

Translate this page into:
Design, synthesis, biological evaluation, and in silico study of quinazoline-based dual inhibitors targeting EGFR and VEGFR-2 for cancer therapy
* Corresponding author: E-mail address: a.altharawi@psau.edu.sa (A. Altharawi)
-
Received: ,
Accepted: ,
Abstract
Quinazoline derivatives with substitution at the N3 and C2 positions, were synthesized for targeting a dual inhibition of vascular endothelial growth factor receptor 2 (VEGFR) and epidermal growth factor receptor (EGFR) kinases. The final derivatives 4a-e were characterized using various spectroscopic techniques, and their antiproliferative activity against A-549, MDA, and HeLa cancer cell lines was evaluated in vitro. Among the compounds, 4e demonstrated the most significant cytotoxicity, with an IC50 value of 0.59 ± 0.01 µM against A-549 cells. The SAR analysis suggested that the presence of the ethyl acetate fragment plays a crucial role in enhancing anticancer efficacy, while the ethyl group did not notably impact its antiproliferative activity. Further investigation of compound 4e revealed its high binding affinity to EGFR, with an IC50 of 69.4 ± 1.55 nM compared to Docetaxel (IC50 = 56.1±1.17 nM). Molecular docking and molecular dynamics (MD) simulations provided valuable insights into the interactions between quinazoline derivatives, EGFR, and VEGFR-2. However, these studies suggest that 4e exhibits a good binding affinity toward EGFR and VEGFR-2 with docking scores of -4.46 kcal/mol and -4.41 kcal/mol, respectively. Ligand 4e and VEGFR-2 formed a stable complex, indicating its potential as promising anticancer drug over docetaxel.
Keywords
Cytotoxicity
EGFR
Kinases assay
Molecular dynamics
Quinazoline
VEGFR-2

1. Introduction
Cancer is currently recognized as one of the leading causes of global mortality, second only to cardiovascular diseases. The World Health Organization (WHO) predicts a significant surge in incident cancer cases, which is expected to reach ∼ 20 million by 2025 [1,2]. Despite the availability of multiple chemotherapeutic drugs, their effectiveness is often outweighed by adverse effects, particularly toxicity, which hinders their widespread use [3,4]. Consequently, there is a pressing need to develop alternative strategies to combat cancer. One promising approach is targeted chemotherapy, which aims to identify and develop chemical agents that can treat cancer with fewer side effects. In recent years, considerable advancements have been made in medicinal chemistry [5], especially with the integration of targeted therapies and personalized treatments, improving the effectiveness of cancer treatments.
The inclusion of heteroatoms, particularly nitrogen [6], in cyclic structures, has gained prominence in medicinal chemistry due to their role in the bioactive compounds, including those with antitumor and anti-inflammatory properties [7]. Quinazoline derivatives have shown great potential as therapeutic agents in cancer treatment, demonstrating efficacy against solid tumors [8]. This chemical scaffold has become a focal point in anticancer research, with substantial efforts dedicated to modifying the quinazoline structure to enhance its antitumor activity. These modifications have resulted in a diverse range of compounds with various pharmacological effects.
Quinazoline derivatives, particularly those bearing a 4-ylamino substituent, have garnered attention due to their strong and selective inhibition of the human Pin1 enzyme, positioning them as promising candidates for the development of more effective cancer therapies [9]. Additionally, compounds with a 2,4-disubstituted quinazoline structure have exhibited potent inhibition of key biological targets such as dihydrofolate reductase, aurora kinases, and tyrosine kinases [10-13]. These derivatives are associated with a wide range of biological activities, giving them versatile therapeutic applications [14-16]. Several modified quinazoline compounds, including thiazolo-, furano-, and thieno-quinazoline, have demonstrated remarkable in-vivo and in vitro anticancer effects against various human cancer cell lines [17,18]. Furthermore, quinazoline derivatives have been implicated in anticancer mechanisms through their inhibition of DNA repair enzymes [19].
Quinazoline derivatives have emerged as important components in the development of multi-kinase inhibitors, with several compounds receiving FDA approval for the treatment of various cancers. These inhibitors primarily work by interfering with key cellular processes such as transcription, DNA replication, and the activity of critical protein kinases involved in tumor progression. Among their targets, vascular endothelial growth factor receptor 2 (VEGFR) and epidermal growth factor receptor (EGFR) tyrosine kinases play essential roles in regulating processes like cell adhesion, migration, and differentiation. Mutations and overexpression of EGFR are implicated in a variety of cancers, which has led to a strong focus on EGFR inhibitors in cancer therapy development [20]. VEGFR-2, located on endothelial cells, represents another significant target for antiangiogenic treatments (disrupting the blood vessel formation required for tumor growth), thereby enhancing the efficacy of cancer therapies [21-23]. Quinazoline-based compounds remain central to cancer chemotherapy, with several FDA-approved agents such as lapatinib, gefitinib, and erlotinib playing a critical role in oncology [24] (Figure 1).

- Overview of the molecular design strategy for developing new VEGFR-2/EGFR inhibitors based on a quinazoline core framework. VEGFR-2: Vascular endothelial growth factor receptor-2, EGFR: Epidermal growth factor receptor.
Molecular hybridization has become an innovative approach in drug discovery, aiming to combine two or more active pharmacophoric elements from known molecular structures to create new compounds with enhanced activity and reduced side effects. This strategy seeks to merge the beneficial features of existing molecules while improving their therapeutic profiles. It has increasingly gained attention in the scientific community as a promising method for developing compounds with superior biological efficacy. Researchers are using this approach to design novel analogs that hold significant potential for therapeutic advancement, making them ideal candidates for further evaluation [25].
This research is centered on quinazolinones, which have shown promise as inhibitors of VEGFR-2 and EGFR enzymes, as highlighted by previous studies. The quinazoline framework was selected as the basis for developing novel anticancer agents. The design process begins with an extensive review of existing compounds that target VEGFR-2 and EGFR. Following this, we focused on modifying erlotinib derivatives by introducing a nitro group and phenyl group at the C-7 position of the quinazoline ring and N-3 position, respectively, to enhance interactions through hydrogen bonding.
To further optimize the compounds, this study builds upon our previous work on the same heterocyclic core and the same cancer cells overexpressing EGFR and VEGFR-2 genes. With the aim of expanding our understanding of the directions to pursue in future projects, we investigated the impact of incorporating different para-aryl groups at the C-2 position to evaluate the structure-activity relationship. An in-depth in silico study was conducted to refine the design and support the experimental strategy (Figure 1).
