

Translate this page into:
Design of novel anti-cancer agents targeting COX-2 inhibitors based on computational studies
⁎Corresponding author. hamada.tarayrah@gmail.com (Hamada Imtara) hamada.imtara@aaup.edu (Hamada Imtara)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The overexpression of cyclooxygenase-2 (COX-2) was clearly associated with carcinogenesis, and COX-2 as a possible target has long been exploited for cancer therapy. A group of 29 derivatives of 1, 5-diarylpyrazole was used to study its structural requirements using three-dimensional quantitative structure–activity relationship (3D-QSAR), the density functional theory method, molecular docking, and molecular dynamics. Four 3D-QSAR models were developed, and the predictive capability of the four selected models was also successfully tested using different validation methods. The contribution contours of the comparative molecular field analysis (CoMFA) and comparative molecular similarity index analysis (CoMSIA) models effectively illustrate the relationships between the various chemical characteristics and their biological activities. Using the density functional theory method with the 6-31G (d, p) basis set and the Becke, 3-parameter, Lee-Yang-Parr (B3LYP) function to evaluate chemical reactivity properties, the results obtained from energy gaps of 3.431, 3.446, and 2.727 ev for molecules numbers 21, 22, and 23 indicate that these three molecules have good chemical stability and reactivity and select the most reactive regions in the three molecules studied. Molecular docking results revealed that the active sites of the COX-2 protein (PDB code: 3PGH) were residues ARG222, THR212, HIS386, HIS207, TYR148, and ASP382, in which the most active ligands and now ligands can inhibit the COX-2 enzyme. Based on the various results obtained by molecular modeling, four new compounds (N1, N2, N3, and N4) were proposed with significant predicted activity by different 3D-QSAR models. A molecular docking study and molecular dynamics simulations of the proposed new molecules (N1 and N2) and the most active molecule over 100 ns revealed that all three molecules establish multiple hydrogen interactions with several residues and also exhibit frequent stability throughout the simulation period. As a result, it is strongly recommended to consider the two newly proposed molecules, N1 and N4, as promising candidates for novel anti-cancer agents specifically designed to target COX-2 inhibition.
Keywords
Anti-cancer
COX-2 inhibitors
3D-QSAR
DFT reactivity
Molecular docking
Molecular dynamic

1 Introduction
As illustrated today, diseases are becoming a great challenge for humanity at the moment. Cancer remains the second-most common disease and one of the most serious health challenges in the world (Wang et al., 2014). Despite the development of materials and methods used to discover drugs, the problem of mortality from this disease continues to increase in recent years (Rani et al., 2022; Whiteford et al., 2013). Traditional nonsteroidal anti-inflammatory drugs (tNSAIDs) are commonly used for managing pain and mitigating the symptoms of inflammatory conditions (Fiorucci et al., 2001; Shah and Mujwar, 2022). Furthermore, several laboratory and animal studies have demonstrated that these tNSAIDs have the potential to halt tumor growth by promoting apoptosis and altering the tumor microenvironment (B. Vendramini-Costa and E. Carvalho, 2012). The efficacy of tNSAIDs in treating diseases stems from their ability to inhibit the cyclooxygenase (COX) enzyme (Díaz-González and Sánchez-Madrid, 2015). COX plays a vital role in the synthesis of prostanoids, including prostaglandins, which are implicated in the inflammatory process (Wang and DuBois, 2008). Specifically, COX catalyzes the conversion of arachidonic acid into prostaglandins (Abbas et al., 2012; Rani and Goyal, 2019). Prostaglandins are essential mediators of signal transmission pathways and are involved in various cellular processes such as cell adhesion, growth, and differentiation. It is important to note that COX exists in three different isoforms: COX-1, COX-2, and COX-3 (Khan et al., 2022; Shah et al., 2020). Although the three enzymes are comparable in size and structure, COX enzymes are controlled by different mechanisms and have varying functional roles (Arora et al., 2020). COX-2 selective inhibitors are excellent examples of a new class of drugs that could be used as adjunctive therapy to treat cancer with fewer side effects and greater efficacy (Shah et al., 2019a). These facts explain the importance of studying this molecule and proposing new selective COX-2 inhibitors. COX-2 is an enzyme that can be induced, and its expression is increased in various types of solid tumors, including breast, prostate, and colorectal cancer (Agrawal et al., 2020; Ravi Kiran Ammu et al., 2019). Numerous studies have shown that the overexpression of COX-2 is linked to the development of carcinogens, tumor growth, spread of the cancer, and its ability to invade other tissues (Han et al., 2019). Over the past few decades, several COX-2 inhibitors, known as coxibs (e.g., Celecoxib, Valdecoxib, Parecoxib, and Rofecoxib), have been discovered. These inhibitors have been shown in multiple studies to reduce the side effects of traditional NSAIDs while exhibiting efficacy in terms of inhibiting cell proliferation, promoting apoptosis, and preventing metastasis and invasion in cancer (Abouzid et al., 2010). Regrettably, numerous clinical studies have indicated that the use of COX-2 inhibitors is associated with an elevated risk of cardiovascular events and myocardial infarction (Grover et al., 2014). Consequently, certain coxibs, including Rofecoxib and Valdecoxib, have been withdrawn from the market due to the increased cardiovascular risk (Kciuk et al., 2022a; Regulski et al., 2018).
The comparative molecular field analysis (CoMFA) and comparative similarity index analysis (CoMSIA) methodologies are employed to connect changes in dependent variables (biological activity) with various molecular characteristics, such as steric and electrostatic fields, in order to build a robust mathematical model (El fadili et al., 2023a). External and internal validation was used to validate the predictive capacity and robustness of the various models proposed (Er-rajy et al., 2022). Molecular docking analysis was performed to predict the optimized binding configuration of a ligand and also to identify the different active sites and interaction modes between a more active ligand and its target protein (Agrawal et al., 2021a; Er-rajy et al., 2023b; Mujwar and Pardasani, 2022; Zhang et al., 2022). In recent years, density functional theory (DFT) has emerged as the most commonly used quantum chemical method for computing various molecular properties in chemical, physical, and biological systems (Kciuk et al., 2022d; Yari et al., 2020). In order to comprehend the intricacy of the three-dimensional quantitative structure–activity relationship (3D-QSAR) results, chemical reactivity descriptors and a molecular quantum similarity approach were applied within the framework of conceptual DFT to study the impact of substitutions (Gholivand et al., 2022; Mohapatra et al., 2021). At that point, molecular dynamics (MD) simulations would allow us to analyze and investigate the details of the interaction and stability of the selected ligands with the selected protein. A MD simulation was carried out to assess the stability of the protein with the newly designed molecules and the most active molecules over a period of 100 ns (ns) (Amin et al., 2022). This research will undoubtedly improve the scientific community's capacity to create selective COX-2 inhibitors in the future.
2 Materiel and methods
Based on an experimental dataset of 29 derivatives synthesized by Shen-Zhen Ren et al. (Ren et al., 2018) and evaluated for their anticancer biological activity (
), different CoMFA and CoMSIA models were built. The experimental dataset was divided into two groups: a training set used for model construction and a test set used to evaluate the model's performance. The training set comprised 24 molecules, while the test set consisted of 5 molecules (Table 1).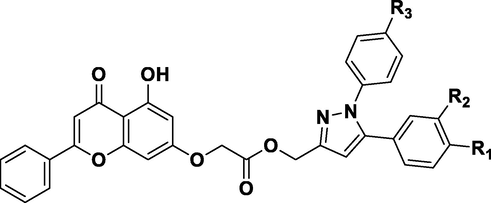
N° a
R1
R2
R3
IC50
pIC50 b
N°
R1
R2
R3
IC50
pIC50
1
-H
-H
-H
2.2
5.658
16
-Cl
-H
–CH3
2.12
5.674
2
–CH3
-H
-H
0.93
6.032
17
-H
-Cl
-H
4.43
5.354
3*
–CH3
-H
–CH3
0.42
6.377
18
-H
-Cl
–CH3
4.8
5.319
4
–CH3
-H
-Cl
1.44
5.842
19
-Br
-H
-H
2.12
5.674
5*
-H
–CH3
-H
1.36
5.866
20
-Br
-H
–CH3
1.08
5.967
6
-H
–CH3
–CH3
1.19
5.924
21
–NO2
-H
-H
1.66
5.780
7
-H
–CH3
-Cl
1.77
5.752
22*
–NO2
-H
–CH3
1.39
5.857
8
–OCH3
-H
-H
0.54
6.268
23
-H
–NO2
-H
5.94
5.226
9
–OCH3
-H
–CH3
0.23
6.638
24
-Cl
-Cl
-H
6.83
5.166
10
–OCH3
-H
-Cl
0.35
6.456
25*
-Cl
-Cl
–CH3
5.02
5.299
11
-H
–OCH3
-H
1.21
5.917
26
-Cl
-Cl
-Cl
8.5
5.071
12
-F
-H
-H
2.54
5.595
27
-CF3
-H
-H
4.33
5.364
13
-F
-H
–CH3
2.84
5.547
28
-CF3
-H
-–CH3
5.02
5.299
14
-H
-F
-H
3.53
5.452
29*
-H
-CF3
-H
4.79
5.320
15
-Cl
-H
-H
1.41
5.851
2.1 Molecular modeling and alignment
The most important and delicate step in CoMFA and CoMSIA analysis is molecular alignment. In this paper, a rigid distillation alignment was established using the SYBYL-X 2.1 program (Meng et al., 2011). All molecules were optimized using the appropriate Gasteiger-Huckel atomic partial charges within the SYBYL-X 2.1 platform and subjected to the standard Tripos force field (Tsai et al., 2010). Furthermore, the convergence criteria for the Powell gradient algorithm were set at 0.005 kcal/ mol, and a stable conformation was achieved after 1000 iterations (Gupta et al., 2022a).
