greater productivity of hydrocarbons C4–C18+ with increasing ethanol flow (while keeping the catalyst mass constant). Nevertheless, neither the composition of reaction products nor the conversion of ethanol were considerably impacted by the change in modified residence time. All of the W/FEthanol values' curves (Fig. 15 F-I) were almost parallel and equally spaced. The range of 20 to 40 g h mol−1produced the best yield (about 97 %) of C4–C18+, with the EtOH productivity being most stable at 40 g h mol−1. When W/F Ethanol was increased from 80 to 160 g h mol−1, the selectivity of C4–C18+ decends to almost 90 %. For the majority of hydrocarbon groups and ethanol conversion, the calculated reaction order (n) for the ethanol input was 1.0 ± 0.2. Aldehydes displayed a reaction order of roughly 1.3, but C1–C3, olefins, and dienes displayed a lesser order of 0.6 ± 0.1. All of the reactions that resulted in the synthesis of hydrocarbon groups, in summary, had rates that were directly correlated with the ethanol feed, with certain variances observed at various reaction temperatures and product compositions.
6 Techno economic analysis
A Techno-Economic Analysis (TEA) for the catalytic conversion of bioethanol into value-added chemicals and fuels is a critical step in assessing the feasibility, economic viability, and potential profitability of such a process. Commercialization efforts often hinge on the outcomes of the economic analysis (Mohsenzadeh et al., 2017a). Several TEAs have been conducted to evaluate the economic viability of converting bioethanol through catalytic processes. One instance of an economic analysis, conducted by Jamil et al. (2019) and (Jamil et al., 2022), centers on the sustainable manufacturing of bioethylene derived from bioethanol. They calculated an annual bioethylene yield of 17,300 tons, with a total expenses used for bioethanol production at 3.60 USD per ton, using wood-based organic matter. After factoring in various cost components such as chemicals, labor, and others, the final estimated price for bioethylene, as determined by American Halcon/Scientific Design, is approximately 9.75 USD per ton (U.S. Billion Ton (2011)). It is important to highlight that the overall expenditure associated with the production of bioethylene from bioethanol dehydration is comparatively lower than that of generating ethylene through the flaking of petroleum fractions. Moreover, this method presents various benefits such as a straightforward process, easily accessible raw materials, and the attainment of high-purity ethylene as the ultimate output (Avilés Martínez et al., 2012). Within this particular context, the financial aspect of bioethanol assumes a substantial role, constituting around 60–70 % of the overall expenses associated with bioethylene production (Mohsenzadeh et al., 2017b). Nevertheless, the total expense of bioethene production exhibits regional variations, which are contingent upon factors like as the accessibility and pricing of biomass feedstocks. Countries like India and Brazil, which have abundant and cost-effective biomass feedstocks, have a comparative advantage in the manufacture of bioethylene, resulting in lower expenses for manufacturing when compared to regions like America and Europe.
Saha et al. (2017) proposed a number of strategies to make bioethanol production more cost-effective using lignocellulosic biomass, including the use of inexpensive raw materials, identifying sites near feedstock derived from biomass sources to minimize shipping expenses, implementing an a multitude of intermediate goods (e.g., fructose), reducing enzyme loading, and using simultaneous saccharification and fermentation. They also proposed the use of green solvents in pretreatment steps for environmental and cost-effective benefits, as these solvents can be easily recycled. Additionally, technological enhancements can be achieved through the application of microfiltration and nanofiltration in fermentation processes to retain unreacted sugars. Bioethylene synthesis from biomass feedstocks has significant potential, but its future adoption is dependent on issues such as biomass feedstock accessibility, which currently faces rivalry for use in food, heat, and energy applications in several different nations. Additionally, the slightly higher cost of bioethylene production can be offset by the substantial reduction in greenhouse gas emissions and the conservation of fossil fuels, offering a compelling environmental and economic case. However, the acceptance of bioethylene, even at slightly higher prices, will largely depend on local policies related to the bio-based economy and their practical implementation. Furthermore, ongoing research and the development of sustainable ethylene production processes, based on detailed computational analyses, are expected to further enhance the prospects for bioethanol production from lignocellulosic biomass (Tripodi et al., 2019). The future outlook for bioethanol production hinges on factors such as pretreatment methods and the development of cost-effective technologies for an economically viable pathway (Anekwe and Isa, 2023).
6.1 Process modeling and description
Advances in catalyst and process development have informed technoeconomic and life-cycle assessments for the chemical transformation of ethanol into fungible hydrocarbon fuel blendstocks (Hannon et al., 2020a). Historical endeavors in this domain predominantly centered on trilateral procedures encompassing dehydration, oligomerization, and hydrogenation. Nevertheless, the consolidated alcohol dehydration and oligomerization (CADO) technique is increasingly becoming prominent. The majority of technologies employed for catalytic conversion of ethanol to hydrocarbons generally encompass three essential stages prior to fractionation in order to comply with fuel standards. It involves the manufacturing of ethene the degradation of ethanol is utilized. Then, the process of oligomerization which involves the conversion of ethylene molecules into hydrocarbons with larger molecular weights. At last, hydrogenation is employed to achieve saturation of these oligomers, yielding a refined renewable fuel that is appropriate for incorporation into traditional fuel blends. Recent comprehensive research has examined the chemistries and processes involved in the conversion of ethanol into midrange distillate fuels. Typically, published designs necessitate elevated temperatures in the input reactor, exceeding 400 °C, together with pressures within the range of 30 to 40 atm, and the provision of hydrogen from an external source. Companies such as Lanza Tech are currently researching techniques based on this principle in partnership with Pacific Northwest National Laboratory (“Byogy renewables,” n.d.). The anticipated costs for converting ethanol to jet fuel using these three ways range from $3.38/GJ to $7.98/GJ ($0.11 to $0.26/L jet fuel), with comparable figures for diesel. When ethanol prices vary from $17.4/GJ to $52.1/GJ ($0.37 to $1.11/L ethanol) and reported yields are considered, the cost range for jet fuel produced by the three-step process is $25.5/GJ to $86.7/GJ ($0.83 to $2.84/L). In the United States, prices for petroleum-derived jet fuel have ranged from $7.9/GJ to $26.2/GJ ($0.26 to $0.80/L) over the last five years.