2. Materials and Methods
2.1. Chemistry
2.1.1. Materials & methods
The intermediate compound 3 and its final derivatives 4a-e were characterized using a Bruker Avance III nuclear magnetic resonance (NMR) spectrometer with a 500 MHz resolution (500 MHz for 1H & 125 MHz for 13C) in the Department of Pharmaceutical Chemistry, College of Pharmacy, Prince Sattam bin Abdulaziz University. DMSO-d6 was used as the solvent for the analysis. The peaks of deuterated DMSO and H2O appear at approximately 2.5 and 3.5 ppm, respectively. Additionally, thin-layer chromatography (TLC) was employed to monitor the reactions, with detection under UV light at 365 nm.
2.1.2. Synthesis of starting material 3
A solution of 30 mmol of 2-aminobenzoic acid (1) (20 mmol, 3.6 g) of the desired aryl isothiocyanate (2), and Et3N in 50 mL of ethanol was prepared. The reaction mixture was heated under reflux for approximately 5 hrs. After completion of the reaction, the mixture was neutralized with dilute HCl, followed by filtration and rapid column purification using a mixture of hexane and ethyl acetate [26].
Benzyl-mercapto-7-nitroquinazolin-4-one (3). Yield 73%. 1H NMR-δ: 7.59-7.61 (m, 1H, HC), 7.46-7.47 (m, 2H, HC), 2.91 (s, 1H, HC), 7.41-7.45 (m, 2H, HC), 7.22-7.41 (m, 2H, HC), 5.78 (s, 2H, CH2). 13C NMR-δ: 176.05 (C), 159.57 (C), 137.27 (C), 137.10 (C), 136.34 (C), 134.51 (C), 128.07 (HC), 127.76 (HC), 127.17 (HC), 126.99 (HC), 115.81 (CA, 115.27 (C), 48.47 (CH2).
2.1.3. Synthesis of final derivatives 4a-e
The reaction was performed by mixing (17 mmol, 5.32 g) of compound 3 with 17 mmol of the appropriate 1-halogen-4-(chloromethyl) benzene derivative in 45 mL of anhydrous acetone. Potassium carbonate was added in equimolar amount (1 equivalent), and the mixture was stirred at room temperature (25°C) for 10 hrs. At the end of the reaction, each product was subjected to rapid column purification using a mixture of hexane and ethyl acetate [18].
Benzyl-nitro-2-((4-nitrobenzyl)thio)quinazolin-4-one (4a). Yield 68%. 1H NMR-δ : 8.26-8.29 (m, 2H, HC), 8.01-8.04 (m, 1H, HC), 7.14-7.25 (m, 9H, HC), 5.24 (s, 2H, CH2), 4.36 (s, 2H, CH2). 13C NMR-δ : 160.76 (C), 159.75 (C), 151.75 (C), 147.61 (C), 134.59 (C), 130.67 (C), 129.27 (HC), 128.89 (HC), 128.77 (HC), 128.12 (HC), 127.65 (HC), 123.44 (HC), 121.61 (C), 119.49 (C), 47.80 (CH2), 36.25 (CH2).
Benzyl-nitro-2-((4-(fluoromethyl)benzyl)thio)quinazolin-4-one (4b). Yield 85%. 1H NMR-δ : 8.33-8.41 (m, 1H, HC), 8.31-8.32 (m, 1H, HC), 8.18-8.20 (m, 1H, HC), 7.61-7.65 (m, 2H, HC), 7.22-7.41 (m, 7H, HC), 5.30 (s, 2H, CH2), 4.59 (s, 2H, CH2). 13C NMR-δ : 160.59 (C), 158.27 (C), 151.80 (C), 147.37 (C), 139.11 (C), 134.38 (C), 131.94 (HC), 131.68 (HC), 129.70 (HC), 129.34 (HC), 128.84 (HC), 128.24 (HC), 127.54 (HC), 124.21 (HC), 123.45 (HC), 122.04 (HC), 121.69 (C), 121.35 (C), 119.79 (C), 47.81 (CH2), 35.62 (CH2).
Benzyl-2-(benzylthio)-nitroquinazolin-4-one (4c). Yield 82%. 1H NMR-δ : 7.98 (s, 1H, HC), 7.54-7.67 (m, 4H, HC), 7.25-7.35 (m, 8H, HC), 5.39 (s, 2H, CH2), 4.55 (s, 2H, CH2). 13C NMR-δ : 161.45 (C), 158.32 (C), 145.50 (C), 145.42 (C), 136.22 (C), 135.89 (C), 131.10 (HC), 129.38 (HC), 128.46 (HC), 128.44 (HC), 127.38 (HC), 127.04 (HC), 126.17 (HC), 126.17 (HC), 126.05 (HC), 119.16 (C), 46.78 (CH2), 20.30 (CH2).
((Benzyl-nitro-4-oxo-quinazolin-2-yl)thio)methyl)benzonitrile (4d). Yield 75%. 1H NMR-δ : 8.35-8.25 (s, 2H, HC), 8.00-8.19 (m, 1H, HC), 7.70-7.10 (m, 9H, HC), 5.19 (s, 2H, CH2), 4.41 (s, 2H, CH2). 13C NMR-δ : 160.53 (C), 158.70 (C), 151.75 (C), 147.45 (C), 141.83 (C), 134.50 (C), 132.46 (HC), 129.99 (HC), 129.33 (HC), 128.82 (HC), 128.19 (HC), 127.62 (HC), 123.46 (HC), 121.53 (HC), 119.66 (HC), 118.55 (HC), 111.62 (HC), 47.80 (CH2), 36.18 (CH2).
Ethyl ((benzyl-7-nitro-4-oxo-quinazolin-2-yl)thio)acetate (4e). Yield 83%. 1H NMR-δ : 8.32-8.33 (m, 1H, HC), 8.17-8.19 (m, 1H, HC), 8.10-8.12 (m, 1H, HC), 7.28-7.37 (m, 5H, HC), 5.35 (s, 2H, CH2), 4.02 (s, 2H, CH2), 4.09 (q, 2H, J = 7.2 Hz, CH2), 1.22 (t, 3H, CH2). 13C NMR-δ : 168.57 (C), 160.30 (C), 159.80 (C), 151.82 (C), 147.22 (C), 135.37 (HC), 129.56 (HC), 129.12 (HC), 128.12 (HC), 127.31 (HC), 123.40 (HC), 121.12 (HC), 120.19 (HC), 61.70 (CH2), 47.89 (CH2), 34.84 (CH2), 14.60 (CH2).
