

Translate this page into:
Diastereoselective synthesis of a novel phosphinic peptide as ACE inhibitor: Fragment-based design approach
⁎Corresponding authors. moaz.chem@gmail.com (Moaz M. Abdou)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In medicinal chemistry for the purpose of lead optimization, hit selection of new isofunctional chemotypes are crucial for the success of identifying novel chemical entities of increased potency. Using fragment-based design approach with the N-selective inhibitor RXP407, a novel phosphinic peptide scaffold that consisted of modified RXP407 fragments was generated. The presented synthetic route is straightforward and produces the desired product Z-RXP407 in moderate yield. The (S,R,S,S)-Z-RXP407 analog has been evaluated for the C- and N-domain constructs of angiotensin-converting enzyme. The potency of this analog has been much lower compared to the parent compound RXP407, providing thus valuable insights regarding further design based on structure–activity relationships.
Keywords
Fragment-based design
RXP407
Phosphinic peptide
Angiotensin-converting enzyme

1 Introduction
Angiotensin-converting enzyme (ACE) is one of the major therapeutic targets for controlling hypertension and related cardiovascular diseases. The somatic, membrane-bound angiotensin-converting enzyme contains two extracellular domains (N and C) of high homology but distinct ligand specificity (Barinka et al., 2008; Bernstein et al., 2011; Georgiadis et al., 2004; Anthony et al., 2010; Georgiadis et al., 2003; Dive et al., 1999; Kröger et al., 2009; Junot et al., 2001) (Fig. 1).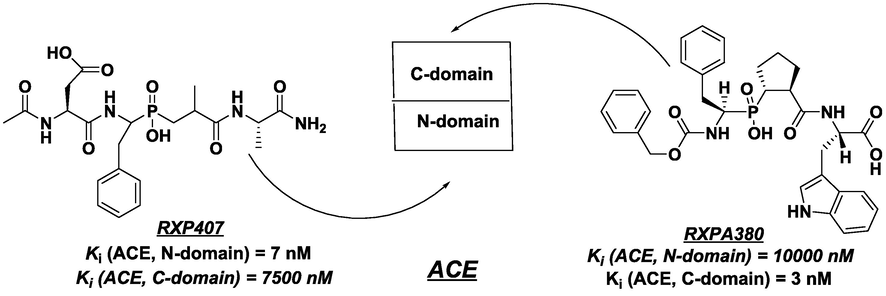
Structures of phosphinic peptide inhibitors RXP407 and RXPA380 that discriminate the 2 active sites of ACE.
RXP407 is currently the only compound known to display marked N-selectivity of more than 3 orders of magnitude, owing its potency to several unique structural features that fulfill the polar interactions with the S2 pocket of the N domain active site (Anthony et al., 2010; Georgiadis et al., 2003; Dive et al., 1999; Kröger et al., 2009; Junot et al., 2001). In addition to this, the phosphoryl oxygen of RXP407 interacts with Tyr501, whereas its phosphinyl group is implicated in zinc coordination. RXP407 still remains the most promising N-domain-selective angiotensin-1-converting enzyme inhibitor, but unfortunately, a poor bioavailability and unfavorable proportions preclude its utilization as a drug candidate (Anthony et al., 2010).
Recently, fragment-based drug design (FBDD) encompasses a broad range of methodologies and strategies for developing a new drug candidate with desired bioactivity properties. Significant progress has been achieved in many drug discovery projects with one approved drug and many more compounds in clinical trials (Romasanta et al., 2018; Śledź and Caflisch, 2018). Prompted by these promising results for RXP407, many research groups tried to optimize the RXP407 scaffold by substituting the central core by different appropriate functional groups and led to the identification of additional series of candidates that exhibited potent ACE inhibition in the low nanomolar range (Fig. 2) (Dive et al., 1999; Douglas et al., 2014). Detailed structure–activity studies revealed the importance of the N-acetyl group, P2 aspartate residue and C-terminal amide as contributors to the observed N-selectivity (Dive et al., 1999; Sharma et al., 2012; Natesh et al., 2003; Corradi et al., 2006).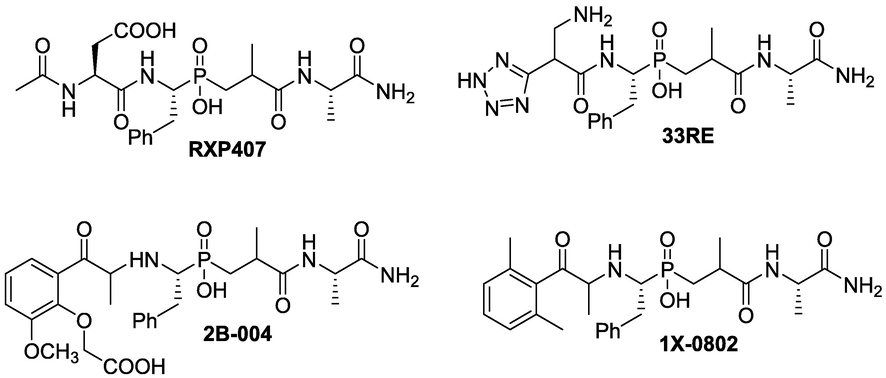
Well-known ACE inhibitors.
Encouraged by these promising results, and in pursuit of our studies on the chemistry of phosphinic acids (Abdou, 2020; Abdou et al., 2019, 2022; Abdou and El-Saeed, 2019; Abdou et al., 2022b, 2022a) towards further development of the RXP407 scaffold, the fragment-based design strategy was employed for designing a novel analogue Z-RXP407, in which the N-terminal acetyl group has been replaced by carboxybenzyl (Z) group, so as to explore this specific N-domain residue interaction (Fig. 3). The Cbz group has been chosen based on other potent phosphinic peptide inhibitors of angiotensin converting enzyme, that incorporate the Cbz group at this position, such as 2 and 3 (Fig. 4) (Arora and Chauhan, 2013; Zhao et al., 2014). Moreover, the increased lipophilicity of Z-RXP407 due to the presence of the phenyl group could favorably interact with the hydrophobic S1 pocket and enhance the inhibitor/enzyme interaction via n-π accumulation. Furthermore, this approach has allowed for the identification of potentially useful fragments that are not limited to natural amino acid residues (Zhao et al., 2014).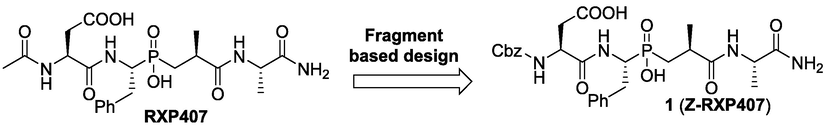
Chemical modification strategy for design of a novel ACE inhibitor Z-RXP407.
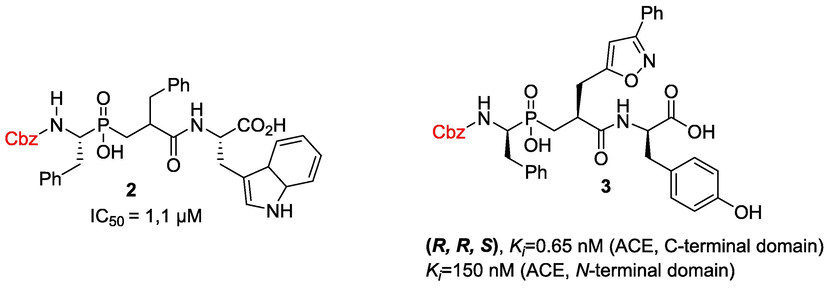
Representative examples of Cbz-phosphinic peptide inhibitors of angiotensin converting enzyme (ACE).