2.2 Building CoMFA and CoMSIA models
Using the Sybyl X-2.1 developed created by Tripos, Inc. in the United States, research was conducted on the fields of CoMFA and CoMSIA (Yadav et al., 2018). The CoMFA descriptors were generated on a grid with a spacing of 1 and covering a three-dimensional region extending up to 4 units (Böhm et al., 1999). CoMFA calculates steric (S) and electrostatic (E) properties. The interaction energies between each molecule and the “probe” atoms were calculated for each grid point based on the energy of the Lennard-Jones and Coulombic fields. CoMFA descriptors with a distance-dependent dielectric at each grid point were calculated using a sp3 hybrid carbon atom probe with +1.0 charges. Column filtering was set to 2.0 kcal/ mol, to achieve a better signal-to-noise ratio by excluding grid points whose energy variation was below this level. The cut-off for steric and electrostatic fields was set at 30 kcal/ mol (Song et al., 2010). For CoMSIA, a Gaussian-type distance-dependent physicochemical characteristic was chosen to avoid singularities at atomic positions. The standard parameters for CoMSIA field calculations, including steric, electrostatic, hydrophobic (H), hydrogen bond acceptor (A), and hydrogen bond donor (D) effects, were similar to those used for CoMFA. However, there were no arbitrary constraints imposed on the CoMSIA field calculation. The column filtering and attenuation factors were set to their default values of 0.3 and 2.0 kcal/mol, respectively (Xu et al., 2017).
2.3 Partial least squares (PLS) analysis and validation
The inhibitory activity of COX-2 was evaluated with respect to the different fields using PLS analysis (Clark and Cramer, 1993). The leave-one-out (LOO) cross-validation technique was applied to the PLS analysis to determine the optimal number of components (NC) and derive the final CoMFA and CoMSIA models (Er-rajy et al., 2023a; Liu et al., 2014). The cross-validation correlation coefficient (Q2), the coefficient of determination (R2), and the standard error of estimate (SEE) were used to assess the models' validity (Jawarkar et al., 2022). Finally, the various CoMFA and CoMSIA models were generated through non-validated PLS analysis. The best model was selected based on the highest statistical values of Q2 and R2 and the lowest value of SEE (El fadili et al., 2023b; Roy and Pratim Roy, 2009). To evaluate the predictive power of the models, a set of molecules was selected as the external test set, and the models were used to make predictions for these molecules (Roy et al., 2012). To evaluate the predictive power of the models, a set of molecules was selected as the test set, and the models were used to make predictions for these molecules (El fadili et al., 2022; Rinnie et al., 2019).
2.4 Molecular docking
Molecular docking involves simulating the optimal conformation based on complementarity and pre-organization, enabling us to predict and obtain the binding affinity and interaction mode between the ligand and the receptor (Gupta et al., 2022b, 2022c; Morris and Lim-Wilby, 2008). To validate the results of the CoMFA and CoMSIA contour maps, a molecular docking study was performed using AutoDock 4.2 (Gupta et al., 2023; Kciuk et al., 2022b; Norgan et al., 2011; Shah et al., 2019b; Sharma et al., 2023; Shinu et al., 2022). In order to observe the ligand–protein interaction and eliminate water molecules, Discovery 2020 was used (“Free Download: BIOVIA Discovery Studio Visualizer - Dassault Systèmes,”). To explain the anticancer activities of the two most potent molecules, N° 3 and N° 9, in the three-dimensional X-ray structure of Matrix COX-2 (PDB code: 3PGH). The crystal structure was obtained from the RCSB Protein Data Bank (https://www.rcsb.org/pdb/ home/home.do) (Kurumbail et al., 1996). The active site is surrounded by the three-dimensional configuration (X = 32.622, Y = 32.033, Z = -2.592), which has a grid with a size of 60*60*60 points in the x, y, and z directions (Emon et al., 2021). Following the production of the ligand and receptor, a molecular docking procedure was carried out on two of the most active compounds as well as on the proposed new compounds.
2.5 Molecular dynamics simulation
MD simulation of the human COX-2 enzyme complexed with ligands N1, N4, and N3 was performed for 100 ns with the Desmond module of the Schrödinger programmed to assess the binding stability and orientation patterns of the acquired leads from virtual screening (Agrawal et al., 2021b; Kciuk et al., 2022c; Mujwar, 2021; Mujwar et al., 2022; Mujwar and Harwansh, 2022). For the orthorhombic-shaped simulation box, a 10 Å gap was left between the wall and the ligand–protein complex, and counter ions were injected using 0.15 M NaCl to neutralize the existing charge. Following that, 2000 iterations were performed using a merging threshold of 1 kcal/mol to decrease the system's energy. Then, over the next 100 ns, a constant temperature of 300 K and pressure of 1.013 bar were maintained. To create simulation interaction graphs, the trajectory route was set to 9.6 with an energy interval of 1.2 ps after the simulation process was completed (Gupta et al., 2022d, 2022a; Kciuk et al., 2022e; Mujwar et al., 2021; Mujwar and Tripathi, 2022; Yoshida et al., 2000). The same parameters were utilized to perform the simulation study of all three complexes considered in the current study. Finally, conformational changes in root mean square deviation (RMSD) were observed and recorded over a molecular dynamics simulation time of 100 ns.
2.6 DFT calculation studies
Computational techniques, such as DFT, are a very important method to study molecular systems of medium or larger size, with excellent performances over semi-empirical or other ab initio methods; they also offer low computational cost and reliable results for complex molecules in quantum chemistry (Horchani et al., 2020). DFT calculations at the level of the B3LYP density functional with base 6-31G (d, p) were performed to better understand the molecular structure and chemical reactivity of the 29 novel hybrids of 1, 5-diarylpyrazole and Chrysin derivatives recently synthesized by Shen-Zhen Ren et al. (Ren et al., 2018; Zhao and Truhlar, 2008). Several quantum chemical descriptors were calculated, such as total energy, the highest occupied molecular orbital energy (HOMO), the lowest unoccupied molecular orbital energy (LUMO), and the dipole moment. To visualize the results obtained, the software Gaussian 09 and GaussView were used (Şahiner et al., 2022). An analysis of the locations of the molecular frontier orbitals and the molecular electrostatic potential (MESP) has been realized (Chattaraj and Roy, 2007).
3 Results and discussion
3.1 Alignments rules
To build a very robust and reliable model, all the molecules must be superimposed in a very sensitive manner (Pandey et al., 2022). All molecule derivatives were included in a database and superimposed on the common core by the Sybyl X-2.1 rigid distillation method, using molecule number 9 (the most active) as a template (Mahdi et al., 2022). Fig. 1 depicts that all compounds are well aligned on the core (molecules on the left; the core is the molecule on the right).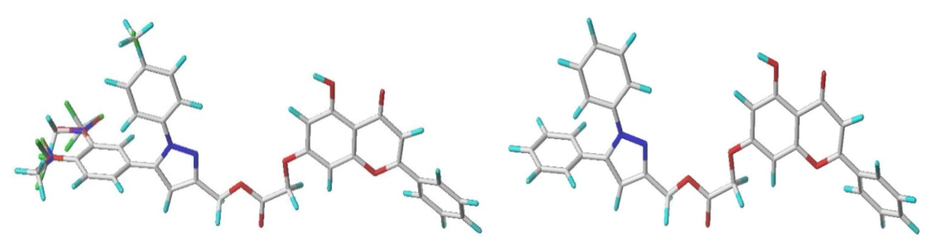
Structure superposition and training and testing set alignment.
3.2 CoMFA and CoMSIA studies
Observed and predicted activity and its residuals according to the different models are presented in supplementary table S1. To quantitatively explain and forecast the anticancer properties of a series of 1, 5-diarylpyrazole and Chrysin derivatives, QSAR models were proposed. Table S1 shows that there are very important residues at the level of molecules 8, 15, 20, 3, and 29, implying that additional research is required to assess the reliability of the proposed model. To establish the appropriate statistical parameters for each model, a PLS cross-validation analysis was performed on the independent variables collected in the training set. The outcomes for the various statistical models and their unique statistical parameters are displayed in the Table 2.
SEE
NC
Fraction
Steric
Electro
Hydrophobic
Donor
CoMFA
0.721
0.933
0.127
8
0.771
0.514
0.486
–
–
CoMSIA/SE
0.574
0.826
0.173
2
0.728
0.158
0.842
CoMSIA/SEA
0.6
0.199
0.127
5
0.837
0.173
0.680
–
–
CoMSIA/SHE
0.765
0.923
0.118
3
0.938
0.087
0.460
0.454
CoMSIA/SED
0.574
0.826
0.173
2
0.728
0.158
0.842
0.000005
CoMSIA/SEHDA
0.750
0.925
0.116
3
0.950
0.076
0.443
0.436
0.000.0.15
Based on Table 2, the CoMFA model contributes steric and electrostatic fields, which account for 48.6% and 51.4% of the variance, respectively. The values of the cross-validated correlation coefficient of the training set and the non-validated correlation coefficient R2 are 0.721 and 0.933, respectively, with an external prediction value of 0.771. The optimal number of principal components used is 8, and a lower value of SEE equals 0.127. Table 2 shows that the CoMSIA models were better models because of the values of the parameters Q2 and R2 which have larger and lower SEE. The CoMSIA/SEHDA model contributes steric, electrostatic, hydrophobic, donor, and acceptor of liaison hydrogen fields, which account for 7.6%, 44.3%, 43.6%, 0.015%, and 4.6% of the variance, respectively. The values of the cross-validated correlation coefficient and the non-validated correlation coefficient R2 are 0.75 and 0.925, respectively. The optimal number of principal components used is 3, and a lower value of SEE equals 0.116. In the CoMSIA study, the evaluation analysis of the three selected models was conducted with various champs’ combinations. The results in Table 2 indicate that the CoMSIA/SEA, CoMSIA/SEH, and CoMSIA/SEHDA models have the best external ( prediction of anti-cancer activity, with values of 0.837, 0.938, and 0.950, respectively. To ensure the robustness of the proposed model, the obtained results are subject to external validation (Alexander et al., 2015; Roy and Pratim Roy, 2009).
3.2.1 External validation
External validation allows one to evaluate and judge the predictive capacities of the obtained model; the following table groups the different external validation statistical parameters.
Using the results in Table 3, the four models, CoMFA, CoMSIA/SEA, CoMSIA/SHE, and CoMSIA/SEHDA, have better external prediction coefficients, which were used to validate the external predictive ability. As observed in Table 3, all the models have Q2 values greater than 0.5 (Golbraikh et al., 2003). The predictive ability of the external validation confirmed the favorable stability estimate and high predictive quality of the four QSAR models.
