

Translate this page into:
Garlic as an effective antifungal inhibitor: A combination of reverse docking, molecular dynamics simulation, ADMET screening, DFT, and retrosynthesis studies
⁎Corresponding authors. s.bouamrane@edu.umi.ac.ma (Soukaina Bouamrane), h.maghat@umi.ac.ma (Hamid Maghat)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Fungal infections profoundly affect human health, causing a substantial number of infections and millions of fatalities annually on a global scale. The identification of new drugs targeting this infection is a challenge that is not yet complete. Natural products, including medicinal and aromatic plants, substances that act as sources of beneficial chemical compounds for the development of efficient therapies, are among the medicines that can be used to combat this type of infection. In this study, seven bioactive molecules derived from garlic plant as potential antifungal inhibitors were investigated using computational methods. Alliin and S-allyl-cysteine, bioactive molecules generated from garlic, showed good stability at the active site of the studied receptor (PDB code: 5TZ1). They provided binding energies of −4.80 and −4.90 Kcal/mol, and inhibition constant (Ki) values of 303.78 and 253.68 µM, respectively. Similarly, alliin and S-allyl-cysteine were stabilized in the active site of the target receptor by conventional hydrogen bonds with residues Ser507 (2.47 Å), Ser378 (3.01 Å), Met508 (2.62 Å, 3.46 Å), and His377 (3.00 Å), Ser378 (3.09 Å), Met508 (2.01 Å), Ser507 (2.26 Å), respectively. These results were confirmed by molecular dynamic simulation. The selected molecules comply with the most important drug rules such as Lipinski, Veber and Egan, have good ADME properties and are not toxic; therefore, these bioactive molecules have good pharmacokinetic properties and bioavailability. The retrosynthesis method has created a pathway for the synthesis of these candidate inhibitors. As a result, the outcomes of this study strongly suggest that Alliin, S-allyl-cysteine, are potential antifungal inhibitors in the future.
Keywords
Garlic
Molecular docking
MD simulation
ADMET
DFT
Retrosynthesis

1 Introduction
The incidence of fungal infections is increasing year by year in humans due to the increasing number of immunocompromised patients, cancer, chemotherapy, and marrow transplantation (Yang et al., 2022). It is therefore necessary to study antifungals or fungicides, which are compounds with the ability to treat mycoses caused by microscopic fungi and yeasts. The antifungal therapy is divided into two parts: cutaneous-mucosal mycoses (local treatment) and deep mycoses (systemic treatment). Fungal infections are thus mainly opportunistic infections, resulting from one or more of these risk factors. They are a major cause of morbidity and mortality. The most common pathogens include yeasts (Candida, Cryptococcus, etc.) and filamentous fungi (Aspergillus, Mucorales, etc.). Local treatments are performed for fungi superficial that attack the skin, scalp, and nails (Zhang et al., 2021).
Spices are considered one of the ancient natural substances approved for the treatment of a series of diseases. Garlic is among the natural product that has long been used in traditional treatments. Moreover, the biological effects of garlic have been shown in several investigations; it has long been used in traditional medicine as an antioxidant, cardiovascular protector, anticancer, anti-inflammatory, immunomodulation, anti-diabetic, anti-obesity (Diretto et al., 2017), antibacterial and antifungal (Wei et al., 2022).
Molecular docking simulation is a computational technique used to predict and analyze interactions between ligands and a protein to predict their binding modes and affinity (Jarad et al., 2023). It plays a crucial role in drug discovery, aiding researchers in identifying potential drug candidates by studying how molecules interact and bind to specific biological targets (Shaaban et al., 2023).
The Density Functional Theory (DFT) is a quantum mechanical modeling method widely used in computational chemistry to understand the electronic structure of molecules and predict their properties (Hrichi et al., 2023). In the context of drug design, DFT can be a valuable tool for studying the molecular and electronic properties of drug candidates. DFT provides information about the electron density distribution within a molecule and the molecular orbitals. This information is vital for understanding how a drug interacts with its target at the atomic level. Identifying regions of high electron density can help predict potential binding sites and interactions (Latif et al., 2023).
In the current investigation, we conducted a computational study on the bioactive molecules derived from garlic as an antifungal plant. First, molecular docking was conducted using three different receptors to examine their interactions with bioactive molecules extracted from the studied plant. Second, the obtained outcome was further confirmed by molecular dynamic simulation. Third, Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) parameters of the studied molecules were predicted to assess their bioavailability, pharmacokinetics, and ability to behave as drugs. Finally, a retrosynthesis study was performed to design a synthesis pathway for the best-selected molecules.
2 Material and methods
2.1 Dataset selection
A database of 7 compounds derived from garlic plants as antifungal inhibitors was studied using molecular docking. The antifungal activity of the garlic plant was evaluated by Shang et al. (Shang et al., 2019). Table 1 displays the compounds under investigation's chemical structures.
Plant
Bioactive molecule
Chemical structure

GarlicDiallyl sulfide (DAS)
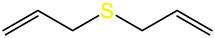
Diallyl disulfide (DADS)
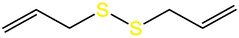
Diallyl trisulfide (DATS)
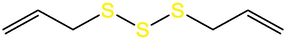
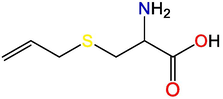
Alliin
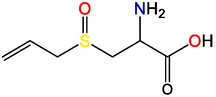
S-allyl-cysteine
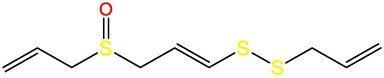
E-ajoune
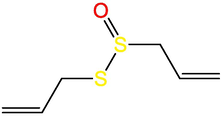
Allicin
2.2 Chemical composition of the studied plant
The chemical composition of the garlic plant shows a range of bioactive molecules, including organosulphides, saponins, phenols and polysaccharides (Kodera et al., 2017, Yoo, et al., 2014). Organosulphur compounds, including diallyl thiosulphonate (allicin), diallyl sulphide, diallyl disulphide, diallyl trisulphide, ajoune, S-allyl-cysteine and S − allyl-cysteine sulphoxide (alliin), are the primary active constituents of garlic (Wang et al., 2018). In addition to the above, garlic was found to contain more than 20 phenolic molecules, at levels higher than those found in many common vegetables. The most important phenolic compound was β-resorcylic acid, followed by pyrogallol, gallic acid, rutin, protocatechuic acid and quercetin. Garlic polysaccharides are reported to have 85 % fructose, 14 % glucose and 1 % galactose (Bradley et al., 2016). In vitro studies show that organosulfur molecules have good antifungal activity (Mansingh, et al., 2018).
2.3 Molecular docking
In molecular modeling, molecular docking has become one of the most widely used methods to carefully examine ligand-receptor interactions and to identify the preferential orientation of this ligand towards a target receptor (Bouamrane et al., 2022; Khaldan et al., 2022; Khaldan et al., 2021). Three proteins (PDB: 5TZ1, PDB: 4UYM and PDB: 2Y7L) were selected as templates using the Protein Data Bank available at https://www.rcsb.org/, and molecular docking was simulated using two programs; Autodock Vina (Trott et al., 2010) and Autodock Tools 1.5.6 (Hunter et al., 2001). In molecular docking protocol, all water residues and default ligands were deleted from the concerned proteins and then hydrogen atoms were added using the Discovery Studio 2016 software program (Dassault Systemes BIOVIA, 2017). The active sites of the target receptors were identified, and their coordinates are as follows (x = 64.326, y = 71.200 and z = 3.133 for 5TZ1), (x = 131.34, y = 194.572 and z = 1.239 for 4UYM), and (x = -0.427, y = 35.34 and z = -5.023 for 2Y7L). The grid parameters were set as follows (x = 38, y = 30 and z = 30 for 5TZ1), (x = 28, y = 28 and z = 28 for 4UYM), and (x = 30, y = 40 and z = 30 for 2Y7L) inside the receptor pocket with a grid point spacing of 1 Å. The ligands extracted from garlic were sketched and optimized using the DFT technique with the B3LYP/6-31G* basis and using Gaussian G09 program (Frisch, 2009). Next, an extended PDB format called PDBQT was used for the targeted ligands to dock them to the active sites of the receptors studied using the Autodock Vina tool to determine in which active site they are stable (Bouamrane et al., 2021). The receptor protein remained rigid throughout the docking process while the small compounds ligands were flexible (Khaldan et al., 2021, Bouamrane et al., 2020). The molecular docking outputs between ligands and proteins were visualized using PyMol (DeLano, 2002) and Discovery Studio 2016 (Dassault Systemes BIOVIA, 2017).