6.2 Cost estimation methodology
When examining the Capital Expenditure (CAPEX) and Operating Expenditure (OPEX) aspects of this conversion process, several key factors must be considered. CAPEX entails the initial investment in infrastructure, which includes reactors, catalysts, and facilities. These costs can be substantial, particularly when specialized equipment is required for catalytic processes (Becerra et al., 2017), (Gozan, 2020). Conversely, OPEX encompasses recurring operational costs, including energy, labor, maintenance, and raw materials such as the bioethanol feedstock (Becerra et al., 2018). The efficiency and stability of the catalyst play a crucial role in managing OPEX, as higher catalyst turnover and maintenance needs can significantly impact ongoing operational expenses. The TEA's outcome is vital for determining the overall economic feasibility of the process, taking into account factors like revenue generated from value-added chemicals and fuels, potential byproducts, and market conditions. It assesses the balance between CAPEX and OPEX to ensure cost-effectiveness and long-term sustainability (Cabrera Camacho et al., 2022). In an industrial setting where ethanol is produced and recovered through distillation, a beer column is employed to separate the ethanol-rich broth resulting from fermentation. This separation process involves the release of moist ethanol vapor from the top of the column, while water and any solid wastes are discharged from the bottom. In general, the moist vapor concentration is commonly seen to be roughly 40 wt% (wt%), which corresponds to a feed solution of about 6.4 wt% (8.1 vol/vol%). The aforementioned concentration exemplifies a procedure that yields second-generation ethanol. It is anticipated that the influence of cost estimates, which are dependent on the ethanol concentration exiting the beer column, on the consolidated alcohol dehydration and oligomerization (CADO) process will be negligible. This primarily affects capital costs (CAPEX). However, the sensitivity to cost estimates is expected to be somewhat greater for the production of anhydrous ethanol from 40 wt% ethanol vapor. In the preceding example, both operational expenditures (OPEX) and capital expenditures (CAPEX) are impacted, specifically in relation to the demand for distillation steam.
In this study, we conduct cost comparisons to evaluate the conversion of wet ethanol vapor into anhydrous ethanol or fungible blendstocks using the CADO method, considering several process configurations. Both instances are predicated on an annual ethanol manufacturing capacity of 61 million gallons per year (equivalent to 231 million liters per year), a common figure for small-scale ethanol facilities in the United States and Brazil, as posited by the National Renewable Energy Laboratory (NREL) in their Techno-Economic Analysis of cellulosic ethanol production. In the case of anhydrous ethanol, moisture-containing ethanol vapor traverses a rectifying column before being dehydrated via molecular sieve. During the CADO process, the gaseous form of ethanol is introduced into a catalytic reactor where it undergoes a chemical transformation, resulting in the formation of liquid hydrocarbons, light gases of ethylene, and water. The mixture is subsequently cooled to the surrounding temperature and introduced into a 3-phase separator. Within this separator, water is extracted from the lower section, less-dense liquid hydrocarbons are obtained from the middle section, and noncondensable gases are collected from the upper section. Approximately 73 % of the gas, by mass, is returned to the reactor for the purpose of converting it into liquid fuels. The remaining portion is combusted in order to decrease the reliance on natural gas for process heat (Hannon et al., 2020b).
6.3 Economic evaluation of bioethanol conversion
The estimation of the production cost of fuel-grade ethanol derived from moist ethanol vapor was conducted utilizing the 2011 NREL model (36). The expected prices for catalyst purchases, specifically for the process of transformation of wet ethanol into fungible blendstocks, were determined based on the NREL catalyst cost estimator (37). The current and future catalyst purchase prices were projected to be $70/kg and $30/kg, respectively, taking into account the current and anticipated metal loadings. The costs mentioned were confirmed by conversation with a producer specializing in commercial catalysts. The expected catalytic lifespan for experimental powder and pilot pellets catalysts was 6 months, however it was increased to 9 months for current commercial and future catalysts. This extension was calculated using half-lives and extrapolation from 200-hour aging studies. Longer-term aging studies might lend additional credibility to these statistics. A standard electricity rate of $0.05 per kilowatt-hour (kWh), which is commonly observed in the midwestern region of the United States, was utilized. The collective expenses associated with catalyst installation and electricity consumption constituted nearly 60 % of the overall operational expenditures. A supplementary fee of $1.85 per 1,000 L of water that has been treated was incorporated to account for the expenses associated with chemical wastewater treatment additives utilized in the elimination of dissolved hydrocarbons. At a unit cost of $2,000 per ton, catalyst disposal costs covered the cost of sending used catalyst back to the supplier. To be consistent with the NREL anhydrous ethanol model, additional expenses were incorporated, such as insurance, taxes, and maintenance (36). The labor demands associated with the transformation of fungible blendstocks into ethanol were presumed to be comparable to those required for ethanol dehydration, owing to the same intricacy of the unit activities entailed.
