

Translate this page into:
Evaluating the anticorrosive properties of Camellia chrysantha flower extract on carbon steel in acidic solutions: A computational approach
*Corresponding author: E-mail address: nwazzan@kau.edu.sa (N. Wazzan)
-
Received: ,
Accepted: ,
Abstract
Plant extracts are emerging as viable alternatives to traditional corrosion inhibitors, and their inhibitory mechanisms require further investigation. This study examined the adsorption properties of phytochemicals from Camellia chrysantha flower extract on carbon steel in acidic solutions, utilizing both periodic and non-periodic computational methods. The primary phytochemicals, (+)-catechin (CTCH), quercetin (QRCT), quercetin-3-O-methyl ether (QCTO), and kaempferol (KMFR), were analyzed using the Density Functional Theory (DFT) and Density Functional Tight Binding (DFTB) computations. The PCM/B3LYP/6-311+G(2d,p) method revealed that the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of these compounds closely align with those of iron, promoting effective electron donation and acceptance during adsorption. Among the investigated compounds, QRCT and KMFR demonstrated the strongest adsorption energies, particularly in parallel configurations, with interaction strengths of -2.206 eV and -2.784 eV, respectively. This study highlights the potential of Camellia chrysantha phytochemicals as environmentally friendly corrosion inhibitors, providing detailed insights into their electronic and adsorption properties.
Keywords
Camellia chrysantha flower
Corrosion inhibitor
Density functional theory
Density functional tight binding
Green inhibitor
Plant extract

1. Introduction
Corrosion of metals is a serious problem that affects various industries, leading to significant structural damage and economic losses [1]. Metals exposed to environmental factors, particularly in the presence of moisture and acids, undergo chemical reactions that result in their deterioration [2]. Corrosion leads to metal degradation and significant economic losses, mitigated through coatings, cathodic protection, and corrosion inhibitors [3].
Corrosion inhibitors are particularly effective for protecting metals in environments where acids are used, such as in industrial acidization processes. Organic inhibitors primarily work by adhering to the metal surface and creating a protective layer that blocks corrosive substances from interacting with the metal directly [4]. Traditional corrosion inhibitors are often organic compounds with specific structural and electronic features that facilitate strong adsorption onto the metal surface. These compounds typically contain heteroatoms such as nitrogen, oxygen, and sulfur, which possess lone-pair electrons that can interact with the metal atoms [5].
Recently, plant-derived green corrosion inhibitors have emerged as promising substitutes for traditional inhibitors, which are frequently linked to environmental hazards and toxicity concerns [5]. Plant extracts are rich in phytochemicals, which are natural compounds with molecular structures similar to traditional inhibitors. These phytochemicals include flavonoids, alkaloids, and terpenoids, which can offer synergistic effects through multiple adsorption sites and mechanisms [6]. Employing plant-derived inhibitors helps meet the increasing demand for environmentally sustainable and green approaches.
However, the complexity of plant extracts’ composition makes understanding the metal protection mechanisms a challenging task [7]. This complexity makes it difficult to ascertain which components are most effective in preventing corrosion. Computational studies, particularly those using density functional theory (DFT), can provide valuable insights into the electronic properties and interaction mechanisms of individual phytochemicals with metal surfaces [8,9]. These studies can help identify the most potent compounds and elucidate their adsorption behaviors, thereby enhancing our understanding of how plant extracts inhibit corrosion [10].
While studying the electronic properties and reactivity of standalone molecules is useful, it has limitations [11]. Such studies may not fully capture the interactions between phytochemicals and metal surfaces. Therefore, it is necessary to incorporate periodic DFT calculations, which provide a more accurate representation of adsorption processes [12]. In this regard, density functional based tight binding (DFTB) offers a promising and time-efficient method for such calculations, allowing the study of larger systems and more complex interactions [10].
Recent studies have demonstrated the utility of computational methods in identifying and characterizing the most effective phytochemicals for corrosion inhibition. Wazzan [13] demonstrated the superior adsorption and inhibition properties of Allium jesdianum components. Similarly, Al-Qurashi and Wazzan [14] revealed mixed adsorption mechanisms for phytochemicals from Aerva lanata, identifying catechin as particularly effective. Lgaz and Lee [15] emphasized the role of orbital hybridization in the strong adsorption of hydroxycinnamic acids. These studies underscore the value of computational approaches in predicting inhibitor performance.
The objective of this study is to computationally investigate the reactivity and adsorption characteristics of phytochemicals from Camellia chrysantha flower extract on Q235 steel in a 1 M HCl solution. While experimental studies by Dai and An [16] have validated the extract’s corrosion inhibition efficacy using electrochemical impedance spectroscopy, polarization curves, and weight loss methods, computational analysis of the individual phytochemicals responsible for this inhibition remains unexplored. To address this gap, we employed DFT calculations to analyze the reactivity behavior and adsorption mechanisms of the key phytochemicals: (+)-catechin (CTCH), quercetin (QRCT), quercetin-3-O-methyl ether (QCTO), and kaempferol (KMFR). Additionally, self-consistent-charge density-functional tight-binding method (SCC-DFTB) simulations were conducted to investigate the electronic properties and adsorption interactions between these phytochemicals and iron surfaces, providing deeper insights into their inhibition mechanisms. This integrated computational approach aims to identify the most effective corrosion inhibitors within the extract and support the development of environmentally friendly corrosion protection strategies. The general and IUPAC names of the four phytochemicals are: (+)-catechin (2R,3S)-2-(3,4-dihydroxyphenyl)chromane-3,5,7-triol, quercetin (2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-4H-chromen-4-one), quercetin-3-O-methyl ether (2-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-methoxy-4H-chromen-4-one), and kaempferol (3,5,7-trihydroxy-2-(4-hydroxyphenyl)-4H-chromen-4-one), as illustrated in Figure 1.
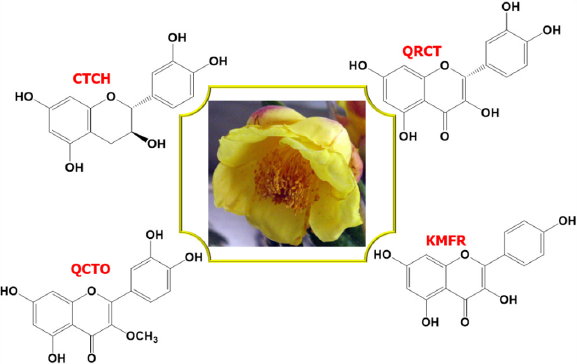
-
Camellia chrysantha flower and the chemical structures of its four phytochemicals; (+)-catechin (CTCH), quercetin (QRCT), quercetin-3-O-methyl ether (QCTO), and kaempferol (KMFR).
2. Materials and Methods
2.1. Computational details
2.1.1. Predicting microspecies of the phytochemicals
The determination of the predominant microspecies of the four inhibitors across various pH levels was performed using the MarvinSketch software. The pH range spanned from 0.0 to 14.0. Based on the output from MarvinSketch, it was observed that CTCH, QRCT, and QCTO exhibited 18 distinct forms, while KMFR showed 14 forms. In an experimental setup [16], HCl was chosen as the corrosive medium, with a pH of approximately 0.00. As a result, the dominant form of each inhibitor molecule at pH = 0.00 was identified and has been illustrated in Figure 2. It was found that the predominant microspecies of these inhibitors were largely in their neutral forms, with CTCH displaying 100% neutral species, while the other compounds were around 98%. These forms were subsequently used for theoretical calculations.
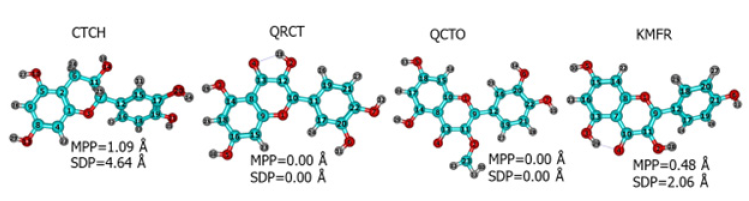
- Optimized geometrical structures of the four investigated phytochemicals. The Deviation from the Plane (SDP) and molecular planarity parameters (MPP) values are superimposed.