2.4 Molecular dynamic (MD) simulation
MD computation on the ideal docking pose was conducted to gain a deeper insight into the stability of the protein–ligand interaction of the 5TZ1-S_allyl_cysteine. The web-based CHARMM-GUI was executed to build the system (Jo et al., 2008; Lee et al., 2016) interface with the CHARMM36 force field (Best et al., 2012). The ligand topology was created by the general CHARMM force field (Yu et al., 2012) via the Param-Chem server. The CHARMM-GUI solution constructor consists of five stages. In the first stage, the tool reads the protein–ligand complex's coordinates. In the second stage, the protein–ligand complex is solved, and the system's size and structure are determined. In this stage, the system used was neutralized with Na+ and Cl- ions. The third stage establishes periodic boundary conditions (PBC), which are used to approximate a vast system by replicating a unit cell in all directions. Only the atoms existing inside the PBC box are subjected to the MD simulation. In this stage, quick minimization is used to eliminate bad contacts. The fifth step involves equilibrating the system and production. To ensure that the system has attained the required temperature and pressure, a two-phase equilibration process is performed, which involves the NVT ensemble and the NPT ensemble. This step-by-step approach allows for the system to achieve a state of thermal and pressure equilibrium, providing confidence in the accuracy and reliability of the results. Afterwards, the necessary modifications are made to the input files for both the equilibration and production phases. These adjustments may include altering the number of steps in a molecular dynamics (MD) run, adjusting the frequency of trajectory saves, and specifying the calculation of energy, among other parameters. These changes ensure that the simulation runs smoothly and efficiently, allowing for accurate and meaningful data generation during both the balancing and production stages. GROMACS 2020.2, a widely used software package, was employed consistently for the MD calculation, encompassing both the equilibration and production simulation. When the complex was previously solvated in a TIP3P water cube box, Na+ and Cl- ions were given to neutralize the system's overall atomic charge by randomly exchanging water molecules (Jorgensen et al., 1983; Balupuri et al., 2020; Li et al., 2022, El-Mernissi et al., 2023). The size and shape of the system were considered when the periodic boundary conditions (PBC) were imposed. Unbound interactions were handled using a cut-off distance of 12 Å, and the Verlet cut-off strategy was used to buffer the neighbors search list. The Particle-Mesh Ewald (PME) method (Darden et al., 2017; Essmann et al., 1995; Shi et al., 2022) was utilized to address long-range electrostatic interactions. The studied complex was subjected to the CHARMM36 force field (Best et al., 2012). Before initiating the production simulation, the system's energy was minimized by employing the steepest descent algorithm, which involved a total of 5000 steps. The chosen complex was subsequently subjected to NVT (constant number of particles, volume, and temperature) and NPT (constant number of particles, pressure, and temperature) simulation. This simulation was carried out for a duration of 125 picoseconds at a temperature of 300.15 Kelvin. During the equilibration process, positional restraints were applied to the backbone and side chains, with values of 400 kJ mol−1 nm2 and 40 kJ mol−1 nm2, respectively. This approach aimed to facilitate the equilibration of the complex, ensuring temperature and pressure stabilization for accurate subsequent analysis. Then, the complex was subjected to a production simulation that spans a duration of 100 ns within an NPT ensemble. Throughout this simulation, the temperature is maintained at 300.15 Kelvin, and the pressure is set to 1 bar. The temperature was regulated by the Nose-Hoover thermostat, while the pressure was maintained by the Parrinello-Rahman barostat. The inputs provided by CHARMM-GUI were employed, and the LINCS algorithm was utilized to impose constraints on hydrogen bonds within the system. The V-rescale thermostat was employed to maintain a temperature of 300 Kelvin, with a coupling constant of 1 picosecond. Trajectories were saved at intervals of every two picoseconds, allowing for the recording of system dynamics and analysis at various time points during the simulation. Finally, a sequence of simulations were performed in the NPT ensemble and each simulation lasting for 100 ns.
2.5 Trajectory analysis
The analysis of the MD simulation was carried out using the GROMACS utilities. By adapting the protein backbone atom using the gmx_rms subprogram, the root mean square deviation (RMSD) of the protein atom locations and ligand was calculated. Also, root mean square fluctuations (RMSF) based on the C-alpha atoms of the protein were computed by means of gmx_rmsf subprogram. The number of hydrogen bonds (at the protein–ligand interface) was determined by means of gmx hbond and the radius of gyration (Rg) of all protein atoms was determined utilizing gmx gyrate. Throughout the simulation, the function gmx distance was employed to determine the center of mass distance between the ligand and the protein. Protein-ligand interaction frequency analysis and trajectory visualization were performed using the VMD molecular graphics tool.
2.6 Binding free energy estimation using the MM/PBSA method
The GROMACS tool g_mmpbsa was utilized to conduct MM/PBSA (Molecular Mechanics/Poisson-Boltzmann Surface Area) calculations and estimate the binding affinity for the systems that were selected for additional investigation. The formula (1) below was used to identify the free energy of binding of the protein to the ligand in the solvent:
Where, is the total free energy of the ligand- protein complex, and and are total free energies of the isolated protein and ligand in solvent, respectively. Additionally, the energy contribution per residue to the binding energy can be calculated using g mmpbsa. , and were initially computed independently for each residue in order to decompose the binding energy, and they were then added to ascertain how each residue affects the binding energy. Given that g mmpbsa can only read files from a select few GROMACS versions, GROMACS 5.1.4 developed a new binary run input file (.tpr) necessary for MM-PBSA computation over g mmpbsa. The MD procedure produced the files needed to create the binary run input file: the molecular structure file (.gro), topology file (.top), and MD-parameter file (.mdp).
2.7 Lipinski’s rule and ADMET properties
Using the SwissADME (Daina et al., 2017) and pkCSM (Pires, et al., 2015) online servers, Lipinski’s rules and pharmacokinetics properties of the bioactive molecules derived from the garlic plant were determined. LogP, number of donor hydrogen bonds (HBD), number of acceptor hydrogen bonds (HBA), number of rotational bonds (nrotb) and molecular weight (MW) known as Lipinski's rule of five are calculated to determine the bioavailability and permeability of the seven molecules studied. Theoretically, a molecule that fails to meet more than one of these criteria could have issues with bioavailability and is highly likely to not be drug-like (Lipinski et al., 1997).
ADMET parameters of the molecules under study were predicted using pkCSM (Pires et al., 2015) program. ADME refers to the disposition and fate of pharmaceutical compounds in an organism, in particular in the human body. Poor pharmacokinetics and toxicity, rather than ineffectiveness of the candidate molecule, are the main reasons for drug development failure. In fact, pharmacokinetics has been a major source of failure in drug development. Consequently, ADMET and pharmacological properties could help to evaluate the pharmacological properties of selected compounds.
2.8 DFT analysis
The minimum-energy geometries of the investigated compounds were achieved using the Density Functional Theory (DFT) technique and the Gaussian G09 package (Frisch, 2009) by using B3LYP/6–311++G (d,p) basis set. The chemical potential (μ), global electrophilicity (ω), and global nucleophilicity (N) chemical hardness (η), and chemical softness (S), called global reactivity indices, were determined for the selected bioactive molecules. They were used to figure out the most nucleophilic and electrophilic inhibitors for the compounds under study. Practically, μ index was derived according to the frontier molecular orbital LUMO and HOMO by using the equation (2). Using the expressions (3) and (4) (Pearson and Songstad, 1967), respectively, the chemical hardness (η) and chemical softness (S) were computed. The global electrophilicity (ω) (Parr et al., 1999) and global nucleophilicity (N) (Domingo et al., 2008) were determined by means the equations (5) and (6), respectively.
Since it has a lower HOMO energy value, tetracyanoethylene (TCE) was chosen as a computation reference (Domingo et al., 2008; Domingo and Pérez, 2011).
2.9 Retrosynthesis study
The retrosynthesis method was performed to propose a route for the synthesis of bioactive molecules extracted from garlic plant. This method is considered useful for the synthesis of modern products. It gives a diversity of choices based on the structural dissociation of focus molecules using a series of suitable reaction steps from available bioactive starting molecules by applying E. J. Corey's retrosynthesis (Corey's et al., 1991). In this work, we took advantage of one of the most prevalent databases (spaya.ai) and focused on five bioactive compounds from garlic in order to suggest a synthetic pathway for these molecules and to give a clear view to experimenters.
