

Translate this page into:
Forging of nicotine for the effective management of diabetic wounds: A hybrid of scaffold hopping and molecular dynamics simulation approaches
⁎Corresponding authors at: Department of Pharmacology, Faculty of Pharmacy, Marwadi University, Rajkot, Gujarat 360003, India (BKR Sanapalli). College of Pharmacy & Health Sciences, Ajman University, PO Box 340, Ajman, United Arab Emirates (R.R. Bhandare). College of Pharmacy, King Khalid University, Abha 61421, Saudi Arabia (SK Annadurai). Department of Pharmaceutics, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Ooty, Tamil Nadu 643001, India (VVSR Karri). bharathsanapalli@yahoo.in (Bharat Kumar Reddy. Sanapalli), r.bhandareh@ajman.ac.ae (Richie R. Bhandare), sannadurai@kku.edu.sa (Sivakumar. Annadurai),
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
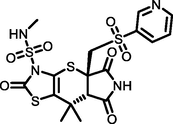
Diabetic wound (DW) is a huge threat to the health care community and is always challenging to treat. The main biochemical culprits in DW recalcitrance are elevated inflammatory mediators, proteases, cell proliferation and migration suppressors, anti-angiogenic factors, and bacterial infections. In this scenario, using a scaffold to target important factors at each stage of pathogenesis can accelerate the healing process. Many shreds of evidence disclosed the role of nicotine scaffold in handling inflammation, infection, proliferation, migration, and angiogenesis. All these factors made us forge nicotine by employing a scaffold hopping approach. The hops were then subjected to molecular docking and binding free energy calculations against Matrix metallopeptidase 9, Glycogen synthase kinase 3 beta, Tumor necrosis factor alpha, MurC and ParE enzymes. Gratifyingly, molecule H1 was found to possess significant inhibitory activity against the selected receptors as evidenced by their high negative glide score and binding energy. Furthermore, 100 ns of molecular dynamics simulation studies (MD) was performed for the five H1/4XCT, H1/5F95, H1/2AZ5, H1/4C13 and H1/4MOT complexes to get insight into the binding modes and stability. The MD results showed significant stability as evidenced by the low conformational changes of the H1 with the chosen receptors. Hence, H1 might be a druggable candidate in the therapeutic management of DW. However, further research is strongly recommended to advance the drug into the therapeutic pipeline.
Keywords
Diabetic wound
Nicotine
Scaffold hopping
Molecular docking
Molecular dynamics simulation

1 Introduction
Diabetes mellitus (DM) greatly impacts the quality of life and survival expectation of patients (Belardinelli et al., 1999). In fact, as a consequence of hyperglycemia, diabetic patients are at increased risk for comorbid conditions affecting several organs (Vigneri et al., 2009). One of the main consequences of diabetes is the impairment of self-repairing abilities (Spampinato et al., 2020). Diabetic wound (DW) is a very common secondary complication of DM that poses a significant challenge to the health care system (Sen, 2019). The major biochemical culprits in the recalcitrance of DWs are elevated inflammatory mediators, proteases, cell proliferation and migration suppressors, anti-angiogenic factors, and bacterial infections (Acosta et al., 2008). All these factors are being considered as the hallmarks of the delayed healing of DWs. The late inflammatory phases of wound healing in diabetes are characterized by altered leukocyte infiltration and Interleukin-6, according to Fahey et al. As a result, a changed cytokine pattern in the wound milieu may play a role in prolonged inflammation of DW (Fahey et al., 1991). Pathogenic markers (overexpression of c-myc and localization of β-catenin) that correlate with delayed healing have been found by molecular studies from the epidermal biopsies of DW patients (Stojadinovic et al., 2005; Brem et al., 2007). In this context, targeting the important factors at every stage of pathogenesis with a scaffold may potentiate the healing process. With this motive, we exemplified the forging of nicotine using in silico approaches.
Nicotine, a chiral alkaloid, possesses anti-inflammatory properties both in vitro and in vivo (Piao et al., 2009). Nicotine exerts its action in both humoral and cell-mediated immunity, leading to altered immune response characterized by reduced inflammation, antibody response and T-cell receptor mediated signaling (Johnson et al., 1990; Shi et al., 2009; Kalra et al., 2000; Sopori et al., 1998; Basta et al., 2000). It also plays a role in humoral immunity by regulating B lymphocytes via nicotinic acetylcholine receptors. There are many shreds of evidences for anti-inflammatory properties of nicotine. A study by Takahashi HK et al., 2006 convinced the inhibition of IL-12, IL-18 by nicotine through upregulation of COX-2 expression (Takahashi et al., 2006; Takahashi et al., 2006). Further, low-dose of nicotine also proved to inhibit the production of IL-12, prostaglandin E2, IFN-γ, TNF-α and macrophage inflammatory protein (MIP)-1 (Wang et al., 2004). It also inhibits transcriptional activity of NF-κB by suppressing the I-κB phosphorylation (Vassallo et al., 2005). However, all these suppressive effects of nicotine were mediated through α7nAChR at transcription level (Utsuki et al., 2002). Besides, nicotine also plays a key role in inhibition (500 ng/mL) of matrix metalloproteinase-9 (MMP-9) expression through the α7-nAChR-JNK pathway in dose-dependent fashion (Renò et al., 2011). Further, nicotine acts on α7-nAChRs to inhibit MMP-9 production by macrophages through modulation of the PI3K/Akt–NF-κB pathway. The other receptor namely GSK-3β whose inhibition promotes wound healing through β-catenin dependent Wnt signaling pathway (Harish et al., 2008). Although, no studies were reported for the inhibitory activity of GSK-3β by nicotine, the scaffold present in the nicotine (pyridine) has been profoundly used in the designing and synthesis of GSK-3β inhibitors (Tantray et al., 2016; Ramurthy et al., 2020). Nicotine has also shown to bring about tissue healing through release of growth factors such as basic fibroblast growth factor (bFGF) and transforming growth factor beta (TGF-β), vascular endothelial growth factor (VEGF) and matrix metalloproteinases in endothelial cells. These cytokines/ factors are responsible for migration; fibrin and collagen synthesis; and neovascularization which form key steps in wound healing (Cucina et al., 1999; Carty et al., 1996; Conklin et al., 2002; Jacobi et al., 2002). These studies demonstrated that nicotine enhanced angiogenesis by activation of nAChR leading to acceleration of wound healing in a murine excisional wound model.
It is shown by multiple reports that cigarette smoke causes delay in wound healing and nicotine is the toxic constituent of cigarette smoke. Nicotine increases vasoconstriction, reduces blood supply to the skin, increases the platelet adhesion and causes damage to the endothelial lining of the vessels, resulting in tissue ischemia. However, Morimoto et al, (2008) have shown at low concentrations nicotine had beneficial effects on wound healing by accelerating angiogenesis and simulating nAChR (Morimoto et al., 2008).
The most common infection causing bacteria observed in DW are Staphylococcus aureus, Streptococcus sp., Escherichia coli and Pseudomonas aeruginosa (Serra et al., 2015). Although many antibacterial targets are available for the infection control, the MurC (Ishibashi et al., 2007) and ParE (Drlica and Zhao, 1997) were considered as effective druggable targets as they are conserved in all species of bacteria and has no resistance reported so far. According to the available literature, (Pavia et al., 2000) nicotine causes a dose-dependent growth inhibition of a broad spectrum of both gram-positive and gram-negative bacteria (Shi et al., 2019).
Discovering novel bio-active compounds with iso-functional activity is a key challenge in medicinal chemistry, to enhance synthetic accessibility, potency, explore unmapped regions in the chemical space and druggability of hits and leads (Böhm et al., 2004). Further, ligand-based drug discovery has been advanced with the introduction of molecular descriptors such as distribution of pharmacophoric sites, steric, atomic and electronic environment, which in turn provided a strong basis for the ligand-based screening, efforts in target identification and de novo design of small compounds. Scaffold or core hopping (Schneider et al., 1999) is a widely-used strategy for the development of novel small molecules inhibitors from the existing drugs against a panel of targets (Li et al., 2011; Ma et al., 2012). Li et al. 2011 designed a series of novel inhibitors for hemagglutinin against H1N1 influenza virus using the core hopping approach (Li et al., 2011). Later Jin et al. 2014 had designed specific inhibitors against protein tyrosine phosphatase SHP-2 by virtual screening and core hopping methods (Jin et al., 2014). In the same year, Zhang et al. also designed a SAHA-based novel HDAC inhibitor by core hopping technique (Zang et al., 2014).
In addition, molecular docking study has been profoundly used in drug design and discovery (Ooms, 2000). To enhance the activity of a lead molecule, we usually modify the side chains attached to the core part of the compound, because in various cases it is the side chain of the compound that forms an interaction with the receptor residues. However, core structure may also vary to explore novel compounds with great diversity (Sun et al., 2012; Jiang et al., 2013).
In this paper, we used molecular modeling techniques with a core hopping approach by screening fragment database (Vainio et al., 2013; Flood et al., 2017), with an aim to identify novel nicotine analogues for the management of DW. Furthermore, Molecular dynamics simulation (MD) studies were employed to get insights into the binding poses of protein–ligand complexes.
2 Materials and Methods:
2.1 Protein preparation
The initial three-dimensional crystal structures of various receptors MMP-9, GSK-3β, TNF-α, and antibacterial targets (MurC and ParE) that are involved in the pathogenesis of DW were retrieved from RCSB-protein data bank with accession IDs of 4XCT (1.30 Å) (Nuti et al., 2015), 5F95 (2.52 Å) Luo et al. (2016), 2AZ5 (2.10 Å) (Silvian et al., 2011; He et al., 2005), 4C13 (1.90 Å) (Wang et al., 2021) and 4MOT (1.75 Å) (Kale et al., 2014) respectively. The crystals were selected based on their resolution, expression system, completeness, co-crystallized ligand etc. The dossier of the receptors was provided in Supplementary Table S1. Preparation of the protein was performed using ‘protein preparation wizard’ available in Schrödinger suite 2020–3. All the water molecules were removed; missing side chains and hydrogen bonds were added prior to protein optimization (Sastry et al., 2013; Iwaloye et al., 2020). The same procedure was employed for the preparation of apoprotein. A grid within a radius of 10 Å was generated using centroid of the active site of the co-crystallized ligand.
2.2 Molecular docking and scaffold hopping approach
Various useful insights for drug design could be obtained using molecular docking studies (Wang and Chou, 2012; Wang and Chou, 2011). However, to acquire even more useful information for drug design, a new computational approach called ‘scaffold hopping’ or lead hopping or core hopping was adopted in this study which is featured by having the functions to perform both the fragment-based replacement and molecular docking.
Scaffold hopping is a powerful and cutting-edge tool for de novo drug design because it can significantly improve the receptors binding affinity with its ligands. The minimized binding poses thus obtained was used as an initial structure for further optimization by searching the fragment database to determine the optimal hops that are attached to other parts of the parent compound.
In the current study, core hopping algorithm (CombiGlide 2.5 Schrödinger suite 2019–2) was employed during docking procedure with the functions to perform both fragment-based replacement and docking. The scaffold hopping strategy is adopted to screen multiple scaffolds or hops against a guiding structure, identifying for alignments of potential attachment sites on the hops with the attachment sites on the guiding structure. In general, the core hopping process contains 4 steps (www.schrodinger.com):
-
The first and foremost step is to define some probable sites at which the cores are attached to the hop. It also defines combination steps from the Combinatorial screening package available Schrödinger suite 2019–2.
-
The second step includes the generation of grid file in the “Receptor Preparation”wizard in Schrödinger suite 2019–2.
-
The third step involves the preparation of core using Protocore Preparation module in Schrödinger suite 2019–2 to determine the core attaching to the hop employing fragments available in ZINC database.