2.1.2. Molecular DFT details
In this study, DFT was applied to analyze four phytochemical compounds, CTCH, QRCT, QCTO, and KMFR, as shown in Figure 1. The molecular geometries of these species, in their neutral and isolated states, were optimized using the B3LYP functional within the DFT framework [17,18] in conjunction with the 6-311+G(2d,p) basis set. B3LYP is recognized for providing accurate geometries across numerous chemical systems. Although functionals like M06-2X or ωB97XD were considered, B3LYP was chosen to balance computational efficiency with accuracy and ensure compatibility with prior studies [19,20]. All calculations were performed in an aqueous environment using the Polarized Continuum Model (PCM) to account for solvation effects [21]. Following the geometry calculations, harmonic frequency calculations were performed at the same level of theory to ensure that all structures correspond to true minima on the potential energy surface. The molecules, along with their 3D iso-surfaces representing Frontier molecular orbitals and molecular electrostatic potential maps, were constructed, visualized, and plotted using the GaussView 6.16 software [22]. All computational tasks were executed using the Gaussian09 program suite [23]. Additionally, single-point energy calculations incorporating Natural Bond Orbital (NBO) analysis [24] were carried out to evaluate the Mulliken charges for systems with N, N-1, and N+1 electrons.
2.1.3. SCC-DFTB simulations
The SCC-DFTB method allows for efficient and accurate modeling of molecular interactions at the quantum level, making it suitable for studying complex systems with reduced computational cost. This method is well-regarded for its effectiveness in modeling adsorption systems involving organic molecules and transition metals, as supported by previous studies [25,26]. It provides the advantage of accurately predicting electronic and structural properties while being computationally more efficient than first-principles DFT simulations. The current work implemented the spin-polarized SCC-DFTB method, which enabled a detailed understanding of the molecular dynamics and electronic behavior that govern the adsorption process [27].
Thermal effects were managed using the Methfessel-Paxton method, and the Broyden mixing scheme was employed for improved convergence, while the adsorption systems were optimized with an SCC tolerance of 10−8 atomic units. All convergence parameters were carefully selected to meet the high precision demands of DFTB+. To achieve precise modeling, the lattice parameters were refined using an 8×8×8 k-point mesh, yielding a lattice constant of 2.877 Å, which showed close agreement with the experimentally determined value of 2.862 Å. For optimizing the interactions between the phytochemicals and the Fe(110) surface, a 2×2×1 k-point grid was employed. Adsorption simulations were conducted on the Fe(110) surface within a 5×5 supercell, and a 20 Å vacuum gap was introduced along the z-axis to avoid interference from periodic boundary conditions. The final simulation box dimensions were 15.07×15.07×26.15 Å3. The (110) iron structure was chosen due to its high thermodynamic stability. The densely packed atomic configuration of this surface minimizes its surface energy, making it more energetically favorable when compared to other orientations such as the (111) and (001) planes [28].
The investigated phytochemicals were placed on the topmost layer of the iron slab, where structural relaxation was applied to all atomic layers except for the two deepest ones. The simulations were conducted with a surface coverage of 1/25 monolayer (ML) for each phytochemical. In addition, the isolated molecules were optimized in a cubic simulation box with dimensions of 40 Å using the SCC-DFTB method. The interaction energy is a key factor for evaluating the adsorption strength of the phytochemicals, was calculated using the Eq. (1):
where Emol, Esurf, and Emol/surf represent the total energies associated with the isolated molecules, the clean Fe(110) surface, and the adsorption system consisting of both the molecule and the Fe(110) surface, respectively.
3. Results and Discussion
3.1. Molecular DFT results
3.1.1. Optimized geometries (planarity, ESPMs, and FMOs distribution)
The Span of Deviation from the Plane (SDP) and Molecular Planarity Parameters (MPP) were calculated using the Multiwfn software [29]. This was performed to determine the degree of planarity of each molecule since this will affect the adsorption mechanism of the phytochemical on the metal surface, especially in the case of parallel orientation. The MPP assesses the overall deviation of the entire structure from the reference plane, whereas the SDP quantifies the deviation of individual structural segments from the same plane [30]. Lower values of MPP indicate greater planarity, while a lower SDP signifies reduced deviation from the fitting plane. The planarity of the four molecules according to MPP and SDP values can be arranged as follows: QRCT = QCTO > KMFR > CTCH. Since QRCT and QCTQ are totally in-plane molecules, see Figure 2.
The planarity of a molecule directly impacts its adsorption behavior by influencing the extent of contact with the metal surface. Molecules with higher planarity, such as QRCT and KMFR, can achieve more effective parallel adsorption on the Fe(110) surface, maximizing the overlap of their π-electrons with the d-orbitals of iron. This increased contact area enhances electronic interactions, leading to stronger adsorption energies. In contrast, less planar molecules, such as CTCH, exhibit reduced overlap and weaker adsorption interactions, as evidenced by their lower adsorption energies.
The interaction between inhibitor molecules and metal surfaces is primarily influenced by molecular orbitals, especially the HOMO and LUMO. This interaction is facilitated through two primary mechanisms: (i) donation of electrons from the inhibitor’s HOMO to the vacant d-orbitals of the metal, and (ii) the reverse process, where electrons are donated back from the filled d-orbitals of the metal to the inhibitor’s LUMO. These complementary electron exchange processes work synergistically to enhance the adsorption strength of the inhibitor on the metal surface.
Figure 3 illustrate the 3d-isosurfaces of HOMOs, LUMOs, electrostatic potential maps (ESPMs), energies of HOMOs and LUMOs, and the energy gaps of the four investigated phytochemicals. Figure 3 indicates that the HOMOs and LUMOs of investigated molecules are distributed over the entire moiety of the investigated phytochemicals, indicating the possibility of using these inhibitors to be adsorbed on the Fe surface. Exception should be noticed here for the HOMO of the CTCH molecule, which is distributed on the pyrocatechol unit of the molecule, reflecting that this part is reasonable for electron’s donation during the adsorption process. These results are also confirmed by plotting the 3D-isosurfaces of the total density at HOMOs and LUMOs surfaces (TD-HOMO and TD-LUMO) as shown in Figure 4. The strong red color on the oxygen atoms and the phenyl groups marks these regions as favourable for electrophilic attacks , while the strong blue color on some C and H atoms reflect the ability of these site to undergo nucleophilic attacks.
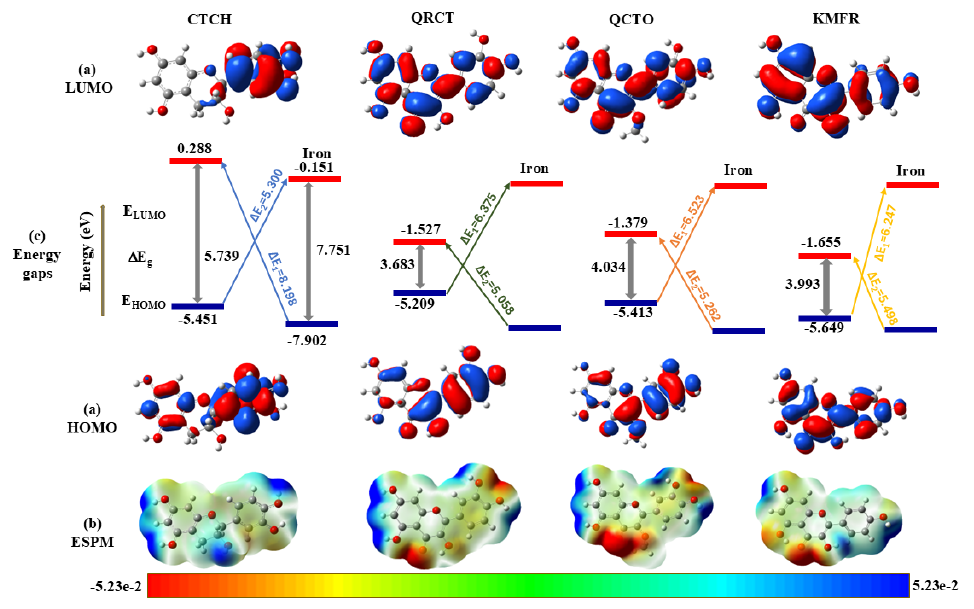
- 3D iso-surface representations of (a) frontier molecular orbitals (FMOs), (b) energy gap diagrams (energies are in eV units), and (c) electrostatic potential maps (ESPMs) in aqueous solution. The parameters used for these visualizations include an iso-value of 0.02 au, a density of 0.0004 au, and a Laplacian value of 0.00 au.
