

Translate this page into:
Green synthesis, density functional theory (DFT), molecular docking, molecular dynamics simulation and biological activity of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives as PLA2 and proteinase K inhibitors
⁎Corresponding author. m.bakht@psau.edu.sa (Md. Afroz Bakht)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
In diseases like atherosclerosis, rheumatoid arthritis, and sepsis, phospholipase A2 (PLA2) and proteinase K play a role in inflammation by releasing arachidonic acid (AA). The crucial step in the inflammatory process is believed to be the release of prostaglandins after PLA2 mobilizes AA. Drugs obstructing the COX and LOX pathways in the arachidonic acid cascade, drugs that inhibit these enzymes can treat inflammatory processes. To combat against these inflammatory promoters,the authors herein report an effective method for the synthesis of a series of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives (4a–h) under the effect of TiO2 as a photocatalyst in ethanolic medium, and their effect against phospholipase A2, as well as proteinase K, was evaluated. The results confirmed that using TiO2 (20 mg) as a photocatalyst exhibits excellent performance in terms of yield and reaction time. The yield of all the synthesized compounds is between 79 and 91 % in just 60 min. After closely evaluating the SAR of the examined compounds against both enzymes, it was discovered that compounds (4b, 4f) with electron-donating substituents have higher PLA2 inhibitory (%) activity than those with electron-removing substituents (4a, 4e). Compounds (4c, 4d) containing heterocyclic rings showed significantly higher proteinase inhibitory (%) activity when compared to other electron-donating or withdrawing compounds. It was further confirmed that studied compounds against both enzymes exhibited dose-dependent behavior. We studied the intermolecular interactions of the compounds by molecular docking with human non-pancreatic secretory phospholipase A2 and proteinase K. We evaluated the dynamical stability of the docked complexes using a 100 ns molecular dynamics simulation, which revealed stable interactions based on root mean square deviation/fluctuation (RMSD/RMSF), radius of gyration, Gibbs free energy landscapes, and principal component analyses (PCA). Moreover, the ADME properties of the compounds align with Lipinski’s rule of five, suggesting their potential as viable candidates for the development of therapies against inflammatory diseases. All compounds tested inhibited PLA2 more than proteinase K. However, compounds 4b and 4f better inhibited phospholipase A2, whereas compounds 4c and 4d showed better activity against proteinase K.
Keywords
Pyrimidine
Phospholipase A2 inhibitor
Proteinase K activity inhibitor
DFT study
Molecular docking
Molecular dynamics simulation

1 Introduction
Phospholipases A2 (PLA2) are a superfamily of enzymes that are actively involved in the hydrolysis of the ester bond of the membrane of glycerophospholipids at the sn-2 position. Various clinical conditions use the majority of the unsaturated fatty acids produced by this enzyme activity as bioactive mediators (Vasilakaki et al., 2016). Mammals contain the enzyme phospholipase A2 (PLA2), which varies in molecular weight, calcium sensitivity, and amino acid sequence. There are six different kinds of PLA2 in mammals: cPLA2, iPLA2, sPLA2, PAF-AHS, GXV PLA2, and GXVI PLA2 (Dennis et al., 2011). cPLA2 is the only type of phospholipase enzyme used for the hydrolysis of arachidonic acid (Kokotou et al., 2017). Activation of cPLA2 releases arachidonic acid and lysophospholipids, leading to the production of various lipid mediators such as leukotrienes and prostaglandins (Psarra et al., 2018).
Proteinases are hydrolytic enzymes that help breakdown proteins, and as a result, a variety of peptides and proteins are formed. These enzymes are categorized as proteinases K-esperase, proteinase, and Bacillus proteinase, depending on their source and functions (Garcia-Carreon, 1997). Enzymes originating from plants, animals, and microorganisms are responsible for a number of pathogenic illnesses, including inflammation, cancer, hypertension, and AIDS (El-Sayed et al., 2016). Therefore, we can utilize proteinases as therapeutic targets for drug discovery, development, and disease prevention. Human-secreted phospholipase A2 enzymes are involved in various oxidative and inflammatory processes, according to several studies (Cesar et al., 2021; Giresha et al., 2022). Chronic inflammatory illnesses are often caused by Group II secreted phospholipase A2 (GIIA), one of nine catalytically active human sPLA2 enzymes, an inhibitor that inhibits SARS-CoV-2 Mpro, and PLA2, an enzyme involved with inflammatory disorders (Theodoropoulou et al., 2022; Oh and Jang, 2023).
From the perspective of green chemistry, multicomponent reactions (MCRs) are one of the more prevalent and useful approaches in organic synthesis for the development of pharmacologically relevant frameworks due to their time-energy, environmental-friendly, atom-efficient, and quick implementation (Jadhav et al., 2019). Particularly in the fields of drug discovery, organic synthesis, and material science, the construction of MCRs using the green approach has garnered a lot of interest (Ryzhkova et al., 2021). Chemical research greatly benefits from green chemistry, consistently favoring 'greener' reaction conditions (Ryzhkova et al., 2021). Pyrimidine is considered one of the most essential structural cores in chemistry, with medical implications for its antimalarial, anthelmintic, anticancer, antibacterial, anti-convulsion, and anti-inflammatory properties (Xian et al., 2020; Jeelan and Goudgaon, 2021; Patil, 2023). Recently, pyrimidine scaffolds have been extensively used in different pharmacological activities such as SARS-CoV-2 (Hosen et al., 2022; Amin et al., 2022; Yasmin et al., 2021), antimicrobial (Moawa et al., 2021; Kawsar et al., 2022), and anticancer (Amin et al., 2021).
Despite the development of numerous multicomponent techniques for pyrimidine derivative synthesis (Goel et al., 2015), their long reaction times, complex procedures, high reaction temperatures, and low yields (Vitório et al., 2015) often prevent their practical use. To resolve these issues, condensation of aldehydes and cynoacetate with urea or thiourea is used to synthesize cynopyrimidine derivatives through a multicomponent reaction (Vitório et al., 2015). Some other reported 1,2,3,4,-tetrahydropyrimidines were also synthesized using Biginelli-type MCR reactions, considering environmentally safe conditions such as [Al(H2O)6](BF4)3 (Mandhane et al., 2010), HClO4-SiO2 (Narahari et al., 2012), SnCl2⋅H2O (Karade et al., 2012), and H3BO3 (Bakht et al., 1944).
Visible light-responsive photocatalysts such as titanium dioxide (TiO2) nanoparticles allow moderate organic reactions, lowering the risk of unsafe reagents and environmental impact. Visible light photocatalysis has many applications in heterocyclic chemical synthesis, medicinal chemistry, drug discovery, late-stage C–H functionalization, bio-conjugation, and isotopic labeling (Srivastava et al., 2022). Nano-sized metal-oxide semiconductors, particularly titanium dioxide (TiO2), are leading in numerous fields (Srivastava et al., 2022; Zhou et al., 2015). This work, in continuation of our earlier work, aims to synthesize 1,2,3,4-tetrahydrocynopyrimidines employing a one-pot, three-component synthesis comprising aldehydes, cynoacetate, and urea/thiourea at room temperature and achieving excellent yields under environmentally benign conditions (Alharthi et al., 2023). The study uses visible-light photocatalysis to improve greener chemistry. TiO2 photocatalyst pyrimidine synthesis is efficient and environmentally friendly, which coincides with global eco-friendly initiatives.
To continue with the same catalyst and using visible light as an energy source, we developed a few more derivatives and biologically assessed them against phospholipase A2 and proteinase K. The study also focused on using various computational tools like DFT analysis, molecular docking, and molecular dynamics simulation to develop a quantum mechanical framework for predicting the molecular properties of the designed ligands. This helps to better understand their binding interactions with the biological target proteins (Phospholipase A2 and Proteinase K) using a molecular docking approach. The stability of the protein–ligand complexes was assessed using molecular dynamics simulation, binding free-energy calculations, principal component analysis (PCA), and free-energy landscape (FEL) analyses. This study employed a combination of computational chemistry, a green-chemistry synthesis approach, and in-vitro enzymatic activity assays to design, synthesize, predict, and validate the activity of a series of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives.
2 Experimental technique
2.1 Chemistry
2.1.1 General procedure for the synthesis of tetrahydropyrimidine-5-carbonitrile derivatives (4a-h)
In a 50-mL beaker, a mixture of aldehydes (1a-h) (1 mmol), ethylcynoacetate (2) (1 mmol), urea/thiourea (3) (1.2 mmol), and 20 mg of TiO2 was stirred after adding absolute ethanol (10 mL) (Yasmin et al., 2021). We added ethanol (98 %) (10 mL) to the reaction mixture and exposed it to a visible light source (100 mW cm-2). Thin-layer chromatography (TLC), using toluene, ethyl acetate, and formic acid in a 5:4:1 ratio, continuously monitored the reaction. At the end of the process, we added ethyl acetate and 20 % aqueous NaOH, and used centrifugation to extract TiO2 (Yasmin et al., 2021). A solid product was obtained after being evaporated and purified by recrystallization with ethanol. The synthesized compounds were further characterized by FT-IR, 1H NMR, and 13C NMR and are presented below.