2 Results and discussion
2.1 Chemistry
The synthetic procedure of (S,R,S,S)-Z-RXP407 started with the synthesis of Z-PhePO2H2 which was achieved after several steps, starting from a three-component condensation of hypophosphorous acid to the in-situ formed imine of 4 and phenylacetaldehyde 5 (Scheme 1) (Baylis, 1984; Grobelny, 1987). Removal of the diphenyl group from the adduct 6 by using strong acid led to the formation of free amine 7 followed by the protection of the amino group of the obtained product with benzyl chloroformate to produce the product (R/S)-8 in the form of the racemic mixture (Scheme 1). Then, the resolution through the formation of diastereomeric salts using (R)-phenylethyl amine 9 led to the desired (R) isomer (R)-8. This salt formed contained a mixture of two diastereomers, which have different physical properties (different solubility in ethanol), which in turn, facilitates the separation by crystallization. The main advantages of this approach are simplicity and low cost.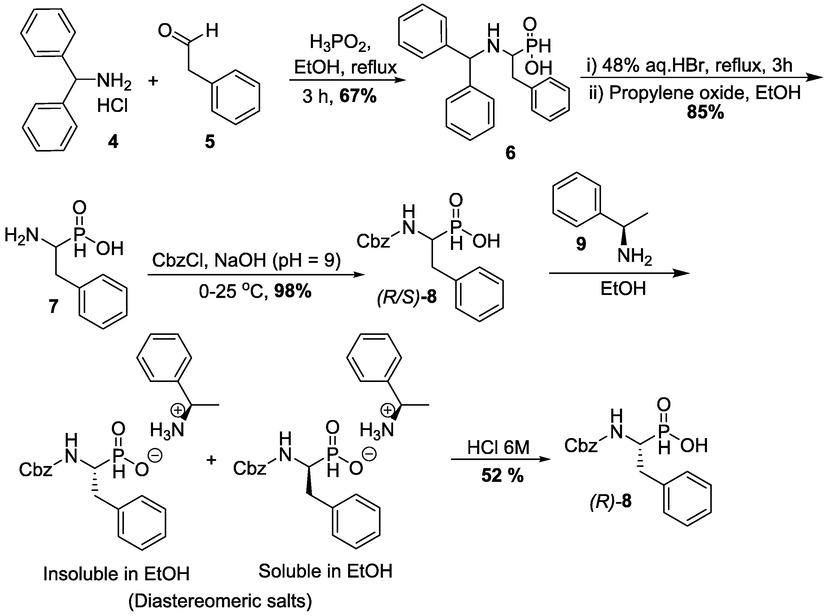
Synthesis of Z-PhePO2H2 and chemical resolution towards (R)-8 enantiomer.
The diastereoselective synthetic procedure of compound Z-RXP407 continued with the activation of Z(R)-PhePO2H2 (R)-8 into its ether by silylation with hexamethyldisilazane followed by addition to ethyl methylacrylate 10 to produce 11 which was then subjected to saponification with sodium hydroxide in methanol, affording the carboxylic acid 12 (Scheme 2) (Dive et al., 1999). This in turn was used in the coupling of alaninamide in the presence of 1-ethyl-3-(3-dimethyllaminopropyl)carbodiimide hydrochloride (EDC.HCl) and 1-hydroxybenzotriazole hydrate (HOBT) to afford amide 13. The Cbz-protecting group of 13 was removed under hydrogenation conditions to give 14 using hydrogen gas and palladium-charcoal (10 %) in methanol at room temperature. This analogue 14 was further used to access the phosphinic acid 16 via coupling with CbzAsp(OtBu)ONP 15. The tert-butyl ester group of the resulting 16 was selectively deprotected under acidic conditions to obtain the (S,R,R/S,S)-Z-RXP407.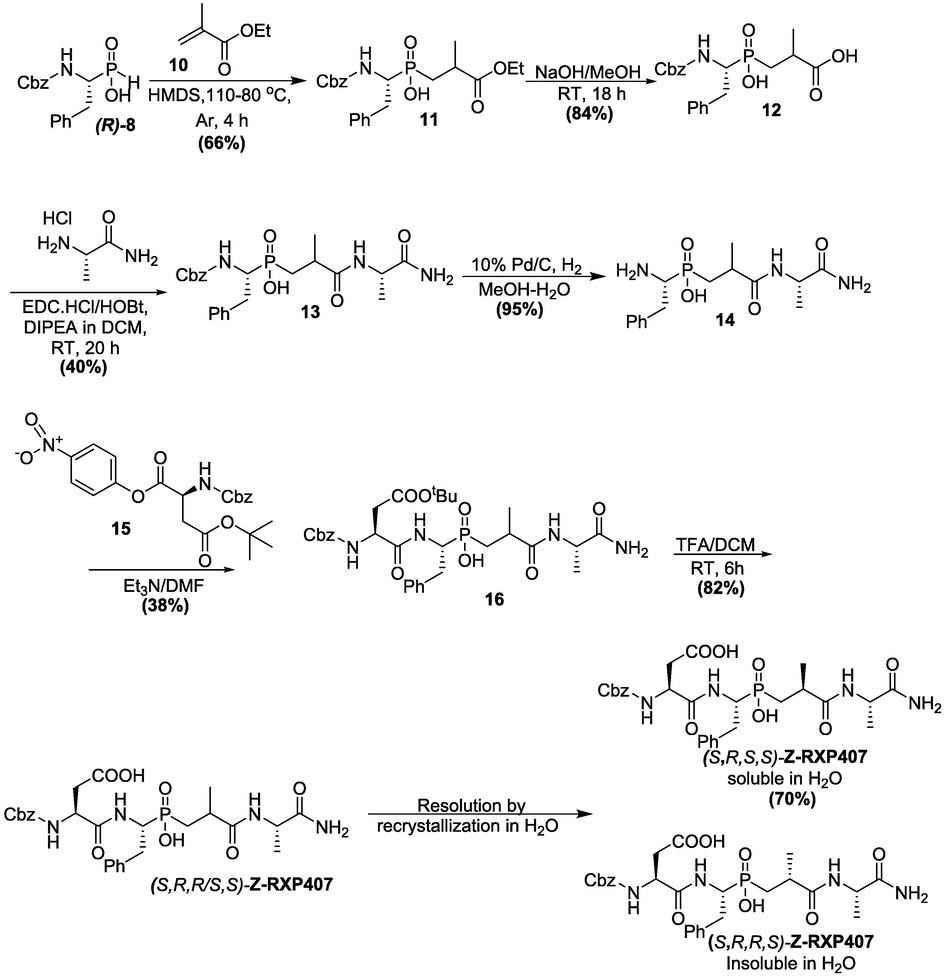
Diastereoselective synthetic protocol for the novel compound (S,R,S,S)-Z-RXP407.
Although the 2 diastereoisomers of (S,R,R/S,S)-Z-RXP407 can be separated by RP-HPLC (Fig. 5), we were able to succeed in a more practical process and achieved the initial goal for the diastereomer separation via simple crystallization from water and the soluble isomer assigned as the (S,R,S,S)-Z-RXP407, monitoring by HPLC analysis, was recovered from the filtrate in high optical purity (Fig. 6).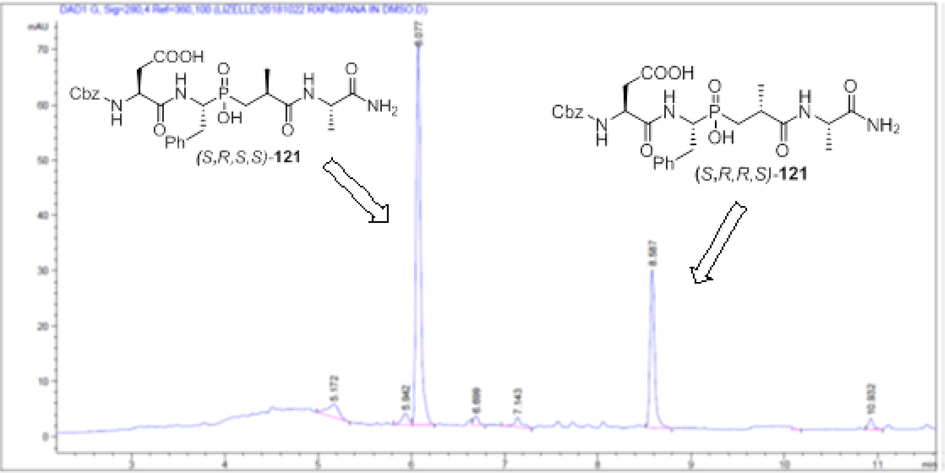
HPLC profile of the racemic form (S,R,R/S,S)-Z-RXP407.
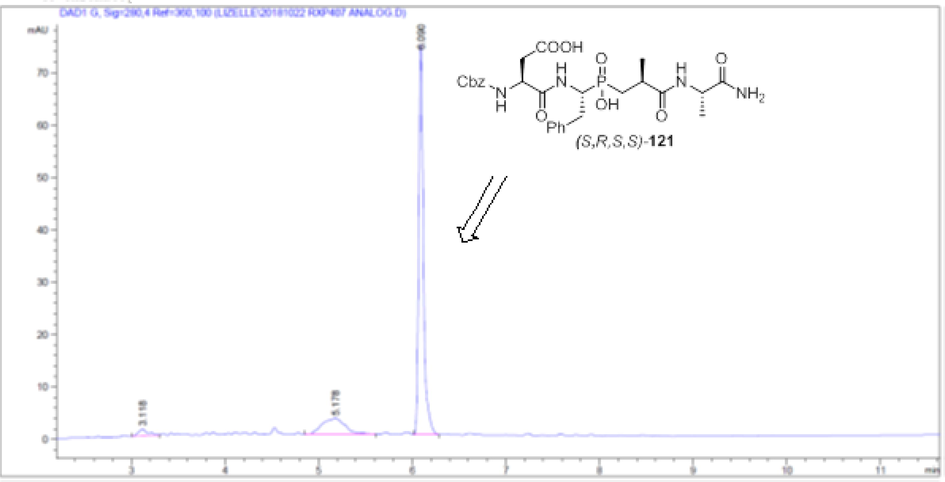
HPLC profile of the diastereoisomer (S,R,S,S)-Z-RXP407.