3 Results and discussions
3.1 Molecular docking result
3.1.1 Binding energy and inhibition constant (Ki)
Bioactive molecules from garlic plant were docked to the active sites of the three studied proteins (PDB: 5TZ1, PDB: 2Y7L, and PDB: 4UYM). The docked molecules' binding energies and inhibition constant (Ki) are shown in Table 2. The results listed in Table 2 show that Alliin and S-allyl-cysteine molecules are stable with the 5TZ1 receptor and have the best binding energy values of −4.80 and −4.90 kcal/mol, respectively. Also, it is noted that the Alliin molecule exhibits a favorable binding energy value of −5.20 kcal/mol when interacting with the 4UYM protein. Furthermore, the concentration needed to reduce the activity of the cytochrome P450 (CYP) enzyme by half to inhibit it is known as the inhibition constant (Ki) of a medication. The Ki was determined by using the following formula:
, where ΔG is the binding energy, R is the universal gas constant (
), and T represents the temperature at a value of 298.15 K. Practically, the lower the Ki value, the better the efficacy of the active agent (Ortiz et al., 2019). The results of Table 2 indicate that Alliin and S-allyl-cysteine molecules have the lowest Ki inhibition constant, so they are more effective. On the other hand, the seven studied molecules demonstrated a strong binding energy with the 5TZ1 receptor. Additionally, the data in Table 2 indicate that Alliin exhibits better binding and Ki values of −5.20 and 152.80, respectively, with the 4UYM receptor. Therefore, in our future analyses, we will present and focus on the interactions of the studied molecules with the 5TZ1 receptor and on the interaction between the Alliin molecule and the 4UYM receptor.
Plant
Bioactive molecule
Receptor
5TZ1
4UYM
2Y7L
Binding Energy (Kcal/mol)
Inhibition constant (µM)
Binding Energy (Kcal/mol)
Inhibition constant (µM)
Binding Energy (Kcal/mol)
Inhibition constant (µM)
Garlic
DAS
−3.90
1374.37
−3.30
3787.88
−3.50
2701.67
DADS
−3.70
1926.94
−3.60
2281.66
−3.60
2281.66
DATS
−3.80
1627.37
−3.80
1627.37
−3.50
2701.67
Alliin
−4.80
303.78
−5.20
152.80
−4.4
590.46
S-allyl-cysteine
−4.90
253.68
−4.40
590.46
−4.1
980.26
Ajoene
−4.40
590.46
−4.40
590.46
−3.80
1627.37
Allicin
−4.700
355.67
−3.80
1627.37
−3.90
1374.37
3.1.2 Molecular docking interaction
The two- and three-dimensional docking interaction of the seven molecules studied and their residues with 5TZ1 are presented in Table 3. As mentioned above, the Alliin and S-allyl-cysteine molecules exhibit the best binding energy values with the 5TZ1 receptor. Alliin has also demonstrated good binding energy with the 4UYM protein. These molecules will be discussed in terms of molecular docking interactions. The Alliin molecule interacts very well with the active site of the 5TZ1 protein and shows two types of interactions with varying residues and distances. The aminyl group (–NH2) forms three conventional hydrogen bonds with residues Ser507 (2.47 Å) and Met508 (2.62 Å, 3.46 Å). The oxygen atom of the ester group made a hydrogen bond with Ser378 residue at distance of 3.01 Å. The other oxygen atom of the same group (ester) provides a carbon hydrogen bond with Ser378 (3.78 Å). Thus, the presence of a large number of hydrogen bonds could account for the good stability of the Alliin molecule. Continuously, the docking interaction of the S-allyl-cysteine molecule with 5TZ1 receptor shows important interaction types, especially those that are favourable. Two conventional hydrogen bonding interactions were identified for the aminyl group with residues Ser507 (2.26 Å) and Met508 (2.01 Å), and two more were found for the ester oxygen atom with residues His377 (3.00 Å) and Ser378 (3.09). On the other hand, the docking interaction between Alliin and the 4UYM receptor has resulted in various types of interactions as shown in Fig. 1. A conventional hydrogen bond was formed between the aminyl group and the amino acid threonine122 at a distance of 2.42 Å. The hydroxyl group provides a conventional hydrogen bond with the residue Tyr68. The other two conventional hydrogen bonds were established by the thionyl group with residues His374 (2.79 Å) and Ser375 (2.85 Å). The same group offers a pi-sulfur interaction with His374 (4.12 Å). It should be noted that the molecule Alliin forms a carbon hydrogen bond with Leu503 residue. These interactions demonstrate the good stability of the Alliin molecule in the active site of the 4UYM receptor.
Bioactive molecule
3D interaction
2D interaction
Residues
DAS
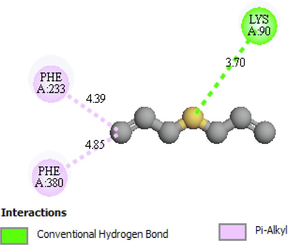
Lys90, Phe233, Phe380
DADS
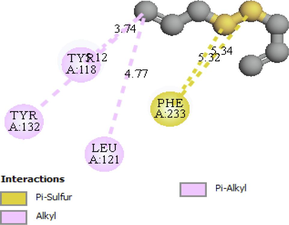
Tyr118, Tyr132, Leu12, Phe233
DATS
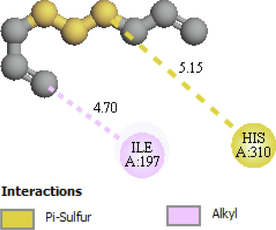
His310, Ile197
Alliin
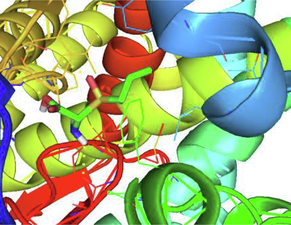
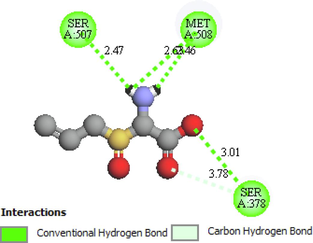
Se507, Met508, Ser378
S-allyl-cysteine
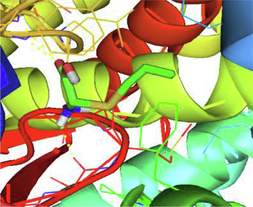
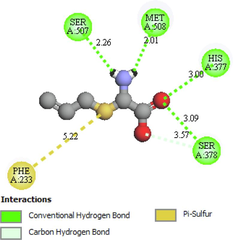
Ser507, Met508, His377, Ser378, Phe233
Ajoene
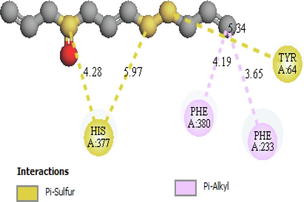
His377,Tyr64 Phe380, Phe233
Allicin
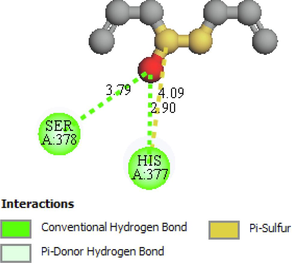
Ser378, His377
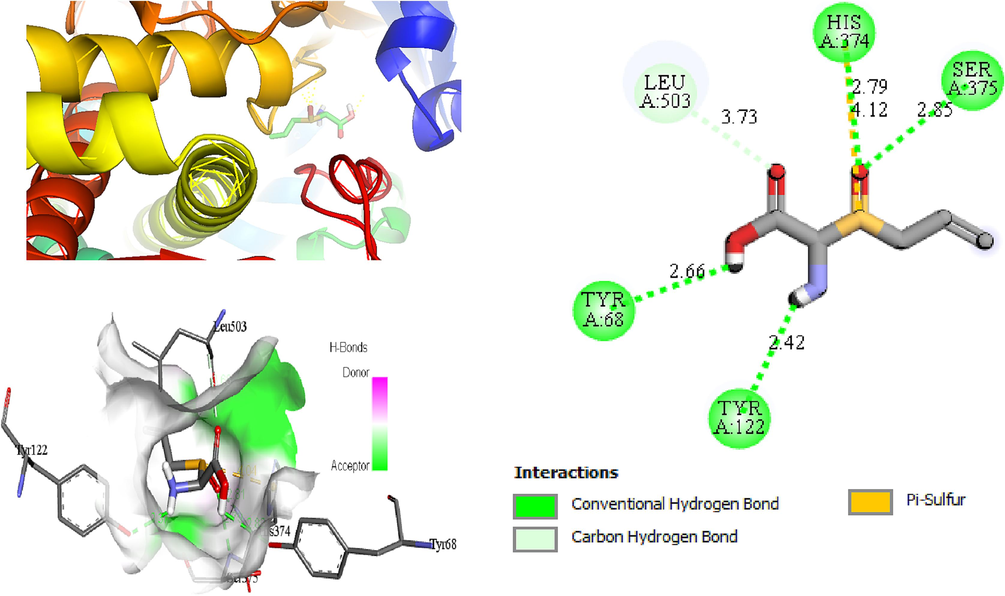
2D (left) and 3D (right) interactions of Alliin and 4UYM protein.