2.1.2 2,4-dioxo-6-(3,4,5-trimethoxyphenyl)-1,2,3,4-tetrahydro-5-pyrimidinecarbonitrile(4a)
White solid; Yield: 91 %; M.P 236–238 0C; FT-IR (cm−1, ATR); 3445, 3329 (2 N-H), 2202 (CN), 1694 (C = O), 1449 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.33 (s, 1H, NH), 7.51 (s, 2H, Ar-H), 5.40 (s, 1H, NH), 3.83–3.34(s, 9H, 3x OCH3); 13C NMR (DMSO–d6, 100 MHz): 162.51, 160.00, 155.43, 153.33, 142.50, 126.93, 116.53, 109.30, 101.24, 62.74, 60.81, 56.51.
2.1.3 1,2,3,4-tetrahydro-2,4-dioxo-6-p-tolylprimidine-5-carbonitrile (4b)
White solid; Yield: 89 %; M.P 223–224 0C FT-IR (cm−1, ATR); 3431, 3280 (2 N-H), 2227(CN), 1668 (C = O), 1449 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.4 (s, 2H, 2xNH), 8.04–6.75 (m, 4H, Ar −H), 2.48(s, 3H, CH3); 13C NMR (DMSO–d6, 100 MHz): 160.21, 155.54, 144.99, 131.53, 130.53, 126.41, 116.37, 101.70, 62.83, 21.91.
2.1.4 6-(furan-2-yl)-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (4c)
Light brown solid; Yield: 88 %; M.P 196–198 0C FT-IR (cm−1, ATR); 3369, 3166(2 N-H), 2218(CN), 1710(C = O), 1410(C = C); 1H NMR (DMSO–d6, 400 MHz): 8.21(s, 1H, NH), 8.13(s, 1H, Ar –H), 7.53–7.52 (d, 1H, J = 3.6, Ar −H), 6.87–6.86(t, 1H, J = 3.4, Ar −H), 5.42(s, 1H, NH); 13C NMR (DMSO–d6, 100 MHz): 162.68, 160.07, 150.57, 148.64, 139.59, 125.15, 114.80, 97.39, 97.36, 62.61.
2.1.5 6-[5-(hydroxylmethyl)furan-2-yl]-2,4-dioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitrile (4d)
White solid; 85 %; M.P 204–206 0C FT-IR (cm−1, ATR); 3424 (OH), 3290 (2 N-H), 2226(CN), 1651 (C = O), 1443 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.08 (s, 1H, NH), 7.49 (s, 1H, Ar-H), 6.71(s, 1H, Ar-H), 5.41(s, 1H, NH) 4.53(s, 1H, CH2OH); 13C NMR (DMSO–d6, 100 MHz): 178.45, 163.86, 160.05, 152.19, 147.84, 139.44, 111.79, 96.21, 62.54, 56.57.
2.1.6 1,2,3,4-tetrahydro-6-(3,4,5-trimethoxyphenyl)-4-oxo-2-thioxopyrimidine-5-carbonitrile (4e)
Yellow solid; 87 %; 242–243 0C FT-IR (cm−1, ATR); 3351, 3140 (2 N-H), 2220 (CN),1719 (C = O), 1401 (C = C); 1H NMR (DMSO–d6, 400 MHz): 9.88(s, 1H, NH), 7.25 (s, 2H, Ar-H), 4.31(s, 1H, NH) 3.86–3.36(s, 9H, 3x OCH3); 13C NMR (DMSO–d6, 100 MHz): 192.36, 184.30,162.52, 153.79, 143.28, 132.11, 107.19, 101.27, 62.75, 60.68, 56.52.
2.1.7 1,2,3,4-tetrahydro-4-oxo-2-thioxo-6-p-tolylpyrimidine-5-carbonitrile (4f)
Brown solid; 84 %; 228–229 0C; FT-IR (cm−1, ATR); 3363, 3260 (2 N-H), 2211 (CN), 1719 (C = O), 1401 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.33(s, 1H, NH), 7.96–7.94 (d, 1H, J = 8.12, Ar −H), 7.41–7.39(d, 1H, J = 8.04, Ar −H),7.18(s, 1H, Ar −H), 7.07–7.05(d, 1H, J = 8.24, Ar −H), 4.29(s, 1H, NH), 2.39(s, 3H, CH3); 13C NMR (DMSO–d6, 100 MHz): 169.02, 162.92, 149.66, 146.66, 138.29, 132.11, 128.09, 115.31, 74.18. 21.10.
2.1.8 6-(furan-2-yl)-1,2,3,4-tetrahydro-4-oxo-2-thioxopyrimidine-5-carbonitrile (4g)
Yellow solid; 80 %; M.P 212–214 0C; FT-IR (cm−1, ATR); 3130, 3041 (2 N-H), 2217 (CN), 1709 (C = O), 1529 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.21(s, 1H, NH), 8.13 (s, 1H, Ar-H), 7.52–7.51 (d, 1H, J = 3.3, Ar −H), 6.87 (s, 1H, Ar −H), 4.29(s, 1H, NH); 13C NMR (DMSO–d6, 100 MHz): 162.68, 150.57, 148.65, 139.59, 125.13, 115.79, 114.79, 97.37, 62.60.
2.1.9 1,2,3,4-tetrahydro-6-(5-(hydroxymethyl)furan-2-yl)-4-oxo-2-thioxopyrimidine-5-carbonitrile (4h)
Yellow solid; 79 %; M.P 221–223 0C FT-IR (cm−1, ATR); 3410 (OH), 3319, 3215 (2 N-H), 2211(CN), 1611 (C = O), 1665 (C = C); 1H NMR (DMSO–d6, 400 MHz): 8.21 (s, 1H, NH), 6.83 (s, 1H, Ar-H), 6.46–6.38 (d, 1H, J = 29.9, Ar −H),5.44 (s, 1H, NH), 4.47(s, 1H, CH2OH); 13C NMR (DMSO–d6, 100 MHz): 165.71, 158.11, 150.48, 149.32, 132.74, 128.74, 116.85, 115.11, 109.65, 56.21.
2.2 In silico-studies
2.2.1 Density functional theory computations
The Orca 5.0.3 program was used to compute density functional theory (DFT) (Neese, 2012; Neese, 2018). Compounds 4a–4 h were optimized using the Lee-Yang-Parr correlation functional (B3LYP) (Becke, 1988; Lee et al., 1988). Chemcraft, a graphical software for quantum chemistry computations (https://www.chemcraftprog.com), visualized the output file of the DFT-optimized structure. We utilized the Avogadro program (Hanwell et al., 2012) to illustrate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). Additionally, we calculated global reactivity characteristics like electronegativity using the previously reported HOMO and LUMO energies (Azam et al., 2018).
2.2.2 Molecular docking
2.2.2.1 Protein preparation
Phospholipase A2 (PDB ID: 1DB4) and proteinase K (PDB ID: 2PWB) three-dimensional crystal structures We retrieved these data from the RCSB Protein Data Bank (https://www.rcsb.org/pdb/home/home.do). In Biovia Discovery Studio Visualizer 2021, the proteins were evaluated for any irregularities, such as missing residues or atoms. We applied Gasteiger charges to each atom in MGL Tools 1.5.7 and saved the receptor file in pdbqt format for use in the next phase. (Azam et al., 2022). The proteins were evaluated for their structural integrity by generating the Ramachandran plot using the MolProbity webserver (https://molprobity.biochem.duke.edu/).
2.2.2.2 Ligand preparation
Avogadro was used to transform the chemical structures of compounds 4a–4 h acquired from DFT optimization to PDB format. MGLTools 1.5.7 was used to merge all non-polar hydrogens and specify the number of rotatable bonds and the torsion tree. The Gasteiger method was used to add partial atomic charges, which were then saved in pdbqt format. The next step involved making the synthesized ligands flexible for molecular docking in the active sites of phospholipase A2 and proteinase K.
2.2.2.3 ADMET analysis of the ligands
The ADME parameters for compounds 4a-4 h were predicted with the SwissADME server (https://www.swissadme.ch/) and the toxicity prediction of the compounds was done using admetSAR webserver (https://lmmd.ecust.edu.cn/admetsar2/).
2.2.2.4 Structural similarity analysis between phospholipase A2 and proteinase K
The structural similarity between the target proteins phospholipase A2 and proteinase K in terms of the charge of the ligand binding cavity and RMSD of alignment was calculated using the APBS plugin and align tool of PyMol 2.5.