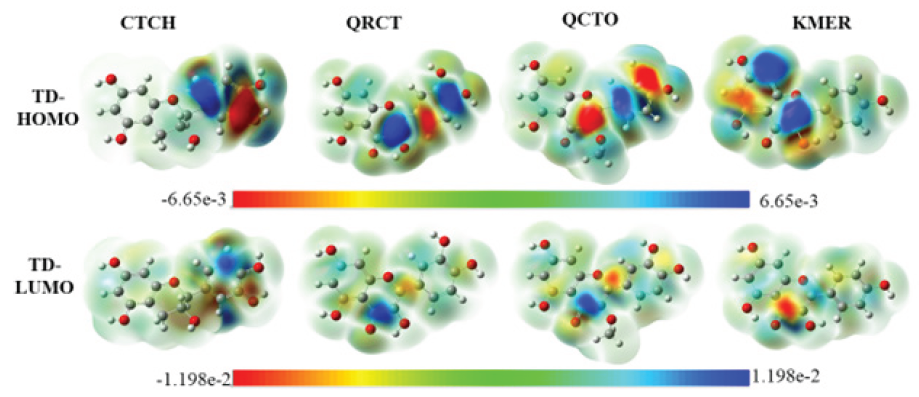
- 3D-Iso-surface total densities of HOMO and LUMO (TD-HOMO and TD-LUMO) of the investigated phytochemicals in aqueous solution (Iso-value: 0.02 au; Density: 0.0004 au; Laplacian = 0.00 au).
In addition, the ESPMs indicate the site of electronic accumulation (red-colored regions, negative potential) and sites of electronic depletion (blue-colored regions, positive potential). From, the heteroatom sites or functional group regions such as O atoms and hydroxyl or ketonic regions are colored red, while C and H atoms are surrounded by blue color, see Figure 3.
The electron-donating capability of an inhibitor is determined by the energy of its HOMO. A higher (less negative) HOMO energy (EHOMO) indicates a greater ease of electron donation, corresponding to a lower ionization energy (I = -EHOMO). On the other hand, the inhibitor’s capacity to accept electrons depends on the energy of LUMO. A lower (more negative) LUMO energy (ELUMO) reflects a higher electron affinity (A = -ELUMO), facilitating easier electron acceptance. When a molecule displays a favorable alignment between its HOMO and LUMO, it is considered chemically soft. According to hard and soft acids and bases (HSAB) theory, soft materials tend to form more stable interactions with each other, a characteristic that holds particular importance for bulk metals, which are chemically classified as soft. Figure 3 presents the HOMO and LUMO energies, along with the energy gaps, for the studied phytochemicals. Based on the energies of HOMOs, our results in Figure 3 indicate that the order of the inhibition efficiencies of the investigated phytochemicals in their isolated form would be: QRCT > QCTO > CTCH > KMFR. However, from the results of ELUMO, the trend would follow the order CTCH > QCTO > QRCT > KMFR.
Furthermore, to enhance the reactivity of the inhibitor toward anti-corrosive behavior, minimization ofthe HOMO-LUMO energy gap (ΔEg= ELUMO- EHOMO) is desirable. Figure 3 illustrates the energy gas values and the energy gap diagram of the investigated inhibitors. Our results in Table 1 and Figure 2 indicate that the energy gaps of the investigated inhibitors have the following theoretical trend: CTCH (5.739 eV) > QCTO (4.034 eV) > KMFR (3.993 eV) > QRCT (3.683 eV). These results reflect that QRCT with the lowest ΔEg value has the lowest energy gap giving it the most powerful anticorrosive effect, while the CTCH molecule with the highest energy gap could have the least chemical reactivity, and hence, could have the least inhibition performance.
| QCPs | CTCH | QRCT | QCTO | KMFR |
|---|---|---|---|---|
| Dipole moment (DM) (/Debye) | 3.579 | 4.256 | 3.247 | 4.667 |
| Adiabatic ionization energy (AIP) | 6.854 | 6.547 | 6.499 | 7.019 |
| Adiabatic electron affinity (AEA) | -0.173 | 0.214 | 0.149 | 0.455 |
| Chemical potential, μ | -3.341 | -3.381 | -3.324 | -3.737 |
| Electronegativity, χ | 3.341 | 3.381 | 3.324 | 3.737 |
| Hardness, η | 3.514 | 3.167 | 3.175 | 3.282 |
| Softness, (/eV-1) | 0.285 | 0.316 | 0.315 | 0.305 |
| Electrophilicity, | 1.588 | 1.804 | 1.740 | 2.128 |
| Nucleophilicity, (eV-1) | 0.630 | 0.554 | 0.575 | 0.470 |
| Electron accepting power, | 3.697 | 3.891 | 3.799 | 4.406 |
| Electron donating power, | 0.357 | 0.510 | 0.475 | 0.669 |
| Net electrophilicity, | 4.054 | 4.401 | 4.274 | 5.076 |
| Fraction of electrons transferred, | 0.211 | 0.227 | 0.236 | 0.165 |
| Back-donation index, | -0.878 | -0.792 | -0.794 | -0.821 |
| 0.156 | 0.164 | 0.176 | 0.089 |
, , , , , , , , , , , , , . of Fe(110) plane = 4.82 eV and ηmetal=0.0 eV at AIP=AEA in bulk.
As the metal and the inhibitor come close to each other, the electrons start to flow. Therefore, the electron’s flow from phytochemical to Fe or from Fe to phytochemical can be energetically described by estimating the differences between the HOMOs and LUMOs of the Fe and phytochemicals (ΔE1 and ΔE2). The following Eq. (2) are employed to calculate each parameter listed in Table 1 based on previous works [31,32]:
Here, the HOMO and LUMO energy levels of iron, denoted as and are considered to be -7.902 eV and -0.151 eV, respectively [33]. In this contest, ΔE1 is the energy required to flow the electron from the HOMO of Fe to the LUMO of the phytochemical. Whereas ΔE2 denotes the energy required to flow the electrons from the HOMO of the phytochemicals to the metal. The results are graphically shown in Figures 3 and 5.
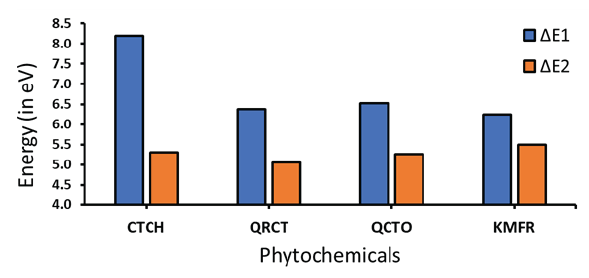
- Energy gaps of the Fe-inhibitors of the four investigated phytochemicals (ΔE1 and ΔE2).
Our results show that for the four investigated inhibitors, ΔE1 is larger than ΔE2, reflecting that electron transfer from inhibitors to Fe metal is energetically favored than the opposite process. This means that during the adsorption process, the investigated phytochemicals act as Lewis’s base and the Fe metal acts as Lewis acid. Comparison of ΔE1s of the investigated inhibitors show that the energetic trend of the electron’s flow from the HOMO of the inhibitors to the Fe metal exhibits the following order: KMER (6.247 eV) > QRCT (6.375 eV) > QCTO (6.523 eV) > CTCH (8.190 eV). On the other hand, when the electrons flow from the HOMO of Fe metal to the LUMO of the investigated inhibitors, the required energy follows the order as follows: QRCT (5.058 eV) > QCTO (5.262 eV) > CTCH (5.30 eV) > KMFR (5.498 eV).
3.1.2. Quantum global reactivity parameters
It should be mentioned that the adiabatic ionization potentials (AIP) and adiabatic electron affinities (AEA) were calculated and used to extract the most relevant global chemical reactivity parameters, as indicated in Table 1. The applied equations are indicated in the last row of the table.
Previous studies reported that the inhibitor molecule with a low electronegativity value has a good corrosion inhibition performance [34]. The obtained results indicate that the order of electronegativity follows the trend QRCT (3.167) < QCTO (3.175 eV) < KMER (3.282 eV) < CTCH (3.341). By taking into account the electronegativity of Fe-metal as 7.0 eV, therefore, the difference between the electronegativity of inhibitor and metal exhibits the trend QRCT (3.833 eV) > QCTO (3.825 eV) > KMER (3.718 eV) > CTCH (3.487 eV). According to Sanderson’s electronegativity equalization principle [35], QCTO, with the lowest electronegativity and the highest difference of electronegativity, could have the highest anti-corrosion performance, while CTCH exhibits the least corrosion inhibition efficiency.
Hardness (η) and softness (σ) are key molecular reactivity indicators that provide insights into the stability and chemical behavior of molecules, particularly within the framework of HSAB theory [36]. Lower values of hardness (η) and higher values of softness (σ) correlate with increased chemical reactivity, which enhances the efficiency of corrosion inhibition. The results presented in Table 1 demonstrate a predicted order of corrosion inhibition effectiveness for the studied phytochemicals, following the trend QRCT > QCTO > KMFR > CTCH.