In order to unambiguously establish the absolute configuration of the P1′ stereogenic center of (S,R,S,S)-Z-RXP407, a literature survey concerning RP-HPLC analysis of phosphinic peptides has been performed. According to these studies, when phosphinic pseudopeptides incorporate an (S)-amino acid at P2‘ position the isomer with the shorter retention time is the one possessing the (S) absolute configuration at the P1‘ position (Tholander et al., 2008; Georgiadis et al., 2000; Reiter et al., 1999; Liu et al., 2002; Reiter and Jones, 1997; Makaritis et al., 2003; Matziari et al., 2010). In the light of the aforesaid literature findings, it becomes apparent that the first fraction (Fig. 5) is the target ACE inhibitor (S,R,S,S)-Z-RXP407.
To unambiguously establish this hypothesis, additional evidence has been obtained. In this context, this pure diastereomer (S,R,S,S)-Z-RXP407 was used to synthesize the known ACE inhibitor RXP407 (Scheme 3). Catalytic (10 % Pd/C) hydrogenation of (S,R,S,S)-Z-RXP407 in methanol at room temperature followed by acetylation with acetic anhydride in the presence of pyridine afforded the corresponding RXP407. Comparison of the spectra of this formed compound with the published one for RXP407 (Dive et al., 2002) demonstrated unequivocally their configurational identity and this confirmed that pure diastereomer (S,R,S,S)-Z-RXP407 was used as precursor for its synthesis. Another interesting feature lies on its use for diastereoselective synthesis of RXP407 via the solution-phase which has not been reported before as proposed here.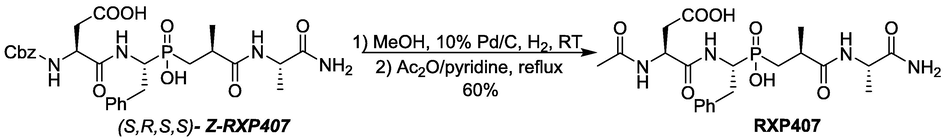
Synthesis of RXP407 as an evidence of the configuration (S,R,S,S)-Z-RXP407.
2.2 Biological evaluation
A dilution of (S,R,S,S)-Z-RXP407 at two concentrations was prepared in assay buffer (50 mM HEPES pH 6.8, 200 mM NaCl and 10 µM ZnCl2). Since this compound was dissolved in water, buffer blanks without DMSO were used in this case. The ACE inhibitory activity of both of them was determined by fluorimetric method at two concentrations (200 uM and 100 uM).
2.2.1 In vitro assay results for (S,R,S,S)-Z-RXP407
Inhibition of both domains by this inhibitor preparation was at 200 µM, the N-domain retained only 20 % activity and the C-domain 50 % activity. This analogue is, however, much less potent than the parent compound RXP407 for binding to the N-domain and the previously reported 1000_fold selectivity of RXP407 is decreased to a mere 2-fold N-selectivity. There is perhaps not enough space in the N-domain active site to accommodate the added P2 carboxybenzyl group. Future docking or co-crystal structures of (S,R,S,S)-Z-RXP407 may shed light on the cause of its decreased binding affinity.
3 Experimental section
3.1 Synthesis
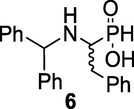 [1-[(Diphenylmethyl)amino]-2-phenylethyl]phosphinic acid (6): 15 A mixture of diphenylmethanamine hydrochloride 4 (114 g, 0.52 mol) and 50 % H3PO2 (53.5 ml, 0.52 mol) was dissolved in EtOH (750 ml) and the resulting mixture was allowed to reflux at 85–90 °C until the dissolution was complete. At this temperature, a solution of phenylacetaldehyde 5 (57.8 ml, 0.52 mol) in EtOH (175 ml) was added dropwise over 3 h then the reaction was left under stirring for another 3 h and at ambient temperature for 16 h. The white solid product formed was filtered, washed with cold EtOH and then Et2O and dried to give white crystals of 6 (122 g, 67 %), Rf = 0.44 (CHCl3/MeOH/AcOH, v/v, 7/0.5/0.5). M.p 209–210 °C (lit. 211 °C); IR (KBr) ν (cm−1) 2330, 1190, 1035; 1H NMR (400 MHz, D2O/Na2CO3) δ 2.19–2.57 (m, 2H), 2.82–2.98 (m, 1H), 4.55 (d, 1H), 6.55–7.99 (m, 15H), 7.42 (d, JP-H = 508 Hz, PH); 13C NMR (100 MHz, D2O/Na2CO3) δ 34.97, 55.87, 57.31, 64.37, 126.35, 126.93, 128.43, 129.34, 138.89, 139.06, 142.44, 142.70. 31P NMR (162 MHz, D2O/Na2CO3) δ 27.88, 31.06. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C21H22NO2PNa 374.1286; Found 374.1277.
[1-[(Diphenylmethyl)amino]-2-phenylethyl]phosphinic acid (6): 15 A mixture of diphenylmethanamine hydrochloride 4 (114 g, 0.52 mol) and 50 % H3PO2 (53.5 ml, 0.52 mol) was dissolved in EtOH (750 ml) and the resulting mixture was allowed to reflux at 85–90 °C until the dissolution was complete. At this temperature, a solution of phenylacetaldehyde 5 (57.8 ml, 0.52 mol) in EtOH (175 ml) was added dropwise over 3 h then the reaction was left under stirring for another 3 h and at ambient temperature for 16 h. The white solid product formed was filtered, washed with cold EtOH and then Et2O and dried to give white crystals of 6 (122 g, 67 %), Rf = 0.44 (CHCl3/MeOH/AcOH, v/v, 7/0.5/0.5). M.p 209–210 °C (lit. 211 °C); IR (KBr) ν (cm−1) 2330, 1190, 1035; 1H NMR (400 MHz, D2O/Na2CO3) δ 2.19–2.57 (m, 2H), 2.82–2.98 (m, 1H), 4.55 (d, 1H), 6.55–7.99 (m, 15H), 7.42 (d, JP-H = 508 Hz, PH); 13C NMR (100 MHz, D2O/Na2CO3) δ 34.97, 55.87, 57.31, 64.37, 126.35, 126.93, 128.43, 129.34, 138.89, 139.06, 142.44, 142.70. 31P NMR (162 MHz, D2O/Na2CO3) δ 27.88, 31.06. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C21H22NO2PNa 374.1286; Found 374.1277.
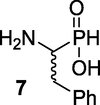 (1-Amino-2-phenylethyl)phosphinic acid (7):15 The diphenylmethylamino-phosphonous acid 6 (176.5 g, 0.5 mol) was heated in 48 % HBr (728 ml) for 3 h at 120 °C. After concentration of the reaction mixture to a small volume, and dilution with H2O, the aqueous layer was washed several times with Et2O to remove Ph2CHBr and then it was evaporated to dryness. The oily residue was dissolved in absolute EtOH (10 ml/g) and propylene oxide (82 ml) added dropwise at 0 °C until precipitation started. The solid was filtered off, washed with cold EtOH and then Et2O, and dried to give white crystals of compound 7 (79 g, 85 %), Rf = 0.32 (CHCl3/MeOH/AcOH, v/v, 7/2/1). M.p 223–224 °C (lit. 227–228 °C). IR (KBr) ν (cm−1) 2400, 1025. 1H NMR (400 MHz, D2O) δ 2.73–2.87 (m, 1H), 3.16–3.27 (m, 1H), 3.31–3.39 (m, 1H), 7.10–7.45 (m, 5H, aromatic), 7.64 (d, JPH = 540 Hz, PH). 13C NMR (100 MHz, D2O) δ 32.04, 51.75/52.62, 127.49, 129.30, 135.26. 31P NMR (162 MHz, D2O) δ 17.06, 20.40. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C8H12NO2PH 186.0684; Found 186.0655.
(1-Amino-2-phenylethyl)phosphinic acid (7):15 The diphenylmethylamino-phosphonous acid 6 (176.5 g, 0.5 mol) was heated in 48 % HBr (728 ml) for 3 h at 120 °C. After concentration of the reaction mixture to a small volume, and dilution with H2O, the aqueous layer was washed several times with Et2O to remove Ph2CHBr and then it was evaporated to dryness. The oily residue was dissolved in absolute EtOH (10 ml/g) and propylene oxide (82 ml) added dropwise at 0 °C until precipitation started. The solid was filtered off, washed with cold EtOH and then Et2O, and dried to give white crystals of compound 7 (79 g, 85 %), Rf = 0.32 (CHCl3/MeOH/AcOH, v/v, 7/2/1). M.p 223–224 °C (lit. 227–228 °C). IR (KBr) ν (cm−1) 2400, 1025. 1H NMR (400 MHz, D2O) δ 2.73–2.87 (m, 1H), 3.16–3.27 (m, 1H), 3.31–3.39 (m, 1H), 7.10–7.45 (m, 5H, aromatic), 7.64 (d, JPH = 540 Hz, PH). 13C NMR (100 MHz, D2O) δ 32.04, 51.75/52.62, 127.49, 129.30, 135.26. 31P NMR (162 MHz, D2O) δ 17.06, 20.40. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C8H12NO2PH 186.0684; Found 186.0655.