2.2.2.5 Molecular docking study
AutoDock Vina 1.1.2 was used for the molecular docking investigation, with the default optimization parameters (Trott and Olson, 2010). The native co-crystallized ligands 8-indole and coumarin were utilized to characterize the docking area in each protein, with dimensions of 18, 18, and 18 points in the x, y, and z directions, respectively. Following the successful completion of docking, the top ten molecular conformations were processed to analyze their corresponding affinity in terms of binding energy and visualized in the Biovia Discovery Studio Visualizer 2020, Ligplot Plus, and PyMol 2.5 programs.
2.2.2.6 Molecular dynamics simulation
The docking analysis only revealed one static intermolecular picture. The compound 4a, 4b, 4d, and 4e complexes and positive control complexes of 8-indole and coumarin were put through molecular dynamics simulations to learn more about how the molecules move and stick together. GROMACS 2022.3 software was used for a 100-ns all-atom molecular dynamics (MD) simulation (Abraham et al., 2015). The study of Saibu et al. (Saibu et al., 2023) served as the basis for preparing all other simulation systems and parameters. To summarize, we prepared the simulation system using LiGRO (Kagami and G.M. das Neves, A.W.S. da Silva, R.A. Caceres, D.F. Kawano, V.L. Eifler-Lima, , 2017), a GUI-based application that automates the system file preparation process necessary for simulation with GROMACS 2022. 3. We utilized the Amber14SB and General Amber Force Field 2 (GAFF2) force field parameters for the protein and ligands, respectively (Lee et al., 2020). The ligand–protein complex was placed in a 3D-cubic box solvated with the TIP3P water model, maintaining a distance of 10 Angstrom from the protein–ligand complex to the cubic box edge. The Monte-Carlo ion-placing method balanced the systems by adding 100 mM Na+ and Cl- ions (Jo et al., 2008), and regular boundary conditions were used to eliminate any surface effects. The Amber14SB forcefield and NPT ensemble were applied. At a temperature of 310.15 K, the first equilibration stage used the Berendsen thermostat for a constant number of particles, volume, and temperature (NVT) ensemble. It went through a single-step protocol for 100,000 steps over a total period of 100 ps. Another 100 ps of second-stage equilibration was then performed with an NPT ensemble using a Nose-Hoover thermostat and a Parrinello-Rahman barostat at 1 bar pressure. For non-bonded interactions (Lennard-Jones and Coulomb potentials), the Verlet cut-off scheme was utilized, with a cut-off radius of 10 Angstrom (1 nm). The Particle-Mesh Ewald (PME) method (Darden et al., 1993) was used to model the long-range electrostatic interactions. The novel Linear Constraint Solver (LINCS) algorithm (Hess et al., 1997) constrained all covalent bonds, including hydrogen atoms. At this stage, we minimized each system, well-equilibrated it at the correct temperature (310.15 K), and prepared it for the 100 ns molecular dynamics experiments. Four independent MD simulations were performed for each ligand, with an integration time step of 2 fs and no restrictions, and the frames were stored every 10 ps (Tuble et al., 2004). The Visual Molecular Dynamics 1.9.3 (VMD) package (University of Illinois at Urbana-Champaign, Champaign, IL, USA) and three built-in trajectory tools of GROMACS 2022.3 were used to analyze the data (Humphrey et al., 1996).
2.2.2.7 Principal component analysis (PCA) with free energy landscape (FEL)
The Geo_Measures v 0.9d program performed the post-simulation ensemble analysis of the complexes (Kagami and G.M. das Neves, L.F.S.M. Timmers, R.A. Caceres, V.L. Eifler-Lima, , 2020). For each ligand–protein complex trajectory, 10,000 frames were included in the study. The MD trajectory vs significant trajectory components motion was shown in 2D using the matplotlib python package (Hunter, 2007) with GeoMeasures, which included a comprehensive library of g sham (Kagami and G.M. das Neves, L.F.S.M. Timmers, R.A. Caceres, V.L. Eifler-Lima, , 2020).
2.3 Biological activity
2.3.1 Phospholipase A2 (PLA2) inhibitory activity
The PLA2 assay was carried out as previously discussed by De Araujo and Radvany (de Araújo and Radvanyi, 1987). Phospholipase A2 is on the open market, purchased from Sigma Aldrich in the United States (P6534). The substrate contained 100 mM NaCl, 10 mM CaCl2, red phenol as a colorimetric indicator of 0.055 mM, 3.5 mM lecithin, 3 mM NaTDC, and 100 mM NaCl. Phosphate buffer assisted in raising the pH of the reaction to 7.6. In 10 % acetonitrile, the sPLA2 protein was solubilized at concentrations ranging from 0.01 to 0.08 g/L. These PLA2 solutions were incubated at room temperature for 20 min in 10 L holding 10 g of each chemical. After adding 1 mL of PLA2 substrate, researchers observed the hydrolysis kinetics for 5 min by measuring the optical density at 558 nm. Comparing the outcomes to a control experiment (one without the compound) allowed researchers to assess the proportion of inhibition.
The PLA2 activity assay was conducted in accordance with De Arajo and Radvany bm's earlier discussion (de Araújo and Radvanyi, 1987). Acquired phospholipase A2 from Sigma Aldrich in the US on the open market (P6534). The substrate was made up of 100 mM NaCl, 10 mM CaCl2, 3.5 mM lecithin, 3 mM NaTDC, and red phenol as a colorimetric indicator of 0.055 mM. Phosphate buffer was used to bring the reaction mixture's pH up to 7.6. The sPLA2 protein was soluble in 10 % acetonitrile at a concentration of 0.01 to 0.08 g/L. After adding 1 mL of PLA2 substrate, the optical density at 558 nm was monitored for 5 min to determine the hydrolysis kinetics. Researchers were able to determine the inhibition percentage by comparing the results to a control experiment (one in which the chemical was not used).
2.3.2 Proteinase K inhibitory activity
The proteinase K utilized in this investigation was purchased commercially (P2308, Sigma Aldrich, USA). Hammerstein casein was used as the substrate for proteinase K testing utilizing the Kunitz caseinolytic method (Kunitz, 1946). The same procedures were used to evaluate the activities of proteinase K inhibitors, with the addition of the inhibitor (0.1 mg/mL) to the reaction mixture and a 10-minute pre-incubation at 37 °C. 2 mL of 1 % casein was added after the remaining enzyme activity test, and the mixture was left to stand for 30 min at 37 °C. The reaction was halted by adding 2.5 mL of a 5 % TCA solution. The reaction mixture's absorbance was assessed at 280 nm following centrifugation (12,000 rpm, 15 min). A proteinase K inhibitor unit is the volume of proteinase K inhibitor that reduces one unit of the related enzyme activity. The assay also contained the proper blanks for the enzyme, inhibitor, and substrate.
In this investigation, proteinase K was procured from commercially accessible sources (P2308, Sigma-Aldrich, Saint Louis, MO, USA). In short, the Kunitz caseinolytic approach was used to perform proteinase K assays using Hammerstein casein as the substrate (Kunitz, 1946). The same procedures were used to assess the proteinase K inhibitory activities: the reaction mixture was pre-incubated for 10 min at 37 °C, and the inhibitor (0.1 mg/mL) was added. After the analysis for remaining enzyme activity, 2 ml of 1 % casein were added, and the mixture was left to stand at 37 ◦C for 30 min. The reaction was halted by adding 2.5 mL of a 5 % TCA solution. After centrifuging the reaction mixture for 15 min at 12,000 rpm, the absorbance was measured at 280 nm. A proteinase K inhibitor unit is the quantity of proteinase K inhibitor that inhibits one unit of related enzyme activity. Appropriate blanks for the substrate, inhibitor, and enzyme were run in parallel with the test. As a positive control, a proteinase K inhibitor cocktail from Sigma was employed. Data are expressed as mean ± standard error of the mean (n = number of experiments in triplicate). P < 0.05 is considered to be statistically significant.