As the electrophilicity increases, the propensity to accept electrons increases, and consequently, the inhibition efficacy increases [37]. From our results of electrophilicity (ω), the anticorrosive performance of the investigated species follows can be arranged as follows: KMFR > QRCT > QCTO > CTCH.
The donor-acceptor capacity of a chemical species can be quantified by calculating the electron-accepting power (denoted as ω-), electron-donating power (denoted as ω+), and net electrophilicity (denoted as Δω±). According to our results, it is found that ω- > ω+ for all species and KMFR phytochemical with the highest ω+, ω- and Δω± values is expected to have the highest ability to accept and donate electron during the adsorption process and the general trend exhibits the order as follows: KMFR > QRCT > QCTO > CTCH.
Δψ, calculated as the square of the difference in electronegativity between the inhibitor and the metal divided by the sum of their chemical hardnesses, quantifies the energy stabilization due to charge transfer. Lower Δψ values indicate stronger charge transfer interactions, enhancing adsorption. From Table 1, Δψ follows the order KMFR (0.089 eV) < QCTO (0.164 eV) < QRCT (0.176 eV) < CTCH (0.156 eV), suggesting KMFR exhibits the most favorable charge transfer conditions among the studied phytochemicals.
The negative values ∆EB-D descriptor indicates that back-donation from molecule to metal is energetically favored. Examination of Table 1 indicates that ∆EB-D are < 0 for all species, and the following order is expected: QRCT > QCTO > KMFR > CTCH, signifying that the process of electron transfer from the metal to the molecule, followed by electron back-donation, is energetically advantageous. Given that enhanced adsorption of the molecule onto the metal surface is likely to improve anticorrosive efficacy, this performance can be further optimized by increasing the stabilization energy between iron and the inhibitor.
The current study calculated the fraction of electrons transferred, ∆N110, and the results are also gathered in Table 1. Investigation of Table 1 indicates that ∆N110 values of all species were positive and less than 3.6, indicating that these inhibitors act as electron donors (Lewis’s base) during the adsorption process. Also, the higher the ∆N110 value, the higher the anticorrosive performance of the investigated inhibitors, and they can be arranged as follows: QRCT > QCTO > CTCH > KMFR.
These trends in global reactivity parameters align with the structural features of the studied phytochemicals. For instance, QRCT and KMFR, which exhibit higher nucleophilicity and lower hardness, have more electron-rich functional groups (e.g., hydroxyl and carbonyl groups) that facilitate stronger interactions with the metal surface. The higher softness (σ) of these molecules also enhances their adaptability to the electronic environment of the Fe(110) surface, improving their adsorption efficiency. Additionally, the negative back-donation index (∆EB−D) values confirm that electron transfer from the inhibitors to the iron surface, followed by back-donation, is energetically favorable. These findings align with established principles of soft acids and bases [38], further supporting the strong inhibitory performance of QRCT and KMFR.
3.1.3. Fukui indices
Kenichi Fukui [39,40] introduced the idea of Fukui functions, which have since become crucial descriptors for understanding local reactivity. These functions take into account structural variations in molecules, providing a more detailed assessment of atomic reactivity [41]. They are particularly useful for identifying the regions within a molecule that are more likely to undergo nucleophilic or electrophilic reactions. Additionally, Fukui functions enable the analysis of electron-donating and electron-accepting tendencies within a molecule. To quantify these characteristics, condensed Fukui descriptors for nucleophilic ( ) and electrophilic ( ) regions, as well as a dual descriptor ( ), were calculated and listed in Table 2. This was accomplished through Mulliken charge analysis within the framework of conceptual density functional theory (CDFT), utilizing the Multiwfn software [29]. The following expressions were employed for these calculations by Morell et al. (Eq. 3) [42]:
| CTCH | QRCT | QCTO | KMFR | ||||
|---|---|---|---|---|---|---|---|
| Atom | Atom | Atom | Atom | ||||
| C1 | -0.041 | O1 | -0.007 | O1 | -0.020 | O1 | 1.006 |
| C2 | 0.045 | O2 | 0.039 | O2 | 0.067 | O2 | -0.093 |
| O3 | 0.004 | O3 | -0.007 | O3 | 0.000 | O3 | -0.214 |
| C4 | 0.006 | O4 | -0.038 | O4 | -0.071 | O4 | 0.152 |
| C5 | -0.015 | O5 | -0.009 | O5 | -0.005 | O5 | -0.133 |
| C6 | 0.003 | O6 | 0.032 | O6 | 0.029 | O6 | -0.432 |
| C7 | -0.019 | O7 | 0.041 | O7 | 0.017 | C7 | 0.204 |
| C8 | 0.000 | C8 | 0.022 | C8 | 0.002 | C8 | -1.041 |
| C9 | -0.032 | C9 | -0.016 | C9 | 0.022 | C9 | -0.521 |
| O10 | 0.009 | C10 | -0.017 | C10 | 0.000 | C10 | -1.175 |
| C11 | -0.002 | C11 | 0.014 | C11 | -0.157 | C11 | 0.864 |
| C12 | -0.010 | C12 | 0.032 | C12 | 0.012 | C12 | 0.182 |
| O13 | 0.035 | C13 | -0.064 | C13 | 0.022 | C13 | 0.096 |
| O14 | 0.003 | C14 | -0.032 | C14 | -0.029 | C14 | 0.437 |
| C15 | -0.047 | C15 | -0.017 | C15 | -0.010 | C15 | 0.242 |
| C16 | 0.038 | C16 | -0.016 | C16 | -0.003 | C16 | 0.192 |
| C17 | 0.012 | C17 | 0.002 | C17 | -0.007 | C17 | 0.030 |
| C18 | -0.045 | C18 | -0.010 | C18 | -0.008 | C18 | -0.137 |
| C19 | -0.012 | C19 | 0.021 | C19 | -0.011 | C19 | 0.087 |
| O20 | 0.046 | C20 | 0.016 | C20 | 0.007 | C20 | 0.058 |
| O21 | 0.023 | C21 | 0.017 | C21 | 0.006 | C21 | 0.195 |
| C22 | -0.003 | C22 | -0.012 | ||||
| C23 | 0.151 | ||||||
A high value of the nucleophilic attack descriptor ( ) for a given atom k suggests that the atom exhibits strong electrophilic characteristics, whereas a high value of the electrophilic attack descriptor ( ) indicates nucleophilic tendencies [43]. Morell et al. [42] expanded on this concept by introducing a more refined descriptor known as the dual local Fukui index ( ), which integrates both and values. This descriptor is defined by the following equation (Eq. 4):
The purpose of is to simultaneously assess the nucleophilic and electrophilic behavior of an atom.
A positive value indicates that the atom is more likely to undergo nucleophilic attack, identifying it as a favorable site for electron acceptance. In contrast, a negative value suggests the atom is more vulnerable to electrophilic attack, designating it as an ideal center for electron donation [44]. In this study, the most significant results of for the phytochemical compounds under investigation are presented graphically in Figure 6.
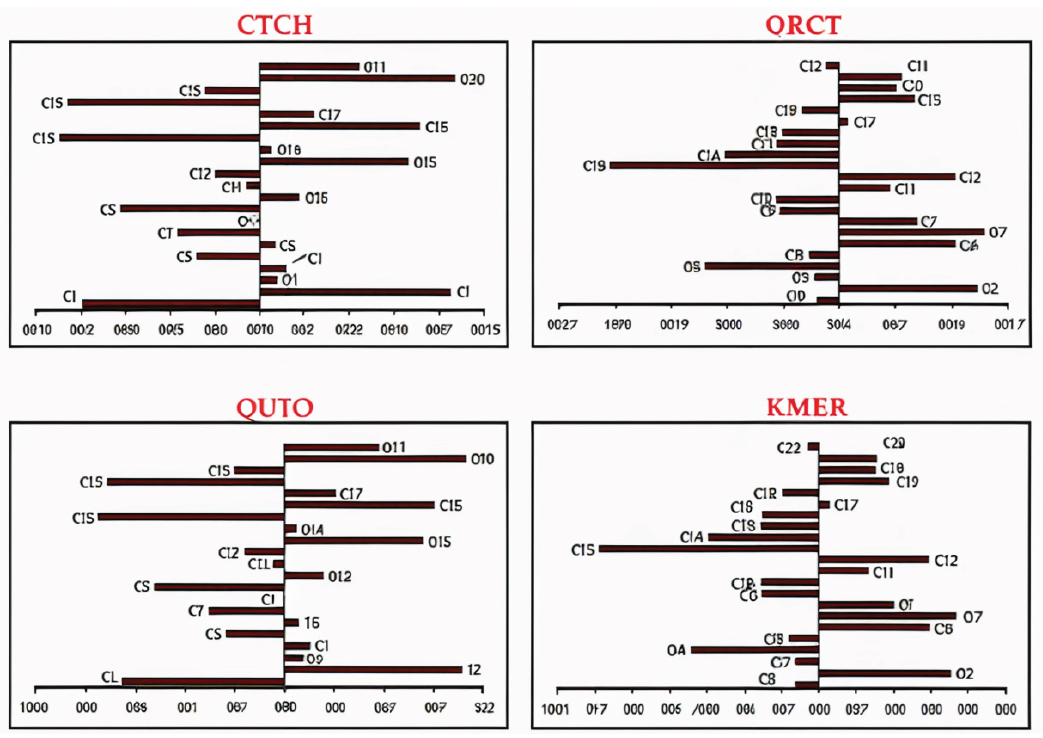
- Dual local Fukui indices of the investigated phytochemicals (fk2). Note: for atomic numbering refer to Figure 2.