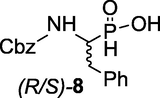 (R/S)-(1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)phosphinic acid ((R/S)- 8): 15 (1-Amino-2-phenylethyl)phosphinic acid 7 (49.4 g, 267 mmol) was dissolved in 120 ml of H2O, the pH of the solution was adjusted to ∼ 9–10 with 4 M NaOH(aq) and the mixture cooled to 0 °C. Benzyl chloroformate was added over 1 h at 0 °C and the mixture was allowed to stir a further 18 h at RT while the pH was maintained at 9.0–10.0. The mixture was extracted with Et2O and the aqueous phase was neutralized with 6 M HCl at 0 °C. White crystalline solid 8 precipitated, filtered and dried well over P2O5 to afford 8 (83.4 g, 98 %) with Rf = 0.43 (CHCl3/MeOH/AcOH, v/v, 7/2/1). M.p 133–135 °C (lit. 137 °C); 1H NMR (400 MHz, CD3OD) δ 2.80–2.89 (m, 1H), 3.07–3.18 (m, 1H), 4.02–4.08 (m, 1H), 4.84–4.89 (d, 2H), 7.14–7.32 (m, 10H); 7.63 (d, JPH = 548 Hz, PH); I3C NMR (100 MHz, CD3OD) δ 32.31, 52.17, 66.35, 126.33, 127.15, 127.50, 128.10, 128.84, 136.74, 137.16, 157.04. 31P NMR (162 MHz, CD3OD) δ 30.38, 30.83. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C16H18NO4PH 320.1052; Found 320.1010.
(R/S)-(1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)phosphinic acid ((R/S)- 8): 15 (1-Amino-2-phenylethyl)phosphinic acid 7 (49.4 g, 267 mmol) was dissolved in 120 ml of H2O, the pH of the solution was adjusted to ∼ 9–10 with 4 M NaOH(aq) and the mixture cooled to 0 °C. Benzyl chloroformate was added over 1 h at 0 °C and the mixture was allowed to stir a further 18 h at RT while the pH was maintained at 9.0–10.0. The mixture was extracted with Et2O and the aqueous phase was neutralized with 6 M HCl at 0 °C. White crystalline solid 8 precipitated, filtered and dried well over P2O5 to afford 8 (83.4 g, 98 %) with Rf = 0.43 (CHCl3/MeOH/AcOH, v/v, 7/2/1). M.p 133–135 °C (lit. 137 °C); 1H NMR (400 MHz, CD3OD) δ 2.80–2.89 (m, 1H), 3.07–3.18 (m, 1H), 4.02–4.08 (m, 1H), 4.84–4.89 (d, 2H), 7.14–7.32 (m, 10H); 7.63 (d, JPH = 548 Hz, PH); I3C NMR (100 MHz, CD3OD) δ 32.31, 52.17, 66.35, 126.33, 127.15, 127.50, 128.10, 128.84, 136.74, 137.16, 157.04. 31P NMR (162 MHz, CD3OD) δ 30.38, 30.83. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C16H18NO4PH 320.1052; Found 320.1010.
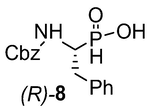 (R)-(1-(((benzyloxy)carbonyl)amino)-phenylethyl)phosphinic acid ((R)-8): 15 A magnetically stirred solution of a racemic mixture of phosphinic analog (83.4 g, 0.26 mol) in absolute EtOH (1000 ml) was allowed to reflux. A solution of R-(+)-N,N-phenylethylamine (34.4 ml, 0.26 mol) was added dropwise and the mixture was allowed to reach room temperature and then cooled at 4 °C overnight. After four days at this temperature, the white solid precipitate which mainly consists of the salt of the enantiomer (R)-8 was filtered and washed with cold absolute EtOH and dry Et2O. The solid was dried over P2O5 followed by recrystallization from absolute EtOH (445 ml). The optical rotation for the salt of (R)-8 [α]24D = -46.7 (c = 1.0, EtOH). Dispersing the salt in solution 6 M HCl (300 ml) with vigorous stirring for 8 h, filtering off, washing with distilled H2O and drying over P2O5, afforded (R)- 8 (21.7 g, 52 %) as a white solid.
(R)-(1-(((benzyloxy)carbonyl)amino)-phenylethyl)phosphinic acid ((R)-8): 15 A magnetically stirred solution of a racemic mixture of phosphinic analog (83.4 g, 0.26 mol) in absolute EtOH (1000 ml) was allowed to reflux. A solution of R-(+)-N,N-phenylethylamine (34.4 ml, 0.26 mol) was added dropwise and the mixture was allowed to reach room temperature and then cooled at 4 °C overnight. After four days at this temperature, the white solid precipitate which mainly consists of the salt of the enantiomer (R)-8 was filtered and washed with cold absolute EtOH and dry Et2O. The solid was dried over P2O5 followed by recrystallization from absolute EtOH (445 ml). The optical rotation for the salt of (R)-8 [α]24D = -46.7 (c = 1.0, EtOH). Dispersing the salt in solution 6 M HCl (300 ml) with vigorous stirring for 8 h, filtering off, washing with distilled H2O and drying over P2O5, afforded (R)- 8 (21.7 g, 52 %) as a white solid.
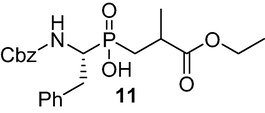 ((R)-1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)(3-ethoxy-2-methyl-3-oxopropyl) phosphinic acid (11): 3a A mixture of (R)-aminophosphinic acid 8 (2 g, 6.26 mmol) and HMDS (4 ml, 18.78 mmol) was flushed by Ar and heated at 110 °C for 3 h. The temperature was reduced to 80 °C, and ethyl methacrylate 10 (7 ml, 7.5 mmol) was added, and the resulting solution was allowed to stir for 4 h at 80 °C. Then, the mixture was cooled at 70 °C, absolute EtOH (6 ml) was added dropwise, and allowed to stir at 25 ◦C for 30 min. The mixture was concentrated and the residue was dissolved in a mixture of Et2O and washed with 5 % NaHCO3. The organic phase was separated, and the aqueous phase was washed twice with Et2O. The aqueous phase was acidified with 1 M HCl to pH 1 and extracted twice with AcOEt followed, and the crude product was obtained after evaporation of the solvents. Flash chromatography using (DCM/MeOH/AcOH, v/v, 7/0.3/0.3) as eluent provided 11 as a white solid (1.79 g, 66 %); 1H NMR (400 MHz, DMSO‑d6) δ 1.18 (t, J = 6.9 Hz, 6H), 1.60–1.72 (m, 1H), 2.05–2.13 (m, 1H), 2.68–2.81 (m, 2H), 3.10 (d, J = 7.2 Hz, 1H), 3.81–4.11 (m, 3H), 4.90 (s, 2H), 7.11–7.35 (m, 10H), 7.68 (d, J = 9.1 Hz, 1H); 13C NMR (100 MHz, DMSO‑d6) δ 14.38, 18.81, 29.60, 30.55, 33.01, 52.67, 60.86, 65.43, 126.64, 127.49, 128.58, 128.70, 129.41, 137.71, 138.91, 156.61, 175.43; 31P NMR (162 MHz, DMSO‑d6) δ 44.25, 44.79. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H28NO6Na 456.1552; Found 456.1552.
((R)-1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)(3-ethoxy-2-methyl-3-oxopropyl) phosphinic acid (11): 3a A mixture of (R)-aminophosphinic acid 8 (2 g, 6.26 mmol) and HMDS (4 ml, 18.78 mmol) was flushed by Ar and heated at 110 °C for 3 h. The temperature was reduced to 80 °C, and ethyl methacrylate 10 (7 ml, 7.5 mmol) was added, and the resulting solution was allowed to stir for 4 h at 80 °C. Then, the mixture was cooled at 70 °C, absolute EtOH (6 ml) was added dropwise, and allowed to stir at 25 ◦C for 30 min. The mixture was concentrated and the residue was dissolved in a mixture of Et2O and washed with 5 % NaHCO3. The organic phase was separated, and the aqueous phase was washed twice with Et2O. The aqueous phase was acidified with 1 M HCl to pH 1 and extracted twice with AcOEt followed, and the crude product was obtained after evaporation of the solvents. Flash chromatography using (DCM/MeOH/AcOH, v/v, 7/0.3/0.3) as eluent provided 11 as a white solid (1.79 g, 66 %); 1H NMR (400 MHz, DMSO‑d6) δ 1.18 (t, J = 6.9 Hz, 6H), 1.60–1.72 (m, 1H), 2.05–2.13 (m, 1H), 2.68–2.81 (m, 2H), 3.10 (d, J = 7.2 Hz, 1H), 3.81–4.11 (m, 3H), 4.90 (s, 2H), 7.11–7.35 (m, 10H), 7.68 (d, J = 9.1 Hz, 1H); 13C NMR (100 MHz, DMSO‑d6) δ 14.38, 18.81, 29.60, 30.55, 33.01, 52.67, 60.86, 65.43, 126.64, 127.49, 128.58, 128.70, 129.41, 137.71, 138.91, 156.61, 175.43; 31P NMR (162 MHz, DMSO‑d6) δ 44.25, 44.79. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H28NO6Na 456.1552; Found 456.1552.