2.2. Biological evaluation
2.2.1. Cell culture
The cancer cell lines utilized in this study were obtained from Sigma-Aldrich. The cell culture medium consisted of MEM, 1% penicillin-streptomycin, and 11% fetal bovine serum (FBS). The cells were cultured at 37°C in a humidified environment with 5% CO2. After treatment, the cells continued to be incubated under the same conditions (5% CO2 & 37°C).
2.2.2. Tyrosine kinase EGFR/VEGFR-2 inhibitory activity
The EGFR/VEGFR-2 kinase activity was assessed using the EGFR/VEGFR-2 Kinase Assay Kit (PBS Bioscience, catalog #40,321), with erlotinib serving as a reference standard. All experimental measurements for the compounds were conducted in triplicates. The IC50 values for the compounds and the reference standard were determined by averaging the results from the three independent experiments, with the standard deviation calculated to assess the variability of the data.
2.2.3. Molecular docking studies
Molecular docking was performed using AutoDock 4.2 with EGFR (PDB ID: 1M17) and VEGFR (PDB ID: 4AG8) structures from RCSB [27]. The Lamarckian genetic algorithm was applied for optimization, and the docking results were saved as .dpf files [28].
2.3. Molecular dynamics simulation
The protein-ligand complex from docking was analyzed using MD simulations in Desmond v2022 [29] with OPLS3e force field and SPC water model [30]. Sodium and chloride ions were added for neutralization. After energy minimization and applying the SHAKE algorithm [31], periodic boundary conditions and Particle mesh Ewald (PME) were used for electrostatics [32]. The system was equilibrated at 300 K and 1.0 bar, followed by a 100 ns production run. Post-simulation analysis included Root-mean-square deviation (RMSD), Root mean square fluctuation (RMSF), and protein-ligand interactions [33].
3. Results and Discussion
3.1. Chemistry
This research details the synthetic approach employed to create the target hybrid compounds 4a-e, as shown in Scheme 1. The process started with the synthesis of quinazoline derivative 3 by reacting anthranilic acid 1 with (isothiocyanatomethyl)-benzene 2 in ethanol, catalyzed by triethylamine. In the subsequent step, compound 3 underwent S-alkylation with various (chloromethyl)-4-R-benzenes and ethyl-2-bromoacetate in acetone, using K2CO3 as a base. This led to the formation of quinazoline-based hybrid molecules 4a-e, following a previously established method [34] (Scheme 1). The compounds were synthesized in high yields (75-85%), purity, and purified by silica gel column chromatography. The design of these compounds involved introducing lipophilic and electronic substituents at the C2 position of the quinazoline ring to enhance the biological activity, as discussed in the introduction. This strategy amis to broaden the SAR profile of the compounds, particularly to VEGFR-2 and EGFR.
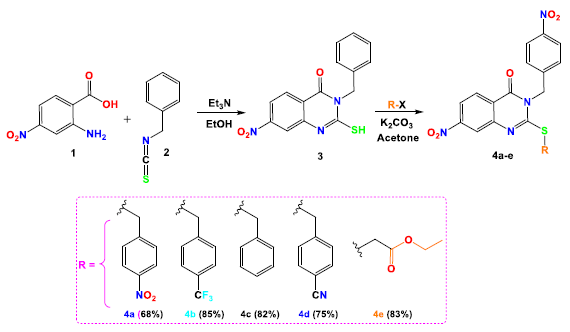
- Illustration of the synthetic pathway leading to the target Quinazoline-Based compounds 4a-e.
The chemical identities of the newly synthesized quinazoline derivatives 4a-e were rigorously verified using a range of analytical techniques. In the case of the NMR analysis of compounds 4a-e, the proton NMR (1H-NMR) spectra displayed a distinct singlet associated with the -CH2-Aryl group, which is directly bonded to the nitrogen atom (N3) in the quinazoline ring, appearing at approximately 5.40 ppm. In the corresponding carbon NMR (13C-NMR) spectra, the same carbon (N3-CH2) yielded a signal typically observed around 47 ppm across all compounds 4a-e. For the variable group introduced during the second synthetic step (R), a singlet was detected between 4.30 and 4.50 ppm, corresponding to the methylene protons (CH2) at the C2 position of the quinazoline core. The 1H-NMR spectra further revealed a triplet at δ 1.22 ppm, indicative of the methyl group, and a quadruplet at δ 4.15 ppm (J = 7.2 Hz), associated with the CH2 group in compound 4e. Additionally, aromatic proton signals appeared as a multiplet in the range of 7.00-8.75 ppm. The 13C-NMR spectra also validated the proposed structures, with a distinct peak at 168 ppm corresponding to the C=O group in compound 4e. Additionally, the 13C-NMR spectrum of compound 4e revealed two characteristic peaks at 14 and 34 ppm, corresponding to the methyl and methylene groups of the ester moiety. It is worth noting that the presence of a peak in the 13C-NMR spectrum of compound 3 around 145 ppm corresponds to the C=N group. In contrast, the characteristic peak of the C=S group typically appears around 178 ppm, which indicates that the absence of this peak (C=S, 178 ppm) suggests that the alkylation reaction occurs on the sulfur atom rather than the nitrogen atom.
3.2. Biological evaluation
3.2.1. In vitro cytotoxicity
The anticancer activity of the newly synthesized quinazoline derivatives was evaluated in vitro against several human cancer cell lines, including HeLa (cervical carcinoma), A-549 (non-small cell lung cancer), and MDA-MB-231 (breast adenocarcinoma). Cytotoxicity was assessed using standard MTT assays, where different concentrations of the compounds were tested to determine their half-maximal inhibitory concentration (IC50). For comparison, docetaxel, a well-established chemotherapy agent, was used as a positive control in these assays (Table 1). The results obtained from these assays allowed for the determination of the potency of the quinazoline derivatives and provided insights into their potential as therapeutic agents in the treatment of various cancer types.