In order to validate the molecular docking protocol, the re-docking of the co-crystallized ligand was done using the RMSD value in the range of 0–2 Å (Wanget al., 2022). This finding was further verified by the superposition of the co-crystallized ligand with the best conformation after re-docking (Fig. 2). One can see from the result in Fig. 1 that the two ligands are completely superimposed and that the RMSD value was 0.982, which is in the range of 0 to 2 Å. Therefore, the docking protocol carried out by Autodock Vina can be considered robust and reliable.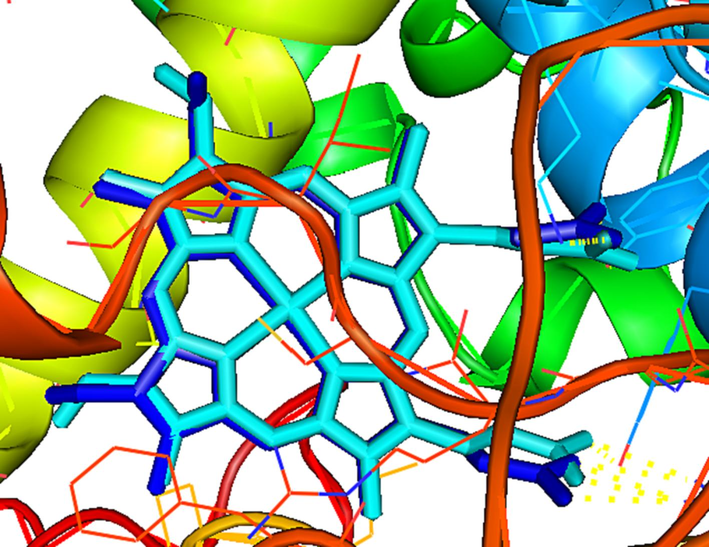
Re-docking of the co-crystallized ligand (cyan) and the docked protoporphyrin IX ligand (blue) with the RMSD value of 0.982.
3.2 MD simulation result
To assess the stability of the binding, the best complex of 5TZ1 with the ligand S-allyl-cysteine was subjected to MD calculation. This simulation was conducted for a duration of 100 ns, simulating normal ambient temperature levels. The present ligand is still coupled to the protein pocket's ligand binding groove, according to the visualization of the trajectories after the simulation run. To evaluate the stability of each structure, various calculations were conducted, which included measuring the RMSD, radius of gyration (Rg), RMSF, hydrogen bonding analysis, average center of mass distance between the ligand and protein and estimating the binding free energy utilizing the MMPBSA approach.
RMSD graph in (Fig. 3) illustrates backbone, the complex, and ligand RMSD for the structure. The studied complex and protein backbone RMSD plot shows little to no fluctuation after 20 ns of simulation time, with average values of less than 2 Angstroms. Ligand RMSD shows fewer fluctuations during 100 ns simulation time. The Rg analysis (Fig. 5) aligns with the RMSD outcomes for the complex, showing very little fluctuations for the compound (less than 0.5 Å) with values between 22.7 and 23.1 Å throughout the whole simulation, which indicates the solidity and the stability of the protein ligand system.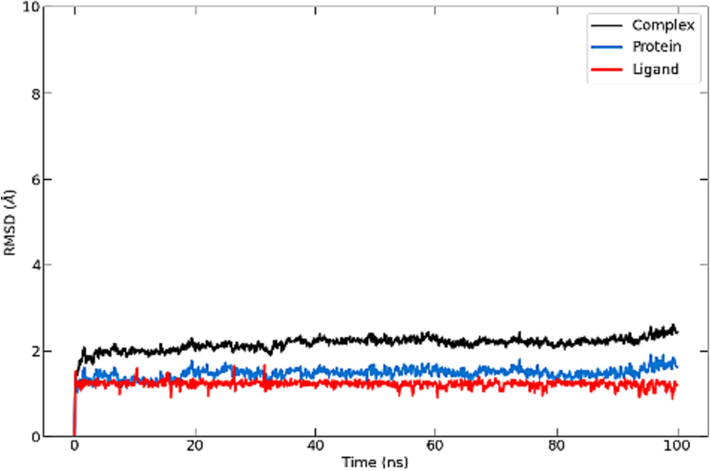
RMSD of S_allyl_cysteine molecule.
Based on the “C-alpha” atoms in the protein complex, the RMSF was calculated via the GROMACS algorithm. Except for specific residues that form turns or loops in the protein, the overall fluctuation intensity of the studied ligand typically remains at approximately 2.0 Å. This indicates that, on average, the atoms in the system exhibit a moderate level of structural variability, while certain regions with defined secondary structures show reduced fluctuations (Fig. 4).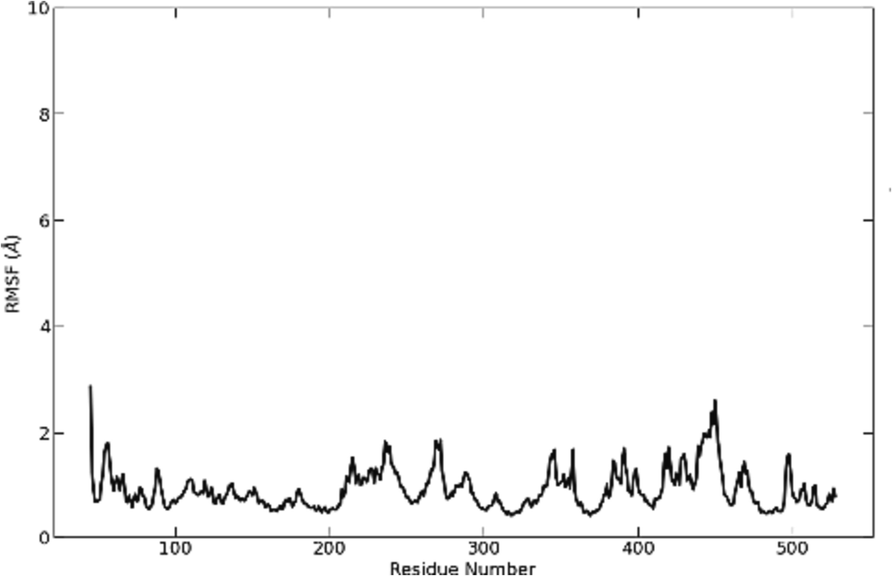
RMSF of S_allyl_cysteine molecule.
The total number of hydrogen bonds made by the S-allyl-cysteine ligand and 5TZ1 protein over the simulation's 100 ns run are displayed in (Fig. 6, A). The ligand maintains at least one intact hydrogen bond with the protein throughout most of simulation time. (Fig. 6, B) shows the average center-of-sass distance between the S-allyl-cysteine compound and the 5TZ1 protein during 100 ns of simulation time. S-allyl-cysteine compound presents an important hydrogen bond number. The complex's Principal Component Analysis (PCA) was computed using the Bio3D program of R (Fig. 7, Column A). The dynamic cross-correlated motions (DCCM) of protein residues were also determined using the Bio3D program. The strength of correlated motion is indicated by colors ranging from red to white to blue, with blue colors denoting a negative relationship, white showing no relationship, and red colors demonstrating highly related movements between the residues (Fig. 7, Column B).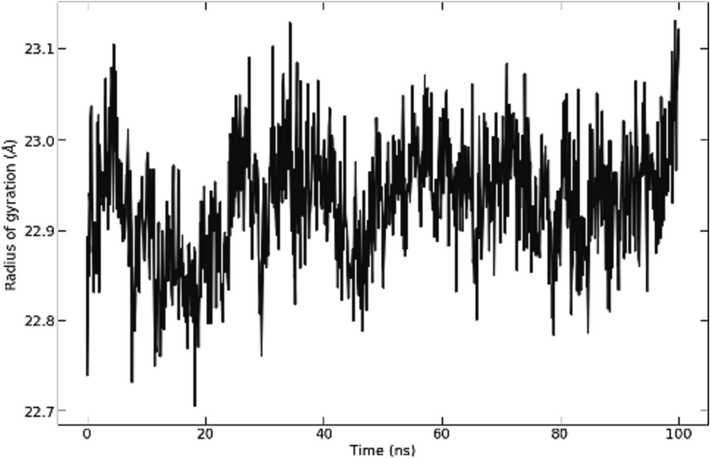
Radius of gyration of S_allyl_cysteine molecule.