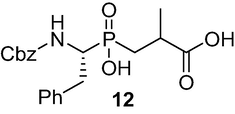 3-(((R)-1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)(hydroxy) phosphoryl)-2-methyl propanoic acid (12): 3a The solution of pseudo dipeptide 11 (2.67 g, 6.16 mmol) in MeOH (38 ml) was treated with 3 M NaOH(aq) (5.4 ml) at 0 °C and the reaction mixture was allowed to stir for 18 h at RT. After removal of the volatiles in vacuo, the residue was suspended in H2O and acidified with 4 M HCl in an ice-water bath to pH 1. The aqueous phase was extracted with AcOEt and the combined organic layers were dried over MgSO4 and concentrated. Purification by column chromatography, using (DCM/MeOH/AcOH, v/v, 9.7/0.4/0.1) as eluent system, afforded the pure products as a white solid 12 (2.10 g, 84 %). 1H NMR (400 MHz, D2O/Na2CO3) δ 1.10 (s, 3H), 1.37–1.57 (m, 1H), 1.76–2.00 (m, 1H), 2.38–2.61 (m, 2H), 2.94–3.15 (m, 1H), 3.60–3.88 (m, 1H), 4.97 (s, 2H), 6.67–7.37 (m, 10H);13C NMR (100 MHz, D2O/Na2CO3) δ 18.96, 31.16, 34.06, 36.80, 53.09, 66.14, 126.47, 126.96,128.96, 128.47, 128.71, 129.20, 136.65, 138.61, 156.89, 185.62; 31P NMR (162 MHz, D2O/Na2CO3) δ 39.69, 39.56, 39.13. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C20H24NO6PNa 428.1239; Found 428.1211.
3-(((R)-1-(((benzyloxy)carbonyl)amino)-2-phenylethyl)(hydroxy) phosphoryl)-2-methyl propanoic acid (12): 3a The solution of pseudo dipeptide 11 (2.67 g, 6.16 mmol) in MeOH (38 ml) was treated with 3 M NaOH(aq) (5.4 ml) at 0 °C and the reaction mixture was allowed to stir for 18 h at RT. After removal of the volatiles in vacuo, the residue was suspended in H2O and acidified with 4 M HCl in an ice-water bath to pH 1. The aqueous phase was extracted with AcOEt and the combined organic layers were dried over MgSO4 and concentrated. Purification by column chromatography, using (DCM/MeOH/AcOH, v/v, 9.7/0.4/0.1) as eluent system, afforded the pure products as a white solid 12 (2.10 g, 84 %). 1H NMR (400 MHz, D2O/Na2CO3) δ 1.10 (s, 3H), 1.37–1.57 (m, 1H), 1.76–2.00 (m, 1H), 2.38–2.61 (m, 2H), 2.94–3.15 (m, 1H), 3.60–3.88 (m, 1H), 4.97 (s, 2H), 6.67–7.37 (m, 10H);13C NMR (100 MHz, D2O/Na2CO3) δ 18.96, 31.16, 34.06, 36.80, 53.09, 66.14, 126.47, 126.96,128.96, 128.47, 128.71, 129.20, 136.65, 138.61, 156.89, 185.62; 31P NMR (162 MHz, D2O/Na2CO3) δ 39.69, 39.56, 39.13. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C20H24NO6PNa 428.1239; Found 428.1211.
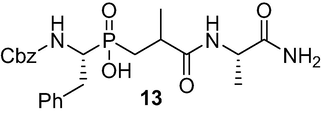 (3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxo propyl)((R)-1-(((benzyloxy)carbonyl) amino)-2-phenylethyl)phosphinic acid (13): To a chilled solution of 12 (2.1 g, 5.18 mmol) in DCM (12 ml) containing DIPEA (1.75 ml, 10.04 mmol), l-alaninamide. HCl (0.64 g, 5.18 mmol), HOBt (0.68 g, 5.07 mmol) and EDC.HCl (3.97 g, 20.69 mmol) were added. The reaction mixture was allowed to stir for 30 min at 0 °C and 20 h at RT. After completion of the reaction, the reaction mixture was then diluted with DCM and washed with a solution of 1MHCl. The organic layer was separated and washed with a sat. aq. NH4HCO3 (3x1 ml), 1 M HCl to pH 1 and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo and the residue was purified over silica gel chromatography using (CHCl3/MeOH/AcOH, v/v, 9.7/0.4/0.1) as eluent afforded the product 13 (1 g, 40 %) as a white solid; 1H NMR (400 MHz, D2O/Na2CO3) δ 0.99–1.16 (m, 6H), 1.36–1.56 (m, 1H), 1.75–1.96 (m, 1H), 2.49 (s, 2H), 2.92–3.12 (m, 1H), 3.57–3.80 (m, 2H), 4.88 (d, J = 7.1 Hz, 2H), 6.74–7.26 (m, 11H), 7.59 (d, J = 9.3 Hz, 2H); 13C NMR (100 MHz, D2O/Na2CO3) δ 16.98, 19.08, 31.44, 34.34, 37.07, 52.75, 66.63, 125.04, 125.10, 128.46, 128.70, 129.17, 136.66, 138.50, 143.07, 157.78, 185.60, 185.78; 31P NMR (162 MHz, D2O/Na2CO3) δ 39.69, 39.54, 39.14. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C23H30N3O6PNa 498.1770; Found 498.1744.
(3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxo propyl)((R)-1-(((benzyloxy)carbonyl) amino)-2-phenylethyl)phosphinic acid (13): To a chilled solution of 12 (2.1 g, 5.18 mmol) in DCM (12 ml) containing DIPEA (1.75 ml, 10.04 mmol), l-alaninamide. HCl (0.64 g, 5.18 mmol), HOBt (0.68 g, 5.07 mmol) and EDC.HCl (3.97 g, 20.69 mmol) were added. The reaction mixture was allowed to stir for 30 min at 0 °C and 20 h at RT. After completion of the reaction, the reaction mixture was then diluted with DCM and washed with a solution of 1MHCl. The organic layer was separated and washed with a sat. aq. NH4HCO3 (3x1 ml), 1 M HCl to pH 1 and brine. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo and the residue was purified over silica gel chromatography using (CHCl3/MeOH/AcOH, v/v, 9.7/0.4/0.1) as eluent afforded the product 13 (1 g, 40 %) as a white solid; 1H NMR (400 MHz, D2O/Na2CO3) δ 0.99–1.16 (m, 6H), 1.36–1.56 (m, 1H), 1.75–1.96 (m, 1H), 2.49 (s, 2H), 2.92–3.12 (m, 1H), 3.57–3.80 (m, 2H), 4.88 (d, J = 7.1 Hz, 2H), 6.74–7.26 (m, 11H), 7.59 (d, J = 9.3 Hz, 2H); 13C NMR (100 MHz, D2O/Na2CO3) δ 16.98, 19.08, 31.44, 34.34, 37.07, 52.75, 66.63, 125.04, 125.10, 128.46, 128.70, 129.17, 136.66, 138.50, 143.07, 157.78, 185.60, 185.78; 31P NMR (162 MHz, D2O/Na2CO3) δ 39.69, 39.54, 39.14. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C23H30N3O6PNa 498.1770; Found 498.1744.
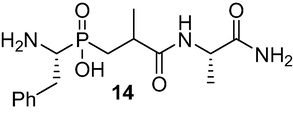 (3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxo propyl)((R)-1-amino-2-phenylethyl) phosphinic acid (14): Dissolve the phosphinic peptide 13 (950 mg, 2 mmol) in MeOH/H2O, v/v, 16/4 ml, add 2 drops AcOH, cool to 0 °C, then add added 10 % Pd/C (0.48 g, 0.45 mmol), apply vacuum, then connect to a balloon filled with H2, while the apparatus is connected to a flask containing sat. Ba(OH)2 solution to trap the generated CO2. The catalyst was removed by filtration through Celite, and the filtrate was evaporated to give the product 14 as white solid (646 mg, 95 %). This compound was used in the next step without further purification. 1H NMR (400 MHz, CD3OD) δ 1.22 (d, J = 6.5 Hz, 3H), 1.44 (d, J = 7.3 Hz, 3H), 1.50–1.61 (m, 2H), 2.06–2.18 (m, 1H), 2.84–2.92 (s, 3H), 3.37–3.39 (m, 1H), 7.25–7.53 (m, 8H) 8.11 (d, J = 9.1 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.55, 18.71, 31.90, 33.74, 34.39, 51.93, 78.22, 126.96, 128.68, 129.04, 136.52, 136.62, 178.99, 179.15; 31P NMR (162 MHz, CD3OD) δ 31.16, 30.27, 29.48. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C15H24N3O4PNa 364.1402; Found 364.1486.