Parameter
Threshold
CoMFA
CoMSIA/SEA
CoMSIA/SHE
CoMSIA/SEHDA
> 0.5
0.721
0.600
0.765
0.750
> 0.6
0.771
0.837
0.938
0.950
–
0.771
0.833
0.937
0.947
–
0.702
0.826
0.927
0.938
|r02- r’02|
< 0.3
0.069
0.007
0.01
0.009
k
0.85 < k < 1.15
1.016
1.019
1.018
1.016
0.0001
0.004
0.001
0.0003
0.85 < k’ <1.15
0.983
0.980
0.981
0.983
0.089
0.013
0.011
0.012
r2m
r2m > 0.5
0.758
0.784
0.908
0.897
ŕ2m
ŕ2m > 0.5
0.568
0.749
0.839
0.845
To validate these results, an assessment of the robustness and predictability of the best-selected models was conducted. The computational results of the four models for the external validation test are presented in Table 3. The results in Table 3 demonstrate that all four models meet the criteria outlined by Golbraikh and Tropsha (Tropsha et al., 2003), indicating their excellent predictive ability for new chemical compounds.
3.2.2 CoMFA contour map study
To visualize the contour maps of the CoMFA model, the most active molecule, N° 9, is used. The different contour maps of the CoMFA model are presented in Fig. 2.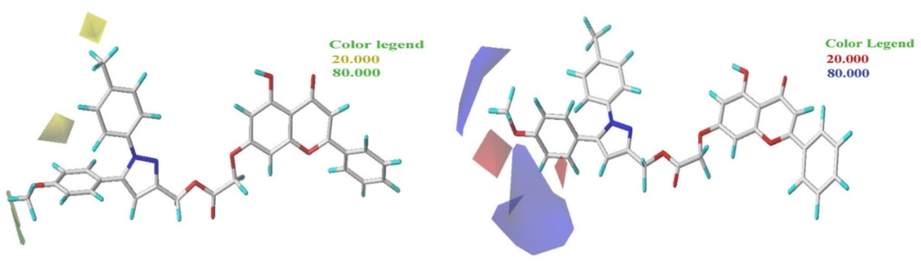
The two contour maps of the CoMFA analysis of molecular N° 9.
The map of electrostatic contours in CoMFA is represented in Fig. 2. The green parts indicate that steric contribution increases the potency of the molecule with a contribution of 80%, while the yellow parts indicate steric hindrance, which decreases the potency of the compound with a contribution of 20%. Fig. 2 shows a small part of the green contours of the steric field surrounding the R3 substituents, which increases the potency of the most active molecule.
The electrostatic contour map of CoMFA is presented in Fig. 2. The red and blue contours represent, respectively, 20% and 80% of electrostatic clutter. A blue colored region surrounds the R2 substituent, i.e., the R2 group is favorable for charging positively. The other part, a small red region located next to the oxygen molecule of substituent R1, is favorable for negatively charging.
3.2.3 CoMSIA/SEHAD contour map study
The CoMSIA/SEHAD model was utilized to view the contour map of the highest active molecule 9. The various contour maps of the CoMSIA/SEHAD model are displayed in Figs. 3 and 4.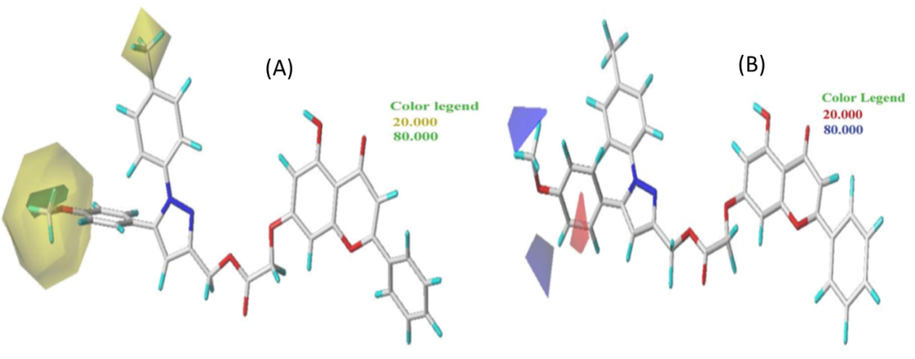
Contour maps of the CoMSIA/SE model analysis with 2 grid spacing in conjunction with molecule N° 9.
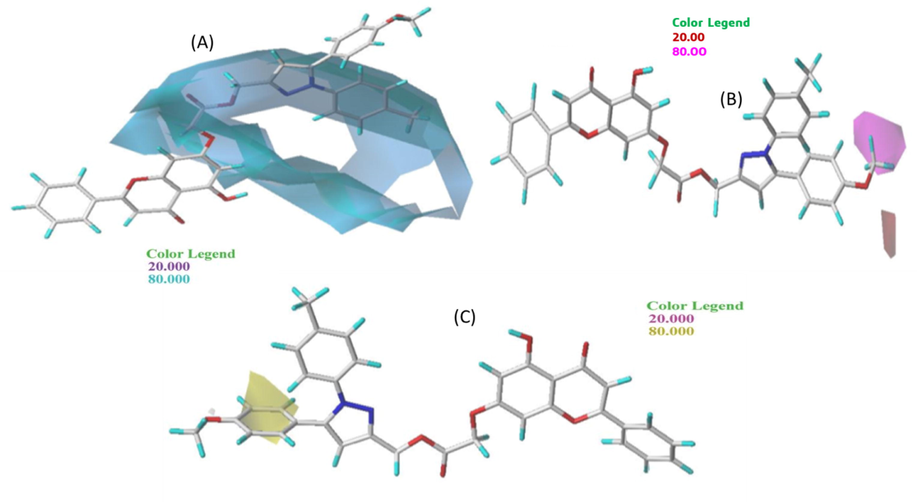
CoMSIA/HDA contour maps with 2 grid spacing in conjunction with molecule N° 9.
In Fig. 3 (A), the green parts indicate that steric contribution increases the potency of the molecule with a contribution of 80%, while the yellow parts indicate steric hindrance that decreases the potency of the compound with a contribution of 20%. A small portion of the green contours of the steric field surrounding the R1 substitution oxygen molecule can be seen, allowing an increase in the potency of the most active molecule. On the other hand, a large proportion of the yellow contours surround the R1 and R2 groups, so that the steric hindrance that decreases the potency of the most active molecule surrounds the R1 substituent. The R1 group is therefore favorable to the addition of bulky groups to COX-2 to make it more effective in stopping inflammation.
Fig. 3 (B) displays the electrostatic contour map of CoMFA. The red and blue contours represent 20% and 80% of the electrostatic bulk, respectively. A blue region surrounds the R1 substituent next to methyl, i.e., the positively charged region favorable to the methyl group. The other part, a small red region next to the benzene group, is favorable for negatively charged groups. After looking at the two steric and electrostatic contour maps, the oxygen molecule can be replaced by an atom with a stronger positive charge to make the more active molecule inhibitory.
Based on Fig. 4(A), the hydrogen bond donor contours of the selected molecule groups show that the cyan contour (80%) shows the favorable hydrogen bond donor regions, and the purple contour (20%) represents the regions where the hydrogen bond donor groups are unfavorable. A large cyan outline can be seen in the middle of the molecule and at the R3 substituents. Thus, the hydrogen bond donor has a significant influence on the inhibitory activity of the most active molecule. Therefore, in the R3 substituents, it is better to add groups that have a hydrogen bond donor to increase the inhibitory activity of COX-2.
Hydrogen bond acceptor contours of chosen compound groups: the magenta contour (80%) shows the areas favorable to hydrogen bond acceptors, and the red contours (20%) represent the areas where hydrogen bond acceptor groups are disadvantaged. In Fig. 4(B), a small magenta contour was observed next to the R3 substituents. Thus, the hydrogen bond acceptor has a significant influence on the inhibitory activity of the most active molecule.
In the hydrophobic bond contour map, the hydrophobic bond contour map of CoMSIA showed a combination of 80% yellow (favorable) and 20% pink (unfavorable) contributions in the contour map. In Fig. 4(C), a small yellow outline can be seen next to the R1 substitute. In summary, the results of the CoMSIA/SEHDA model contour analysis indicate that the R1 group benefits from the incorporation of bulky groups, while the R3 derivative favors a donor hydrogen bond to exhibit better COX-2 inhibitory activity.
3.3 Molecular docking
To identify the active site and gain a deeper understanding of the interaction between the most active molecule and its target protein, molecular docking was performed on the two most active molecules. A molecular docking study for the two most active molecules with the target COX-2 was carried out to clarify how the most active ligand interacts with its protein. The results of the molecular docking study for the two selected molecules (molecules 3 and 9) are depicted in Figs. 5 and 6.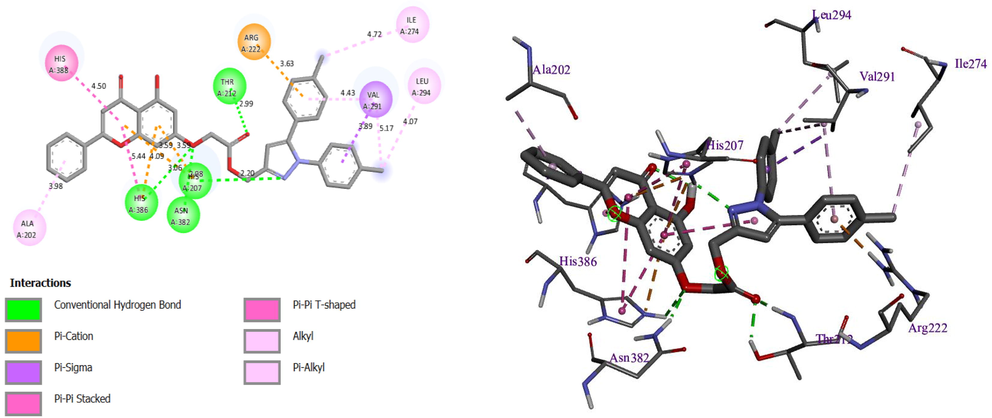
Resulted molecular docking of molecule N° 3.