The NREL model, modified to 2019 USD, was utilized to estimate the Capital Expenditures (CAPEX) amounting to $19.7 million for the manufacture of anhydrous ethanol from wet ethanol vapor (36). The CAPEX numbers were annualized using a multiplication by 0.15, which represents the amortization over a 10-year duration at a 12 % interest rate. In accordance with the prescribed approach, the yearly cost of capital investment amounted to $0.031 per liter of anhydrous ethanol, with a lower heating value (LHV) of $1.46 per gigajoule. The production of fungible blendstocks from wet ethanol vapor is achieved through the utilization of the Consolidated Alcohol Dehydration and Oligomerization (CADO) process. This process involves several key equipment components, including two catalytic reactors that facilitate catalyst regeneration, a three-phase decanter, and four heat exchangers. Additionally, ancillary equipment such as pumps and load-out systems are employed to support the overall operation. The assumption was made that the requirements for storage tanks of fungible blendstock and water-products would be identical to those for ethanol storage. The aqueous solution containing relatively small amounts of dissolved hydrocarbons (usually ranging from 600 to 1,500 parts per million) that remained after the separation process was directed into the wastewater treatment system. The experimental setup consisted of two compressors: one was utilized to generate the required pressure of 0.417 MPa for the passage of wet ethanol through the reactor, while the other was employed for gas recycling purposes. It should be noted that the use of compressors may not be essential in scenarios where distillation proceeds under pressure. Given that a back pressure-controlled flash system might potentially be used in practical applications for the development of a new factory with environmental considerations, the decision to add a compressor into the design can be regarded cautious. As a result, the total installed CAPEX was expected to be around $15.3 million, which is comparable to $0.067/L of ethanol supply or $0.121/L of fungible blendstocks. For both current and future technology, CAPEX for processing via CADO was considered to remain the same. Using the identical 0.15 capital recovery factor as for anhydrous ethanol, the annualized CAPEX contribution for ethanol conversion to fungible blendstocks via CADO was $0.018/L ($0.56/GJ LHV) (Hannon et al., 2020b).
7 Challenges and future perspectives
The production of liquid fuels through various methods, although often more expensive than extracting fossil fuels directly, can be made economically viable by employing available technologies and innovative solutions. The focus should be on finding cost-effective approaches rather than just the technologies themselves. It's crucial to adopt methods that are both financially sustainable and capable of large-scale production. Bioalcohol production, for example, can be cost-competitive if produced at a sufficiently large scale. Catalytic processes have great potential for green alcohol conversion, but challenges like effective catalyst design and the impact of catalyst synthesis techniques must be addressed. Catalyst deactivation, often caused by sintering, leads to reduced catalytic performance. Pore blockage by heavy hydrocarbons and product adsorption on active sites also contribute to deactivation. Pore size and process conditions play a role in catalyst deactivation and lifespan. Recovering catalysts can be challenging, especially in asymmetric homogeneous catalysis. Catalyst leaching and dissolution can occur during recovery, impacting catalyst activity. High costs, leaching rates, solvent expenses, and operating pressures can hinder catalyst recovery. Currently, activated Al2O3-based catalysts are widely used in industrial plants, but there's growing interest in zeolites for bioethanol conversion due to lower operating temperatures. However, zeolites face challenges like stability, carbon deposition, cost, and fabrication complexity. Future research will focus on improving the Al2O3 catalyst and enhancing zeolite stability, potentially revolutionizing catalytic low-alcohol conversion systems. This will require systematic research, including kinetic investigations and modeling, to optimize process parameters and develop more efficient catalytic systems.
8 Conclusion
Bioethanol valorisation has become appealing and promising due to its growing availability and cheap pricing. The catalytic upgrading of bioethanol can provide a broad spectrum of commodity chemicals, therefore mitigating the environmental effect and reliance on fossil fuels. Catalysts are important in bioethanol conversion because they control the product selectivity and distribution during the conversion processes, in addition to the process factors (such reaction temperature, feed composition, and residence duration). Due to the limits of bioethanol as a viable fuel for transportation, low alcohol can be converted by catalytic conversion into unique chemicals and fuel alternatives. The “blending wall” of ethanol, increased feedstock diversity, and increased production efficiency might lead to surplus bioethanol that could be bought at competitive prices and utilized to make a range of commodity chemicals. The relationships between the catalyst activity and structure and the appropriate process parameters were the main focus of this study. For the long-term, sustainable synthesis of chemicals from renewable bioethanol, efficient catalyst development is crucial. Even though a lot of basic research has been done to improve ethanol conversion, selectivity towards the desired product, and catalyst stability, more studies into the structure–activity relationships of catalysts and the related reaction mechanisms are still needed for the efficient upgrading of bioethanol to value-added chemicals and fuels and for the rational design of catalysts. Owing to the elevated water content in bioethanol crude, it is imperative to scrutinize the impact of water on the configuration and functionality of catalysts as well as catalytic processes. Furthermore, in order to facilitate the commercialization of the established bioethanol conversions, the related economic consequences of bioethanol valorisation should also be taken into account and evaluated in comparison with other production pathways. Nonetheless, each of these contributions demonstrates the possibility of reducing greenhouse gas emissions, expanding the variety of energy sources, and promoting a cleaner, more sustainable future.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Abdulrazzaq, H.T., Schwartz, T.J., 2019a. Catalytic Conversion of Ethanol to Commodity and Specialty Chemicals, in: Ethanol. Elsevier, pp. 3–24. 10.1016/B978-0-12-811458-2.00001-8.
- Abdulrazzaq, H.T., Schwartz, T.J., 2019b. Catalytic Conversion of Ethanol to Commodity and Specialty Chemicals, in: Ethanol. Elsevier, pp. 3–24. 10.1016/B978-0-12-811458-2.00001-8.
- Homogeneous Ethanol to Butanol Catalysis—Guerbet Renewed. ACS Catal. 2016;6:7125-7132.