Some of the recently reported anti-phospholipase A2 and anti-proteinase K compounds are presented as in Fig. 1.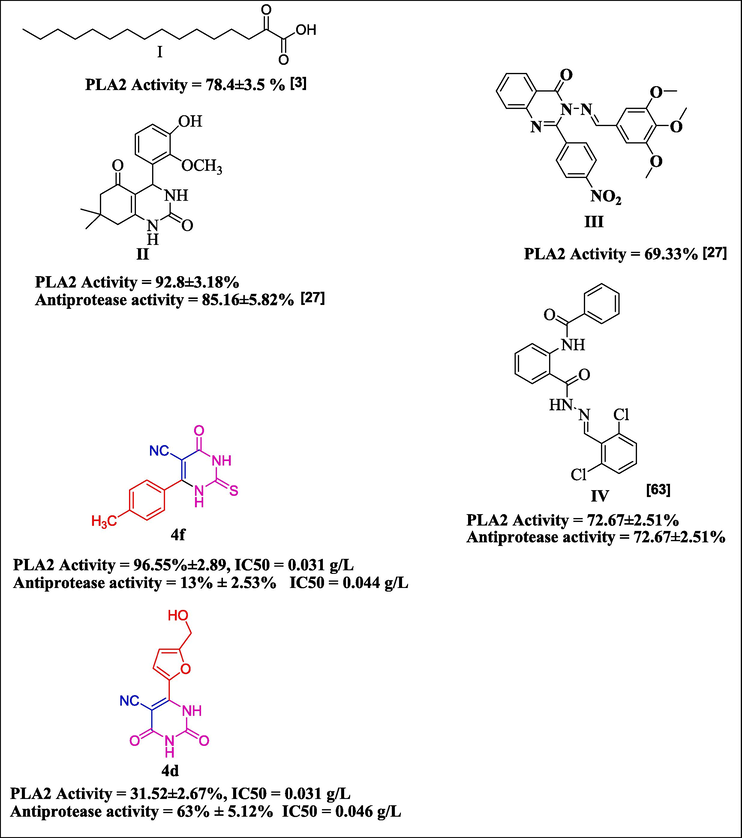
Compounds that have been reported as examples (I-IV) compared with compounds 4d and 4f as lowest and highest antiphospholipase A2, proteinase k activities in present studies.
3 Results and discussion
3.1 In silico-studies
3.1.1 Density functional theory (DFT)
The HOMO-LUMO gap and global reactivity descriptors are important parts of understanding how chemicals react and what molecules are made of (Ramírez-Martínez et al., 2023). Density Functional Theory (DFT) gives us a way to study them. The DFT-optimized structures are shown in Fig. 2. The HOMO-LUMO gap is the energy difference between the highest occupied molecular orbital (HOMO) and the lowest occupied molecular orbital (LUMO) in a molecule. The HOMO-LUMO gap closely relates to the electronic properties of molecules, reflecting the energy required for electron excitation or transfer. A smaller gap indicates easier electron transfer and greater reactivity. The synthesized compounds' HOMO-LUMO gap fell within the range of 3.82–4.61 eV. Interestingly, thione derivatives (4e–4 h) exhibited a lower energy gap in comparison to keto derivatives. Regarding electrophilicity, the index value was at its lowest for the compound 4a, with 4b, 4d, 4e, 4f, 4c, 4 h, and 4 g following in increasing order. However, minimum chemical potential was noted with 4 g, while 4 a displayed the maximum value. Similarly, other DFT-based HOMO and LUMO were also used to compute global reactivity characteristics such as hardness, softness, and electronegativity, as presented in Table 1. The disparities in these values can be attributed to an alteration in chemical structures through diverse substitutions. Red is considered statistically significant. a All values are given in eV.
Optimized geometries of the compounds 4a-4 h.
Comp
HOMO
LUMO
HOMO − LUMO gap
Hardness (η)
Softness (S)
Chemical potential (μ)
Electronegativity (χ)
Electrophilicity index (ω)
4a
−6.54
−2.32
4.22
2.11
0.47
−4.43
4.43
4.65
4b
−6.98
−2.37
4.61
2.30
0.43
−4.67
4.67
4.74
4c
−6.90
−2.73
4.17
2.09
0.48
−4.81
4.81
5.55
4d
−6.68
−2.59
4.08
2.04
0.49
−4.63
4.63
5.26
4e
−6.57
−2.59
3.98
1.99
0.50
−4.58
4.58
5.27
4f
−6.73
−2.65
4.08
2.04
0.49
−4.69
4.69
5.39
4 g
−6.78
−2.96
3.82
1.91
0.52
−4.87
4.87
6.22
4 h
−6.66
−2.83
3.83
1.91
0.52
−4.75
4.75
5.89
Fig. 3 depicts the visualization of calculated HOMO/LUMO, which indicates the delocalization of electrons between HOMO and LUMO, which is regarded as critical for determining the possibility of molecular reactivity. Consequently, they are thought to be the basic orbitals that help determine how a molecule interacts with other species, such as the interaction between a drug and its macromolecular target (Azam et al., 2018; Ahmad et al., 2023). The HOMO and LUMO orbitals on the molecules are staggered with respect to one another, where cyan represents negative surfaces and purple signifies positive surfaces (Fig. 3). The thione group of pyrimidines was mostly covered by HOMO in compounds 4e–4 h, whereas HOMO engulfed the cyano group in compounds bearing the keto group (4a–4d). Compounds 4f and 4 g were dominated by LUMO, sparing the thione group.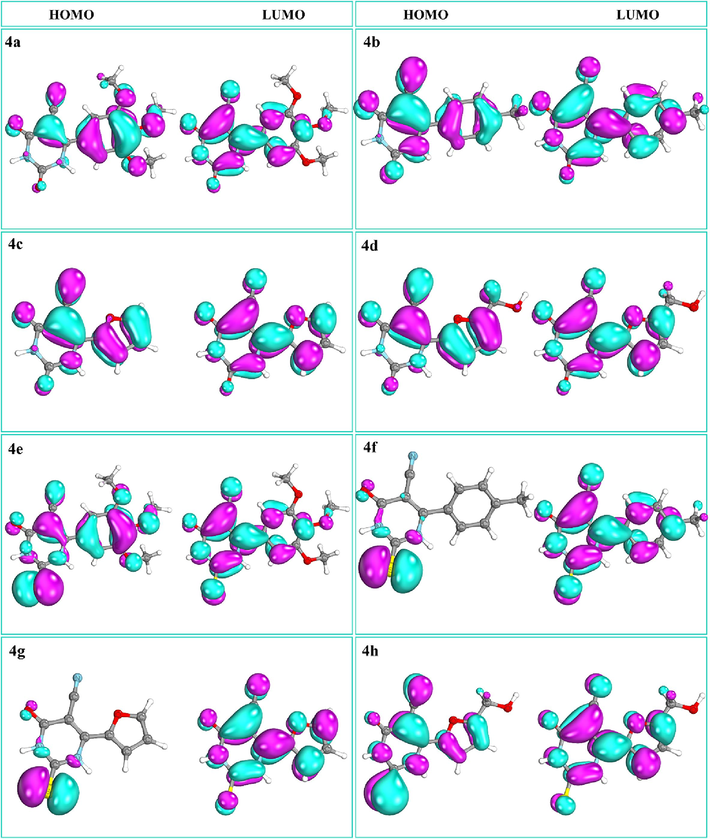
Compounds 4a-4 h displaying HOMO and LUMO.
3.1.2 ADMET analysis of the ligands
As evident from supplementary table 1, all the compounds are following Lipinsky’s rule of five and show high GI absorption with moderate to high solubility. None of them showed potential to cross the blood–brain barrier and bind as a Pgp substrate (except compound 4f). These properties make them suitable for lead-like candidates. The toxicity prediction from supplementary table 2 shows that all the compounds are non-mutagenic and non-carcinogenic. Furthermore, they showed low honey bee toxicity, with the exception of compound 4e.
3.1.3 Structural similarity of phospholipase A2 and proteinase K
The electrostatic analysis of target proteins Proteinase K and Phospholipase A2 was done using Adaptive Poisson-Boltzmann Solver (APBS) plugin in PyMol. The blue color in the color bar shows a positive charge, and the red color indicates a negative charge. As evident from supplementary Fig. 2, the ligand-binding cavities in both proteins were found to be negatively charged. In the overall structural alignment (supplementary Fig. 3) of phospholipase A2 on proteinase K, a significant amount of good alignment was found in terms of lower RMSD, as marked by the blue color, while the region in red shows a higher RMSD in alignment, and the region in grey shows the residues not used during alignment. As evident from the above data, both target proteins share negatively charged ligand binding pockets and align well over the binding pocket residues. This might be the probable reason for both target proteins to bind similar ligands.
3.1.4 Molecular docking
Before moving on to the docking, the structural integrity of the target proteins was assessed using the Ramachandran plot, where both target proteins were shown to have 100 % residues in the allowed region with no outliers, as shown in supplementary Fig. 1. The primary objective of this study was to elucidate the binding mode and energetics of compounds 4a–4 h with the target proteins phospholipase A2 (PDB ID: 1DB4) and proteinase K (PDB ID: 2PWB) using molecular docking simulations. First of all, the native co-crystallized ligands 8-indole ([3-(1-benzyl-3-carbamoylmethyl-2-methyl-1H-indol-5-yloxy)-propyl-]-phosphonic acid) and coumarin were redocked to their respective proteins in order to validate the docking protocol. The crystal structures of the docked conformations were well aligned.