|
||||
|---|---|---|---|---|
| Product | Nature of R | IC50 (µM) | ||
| Hela | A-549 | MDA | ||
| 4a |
|
3.48 ± 0.07 | 1.07 ± 0.02 | 4.58 ± 0.1 |
| 4b |
|
5.19 ± 0.11 | 7.99 ± 0.17 | 8.81 ± 0.18 |
| 4c |
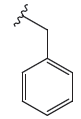
|
48.9 ± 1.02 | 47.1 ± 0.97 | 9.28 ± 0.19 |
| 4d |
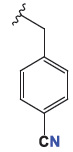
|
4.92 ± 0.1 | 5.48 ± 0.12 | 19.1 ± 0.4 |
| 4e |
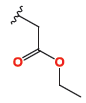
|
1.12 ± 0.03 | 0.59 ± 0.01 | 1.53 ± 0.03 |
| Docetaxel (reference) | 9.65 ± 0.2 | 10.8 ± 0.23 | 3.98 ± 0.08 | |
IC50: Inhibitory concentration 50
The evaluation of the cytotoxic properties of quinazolinone derivatives 4a-e against several distinct cancer cell lines (HeLa, A-549, and MDA) revealed promising results. Among these compounds, derivative 4e, featuring an ethoxy group at the C2 position, was the most potent, exhibiting substantial cytotoxic effects against both HeLa and MDA cell lines, with IC50 values of 1.12 µM and 1.53 µM, respectively. Additionally, 4e demonstrated significant activity against A-549 cells, with a notably lower IC50 value of 0.59 µM. In comparison, product 4d displayed anticancer activity approximately three times more potent than the reference drug docetaxel, with IC50 values ranging from 1.07 µM to 4.58 µM, depending on the cell line tested. Compound 4b showed considerable and nearly consistent cytotoxic effects across all tested cell lines, with IC50 values ranging from 5.19 µM to 8.81 µM. Conversely, compound 4c exhibited lower efficacy, particularly against HeLa and A-549 cells, where IC50 values were as high as 48 µM. Notably, compound 4d was twice as effective as docetaxel for both A-549 and HeLa cells, demonstrating significant anticancer potential, although it was relatively less effective against MDA cells.
3.2.2. Structure-activity correlation
The cytotoxic profiles of the synthesized quinazoline derivatives were evaluated against three human cancer cell lines: A-549 (lung cancer), HeLa (cervical carcinoma), and MDA (breast adenocarcinoma). Among the tested compounds, the unsubstituted quinazolinone derivative 4c, characterized by a phenyl group without any electron-donating or electron-withdrawing substituents, exhibited the least cytotoxicity across all three cell lines (Figure 2). In contrast, the introduction of a trifluoromethyl group a bulky electron-donating group (EDG) at the para position of compound 4b improved the cytotoxicity to a moderate extent, particularly in HeLa cells, where it displayed an IC50 of 5.19 ± 0.11 µM. This increased activity may be attributed to the higher electron density imparted by the fluorine atom. Further modification of the molecule by replacing the para halogen group with a 4-nitrophenyl group (an electron-withdrawing group, EWG) in compound 4a resulted in a significant enhancement in activity, with IC50 values of 3.48 µM and 1.07 µM against HeLa and A-549 cells, respectively. The improvement in cytotoxicity could be attributed to the formation of hydrogen bonds between the electron-withdrawing nitro group (-NO2) and the biological target. On the other hand, replacing the 4-nitrophenyl group with a cyano group (-CN) in compound 4d, another electron-withdrawing substituent, led to a slight reduction in activity compared to 4a. However, compound 4e displayed the most potent anticancer activity among all derivatives, surpassing even the reference drug docetaxel. This enhanced potency is likely due to the structural modification of compound 4e, which features an ethyl acetate group in place of an aryl group found in the other derivatives 4a-d. The IC50 values of 4e ranged from 0.59 to 1.53 µM, with the presence of oxygen atoms in the ethyl acetate group playing a crucial role in facilitating hydrogen bond interactions with the target biomolecule, thereby contributing to its superior anticancer effects.
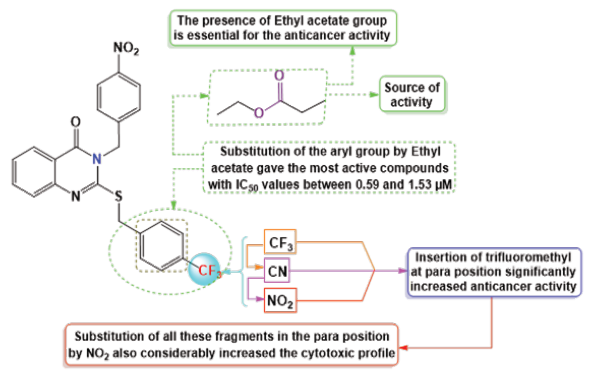
- RSA of target quinazolinone derivatives 4a-e. RSA: Receptor surface area.
In comparison with earlier studies, similar quinazoline-based compounds featuring the same fragment at the C2 position also displayed notable cytotoxic effects and binding affinity. However, the compound with a carbonyl group demonstrated particularly significant activity [18]. In a separate study conducted by our team, a series of compounds was synthesized and tested on cancer cells, revealing that those containing carbonyl groups exhibited a promising anticancer profile by inhibiting cancer cell growth [35]. Upon reviewing these findings and correlating them with the structural features, it can be concluded that the carbonyl group likely plays a crucial role in the observed anticancer activity.
3.2.3. Inhibition of VEGFR-2 and EGFR enzymes
To assess the inhibitory effects of the newly synthesized quinazoline derivatives on the enzymatic activities of EGFR and VEGFR-2 kinases, we utilized the AlphaScreen assay technology (PerkinElmer, USA). This innovative detection method involves the use of an anti-phosphotyrosine antibody to measure the phosphorylation levels of tyrosine residues, which is a key indicator of kinase activity. The AlphaScreen assay is a highly sensitive, bead-based proximity detection system that allows for the precise quantification of kinase inhibition. By employing this technique, we were able to accurately evaluate how the synthesized compounds modulated the phosphorylation of EGFR and VEGFR-2, shedding light on their potential to act as effective therapeutic agents in the treatment of cancers associated with these kinases. This method offers a reliable means of detecting even minor alterations in kinase activity, thus providing valuable data for the development of new anticancer therapies targeting EGFR and VEGFR-2.
In this study, product 4e was identified as the most potent inhibitor among the synthesized quinazoline derivatives. To benchmark its activity, docetaxel was employed as a reference drug, which exhibited IC50 values of 89.3 ± 2.67 and 56.1 ± 1.17 nM against the VEGFR-2 and EGFR, respectively. As shown in Table 2, the IC50 values of derivative 4e against VEGFR-2 and EGFR were measured, confirming its significant inhibitory activity. Compound 4e, on the other hand, demonstrated a reduced efficacy against VEGFR-2, with an IC50 of 189 ± 5.66 nM, roughly half that of docetaxel. However, compound 4e showed moderate inhibitory activity against EGFR, with an IC50 of 69.4 ± 1.55 nM, representing about 80% of docetaxel’s inhibitory potency. These results highlight the varying degrees of efficacy of different quinazoline derivatives in inhibiting key kinases involved in cancer progression.
| Compound | Nature of R | IC50 (nM) | |
|---|---|---|---|
| VEGFR-2 | EGFR | ||
| 4e |
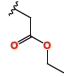
|
189 ± 5.66 | 69.4 ± 1.55 |
| Docetaxel (reference) | 89.3 ± 2.67 | 56.1 ± 1.17 | |
IC50: Inhibitory concentration 50, VEGFR-2: Vascular endothelial growth factor receptor-2, EGFR: Epidermal growth factor receptor.