(3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxo propyl)((R)-1-amino-2-phenylethyl) phosphinic acid (14): Dissolve the phosphinic peptide 13 (950 mg, 2 mmol) in MeOH/H2O, v/v, 16/4 ml, add 2 drops AcOH, cool to 0 °C, then add added 10 % Pd/C (0.48 g, 0.45 mmol), apply vacuum, then connect to a balloon filled with H2, while the apparatus is connected to a flask containing sat. Ba(OH)2 solution to trap the generated CO2. The catalyst was removed by filtration through Celite, and the filtrate was evaporated to give the product 14 as white solid (646 mg, 95 %). This compound was used in the next step without further purification. 1H NMR (400 MHz, CD3OD) δ 1.22 (d, J = 6.5 Hz, 3H), 1.44 (d, J = 7.3 Hz, 3H), 1.50–1.61 (m, 2H), 2.06–2.18 (m, 1H), 2.84–2.92 (s, 3H), 3.37–3.39 (m, 1H), 7.25–7.53 (m, 8H) 8.11 (d, J = 9.1 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.55, 18.71, 31.90, 33.74, 34.39, 51.93, 78.22, 126.96, 128.68, 129.04, 136.52, 136.62, 178.99, 179.15; 31P NMR (162 MHz, CD3OD) δ 31.16, 30.27, 29.48. HRMS (ESI/QTOF) m/z: [M + Na]+ Calcd for C15H24N3O4PNa 364.1402; Found 364.1486.
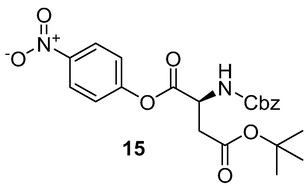 (Benzyloxycarbonyl)-l-aspartic acid β-tert-butyl-α-p-nitro-phenyl ester (Z-Asp(OtBu)-ONP) (15): Z-Asp(OtBu)–OH (0.5 g, 1.55 mmol), and p-nitrophenol (0.28 g, 1.99 mmol) were dissolved in AcOEt (4 ml). The solution was allowed to stir and cooled in an ice- H2O bath while DCC (0.42 g, 2.61 mmol) in AcOEt (1 ml) was added. Stirring at 0 °C was continued for 1 h and then at room temperature overnight. The N,N‘-dicyclohexylurea was removed and the filtrate was evaporated to give an oil, which solidified on seeding and was crystallized from AcOEt and hexane to give white needles of Z-Asp(OtBu)-ONP 15 (0.38 g, 56 %); 1H NMR (400 MHz, DMSO‑d6) δ 1.41 (s, 9H), 2.76–2.82 (m, 1H), 2.90–2.94 (m, 1H), 4.68–2.74 (m, 1H), 5.07 (t, 1H), 7.32–7.44 (m, 6H), 8.13 (d, 1H), 8.32 (d, 2H);13C NMR (100 MHz, DMSO‑d6) δ 28.26, 37.31, 51.22, 66.35, 80.96, 123.26, 126.01, 128.26, 128.84, 137.23, 145.69, 155.61, 156.42, 169.43, 169.82. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H24N2O8H 445.1611; Found 445.1600.
(Benzyloxycarbonyl)-l-aspartic acid β-tert-butyl-α-p-nitro-phenyl ester (Z-Asp(OtBu)-ONP) (15): Z-Asp(OtBu)–OH (0.5 g, 1.55 mmol), and p-nitrophenol (0.28 g, 1.99 mmol) were dissolved in AcOEt (4 ml). The solution was allowed to stir and cooled in an ice- H2O bath while DCC (0.42 g, 2.61 mmol) in AcOEt (1 ml) was added. Stirring at 0 °C was continued for 1 h and then at room temperature overnight. The N,N‘-dicyclohexylurea was removed and the filtrate was evaporated to give an oil, which solidified on seeding and was crystallized from AcOEt and hexane to give white needles of Z-Asp(OtBu)-ONP 15 (0.38 g, 56 %); 1H NMR (400 MHz, DMSO‑d6) δ 1.41 (s, 9H), 2.76–2.82 (m, 1H), 2.90–2.94 (m, 1H), 4.68–2.74 (m, 1H), 5.07 (t, 1H), 7.32–7.44 (m, 6H), 8.13 (d, 1H), 8.32 (d, 2H);13C NMR (100 MHz, DMSO‑d6) δ 28.26, 37.31, 51.22, 66.35, 80.96, 123.26, 126.01, 128.26, 128.84, 137.23, 145.69, 155.61, 156.42, 169.43, 169.82. HRMS (ESI/QTOF) m/z: [M + H]+ Calcd for C22H24N2O8H 445.1611; Found 445.1600.
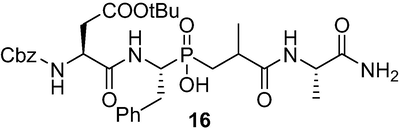 (3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxopropyl)((R)-1-((S)-2-(((benzyloxy)carbonyl)amino)-4-(tert-butoxy)-4-oxobutan amido)-2-phenylethyl)phosphinic acid (16): Phosphinic acid 14 (341 mg, 1 mmol,) was dissolved in dry DMF (5 ml), the pH of the solution was adjusted to ∼ 8–9 with Et3N. CbzAsp(OtBu)ONP 15 (577 mg, 1.3 mmol) was added and the mixture was allowed to stir at RT overnight while the pH was maintained at 8.0. After removal of the volatiles using an oil pump, the residue was dissolved in 5 % NaHCO3 to pH∼9 and extract with AcOEt until Et3N and CbzAsp(OtBu)ONp were removed. Neutralization with 0.5 M HCl to pH ∼6 and extract with AcOEt to remove the NpOH until the aqueous phase lose the yellow color of NpOH. After NpOH removal, AcOEt and 3 M HCl was added at 0 °C under vigorous stirring until pH = 1. The white solid was filtered, washed with H2O and AcOEt, dryied over P2O5 and used in the next step without purification. 1H NMR (400 MHz, CD3OD) δ 1.20 (d, J = 6.4 Hz, 3H), 1.40 (s, 9H), 1.53 (d, J = 7.2 Hz, 3H), 1.68–1.86 (m, 1H), 2.06–2.14 (m, 1H), 2.28–2.35 (m, 1H), 2.59–2.71 (m, 2H), 3.30 (m, 1H), 4.37(m, 1H), 4.47 (m, 1H), 5.09 (m, 2H), 5.17 (m, 1H), 7.09–7.22 (m, 5H), 7.32–7.88 (m, 9H), 8.11 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.04, 27.20, 30.25, 31.02, 34.06, 37.23, 50.33, 51.69, 66.48, 80.94, 126.12, 127.65, 127.74, 127.92, 128.13, 128.84, 136.37, 138.19, 165.80, 169.90, 171.52, 178.49, 179.59; 31P NMR (162 MHz, CD3OD) δ 41.19, 41.59, 41.80. HRMS (ESI/QTOF) m/z: [M + K]+ Calcd for C31H43N4O9PK 685.2405; Found 685.2391.
(3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxopropyl)((R)-1-((S)-2-(((benzyloxy)carbonyl)amino)-4-(tert-butoxy)-4-oxobutan amido)-2-phenylethyl)phosphinic acid (16): Phosphinic acid 14 (341 mg, 1 mmol,) was dissolved in dry DMF (5 ml), the pH of the solution was adjusted to ∼ 8–9 with Et3N. CbzAsp(OtBu)ONP 15 (577 mg, 1.3 mmol) was added and the mixture was allowed to stir at RT overnight while the pH was maintained at 8.0. After removal of the volatiles using an oil pump, the residue was dissolved in 5 % NaHCO3 to pH∼9 and extract with AcOEt until Et3N and CbzAsp(OtBu)ONp were removed. Neutralization with 0.5 M HCl to pH ∼6 and extract with AcOEt to remove the NpOH until the aqueous phase lose the yellow color of NpOH. After NpOH removal, AcOEt and 3 M HCl was added at 0 °C under vigorous stirring until pH = 1. The white solid was filtered, washed with H2O and AcOEt, dryied over P2O5 and used in the next step without purification. 1H NMR (400 MHz, CD3OD) δ 1.20 (d, J = 6.4 Hz, 3H), 1.40 (s, 9H), 1.53 (d, J = 7.2 Hz, 3H), 1.68–1.86 (m, 1H), 2.06–2.14 (m, 1H), 2.28–2.35 (m, 1H), 2.59–2.71 (m, 2H), 3.30 (m, 1H), 4.37(m, 1H), 4.47 (m, 1H), 5.09 (m, 2H), 5.17 (m, 1H), 7.09–7.22 (m, 5H), 7.32–7.88 (m, 9H), 8.11 (d, J = 8.8 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.04, 27.20, 30.25, 31.02, 34.06, 37.23, 50.33, 51.69, 66.48, 80.94, 126.12, 127.65, 127.74, 127.92, 128.13, 128.84, 136.37, 138.19, 165.80, 169.90, 171.52, 178.49, 179.59; 31P NMR (162 MHz, CD3OD) δ 41.19, 41.59, 41.80. HRMS (ESI/QTOF) m/z: [M + K]+ Calcd for C31H43N4O9PK 685.2405; Found 685.2391.