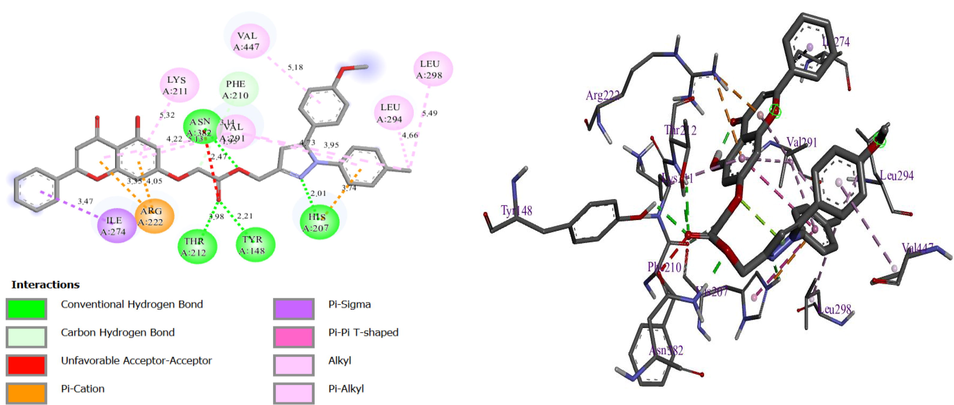
Resulted molecular docking of molecule N° 9.
The first display of these results shows that there is a four hydrogen bond with the residues histidine (HIS) 386, HIS207, threonine (THR) 212, and asparagine (ASN) 382, with distances equal to 3.06, 2.20, 2.99, and 2.08 Ã…, respectively, and a Pi-cation bond with residue arginine (ARG) 222, with a distance equal to 3.63, and Pi-Sigma bonds with residue valine (VAL) 291 and other Pi-Pi binding for molecule N° 3 (Fig. 5).
In the first display of these results for molecule N° 9, there are four hydrogen bonds with residues THR212, HIS207, tyrosine (TYR) 148, and ASN382, with a distance equal to 3.98, 2.01, 2.21, and 1.95 Ã…, respectively, and one Pi cation bond with residue ARG222, with a distance equal to 4.05 Ã…, and other Pi binding. Thus, both molecules are attacked at almost the same site, which means that both molecules are inhibitors of the selected protein. In conclusion, the two molecules demonstrated the ability to form multiple interactions, including hydrogen bonding and halogen bonding, with significant amino acids. As a result, these findings suggest that the molecules may hold promise as a potential solution for overcoming drug resistance in cancer chemotherapy.
Finally, the two compounds N° 3 and 9 have formed several interactions with important amino acids, including hydrogen bonds, alkyl bonds, and Pi bonds. As our observation shows, the ester part of both molecules interacted with very important residues, signifying the importance of this central part of the molecule. These results provide further evidence to clarify the different anticancer activities exhibited by the compounds studied as well as criteria for predicting the inhibitory activity of new molecules.
In addition, molecular dynamics and quantum computational analysis were employed to gain a comprehensive understanding of the stability of the anchored molecules, as well as exploring the molecular orbitals (HOMO and LUMO) and energy gap, enabling the influence of each group attached to the selected molecule on inhibitory activity to be identified.
3.4 Molecular quantum set analysis
3.4.1 Frontier molecular orbital analysis (FMO)
The analysis of frontier molecular orbitals at the boundary provides a qualitative calculation of the excitation capacity and electron transport characteristics of molecules. The HOMO and LUMO energies are electronic parameters that measure the nucleophilic and electrophilic characteristics of the molecules. The HOMO energy represents the ability to donate electrons, while the LUMO energy represents the ability to accept an electron. The chemical reactivity of molecules has been explained by the HOMO-LUMO energy gap, and a small energy gap denotes less kinetic stability. The low HOMO-LUMO energy gap leads to efficient electronic charge transfer from HOMO to LUMO, making the molecule highly polarizable, and molecules with a high energy gap indicate high stability with less reactivity than those with a lower energy gap. To gain a better understanding of the stability and chemical reactivity of the selected molecular structures, various chemical reactivity properties will be investigated. Different chemical properties calculated, such as:
,
energy, total energy (
), energy gap (ΔE), dipole moment, chemical hardness (η), electronegativity (χ), and electron chemical potential (μ), are shown in Table 4. The different properties of energy measured in electro volt (ev). *pIC50 observed.
N
pIC50 obs*
Et (ev)
Dipole moment
LUMO (ev)
HOMO (ev)
ΔE (ev)
χ
µ
η
1
5.6576
-59044.882
5.785
-1.531
-6.058
4.527
3.140
-3.140
2.918
2
6.0315
-50943.836
0.674
-1.611
-5.978
4.366
3.099
-3.099
2.879
3
6.3768
-52013.654
4.231
-1.521
-6.034
4.513
3.128
-3.128
2.906
4
5.8416
-63449.838
3.444
-1.556
-6.082
4.526
3.153
-3.153
2.929
5
5.8665
-50943.676
4.301
-1.531
-6.051
4.520
3.137
-3.137
2.914
6
5.9245
-52013.649
4.213
-1.518
-6.034
4.517
3.128
-3.128
2.906
7
5.7520
-63449.834
4.254
-1.547
-6.077
4.530
3.150
-3.150
2.927
8
6.2676
-52990.103
5.519
-1.516
-5.876
4.360
3.046
-3.046
2.830
9
6.6383
-54060.073
3.675
-1.533
-5.840
4.307
3.027
-3.027
2.813
10
6.4559
-65496.264
4.080
-1.552
-6.006
4.454
3.113
-3.113
2.892
11
5.9172
-52990.089
4.671
-1.505
-6.026
4.520
3.124
-3.124
2.902
12
5.5952
-52573.924
3.717
-1.548
-6.075
4.527
3.149
-3.149
2.926
13
5.5467
-53643.896
3.940
-1.543
-6.064
4.521
3.144
-3.144
2.921
14
5.4522
-52573.919
2.745
-1.555
-6.080
4.525
3.152
-3.152
2.928
15
5.8508
-62379.871
3.702
-1.562
-6.085
4.523
3.154
-3.154
2.931
16
5.6737
-63449.843
3.986
-1.561
-6.082
4.521
3.153
-3.153
2.929
17
5.3536
-62379.864
5.240
-1.533
-6.056
4.524
3.139
-3.139
2.917
18
5.3188
-63449.839
4.485
-1.536
-6.056
4.520
3.139
-3.139
2.917
19
5.6737
-119836.854
3.701
-1.556
-6.086
4.530
3.155
-3.155
2.931
20
5.9666
-120906.828
3.933
-1.543
-6.077
4.533
3.150
-3.150
2.927
21
5.7799
-55438.394
5.422
-2.699
-6.131
3.431
3.178
-3.178
2.953
22
5.8570
-56508.370
5.356
-2.672
-6.119
3.446
3.172
-3.172
2.947
23
5.2262
-55438.531
7.724
-3.101
-5.828
2.727
3.021
-3.021
2.807
24
5.1656
-74885.891
5.207
-1.552
-6.086
4.534
3.155
-3.155
2.931
25
5.2993
-75955.867
4.643
-1.553
-6.083
4.530
3.153
-3.153
2.930
26
5.0706
-75955.867
4.643
-1.553
-6.083
4.530
3.153
-3.153
2.930
27
5.3635
-59044.889
4.420
-1.565
-6.091
4.525
3.157
-3.157
2.933
28
5.2993
-60114.864
4.636
-1.553
-6.079
4.526
3.151
-3.151
2.928
29
5.3197
-59044.882
5.785
-1.531
-6.058
4.527
3.140
-3.140
2.918
HOMO and LUMO energies are crucial in the study of molecular biological activity as they provide information on molecular reactivity, electronic transitions, and intermolecular interactions, which can give insight into a molecule's bioactivity.
According to Table 4, compounds numbers 21, 22, and 23 have a lower energy ΔE value of 3.431, 3.446, and 2.727 ev, respectively, and possess excellent anti-cancer activity compared to all other derivative molecules. The small difference between HOMO and LUMO values supports the bioactivity of a molecule. In this case, compound 23 has a lower gap energy value compared to molecules 21 and 23, indicating its potential for significant anticancer activity.
Chemical hardness is a valuable parameter that is closely associated with the stability of molecular geometries. It represents the energy gap between the HOMO and LUMO orbitals and is an indicator of the molecular system's stability and reactivity. A higher HOMO-LUMO energy value suggests a more stable and less reactive system, while a lower energy gap indicates a higher reactivity and potential for chemical reactions. Therefore, chemical hardness provides valuable insights into the stability and reactivity of molecules in a molecular system.
The value of the electron chemical potential provides an explanation of the ability of electrons to escape from the ground state. According to Table 4, it is also observed that molecules 21 and 22 have the highest electron chemical potential values, with a value of -3.178 and -3.172, respectively, so these two molecules have a higher electron flow than all other molecules. However, compound 23 had a stronger dipole moment (7.724), indicating that it had the greatest dipole–dipole interaction, indicating that these compounds are highly reactive and less stable than all other synthesized molecules. Therefore, based on these results, a molecular electrostatic potential analysis was carried out on these three most reactive molecules, in order to identify the electrophilic and nucleophilic reactive sites of each molecule.
3.4.2 Molecular electrostatic potential (MEP) analysis
Based on the theory of molecular boundary orbitals, HOMO and LUMO are the most important factors because these orbitals are indicators of the reactivity and properties of molecules. The HOMO orbital is capable of donating electrons, while the LUMO can take electrons.
MEP mapping is a tool that can provide insight into the biological recognition process and hydrogen bonding interactions. It can also aid in the interpretation of electrophilic and nucleophilic reactions. The molecular electrostatic potential surfaces of the more active and reactive synthesized compounds were analyzed. Fig. 7 displays the dispersion and energy states of the HOMO and LUMO orbitals as measured by the DFT method with the 6-31G (d, p) basis set and B3LYP functional.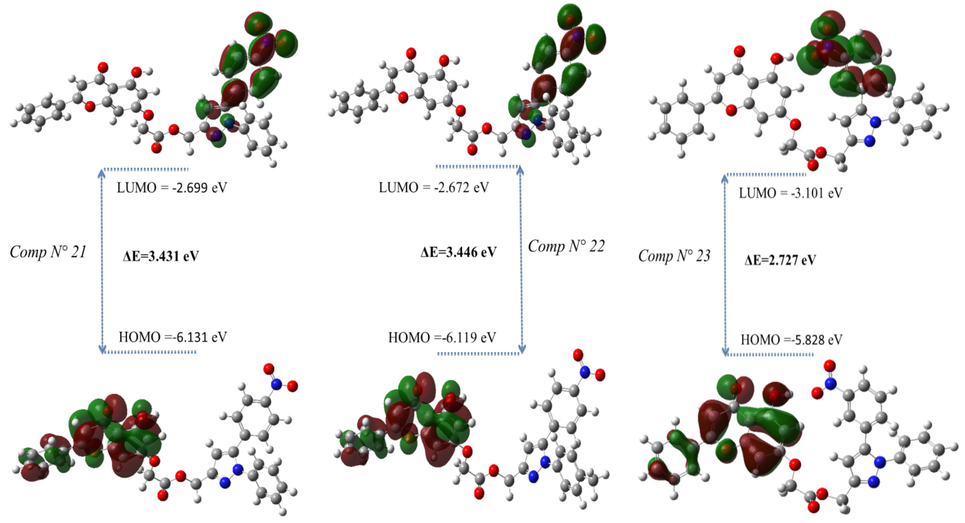
MEP of compounds the most reactive.