3.4. Molecular docking
3.4.1. Docking studies on tEGFR: Analysis of molecular interactions
The results of the molecular docking studies for derivative 4e, in comparison with the reference drug docetaxel, are summarized in Table 3. The 2/3D binding interactions between compound 4e and the EGFR target are depicted in Figure 3. The docking analysis revealed that derivative 4e exhibits superior binding affinity for the EGFR target relative to docetaxel. Specifically, derivative 4e achieved a docking score of -4.46 kcal/mol, significantly better than docetaxel’s score of -2.82 kcal/mol. This enhanced binding was attributed to the formation of two hydrogen bonds between compound 4e and the EGFR residues LYS689 and LYS715. Additionally, compound 4e formed four hydrophobic interactions, including π-alkyl interactions with LYS713 and ILE708, as well as van der Waals interactions with PRO709 and GLU712. On the other hand, the reference drug formed three hydrophobic interactions with specific residues of EGFR, including ILE941, PRO748, and ARG938.
| Target EGFR | BS | NHBI | THBI | NHI | THI | IC50 (nM) |
|---|---|---|---|---|---|---|
| 4e | -4.46 | 2 | LYS689, LYS715 | 4 |
LYS713, ILE708-π alkyl PRO709, GLU712- Van der Waals |
69.4 ± 1.55 |
| Docetaxel (reference) | -2.82 | 2 | ASN747, ASN808 | 3 |
PRO748, ARG938, ILE941 – π Alkyl |
56.1 ± 1.17 |
NHI: Number of Hydrophobic Interactions; NHBI: Number of H-Bond Interactions; THI: Type of Hydrophobic Interactions; BS: Binding Score In Kcal/mol; THBI: Type of H-Bond interactions.
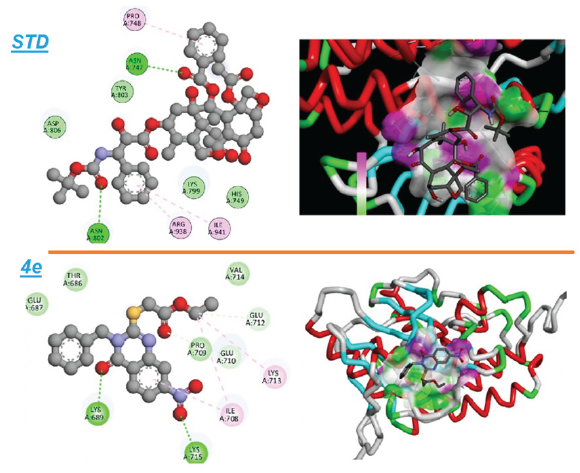
- An illustration of both the 2D and 3D binding interactions between compound 4e and the EGFR target.
The docking studies reveal that derivative 4e establishes favorable interactions within the EGFR binding pocket. primarily driven by its stronger binding affinity and the presence of key hydrogen and hydrophobic interactions. The higher number of hydrophobic interactions in compound 4e, particularly with critical residues such as LYS713 and ILE708, is likely to contribute to its enhanced docking score and suggests a more stable binding conformation within the EGFR binding pocket. These results underscore the potential of compound 4e as a promising EGFR inhibitor, which may offer therapeutic advantages over docetaxel. Future experimental studies will be essential to assess the inhibitory efficacy of compound 4e in biological models.
3.4.2. Findings from molecular docking targeting VEGFR-2
The inhibition results for VEGFR-2 are detailed in Table 4, which includes the reference drug docetaxel for comparison. The 2D and 3D representations of the binding interactions between derivative 4e and VEGFR-2, as well as docetaxel, are illustrated in Figure 4. Molecular docking simulations of derivative 4e with VEGFR-2 (PDB ID: 4AG8) revealed that derivative 4e demonstrates a higher binding affinity than docetaxel. Specifically, derivative 4e obtained a binding score of -4.41 kcal/mol, surpassing docetaxel’s score of -4.24 kcal/mol. The docking analysis showed that derivative 4e forms two hydrogen bonds with the residues GLY909 and LEU912. Additionally, it establishes three hydrophobic interactions: π-alkyl with ALA874, carbon-hydrogen bonding with PRO911, and π-cation interaction with HIS879. On the other hand, docetaxel forms a single hydrogen bond with GLY1108 and two hydrophobic interactions with ILE1111 (π-alkyl) and LYS1110.
|
Target VEGFR-2 |
BS | NHBI | THBI | NHI | THI | IC50 (nM) |
|---|---|---|---|---|---|---|
| 4e | -4.41 | 2 |
GLY909, LEU912 |
3 |
ALA874 -π alkyl PRO911- carbon-hydrogen bond HIS879- π cation |
189 ± 5.66 |
| Docetaxel | -4.24 | 1 | GLY1108 | 2 | LYS1110, ILE1111 – π Alkyl | 89.3 ± 2.67 |
NHI: Number of Hydrophobic Interactions; NHBI: Number of H-Bond Interactions; THI: Type of Hydrophobic Interactions; BS: Binding Score In Kcal/mol; THBI: Type of H-Bond interactions.
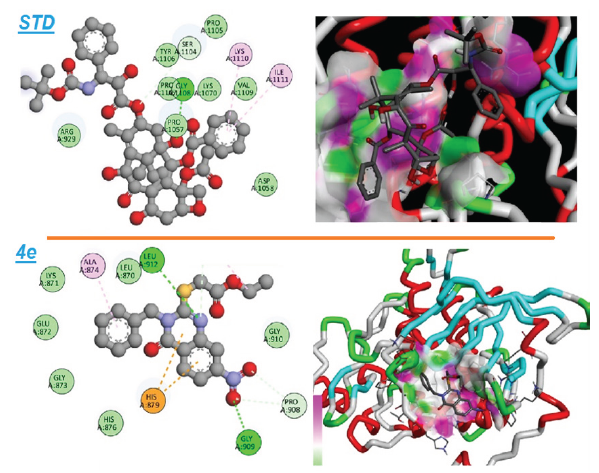
- An illustration of both the 2D and 3D binding interactions between compound 4e and the VEGFR-2 target.