The docking scores in kcal/mol are shown in Supplementary table 3. Our results revealed that ligands 4a–4 h interact with the specific binding sites on both proteins, phospholipase A2 and proteinase K, through a network of hydrogen bonds and hydrophobic interactions. The intermolecular interactions of the selected compounds have been presented in Figs. 4a and 4b. In general, the interactions are in good agreement with known binding modes of native ligands, bolstering the reliability of docking predictions. In the phospholipase A2 binding site, Gly29, Gly31, His47, Asp48, and Asp91 were the most important residues for polar interactions. However, residues such as Leu2, Phe5, Ala18, Tyr21, and Gly22 were contributors to hydrophobic contacts. In proteinase K, hydrogen bonds were formed with His69, Leu133, Gly134, Ala158, and Ser224, whereas hydrophobic interactions were afforded by Leu96, Gly100, Ala158, Asn161, and Tyr169. However, His69, Leu133, and Gly134 were involved in the polar and non-polar interactions. Overall, these results give us useful information about the structure of the ligand–protein complex. They help us understand how the molecules interact and what their possible functional effects might be.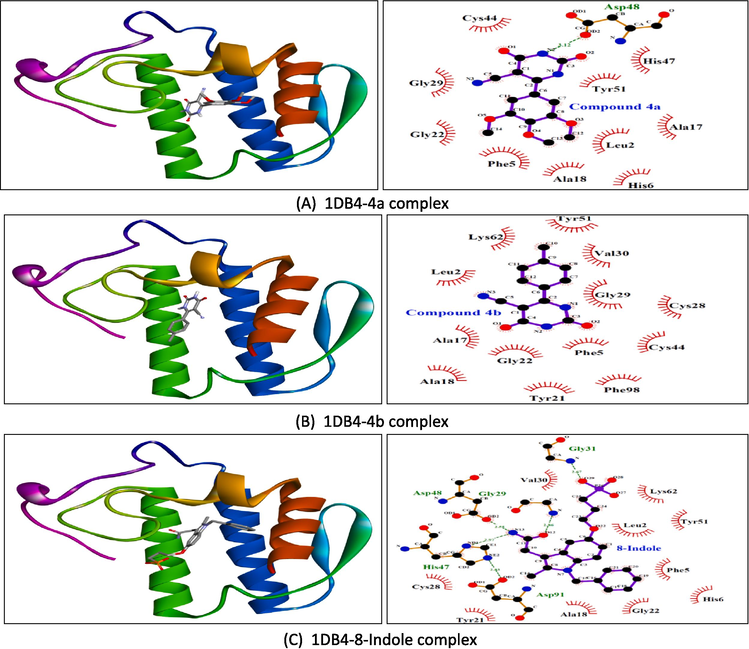
Molecular docking results showing 3D docked pose and two-dimensional binding interactions of phospholipase A2 (PDBID: 1DB4) with compound 4a (A), compound 4b (B), and 8-indole (C).
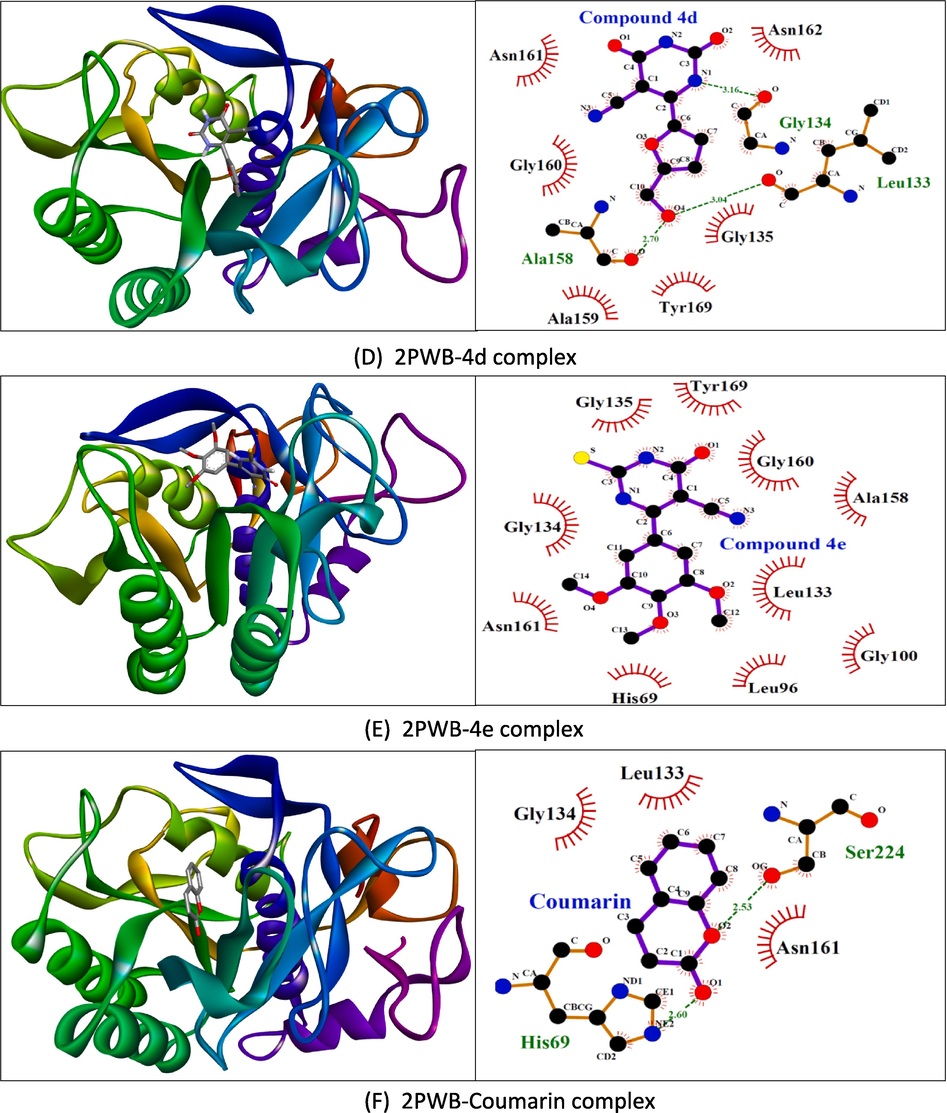
Molecular docking results showing 3D docked pose and two-dimensional binding interactions of proteinase K (PDBID: 2PWB) with compound 4d (D), compound 4e (E), and coumarin (F).
3.1.5 Molecular dynamics (MD) simulation
Since MD simulation mimics the actual biological phenomenon of protein–ligand interaction in a biological environment, this study is thought to be more relevant than docking (Durrant and McCammon, 2011; Singh et al., 2023). In order to provide information regarding the dynamic and thermodynamic properties and structure of molecules, MD simulations are now regarded as an integral part of the modern drug development process (Karplus and McCammon, 2002; Parveen et al., 2023; Hospital et al., 2015). In all-atom MD simulation, the modeled system comprises interacting particles placed in a giant simulation box, and Newton’s law of motion solves the movements. The simulation trajectories have been analyzed for structure stability using carbon-α atoms. To assess this, the root-mean-square deviation (RMSD) analysis was conducted, which measures the average distance between the superimposed protein atoms (Maiorov and Crippen, 1994). A high RMSD value throughout the simulation suggests larger structure deviations, while a low RMSD value indicates fewer structural changes. Protein-ligand complexes with reduced RMSD values show strong intermolecular contacts and stable docked conformation since large carbon alpha fluctuations in this context lead to the dissociation of ligand-bound receptors (Fusani et al., 2020). The RMSD values for the phospholipase A2-bound complexes of compounds 4a, 4b, and 8-indole (as a positive control) were 2.02 Å, 1.90 Å, and 2.04 Å, in that order (Fig. 5A). It seems that the compounds were effectively attached to the phospholipase A2 binding site, and according to the VMD analysis, there was no noticeable change in the binding mode. The minor RMSD jumps in the systems were due to the flexible loops in the enzyme that are naturally present to aid in enzyme catalytic mechanisms. The proteinase K-bound systems, including compounds 4d, 4e, and coumarin, displayed average RMSDs of 1.31 Å, 1.41 Å, and 1.25 Å, respectively (Fig. 5B).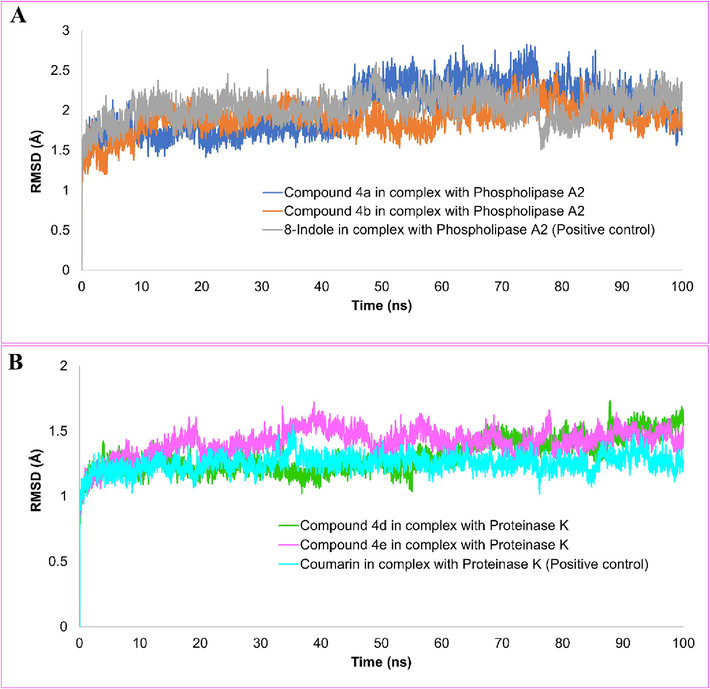
The molecular dynamics simulation trajectories have been analyzed to investigate the stability of the structure showing root-mean square deviation (RMSD) of compounds 4a, 4b and 8-indole in complex with phospholipase A2 (A). RMSD plot of the compounds 4d, 4e and coumarin (B).