For molecules N° 21 and N° 22, as shown in Fig. 7, the most occupied molecular orbital is located on the chrysin fragment, and the lowest unoccupied molecular orbital is localized on the 5-(4-nitrophenyl)-1H-pyrazole group. Alternatively, the HOMO orbital of molecule 23 is confined to the chrysin fragment, except that the overlap is dark, especially for the benzene group. The same is true for the LUMO orbital, which is located on 5-(4-nitrophenyl)-1H-pyrazole.
3.4.3 Molecular electrostatic potential
The MEP is a valuable tool in molecular biology and chemistry for understanding the distribution of electric charge within a molecule and its potential interactions with other molecules. The MEP map of molecules N° 21, 22, and 23 (as shown in Fig. 8) provides insight into biological recognition processes and hydrogen bond interactions and also aids in the analysis of electrophilic and nucleophilic reactions.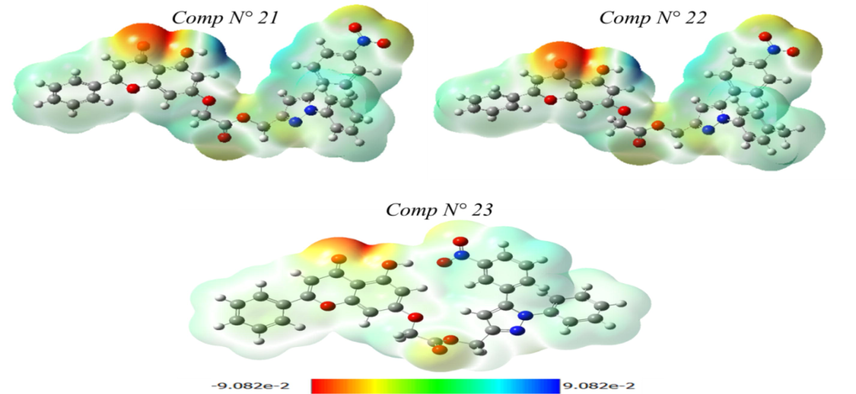
FMO of compounds the most reactive.
This characteristic is used to predict the behavior and reactivity of these molecules. Using the theoretical calculation for the optimized geometry with the 6-31G (d, p) basis set and B3LYP functional, we use the electrostatic potential to predict the reactive sites of electrophilic and nucleophilic attack for the molecules studied. The blue districts of MEP, which are positive, are associated with nucleophilic reactivity. On the other hand, the red and yellow regions, which are negative, account for electrophilic reactivity.
As shown in Fig. 8, the MEP of molecules N° 21 and 22 is a single dark blue area that is observed around the oxygen atom bound to a hydrogen atom. As a result of its high electron density, its free doublet can preferentially attack electrophilic sites. The MEP of molecule N° 21, on the other hand, reveals that the ketone (of the chrysin group) is electron-withdrawn and is surrounded by the most negative regions. For molecule N° 23, we observe that light blue regions (electropositive sites) are mainly located on the two phenyl groups. On the ketone group, there were also sizeable light-red areas (electronegative sites).
3.5 Design of new compound and study reactivity
To enrich our work, we rely on the results obtained by molecular docking and the analysis of CoMFA and CoMSIA contribution diagrams to propose new compounds similar to those synthesized. Resistance to anti-cancer drugs is reaching dangerously high levels in all regions of the world. Researchers urgently need to develop new drugs to combat this health threat. With this in mind, we have developed new anti-cancer drug candidates based on 3D-QSAR and molecular docking analysis guidelines obtained from the structural properties of the most active molecule, molecule N° 9. Based on the information obtained by different methods such as the 3D-QSAR contour map, molecular docking, and reactivity study, Fig. 9 summarizes the structural requirements based on these different methods.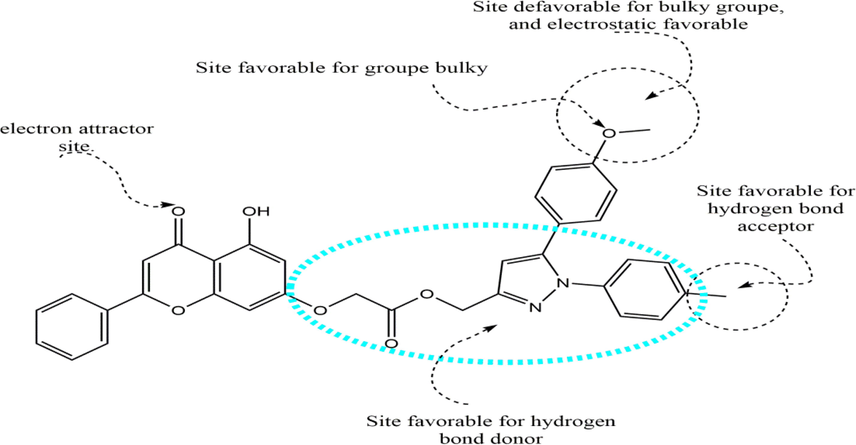
A brief description of the structural requirements based on the analysis of the 3D-QSAR and DFT studies.
The more new molecules were minimized and aligned with the studied database with the same minimization parameter as in the previous section. Next, the CoMFA and CoMSIA models were used to predict the activity of the new molecules (Table 5), along with a reactivity study for each molecule.
ID
Reactivity descriptors
Pred pIC50
Et (ev)
Dipole moment
LUMO (ev)
HOMO (ev)
Gap (ev)
CoMFA
CoMSIA/ SEA
CoMSIA/ SEH
CoMSIA/ SEHDA
N1
-54713.702
3.985
-1.540
-6.062
4.522
6.1686
5.5673
5.7085
5.7107
N2
-54589.537
5.677
-1.499
-5.325
3.826
6.1083
5.4648
5.6279
5.6300
N3
-53519.732
5.862
-1.497
-5.326
3.829
6.1086
5.4648
5.6278
5.6299
N4
-54589.310
5.767
-1.500
-5.202
5.011
6.1201
5.4806
5.6536
5.6568
According to Table 5, compounds numbered N1, N2, and N3 have a lower value ΔE of 4.522, 3.826, and 3.829 ev, so they are more reactive and also possess excellent predicted anticancer activity. To better understand the geometry of the new proposed molecules and also to identify the interaction mode with the target protein, a molecular docking study was carried out for the four new compounds.
3.5.1 Molecular docking of now molecules
In order to better understand the interaction between the newly proposed molecules and their proteins. We performed a molecular docking study for the four new molecules with the target protein COX-2 in order to clarify how the most active ligand interacts with its protein. In order to better understand the interaction between the newly proposed molecules and their proteins. A molecular docking study was carried out for the four new molecules with the targeted COX-2 protein to clarify how the new ligand interacts with its protein. The results achieved by molecular docking for the four selected new molecules are presented in Fig. 10 and Table 6. The results of the docking study for the newly designed molecules and the reference ligand for QSAR modeling against the human COX-2 enzyme have been obtained.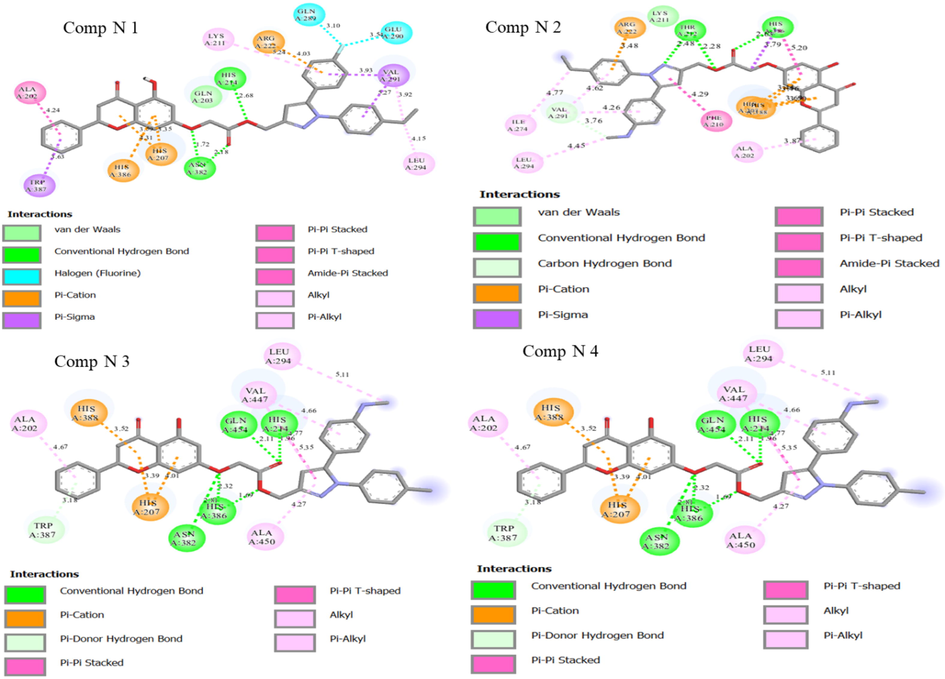
Two-dimensional visualizations were created to display the key potential interactions between the four novel ligands and active residue sites located in the COX-2 protein pocket (PDB code: 3PGH).