For the CTCH molecule, nucleophilic attack is most likely at C15, C18, and C1, as indicated by their positive values, while electrophilic attack is favored at O20, C2, C16, and O13 due to their negative values. In QRCT, nucleophilic interactions are primarily associated with the C13, C4, and O4 centers, whereas electrophilic interactions predominantly occur at O7, O2, and O6 atoms. Similarly, in QCTO, the nucleophilic sites with the highest reactivity are C15, C1, and C18, while O20, C2, C16, and O13 exhibit the strongest propensity for electrophilic interactions. For KMFR, nucleophilic attack centers are identified at C13, O4, and C14, whereas O7, O2, and O6 demonstrate the highest capability for electron donation to the Fe surface, facilitating electrophilic interactions. These differences in reactive sites across the molecules are strongly influenced by their structural features, with hydroxyl and carbonyl groups consistently playing key roles in adsorption interactions.
3.2. Simulations of molecule-Fe interactions by SCC-DFTB
3.2.1. Geometrical and energetic aspects of adsorption
The SCC-DFTB calculations can provide a deeper understanding of how the investigated phytochemicals interact with the iron surface [10]. In this section, the adsorption behavior of the four investigated phytochemicals on the Fe(110) surface is explored using SCC-DFTB simulations. Figures 7 and 8 present graphical depictions of the adsorption configurations of CTCH, QRCT, KMFR, and QCTO on the Fe(110) surface, optimized using the SCC-DFTB method. The configurations include both parallel and perpendicular orientations relative to the surface.
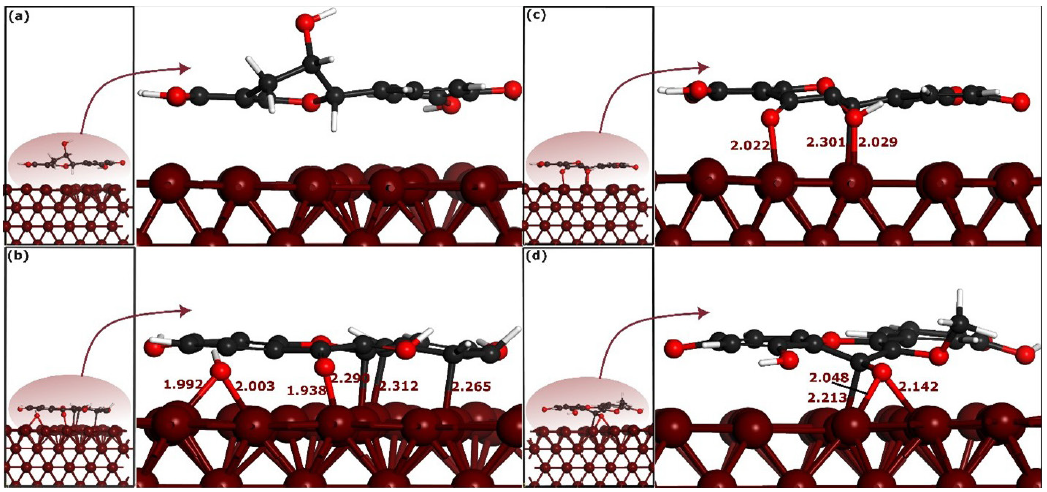
- Self-consistent-charge density-functional tight-binding method (SCC-DFTB) optimized parallel adsorption configurations of selected phytochemicals on the Fe(110) surface. (a), (b), (c) and (d) represent CTCH, QRCT, KMFR and QCTO, respectively. The bond distances are measured in angstroms (Å).
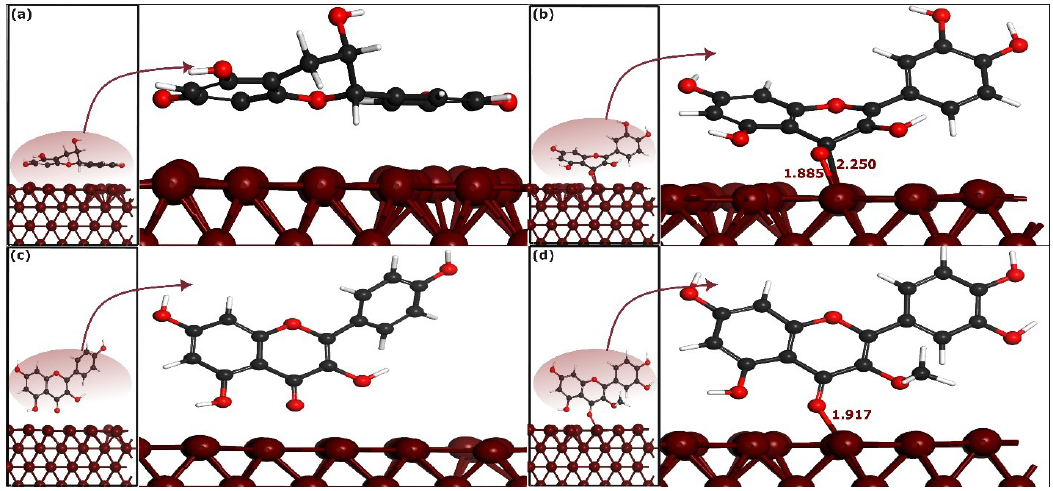
- Optimized perpendicular adsorption configurations of selected phytochemicals on Fe(110) surface by self-consistent-charge density-functional tight-binding method (SCC-DFTB). (a), (b), (c) and (d) represent CTCH, QRCT, KMFR and QCTO, respectively. The bond distances are measured in angstroms (Å).
Organic inhibitors in acidic media typically mitigate metal corrosion through physicochemical adsorption, beginning with physisorption followed by chemisorption [4]. It is useful to categorize molecule-metal bonds by comparing the sum of the covalent radii of the interacting atoms. Bonds classified as covalent typically fall within this sum, whereas bonds considered physical are characterized by distances exceeding 3 Å [45].
Visual inspection of Figures 7 and 8 shows that CTCH molecules do not bond with iron atoms in their stable parallel and perpendicular adsorption geometries. On the other hand, QRCT establishes connections with iron atoms through multiple carbon and oxygen atoms in both parallel and perpendicular adsorption configurations, Figure 7(b) and 7(c). In the parallel adsorption mode, QRCT forms three bonds with iron atoms: one Fe-C bond (2.301 Å) and two Fe-O bonds (2.022 and 2.029 Å). The perpendicular mode forms one Fe-O bond (1.885 Å) and one Fe-C bond (2.250 Å), indicating QRCT’s high affinity for iron atoms. KMFR exhibits strong bonding with iron atoms in its stable parallel geometry, forming three Fe-C bonds (2.265 to 2.312 Å) and three Fe-O bonds, with distances of 1.992, 2.003, and 1.938 Å. However, KMFR does not bond in its perpendicular geometry, suggesting its stability is higher in the parallel form. QCTO, in its stable parallel mode, forms two Fe-O bonds (2.142 and 2.048 Å) and one Fe-C bond (2.213 Å), while the perpendicular mode forms a single Fe-O bond (1.917 Å). These differences in bonding highlight the varying affinities of the phytochemicals for iron atoms: KMFR shows strong affinity in the parallel mode, while QRCT and QCTO bond in both geometries. CTCH shows no bonding, indicating a weak affinity for the iron surface.