Root mean square fluctuation (RMSF) on carbon-α atoms recognized the local residue level fluctuations. The mean RMSF of the compounds 4a, 4b, and 8-indole, in complex with phospholipase A2, was noted as 1.30 Å, 1.08 Å, and 1.23 Å, respectively (Fig. 6A). It can be see that the figure depicts a high stability of protein residues in the presence of the compound, with only a few noticeable exceptions where there are some jumps in the upper region. Likewise, an excellent RMSF profile was shown by the docked compounds in complex with proteinase K. Compounds 4d, 4e and coumarin demonstrated average RMSF of 0.71 Å, 0.72 Å, and 0.71 Å, respectively (Fig. 6B).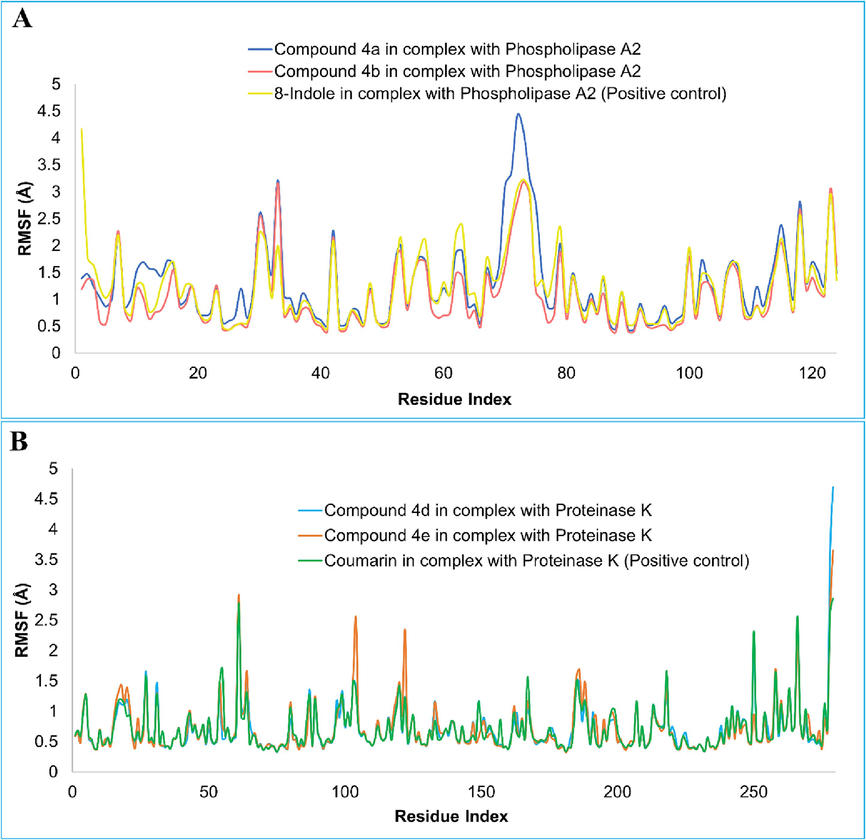
The root-mean square fluctuation (RMSF) of compounds 4a, 4b and 8-indole in complex with phospholipase A2 (A). RMSF plot of the compounds 4d, 4e and coumarin (B).
The radius of gyration (RoG) analysis is an effective method for describing the compactness and relaxed nature of protein 3D structure during simulation time. (Lobanov et al., 2008). The compact nature of the protein is necessary for solid compounds to bind to phospholipase A2 and proteinase K where getting relax confers loose ligand binding. The mean RoG values of compounds 4a (14.64 Å), 4b (14.40 Å), 8-indole (14.59 Å) in complex with phospholipase A2 has been presented in Fig. 7A. A slight expansion in the Phospholipase A structure was observed in complex compound 4a as compared to compound 4b and 8-indole. Fig. 7B displays the RoG plot of compounds 4d (16.96 Å), 4e (16.95 Å), and coumarin (16.97 Å) with Proteinase K where a similar trend in of RoG values were observed in each complex. The low RoG value of complexes determines stable secondary element positioning of the protein in the compound’s presence, exerting less pressure on the compound to dissociate from the binding sites.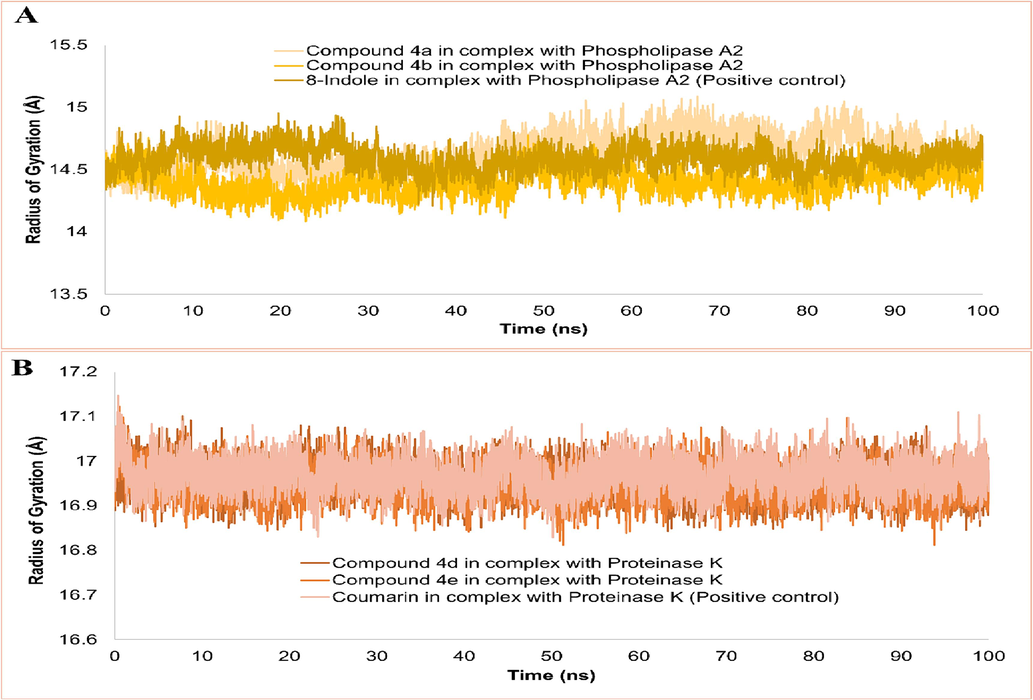
Radius of gyration analysis of complexes done on carbon-α atoms.
The number of hydrogen bonds formed between the compounds 4a, 4b and 8-indole with phospholipase A2 and compounds 4d, 4e and coumarin with proteinase K were calculated as shown in Fig. 8A and 8B respectively. Compound 4a and 8-indole maintained only one hydrogen bond while compound 4b formed 1–2 hydrogen bonds with phospholipase A2 over the trajectory. In case of proteinase K, coumarin formed only one hydrogen bond intermittently while compound 4d and 4e formed 2–4 hydrogen bonds over the simulation trajectory indicating more stability with proteinase K as compared to the coumarin (positive control).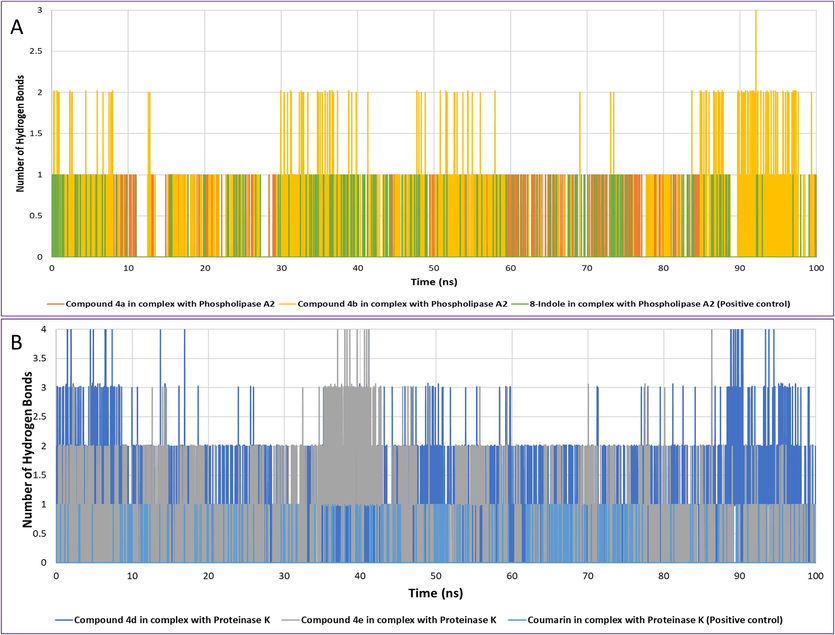
The number of hydrogen bonds formed between compounds 4a, 4b and 8-indole in complex with phospholipase A2 (A). The number of hydrogen bonds formed between compounds 4d, 4e and coumarin with Proteinase K (B).