Ligands
Chemical Structure
Bending Energy (Kcal/Mol)
Interacting Residues
Comp N3
-10.89
THR212, ASN382, HIS388
Comp N9
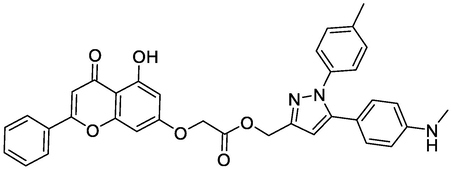
-7.52
HIS207, THR212, GLN289, ASN382
N1
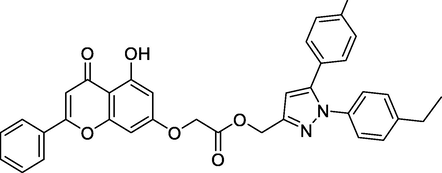
-10.93
ASN382, HIS388
N2
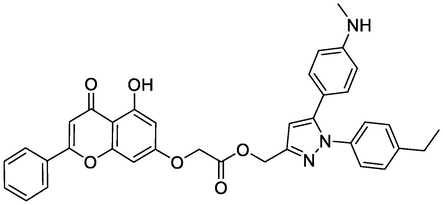
-9.62
THR212, HIS214, GLN289, ASN382
N3
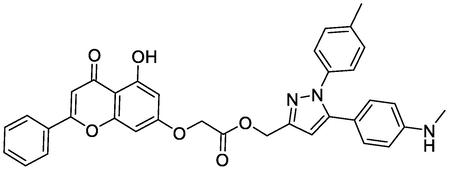
-8.84
HIS214, HIS386, GLN554
N4
-10.95
THR212, HIS388, HIS386
The binding energies of the two most active molecules and the four new compounds proposed showed that the two new compounds (N1 and N4) and compound N° 3 have a good binding affinity (-10.89, -10.93, and -10.95, respectively) for COX-2 enzymes.
Fig. 10 shows two-dimensional visualizations of the main potential interactions between the four candidate drug ligands and active residue sites in the COX-2 protein pocket (PDB code: 3PGH). A summary of the results from the molecular docking simulations is presented in Table 6. The results of this study seem very promising, as the four new compounds showed important interactions with different residues and can also serve as a basis for further studies on the design of drug candidates against cancer. Therefore, the three molecules (N1, N2, and molecule N° 3) that have a good binding affinity have the objective of a 100 ns molecular dynamics study to identify their stability in the target protein for 100 ns.
3.5.2 MD simulation of now molecules
The human COX-2 enzyme was made up of 552 amino acids, 4473 heavy atoms, and 8865 total atoms. The macromolecular secondary structure is conserved in all three simulation processes, with 32% -helices and 4% -sheets present. The results of MD are grouped in Fig. 11 and 12.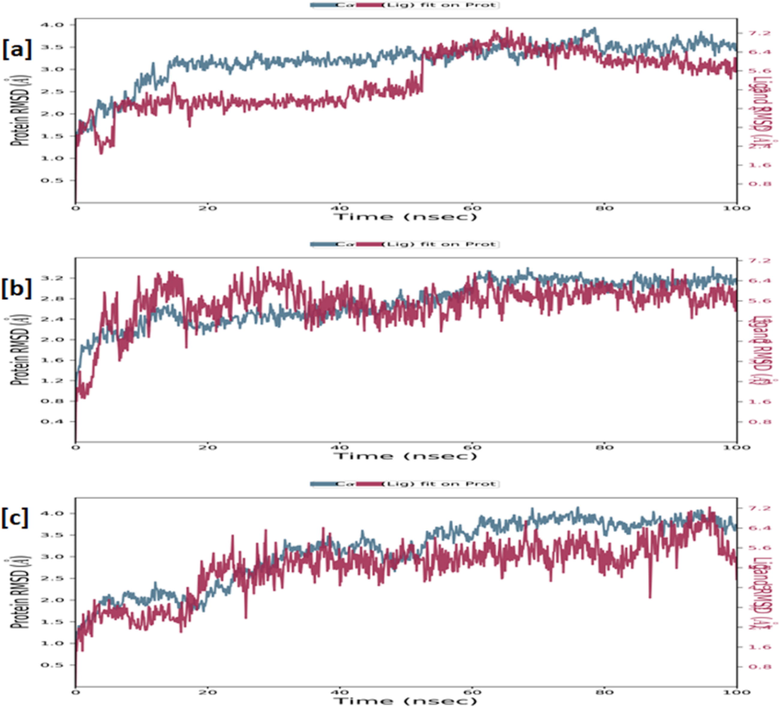
RMSD of the ligand as well as Cα-backbone of the human COX-2 enzyme complexed with ligand N1 (a), N4 (b) and N° 3 (c) measured over 100 ns of MD simulations.
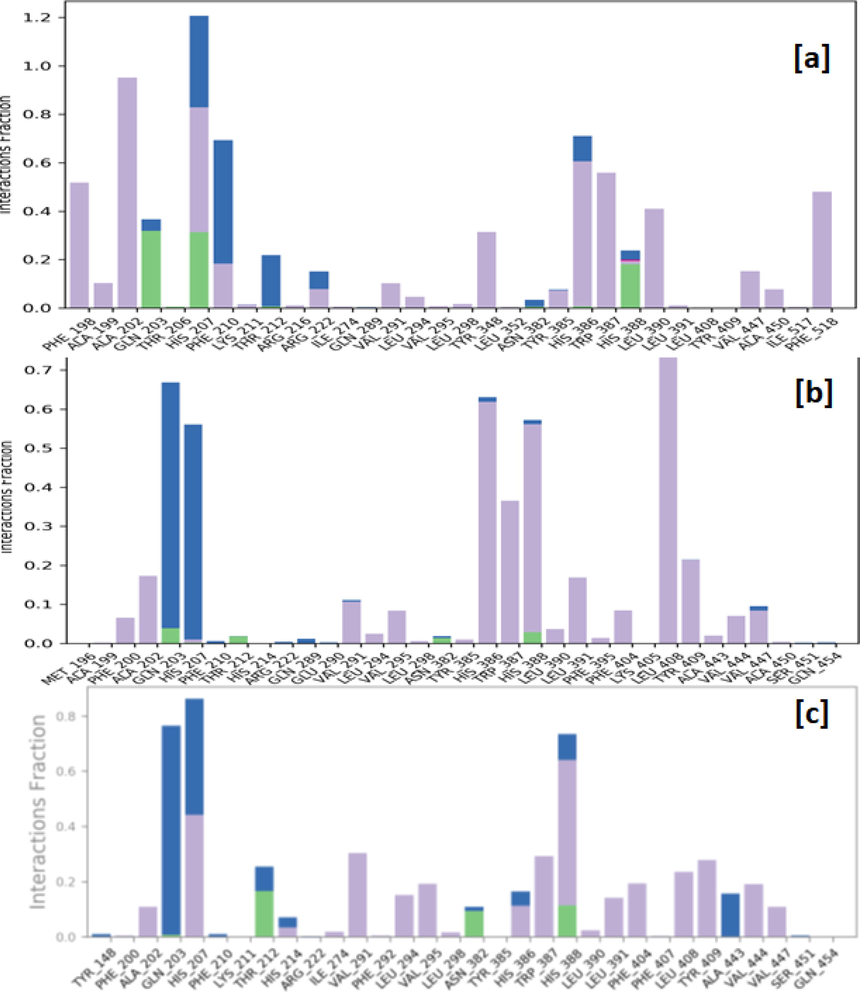
Protein-ligand contacts observed between the human COX-2 enzyme and complexed ligand N1 (a), N4 (b) and N° 3 (c) during a 100 ns MD simulation. Hydrogen bonds are shown as green bars, water bridges as blue bars, and hydrophobic interactions as purple bars.
The macromolecular complex with ligand N1 shows sufficient stability throughout the simulation. The complexed ligand N1 has eleven rotatable bonds and 44 heavy atoms out of a total of 71 atoms. The RMSD was used to calculate the stability and structural changes of the protein backbone. During the simulations, the complex was shown to be stable, with an average RMSD for the macromolecular backbone ranging from 1.5 to 3.5 Å, while the ligand remained stable for the first 50 ns with a RMSD in the range of 2.4–4.5 Å, while at 50 ns the ligand takes a conformational change within the binding cavity and remains stable throughout the simulation with RMSD in the range of 5.6–7.2 Å. The results have revealed that the ligand N1 was hydrophobically interacting with the amino acids phenylalanine (PHE) 198, alanine (ALA) 199 and 202, VAL291, TYR348 and 385, HIS386, tryptophan (TRP) 387, leucine (LEU) 390; whereas glutamine (GLN) 203, HIS388 via forming hydrogen bonds, and residues HIS 207, PHE210, THR212 were engaging with complexed ligand N1 through forming a water bridge.
The macromolecular complex with ligand N4 shows sufficient stability throughout the simulation. The complexed ligand N4 has eleven rotatable bonds and 45 heavy atoms out of a total of 76 atoms. During the simulations, the complex was shown to be stable, with an average RMSD for the macromolecular backbone ranging from 2.0 to 3.2 Å, while the ligand takes a couple of moves within the very early phase of the simulation to achieve the most stabilized conformation within the macromolecular cavity and remains stable throughout the simulation with RMSD in the range of 4.0–6.4 Å. The results showed that the ligand N4 was hydrophobically interacting with the amino acids PHE200, ALA202, VAL291, VAL295, HIS386, TRP387, HIS388, LEU391, PHE404, LEU408, TYR409, whereas GLN203 and HIS207 were forming a water bridge with the complexed ligand N4 and no stable hydrogen bonds were observed throughout the simulation.
The macromolecular complex with ligand N°3 shows sufficient stability throughout the simulation. The complexed ligand N° 3 has ten rotatable bonds and 43 heavy atoms out of a total of 71 atoms. During the simulations, the complex was shown to be stable, with an average RMSD for the macromolecular backbone ranging from 1.5 to 4.0 Å, while the ligand takes certain moves within the first 20 ns timeframe with RMSD in the range of 1.6–3.2 Å to achieve the most stabilized conformation and remains stable throughout the simulation with RMSD in the range of 5.0–7.0 Å. The results have revealed that the ligand N° 3 was hydrophobically interacting with the amino acids ALA202, HIS207, VAL291, LEU294, VAL295, HIS386, TRP387, HIS388, LEU391, PHE404, LEU408, TYR409, VAL444, VAL447; whereas THR212, ASN382, HIS388 via hydrogen bonds, and residues GLN203, HIS207, ALA443 were engaged in interacting with the complexed ligand N° 3 through the formation of a water bridge.