- [CrossRef] [Google Scholar]
- Integration of chemical catalysis with extractive fermentation to produce fuels. Nature. 2012;491:235-239.
- [CrossRef] [Google Scholar]
- Anderson, J.E., Leone, T.G., Shelby, M.H., Wallington, T.J., Bizub, J.J., Foster, M., Lynskey, M.G., Polovina, D., 2012. Octane Numbers of Ethanol-Gasoline Blends: Measurements and Novel Estimation Method from Molar Composition. 10.4271/2012-01-1274.
- Catalytic Conversion of Low Alcohol to Hydrocarbons: Challenges, Prospects, and Future Work Considerations. Int J Energy Res. 2023;2023:1-28.
- [CrossRef] [Google Scholar]
- Araújo, W.A., 2016a. Ethanol Industry: Surpassing Uncertainties and Looking Forward, in: Global Bioethanol. Elsevier, pp. 1–33. 10.1016/B978-0-12-803141-4.00001-0.
- Araújo, W.A., 2016b. Ethanol Industry: Surpassing Uncertainties and Looking Forward, in: Global Bioethanol. Elsevier, pp. 1–33. 10.1016/B978-0-12-803141-4.00001-0.
- Dehydration of Bioethanol by Hybrid Process Liquid-Liquid Extraction/Extractive Distillation. Ind Eng Chem Res. 2012;51:5847-5855.
- [CrossRef] [Google Scholar]
- Novel pathways for fuels and lubricants from biomass optimized using life-cycle greenhouse gas assessment. Proceedings of the National Academy of Sciences. 2015;112:7645-7649.
- [CrossRef] [Google Scholar]
- Ethanol-to-ethylene dehydration on acid-modified ring-shaped alumina catalyst in a tubular reactor. Chemical Engineering Journal. 2019;374:605-618.
- [CrossRef] [Google Scholar]
- Thermodynamic and economic assessment of the production of light olefins from bioethanol. J Environ Chem Eng. 2017;5:1554-1564.
- [CrossRef] [Google Scholar]
- Kinetic modeling of polymer-grade ethylene production by diluted ethanol dehydration over H-ZSM-5 for industrial design. J Environ Chem Eng. 2018;6:6165-6174.
- [CrossRef] [Google Scholar]
- Comparative study of NiLaZr and CoLaZr catalysts for hydrogen production by ethanol steam reforming: Effect of CO2 injection to the gas reactants. Evidence of Rh role as a promoter. Appl Catal A Gen. 2011;407:204-210.
- [CrossRef] [Google Scholar]
- Bio energy (2020), http:/demoplants.bioenergy2020.eu/.
- Development of renewable energy sources market and biofuels in The European Union. J Clean Prod. 2019;228:467-484.
- [CrossRef] [Google Scholar]
- Enzyme-like Specificity in Zeolites: A Unique Site Position in Mordenite for Selective Carbonylation of Methanol and Dimethyl Ether with CO. J Am Chem Soc. 2008;130:16316-16323.
- [CrossRef] [Google Scholar]
- Ethanol conversion over ion-exchanged ZSM-5 zeolites. Appl Catal. 1990;59:13-29.
- [CrossRef] [Google Scholar]
- Assessing the economic and environmental sustainability of bio-olefins: The case of 1,3-butadiene production from bioethanol. J Clean Prod. 2022;374:133963
- [CrossRef] [Google Scholar]
- Butadiene from ethanol employing doped t-ZrO2. Molecular Catalysis. 2018;458:272-279.
- [CrossRef] [Google Scholar]
- Catalytic dehydration of ethanol to ethylene and diethyl ether over alumina catalysts containing different phases with boron modification. Journal of Porous Materials. 2019;26:599-610.
- [CrossRef] [Google Scholar]
- Reactions of ethanol over HZSM-5. Journal of Molecular Catalysis. 1990;62:289-295.
- [CrossRef] [Google Scholar]
- The biorefinery concept: Using biomass instead of oil for producing energy and chemicals. Energy Convers Manag. 2010;51:1412-1421.
- [CrossRef] [Google Scholar]
- Catalytic consequences of hydroxyl group location on the rate and mechanism of parallel dehydration reactions of ethanol over acidic zeolites. J Catal. 2010;271:251-261.
- [CrossRef] [Google Scholar]
- Challenges and Opportunities for the Encapsulation of Enzymes over Porous Solids for Biodiesel Production and Cellulose Valorization into Glucose. ChemCatChem. 2021;13:4679-4693.
- [CrossRef] [Google Scholar]
- Engineering of Active Sites in Metal-Organic Frameworks for Biodiesel Production. Adv Sustain Syst. 2021;5
- [CrossRef] [Google Scholar]
- Homogeneous Catalysis-New Approaches to Catalyst Separation, Recovery, and Recycling. Science. 2003;1979(299):1702-1706.
- [CrossRef] [Google Scholar]
- PdZnAl catalysts for the reactions of water-gas-shift, methanol steam reforming, and reverse-water-gas-shift. Appl Catal A Gen. 2008;342:63-68.
- [CrossRef] [Google Scholar]
- Single-Step Conversion of Ethanol to n -Butene over Ag-ZrO 2 /SiO 2 Catalysts. ACS Catal. 2020;10:10602-10613.
- [CrossRef] [Google Scholar]
- The first step of the propylene generation from renewable raw material: Acetone from ethanol employing CeO 2 doped by Ag. Catal Today. 2017;279:252-259.