The characteristics of the bonds formed between the adsorbed molecules and the iron surface can be analyzed by comparing the measured bond distances with the combined covalent radii of the interacting atoms. For Fe-C and Fe-O bonds, previous research has determined these sums to be approximately 2.08 Å and 1.98 Å, respectively [46]. In this study, the calculated bond distances fall within these thresholds, suggesting the possibility of chemical interactions.
The nature of these bonds can be determined by comparing the bond lengths to the total covalent radii of the atoms involved [47]. Previous studies indicate that the covalent radii for Fe-C and Fe-O interactions are approximately 2.08 Å and 1.98 Å, respectively [46]. The calculated bond lengths align with these values, indicating that the interactions between the molecules and the iron surface are likely to involve chemical bonding rather than mere physical adsorption.
For further assessment of the molecules’ interactive force, interaction energies were analyzed. Table 3 presents these energies, revealing that KMFR, when adsorbed in a parallel orientation, exhibits the most negative interaction energy (-2.784 eV), indicative of the strongest adsorption among the compounds studied. QRCT follows with an interaction energy of -2.206 eV, and QCTO registers at -1.974 eV. CTCH, however, shows the weakest interaction, with an energy of -0.165 eV. The consistently negative interaction energies across all molecules point to the thermodynamic favorability of their adsorption onto the iron surface, affirming the stability of these interactions [48]. The perpendicular geometries show significantly lower interaction energies, with QRCT displaying the strongest interaction (-1.342 eV), followed by QCTO (-0.345 eV), KMFR (-0.103 eV), and CTCH (-0.132 eV).
| Molecule | Einter(eV) | |
|---|---|---|
| Parallel | Perpendicular | |
| CTCH | -0.165 | -0.132 |
| QRCT | -2.206 | -1.342 |
| QCTO | -1.974 | -0.345 |
| KMFR | -2.784 | -0.103 |
DFTB: Density functional based tight binding.
These results suggest that the number and nature of functional groups in each phytochemical do not strongly influence adsorption strength. Despite having fewer hydroxy groups than QRCT, KMFR shows a stronger affinity for the iron surface. QCTO, with a methoxy group instead of a hydroxy group as in QRCT, still shows a different adsorption strength. This indicates that the adsorption strength is likely associated with the nature of the bonding, its strength, and structural factors such as steric effects, explaining the observed interaction magnitudes.
3.2.2. PDOS analysis
The PDOS analysis offers valuable insights into the electronic structures corresponding to the optimized adsorption configurations of the phytochemicals on the Fe(110) surface [49]. This method allows for the mapping of electronic states across the s and p orbitals of the phytochemicals and the 3d orbitals of the iron atoms. Figure 9 illustrates the PDOS for the isolated molecules situated at a distance of 7 Å from the iron surface, providing a baseline for comparison. In contrast, Figure 10 displays the PDOS after the molecules have adsorbed onto the iron surface, capturing the changes in electronic interactions due to bonding.
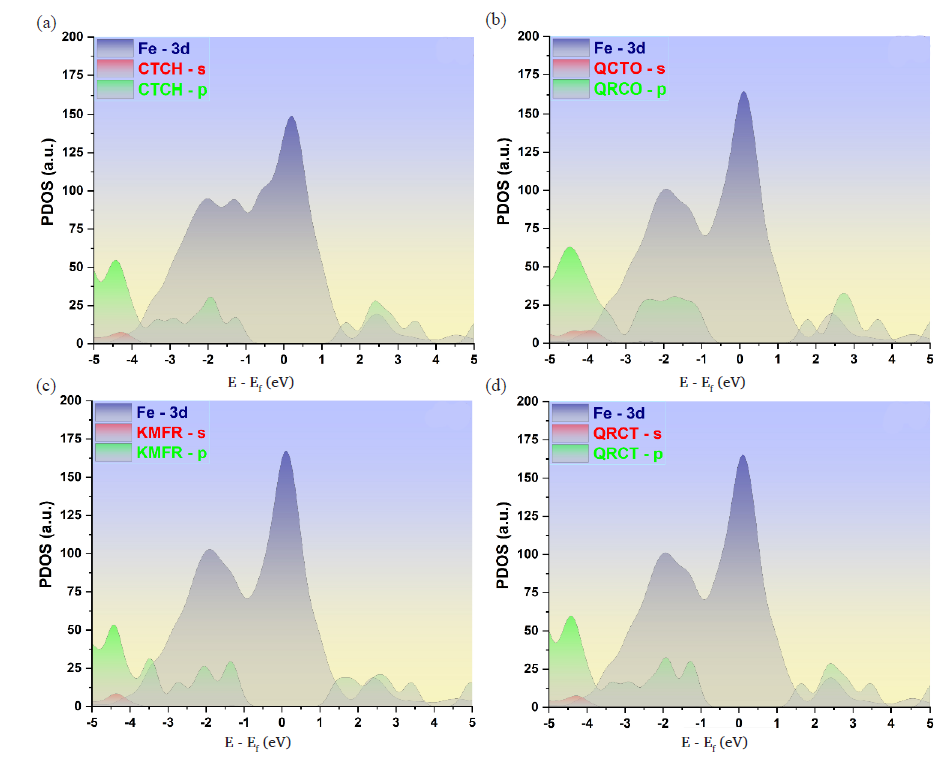
- PDOSs for the adsorption of selected phytochemicals on the Fe(110) surface. Panels (a)-(d) show the PDOS for molecules located at a distance of 7 Å above the topmost layer of the Fe(110) surface.
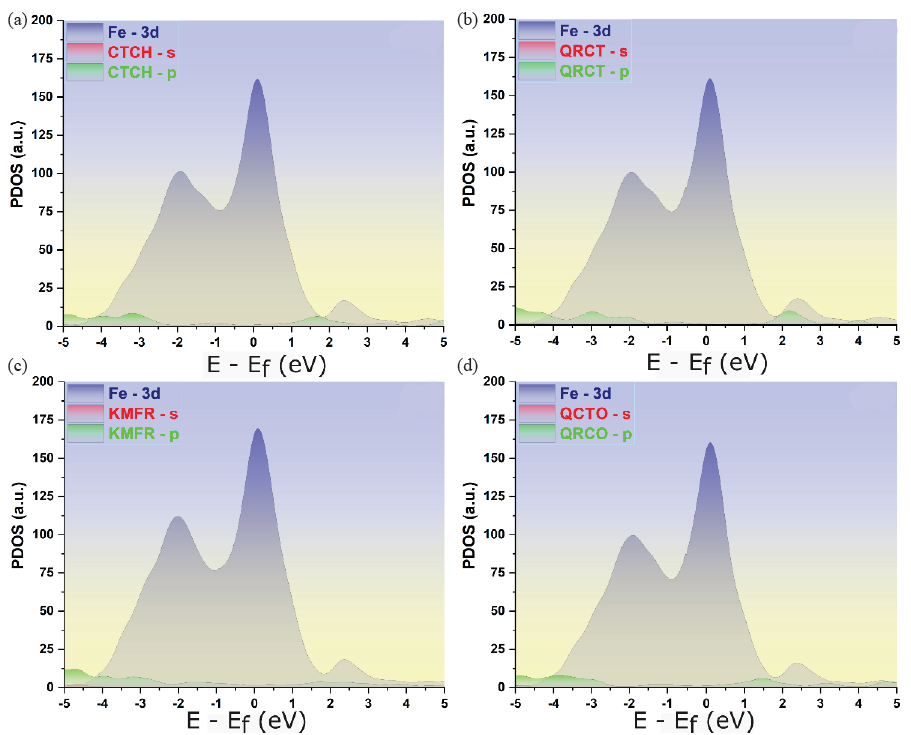
- PDOSs for the adsorption of selected phytochemicals on the Fe(110) surface. (a)-(d) represent the PDOSs of adsorbed molecules on the Fe(110) surface.
To ensure focus on significant adsorption behavior, only configurations where bonding occured, were considered for the PDOS analysis . Both figures also include the PDOS for the iron’s 3d orbitals, consistent with earlier findings [45], allowing for a clear comparison between the pre-adsorption and post-adsorption electronic states.
As shown in Figure 9, the PDOS patterns for the phytochemicals prior to adsorption display noticeable peaks, several of which lie in the energy range between -5 and 5 eV. This range when compared with that of iron, indicates a possible orbital overlap and hybridization between the s and p orbitals of the molecules and the vacant d-orbitals of the iron atoms. Such an overlap suggests the presence of substantial bonding interactions [50].