The molecular docking analysis suggests that derivative 4e demonstrates significantly stronger interactions with VEGFR-2 than docetaxel, as reflected by its lower binding energy score. The presence of hydrogen bonds with GLY909 and LEU912, coupled with three hydrophobic interactions, contributes to the stability of compound 4e within the VEGFR-2 binding pocket. While docetaxel also forms hydrogen bonds and hydrophobic interactions, its weaker binding affinity indicates that derivative 4e may offer superior inhibition of VEGFR-2. Based on these results, derivative 4e shows significant potential as a candidate for further development, with the possibility of providing enhanced therapeutic effects when compared to current VEGFR-2 inhibitors like docetaxel.
3.4.3. MD simulation of VEGFR-2 complexed with derivative 4e
Protein-ligand complexes, exhibiting strong binding affinity (VEGFR-4e), underwent a 100 ns molecular dynamics (MD) simulation to assess their stability and behavior over time. The Root Mean Square Deviation (RMSD) parameter, which quantifies the average deviation in atomic positions over the simulation period, plays a key role in analyzing the equilibration process and the structural integrity of the protein-ligand complex. The RMSD plot for the 4e-VEGFR complex has been illustrated in Figure 5. In the initial phase of the simulation, the RMSD for the protein complex showed a noticeable spike, reaching a maximum of 7.5 Å at approximately 6 ns. After this peak, the protein RMSD gradually stabilized, maintaining a consistent average of 7.1 Å from 10 to 96 ns, indicating that the complex had achieved structural equilibrium. In comparison, the ligand RMSD displayed an initial peak of 6 Å at 4 ns, followed by stabilization at an average value of 3 Å up until 84 ns. However, after this point, the ligand RMSD gradually increased to an average of 5 Å, persisting for the remainder of the simulation. These fluctuations in ligand RMSD suggest that higher RMSD values are associated with significant deviations in the ligand’s position relative to its original binding site, signaling a degree of conformational movement or instability in the ligand within the binding pocket.
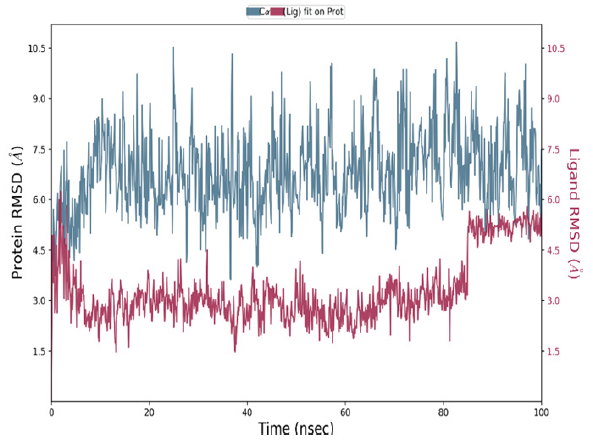
- The RMSD plots of the VEGFR-2 and 4e complex. RMSD: Root mean square deviation.
Root Mean Square Fluctuation (RMSF) is a key parameter used to quantify the positional deviations of amino acid residues over the MD simulation time. It serves as an indicator of the flexibility of specific residues within the protein-ligand complex [36]. The RMSF graph, shown in Figure 6, plots the residue indices along the x-axis and the corresponding RMSF values (in Å) along the y-axis. In the case of the 4e-VEGFR complex (Figure 6), certain residues, including His816, His891, His895, His1026, His1144, Cys817, Glu818, and Ala1168, exhibit noticeably higher RMSF values. This suggests that these residues possess greater flexibility, likely due to their structural properties and the absence of strong stabilizing interactions, which allows them to undergo more movement in the solvent. On the other hand, residues involved in secondary structures, such as α-helices and β-sheets, display lower RMSF values, indicating reduced fluctuations and increased structural stability. Interestingly, the ligand appears to avoid interacting with the residues exhibiting the most significant fluctuations, indicating a preference for binding to more stable regions of the protein, which may contribute to a more robust and specific binding interaction.
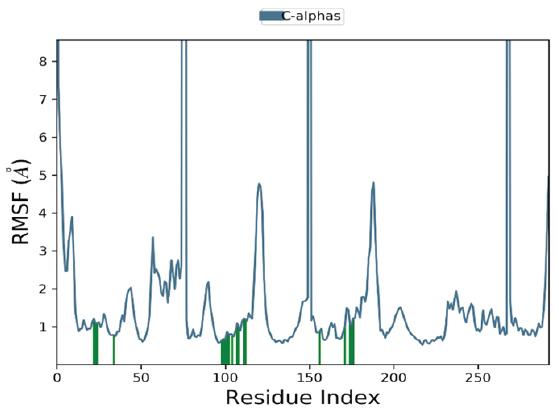
- The RMSF plots of the VEGFR-2 and 4e complex. RMSF: Root mean square fluctuation.
The intermolecular interactions between compound 4e and the VEGFR, observed throughout the MD simulation, have been summarized in the histogram shown in Figure 7. Several key residues were identified to maintain persistent interactions during the simulation. Cys919 consistently formed stable hydrogen bonds with the ligand, with an interaction fraction exceeding 0.75. Water-mediated bridges were formed by residues Lys838, Pro839, Glu850, and Lys920, further stabilizing the complex. Additionally, hydrophobic interactions were observed between 4e and residues Phe918, Leu840, and Phe1047, which likely contribute to the stability of the ligand binding. Ionic interactions involving Lys838, Asn923, Lys931, and Arg1051 residues, also played a significant role. Furthermore, the complex formed a persistent π-cation bond and salt bridges with Cys919 and Phe918, respectively. These interactions were present throughout the simulation, suggesting their importance in maintaining the stability of the 4e-VEGFR complex. A 2D representation of the interactions, which lasted for more than 30% of the total simulation time, has been shown in Figure 7 for both complexes.