-
The final step involves the alignment and docking of structures generated by the core and hop into the selected receptors.
From the products of the scaffold hopping approach, a total of 100 molecules were sorted and filtered by goodness of alignment, and then redocked against the catalytic pocket of the receptors after attaining the hops (Friesner et al., 2006) (Supplementary Table S2). The final molecules were filtered using glide score (Gscore), glide emodel, etc. We performed docking for the obtained scaffolds in standard precision mode against all the selected receptors. The top 50 molecules were selected based on their Gscore, glide emodel, glide eVan der Waals, glide ecoulomb, and glide energy for redocking in extra precision (XP) mode. All these 50 molecules were further subjected to binding free energy calculations. The top 10 molecules were selected based on the Gscore, binding energy and protein–ligand interactions that were observed in XP docking and binding free energy calculation, and subjected to further analysis.
In addition, XP-Glide docking protocol was validated by removing the co-crystallized ligand from the catalytic pocket and redocked it to the active sites of MMP-9, GSK-3β, TNF-α, MurC and ParE enzymes. Similar orientations between the docked pose and the pose of co-crystallized ligand conformations were observed with a low root-mean square deviation (RMSD) (4XCT: 1.26 Å; 5F95: 0.98 Å; 2AZ5: 0.29 Å; 4C13: 1.89 Å; 4MOT: 0.72 Å) (Supplementary Figure S1(a-e)). This recommends the reliability of our XP-docking protocol in reproducing the experimentally observed binding pose.
2.3 Binding free energy calculations
Molecular Mechanics Generalized Born Surface Area (MM-GBSA) approach was used for calculating binding free energy of the final molecules by Prime module available in Schrödinger suite 2020–3. Energy minimization and simulation was performed using VSGB 2.0 energy model (Li et al., 2011) by keeping input partial charges for ligand complexes, without applying any restriction on flexible residues. OPLS3e force field (Roos et al., 2019) was used for energy calculation and optimization which incorporate physics-based corrections for self-contact, π-π, π-cationic, hydrogen bonding and hydrophobic interactions along with an optimized implicit solvation model. All 10 molecules were subjected to binding free energy calculation against the selected receptors (Jacobson et al., 2004) (Li et al., 2011).
2.4 Prediction of pharmacological and toxicological properties
In recent years, the significance of favorable pharmacokinetic features for the discovery of successful drug candidates has been widely acknowledged. Thus, ADMET evaluations are integrated earlier into the field of drug design and discovery. QikProp (Duffy and Jorgensen, 2000) has been proved to be a novel tool in computing and optimizing the pharmacokinetic profile or ADMET relevant descriptors for pharmaceutically acceptable compounds. Further, this algorithm is also used to correlate physicochemical and pharmacokinetic properties with 3D molecular structure of the test ligands. Neutralization of compounds is an essential step that is performed before being used by QikProp, as this tool is incapable to neutralize a compound. A total of 44 predicted properties such as physicochemical properties, principal descriptors, LogP, QP% and log HERG are generated in normal mode. It also determines the acceptability of the ligands, based on Lipinski’s rule of five (Lipinski et al., 1997) and three, that are crucial for rational drug design.
2.5 Molecular dynamics simulation
Conformational stability is crucially important for potent inhibition of any receptor and an MD study provides conformational landscape of protein–ligand complexes at given temperature. In order to understand the dynamics interaction mechanism between the receptor and the ligand, we have performed a molecular dynamics simulation study. The XP-docked poses of H1/4XCT, H1/5F95, H1/2AZ5, H1/4C13, H1/4MOT complexes, and apoproteins were used as initial structures for MD simulation using the Desmond module with OPLS3e force field available in Schrödinger suite (Jacobson et al., 2004). The complexes were solvated explicitly with TIP4P water molecules (Duffy and Jorgensen, 2000; Lawrence and Skinner, 2003; Jorgensen and Madura, 1985) in an orthorhombic box permitting 10 Å buffer region between amino acid residues or atoms and box sides. The solvated systems were neutralized by adding (11Na+-H1/4XCT; 8Cl−1- H1/5F95; 3Na+-H1/2AZ5; 15Na+-H1/4C13; 3Na+-H1/4MOT) counter ions. The systems comprise of water molecules (5838-H1/4XCT;15419-H1/5F95; 11329-H1/2AZ5; 17414-H1/4C13; 8408-H1/4MOT), atoms (19997-H1/4XCT; 51936-H1/5F95; 38689-H1/2AZ5; 59887-H1/4C13; 28474-H1/4MOT). Minimization of systems were accomplished with OPLS3e force field using 200 steepest descent step algorithms, followed by 1000 steps of conjugate gradient method until a gradient threshold of 25 kcal/mol was achieved. Particle-mesh Ewald method at a tolerance of 1e-09 was used for long-range electrostatic interactions treatment (Essmann et al., 1995). Whereas for Coulombic and Van der Waals interactions, a cut-off radius of 9 Å was applied. The minimized system was then simulated for 100 ns at constant temperature and pressure using isothermal and isobaric ensemble (NPT). A Martyna Tobias Klein barostat and Nose-Hoover thermostat were used to maintain constant pressure and temperature, respectively. RESPA integration algorithm was used for all bonded (2 fs time steps), near nonbonded (2 fs time steps) and far nonbonded (6 fs time steps) (Böhm et al., 2004; Martyna et al., 1992; Martyna et al., 1994; Martyna et al., 1996). The 3D-structures and obtained trajectories for all the complexes were visualized using Maestro-graphical interface.
3 Results and discussion
3.1 Scaffold hopping and molecular docking
Scaffold hopping refers to a type of bioisosteric replacement where a key core of a template structure is kept constant while replacing some of the key features. In general, the main aim of scaffold hopping is to discover equipotent derivatives with different backbone which have improved physicochemical properties and enhanced biological activity than the compounds that are active against a preselected target. The alterations of a template structure with desired biological activity may include ring replacement by heterocyclic scaffolds.
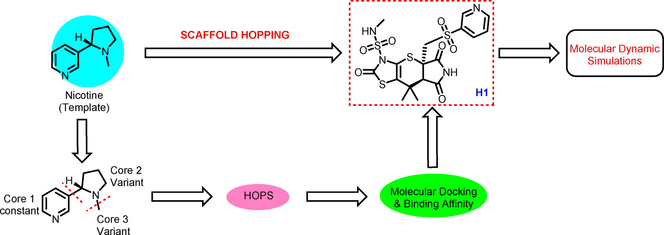
The structure of nicotine, which is conceived as a therapeutic model to develop novel bioactive molecules for the management of DW. Considering the significant importance of pyridine moiety (Core 1) for inhibiting the MMP-9, GSK-3β, TNF-α, MurC and ParE receptors, retained the Core 1 part during the scaffold hopping calculation. The first scaffold hopping operation was aimed at the Core 3 part followed by Core 2 by replacing with different moieties (Fig. 1).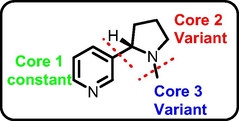
General scaffold hopping of nicotine.
Consequently, a total of 100 molecules were sorted and filtered by goodness of alignment obtained through scaffold hopping of nicotine. These compounds were SP-docked into catalytic pockets of MMP-9, GSK-3β, TNF-α, and antibacterial targets (MurC and ParE), respectively, screening out 50 molecules with higher Gscore and better binding affinity than the parent compound. These 50 molecules subjected to XP-docking and free energy analysis. The 10 top-ranked molecules (High negative Gscore, binding free energy and excellent protein–ligand interactions) were used for further analysis. The Gscore and ΔGbind of these compounds with selected receptors (Table 1) were higher than the original compound.
S. No.
HOPS
MMP-9 (pdb.4xct)
GSK-3β (pdb.5f95)
TNF-α (pdb.2az5)
MurC (pdb.4c13)
ParE (pdb.4mot)
Gscore
ΔGbind
Gscore
ΔGbind
Gscore
ΔGbind
Gscore
ΔGbind
Gscore
ΔGbind
1
−7.78
−74.32
−8.14
−55.39
−5.78
−58.94
−4.13
−68.49
−5.93
−78.79
2
−7.76
−64.73
−7.77
−46.18
−5.71
−57.62
−4.07
−65.70
−5.31
−62.89
3
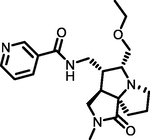
−7.75
−61.42
−7.76
−45.83
−5.70
−54.86
−4.05
−55.33
−4.89
−61.12
4
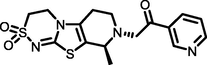
−7.59
−59.70
−7.27
−39.32
−5.58
−39.77
−3.89
−55.14
−4.39
−58.10
5
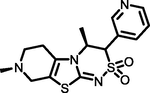
−7.54
−48.23
−6.68
−37.12
−5.52
−38.12
−3.71
−51.67
−4.36
−56.30
6
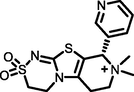
−6.23
−41.89
−6.65
–32.37
−5.45
−29.59
−3.64
−48.66
−3.97
−43.44
7
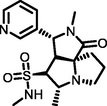
−5.61
−39.23
−6.55
−31.91
−5.38
−29.45
−3.52
−36.79
−3.85
−25.75
8
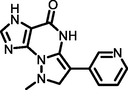
−5.25
−35.63
−6.49
−30.46
−5.35
–22.65
−3.23
−25.98
−3.85
−18.82
9
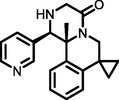
−5.01
–32.78
−5.88
−28.96
−5.34
−14.84
−3.10
−21.59
−3.84
−5.79
10
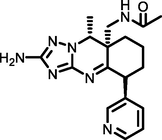
−4.99
−30.11
−5.52
−26.63
−5.34
−14.65
−3.07
−20.70
−3.68
−4.39
11
Nicotine
−4.58
−48.56
−5.63
–22.64
−3.75
−9.96
−2.43
−12.35
−3.18
−55.15
12
Co-crystal
−5.20
−41.17
−9.98
−45.23
−6.58
−62.03
−13.97
−100.11
−5.34
−57.46
Of the top ten hops, the hop 1 (H1) has the strongest binding affinity with all the selected receptors and hence it was singled out for further investigation. The H1 complexed with MMP-9 receptor exhibited potent binding affinity (Gscore −7.78 kcal/mol and ΔGbind −74.32 kcal/mol) when compared with its parent compound (Gscore −4.58 kcal/mol and ΔGbind −48.56 kcal/mol) and co-crystal (Gscore −5.20 kcal/mol and ΔGbind −41.17 kcal/mol). The H1 displayed hydrogen bonding interactions with conserved amino acid residues of MMP-9 crystal structure (Fig. 2). The common amino acid residue that is conserved in the co-crystal, nicotine and H1 is Ala189. The carbonyl group present at position two of tetrahydropyyrole ring showed a key hydrogen bonding interaction with the backbone –NH moiety of Ala189 (>C = O⋯HN, 2.03 Å). One more hydrogen bonding interaction was exhibited by Ala189 with NH moiety of tetrahydropyyrole ring (NH⋯O = C<, 2.15 Å). The sulphonyl oxygen displayed a hydrogen bonding interaction with the –OH moiety of Glu227 (>S = O⋯HO, 2.93 Å). Further, Glu227 accepting a hydrogen bonding interaction from the –NH moiety of tetrahydropyyrole ring (NH⋯O = C<, 2.44 Å). In addition, side chain of H1 donating a hydrogen bonding interaction to the carbonyl group of Gly186 (NH⋯O = C<, 2.30 Å). Moreover, the H1 was stabilized within the catalytic pocket of MMP-9 by forming π-π interaction with the electron cloud of pyridine molecule and Tyr248. From this, it is evident that the hydrogen bonding network played a role in stabilizing the conformation of H1 in the catalytic pocket of MMP-9, which is indispensably important for binding affinity and activation.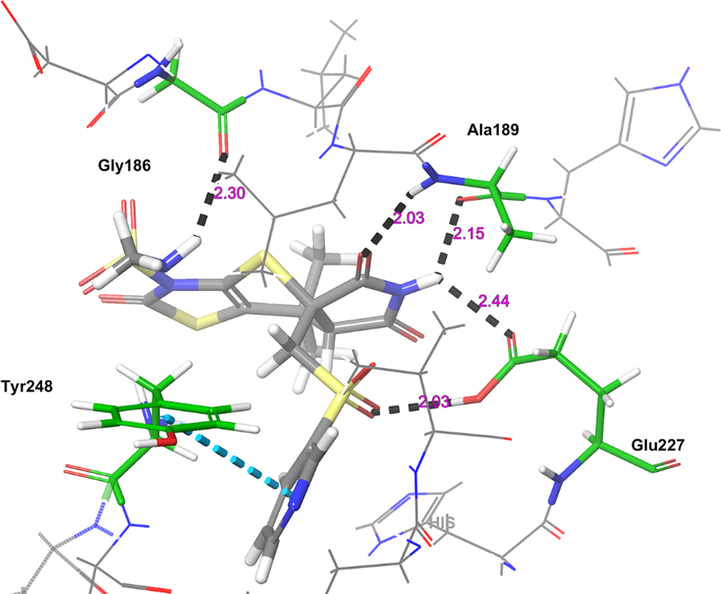
The 3D-interaction diagram of H1 in the catalytic pocket of MMP-9 (PDB ID: 4XCT) exposing key interactions.