- [CrossRef] [Google Scholar]
- de Souza Abud, A.K., de Farias Silva, C.E., 2019. Bioethanol in Brazil: Status, Challenges and Perspectives to Improve the Production, in: Bioethanol Production from Food Crops. Elsevier, pp. 417–443. https://doi.org/10.1016/B978-0-12-813766-6.00021-7.
- Selective Gas-Phase Hydrogenation of CO 2 to Methanol Catalysed by Metal-Organic Frameworks. Angewandte Chemie International Edition 2023
- [CrossRef] [Google Scholar]
- Continuous catalytic upgrading of ethanol to n-butanol and >C 4 products over Cu/CeO 2 catalysts in supercritical CO 2. Green Chemistry. 2015;17:3018-3025.
- [CrossRef] [Google Scholar]
- Structural characterization and catalytic activity of molybdenum oxide supported zirconia catalysts. Microporous and Mesoporous Materials. 2007;102:128-137.
- [CrossRef] [Google Scholar]
- Ethanol exports and trade (2020), https://ethanolrfa.org/exports-and-trade/.
- Hydrogen production from bioethanol catalyzed by NiXMg2AlOY ex-hydrotalcite catalysts. Appl Catal B. 2014;152–153:370-382.
- [CrossRef] [Google Scholar]
- Review of catalytic systems and thermodynamics for the Guerbet condensation reaction and challenges for biomass valorization. Catal Sci Technol. 2015;5:3876-3902.
- [CrossRef] [Google Scholar]
- Catalytic Upgrading of Bioethanol to Fuel Grade Biobutanol: A Review. Ind Eng Chem Res. 2015;54:7181-7194.
- [CrossRef] [Google Scholar]
- Catalytic Transformations of Ethanol for Biorefineries. J Braz Chem Soc 2014
- [CrossRef] [Google Scholar]
- A study of ethanol dehydrogenation to acetaldehyde over copper/zinc aluminate catalysts. Catal Today. 2020;354:167-175.
- [CrossRef] [Google Scholar]
- Kinetic Modelling of the Transformation of Aqueous Ethanol into Hydrocarbons on a HZSM-5 Zeolite. Ind Eng Chem Res. 2001;40:3467-3474.
- [CrossRef] [Google Scholar]
- Metal-organic framework as a heterogeneous catalyst for biodiesel production: A review. Chemical Engineering Journal Advances. 2022;12:100415
- [CrossRef] [Google Scholar]
- Synergistic Effects in Bimetallic Palladium-Copper Catalysts Improve Selectivity in Oxygenate Coupling Reactions. J Am Chem Soc. 2016;138:6805-6812.
- [CrossRef] [Google Scholar]
- Combining microbial production with chemical upgrading. Curr Opin Biotechnol. 2016;38:47-53.
- [CrossRef] [Google Scholar]
- Technology selection for rice straw-based second-generation bioethanol production in West Java. IOP Conf Ser Earth Environ Sci. 2020;599:012095
- [CrossRef] [Google Scholar]
- Emissions from light duty gasoline vehicles operating on low blend ethanol gasoline and E85. Atmos Environ. 2008;42:4498-4516.
- [CrossRef] [Google Scholar]
- Technoeconomic and life-cycle analysis of single-step catalytic conversion of wet ethanol into fungible fuel blendstocks. Proceedings of the National Academy of Sciences. 2020;117:12576-12583.
- [CrossRef] [Google Scholar]
- Yttrium-Modified Ceria As a Highly Durable Catalyst for the Selective Conversion of Ethanol to Propene and Ethene. ACS Catal. 2013;3:14-17.
- [CrossRef] [Google Scholar]
- One-step synthesis of 2-pentanone from ethanol over K-Pd/MnOx-ZrO2-ZnO catalyst. J Mol Catal A Chem. 2005;226:89-92.
- [CrossRef] [Google Scholar]
- Hydrogen production from ethanol steam reforming over Rh/CeO2 catalyst. Catal Commun. 2015;58:137-140.
- [CrossRef] [Google Scholar]
- Remarkably enhanced stability of HZSM-5 zeolite co-modified with alkaline and phosphorous for the selective conversion of bio-ethanol to propylene. Appl Catal A Gen. 2016;520:99-104.
- [CrossRef] [Google Scholar]
- Ethanol conversion to aromatic hydrocarbons over several zeolite catalysts. Reaction Kinetics and Catalysis Letters. 2006;88:135-141.
- [CrossRef] [Google Scholar]
- Production of <math> <mrow> <msub> <mtext>C</mtext> <mrow> <mn mathvariant=“bold”>3</mn> <mo>+</mo> </mrow> </msub> </mrow> </math> Olefins and Propylene from Ethanol by Zr-Modified H-ZSM-5 Zeolite Catalysts. Advances in Materials Science and Engineering. 2012;2012:1-7.
- [CrossRef] [Google Scholar]
- Selective catalytic conversion of bio-ethanol to propene: A review of catalysts and reaction pathways. Catal Today. 2015;242:243-248.
- [CrossRef] [Google Scholar]
- Direct and Selective Production of Propene from Bio-Ethanol on Sc-Loaded In 2 O 3 Catalysts. Chemistry - A European Journal. 2013;19:7214-7220.
- [CrossRef] [Google Scholar]
- Hydrogen production from ethanol steam reforming over cerium and nickel based oxyhydrides. Int J Hydrogen Energy. 2010;35:12741-12750.
- [CrossRef] [Google Scholar]
- Greener and sustainable production of bioethylene from bioethanol: current status, opportunities and perspectives. Reviews in Chemical Engineering. 2022;38:185-207.
- [CrossRef] [Google Scholar]
- Ethylene oligomerization over H- and Ni-form aluminosilicate composite with ZSM-5 and MCM-41 structure: Effect of acidity strength, nickel site and porosity. Catal Today. 2020;355:148-161.