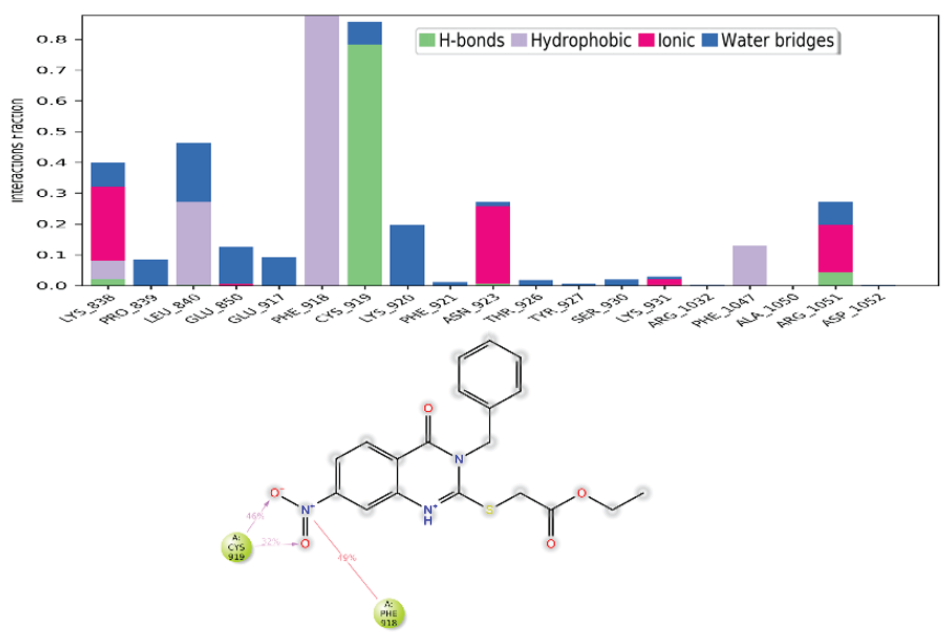
- Analysis of the intermolecular interactions between the target protein and the ligand throughout the MD simulation. A histogram depicting the frequency of various interactions over time for the VEGFR-2 and 4e complex was generated. A 2D representation was created to visualize interactions that persisted for more than 30% of the simulation duration for VEGFR-2 and 4e complex. MD: Molecular dynamics, VEGFR-2: Vascular endothelial growth factor receptor 2.
4. Conclusions
In the quest for potent dual inhibitors of both EGFR and VEGFR-2, a series of five quinazoline derivatives was synthesized, each featuring specific substitutions at the C2 and N3 positions. The synthesized compounds, labeled 4a-e, were meticulously characterized through NMR spectroscopy to confirm their molecular structures. To evaluate their anticancer potential, these derivatives were tested against three different cancer cell lines: A-549, HeLa, and MDA. The results of the cytotoxicity assays demonstrated that most of the compounds exhibited promising anticancer effects, with IC50 values ranging from 0.59 ± 0.01 µM to 48.9 ± 1.02 µM. Notably, compound 4e showed the highest potency across all cell lines, with IC50 values ranging from 0.59 ± 0.01 µM to 4.58 ± 0.1 µM. A detailed SAR analysis identified the ethyl acetate group as a key feature contributing to the enhanced activity of these derivatives. Given the promising in vitro results, compound 4e was selected for further in-depth analysis of its binding affinity to EGFR and VEGFR-2. The compound demonstrated superior activity against VEGFR-2, with an IC50 value of 189.4 ± 5.66 nM. In addition, in silico simulations were performed to investigate the binding interactions of compound 4e with EGFR and VEGFR-2. The results indicate that compound 4e demonstrates strong binding affinity for EGFR, with a docking score of -4.46 kcal/mol, and for VEGFR-2, with a docking score of -4.41 kcal/mol. A stable complex between ligand 4e and VEGFR-2 was identified, suggesting that this compound may hold significant anticancer potential, potentially surpassing docetaxel in terms of efficacy.
Acknowledgement
This study is supported via funding from Prince sattam bin Abdulaziz University project number (PSAU/2025/R/1446). This research work through the project number (PSAU/2024/03/31735).
CRediT authorship contribution statement
Ali Altharawi; Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Supervision; Writing – original draft; Writing – review & editing. Manal A. Alossaimi; Conceptualization; Data curation; Formal analysis; Methodology; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing. Taibah Aldakhil; Conceptualization; Data curation; Formal analysis; Funding acquisition; Methodology; Project administration; Resources; Software; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- How cancer arises. Scientific American. 1996;275:62-70. https://doi.org/10.1038/scientificamerican0996-62
- [CrossRef] [PubMed] [Google Scholar]
- Cristofanilli, Molecular mechanisms of metastasis in breast cancer-clinical applications. Nature Reviews Clinical Oncology. 2010;7:693-701. https://doi.org/10.1038/nrclinonc.2010.171
- [CrossRef] [PubMed] [Google Scholar]
- Cancer statistics, 2024. CA: A Cancer Journal for Clinicians. 2024;74:12-49. https://doi.org/10.3322/caac.21820.
- [CrossRef] [PubMed] [Google Scholar]
- Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer Journal for Clinicians. 2024;74:229-263. https://doi.org/10.3322/caac.21834
- [CrossRef] [PubMed] [Google Scholar]
- Thiazolidinone-linked1, 2, 3-triazoles with monoterpenic skeleton as new potential anticancer agents: Design, synthesis and molecular docking studies. Bioorganic Chemistry. 2021;115:105184. https://doi.org/10.1016/j.bioorg.2021.105184
- [CrossRef] [PubMed] [Google Scholar]
- Semisynthesis of novel chalcone hybrid compounds linked by 1,2,3-triazole and evaluation of their cytotoxic effects. Journal of Molecular Structure. 2025;1319:139648. https://doi.org/10.1016/j.molstruc.2024.139648
- [Google Scholar]
- Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini Reviews in Medicinal Chemistry. 2005;5:409424. https://doi.org/10.2174/1389557053544047
- [Google Scholar]
- Benzimidazole: a medicinally important heterocyclic moiety. Medicinal Chemistry Research : An International Journal. 2012;21:269-283. https://doi.org/10.1007/s00044-010-9533-9
- [CrossRef] [Google Scholar]
- Synthesis and biological evaluation of novel quinazoline-derived human Pin1 inhibitors. Bioorganic & Medicinal Chemistry. 2011;19:2797-2807. https://doi.org/10.1016/j.bmc.2011.03.058
- [CrossRef] [PubMed] [Google Scholar]
- Selective inhibition of EGFR and VEGFR2 tyrosine kinases controlled by a boronic acid substituent on 4-anilinoquinazolines. Medicinal Chemistry Communication. 2010;1:282-286. https://doi.org/10.1039/C0MD00115E
- [CrossRef] [Google Scholar]
- The discovery of the potent aurora inhibitor MK-0457 (VX-680) Bioorganic & Medicinal Chemistry Letters. 2009;19:3586-3592. https://doi.org/10.1016/j.bmcl.2009.04.136
- [PubMed] [Google Scholar]
- Synthesis, dihydrofolate reductase inhibition, antitumor testing, and molecular modeling study of some new 4(3H)-quinazolinone analogs. Bioorganic & Medicinal Chemistry. 2006;14:8608-8621. https://doi.org/10.1016/j.bmc.2006.08.030
- [CrossRef] [PubMed] [Google Scholar]
- Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. Journal of Medicinal Chemistry. 2011;54:6139-6150. https://doi.org/10.1021/jm200903z
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Synthesis and QSAR of quinazoline sulfonamides as highly potent human histamine H4 receptor inverse agonists. Journal of Medicinal Chemistry. 2010;53:2390-2400. https://doi.org/10.1021/jm901379s.