The next receptor which demonstrated significant binding affinity with H1 molecule is GSK-3β (Fig. 3). The H1 showed higher binding affinity (Gscore −8.14 kcal/mol and ΔGbind −55.39 kcal/mol) than nicotine (Gscore −5.63 kcal/mol and ΔGbind –22.64 kcal/mol). Further, H1 exhibited lower Gscore but showed a higher binding free energy than co-crystal (Gscore −9.98 kcal/mol and ΔGbind −45.23 kcal/mol). The H1 displayed six hydrogen bonding interactions with the key amino acid residues available in the catalytic pocket of GSK-3β. The first hydrogen bonding interaction was found between the carbonyl group present at position two of tetrahydropyrrole ring and the protonated –NH of Arg141 (>C = O⋯HN(H), 2.20 Å). The both sulphonyl oxygens accepting two hydrogens from the side chain –NH of protonated Arg141 (>S = O⋯HN(H), 2.44 Å) and Gln185 (>S = O⋯HN(H), 2.18 Å). The side chain –NH of H1 donating two hydrogen bonding interactions with the carbonyl group of Asn186 (NH⋯O = C<, 1.99 Å) and Asp200 (NH⋯O = C<, 2.52 Å). Further, nitrogen present in the pyridine moiety exhibited a hydrogen bonding interaction with protonated Asn64 ((Ar)N⋯HN, 2.07 Å). All these interactions were involved in the stabilization of H1 in the GSK-3β making the binding steadier.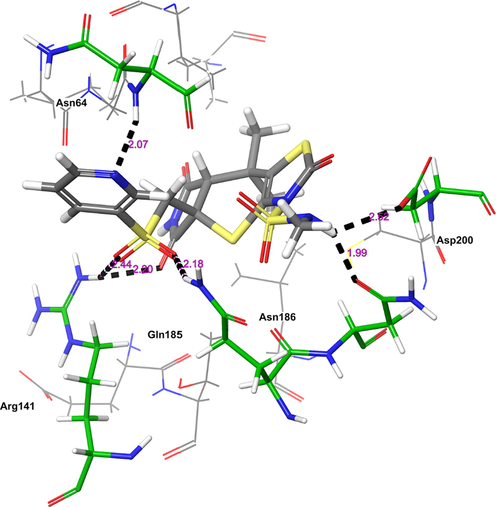
The 3D-interaction diagram of H1 in the catalytic pocket of GSK-3β (PDB ID: 5F95) exposing key interactions.
The other receptor which exhibited moderate binding affinity with H1 molecule is TNF-α (Fig. 4). The H1 showed higher binding affinity (Gscore −5.78 kcal/mol and ΔGbind −58.94 kcal/mol) than nicotine (Gscore −3.75 kcal/mol and ΔGbind −9.96 kcal/mol). Further, H1 exhibited lower binding affinity than co-crystal (Gscore −6.58 kcal/mol and ΔGbind −62.03 kcal/mol). The H1 exhibited only two hydrogen bonding interactions with the key amino acid residues of TNF-α. The –NH moiety of tetrahydropyrrole ring donating a hydrogen bonding interaction to the carbonyl oxygen of Leu120 (NH⋯O = C<, 1.94 Å). The other donating hydrogen bonding interaction was exhibited between the –NH of H1 and the aromatic –OH of Tyr151 (NH⋯O = C<, 1.76 Å). As the interactions are not prominent between the H1 and TNF-α, the complex was found to be moderately stable in contrast to the co-crystal.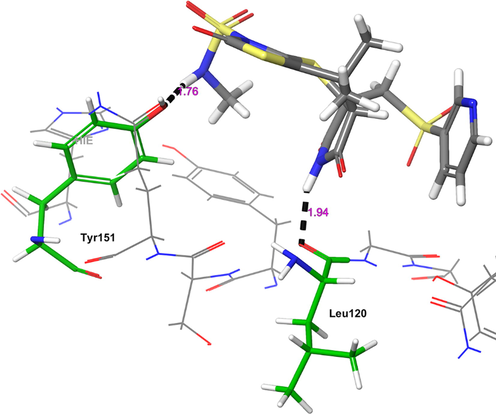
The 3D-interaction diagram of H1 in the catalytic pocket of TNF-α (PDB ID: 2AZ5) exposing key interactions.
The other receptor which exhibited significant binding affinity with H1 molecule is MurC (Fig. 5). The H1 showed higher binding affinity (Gscore −4.13 kcal/mol and ΔGbind −68.49 kcal/mol) than nicotine (Gscore −2.43 kcal/mol and ΔGbind −12.352 kcal/mol). Further, H1 exhibited lower binding affinity than co-crystal (Gscore −13.97 kcal/mol and ΔGbind −100.118 kcal/mol). The H1 displayed five hydrogen bonding interactions with the key amino acid residues available in the catalytic pocket of MurC. The carbonyl oxygen of thiazole ring exhibited two hydrogen bonding interactions with Thr46 (>C = O⋯HN, 1.91 Å) and Val47 (>C = O⋯HN, 2.15 Å). The sulphonyl oxygens of H1 accepting two hydrogen bonding interactions with the Tyr45 (>S = O⋯HN, 2.29 Å) Thr46 (>S = O⋯HN, 2.80 Å). Further, nitrogen of pyridine ring accepts a hydrogen bonding interaction with –NH moiety of protonated Arg187 ((Ar)N⋯HN, 2.57 Å). All these interactions played a pivotal role in stabilizing the H1 in the catalytic domain of MurC.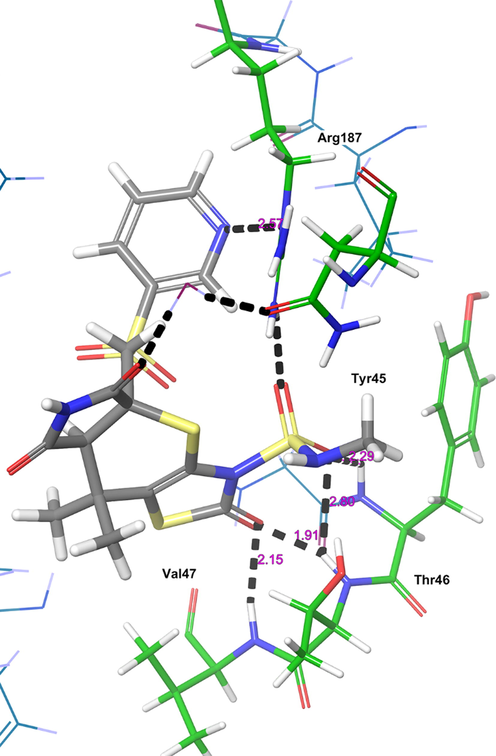
The 3D-interaction diagram of H1 in the catalytic pocket of MurC (PDB ID: 4C13) exposing key interactions.
The final receptor which exhibited potent binding affinity with H1 molecule is ParE (Fig. 6). The H1 showed higher binding affinity (Gscore −5.93 kcal/mol and ΔGbind −78.79 kcal/mol) than nicotine (Gscore −3.18 kcal/mol and ΔGbind −55.15 kcal/mol) and co-crystal (Gscore −5.34 kcal/mol and ΔGbind −57.46 kcal/mol). The H1 exhibited four hydrogen bonding interactions with key amino acid residues responsible for the inhibitory activity of ParE. The carbonyl oxygen of tetrahydropyrrole ring accepting a hydrogen bonding interaction with side chain –NH of Leu119 (>C = O⋯HN, 1.82 Å). Further, –NH group of tetrahydropyrrole ring donating a hydrogen bonding interaction with carbonyl oxygen of Leu119 (NH⋯O = C<, 2.31 Å). The protonated Arg140 exhibited a hydrogen bonding interaction with the carbonyl oxygen of thiazole ring (N(H)H⋯O = C<, 2.63 Å). In addition, the –NH moiety of sulphonamide side chain (NH⋯O = C<, 1.78 Å) donating a hydrogen bonding interaction to the Gly82. The hydrogen bonding network played a pivotal role in stabilizing the complex and inhibitory activity of ParE.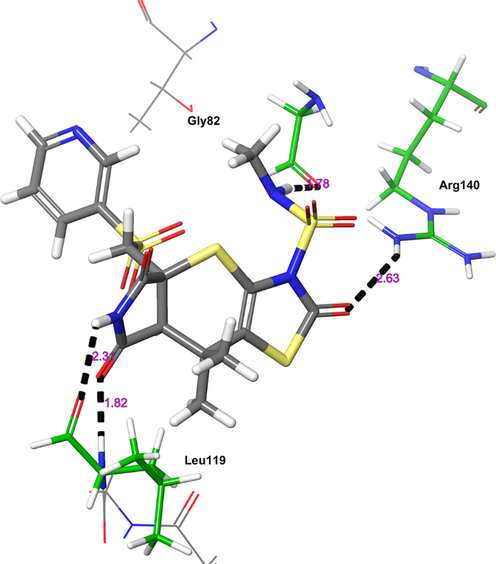
The 3D-interaction diagram of H1 in the catalytic pocket of ParE (PDB ID: 4MOT) exposing key interactions.
3.2 Molecular dynamics simulation analysis
To get insights into the binding modes, MD simulation studies were performed for the H1/4XCT, H1/5F95, H1/2AZ5, H1/4C13, H1/4MOT complexes and compared with their respective apoproteins. The stabilities of the binding pose of complexes were analyzed by investigating the trajectory frames obtained from the MD simulation studies.