- [CrossRef] [Google Scholar]
- The Hydrocarbon Pool in Ethanol-to-Gasoline over HZSM-5 Catalysts. Catal Letters. 2009;127:1-6.
- [CrossRef] [Google Scholar]
- Catalytic transformation of ethanol into 1,3-butadiene. Chem Cent J. 2014;8:53.
- [CrossRef] [Google Scholar]
- Aerobic oxidation of aqueous ethanol using heterogeneous gold catalysts: Efficient routes to acetic acid and ethyl acetate. J Catal. 2007;251:332-337.
- [CrossRef] [Google Scholar]
- Methanol conversion on metal-incorporated SAPO-34s (MeAPSO-34s) J Mol Catal A Chem. 2000;160:437-444.
- [CrossRef] [Google Scholar]
- Synergistic Catalysis by Lewis Acid and Base Sites on ZrO 2 for Meerwein–Ponndorf–Verley Reduction. The Journal of Physical Chemistry C. 2015;119:26540-26546.
- [CrossRef] [Google Scholar]
- Heterogeneous Catalysts for the Guerbet Coupling of Alcohols. ACS Catal. 2013;3:1588-1600.
- [CrossRef] [Google Scholar]
- Vapor-phase catalytic conversion of ethanol into 1,3-butadiene on Cr-Ba/MCM-41 Catalysts. Brazilian Journal of Chemical Engineering. 2015;32:489-500.
- [CrossRef] [Google Scholar]
- Integration of the first and second generation bioethanol processes and the importance of by-products. Bioresour Technol. 2014;165:3-8.
- [CrossRef] [Google Scholar]
- Propylene Oligomerization to Produce Diesel Fuel on Zr-ZSM-5 Catalyst. Chemistry and Technology of Fuels and Oils. 2013;49:156-164.
- [CrossRef] [Google Scholar]
- Potential of acetone-butanol-ethanol (ABE) as a biofuel. Fuel. 2019;242:673-686.
- [CrossRef] [Google Scholar]
- Catalytic conversion of ethanol over ZSM-11 based catalysts. Appl Catal A Gen. 2016;523:118-129.
- [CrossRef] [Google Scholar]
- The Synergistic Effect to Promote the Direct Conversion of Bioethanol into Isobutene over Ternary Multifunctional Cr x Zn y Zr z O n Catalysts. ChemCatChem. 2017;9:1758-1764.
- [CrossRef] [Google Scholar]
- A study of ZnxZryOz mixed oxides for direct conversion of ethanol to isobutene. Appl Catal A Gen. 2013;467:91-97.
- [CrossRef] [Google Scholar]
- Carbonate hydroxyapatite as a catalyst for ethanol conversion to hydrocarbon fuels. Appl Catal A Gen. 2017;542:136-145.
- [CrossRef] [Google Scholar]
- Effects of adding La and Ce to hydrotalcite-type Ni/Mg/Al catalyst precursors on ethanol steam reforming reactions. Appl Catal A Gen. 2010;388:77-85.
- [CrossRef] [Google Scholar]
- Ethanol transformation over HFAU, HBEA and HMFI zeolites presenting similar Brønsted acidity. Appl Catal A Gen. 2009;367:39-46.
- [CrossRef] [Google Scholar]
- Radical Species Detection and Their Nature Evolution with Catalyst Deactivation in the Ethanol-to-Hydrocarbon Reaction over HZSM-5 Zeolite. ACS Catal. 2011;1:417-424.
- [CrossRef] [Google Scholar]
- Catalytic study of the conversion of ethanol into 1,3-butadiene. Catal Today. 2012;198:338-344.
- [CrossRef] [Google Scholar]
- Main catalytic challenges in ethanol chemistry: A review. Catalysis Reviews. 2022;1–40
- [CrossRef] [Google Scholar]
- Production of Hydrogen from Ethanol: Review of Reaction Mechanism and Catalyst Deactivation. Chem Rev. 2012;112:4094-4123.
- [CrossRef] [Google Scholar]
- Megawati, Sediawan, W.B., Sulistyo, H., Hidayat, M., 2011. Kinetics of sequential reaction of hydrolysis and sugar degradation of rice husk in ethanol production: Effect of catalyst concentration. Bioresour Technol 102, 2062–2067. https://doi.org/10.1016/j.biortech.2010.09.084.
- Meiring, P., Hansen, A.C., Vosloo, A.P., Lyne, P.W.L., 1983. High Concentration Ethanol-Diesel Blends for Compression-Ignition Engines. https://doi.org/10.4271/831360.
- The conversion of methanol to hydrocarbons over zeolite H-beta. Microporous and Mesoporous Materials. 1999;29:173-184.
- [CrossRef] [Google Scholar]
- One-path and Selective Conversion of Ethanol to Propene on Scandium-modified Indium Oxide Catalysts. Chem Lett. 2012;41:892-894.
- [CrossRef] [Google Scholar]
- Catalytic oxidative steam reforming of bio-ethanol for hydrogen production over Rh promoted Ni/CeO2–ZrO2 catalyst. Int J Hydrogen Energy. 2015;40:2529-2544.
- [CrossRef] [Google Scholar]
- Oxidative and non-oxidative steam reforming of crude bio-ethanol for hydrogen production over Rh promoted Ni/CeO 2 -ZrO 2 catalyst. Appl Catal A Gen. 2015;499:19-31.
- [CrossRef] [Google Scholar]
- A review of catalytic upgrading of bio-oil to engine fuels. Appl Catal A Gen. 2011;407:1-19.
- [CrossRef] [Google Scholar]
- Silicon-rich H-ZSM-5 catalyzed conversion of aqueous ethanol to ethylene. J Catal. 1989;117:19-32.