4 Conclusion
In conclusion, this study is focused on the design of new drug candidates capable of overcoming the problem of resistance to anti-cancer drugs. The results achieved from three-dimensional quantitative structure–activity relationships show that the three models constructed have a high degree of reliability and strong predictive capacity, which was assessed by several validation methods. Analysis of the contour maps of the CoMFA and CoMSIA models allows us to better understand the relationship between structure and biological activity, which would be an advance in guiding the design of novel and powerful anti-cancer molecules. The molecular docking studies show the importance of hydrogen bonds found with key amino acids of the targeted protein; they help stabilize the ligands in the active sites of the COX-2 receptor and increase the inhibitory efficacy. By the density functional theory method, with 6-31G (d, p) basis set and B3LYP functional, we find that the HOMO-LUMO energy gap of 3.431, 3.446, and 2.727 electro volt of molecules N° 21, 22, and 23 indicates that these three molecules have good chemical stability and reactivity. Based on this use of quantitative three-dimensional structure–activity relationships, molecular docking and molecular reactivity analysis provide more precise recommendations for the development of four (N1, N2, N3 and N4) novel compounds with significant activity.
Molecular docking and molecular dynamics studies at 100 ns of the three molecules (N1 and N4) and the most active molecule (N°3) showed that all three compounds exhibited significant hydrogen bonding to key amino acids of the target protein and also showed frequent stability throughout the molecular dynamics simulation period. As a result, the findings of this research may be useful for designing inhibitors that can overcome resistance to anticancer medications. Consequently, the two new molecules proposed (N1 and N4) are highly recommended as new anti-cancer agents targeting COX-2 inhibitors.
Acknowledgments
The authors extend their appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R165), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. Thankful also to Reserchers Supporting Project number (RSPD2023R732), King Saud University, Riyadh, Saudi Arabia.
Funding
This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R165), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. Thankful also to Reserchers Supporting Project number (RSPD2023R732), King Saud University, Riyadh, Saudi Arabia.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- New quinazolinone–pyrimidine hybrids: Synthesis, anti-inflammatory, and ulcerogenicity studies. Eur. J. Med. Chem.. 2012;53:141-149.
- [CrossRef] [Google Scholar]
- Structure-based molecular design, synthesis, and in vivo anti-inflammatory activity of pyridazinone derivatives as nonclassic COX-2 inhibitors. Med. Chem. Res.. 2010;19:629-642.
- [CrossRef] [Google Scholar]
- Agrawal, N., Upadhyay, P.K., Mishra, P., 2020. Analgesic, anti-inflammatory activity and docking study of 2-(substituted phenyl)-3-(naphthalen-1-yl)thiazolidin-4-ones, J. Indian Chem. Soc.
- Phytoestrogens as potential antiandrogenic agents against prostate cancer: An in silico analysis. Lett. Drug Des. Discov.. 2021;19:69-78.
- [CrossRef] [Google Scholar]
- Phytoestrogens as potential antiandrogenic agents against prostate cancer: An in silico analysis. Lett. Drug Des. Discov.. 2021;19:69-78.
- [CrossRef] [Google Scholar]
- Beware of R2: Simple, unambiguous assessment of the prediction accuracy of QSAR and QSPR models. J. Chem. Inf. Model.. 2015;55:1316-1322.
- [CrossRef] [Google Scholar]
- Binary quantitative activity-activity relationship (QAAR) studies to explore selective HDAC8 inhibitors: In light of mathematical models, DFT-based calculation and molecular dynamic simulation studies. J. Mol. Struct.. 2022;1260:132833
- [CrossRef] [Google Scholar]
- Structural investigation on the selective COX-2 inhibitors mediated cardiotoxicity: A review. Life Sci.. 2020;251:117631
- [CrossRef] [Google Scholar]
- Three-dimensional quantitative structure−activity relationship analyses using comparative molecular field analysis and comparative molecular similarity indices analysis to elucidate selectivity differences of inhibitors binding to trypsin, thrombin, and factor Xa. J. Med. Chem.. 1999;42:458-477.
- [CrossRef] [Google Scholar]
- Chattaraj, P.K., Roy, D.R., 2007. Update 1 of: Electrophilicity index. Chem Rev 107, PR46–PR74. https://doi.org/10.1021/CR078014B/ASSET/CR078014B.FP.PNG_V03.
- The probability of chance correlation using partial least squares (PLS) Quantit. Struct.-Activity Relationships. 1993;12:137-145.
- [CrossRef] [Google Scholar]
- NSAIDs: Learning new tricks from old drugs. Eur. J. Immunol.. 2015;45:679-686.
- [CrossRef] [Google Scholar]
- El fadili, M., Er-rajy, M., Imtara, H., Kara, M., Zarougui, S., Altwaijry, N., Al kamaly, O., Al Sfouk, A., Elhallaoui, M., 2022. 3D-QSAR, ADME-Tox In Silico Prediction and Molecular Docking Studies for Modeling the Analgesic Activity against Neuropathic Pain of Novel NR2B-Selective NMDA Receptor Antagonists. Processes 2022, Vol. 10, Page 1462 10, 1462. https://doi.org/10.3390/PR10081462.
- El fadili, M., Er-rajy, M., Ali Eltayb, W., Kara, M., Assouguem, A., Saleh, A., Al Kamaly, O., Zerougui, S., Elhallaoui, M., 2023a. In-silico screening based on molecular simulations of 3,4-disubstituted pyrrolidine sulfonamides as selective and competitive GlyT1 inhibitors. Arabian Journal of Chemistry 16, 105105. https://doi.org/10.1016/J.ARABJC.2023.105105
- El fadili, M., Er-rajy, M., Imtara, H., Noman, O.M., Mothana, R.A., Abdullah, S., Zerougui, S., Elhallaoui, M., 2023b. QSAR, ADME-Tox, molecular docking and molecular dynamics simulations of novel selective glycine transporter type 1 inhibitors with memory enhancing properties. Heliyon 9. https://doi.org/10.1016/j.heliyon.2023.e13706
- Antipyretic activity of Caesalpinia digyna (Rottl.) leaves extract along with phytoconstituent’s binding affinity to COX-1, COX-2, and mPGES-1 receptors: In vivo and in silico approaches. Saudi J. Biol. Sci.. 2021;28:5302-5309.
- [CrossRef] [Google Scholar]
- 2D-QSAR modeling, drug-likeness studies, ADMET prediction, and molecular docking for anti-lung cancer activity of 3-substituted-5-(phenylamino) indolone derivatives. Struct. Chem.. 2022;33:973-986.
- [CrossRef] [Google Scholar]
- Er-rajy, M., fadili, M. El, Mujwar, S., Zarougui, S., Elhallaoui, M., 2023b. Design of novel anti-cancer drugs targeting TRKs inhibitors based 3D QSAR, molecular docking and molecular dynamics simulation. https://doi.org/10.1080/07391102.2023.2170471 1–14. https://doi.org/10.1080/07391102.2023.2170471.
- QSAR, molecular docking, and molecular dynamics simulation–based design of novel anti-cancer drugs targeting thioredoxin reductase enzyme. Struct. Chem.. 2023;1–17
- [CrossRef] [Google Scholar]
- Dual inhibitors of cyclooxygenase and 5-lipoxygenase. A new avenue in anti-inflammatory therapy? Biochem. Pharmacol.. 2001;62:1433-1438.
- [CrossRef] [Google Scholar]
- Free Download: BIOVIA Discovery Studio Visualizer - Dassault Systèmes [WWW Document], n.d. URL https://discover.3ds.com/discovery-studio-visualizer-download (accessed 3.11.22).
- Evaluating anti-coronavirus activity of some phosphoramides and their influencing inhibitory factors using molecular docking, DFT, QSAR, and NCI-RDG studies. J. Mol. Struct.. 2022;1248:131481
- [CrossRef] [Google Scholar]
- Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des.. 2003;17:241-253.
- [CrossRef] [Google Scholar]
- Synthesis, biological evaluation, molecular docking and theoretical evaluation of ADMET properties of nepodin and chrysophanol derivatives as potential cyclooxygenase (COX-1, COX-2) inhibitors. Eur. J. Med. Chem.. 2014;80:47-56.
- [CrossRef] [Google Scholar]
- Indene-derived hydrazides targeting acetylcholinesterase enzyme in alzheimer’s: Design, synthesis, and biological evaluation. Pharmaceutics. 2023;15:94.
- [CrossRef] [Google Scholar]
- Gupta, N., Qayum, A., Singh, S., Mujwar, S., Sangwan, P.L., 2022c. Isolation, Cytotoxicity Evaluation, Docking, ADMET and Drug Likeness Studies of Secondary Metabolites from the Stem Bark of Anthocephalus cadamba (Roxb.). ChemistrySelect 7, e202202950. https://doi.org/10.1002/SLCT.202202950.
- Gupta, S.M., Behera, A., Jain, N.K., Kumar, D., Tripathi, A., Tripathi, S.M., Mujwar, S., Patra, J., Negi, A., 2023. Indene-Derived Hydrazides Targeting Acetylcholinesterase Enzyme in Alzheimer’s: Design, Synthesis, and Biological Evaluation. Pharmaceutics 15, 94. https://doi.org/10.3390/PHARMACEUTICS15010094/S1.
- Isolation, cytotoxicity evaluation, docking, ADMET and drug likeness studies of secondary metabolites from the stem bark of Anthocephalus cadamba (Roxb.) Chemistry Select. 2022;7:e202202950.
- [Google Scholar]
- Isolation, anticancer evaluation, molecular docking, drug likeness and ADMET studies of secondary metabolites from Psoralea corylifolia seeds. Chemistry Select. 2022;7:e202202115.
- [Google Scholar]
- Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the COX-2/STAT3 axis. Cancer Lett.. 2019;440–441:116-125.
- [CrossRef] [Google Scholar]
- New pyrazolo-triazolo-pyrimidine derivatives as antibacterial agents: Design and synthesis, molecular docking and DFT studies. J. Mol. Struct.. 2020;1199:127007
- [CrossRef] [Google Scholar]
- QSAR based virtual screening derived identification of a novel hit as a SARS CoV-229E 3CLpro Inhibitor: GA-MLR QSAR modeling supported by molecular Docking, molecular dynamics simulation and MMGBSA calculation approaches. Arab. J. Chem.. 2022;15:103499
- [CrossRef] [Google Scholar]
- Kciuk, M., Mujwar, S., Szymanowska, A., Marciniak, B., Bukowski, K., Mojzych, M., Kontek, R., 2022d. Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. International Journal of Molecular Sciences 2022, Vol. 23, Page 5892 23, 5892. https://doi.org/10.3390/IJMS23115892.