The results obtained for MD simulation of all the complexes were compared with the apoproteins which was simulated for 100 ns (Fig. 7a-e). The first protein 4XCT exhibited gradual increase of root-mean square deviation (RMSD) for first 50 ns and then stabilized with 1.89–5.06 of Cα Å and 2.50–5.04 Å of heavy atoms (HA) (Fig. 7a). While the mean RMSD for apoprotein 5F95 was found to be stable throughout MD study with the values of 1.65–2.78 Å and 2.26–3.35 Å for Cα and HA, respectively (Fig. 7b). RMSD of Cα and HA have shown steady increase from 0 to 10 ns and then system equilibrated with values of 1.66–2.46 Å (Cα) and 2.27–2.84 Å (HA) for 2AZ5 apoprotein (Fig. 7c). Whereas, the apoprotein of 4C13 exhibited stable RMSD till 50 ns with value of 1.65–2.01 Å (Cα) and 2.15–2.64 Å (HA). After which a steady increase of RMSD was observed for 20 ns and then system was found to be stable for the remaining time of MD study (Fig. 7d). Besides, the apoprotein 4MOT has also exhibited stable RMSD with the values 2.28–3.50 Å (Cα) and 2.83–3.51 Å (HA) (Fig. 7e).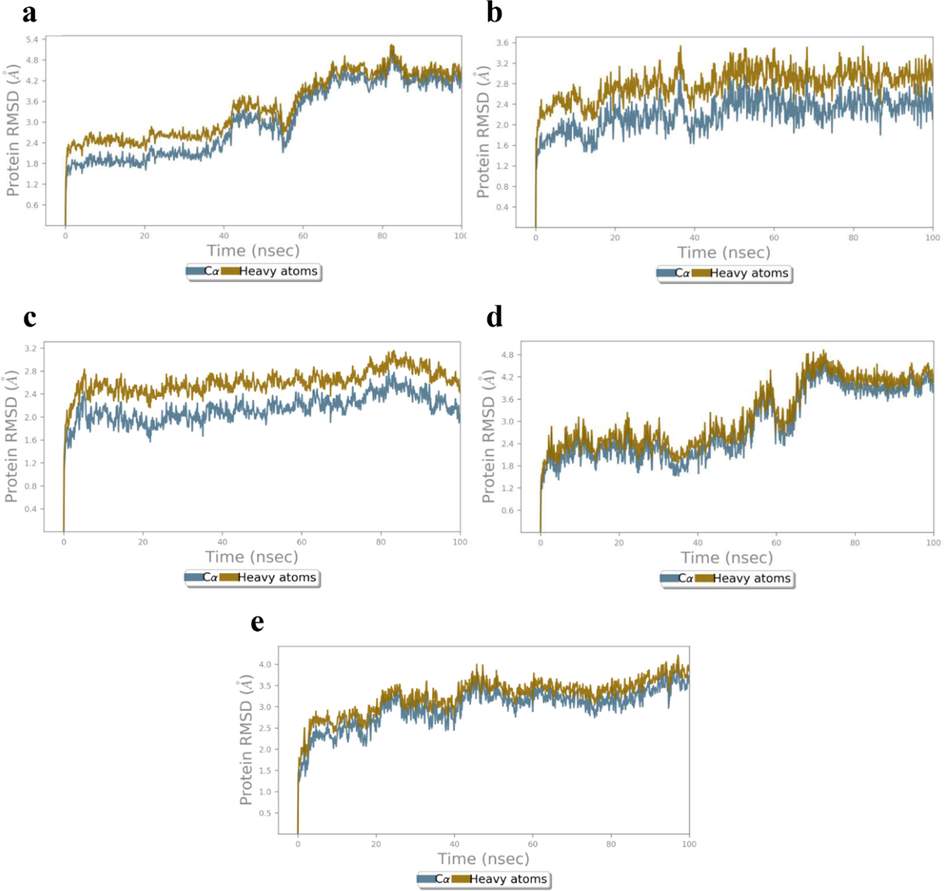
RMSD (Å) of the simulated apoproteins (a) 4XCT (b) 5F95, (c) 2AZ5, (d) 4C13 and (e) 4MOT during 100 ns of MD simulation.
RMSD of initial H1/4XCT complex configurations increased slightly during equilibration and converged till 20 ns of 100 ns simulation (Fig. 8a). After equilibrium, the RMSD values of complex Cα (2.40–3.78 Å) and HA (2.93–4.32 Å), indicated less structural alterations in the protein structure. Whereas, RMSD values of H1/5F95 complex Cα and HA were observed in the range of 1.72–2.79 Å and 2.29–3.50 Å, respectively (Fig. 8b). Moreover, there was a slight increase in the RMSD values after 50 ns which may be attributed to the conformational changes in the protein structure.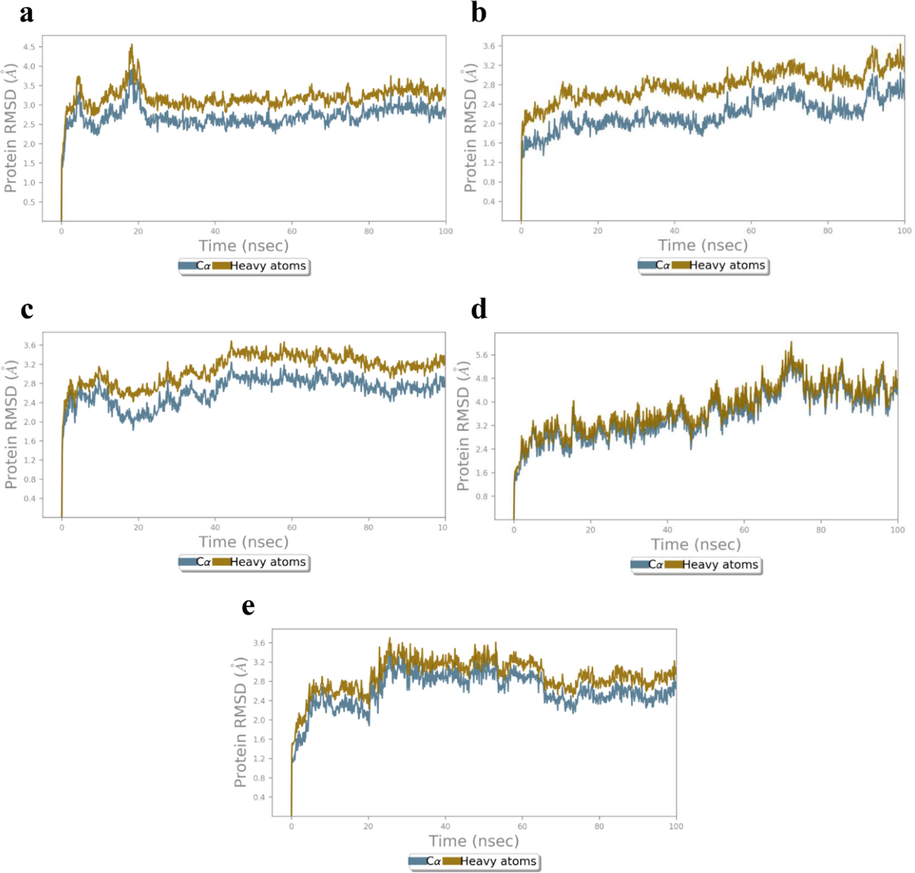
RMSD (Å) of the simulated positions of Cα and heavy atoms (a) H1/4XCT, (b) H1/5F95, (c) H1/2AZ5, (d) H1/4C13 and (e) H1/4MOT complexes during MD simulation.
While, H1/2AZ5 MD simulation of complex, an initial increase in the RMSD was observed during the first 20 ns of MD simulation, which may due to the structure relaxation that are being solvated in the system. The time-dependent RMSD values of H1/2AZ5 complex Cα and HA were observed to be 2.34–3.01 and 2.22–3.03 Å, respectively. This shows the minimal fluctuations in the structure of protein while shifting from its crystal structure to the protein structure in solvation system (Fig. 8c). RMSD of both antibacterial targets were also analyzed to explore the dynamics stability of proteins 4C13 and 4MOT. In case of H1/4C13 complex, RMSD values of Cα and HA are 2.43–4.95 and 2.74–5.49 Å, respectively indicating that the residues did not dramatically alter the protein structure in water (Fig. 8d). Whereas RMSD of initial H1/4MOT complex configurations increased slightly during equilibrium till 26 ns of 100 ns MD simulation (Fig. 8e). Moreover, after equilibrium, the RMSD values of complex Cα (2.42–3.36 Å) and HA (2.50–3.95 Å), which indicates no structural alterations in the protein structure during MD simulation study.
Root-mean square fluctuations (RMSF) which determines the flexibility of protein was calculated for the apoprotein of all receptors (Fig. 9a-e). RMSF values was significantly higher for the amino acid residues Gly178 (Cα: 8.35 Å, HA: 8.02 Å), Asp182 (Cα: 6.35 Å, HA: 6.78 Å) Lys144 (Cα: 6.29 Å, HA: 7.58 Å), and His175 (Cα: 5.74 Å, HA: 6.49 Å) for 4XCT apoprotein (Fig. 9a). In case of 5F95 apoprotein, the Lys123 (Cα: 3.77 Å, HA: 4.61 Å), Arg144 (Cα: 2.09 Å, HA: 3.59 Å), Asn287 (Cα: 2.87 Å, HA: 3.54 Å), Thr289 (Cα: 3.24 Å, HA: 3.41 Å) Glu290 (Cα: 3.01 Å, HA: 3.31 Å), and Ala382 (Cα: 2.87 Å, HA: 3.10 Å) (Fig. 9b) have exhibited greater fluctuations when compared to other residues. Furthermore, the RMSF values was significantly higher for amino acid residues Ala35 (Cα: 2.55 Å, HA:2.50 Å), Ser71 (Cα: 1.97 Å, HA:2.14 Å), Gln102 (Cα: 2.00 Å, HA:3.32 Å), Pro106 (Cα: 3.47 Å, HA:3.55 Å) Tyr87 (Cα: 2.53 Å, HA:3.87 Å), and Gly108 (Cα: 4.04 Å, HA:3.93 Å) of 2AZ5 apoprotein (Fig. 9c). Further, RMSF of 4C13 also exhibited greater fluctuations for amino acids Asn151 (Cα: 3.83 Å, HA:4.49 Å), Pro73 (Cα: 4.77 Å, HA:4.64 Å), Ala57 (Cα: 3.70 Å, HA:3.82 Å), Cys53 (Cα: 3.28 Å, HA:3.22 Å), Tyr45 (Cα: 3.67 Å, HA:4.77 Å), Gly467 (Cα: 3.78 Å, HA:3.77 Å), and Pro341 (Cα: 3.06 Å, HA:3.28 Å) (Fig. 9d). Whereas, the RMSF of 4MOT has shown greater fluctuations for amino acids Met90 (Cα: 4.35 Å, HA: 5.67 Å), Phe108 (Cα: 5.17 Å, HA: 5.46 Å), Gly109 (Cα: 6.45 Å, HA: 6.23 Å), Gly112 (Cα: 5.00 Å, HA: 4.16 Å), Thr115 (Cα: 6.58 Å, HA: 6.61 Å), Tyr113 (Cα: 4.05 Å, HA: 4.26 Å), and Asn226 (Cα: 2.55 Å, HA: 3.14 Å) (Fig. 9e).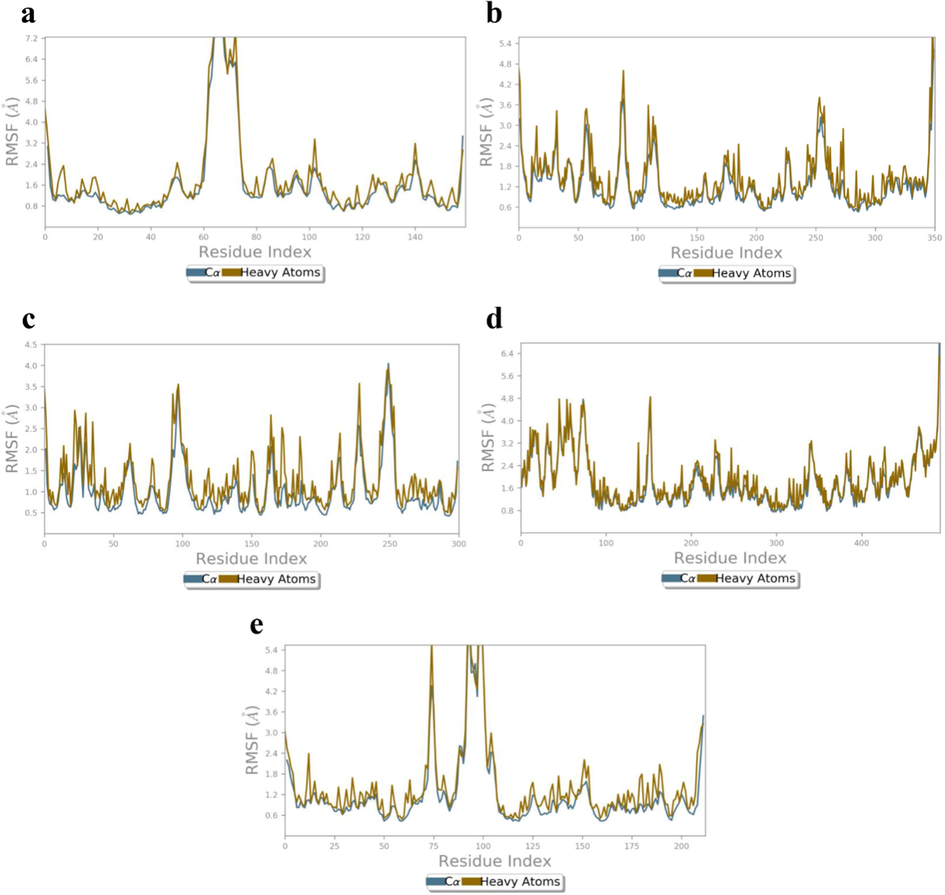
RMSF (Å) of the simulated apoproteins (a) 4XCT (b) 5F95, (c) 2AZ5, (d) 4C13 and (e) 4MOT during 100 ns of MD simulation.