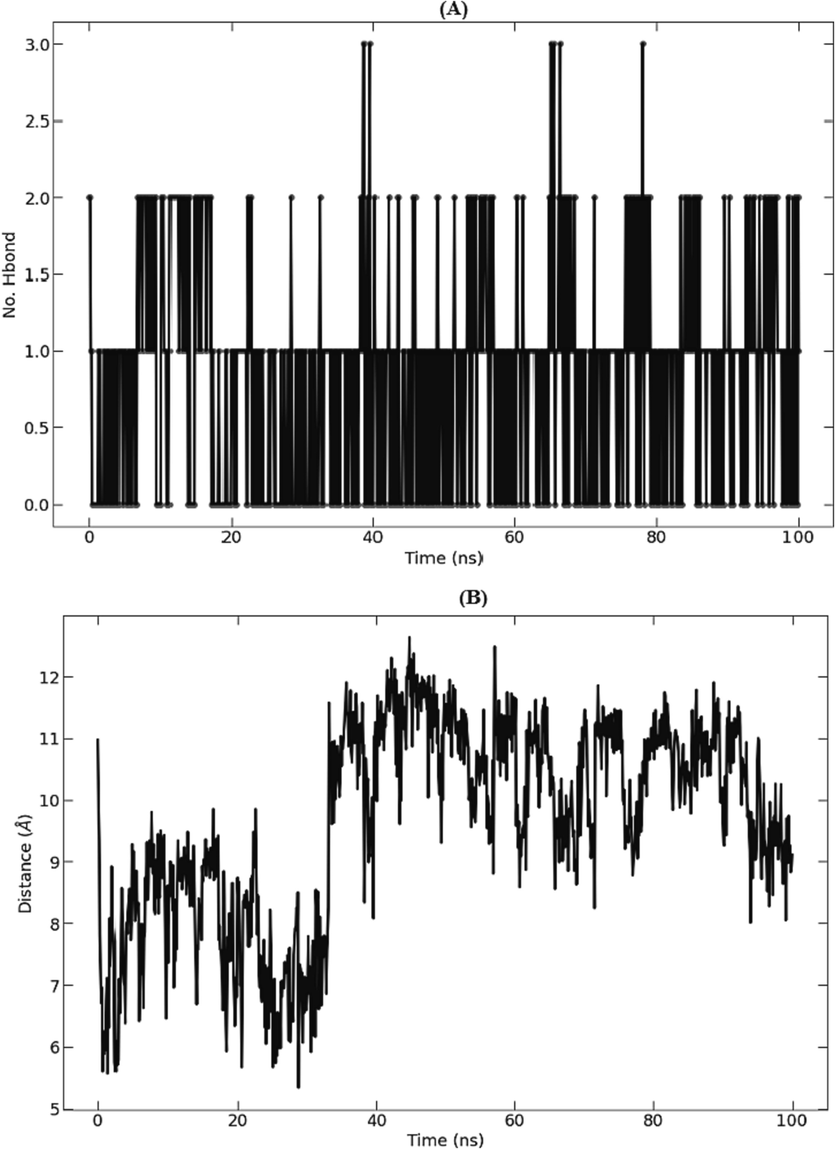
Protein-ligand hydrogen bonds (A) and average distance between protein and ligand (B) of the S_allyl_cysteine-5TZ1 complex studied during the 100 ns simulation.
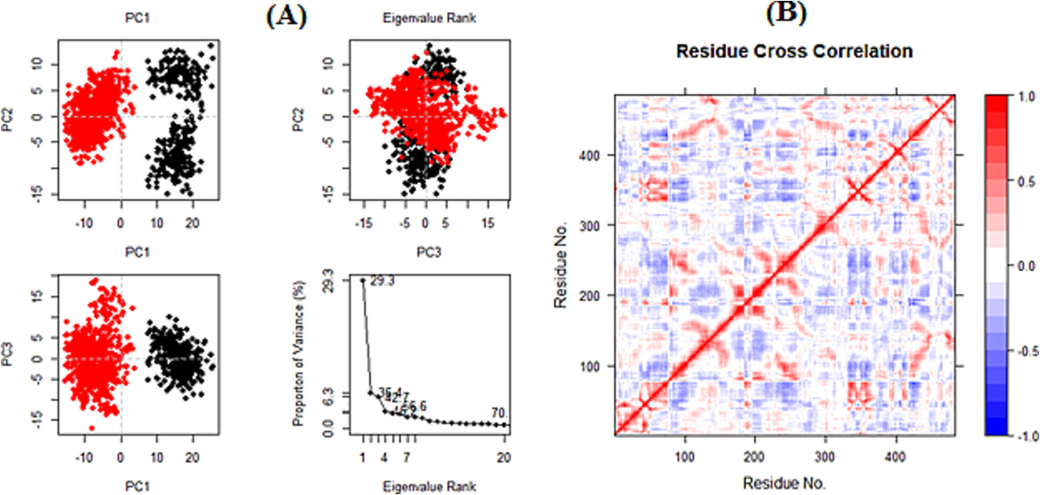
PCA (A) and DCCM (B).
The temperature, potential energy, and pressure of the system as determined from the GROMACS edr file are shown in Fig. 8 for a 100 ns MD simulation. The graph depicts converging temperature, potential energy, and pressure over the 100 ns runs. In order to rescore complex, the Molecular Mechanics/Poisson Boltzman Surface Area (MM/PBSA) technique was applied since it calculates the free energy of binding more rapidly than alternative force field-based methods such the thermodynamic integration (TI) or free energy perturbation (FEP) methods. G-mmpbsa software was applied to determine the MM/PBSA. Table 4 displays the computed binding free energy. Table 5 illustrates the summary of MD simulations. It can be concluded from the result in Table 5 that the complex studied is stable throughout the simulation time of 100 ns.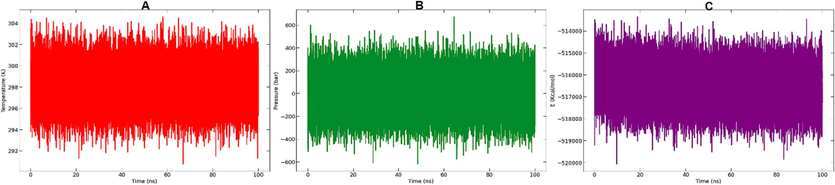
Temperature (A), pressure (B) and potential energy (C) throughout the entire simulation of 100 ns.
Complex
Van der Waal energy
Electrostatic energy
Polar solvation energy
SASA energy
5TZ1-S-allyl-cysteine-
−54.981 +/- 9.761
−68.056 +/- 9.616
−36.147 +/- 20.702
59.055 +/- 26.188
−9.833 +/- 0.805
Compound
Simulation box dimensions
Number of neutralizing ions
Stability
S_Allyl_Cysteine
94
94
941Na+
Stable
HAPC
62
62
621Cl-
Stable
3.3 Lipinski’s rule and ADMET result
3.3.1 Lipinski’s rule and pharmacokinetic properties
The Lipinski rule of five has been applied to the active molecules of garlic plants to determine their potential for oral absorption in humans. Lipinski’s rule including MW, HBA, HBD, nrotb and LogP were calculated for the bioactive compounds using the SwissADME (Daina et al., 2017) software as listed in Table 6. Abbreviations: HBA: number of hydrogen bonds acceptors, HBD: number of hydrogen bonds donors, TPSA: Topological Polar Surface Area, LogP: logarithm of partition coefficient of compound between n-octanol and water, nrotb: number of rotatable bonds, SA: Synthetic accessibility, MW: Molecular Weight.