3.1.6 Principal component analysis (PCA) with free energy landscape (FEL)
The MD simulation trajectories were subjected to principal component analysis (PCA) for 4a, 4b, and 8-indole compounds bound to the Phospholipase A2 to analyze the conformational subspace of the complexes in order to comprehend the stable dynamic behavior. Compared to the positive control (8-indole), higher conformational subspace was covered by 4a and 4b over the PC2. Similarly, the trajectories of compounds 4d, 4e and coumarin in complex with proteinase K were also studied. The plot shown in Fig. 9 displays the displacement of atomic fluctuations and movements in the structures as a function of eigenvectors 1 and 2 (PC1 and PC2). The positive values are indicative of corelated movements while the negative values are indicative of anti-corelated movements. Compared to the positive control (coumarin), complexes of proteinase K with compounds 4d and 4e showed the protein minimum subspaces are covered by the clusters, which are well characterized.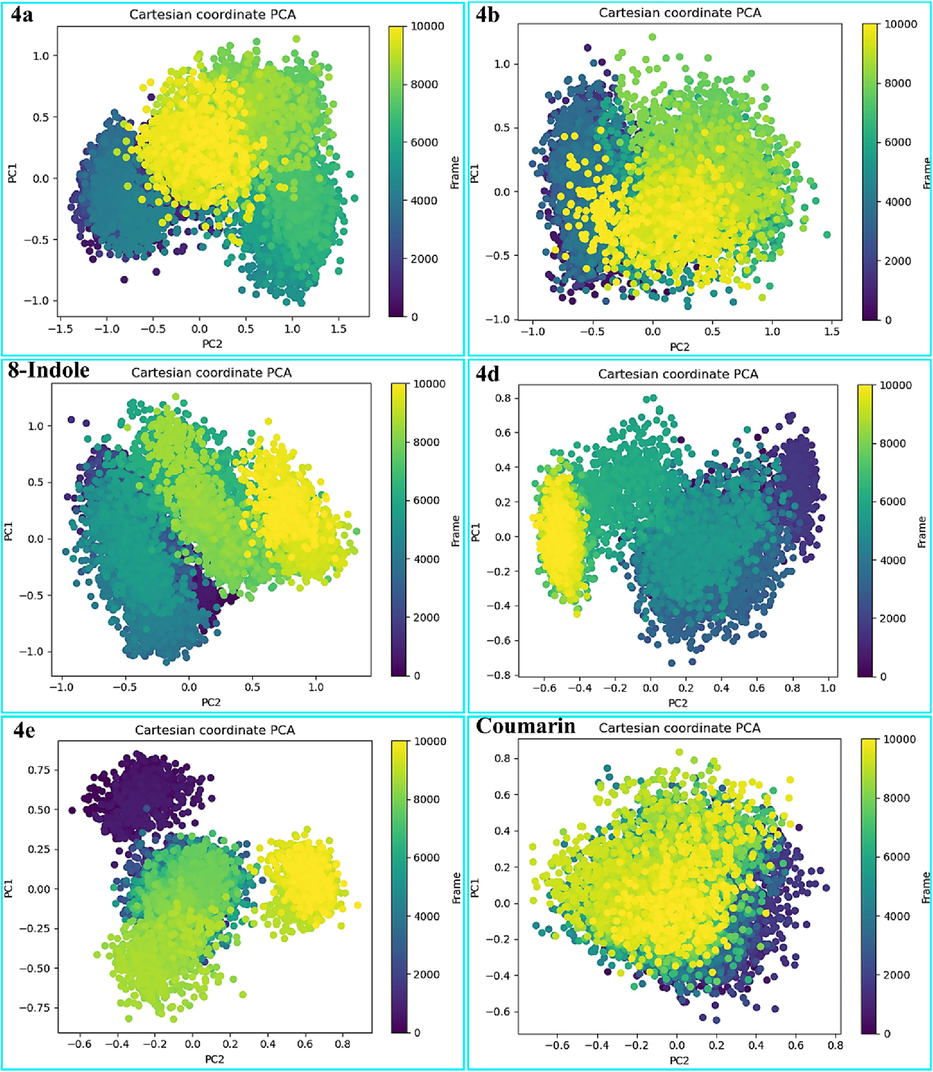
Principle component analysis (PCA) was performed on 10,000 frames from MD simulations of all complexes. (A) 4a-phospholipase A2 (B) 4b-phospholipase A2, (C) 8-indole-phospholipase A2, (D) 4d-proteinase K, (E) 4e-proteinase K, and (F) coumarin-proteinase K complexes.
Gibbs free energy landscape was created using the RMSD and RG values obtained from the MD trajectory. Fig. 10 depicts that the FEL was calculated for several compounds, such as 4a, 4b, and 8-indole, which were found to be bound to phospholipase A2 and 4d, 4e, and coumarin, which were found to be bound to proteinase K. The corresponding Gibbs free energies (kJ/mol) of different conformational states were depicted using a color bar, with blue representing the lowest energy state and dark red representing the highest energy state. The FEL analysis revealed that each compound acquired the least energy, corresponding to the most stable conformations. Additionally, enriched energy minima, indicated by the blue color with ample spacing, were detected in all.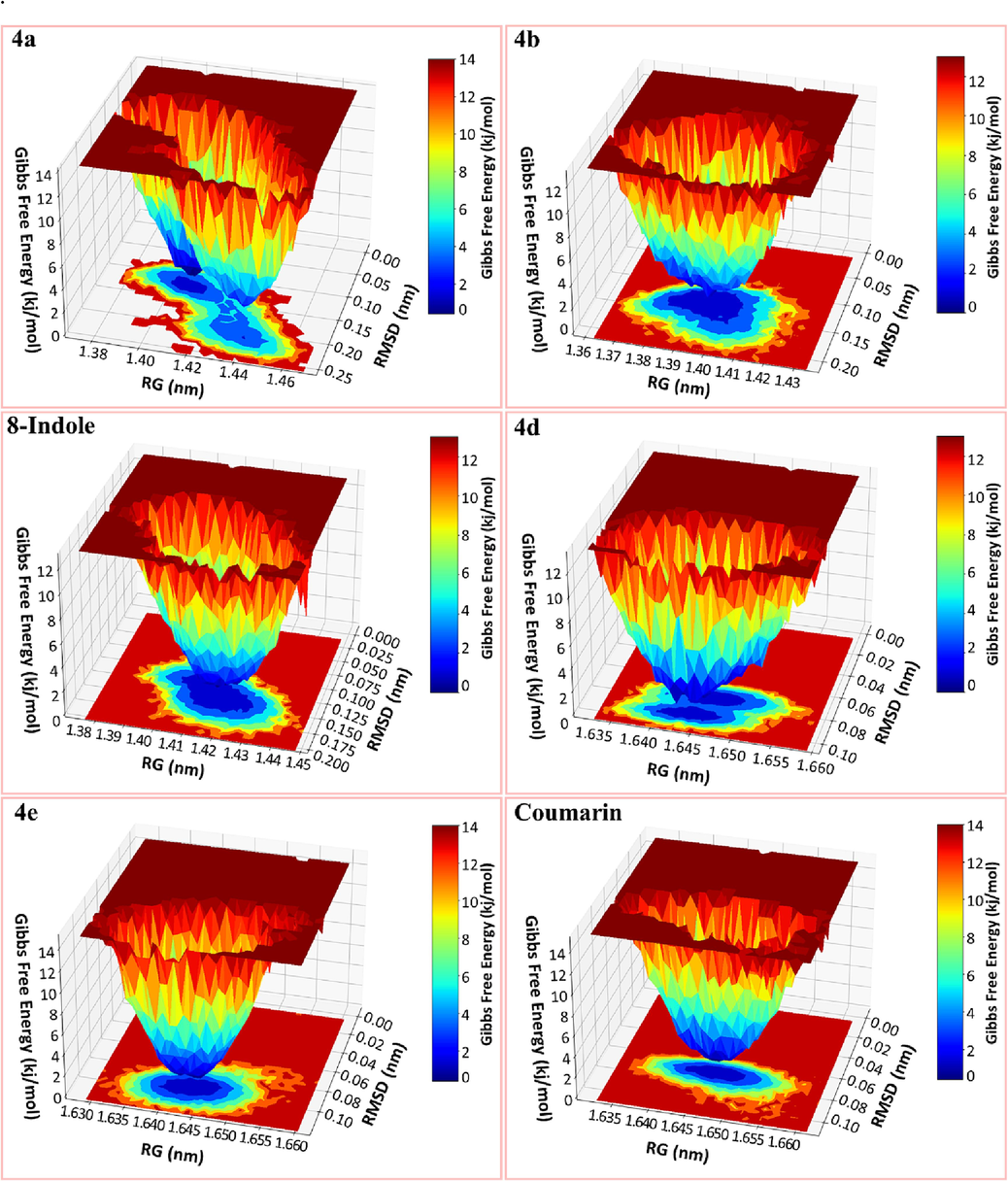
The Gibbs free energy landscape (FEL) was plotted and color-coded using Root Mean Square Deviation (RMSD) and Radius of Gyration (RoG) values.