RMSF of the amino acid residues of proteins 4XCT, 5F95, 2AZ5, 4C13 and 4MOT are represented in Fig. 10a-e. The H1/4XCT complex exhibited lower mean RMSF values Cα and HA at 0.43 and 0.51 Å, respectively demonstrating less structural changes in the protein crystal structure (Fig. 10a).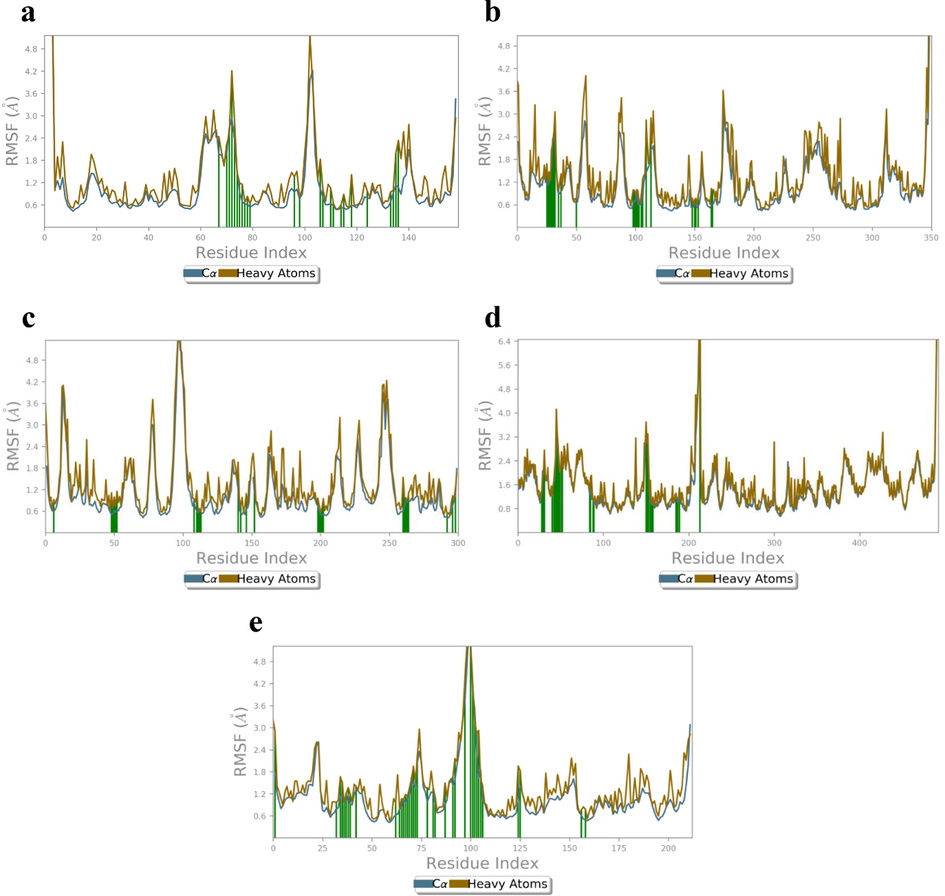
RMSF (Å) of the simulated positions of Cα and heavy atoms (a) H1/4XCT (b) H1/5F95, (c) H1/2AZ5, (d) H1/4C13 and (e) H1/4MOT complexes.
The ligand contacting amino acid stretches are Tyr179 (Cα: 1.97 Å, HA: 2.44 Å), Pro180 (Cα: 1.94 Å, HA: 2.40 Å), Phe181 (Cα: 1.75 Å, HA: 1.65 Å), Asp182 (Cα: 1.97 Å, HA:2.23 Å), Gly183 (Cα: 2.66 Å, HA:2.49 Å), Lys184 (Cα: 2.92 Å, HA: 4.20 Å), Asp185 (Cα: 2.37 Å, HA: 2.90 Å), Leu 187 (Cα: 0.94 Å, HA: 1.19 Å), His190 (Cα: 0.61 Å, HA: 0.83 Å), Pro194 (Cα: 0.61 Å, HA: 0.67 Å), Gln199 (Cα: 0.64 Å, HA: 0.93 Å), Asp206 (Cα: 0.95 Å, HA: 1.28 Å), Phe223 (Cα: 0.48 Å, HA: 0.57 Å), Pro240 (Cα: 0.95 Å, HA: 1.12 Å). However, no ligand contacts were observed at higher fluctuation regions (Cα and HA RMSF values above 3.95 and 5.18 Å). The RMSF of Cα (2.92 Å) and HA (3.12 Å) was observed for the catalytic residue Lys184 which is present in the loop (Gly171-Leu188) connecting two β-sheets on either side (Ile166-Phe170 and Ala189-Ala191).
The catalytic domain of binding residues of H1/5F95 complex exhibited lower mean RMSF values for amino acid residues Lys60 (Cα: 1.22 Å, HA: 1.68 Å), Ile62 (Cα: 1.22 Å, HA: 1.47 Å), Gly63 (Cα: 1.40 Å, HA: 1.23 Å), Asn64 (Cα: 1.60 Å, HA: 2.05 Å), Gly65 (Cα: 2.32 Å, HA: 2.23 Å), Ser66 (Cα: 2.42 Å, HA: 2.53 Å), Val69 (Cα: 0.98 Å, HA: 1.19 Å), Gln72 (Cα: 0.96 Å, HA: 1.42 Å), Lys85 (Cα: 0.64 Å, HA: 0.72 Å), Asp133 (Cα: 0.62 Å, HA: 0.99 Å), Val135 (Cα: 0.71 Å, HA: 0.85 Å), Pro136 (Cα: 0.96 Å, HA: 1.00 Å), Glu137 (Cα: 0.60 Å, HA: 0.96 Å), Val139 (Cα: 0.61 Å, HA: 0.66 Å), Lys183 (Cα: 0.53 Å, HA: 0.77 Å), Gln185 (Cα: 0.64 Å, HA: 0.74 Å), Asn186 (Cα: 0.60 Å, HA: 0.77 Å), Leu (Cα: 0.50 Å, HA: 0.60 Å), Cys199 (Cα: 0.58 Å, HA: 1.03 Å), indicating less structural alterations of these residues (Fig. 10b). The Cα and HA of residues between the amino acid stretches Lys60-Phe67 exhibited high fluctuations in the range of 1.22–2.27 Å and 1.19–2.31 Å, respectively. The amino acid residue Ser66 has showed maximum RMSF values (2.42 and 2.61 Å for both Cα and HA), present within the flexible loop (Gly63-Phe67) connecting two β-sheets on either side (Val60-Lys61 and Gly68-Leu75). From the RMSF plot, it is evident that RMSF values of Cα and HA atoms were in the order of 1–3 Å for connecting residues.
The H1/2AZ5 complex exhibited lower mean RMSF values of 0.43 Å and 0.48 Å for Cα and HA (Fig. 10c). This indicated lower fluctuations in the protein. The maximum RMSF of Cα and HA at 5.75 and 5.80 Å, respectively was observed for Pro106 present on the flexible loop (Ser99-Pro113) connecting two β-sheets Trp114-Leu126 and Thr89-Ile97. The high conformational changes in Pro106 are may be due to the presence of flexible body movement of Ser99-Pro113 residues. The ligand interactions were observed with amino acid residues His15 (Cα: 0.59 Å, HA:0.92 Å), Leu57 (Cα: 0.51 Å, HA: 1.11 Å), Ile58 (Cα: 0.50 Å, HA: 0.69 Å), Tyr59 (Cα: 0.61 Å, HA: 0.99 Å), Ser60 (Cα: 0.56 Å, HA: 0.73 Å), Gln61 (Cα: 0.62 Å, HA: 1.15 Å), Pro117 (Cα: 0.86 Å, HA: 0.94 Å), Tyr119 (Cα: 0.62 Å, HA: 1.17 Å), Leu120 (Cα: 0.55 Å, HA: 0.63 Å), Gly121 (Cα: 0.67 Å, HA: 0.63 Å), Gly122 (Cα: 0.56 Å, HA: 0.56 Å), Gln149 (Cα: 1.20 Å, HA: 1.96 Å), Tyr151 (Cα: 0.54 Å, HA: 0.70 Å) and Ile155 (Cα: 0.67 Å, HA: 1.09 Å). The RMSF values of all the connecting residues were not exceeded 3 Å. Further, RMSF was found to be lower for the contacting residues when compared with apoprotein, signifies the stability of ligand with protein residues.
The protein Cα and HA of 4C13 active site ligand residues exhibited lower RMSF values, indicating minimal fluctuations of catalytic residues (Fig. 10d). The ligand interactions with protein were found between the amino acid stretches Val40-Phe52, Asp83-Ser89, Ala150-Val157, Leu185 and Met212. The contacting residues Thr28 (Cα: 1.34 Å, HA: 1.42 Å), Asp29 (Cα: 1.80 Å, HA: 2.05 Å), Ser30 (Cα: 1.99 Å, HA: 2.10 Å), Phe39 (Cα: 1.38 Å, HA: 1.55 Å), Ser42 (Cα: 1.64 Å, HA: 1.73 Å), Val43 (Cα: 1.89 Å, HA: 2.11 Å), Gly44 (Cα: 2.44 Å, HA: 2.48 Å), Tyr45 (Cα: 3.01 Å, HA: 4.12 Å), Thr46 (Cα: 3.06 Å, HA: 3.33 Å), Val47 (Cα: 2.78 Å, HA: 2.90 Å), Asp48 (Cα: 2.27 Å, HA: 2.44 Å), Lys51 (Cα: 2.47 Å, HA: 2.70 Å), Thr84 (Cα: 1.30 Å, HA: 1.37 Å), Gly186 (Cα: 1.13 Å, HA: 1.11 Å), Arg189, (Cα: 1.11 Å, HA: 1.50 Å), exhibited minimal RMSF values. However, maximum RMSF values was observed for Thr212 (Cα: 5.91 Å, and HA: 6.74 Å). These residues observed between the amino acid stretches Thr27-Arg31 with fluctuations are present in the loop (Asp25-Arg36) connecting a β-sheet (Thr27-Asp25) and α-helix (Thr28-Arg36) on either side.