- [CrossRef] [Google Scholar]
- 27Al-n.m.r. characterization of natural and synthetic zeolites. Zeolites. 1984;4:133-139.
- [CrossRef] [Google Scholar]
- n-Butanol derived from biochemical and chemical routes: A review. Biotechnology Reports. 2015;8:1-9.
- [CrossRef] [Google Scholar]
- Conversion of ethanol in aqueous solution over ZSM-5 zeolites. Appl Catal. 1990;58:119-129.
- [CrossRef] [Google Scholar]
- Atomically Dispersed Pd Supported on Zinc Oxide for Selective Nonoxidative Ethanol Dehydrogenation. Ind Eng Chem Res. 2020;59:2648-2656.
- [CrossRef] [Google Scholar]
- Catalytic Conversion of Bio-ethanol to Ethylene over La-Modified HZSM-5 Catalysts in a Bioreactor. Catal Letters. 2009;132:64-74.
- [CrossRef] [Google Scholar]
- Conversion of ethanol over transition metal oxide catalysts: Effect of tungsta addition on catalytic behaviour of titania and zirconia. Appl Catal A Gen. 2015;489:180-187.
- [CrossRef] [Google Scholar]
- Highly Efficient and Stable CeNiHZOY Nano-Oxyhydride Catalyst for H2 Production from Ethanol at Room Temperature. Angewandte Chemie International Edition. 2011;50:10193-10197.
- [CrossRef] [Google Scholar]
- A review on biofuels for light-duty vehicles in Europe. Renewable and Sustainable Energy Reviews. 2021;137:110398
- [CrossRef] [Google Scholar]
- Tunable catalytic properties of bi-functional mixed oxides in ethanol conversion to high value compounds. Catal Today. 2016;269:82-87.
- [CrossRef] [Google Scholar]
- Zinc oxide-modified mordenite as an effective catalyst for the dehydrogenation of (bio)ethanol to acetaldehyde. Sustain Energy Fuels. 2021;5:2136-2148.
- [CrossRef] [Google Scholar]
- Selective oxidation of ethanol to acetaldehyde over Au–Cu catalysts prepared by a redox method. Catal Today. 2015;241:246-254.
- [CrossRef] [Google Scholar]
- Chemicals from ethanol—The acetone one-pot synthesis. Appl Catal A Gen. 2013;458:111-118.
- [CrossRef] [Google Scholar]
- Saha, K., R, U.M., Sikder, J., Chakraborty, S., da Silva, S.S., dos Santos, J.C., 2017. Membranes as a tool to support biorefineries: Applications in enzymatic hydrolysis, fermentation and dehydration for bioethanol production. Renewable and Sustainable Energy Reviews 74, 873–890. https://doi.org/10.1016/j.rser.2017.03.015.
- Indian scenario of ethanol fuel and its utilization in automotive transportation sector. Resour Conserv Recycl. 2018;132:102-120.
- [CrossRef] [Google Scholar]
- Impact of bioethanol impurities on steam reforming for hydrogen production: A review. Int J Hydrogen Energy. 2020;45:11923-11942.
- [CrossRef] [Google Scholar]
- Sustainable production of ethylene from bioethanol over hierarchical ZSM-5 nanosheets. Sustain Energy Fuels. 2019;3:115-126.
- [CrossRef] [Google Scholar]
- Steam reforming of ethanol over copper-zirconia based catalysts doped with Mn, Ni, Ga. Int J Hydrogen Energy. 2021;46:555-564.
- [CrossRef] [Google Scholar]
- Changing the Oxygen Mobility in Co/Ceria Catalysts by Ca Incorporation: Implications for Ethanol Steam Reforming. J Phys Chem A. 2010;114:3796-3801.
- [CrossRef] [Google Scholar]
- Chemocatalytic Upgrading of Tailored Fermentation Products Toward Biodiesel. ChemSusChem. 2014;7:2445-2448.
- [CrossRef] [Google Scholar]
- Production of an acetone-butanol-ethanol mixture from Clostridium acetobutylicum and its conversion to high-value biofuels. Nat Protoc. 2015;10:528-537.
- [CrossRef] [Google Scholar]
- Enhanced ethanol dehydration on γ-Al2O3 supported cobalt catalyst. J Catal. 2019;373:276-296.
- [CrossRef] [Google Scholar]
- Direct Catalytic Conversion of Ethanol to C 5+ Ketones: Role of Pd–Zn Alloy on Catalytic Activity and Stability. Angewandte Chemie International Edition. 2020;59:14550-14557.
- [CrossRef] [Google Scholar]
- Efficient Catalytic Conversion of Ethanol to 1-Butanol via the Guerbet Reaction over Copper- and Nickel-Doped Porous. ACS Sustain Chem Eng. 2017;5:1738-1746.
- [CrossRef] [Google Scholar]
- Recent Advances in Catalytic Conversion of Ethanol to Chemicals. ACS Catal. 2014;4:1078-1090.
- [CrossRef] [Google Scholar]
- Direct Conversion of Bio-ethanol to Isobutene on Nanosized Zn x Zr y O z Mixed Oxides with Balanced Acid-Base Sites. J Am Chem Soc. 2011;133:11096-11099.
- [CrossRef] [Google Scholar]
- Meerwein–Ponndorf–Verley–Oppenauer reaction of crotonaldehyde with ethanol over Zr-containing catalysts. J Catal. 2014;316:121-129.
- [CrossRef] [Google Scholar]
- Difference between the mechanisms of propylene production from methanol and ethanol over ZSM-5 catalysts. Appl Catal A Gen. 2013;467:380-385.