- [CrossRef] [PubMed] [Google Scholar]
- Novel chemical class of pUL97 protein kinase-specific inhibitors with strong anticytomegaloviral activity. Antimicrobial Agents and Chemotherapy. 2004;48:4154-64162. https://doi.org/10.1128/aac.48.11.4154-4162.2004
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Influence of the replacement of the oxo function with the thioxo group on the antimycobacterial activity of 3-aryl-6,8-dichloro-2H-1,3-benzoxazine-2,4(3H)-diones and 3-arylquinazoline-2,4(1H,3H)-diones. Farmaco (Societa Chimica Italiana : 1989). 2001;56:803-7. https://doi.org/10.1016/s0014-827x(01)01134-x.
- [CrossRef] [PubMed] [Google Scholar]
- Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. European Journal of Medicinal Chemistry. 2009;44:2379-2391. https://doi.org/10.1016/j.ejmech.2008.09.015
- [CrossRef] [PubMed] [Google Scholar]
- Synthesis and in vitro antitumor activity of substituted quinazoline and quinoxaline derivatives: search for anticancer agent. European Journal of Medicinal Chemistry. 2011;46:2327. https://doi.org/10.1016/j.ejmech.2011.03.015
- [CrossRef] [PubMed] [Google Scholar]
- Novel indolo[2,1-b]quinazoline analogues as cytostatic agents: synthesis, biological evaluation and structure–activity relationship. Bioorganic & Medicinal Chemistry Letters. 2002;12:2303-2307. https://doi.org/10.1016/S0960-894X(02)00431-6
- [PubMed] [Google Scholar]
- Entrectinib, a new multi-target inhibitor for cancer therapy. Biomedicine & Pharmacotherapy. 2022;150:112974. https://doi.org/10.1016/j.biopha.2022.112974
- [CrossRef] [PubMed] [Google Scholar]
- New quinoline-based BODIPYs as EGFR/VEGFR-2 inhibitors: Molecular docking, DFT and in vitro cytotoxicity on HeLa cells. Journal of Molecular Structure. 2022;1247:131312. https://doi.org/10.1016/j.molstruc.2021.131312
- [CrossRef] [Google Scholar]
- Synthesis of novel (R)-Carvone-1,2,3-triazole hybrids: Network pharmacology, molecular docking, and dynamics simulation targeting tumor protein markers. Journal of Molecular Structure. 2025;1322:140489. https://doi.org/10.1016/j.molstruc.2024.140489
- [CrossRef] [Google Scholar]
- In vitro cytotoxicity and docking study of novel symmetric and asymmetric dihydropyridines and pyridines as EGFR tyrosine kinase inhibitors. Chemical Biology & Drug Design. 2022;100:121-135. https://doi.org/10.1111/cbdd.14058
- [PubMed] [Google Scholar]
- An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. Medicinal Chemistry Communications. 2017;8:871-885. https://doi.org/10.1039/c7md00097a.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- In-silico study of molecular docking and dynamics simulations for N-substituted thiazolidinones derived from (R)-carvone targeting PPAR-γ protein: Synthesis and characterization. :1-26. https://doi.org/10.1080/10406638.2024.2415351
- [Google Scholar]
- Design, characterization, and DFT exploration of new quinazoline-N-substituted analogs: Anti-cancer activity and molecular docking insights. Journal of Molecular Structure. 2025;1322:140420. https://doi.org/10.1016/j.molstruc.2024.140420
- [CrossRef] [Google Scholar]
- In silico Design and Synthesis of Some New Imidazole Derivatives for Tuberculosis. Indian Journal of Heterocyclic Chemistry. 2023;33:43-9. https://connectjournals.com/01951.2023.33.43
- [CrossRef] [Google Scholar]
- Exploring the cyclization thiosemicarbazone to 1,3,4-thiadiazole: Synthesis, characterization and in-silico study. Journal of Molecular Structure. 2025;1322:15, 140385. https://doi.org/10.1016/j.molstruc.2024.140385
- [Google Scholar]
- Computational and experimental characterisation of a new (R)-camphor-Thiazolidinone derivative: A combined approach for structure optimisation and activity prediction. Journal of Molecular Liquids. 2024;410:125615. https://doi.org/10.1016/j.molliq.2024.125615
- [CrossRef] [Google Scholar]
- Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. The Journal of Physical Chemistry A. 2001;105:9954-9960. https://doi.org/10.1021/jp003020w
- [CrossRef] [Google Scholar]
- A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. Journal of Computational Chemistry. 2001;22:501-8. https://doi.org/10.1002/1096-987X(20010415)22:5%3C501::AID-JCC1021%3E3.0.CO;2-V
- [Google Scholar]
- Accuracy and efficiency of the particle mesh Ewald method. The Journal of Chemical Physics. 1995;103:3668-3679. https://doi.org/10.1063/1.470043
- [CrossRef] [Google Scholar]
- Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics. 1984;81:3684-3690. https://doi.org/10.1063/1.448118
- [CrossRef] [Google Scholar]
- Structural basis of inhibitor selectivity in MAP kinases. Structure (London, England). 1998;6:1117-1128. https://doi.org/10.1016/s0969-2126(98)00113-0
- [CrossRef] [Google Scholar]
- Solvent-free synthesis, anticancer activity and in-silico studies of 7-hydroxy-4-methylquinolin-2(1H)-one analogues. Journal of Molecular Structure. 2024;1313:138654. https://doi.org/10.1016/j.molstruc.2024.138654
- [CrossRef] [Google Scholar]
- A comparison of multiscale methods for the analysis of molecular dynamics simulations. The Journal of Physical Chemistry B. 2012;116:8722-8731. https://doi.org/10.1021/jp302103t
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