The H1/4MOT complex exhibited minimal RMSF values (Fig. 10e) between amino acid stretches Tyr113-Val122 connecting two α-helices Val95-Ala104 and Gly123-Ala129 on each side of the loop. The contacting residues are Ile48 (Cα: 0.79 Å, and HA: 0.96 Å), Asp50 (Cα: 1.14 Å, and HA: 1.67 Å), Asn51 (Cα: 1.19 Å, and HA: 1.51 Å), Ala52 (Cα: 0.93 Å, and HA: 0.93 Å), Val53 (Cα: 1.00 Å, and HA: 1.22 Å), Asp54 (Cα: 1.16 Å, and HA: 1.37 Å), Glu55 (Cα: 0.92 Å, and HA: 1.20 Å), Gly80 (Cα: 0.79 Å, and HA: 0.77 Å), Gly82 (Cα: 1.04 Å, and HA: 1.04 Å), Pro84 (Cα: 1.11 Å, and HA: 1.14 Å), Thr85 (Cα: 1.14 Å, and HA: 1.33 Å), Met87 (Cα: 1.46 Å, and HA: 1.96 Å), Ala89 (Cα: 2.07 Å, and HA: 2.22 Å), Thr94 (Cα: 0.98 Å, and HA: 1.37 Å), His103 (Cα: 0.94 Å, and HA: 1.12 Å) and Phe108 (Cα: 1.57 Å, and HA: 2.16 Å). However, maximum RMSF values were observed for Ser116 (Cα: 5.33 Å, and HA: 5.22 Å), Gly117 (Cα: 3.99 Å, and HA: 4.21 Å), and Gly118 (Cα: 3.54 Å, and HA: 3.15 Å).
Analysis of MD trajectory of the compound H1 exposed hydrogen bonding interactions, π-π stacking interactions and salt-bridges with key amino acid residues within the catalytic pockets of 4XCT, 5F95, 2AZ5, 4C13 and 4MOT, represented in Figs. 11a-e. MD simulation analysis of H1/4XCT complex exhibited similar modes of binding interactions as predicted by the docking study. Among four hydrogen bonding interactions of XP-docking, two were preserved in 8–75% of MD trajectory. As shown in Fig. 11a, two high frequency hydrogen bonding networks were observed between the carbonyl group of tetrahydropyrrole ring and Leu188 (75% MD trajectory) and Ala189 (60% MD trajectory). One more high frequency donating hydrogen bonding interaction was observed between the NH group of tetrahydropyrrole and Ala189 (74% MD trajectory). The remaining hydrogen bonding networks are mediated through water bridge and low in frequency (His190, Ala191, Glu227, Met247, His236). Further, complex was stabilized by π-π stacking interaction between the pyridine molecule of H1 and electron cloud of His226 (22% MD trajectory).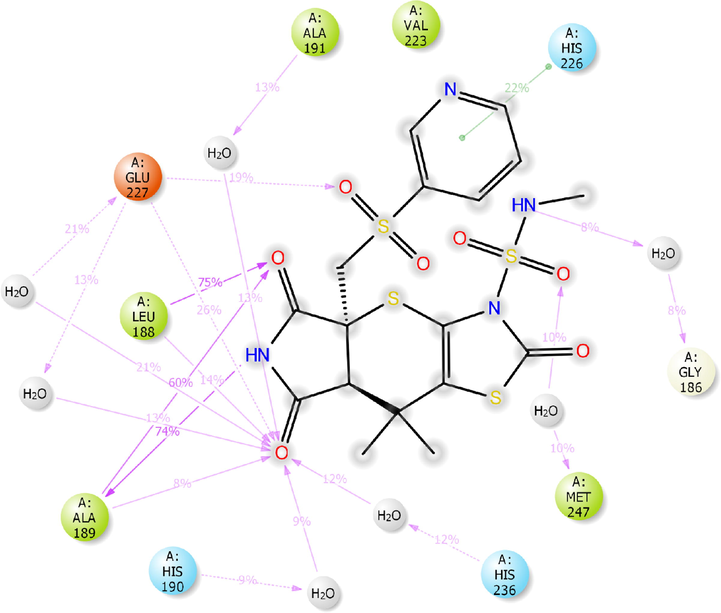
2D-interaction diagram of H1 with the catalytic pocket residues of MMP-9 (PDB ID: 4XCT) proteins throughout the 100 ns simulation trajectory.
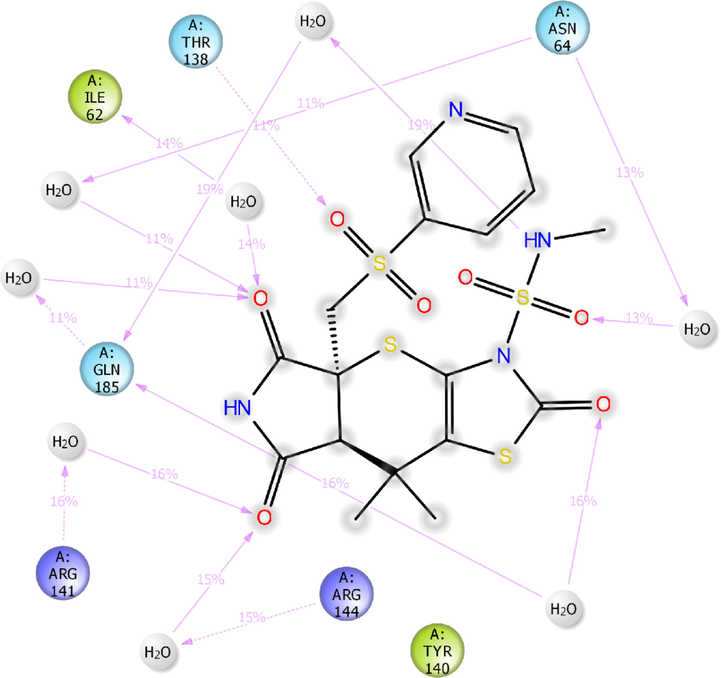
2D-interaction diagram of H1 with the catalytic pocket residues of GSK-3β (PDB ID: 5F95) protein throughout the 100 ns simulation trajectory.
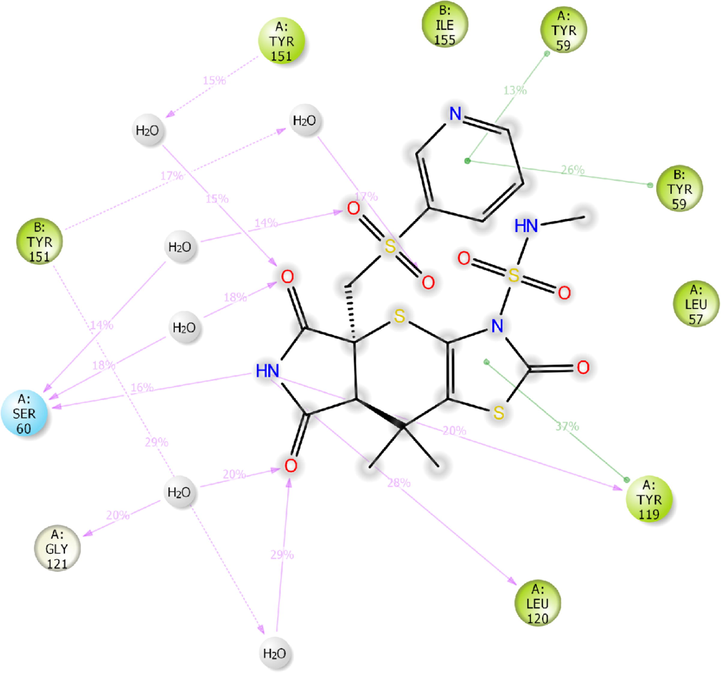
2D-interaction diagram of H1 with the catalytic pocket residues of TNF-α protein (PDB ID: 2AZ5) throughout the 100 ns simulation trajectory.
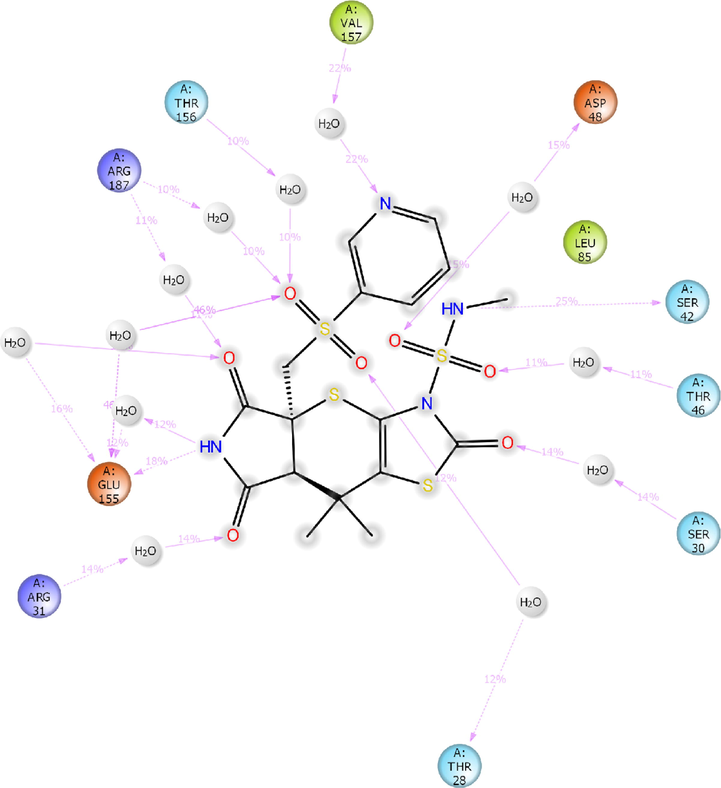
2D-interaction diagram of H1 with the catalytic pocket residues of MurC protein (PDB ID: 4C13) throughout the 100 ns simulation trajectory.
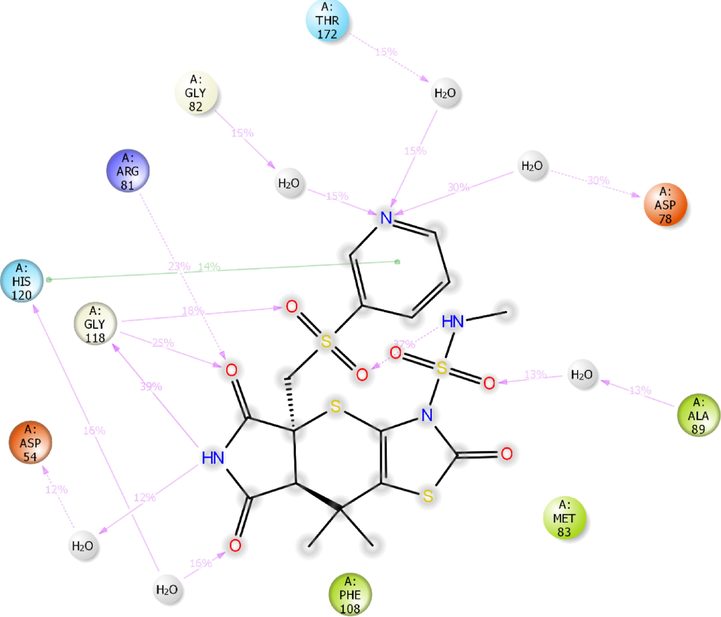
2D-interaction diagram of H1 with catalytic pocket residues of ParE protein (PDB ID: 4MOT) throughout the 100 ns simulation trajectory.