 (3S)-4-(((1R)-1-((3-(((S)-1-amino-1-oxopropan-2-yl) amino)-2-methyl-3-oxopropyl)(hydroxy)phosphoryl)-2-phenylethyl)amino)-3-(((benzyloxy) carbonyl)amino)-4-oxobutanoic acid (S,R,R/S,S)-Z-RXP407: The phosphinic acid 16 (20 mg) was dissolved in 1 ml (TFA/DCM, v/v, 4/1), and the resulting mixture was allowed to stir at RT for 6 h. The solvent was removed in vacuo and the crude was dissolved in Et2O, washed with 5 % NaHCO3 and brine. The organic phase was dried with MgSO4, and concentrated to provide the product as a white solid (77 mg, 82 %). RP-HPLC analyses of final compounds carried out on an Agilent Eclipse XDB-C18 column (4.6 X 250 nm), which thermostated at 25 °C. The flow rate was 1 ml/min, whereas the injection volume was 15 uL. The mobile phase consisted of solvent A [H2O/CH3CN/HCOOH, v/v, 90/10/0.1)] and solvent B [H2O/CH3CN/HCOOH, v/v, 10/90/0.09] with different elution gradients at the wavelength of 254 and 280 nm.
(3S)-4-(((1R)-1-((3-(((S)-1-amino-1-oxopropan-2-yl) amino)-2-methyl-3-oxopropyl)(hydroxy)phosphoryl)-2-phenylethyl)amino)-3-(((benzyloxy) carbonyl)amino)-4-oxobutanoic acid (S,R,R/S,S)-Z-RXP407: The phosphinic acid 16 (20 mg) was dissolved in 1 ml (TFA/DCM, v/v, 4/1), and the resulting mixture was allowed to stir at RT for 6 h. The solvent was removed in vacuo and the crude was dissolved in Et2O, washed with 5 % NaHCO3 and brine. The organic phase was dried with MgSO4, and concentrated to provide the product as a white solid (77 mg, 82 %). RP-HPLC analyses of final compounds carried out on an Agilent Eclipse XDB-C18 column (4.6 X 250 nm), which thermostated at 25 °C. The flow rate was 1 ml/min, whereas the injection volume was 15 uL. The mobile phase consisted of solvent A [H2O/CH3CN/HCOOH, v/v, 90/10/0.1)] and solvent B [H2O/CH3CN/HCOOH, v/v, 10/90/0.09] with different elution gradients at the wavelength of 254 and 280 nm.
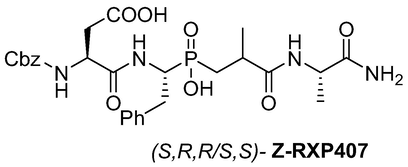
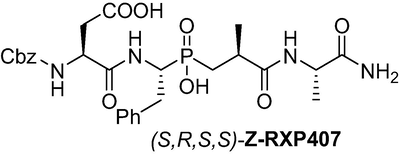 (3S)-4-(((1R)-1-(((S)-3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxopropyl)(hydroxy)phosphoryl)-2-phenylethyl)amino)-3-(((benzyl oxy)carbonyl)amino)-4-oxobutanoic acid (S,R,S,S)-Z-RXP407: A magnetically stirred solution of a racemic mixture of phosphinic analog (S,R,R/S,S)-Z-RXP407 (77 mg, 0.13 mmol) in H2O (2.5 ml) was refluxed for 40 min and then left at RT. At this temperature, the white solid precipitate was filtered. The filtrate was evaporated and dried over P2O5 to give the target compound (S,R,S,S)-Z-RXP407 as a white solid (54 mg, 70 %). 1H NMR (400 MHz, CD3OD) δ 1.23 (d, J = 6.6 Hz, 3H), 1.56 (d, J = 7.5 Hz, 3H), 1.71–1.86 (m, 1H), 2.10–2.18 (m, 1H), 2.30–2.36 (m, 1H), 2.55–2.73 (m, 2H), 3.35–3.50 (m, 1H), 4.33–4.47 (m, 2H), 4.60–4.88 (m, 1H), 5.11–5.31 (m, 2H), 5.46–4.62 (m, 1H), 7.16–7.34 (m, 5H), 7.40–7.75 (m, 9H), 8.06 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.22, 30.33, 31.24, 33.03, 33.82, 37.11, 50.22, 51.24, 51.96, 66.55, 81.11, 126.12, 127.44, 127.65, 127.74, 127.92, 128.13, 128.75, 128.84, 136.53, 137.93, 156.79, 169.95, 171.67, 178.49, 179.59; 31P NMR (162 MHz, CD3OD) δ 44.03. HRMS (ESI/QTOF) m/z: [M]+ Calcd for C27H35N4O9P 590.2142; Found 590.2157.
(3S)-4-(((1R)-1-(((S)-3-(((S)-1-amino-1-oxopropan-2-yl)amino)-2-methyl-3-oxopropyl)(hydroxy)phosphoryl)-2-phenylethyl)amino)-3-(((benzyl oxy)carbonyl)amino)-4-oxobutanoic acid (S,R,S,S)-Z-RXP407: A magnetically stirred solution of a racemic mixture of phosphinic analog (S,R,R/S,S)-Z-RXP407 (77 mg, 0.13 mmol) in H2O (2.5 ml) was refluxed for 40 min and then left at RT. At this temperature, the white solid precipitate was filtered. The filtrate was evaporated and dried over P2O5 to give the target compound (S,R,S,S)-Z-RXP407 as a white solid (54 mg, 70 %). 1H NMR (400 MHz, CD3OD) δ 1.23 (d, J = 6.6 Hz, 3H), 1.56 (d, J = 7.5 Hz, 3H), 1.71–1.86 (m, 1H), 2.10–2.18 (m, 1H), 2.30–2.36 (m, 1H), 2.55–2.73 (m, 2H), 3.35–3.50 (m, 1H), 4.33–4.47 (m, 2H), 4.60–4.88 (m, 1H), 5.11–5.31 (m, 2H), 5.46–4.62 (m, 1H), 7.16–7.34 (m, 5H), 7.40–7.75 (m, 9H), 8.06 (d, J = 8.7 Hz, 2H); 13C NMR (100 MHz, CD3OD) δ 18.22, 30.33, 31.24, 33.03, 33.82, 37.11, 50.22, 51.24, 51.96, 66.55, 81.11, 126.12, 127.44, 127.65, 127.74, 127.92, 128.13, 128.75, 128.84, 136.53, 137.93, 156.79, 169.95, 171.67, 178.49, 179.59; 31P NMR (162 MHz, CD3OD) δ 44.03. HRMS (ESI/QTOF) m/z: [M]+ Calcd for C27H35N4O9P 590.2142; Found 590.2157.
3.1.1 The ACE inhibitory activity
(S,R,S,S)-Z-RXP407 was dissolved in DMSO, to yield a 50 mm stock solution. Aliquots of stock solutions were diluted to 10 mm working stocks with H2O followed by dilution in phosphate incubation buffer (50 mM HEPES pH 6.8, 200 mM NaCl and 10 µM ZnCl2).