Plant
Molecule
Property
LogP
HBD
HBA
TPSA
nrotb
MW
SA
Rule
<=5
<5
<10
<140
<10
<500
0 < SA < 10
Garlic
DAS
2.09
0
1
25.30
4
114.21
2.34
DASD
2.73
0
2
50.60
5
146.26
3.12
DATS
3.38
0
3
75.90
6
178.34
3.58
Alliin
−0.66
2
3
99.60
5
177.22
3.21
S-allyl-cysteine
0.31
2
3
88.62
5
161.22
2.80
E-ajoune
3
0
3
99.60
8
234.41
3.21
Allicin
1.75
0
2
88.62
5
162.28
2.8
As observed in Table 6, the seven molecules derived from garlic meet all the criteria given in Lipinski's rule. In addition, a compound with a TPSA of no more than 140 Å2 and nrotb below than 10 is regarded as more adaptive and more able to bind the particular receptor (Liu, 2022). The TPSA and nrotb results indicate that all bioactive compounds can flexibly interact with the relevant receptor. On top of that, synthetic accessibility (SA) parameter was predicted to whether the studied molecules can be synthetic or not. For a given compound, a SA value close to 1 and far from 10 is likely to be synthesized (Vema et al., 2022, Khaldan et al., 2023). SA parameters result of all molecules under study demonstrates that can be easily synthesized. The results of the pharmacokinetic properties favor the bioactive molecules derived from garlic plant and give them a drug-like behavior.
The data shown in Table 7 demonstrate that there are no issues with oral bioavailability for the seven compounds because they all fulfilled the Lipinski, Veber, and Egan rules.
Plant
Bioactive molecule
Lipinski
Veber
Egan
Garlic
DAS
Yes
Yes
Yes
DASD
Yes
Yes
Yes
DATS
Yes
Yes
Yes
Alliin
Yes
Yes
Yes
S-allyl-cysteine
Yes
Yes
Yes
E-ajoune
Yes
Yes
Yes
Allicin
Yes
Yes
Yes
3.3.2 ADMET result
As mentioned above, all the molecules studied have good pharmacokinetic properties. To ensure that these molecules could be candidate inhibitors to treat fungal infections, they were further investigated using the ADMET evaluation. The derived results are displayed in Table 8.
Bioactive Compound
Absorption
Distribution
Metabolism
Excretion
Toxicity
Intestinal absorption (human)
Blood Brain Barrier Permea Bility
CNS permea bility
CYP
Renal OCT2 substrat e
Clearance
AMES toxicity
Hepatot oxicity
2D6
3A4
2D6
3A4
Substrate
Inhibitor
Numeric (% Absorbed)
Numeric (log BBB)
Numeric (log PS)
Categorical (Yes/No)
Categorical (Yes/No)
Categorical (Yes/No)
DAS
96.26
0.69
−2.102
No
No
No
No
No
0.55
No
No
DASD
94.76
0.78
−2.21
No
No
No
No
No
0.54
No
No
DATS
92.57
0.76
−2.30
No
No
No
No
No
0.44
No
No
Alliin
80.00
−0.42
−3.10
No
No
No
No
No
0.36
No
No
S-allyl-cysteine
81.00
−0.30
−3.05
Yes
No
No
No
No
0.59
No
No
E-ajoune
95.18
0.70
−2.17
No
No
No
No
No
0.53
No
No
Allicin
96.30
0.50
−2.31
No
No
No
No
No
0.71
No
No
Intestinal absorption is an indicator used to describe absorption. In practice, a value above 30 % indicates high absorption (Bouamrane et al., 2022; Naz et al., 2020). As observed in Table 8, all molecules have an intestinal absorption value between 80 and 97 %; indicating that these molecules are well absorbed by the human intestine. Blood Brain Barrier (BBB) and CNS permeability are two parameters that characterized the distribution. BBB permeability deemed well if its value is not less than 0.3 and poor if LogBB < -1 (Brunton et al., 2011). For CNS index, a molecule with LogPS > -2 is considered capable of penetrating the CNS, while molecule with LogPS < -3 is deemed incapable of penetrating the CNS (Kwong, 2017). The BBB result indicates that all molecules have good distribution ability. Regarding the metabolism, cytochrome P450 (CYP) is a fundamental detoxification enzyme found in all human body tissues (Jawarkar et al., 2022, Khaldan et al., 2022). In fact, the CYP enzyme oxidizes foreign germs to make it easier to eliminate them. CYP inhibitors can influence the metabolism of the molecule, and the molecule can also have a reverse influence (Sahin et al., 2022). Therefore, the evaluation of the ability of the molecules derived from garlic plant to inhibit cytochromes (CYP) has become increasingly essential. The most crucial subtype of CYP P450 is CYP3A4 (inhibitor and substrate). The result reported in Table 8 demonstrates that all bioactive compounds are inhibitors and substrates of CYP3A4, which is the ultimate evidence that these compounds can be metabolized in the liver (Bouamrane et al., 2023). Moreover, the excretion of drugs refers to the process by which the drug and its metabolites are eliminated from the body. This is a crucial aspect of pharmacokinetics, the study of how the body handles drugs. The primary organs and routes involved in the excretion of drugs are the kidneys, liver, and bile. The clearance index is the most commonly used parameter to assess the elimination of a drug from the body. In fact, the clearance index refers to how the body removes the drug and reduces its concentration in the body (Zhang et al., 2021; Khaldan et al., 2022). The longer a substance stays in the body, the lower the clearance index is (Khaldan et al., 2022). As can be seen in Table 8, all molecules have a lower clearance index value; these molecules therefore have the ability to persist in the human body.
The toxicity of a drug refers to its potential to cause harm or adverse effects when taken in certain doses. The study of toxicity is an essential step in the process of drug development, prescription and clinical use. It is essential to strike a balance between a drug's therapeutic benefits and its potential for toxicity, in order to guarantee patient safety. In the context of drug development and safety assessment, the Ames test is often utilized to screen for potential mutagenic effects of pharmaceutical compounds. Hepatotoxicity, another parameter used to assess the toxicity of a drug, refers to the adverse effects of drugs on the liver, leading to liver damage. The toxicity of the bioactive molecules derived from garlic was examined using the Ames test and hepatoxicity parameters. A negative Ames test and hepatotoxicity indicate that the molecule is not mutagenic and does not cause any liver damage. Table 8 shows that the studied molecules are neither mutagenic nor hepatotoxic. Therefore, these compounds could be used as a therapeutic target for treating fungal infections.
3.4 DFT analysis result
3.4.1 Global indices and frontier molecular orbitals (FMOs)
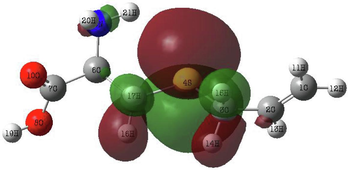
In order to identify the electrophilic and nucleophilic compounds, the global reactivity indices were determined as shown in Table 9. S-allyl-cysteine and Alliin have nucleophilic indexes N of 3.073 and 3.122, respectively. These compounds are considered good nucleophiles according to the nucleophilic scale (Domingo et al., 2002). Moreover, the FMOs of the investigated molecules have been analyzed at B3LYP/6–311++G (d,p) level as shown in Table 10 to obtain information on the location of the electron density.