As shown in Fig. 11b, the compound H1 preserved hydrogen bonding interactions of XP-docking pose of H1/5F95 complex. However, all the hydrogen bonding network observed in moderate frequency with the key amino acid residues of catalytic pocket of 5F95. The carbonyl groups of tetrahydropyrrole formed five water-mediated hydrogen bonding interactions with Ile62 (14% MD trajectory), Asn64 (11% MD trajectory), Gln185 (11% MD trajectory), Arg141 (16% MD trajectory) and Arg144 (15% MD trajectory).
MD trajectory analysis of H1/2AZ5 complex exposed three hydrogen bonding interactions, out of which two were observed in XP-docking analysis. The NH moiety of tetrahydropyrrole and Ser60 (16% MD trajectory), Tyr119 (20% MD trajectory) and Leu120 (28% MD trajectory) (Fig. 11c). The other hydrogen bonding network was observed to be water-mediated and low in frequency. However, the compound H1 was stabilized by the formation of three π-π stacking interaction between the electron cloud of thiazole, pyridine ring and Tyr119 (37% MD trajectory), Tyr59 (26% MD trajectory), respectively.
In addition, the compound H1 has shown significant interactions with both the targets of bacteria (MurC and ParE). The H1/4C13 complex exhibited hydrogen bonding interactions (12–46% MD trajectory) with key amino acid residues within the catalytic pocket of 4C13 (Fig. 11d). Only two hydrogen bonding interactions were preserved from XP-docking analysis of H1/4C13 complex.
The NH moiety of tetrahydropyrrole formed a low frequency hydrogen bonding interaction with Glu155 (18% MD trajectory). The sulphonyl group formed a high frequency water-mediated hydrogen bonding interaction with Glu155 (46% MD trajectory). The NH moiety of sulphonamide linker formed a moderate frequency hydrogen bonding interaction with Ser42 (25% MD trajectory). The nitrogen of pyridine ring formed a water-mediated hydrogen bonding interaction with hydrophobic residue Val157 (22% MD trajectory). Further, carbonyl groups of tetrahydropyrrole ring formed a low frequency hydrogen bonding interaction with protonated Arg31 and Glu155.
Further, the compound H1 has exhibited significant interactions with the critical amino acid residues of 4MOT (Fig. 11e). Out of seven hydrogen bonding interactions observed in MD simulation, only one was preserved from XP-docking studies.
€Gly118 of 4MOT formed two moderate frequency hydrogen bonding interactions with the –NH moiety (39% MD trajectory) and carbonyl group (25% MD trajectory) of tetrahydropyrrole ring. Besides, sulphonyl group formed a low frequency hydrogen bonding interaction with Gly118 (18% MD trajectory). The carbonyl group of tetrahydropyrrole ring formed a hydrogen bonding network with the protonated Arg81 (23% MD trajectory). The nitrogen moiety present in the pyridine ring formed three low frequency hydrogen bonding interactions with Gly82 (15% MD trajectory), Asp78 (30% MD trajectory) and Thr172 (15% MD trajectory). The compound H1 exhibited an intramolecular hydrogen bonding interaction between NH moiety of sulphonamide and carbonyl group of sulphonyl moiety. Moreover, the compound H1 was stabilized by π-π stacking interaction formed between the pyridine nucleus and electron cloud of His120.
Further, MMGBSA calculations were performed for the MD trajectories for the selected complexes. The results signify the H1 compound has significant binding energy and binding affinity against the selected receptors (Supplementary Table S3). Additionally, ligand properties were analyzed with the selected receptors 4XCT, 5F95, 2AZ5, 4C13 and 4MOT (Supplementary Figure S2a-e). Radius of gyration (ROG) (measures the ‘extendedness’ of a ligand) of the H1/4XCT complex was ranged between 3.82 and 4.19 Å, indicating low conformational modifications of the compound H1 and its stability within the catalytic pocket of 4XCT. Further, lower RMSD (0.63–1.25 Å) values during the MD simulation confirms stability and lower conformational flexibility of the compound H1 (Supplementary Figure S2a). Further, stabilization of the H1 was supported by solvent accessible surface area (SASA) (80–323 Å2) and polar surface area (PSA) (245–283 Å2). Molecular surface area (MolSA) values ranging from 358 to 382 Å2 indicating lower fluctuations of the H1 within the catalytic pocket of 4XCT. From the Supplementary Figure S2b, it is observed that the average RMSD (1.23–2.61 Å) and ROG (3.75–4.10 Å) values of H1 in the binding pocket of 5F95 indicates the lower fluctuations and increased stability after 20 ns of MD study. However, stabilization of H1 was achieved through PSA (258–286 Å2), MolSA (355–382 Å2), and SASA (176–352 Å2). Ligand properties of H1 with 2AZ5 also revealed higher stability of the complex as evident by their RMSD (0.4–1.2 Å), ROG (3.75–4.22 Å), PSA (240–286 Å2), MolSA (356–380 Å2), and SASA (190–362 Å2) values (Supplementary Figure S2c). The H1 also stabilized with the antibacterial targets. H1/4C13 (Supplementary Figure S2d) and H1/4MOT (Supplementary Figure S2e) complexes have shown lower fluctuations with RMSD of 0.76–1.45 Å and 0.32–1.57 Å and ROG of 3.25–4.04 and 3.62–4.05 Å, respectively, demonstrating less conformational changes and enhanced stability during 100 ns of MD study. The stabilization of the complexes was achieved by the PSA (238–284 and 236–290 Å2), MolSA (353–378 and 349–380 Å2), and SASA (152–323 and 99–395 Å2).
3.3 Pharmacological and toxicological parameters
QikProp algorithm and Swiss ADME (
http://www.swissadme.ch/) online servers were used to predict the acceptability and ADME properties of the hops 1–10 (Table 2). The results show that the hops possess druggable characteristics and obeyed the Lipinski rule of five (RF) with 0–1 violation. The hops CNS activity was found in the range of −2 as inactiveinto + 2 as active, exemplifying lack of CNS activity. The hopsare well absorbed through phospholipid bilayer as evident by their QPlogO/W values (-0.48 to 1.68). Further, polar surface area of the hops was found in the range of 17.98 to 188.60 Å2, implicating significant van der Waals surface around these hops. The hops are well fitted within the binding pocket may be because of the low conformational changes of protein upon ligand, which was supported by SASA (398.83 to 684.67 Å2). Moreover, the hops also possess apparent Caco-2 cell permeability (50.15 to 256.43 Å2), indicating moderate permeability of the hops. Not only permeability, the hops also found to be safety for clinical testing as evident by their QPlogHERG values (-3.16 to −6.20). [a]CNS: Central nervous system activity (-2 as inactive to + 2 as active); [b]SASA: Solvent accessible surface area (300–1000); [c]Donor HB: Total number of donatable hydrogen bonds (0.0–6.0); [d]Acceptor HB: Total number of acceptable hydrogen bonds (2.0–20.0); [e]QPlogPo/w: Predicted octanol/water partition coefficient (-2 to 6.5); [f]QPlog HERG: Speculated50% inhibitory concentration values of human-ether-a-go-go (HERG) K+ Channels (<-7); [g]QPPCaco: Speculated permeability of Caco-2 cells in nm/sec (<25 poor, >500 great); [h]PSA (Polar Surface Area) (7–200): Total Van der Waals surface area of polar nitrogen and oxygen atoms of the ligands; [i]RF: Rule of 5 indicates the number of violations of Lipinski’s rule of five (Max4); [j]TPSA (<120 Å2): Topological polar surface area indicates the sum of overall polar atoms or molecules; [k]BBB: Blood-brain barrier permeant; [l]P-gp: P-glycoprotein substrate, actively pumped up from the brain or to the gastrointestinal lumen; [m-q]CYP1A2, CYP2c19 CYP2c9 CYP2D6 CYP3A4: Cytochrome P450 involved in metabolism of xenobiotics; [r]LogKp(cm/s): Skin permeation constant indicates the drug absorption through skin membrane; [s]BS: Bioavailability Score, describes the permeability and bioavailability properties; [t]PA: Pains alert, PAN assay interference compounds, indicates thenonspecific interaction with numerous biological targets rather than specifically affecting one desired target.
Compound
[a]CNS
[b]SASA
[c]donorHB
[d]acceptHB
[e]QPlogPo/w
[f]QPlogHERG
[g]QPPCaco
[h]PSA
[i]RF
[j]TPSA
[k]BBB
[l]P-gp
[m]CYP1A2 inhibitor
[n]CYP2c19 inhibitor
[o]CYP2c9 inhibitor
[p]CYP2D6 inhibitor
[q]CYP3A4 inhibitor
[r]Log Kp
[s]BS
[t]PA
1
−2
620.77
1
13
0.30
−3.92
51.99
188.6
2
231.64
no
no
no
no
no
no
no
−9.09
0.17
0
2
−2
649.11
1.5
7.5
1.88
−6.20
56.14
94.56
0
113.24
yes
no
no
no
no
no
yes
--7.47
0.55
0
3
1
684.67
1
9.7
1.26
−4.69
154.43
89.10
0
74.77
no
no
no
no
no
no
no
−8.26
0.55
0
4
0
601.89
0
10.5
−0.14
−5.39
50.15
99.39
0
121.25
no
no
yes
yes
yes
yes
yes
−6.23
0.55
0
5
1
555.81
0
8.5
0.60
−4.94
142.86
71.23
0
104.18
no
no
no
no
no
no
yes
−7.98
0.55
0
6
−2
542.36
0
5
1.28
−3.67
100.89
98.65
0
100.94
no
yes
no
no
no
no
no
−8.17
0.55
0
7
1
583.92
1
10
0.10
−3.46
105.55
91.14
0
90.99
no
yes
no
no
no
no
no
−8.52
0.55
0
8
1
485.88
1
9
−0.48
−5.18
80.048
94.55
0
82.60
no
no
no
yes
no
no
no
−7.71
0.55
0
9
1
541.35
1
6
1.68
−3.74
256.43
53.01
0
45.23
yes
yes
no
no
no
yes
yes
−7.11
0.55
0
10
−2
595.40
3
8
0.96
−3.16
124.72
117.61
0
111.08
no
yes
no
no
no
no
no
−8.07
0.55
0
Nicotine
2
398.83
0
3.5
1.18
−4.42
1269.18
17.98
0
16.13
yes
no
no
no
no
no
no
−6.46
0.55
0
Besides, topological polar surface area (TPSA: 16.13 to 231.64 Å2) of the hops were found within the recommended range. Except hops H2, H9, H10 and nicotine are not permeable through blood–brain barrier. All the hops except, H2, H9 and nicotine are not P-glycoprotein substrates; not actively pumped out from the gastrointestinal lumen and brain. The hops also considered as inhibitors of some of the CYP450s. Further, the hops possess absorption properties through skin as evident by their LogKp (skin permeation indicator) values. The hops also showed significant bioavailability scores, implicating their permeability and bioavailability of these hops. The hops exhibited significant specific interactions with desired target only and has not involved in off-targeteffect as indicated by their zero pains alert.
3.4 Plausible synthetic feasibility of compound H1.
The plausible synthetic scheme was designed using reaction-based enumeration-pathfinder module available in Schrödinger suite 2020–3. The scheme was provided in the supplementary information (Supplementary Figure S3).