After adsorption, as illustrated in Figure 10, the PDOS for the phytochemicals exhibits a noticeable broadening of molecular peaks in comparison to their pre-adsorption state. The reduction in intensity and the vanishing of certain peaks within the energy range of -5 to 5 eV indicate a stronger interaction between the molecules and the iron surface. This broadening could be a result of increased electronic disorder or localized structural changes upon adsorption. Additionally, the PDOS after adsorption shows a shift to lower energy values, indicating that the system has stabilized [45]. It suggests that this energy shift plays a key role in reinforcing the stability of the molecule-surface interactions, leading to a more robust adsorption configuration.
The interaction between the phytochemicals and iron appears to be strongly influenced by orbital hybridization, as evidenced by the notable overlap between the s and p orbitals of the phytochemicals and the 3d orbitals of iron [51]. This hybridization is indicative of the formation of strong covalent bonds between the atoms of phytochemicals and the iron surface. As a consequence, the molecular orbitals of the adsorbed compounds likely fuse with the iron’s d-orbitals, leading to the creation of hybridized orbitals through a charge transfer mechanism. This process results in noticeable shifts and reorganization within the PDOS peaks, highlighting the substantial alterations in the electronic structure induced by the adsorption event.
3.3. Integrated discussion and future perspectives
The results obtained from the DFT, DFTB, and Fukui index analyses, collectively provide a comprehensive understanding of the phytochemicals’ behavior as corrosion inhibitors. Each method offers a unique perspective, and their integration highlights the synergistic roles of electronic properties, reactivity, and adsorption mechanisms in defining the inhibitors’ effectiveness.
The DFT calculations demonstrate that the HOMO-LUMO energy alignments of QRCT and KMFR with the Fe surface are the most favorable, facilitating strong electron donation and back-donation. This interaction is essential for effective adsorption, as shown by the lower HOMO-LUMO gaps in QRCT (3.683 eV) and KMFR (3.993 eV). Fukui indices further pinpoint the specific atomic sites contributing to this reactivity. The oxygen atoms in hydroxyl and carbonyl groups emerge as key sites for nucleophilic and electrophilic interactions, underscoring their role in stabilizing the adsorption process. This alignment of DFT and Fukui findings indicates that electronic and local reactivity descriptors complement each other in identifying the most effective inhibitors.
The DFTB simulations confirm the critical role of molecular geometry and adsorption orientation. QRCT and KMFR exhibit the strongest interaction energies (-2.206 eV and -2.784 eV, respectively) in parallel configurations, highlighting the importance of maximizing contact with the Fe(110) surface. These findings align with the planarity trends observed in DFT calculations, where QRCT and QCTO were found to have higher molecular planarity, promoting effective adsorption. The consistency between localized DFT findings and the periodic DFTB simulations reinforces the conclusion that QRCT and KMFR are the most effective phytochemicals due to their electron-rich structures and favorable adsorption geometries.
While DFT provides insights into electronic behavior and chemical reactivity, Fukui indices refine these findings by identifying site-specific contributions. DFTB complements these results by simulating the adsorption process in a periodic environment, bridging the gap between molecular-scale interactions and macroscopic adsorption behavior. Together, these methods paint a coherent picture of the inhibitors’ performance, showing QRCT and KMFR as the most promising candidates.
To further optimize these findings, future studies should explore synergistic combinations of phytochemicals, leveraging their complementary electronic and adsorption properties. Experimentally validating the computational predictions under real-world conditions will provide valuable insights into their practical applicability. Additionally, extending this approach to other natural extracts could broaden the range of effective green corrosion inhibitors, paving the way for sustainable corrosion protection strategies.
4. Conclusions
This study investigated the specific phytochemicals (CTCH, QRCT, QCTO, and KMFR) responsible for corrosion inhibition and their adsorption mechanisms, from Camellia chrysantha flower. Utilizing DFT calculations, we analyzed their electronic properties and adsorption behaviors. The PCM/B3LYP/6-311+G(2d,p) method revealed that the close alignment of HOMO and LUMO energy levels with those of iron enables these phytochemicals to effectively donate and accept electrons, confirming their role as Lewis bases during adsorption. Despite minor discrepancies in quantum chemical parameters, consistent trends in nucleophilicity and electron transfer capabilities were observed. DFTB results highlighted QRCT and KMFR as the most effective inhibitors, exhibiting strong adsorption energies and robust interactions with the iron surface. The parallel adsorption mode demonstrated the strongest affinity for Fe atoms, with binding energies ranked as KMFR (-2.784 eV) > QRCT (-2.206 eV) > QCTO (-1.974 eV) > CTCH (-0.165 eV). These findings provide valuable insights into the electronic interactions governing the adsorption process, facilitating the identification of effective natural corrosion inhibitors and supporting the development of environmentally friendly corrosion protection strategies.
Acknowledgment
Authors gratefully acknowledge King Abdulaziz University’s High-Performance Computing Centre (Aziz Supercomputer) (http://hpc.kau.edu.sa) for assisting with the calculations for the work of this paper.
CRediT authorship contribution statement
All authors contributed to the study conception and design. Hassane Lgaz: Methodology, Investigation, Resources, Writing - Review & Editing, Project administration; Ohoud Al-Qurashi: Methodology, Investigation, Resources, Writing - Review & Editing, Visualization; Nuha Wazzan: Methodology, Investigation, Resources, Writing - Review & Editing, Project administration. All authors read and approved the final manuscript.
Declaration of competing interest
The authors declare that they have no conflicts of interest.
Data availability
The authors confirm that the data supporting this study’s findings are available within the article.
Declaration of Generative AI and AI-assisted technologies in the writing process
The authors confirm that there was no use of AI-assisted technology for assisting in the writing of the manuscript and no images were manipulated using AI.
References
- Corrosion: Fundamental and economic impact. Carbon allotropes. CRC Press; 2024. https://doi.org/10.1201/97810033239762
- Corrosion in amine treating units. Woodhead Publishing; 2021. https://doi.org/10.1016/C2020-0-04139-3
- CHAPTER 2 - Corrosion prevention and protection methods. In: Guo L., Verma C., Zhang D., eds. Eco-Friendly Corrosion Inhibitors. Elsevier; 2022. https://doi.org/10.1016/B978-0-323-91176-4.00023-4
- [Google Scholar]
- Corrosion inhibitors: physisorbed or chemisorbed? Corrosion Science. 2022;196:109939. https://doi.org/10.1016/j.corsci.2021.109939
- [CrossRef] [Google Scholar]
- An overview on plant extracts as environmental sustainable and green corrosion inhibitors for metals and alloys in aggressive corrosive media. Journal of Molecular Liquids. 2018;266:577-590. https://doi.org/10.1016/j.molliq.2018.06.110
- [CrossRef] [Google Scholar]
- Evaluation of antioxidant and anticorrosive activities of Ceriops tagal plant extract. Applied Sciences. 2021;11:10150. https://doi.org/10.3390/app112110150
- [CrossRef] [Google Scholar]
- Challenges and advantages of using plant extract as inhibitors in modern corrosion inhibition systems: Recent advancements. Journal of Molecular Liquids. 2021;321:114666. https://doi.org/10.1016/j.molliq.2020.114666
- [CrossRef] [Google Scholar]
- Molecular modeling of corrosion inhibitors. In: Wandelt K., ed. Encyclopedia of interfacial chemistry. Oxford: Elsevier; 2018. https://doi.org/10.1016/B978-0-12-409547-2.13444-4
- [Google Scholar]
- Integrated modeling and electrochemical study of Myrobalan extract for mild steel corrosion retardation in acidizing media. Journal of Molecular Liquids. 2020;298:112046. https://doi.org/10.1016/j.molliq.2019.112046
- [CrossRef] [Google Scholar]
- Computational exploration of phenolic compounds in corrosion inhibition: A case study of hydroxytyrosol and tyrosol. Materials. 2023;16:6159. https://doi.org/10.3390/ma16186159
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Simplistic correlations between molecular electronic properties and inhibition efficiencies: Do they really exist? Corrosion Science. 2021;179:108856. https://doi.org/10.1016/j.corsci.2020.108856
- [Google Scholar]
- DFT study of aqueous-phase adsorption of cysteine and penicillamine on Fe(110): Role of bond-breaking upon adsorption. Applied Surface Science. 2020;514:145896. https://doi.org/10.1016/j.apsusc.2020.145896
- [CrossRef] [Google Scholar]
- Phytochemical components of Allium Jesdianum flower as effective corrosion-resistant materials for Fe(110), Al(111), and Cu(111): DFT study. Arabian Journal of Chemistry. 2023;16:104625. https://doi.org/10.1016/j.arabjc.2023.104625
- [Google Scholar]
- Molecular and periodic DFT calculations of the corrosion protection of Fe(110) by individual components of Aerva lanata flower as a green corrosion inhibitor. Journal of Saudi Chemical Society. 2022;26:101566. https://doi.org/10.1016/j.jscs.2022.101566
- [Google Scholar]
- Interfacial adsorption mechanism of hydroxycinnamic acids on iron surfaces: A computational perspective toward eco-friendly corrosion mitigation strategies. Applied Surface Science. 2024;644:158763. https://doi.org/10.1016/j.apsusc.2023.158763
- [CrossRef] [Google Scholar]
- Corrosion inhibition properties of Camellia chrysantha flower extract for Q235 in 1 M HCl solution. International Journal of Electrochemical Science. 2023;18:100080. https://doi.org/10.1016/j.ijoes.2023.100080
- [CrossRef] [Google Scholar]
- Density-functional exchange-energy approximation with correct asymptotic behavior. Physical review A. 1988;38:3098. https://doi.org/10.1103/PhysRevA.38.3098
- [CrossRef] [Google Scholar]
- Density‐functional thermochemistry. IV. A new dynamical correlation functional and implications for exact‐exchange mixing. The Journal of chemical physics. 1996;104:1040-1046. https://doi.org/10.1063/1.470829
- [Google Scholar]
- A computational study on corrosion inhibition performances of novel quinoline derivatives against the corrosion of iron. Journal of Molecular Structure. 2017;1134:751-761. https://doi.org/10.1016/j.molstruc.2017.01.037
- [Google Scholar]
- A theoretical density functional study of association of Zn2+ with oxazolidine and its thio derivatives in the gas phase. Journal of Physical Organic Chemistry. 2010;23:751-758. https://doi.org/10.1002/poc.1648
- [Google Scholar]
- Polarizable continuum model (PCM) calculations of solvent effects on optical rotations of chiral molecules. The Journal of Physical Chemistry A. 2002;106:6102-6113. https://doi.org/10.1021/jp020124t
- [CrossRef] [Google Scholar]
- GaussView, Version 6.1 GaussView, Version 61. Semichem Inc, Shawnee Mission; 2016.