Bioactive molecule
Global indices
HOMO (ev)
LUMO (ev)
µ (ev)
η (ev)
S (ev)
ω (ev)
N (ev)
Alliin
−6.295
−0.533
−3.414
5.762
0.174
1.011
3.073
S-allyl-cysteine
−6.246
−0.284
−3.265
5.962
0.168
0.894
3.122
Bioactive molecule
Orbital
HOMO
LUMO
Alliin
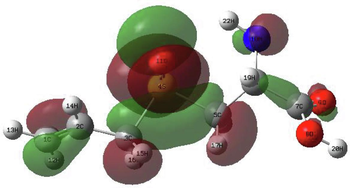
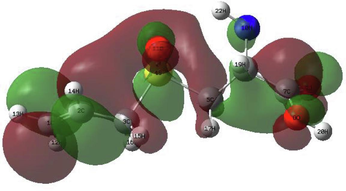
S-allyl-cysteine
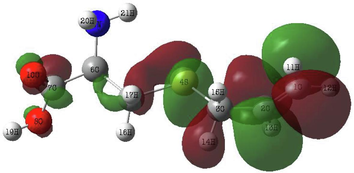
3.4.2 Molecular electrostatic potential (MEP)
MEP maps are used to obtain information on the nucleophilic and electrophilic reactivity zones of a molecule. The MEP surface was built using B3LYP/6–311++G (d,p) basis in the Gaussian software and applied to view the charge distributions of the molecules in three dimensions as shown in Fig. 9. In MEP contours, the blue color shows the positive (electron-poor) region; the light blue indicates the slightly electron-deficient region, the neutral region is denoted by green, and the red color demonstrates the negative (electron-rich) part, and the yellow color represents the part slightly rich in electrons. Fig. 9 shows that the Alliin molecule has a high negative potential (red color) at the sulfinyl group (-SO), indicating that these centers are the most attractive targets for electrophilic attack. The MEP contour of the S-allyl-cysteine molecule shows a slightly negative potential at the amine group (–NH2), which demonstrates that the nitrogen atom is the most attractive target for electrophilic attack.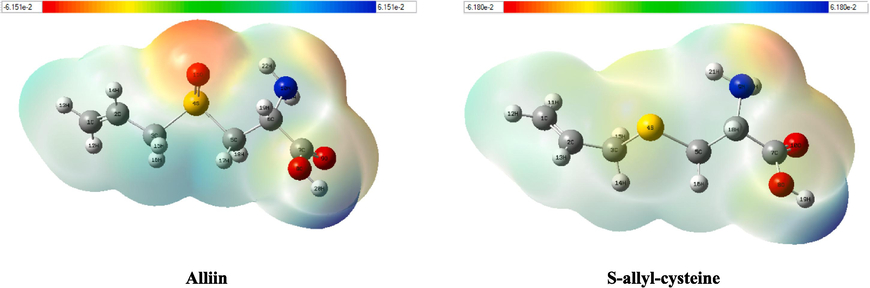
The electrostatic potential surface of the studied compounds.
3.5 Retrosynthesis results
The good findings of the molecular docking and MD simulation of the bioactive molecules (Allicin, Alliin, and S-allyl-cysteine) of the garlic plant encouraged us to make the retrosynthesis of these compounds in order to consider the synthesis of natural molecules with antifungal activity. The obtained results are presented in Table 11 and Table 12. The retrosynthetic analysis of the three bioactive molecules gives the precursor molecules to synthesize these compounds. All molecules provided a score of 1 and the synthesis is done in one step as shown in Table 12.
Compound
Retrosynthesis analysis
Score
Alliin
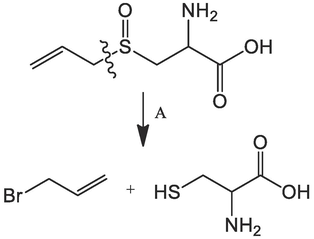
1
S-allyl-cysteine
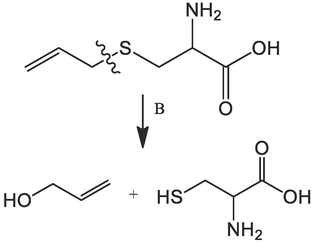
1
Allicin
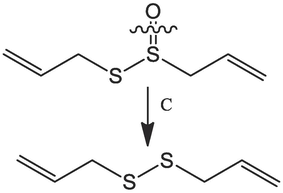
1
Step
The precursors of the synthesis
Similarity
A
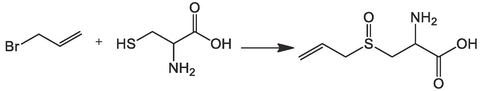
Reference:US06689588B1
Garlic alliinase covalently bound to carrier for continuous production of allician1
B
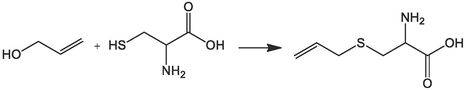
Tanaka, Shinji, Pradhan, Prasun Kanti, Maegawa, Yusuke, Kitamura, Masato Chemical Communications, 2010, vol. 46, # 22p. 3996 – 39981
C

Sulfanyl to sulfinyl
Reference:US20060110472A1_0033
Use of allicin as insect repellent and insecticide in agricultural crops1
4 Conclusion
This study consisted of a molecular docking analysis using three distinct proteins (PDB: 5TZ1, PDB: 4UYM and PDB: 2Y7L) on seven molecules from garlic plant. All bioactive compounds were docked to the three active sites of the receptors studied. The results obtained showed that the studied molecules have good binding energy and interactions with 5TZ1 receptor, especially the Alliin and S-allyl-cysteine molecules. Similarly, the Alliin molecule showed good interaction and binding energy with the 4UYM receptor. The MD simulation result of the S-allyl-cysteine molecule showed good stability throughout the 100 ns. The seven garlic-derived molecules showed good ADME properties and no toxicity. The retrosynthesis results proposed a route to synthesize the Allicin, Alliin, and S-allyl-cysteine molecules. This analysis has successfully allowed us to study the interactions of each molecule from the garlic plant with three different receptors, thus determining the best molecule that exhibits a strong interaction and good binding energy, validating its stability throughout the MD simulation. It also proposes a synthesis pathway for the selected molecules. Furthermore, quantum analysis demonstrated that Alliin and S-allyl-cysteine are considered powerful nucleophiles. Molecular electrostatic potential (MEP) analysis identified preferred sites for electrophilic and nucleophilic attack. Therefore, the results obtained from this study strongly suggest that Alliin and S-allyl-cysteine hold significant promise as potential agents for combating fungal infections. The challenge and limitation of this study lie in practically synthesizing these molecules as potential antifungal drugs, which we will do in collaboration with other scientific teams.
Acknowledgments
We dedicate this work to the “Moroccan Association of Theoretical Chemists” (MATC) for its pertinent help concerning the programs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 3D-QSAR, docking, molecular dynamics simulation and free energy calculation studies of some pyrimidine derivatives as novel JAK3 inhibitors. Arab. J. Chem.. 2020;13(1):1052-1078.
- [CrossRef] [Google Scholar]
- Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput.. 2012;8(9):3257-3273.
- [CrossRef] [Google Scholar]
- Bouamrane, S., Khaldan, A., Maghat, H., Ajana, M.A., Sbai, A., Bouachrine, M., Lakhlifi, T., 2021. In-silico design of new triazole analogs using QSAR and molecular docking models, Rhazes: Green and Applied Chemistry, 11, 223- 236. https://doi.org/10.48419/IMIST.PRSM/rhazes-v11.25084.
- Quantitative Structure-Activity Relationships and Molecular Docking studies of 1.2.4 triazole derivatives as antifungal activity. Rhazes: Green Appl. Chem.. 2020;10:33-48.
- [CrossRef] [Google Scholar]
- combined 3D-QSAR, molecular docking and ADMET properties to identify effective triazole compounds against candida albicans. Malay. J. Biochem. Mol. Biol.. 2022;1:130-140.
- [Google Scholar]
- 3D-QSAR, molecular docking, molecular dynamic simulation, and ADMET study of bioactive compounds against candida albicans. Moroccan J. Chem.. 2022;10(3):523-541.
- [CrossRef] [Google Scholar]
- In silico identification of 1,2,4-triazoles as potential Candida Albicans inhibitors using 3D-QSAR, molecular docking, molecular dynamics simulations, and ADMET profiling. Mol. Divers.. 2023;27:2111-2132.
- [CrossRef] [Google Scholar]
- Garlic-derived organic polysulfides and myocardial protection. Nutr. J.. 2016;146:403S-409S.
- [CrossRef] [Google Scholar]
- Goodman and Gilman’s the pharmacological basis of therapeutics” (12th ed.). McGraw-Hill Education/Medical; 2011.
- Angew. Chem. Int. Ed.. 1991;5:30455-30465.
- Daina, A., Michielin, O., Zoete, V., 2017. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports, 7, 42717. https://doi.org/10.1038/srep42717.
- Darden, T., York, D., & Pedersen, L. 1993. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. The Journal of Chemical Physics, 98, 10089-10092. https://doi.org/10.1063/1.464397.
- Dassault Syst emes BIOVIA, 2017. Discovery Studio Modeling Environment, Release, Dassault Syst emes, San Diego, 2016 [WWW document], http://accelrys.com/products/collaborativescience/biovia-discovery-studio/. (Accessed 25 February 2017).