- Kciuk, M., Mujwar, S., Szymanowska, A., Marciniak, B., Bukowski, K., Mojzych, M., Kontek, R., 2022e. Preparation of Novel Pyrazolo[4,3-e]tetrazolo[1,5-b][1,2,4]triazine Sulfonamides and Their Experimental and Computational Biological Studies. International Journal of Molecular Sciences 2022, Vol. 23, Page 5892 23, 5892. https://doi.org/10.3390/IJMS23115892.
- Cyclin-dependent kinase synthetic lethality partners in DNA damage response. Int. J. Mol. Sci.. 2022;23
- [CrossRef] [Google Scholar]
- Computational bioprospecting guggulsterone against ADP Ri-2 bose phosphatase of SARS-CoV-2. Molecules. 2022;2022
- [CrossRef] [Google Scholar]
- Computational bioprospecting guggulsterone against ADP ribose phosphatase of SARS-CoV-2. Molecules. 2022;27:8287.
- [CrossRef] [Google Scholar]
- Khan, H., Sharma, K., Kumar, A., Kaur, A., Singh, T.G., 2022. Therapeutic implications of cyclooxygenase (COX) inhibitors in ischemic injury. Inflammation Research 2022 71:3 71, 277–292. https://doi.org/10.1007/S00011-022-01546-6.
- Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996;384:644-648.
- [CrossRef] [Google Scholar]
- Receptor and ligand-based 3D-QSAR study on a series of pyrazines/ pyrrolidylquinazolines as inhibitors of PDE10A enzyme. Med. Chem. Res.. 2014;23:775-789.
- [CrossRef] [Google Scholar]
- New organic PVC photo-stabilizers derived from synthesised novel coumarine moieties. Mater. Sci. Energy Technol.. 2022;5:278-293.
- [CrossRef] [Google Scholar]
- Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des.. 2011;7:146.
- [CrossRef] [Google Scholar]
- Computational investigations of three main drugs and their comparison with synthesized compounds as potent inhibitors of SARS-CoV-2 main protease (Mpro): DFT, QSAR, molecular docking, and in silico toxicity analysis. J. King Saud. Univ. Sci.. 2021;33:101315
- [CrossRef] [Google Scholar]
- Computational bioprospecting of andrographolide derivatives as potent cyclooxygenase-2 inhibitors. Biomed. Biotechnol. Res. J. (BBRJ). 2021;5:446.
- [CrossRef] [Google Scholar]
- In silico bioprospecting of taraxerol as a main protease inhibitor of SARS-CoV-2 to develop therapy against COVID-19. Struct. Chem.. 2022;33:1517-1528.
- [CrossRef] [Google Scholar]
- Molecular docking simulation-based pharmacophore modeling to design translation inhibitors targeting c-di-GMP riboswitch of Vibrio cholera. Lett. Drug Des. Discov.. 2022;20:745-754.
- [CrossRef] [Google Scholar]
- Docking based screening of curcumin derivatives: A novel approach in the inhibition of tubercular DHFR. Int. J. Comput. Biol. Drug Des.. 2021;14:297-314.
- [CrossRef] [Google Scholar]
- Repurposing benzbromarone as antifolate to develop novel antifungal therapy for Candida albicans. J. Mol. Model.. 2022;28:1-9.
- [CrossRef] [Google Scholar]
- In silico evaluation of food-derived carotenoids against SARS-CoV-2 drug targets: Crocin is a promising dietary supplement candidate for COVID-19. J. Food Biochem.. 2022;46:e14219.
- [Google Scholar]
- Inhibition of Plasmodium falciparum fatty acid biosynthesis (FAS-II Pathway) by natural flavonoids: A computer-aided drug designing approach. Chem. Afr.. 2022;5:1469-1491.
- [CrossRef] [Google Scholar]
- Rani, I., Goyal, A., 2019. ROLE OF GSK3 (GLYCOGEN SYNTHASE KINASE 3) AS TUMOR PROMOTER AND TUMOR SUPPRESSOR-A REVIEW 19, 1360–1365.
- Computational design of phosphatidylinositol 3-kinase inhibitors. Assay Drug Dev. Technol.. 2022;20:317-337.
- [CrossRef] [Google Scholar]
- Ravi Kiran Ammu, V.V.V., Garikapati, K.K., Krishnamurthy, P.T., Chintamaneni, P.K., Pindiprolu, S.K.S.S., 2019. Possible role of PPAR-γ and COX-2 receptor modulators in the treatment of Non-Small Cell lung carcinoma. Med Hypotheses 124, 98–100. https://doi.org/10.1016/J.MEHY.2019.02.024
- Synthesis, in vitro and in silico evaluation of novel trans-stilbene analogues as potential COX-2 inhibitors. Bioorg. Med. Chem.. 2018;26:141-151.
- [CrossRef] [Google Scholar]
- Design and biological evaluation of novel hybrids of 1, 5-diarylpyrazole and Chrysin for selective COX-2 inhibition. Bioorg. Med. Chem.. 2018;26:4264-4275.
- [CrossRef] [Google Scholar]
- Rinnie, Gaba, V., Rani, K., Shilpa, Gupta, M.K., 2019. QSAR study on 4-alkynyldihydrocinnamic acid analogs as free fatty acid receptor 1 agonists and antidiabetic agents: Rationales to improve activity. Arabian Journal of Chemistry 12, 1758–1764. https://doi.org/10.1016/J.ARABJC.2014.11.047
- Comparative studies on some metrics for external validation of QSPR models. J. Chem. Inf. Model.. 2012;52:396-408.
- [CrossRef] [Google Scholar]
- Comparative chemometric modeling of cytochrome 3A4 inhibitory activity of structurally diverse compounds using stepwise MLR, FA-MLR, PLS, GFA, G/PLS and ANN techniques. Eur. J. Med. Chem.. 2009;44:2913-2922.
- [CrossRef] [Google Scholar]
- Optimization of energy surface of thiophene-benzothiazole derivative Schiff base molecule with fuzzy logic modelling. Comput. Theor. Chem.. 2022;1211:113680
- [CrossRef] [Google Scholar]
- Shah, K., Mujwar, S., Gupta, J.K., Shrivastava, S.K., Mishra, P., 2019a. Molecular Docking and In Silico Cogitation Validate Mefenamic Acid Prodrugs as Human Cyclooxygenase-2 Inhibitor. https://home.liebertpub.com/adt 17, 285–291. https://doi.org/10.1089/ADT.2019.943.
- Shah, K., Mujwar, S., Krishna, G., Gupta, J.K., 2020. Computational Design and Biological Depiction of Novel Naproxen Derivative. https://home.liebertpub.com/adt 18, 308–317. https://doi.org/10.1089/ADT.2020.977
- Delineation of a novel non-steroidal anti-inflammatory drugs derivative using molecular docking and pharmacological assessment. Indian J. Pharm. Sci.. 2022;84:642-653.
- [CrossRef] [Google Scholar]
- Molecular docking and in silico cogitation validate mefenamic acid prodrugs as human cyclooxygenase-2 inhibitor. Assay Drug Dev. Technol.. 2019;17:285-291.
- [CrossRef] [Google Scholar]
- Computational design of plant-based antistress agents targeting nociceptin receptor. Chem. Biodivers.. 2023;20
- [CrossRef] [Google Scholar]
- Shinu, P., Sharma, M., Gupta, G.L., Mujwar, S., Kandeel, M., Kumar, M., Nair, A.B., Goyal, M., Singh, P., Attimarad, M., Venugopala, K.N., Nagaraja, S., Telsang, M., Aldhubiab, B.E., Morsy, M.A., 2022. Computational Design, Synthesis, and Pharmacological Evaluation of Naproxen-Guaiacol Chimera for Gastro-Sparing Anti-Inflammatory Response by Selective COX2 Inhibition. Molecules 2022, Vol. 27, Page 6905 27, 6905. https://doi.org/10.3390/MOLECULES27206905.
- Exploring 3D-QSAR for ketolide derivatives as antibacterial agents using CoMFA and CoMSIA. Lett. Drug Des. Discov.. 2010;7:149-159.
- [CrossRef] [Google Scholar]
- The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb. Sci.. 2003;22:69-77.
- [CrossRef] [Google Scholar]
- A comparison of different electrostatic potentials on prediction accuracy in CoMFA and CoMSIA studies. Eur. J. Med. Chem.. 2010;45:1544-1551.
- [CrossRef] [Google Scholar]
- B. Vendramini-Costa, D., E. Carvalho, J., 2012. Molecular Link Mechanisms between Inflammation and Cancer. Curr Pharm Des 18, 3831–3852. https://doi.org/10.2174/138161212802083707
- Pro-inflammatory prostaglandins and progression of colorectal cancer. Cancer Lett.. 2008;267:197-203.
- [CrossRef] [Google Scholar]
- The global burden of liver disease: The major impact of China. Hepatology. 2014;60:2099-2108.
- [CrossRef] [Google Scholar]
- Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease Study 2010. The Lancet. 2013;382:1575-1586.
- [CrossRef] [Google Scholar]
- Safety and efficacy of fruquintinib in patients with previously treated metastatic colorectal cancer: a phase Ib study and a randomized double-blind phase II study. J. Hematol. Oncol.. 2017;10:1-8.
- [CrossRef] [Google Scholar]
- Yadav, D., Nath Mishra, B., Khan, F., 2018. 3D-QSAR and docking studies on ursolic acid derivatives for anticancer activity based on bladder cell line T24 targeting NF-kB pathway inhibition. https://doi.org/10.1080/07391102.2018.1528888 37, 3822–3837. https://doi.org/10.1080/07391102.2018.1528888.
- Yari, A., Boochani, A., Rezaee, S., 2020. Thermoelectric and optical properties of the SrS graphene by DFT. https://doi.org/10.1080/14786435.2020.1803509 100, 3108–3124. https://doi.org/10.1080/14786435.2020.1803509.
- Density functional vibrational analysis using wavenumber-linear scale factors. Chem. Phys. Lett.. 2000;325:477-483.
- [CrossRef] [Google Scholar]
- Molecular docking-based computational platform for high-throughput virtual screening. CCF Trans. High Perform. Comput.. 2022;4:63-74.
- [CrossRef] [Google Scholar]
- Density functionals with broad applicability in chemistry. Acc. Chem. Res.. 2008;41:157-167.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105193.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