4 Conclusion
Computational docking serves as an indirect strategy to scaffold hopping in the search of bioactive compounds that are structurally different from known active molecules. In summary, the hops obtained through scaffold hopping of nicotine were screened for their binding affinity with the receptors involved in pathogenesis of DW. The H1 molecule possesses high negative Gscore and free energy with the selected receptors MMP-9 (Gscore: −7.780 kcal/mol, ΔGbind:-74.321 kcal/mol), GSK-3β (Gscore: −8.143 kcal/mol, ΔGbind: −55.39 kcal/mol), TNF-α (Gscore: −5.788 kcal/mol, ΔGbind: −58.94 kcal/mol), MurC (Gscore: −4.138 kcal/mol, ΔGbind: −68.49 kcal/mol) and ParE (Gscore: −5.930 kcal/mol, ΔGbind: −78.79 kcal/mol).Docking results revealed similar kinds of interactions of conformationally flexible co-crystallized ligands with the binding residues of respective receptors. Molecular dynamics simulation studies revealed the role of hydrogen-bonding and hydrophobic interactions for the binding of H1 with the selected receptors. Besides, low conformational fluctuations of H1, demonstrates the stability within the catalytic pocket of receptors throughout the 100 ns of MD study. These results suggest that molecule H1 may be a potential druggable candidate for the management of DW. These findings lend strong support to in vitro and in vivo studies that are highly required to confirm the potency designed molecules against DW.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
CRediT authorship contribution statement
Bharat Kumar Reddy. Sanapalli: Conceptualization, Writing – original draft, Supervision. Vidyasrilekha. Yele: Software, Supervision, Validation. Dilep Kumar. Sigalapalli: Writing – review & editing. Nikhil Gadewal: Software. Afzal B. Shaik: Writing – review & editing. Richie R. Bhandare: Supervision. Sivakumar. Annadurai: Supervision. Veera Venkata Satyanarayana Reddy. Karri: Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- The pro-inflammatory environment in recalcitrant diabetic foot wounds. International wound journal.. 2008;5(4):530-539.
- [Google Scholar]
- Gestational nicotine exposure alone or in combination with ethanol down-modulates offspring immune function. International journal of immunopharmacology.. 2000;22(2):159-169.
- [Google Scholar]
- Randomized, controlled trial of long-term moderate exercise training in chronic heart failure: effects on functional capacity, quality of life, and clinical outcome. Circulation.. 1999;99(9):1173-1182.
- [Google Scholar]
- Molecular markers in patients with chronic wounds to guide surgical debridement. Molecular medicine.. 2007;13(1):30-39.
- [Google Scholar]
- Nicotine and cotinine stimulate secretion of basic fibroblast growth factor and affect expression of matrix metalloproteinases in cultured human smooth muscle cells. Journal of Vascular Surgery.. 1996;24(6):927-935.
- [Google Scholar]
- Nicotine and cotinine up-regulate vascular endothelial growth factor expression in endothelial cells. The American journal of pathology.. 2002;160(2):413-418.
- [Google Scholar]
- Nicotine Regulates Basic Fibroblastic Growth Factor and Transforming Growth Factor β1Production in Endothelial Cells. Biochemical and biophysical research communications.. 1999;257(2):306-312.
- [Google Scholar]
- DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiology and molecular biology reviews.. 1997;61(3):377-392.
- [Google Scholar]
- Prediction of properties from simulations: free energies of solvation in hexadecane, octanol, and water. Journal of the American Chemical Society.. 2000;122(12):2878-2888.
- [Google Scholar]
- A smooth particle mesh Ewald method. The Journal of chemical physics.. 1995;103(19):8577-8593.
- [Google Scholar]
- Diabetes impairs the late inflammatory response to wound healing. Journal of Surgical Research.. 1991;50(4):308-313.
- [Google Scholar]
- Thiophene/thiazole-benzene replacement on guanidine derivatives targeting α2-Adrenoceptors. European journal of medicinal chemistry.. 2017;138:38-50.
- [Google Scholar]
- Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein− ligand complexes. Journal of medicinal chemistry.. 2006;49(21):6177-6196.
- [Google Scholar]
- Wound healing activity and docking of glycogen-synthase-kinase-3-β-protein with isolated triterpenoid lupeol in rats. Phytomedicine.. 2008;15(9):763-767.
- [Google Scholar]
- Isolation of temperature-sensitive mutations in murC of Staphylococcus aureus. FEMS microbiology letters.. 2007;274(2):204-209.
- [Google Scholar]
- In silico molecular studies of natural compounds as possible anti-Alzheimer’s agents: ligand-based design. Network Modeling Analysis in Health Informatics and Bioinformatics.. 2020;9(1):1-14.
- [Google Scholar]
- Nicotine accelerates angiogenesis and wound healing in genetically diabetic mice. The American journal of pathology.. 2002;161(1):97-104.
- [Google Scholar]
- A hierarchical approach to all-atom protein loop prediction. Proteins: Structure, Function, and Bioinformatics.. 2004;55(2):351-367.
- [Google Scholar]
- Discovery of highly potent triazole antifungal derivatives by heterocycle-benzene bioisosteric replacement. European journal of medicinal chemistry.. 2013;64:16-22.
- [Google Scholar]
- Design of specific inhibitors of the protein tyrosine phosphatase SHP-2 by virtual screening and core hopping method. Molecular Simulation.. 2014;40(12):904-911.
- [Google Scholar]
- Effects of mainstream and environmental tobacco smoke on the immune system in animals and humans: a review. Critical reviews in toxicology.. 1990;20(5):369-395.
- [Google Scholar]
- Temperature and size dependence for Monte Carlo simulations of TIP4P water. Molecular Physics.. 1985;56(6):1381-1392.
- [Google Scholar]
- Thiazolopyridone ureas as DNA gyrase B inhibitors: optimization of antitubercular activity and efficacy. Bioorganic & medicinal chemistry letters.. 2014;24(3):870-879.
- [Google Scholar]
- Effects of cigarette smoke on immune response: chronic exposure to cigarette smoke impairs antigen-mediated signaling in T cells and depletes IP3-sensitive Ca2+ stores. Journal of Pharmacology and Experimental Therapeutics.. 2000;293(1):166-171.
- [Google Scholar]
- Flexible TIP4P model for molecular dynamics simulation of liquid water. Chemical physics letters.. 2003;372(5–6):842-847.
- [Google Scholar]
- The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins: Structure, Function, and Bioinformatics.. 2011;79(10):2794-2812.
- [Google Scholar]
- Novel inhibitor design for hemagglutinin against H1N1 influenza virus by core hopping method. PloS one.. 2011;6(11):e28111
- [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews.. 1997;23(1–3):3-25.
- [Google Scholar]
- Discovery of isonicotinamides as highly selective, brain penetrable, and orally active glycogen synthase kinase-3 inhibitors. Journal of medicinal chemistry.. 2016;59(3):1041-1051.
- [Google Scholar]
- Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS One.. 2012;7(6):e38546
- [Google Scholar]
- Nosé-Hoover chains: The canonical ensemble via continuous dynamics. The Journal of chemical physics.. 1992;97(4):2635-2643.
- [Google Scholar]
- Constant pressure molecular dynamics algorithms. The Journal of chemical physics.. 1994;101(5):4177-4189.
- [Google Scholar]
- Explicit reversible integrators for extended systems dynamics. Molecular Physics.. 1996;87(5):1117-1157.
- [Google Scholar]
- Nicotine at a low concentration promotes wound healing. Journal of Surgical Research.. 2008;145(2):199-204.
- [Google Scholar]
- N-O-Isopropyl Sulfonamido-Based Hydroxamates as Matrix Metalloproteinase Inhibitors: Hit Selection and in Vivo Antiangiogenic Activity. Journal of medicinal chemistry.. 2015;58(18):7224-7240.
- [Google Scholar]
- Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Current medicinal chemistry.. 2000;7(2):141-158.
- [Google Scholar]
- Antimicrobial activity of nicotine against a spectrum of bacterial and fungal pathogens. Journal of medical microbiology.. 2000;49(7):675-676.
- [Google Scholar]
- Nicotine and inflammatory neurological disorders. Acta Pharmacologica Sinica.. 2009;30(6):715-722.
- [Google Scholar]
- Discovery and optimization of novel pyridines as highly potent and selective glycogen synthase kinase 3 inhibitors. Bioorganic & medicinal chemistry letters.. 2020;30(4):126930
- [Google Scholar]
- Nicotine modulates gelatinase B (MMP-9) and epilysin (MMP-28) expression in reconstituted human oral epithelium. Journal of oral pathology & medicine.. 2011;40(1):33-36.
- [Google Scholar]
- OPLS3e: Extending force field coverage for drug-like small molecules. Journal of chemical theory and computation.. 2019;15(3):1863-1874.
- [Google Scholar]
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. Journal of computer-aided molecular design.. 2013;27(3):221-234.
- [Google Scholar]
- “Scaffold-hopping” by topological pharmacophore search: a contribution to virtual screening. Angewandte Chemie International Edition.. 1999;38(19):2894-2896.
- [Google Scholar]
- Sen CK. Human wounds and its burden: an updated compendium of estimates. Mary Ann Liebert, Inc., publishers 140 Huguenot Street, 3rd Floor New …; 2019.
- Chronic wound infections: the role of Pseudomonas aeruginosa and Staphylococcus aureus. Expert review of anti-infective therapy.. 2015;13(5):605-613.
- [Google Scholar]
- Nicotinic attenuation of central nervous system inflammation and autoimmunity. The Journal of Immunology.. 2009;182(3):1730-1739.
- [Google Scholar]
- Effect of nicotine on Staphylococcus aureus biofilm formation and virulence factors. Scientific reports.. 2019;9(1):1-13.
- [Google Scholar]
- Small molecule inhibition of the TNF family cytokine CD40 ligand through a subunit fracture mechanism. ACS chemical biology.. 2011;6(6):636-647.
- [Google Scholar]
- Effect of nicotine on the immune system: possible regulation of immune responses by central and peripheral mechanisms. Psychoneuroendocrinology.. 1998;23(2):189-204.
- [Google Scholar]
- The treatment of impaired wound healing in diabetes: looking among old drugs. Pharmaceuticals.. 2020;13(4):60.
- [Google Scholar]
- Molecular pathogenesis of chronic wounds: the role of β-catenin and c-myc in the inhibition of epithelialization and wound healing. The American journal of pathology.. 2005;167(1):59-69.
- [Google Scholar]
- Classification of scaffold-hopping approaches. Drug discovery today.. 2012;17(7–8):310-324.
- [Google Scholar]
- α7 Nicotinic acetylcholine receptor stimulation inhibits lipopolysaccharide-induced interleukin-18 and-12 production in monocytes. Journal of pharmacological sciences.. 2006;102(1):143-146.
- [Google Scholar]
- Effect of nicotine on IL-18-initiated immune response in human monocytes. Journal of leukocyte biology.. 2006;80(6):1388-1394.
- [Google Scholar]
- Synthesis of Novel Oxazolo [4, 5-b] pyridine-2-one based 1, 2, 3-triazoles as Glycogen Synthase Kinase-3β Inhibitors with Anti-inflammatory Potential. Chemical biology & drug design.. 2016;87(6):918-926.
- [Google Scholar]
- Nicotine lowers the secretion of the Alzheimer's amyloid β-protein precursor that contains amyloid β-peptide in rat. Journal of Alzheimer's Disease.. 2002;4(5):405-415.
- [Google Scholar]
- Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. The Journal of Immunology.. 2005;175(4):2684-2691.
- [Google Scholar]
- Insights from modeling the 3D structure of New Delhi metallo-β-lactamse and its binding interactions with antibiotic drugs. PLoS One.. 2011;6(4):e18414
- [Google Scholar]
- Insights into the mutation-induced HHH syndrome from modeling human mitochondrial ornithine transporter-1. PLoS One.. 2012;7(1):e31048
- [Google Scholar]
- Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nature medicine.. 2004;10(11):1216-1221.
- [Google Scholar]
- Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nature Chemical Biology.. 2021;1–9
- [Google Scholar]
- SAHA-based novel HDAC inhibitor design by core hopping method. Journal of Molecular Graphics and Modelling.. 2014;54:10-18.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103585.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