- DeLano, W., 2002. The PyMOL Molecular Graphics System DeLano Scientific”, Palo Alto, CA, USA, (2002). http://www.pymol.org. (Accessed 25 February 2017).
- Tissue-specific accumulation of sulfur compounds and saponins in different parts of garlic cloves from purple and white ecotypes. Molecules. 2017;22(8):1359.
- [CrossRef] [Google Scholar]
- Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem.. 2008;73(12):4615-4624.
- [CrossRef] [Google Scholar]
- 3D-QSAR, molecular docking, simulation dynamic and ADMET studies on new quinolines derivatives against colorectal carcinoma activity. Biomol. Struct. Dyn.. 2023;2023
- [CrossRef] [Google Scholar]
- A smooth particle mesh Ewald method. J. Chem. Phys.. 1995;103:8577-8593.
- [CrossRef] [Google Scholar]
- Frisch, M., 2009. GAUSSIAN 09. Revision E. 01, Gaussian Inc.
- A novel colorimetric chemosensor based on 2-[(carbamothioylhydrazono) methyl]phenyl 4-methylbenzenesulfonate (CHMPMBS) for the detection of Cu(II) in aqueous medium. Res. Chem. Intermed.. 2023;49:2257-2276.
- [CrossRef] [Google Scholar]
- Jarad, A. J., Dahi, M. A., Al-Noor, T. H., El‑ajaily, M. M., AL-Ayash, S. R., Abdou, A., 2023. Synthesis, spectral studies, DFT, biological evaluation, molecular docking and dyeing performance of 1-(4-((2-amino-5-methoxy)diazenyl)phenyl) ethanone complexes with some metallic ions, Journal of Molecular Structure, 1287, 135703. https://doi.org/10.1016/j.molstruc.2023.135703.
- QSAR based virtual screening derived identification of a novel hit as a SARS CoV-229E 3CLpro Inhibitor: GA-MLR QSAR modeling supported by molecular Docking, molecular dynamics simulation and MMGBSA calculation approaches. Arab. J. Chem.. 2022;15:103499
- [CrossRef] [Google Scholar]
- CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem.. 2008;29(11):1859-1865.
- [CrossRef] [Google Scholar]
- Comparison of simple potential functions for simulating liquid water. J. Chem. Phys.. 1983;79:926-935.
- [CrossRef] [Google Scholar]
- QSAR study of α-glucosidase inhibitors for benzimidazole bearing Bis-Schiff bases using CoMFA, CoMSIA, and molecular docking. Int. J. Quant. Struct.-Property Relationships. 2021;6:9-24.
- [CrossRef] [Google Scholar]
- Identification of potential α-glucosidase inhibitors: 3D-QSAR modeling, molecular docking approach. Rhazes: Green Appl. Chem.. 2021;12:60-75.
- [CrossRef] [Google Scholar]
- Computational study of quinoline-based thiadiazole compounds as potential antileishmanial inhibitors. New J. Chem.. 2022;46:17554.
- [CrossRef] [Google Scholar]
- In search of new potent α-glucosidase inhibitors: molecular docking and ADMET prediction. Moroccan J. Chem.. 2022;10(4):772-786.
- [CrossRef] [Google Scholar]
- In silico design of new α-glucosidase inhibitors through 3D-QSAR study, molecular docking modeling and ADMET analysis. Moroccan J. Chem.. 2022;10(1):22-36.
- [CrossRef] [Google Scholar]
- In silico study of 2,4,5-trisubstituted thiazoles as inhibitors of tuberculosis using 3D-QSAR, molecular docking, and ADMET analysis. El-Cezeri Fen Ve Mühendislik Dergisi. 2022;9:452-468.
- [CrossRef] [Google Scholar]
- Molecular docking, ADMET prediction and quantum computational on 2-methoxy benzoyl hydrazone compounds as potential antileishmanial inhibitors. Biointerface Res. Appl. Chem.. 2023;4:302.
- [CrossRef] [Google Scholar]
- Chemical and biological properties of S-1-propenyl-l-cysteine in aged garlic extract. Molecules. 2017;22(4):570.
- [CrossRef] [Google Scholar]
- Oral formulation roadmap from early drug discovery to development (1st ed.). John Wiley & Sons Ltd.; 2017.
- Synthesis, spectroscopic characterization, DFT calculations, antibacterial activity, and molecular docking analysis of Ni(II), Zn(II), Sb(III), and U(VI) metal complexes derived from a nitrogen-sulfur Schiff base. Russ. J. Gen. Chem.. 2023;93:389-397.
- [CrossRef] [Google Scholar]
- CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput.. 2016;12(1):405-413.
- [CrossRef] [Google Scholar]
- Identify promising IKK-b inhibitors: A docking-based 3D-QSAR study combining molecular design and molecular dynamics simulation. Arab. J. Chem.. 2022;15:103786
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23(1–3):3-25.
- [CrossRef] [Google Scholar]
- Current perspectives on benzoflavone analogues with potent biological activities: A review. Arab. J. Chem.. 2022;15:104109
- [CrossRef] [Google Scholar]
- Alliin the precursor of allicin in garlic extract mitigates proliferation of gastric adenocarcinoma cells by modulating apoptosis. Pharmacogn. Mag.. 2018;14(55s):s84-s91.
- [CrossRef] [Google Scholar]
- Degradation kinetics of fluvoxamine in buffer solutions: In silico ADMET profiling and identification of degradation products by LC-MS/ESI. Arab. J. Chem.. 2020;13:4134-4146.
- [CrossRef] [Google Scholar]
- Potential inhibitors of galactofuranosyltransferase 2 (GlfT2): Molecular docking, 3D-QSAR, and in silico ADMETox studies. Sci. Rep.. 2019;9:17096.
- [CrossRef] [Google Scholar]
- Application of the principle of hard and soft acids and bases to organic chemistry. J. Am. Chem. Soc.. 1967;89(8):1827-1836.
- [CrossRef] [Google Scholar]
- pkCSM: Predicting smallmolecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58(9):4066-4072.
- [CrossRef] [Google Scholar]
- Synthesis and in silico investigation of organoselenium-clubbed Schiff bases as potential Mpro inhibitors for the SARS-CoV-2 replication. Life. 2023;13:912.
- [CrossRef] [Google Scholar]
- Shang, A., Cao, S.Y., Xu, X.Y., Gan, R.Y., Tang, G.Y., Corke, H., Mavumengwana, V., Li1, H. B., 2019. Bioactive Compounds and Biological Functions of Garlic (Allium sativum L.), Foods, 8(7), 246. https://doi.org/10.3390/foods8070246.
- The novel 4-hydroxyphenylpyruvate dioxygenase inhibitors in vivo and in silico approach: 3D-QSAR analysis, molecular docking, bioassay and molecular dynamics. Arab. J. Chem.. 2022;15:103786
- [CrossRef] [Google Scholar]
- AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [CrossRef] [Google Scholar]
- Identification of novel HDAC8 selective inhibitors through ligand and structure based studies: Exploiting the acetate release channel differences among class I isoforms. Arab. J. Chem.. 2022;15:103863
- [CrossRef] [Google Scholar]
- Effects of garlic polysaccharide on alcoholic liver fibrosis and intestinal microflora in mice. Pharm. Biol.. 2018;56(1):325-332.
- [CrossRef] [Google Scholar]
- Studies on the antibacterial activities and molecular mechanism of GyrB inhibitors by 3D-QSAR, molecular docking and molecular dynamics simulation. Arab. J. Chem.. 2022;15:103872
- [CrossRef] [Google Scholar]
- Yang,J., D.Xie., at al., 2022. Synthesis, antifungal activity and in vitro mechanism of novel 1-substituted-5-trifluoromethyl-1H-pyrazole-4-carboxamide derivatives, Arabian Journal of Chemistry. 15, 103987 https://doi.org/10.1016/j.arabjc.2022.103987.
- Yoo, M., Lee, S., Kim, S., Hwang, J, B., Choe, J., Shin, D., 2014. Composition of organosulfur compounds from cool- and warm-type garlic (Allium sativum L.) in Korea. Food Science and Biotechnology, 23, 337–344. https://doi.org/10.1007/s10068-014-0047-y.
- Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem.. 2012;33(31):2451-2468.
- [CrossRef] [Google Scholar]
- Pharmacophore screening, molecular docking, ADMET prediction and MD simulations for identification of ALK and MEK potential dual inhibitors. J. Mol. Struct.. 2021;1245:131066
- [CrossRef] [Google Scholar]