3.1.2 Data analysis
Graphs of percentage remaining activity of each individual ACE domain in the presence of potential inhibitor were plotted and analyzed using GraphPad Prism 4.01 (GraphPad Software, La Jolla, CA) and/or Microsoft Excel® (Fig. 7). The enzyme without inhibitor was used as a control and deemed to be 100 % enzyme activity. Experiments were repeated and each assay was performed in triplicate.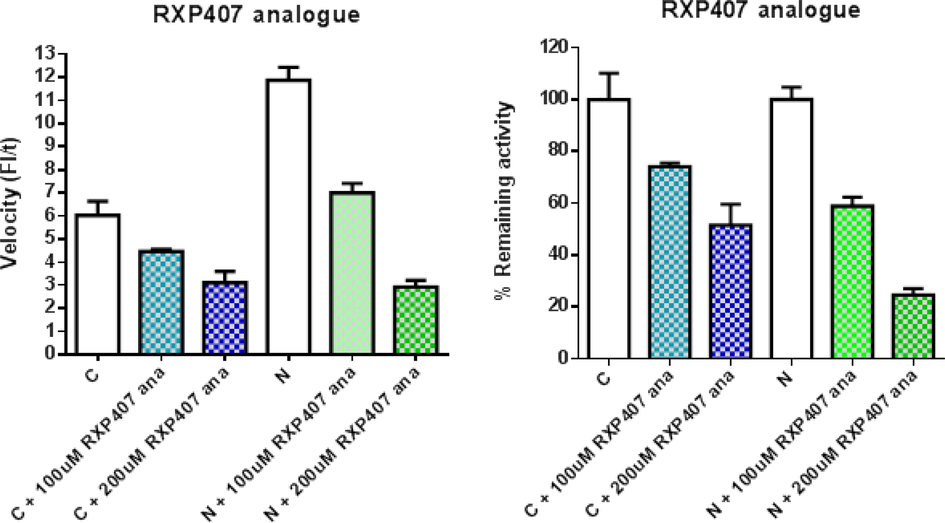
Inhibition screen of (S,R,S,S)-Z-RXP407 (RXP407ana) and their effect on N- and C-domain activity.
4 Conclusions
A high degree of diastereomeric purity and yield of the target phosphinic peptide (S,R,S,S)-Z-RXP407 was achieved via solution-phase synthesis and chemical resolution. The absolute configuration of this enantiomerically pure phosphinic acid was rationally established by different techniques. The applicability of this isomer as backbone in biological oriented targets was exemplified by the preparation of confirmed RXP407 which is a highly potent and selective inhibitor of the N-terminal active site of angiotensin I-converting enzyme. Moreover, it is expected that this identified strategy could expand and open new perspectives to efficient diastereoselective preparation of classical and non-classical pseudopeptidic phosphinic acids bearing more than one asymmetric center for the development of phosphinic peptide inhibitors in the future. The compound (S,R,S,S)-Z-RXP407 has been employed successfully in biological evaluation of the C- and N-domain constructs of angiotensin-converting enzyme and appeared to be less effective inhibitor of ACE as compared to the parent compound RXP407. Co-crystal structures of (S,R,S,S)-Z-RXP407 may shed light on the cause of the decreased binding affinity.
Acknowledgments
This work was financially supported by the Key Program Special Fund in XJTLU (KSF E-52).
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synopsis of recent synthetic methods and biological applications of phosphinic acid derivatives. Tetrahedron. 2020;76(25):131251
- [Google Scholar]
- Design, Synthesis, and Study of a Novel RXPA380–Proline Hybrid (RXPA380-P) as an Antihypertensive Agent. ACS omega. 2022;7(39):35035-35043.
- [Google Scholar]
- Abdou, Moaz M., Paul M. O’Neill, Eric Amigues, and Magdalini Matziari. Structure-based bioisosteric design, synthesis and biological evaluation of novel pyrimidines as antiplasmodial antifolate agents. Journal of Saudi Chemical Society 26 (2022): 101539.
- Innovative and potential chemical transformation of phosphinic acid derivatives and their applications in the synthesis of drugs. Bioorg. Chem.. 2019;90:103039
- [Google Scholar]
- Phosphinic acid based molecules: current status and potential for drug discovery. Drug Discov. Today.. 2019;24(3):916-929.
- [Google Scholar]
- Unprecedented convergent synthesis of sugar-functionalization of phosphinic acids under metal-free conditions. ACS OMEGA. 2022;7(25):21444-21453.
- [Google Scholar]
- The N Domain of Human Angiotensin-I-converting Enzyme the role of n-glycosylation and the crystal structure in complex with an n domain-specific phosphinic inhibitor, RXP407. J. Biol. Chem.. 2010;285(46):35685-35693.
- [Google Scholar]
- Structural basis of interactions between human glutamate carboxypeptidase II and its substrate analogs. J. Mol. Biol.. 2008;376(5):1438-1450.
- [Google Scholar]
- 1-Aminoalkylphosphonous acids. Part 1. Isosteres of the protein amino acids. J. Chem. Soc. Perkin Trans. 1984;1:2845-2853.
- [Google Scholar]
- Different in vivo functions of the two catalytic domains of angiotensin-converting enzyme (ACE) Curr. Opin. Pharmacol.. 2011;11(2):105-111.
- [Google Scholar]
- Crystal structure of the N domain of human somatic angiotensin I-converting enzyme provides a structural basis for domain-specific inhibitor design. J. Mol. Biol.. 2006;357(3):964-974.
- [Google Scholar]
- RXP407, a phosphinic peptide, is a potent inhibitor of angiotensin I converting enzyme able to differentiate between its two active sites. Proc. Natl. Acad. Sci. U.S.A.. 1999;96(8):4330-4335.
- [Google Scholar]
- Dive, V., Cotton, J., Cuniasse, P., Yiotakis, A., Corvol, P., Michaud, A., Chauvet, M.T., Menard, J., Ezan, E., 2002. N-terminal site selective inhibitors of human angiotensin conversion enzyme (ACE). U.S. Patent 6, 482,797.
- Fragment-based design for the development of N-domain-selective angiotensin-1-converting enzyme inhibitors. Clin. Sci.. 2014;126(4):305-313.
- [Google Scholar]
- Potent and selective inhibition of zinc aminopeptidase A (EC 3.4. 11.7, APA) by glutamyl aminophosphinic peptides: Importance of glutamyl aminophosphinic residue in the P1 position. Biochemistry.. 2000;39(5):1152-1155.
- [Google Scholar]
- Roles of the two active sites of somatic angiotensin-converting enzyme in the cleavage of angiotensin I and bradykinin: insights from selective inhibitors. Circ. Res.. 2003;93(2):148-154.
- [Google Scholar]
- Structural determinants of RXPA380, a potent and highly selective inhibitor of the angiotensin-converting enzyme C-domain. Biochemistry. 2004;43(25):8048-8054.
- [Google Scholar]
- A new synthetic route to 1-aminoalkylphosphonous acids. Synthesis.. 1987;10:942-943.
- [Google Scholar]
- RXP407, a selective inhibitor of the N-domain of angiotensin I-converting enzyme, blocks in vivo the degradation of hemoregulatory peptide acetyl-Ser-Asp-Lys-Pro with no effect on angiotensin I hydrolysis. J. Pharmacol. Exp. Ther.. 2001;297(2):606-611.
- [Google Scholar]
- Investigating the domain specificity of phosphinic inhibitors RXPA380 and RXP407 in angiotensin-converting enzyme. Biochemistry. 2009;48(35):8405-8412.
- [Google Scholar]
- Enantioselective synthesis of phosphinyl peptidomimetics via an asymmetric Michael reaction of phosphinic acids with acrylate derivatives. J. Organomet. Chem.. 2002;646(1–2):212-222.
- [Google Scholar]
- Diastereoselective solution and multipin-based combinatorial array synthesis of a novel class of potent phosphinic metalloprotease inhibitors. Chem. Eur. J.. 2003;9(9):2079-2094.
- [Google Scholar]
- Conformational and solvation studies via computer simulation of the novel large scale diastereoselectively synthesized phosphinic MMP inhibitor RXP03 diluted in selected solvents. J. Phys. Chem. B.. 2010;114(1):421-428.
- [Google Scholar]
- Crystal structure of the human angiotensin-converting enzyme–lisinopril complex. Nature. 2003;421(6922):551-554.
- [Google Scholar]
- Amide-assisted hydrolysis of β-carboxamido-substituted phosphinic acid esters. J. Org. Chem.. 1997;62(9):2808-2812.
- [Google Scholar]
- Inhibition of MMP-1 and MMP-13 with phosphinic acids that exploit binding in the S2 pocket. Bioorg. Med. Chem. Lett.. 1999;9(2):127-132.
- [Google Scholar]
- When fragments link: a bibliometric perspective on the development of fragment-based drug discovery. Drug Discov. Today.. 2018;23(9):1596-1609.
- [Google Scholar]
- New ketomethylene inhibitor analogues: synthesis and assessment of structural determinants for N-domain selective inhibition of angiotensin-converting enzyme. Biol. Chem.. 2012;393:485-493.
- [Google Scholar]
- Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol.. 2018;48:93-102.
- [Google Scholar]
- Structure-based dissection of the active site chemistry of leukotriene A4 hydrolase: implications for M1 aminopeptidases and inhibitor design. Chem. Biol.. 2008;15(9):920-929.
- [Google Scholar]
- A rational design strategy of the novel topoisomerase II inhibitors for the synthesis of the 4-O-(2-pyrazinecarboxylic)-4′-demethylepipodophyllotoxin with antitumor activity by diminishing the relaxation reaction of topoisomerase II-DNA decatenation. Bioorg. Med. Chem.. 2014;22(11):2998-3007.
- [Google Scholar]