- 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al.. Vol 121. Wallingford CT: Gaussian. Inc.; 2009. p. :150-166.
- Natural population analysis. The Journal of Chemical Physics. 1985;83:735-746. https://doi.org/10.1063/1.449486
- [CrossRef] [Google Scholar]
- An efficient way to model complex iron carbides: A benchmark study of DFTB2 against DFT. The Journal of Physical Chemistry A. 2023;127:2071-2080. https://doi.org/10.1021/acs.jpca.2c06805
- [CrossRef] [PubMed] [Google Scholar]
- SCC-DFTB parameters for Fe–C interactions. The Journal of Physical Chemistry A. 2020;124:9674-9682. https://doi.org/10.1021/acs.jpca.0c08202
- [CrossRef] [PubMed] [Google Scholar]
- DFTB+, a software package for efficient approximate density functional theory based atomistic simulations. The Journal of Chemical Physics. 2020;152 https://doi.org/10.1063/1.5143190
- [Google Scholar]
- Theoretical insight into an empirical rule about organic corrosion inhibitors containing nitrogen, oxygen, and sulfur atoms. Applied Surface Science. 2017;406:301-306. https://doi.org/10.1016/j.apsusc.2017.02.134
- [CrossRef] [Google Scholar]
- Multiwfn: A multifunctional wavefunction analyzer. Journal of Computational Chemistry. 2012;33:580-592. https://doi.org/10.1002/jcc.22885
- [CrossRef] [PubMed] [Google Scholar]
- Simple, reliable, and universal metrics of molecular planarity. Journal of Molecular Modeling. 2021;27:263. https://doi.org/10.1007/s00894-021-04884-0
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Handbook of chemistry and physics Vol 22. (Vol. 88th edition). CRC Press; 2007. p. :154. https://doi.org/10.1201/b17118
- Absolute hardness: companion parameter to absolute electronegativity. Journal of the American Chemical Society. 1983;105:7512-7516. https://doi.org/10.1021/ja00364a005
- [CrossRef] [Google Scholar]
- Electronegativity: The density functional viewpoint. The Journal of Chemical Physics. 2008;68:3801-3807. https://doi.org/10.1063/1.436185
- [Google Scholar]
- A sustainable approach exploiting Chrysanthemum Coronarium flowers distillation waste as a corrosion suppressor for low carbon steel in monoprotic acid medium through electrochemical and computational evaluations. Materials Chemistry and Physics. 2024;314:128846. https://doi.org/10.1016/j.matchemphys.2023.128846
- [CrossRef] [Google Scholar]
- A computational study on corrosion inhibition performances of novel quinoline derivatives against the corrosion of iron. Journal of Molecular Structure. 2017;1134:751-761.
- [Google Scholar]
- Absolute electronegativity and hardness: Application to inorganic chemistry. Inorganic Chemistry. 1988;27:734-740. https://doi.org/10.1021/ic00277a030
- [CrossRef] [Google Scholar]
- Electrophilicity index. Journal of the American Chemical Society. 1999;121:1922-1924. https://doi.org/10.1021/ja983494x
- [Google Scholar]
- On the consistent use of electrophilicity index and HSAB-based electron transfer and its associated change of energy parameters. Chemical Physics Letters. 2011;507:181-184. https://doi.org/10.1016/j.cplett.2011.03.045
- [Google Scholar]
- A molecular orbital theory of reactivity in aromatic hydrocarbons. The Journal of Chemical Physics. 1952;20:722-725. https://doi.org/10.1063/1.1700523
- [CrossRef] [Google Scholar]
- Frontier orbitals and reaction paths: Selected papers of Kenichi Fukui. World Scientific; 1997. https://doi.org/10.1142/2731
- Exploring the potential role of pyrazoline derivatives in corrosion inhibition of mild steel in hydrochloric acid solution: Insights from experimental and computational studies. Construction and Building Materials. 2020;233:117320. https://doi.org/10.1016/j.conbuildmat.2019.117320
- [CrossRef] [Google Scholar]
- New dual descriptor for chemical reactivity. The Journal of Physical Chemistry A. 2005;109:205-212. https://doi.org/10.1021/jp046577a
- [CrossRef] [PubMed] [Google Scholar]
- Novel Schiff-base molecules as effiient corrosion inhibitors for mild steel surface in 1 M HCl medium: Experimental and theoretical approach. Physical Chemistry Chemical Physics. 2016;18:17898-17911. https://doi.org/10.1039/C6CP01993E
- [CrossRef] [PubMed] [Google Scholar]
- Chemical reactivity descriptors for ambiphilic reagents: dual descriptor, local hypersoftness, and electrostatic potential. The Journal of Physical Chemistry A. 2009;113:8660-8667. https://doi.org/10.1021/jp902792n
- [CrossRef] [PubMed] [Google Scholar]
- Unravelling the mechanisms of corrosion inhibition of iron by henna extract: A density functional theory study. Corrosion Science. 2018;142:102-109. https://doi.org/10.1016/j.corsci.2018.07.011
- [CrossRef] [Google Scholar]
- Covalent radii revisited. Dalton Transactions 2008:2832-2838. https://doi.org/10.1039/B801115J
- [Google Scholar]
- Corrosion inhibition properties of thiazolidinedione derivatives for copper in 3.5 wt.% NaCl Medium. Metals. 2021;11:1861. https://doi.org/10.3390/met11111861
- [Google Scholar]
- Imidazole derivatives as corrosion inhibitors for copper: A DFT and reactive force field study. Corrosion Science. 2020;171:108724. https://doi.org/10.1016/j.corsci.2020.108724
- [CrossRef] [Google Scholar]
- Anticorrosive properties of green-synthetized benzylidene derivatives for mild steel in hydrochloric acid: An experimental study combined with DFTB and molecular dynamics simulations. Journal of Molecular Liquids. 2022;363:119790. https://doi.org/10.1016/j.molliq.2022.119790
- [CrossRef] [Google Scholar]
- Adsorption behaviors of NH3 and HCl molecules on Fe-based crystal planes: A DFT study. Chemical Engineering Science. 2021;246:116976. https://doi.org/10.1016/j.ces.2021.116976
- [CrossRef] [Google Scholar]
- Chapter 4 - Deep understanding of corrosion inhibition mechanism based on first-principle calculations. In: Verma D.K., Verma C., Aslam J., eds. Computational modelling and simulations for designing of corrosion inhibitors. Elsevier; 2023. https://doi.org/10.1016/j.ces.2021.116976
- [Google Scholar]