- [CrossRef] [Google Scholar]
- Effect of crystal size and surface modification of ZSM-5 zeolites on conversion of ethanol to propylene. Journal of Porous Materials. 2014;21:433-440.
- [CrossRef] [Google Scholar]
- Synthesis of Acetoaldehyde, Acetic Acid, and Others by the Dehydrogenation and Oxidation of Ethanol. Catalysis Surveys from Asia. 2011;15:80-88.
- [CrossRef] [Google Scholar]
- HZSM-5 catalysed conversion of aqueous ethanol to hydrocarbons. Appl Catal A Gen. 1997;148:357-371.
- [CrossRef] [Google Scholar]
- Acetic acid production by selective oxidation of ethanol using Au catalysts supported on various metal oxide. Gold Bull. 2009;42:321-327.
- [CrossRef] [Google Scholar]
- Transition Metal B-Site Substitutions in LaAlO3 Perovskites Reorient Bio-Ethanol Conversion Reactions. Catalysts. 2021;11:344.
- [CrossRef] [Google Scholar]
- Bioethylene Production: From Reaction Kinetics to Plant Design. ACS Sustain Chem Eng. 2019;7:13333-13350.
- [CrossRef] [Google Scholar]
- Direct Synthesis of n -Butanol from Ethanol over Nonstoichiometric Hydroxyapatite. Ind Eng Chem Res. 2006;45:8634-8642.
- [CrossRef] [Google Scholar]
- U.S. Billion Ton (2011), biomass supply for a bioenergy and bioproducts industry. R.D. Perlack, and B.J. Stokes (Leads), ORNL/TM-2011/224. Oak Ridge National Laboratory, Oak Ridge TN. p 227.
- Reprint of “Ethanol to higher hydrocarbons over Ni, Ga, Fe-modified ZSM-5: Effect of metal content”. Appl Catal A Gen. 2015;504:621-630.
- [CrossRef] [Google Scholar]
- Study on the formation of 2-pentanone from ethanol over K-CuZrO2 catalysts. Journal of Fuel Chemistry and Technology. 2021;49:80-87.
- [CrossRef] [Google Scholar]
- Catalytic Upgrading of Ethanol to n -Butanol: Progress in Catalyst Development. ChemSusChem. 2018;11:71-85.
- [CrossRef] [Google Scholar]
- Effects of particle size on catalytic conversion of ethanol to propylene over H-ZSM-5 catalysts—Smaller is better. Catal Commun. 2016;73:27-33.
- [CrossRef] [Google Scholar]
- Remarkably enhanced selectivity for conversion of ethanol to propylene over ZrO2 catalysts. Fuel Processing Technology. 2017;166:140-145.
- [CrossRef] [Google Scholar]
- Ethanol conversion reaction over M/ZrO2 (M = Mg, Ca, Sr, and Ba) catalysts: effect of alkaline earth metal introduction. Reaction Kinetics, Mechanisms and Catalysis. 2018;124:363-374.
- [CrossRef] [Google Scholar]
- Highly Selective Lanthanum-Modified Zirconia Catalyst for the Conversion of Ethanol to Propylene: A Combined Experimental and Simulation Study. Catal Letters. 2020;150:150-158.
- [CrossRef] [Google Scholar]
- One-step high-yield production of renewable propene from bioethanol over composite ZnCeOx oxide and HBeta zeolite with balanced Brönsted/Lewis acidity. Appl Catal B. 2020;279:119389
- [CrossRef] [Google Scholar]
- Direct conversion of bio-ethanol to propylene in high yield over the composite of In 2 O 3 and zeolite beta. Green Chemistry. 2017;19:5582-5590.
- [CrossRef] [Google Scholar]
- Effect of the catalyst structure on the formation of carbon nanotubes over Ni/MgO catalyst. Diam Relat Mater. 2013;31:50-57.
- [CrossRef] [Google Scholar]
- Operational Management Implemented in Biofuel Upstream Supply Chain and Downstream International Trading: Current Issues in Southeast Asia. Energies (basel). 2020;13:1799.
- [CrossRef] [Google Scholar]
- Feasibility of bioethanol and biobutanol as transportation fuel in spark-ignition engine: a review. RSC Adv.. 2015;5:100184-100211.
- [CrossRef] [Google Scholar]
- Toward Understanding Metal-Catalyzed Ethanol Reforming. ACS Catal. 2015;5:3841-3863.
- [CrossRef] [Google Scholar]
- Synergy effect of MgO and ZnO in a Ni/Mg–Zn–Al catalyst during ethanol steam reforming for H2-rich gas production. Catal Today. 2011;178:206-213.
- [CrossRef] [Google Scholar]
- Revealing the roles of hydrocarbon pool mechanism in ethanol-to-hydrocarbons reaction. J Catal. 2022;413:517-526.
- [CrossRef] [Google Scholar]
- Selective conversion of bio-ethanol to propene over nano-HZSM-5 zeolite: Remarkably enhanced catalytic performance by fluorine modification. Fuel Processing Technology. 2017;167:50-60.
- [CrossRef] [Google Scholar]
- Research on propene oligomerization reaction over the Fenton’s reagent modified ZSM-5. Journal of Fuel Chemistry and Technology. 2020;48:986-992.
- [CrossRef] [Google Scholar]
- Low temperature growth of carbon nanotubes from methane catalytic decomposition over nickel supported on a zeolite. Nanotechnology. 2010;21:145308
- [CrossRef] [Google Scholar]
- Zimmermann, H., Walzl, R., 2009. Ethylene, in: Ullmann’s Encyclopedia of Industrial Chemistry. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. https://doi.org/10.1002/14356007.a10_045.pub3.