The color bar in the diagram represents the Gibbs free energies (kJ/mol) for the different conformational states. The blue color represents the state with the lowest energy, while the red color represents the state with the highest energy. 4a, 4b and 8-indole were bound to phospholipase A2 while 4d, 4e and coumarin were bound to proteinase K.
3.2 Chemistry
3.2.1 Characterization of synthesized compounds
Synthesis of tetrahydropyrimidine-5-carbonitrile derivatives using TiO2 as photocatalyst in alcoholic medium is represented as scheme1.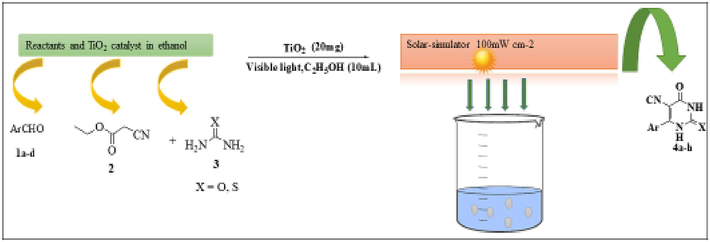
Synthesis of tetrahydropyrimidine-5-carbonitrile derivatives under the effect of TiO2 as photocatalyst in ethanolic medium.
FT-IR analysis revealed a peak at around 2210 cm−1 corresponding to −C≡N, while stretching vibrations of –NH were detected at 3200, 3300, and 3400 cm−1. Most compounds show distinctive peaks in 1H NMR spectroscopy for two NH groups, one at δ 8 ppm (downfield) and the other at 5 ppm (upfield). However, compound 4b displays both NH singlets at δ 8.4 ppm. Each compound justifies the total number of aromatic protons by their representation as a singlet, doublet, or triplet. Reading the 13CNMR readily reveals additional features. The C = C bond in the pyrimidine ring exhibits two significant peaks: one above δ 160 ppm in the downfield region and the other at δ 62–78 ppm in the most upfield region. Compounds 4a–d have 13CNMR peaks between µ 150–160 ppm because they contain a cyclic C = O group. Compounds 4a–h, on the other hand, have C = S peaks at higher chemical shifts, reaching as high as 192.36 ppm (4e) and 162.68 ppm (4 g). In 13CNMR, thiourea compounds show peaks that are farther downfield for the C = C pyrimidine ring. Compounds 4e–h also exhibit C = S peaks that extend further downfield. Furthermore, additional peaks were detected in their original positions, slightly upfield or downfield based on each compound substitution.
3.2.2 Green synthesis of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives
Harvesting of solar light using a lab solar simulator has been used to convert solar energy to chemical energy to synthesize 1, 2, 3, 4-tetrahydropyrimidine-5-carbonitrile derivatives of TiO2 nanoparticles as photocatalysts. The results confirmed that using TiO2 (20 mg) as a photocatalyst exhibits excellent performance in terms of yield and reaction time as compared to the conventional thermal method (Alharthi et al., 2023). The yield of all the synthesized compounds is as high as 91 % (compound 4a) in just 60 min. Our previous experiments yielded similar results (Alharthi et al., 2023).
3.3 Biological evaluation
3.3.1 Inhibitory activity of phospholipase A2 (PLA2)
As presented in Table 2, the 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives 4a-h have been found to exhibit phospholipase inhibitory action at concentrations ranging from 0.01 to 0.08 g/L. For compounds 4a-h, PLA2 activity was seen to increase in proportion to concentration increases. Compounds 4a to 4 h showed 37.98 %±2.66 %, 40 ± 2.54 %, 36.70 ± 1.82 %, 36.49 ± 2.67 %, 38.41 ± 3.11 %, 41.54 ± 2.22 %, 32.42 ± 2.39 %, and 38.60 %±2.78 % at the minimum concentration of 0.01 g/l, while the highest activity was shown at 92.67 %±2.98 %, 95.80 %±3.29, 90.45 %±2.27, 90.46 %±3.33, 92.40 %±3.02, 96.55 %±2.89, 88.67 %±3.66, and 92.45 %±2.74 at concentration of 0.05 g/l, as depicted Table 2.
Conc. g/l
4a
4b
4c
4d
4e
4f
4 g
4 h
0.01
37.98 %±2.66 %
40 ± 2.54 %
36.70 ± 1.82 %
36.49 ± 2.67 %
38.41 ± 3.11 %
41.54 ± 2.22 %
32.42 ± 2.39 %
38.60 %±2.78 %
0.02
42.56 %±2.41 %
42.78 %±1.69 %
41.59 %±2.83 %
37.2 %±1.23 %
42.45 %±1.92 %
45.12 %±1.59 %
34.56 %±1.53 %
41.69 %±2.44 %
0.03
62.45 %±3.06 %
65.5 %±2.22 %
60.49 %±1.88 %
59.39 %±2.02 %
61.78 %±1.12 %
63.34 %±3.09 %
59.23 %±2.78 %
60.23 %±1.42 %
0.04
77.58 %±2.77 %
78.45 %±1.96 %
75.49 %±1.82 %
73.56 %±2.60 %
76.76 %±1.28 %
79.97 %±1.13 %
72.54 %±2.69 %
76.69 %±2.62 %
0.05
92.67 %±2.98 %
95.80 %±3.29
90.45 %±2.27
90.46 %±3.33
92.40 %±3.02
96.55 %±2.89
88.67 %±3.66
92.45 %±2.74
0.06
92.39 %±2.43 %
95.78 %±3.16 %
90.57 %±2.79 %
90.39 %±2.39 %
92.40 %±1.67 %
93.67 %±2.68 %
88.45 %±1.99 %
91.23 %±2.50 %
0.07
87.1 %±2.19 %
92.79 %±2.80 %
86.29 %±2.33 %
88.33 %±3.09 %
88.49 %±2.21 %
88.23 %±1.79 %
80.23 %±2.44 %
89.59 %±1.92 %
0.08
84.87 %±2.32 %
85.79 %±3.06 %
83.79 %±1.44 %
84.35 %±1.77 %
82.86 %±2.34 %
84.64 %±3.16 %
77.39 %±2.26 %
84.42 %±1.61 %
The results are somewhat in agreement with our previous work with the same enzyme, and the PLA2 inhibitory efficiencies of the current molecules are found to be even better than those of other established moieties (El-Sayed et al., 2016; Bakht et al., 1944). Oleonolic acid, as a reference molecule, demonstrated comparatively lower potency (75.58 %±2.48 %) in comparison to compounds at maximum concentration. Given the high levels of PLA2 in many inflammatory disorders (Alafeefy et al., 2015), we will investigate these chemicals for their clinical value in these conditions.
Compounds 4a-h exhibit moderate to high PLA2 inhibitory activities, with IC50 values ranging from 0.023 to 0.031 g/L. Compounds 4b, 4d, 4e, 4f, and 4 h have the highest significant activity; their respective IC50 values are 0.031 g/L (Table 3). We will investigate these compounds in inflammatory conditions because many inflammatory conditions have elevated PLA2 levels (El-Sayed et al., 2016), potentially explaining their clinical significance.
Compounds
IC50
4a
0.023 g/L (0.028 – 0.0321, n = 3)
4b
0.031 g/L (0.028 – 0.033, n = 3)
4c
0.030 g/L (0.029 – 0.032, n = 3)
4d
0.031 g/L (0.029 – 0.033, n = 3)
4e
0.031 g/L (0.028 – 0.033, n = 3)
4f
0.031 g/L (0.028 – 0.033, n = 3)
4 g
0.029 g/L (0.027 – 0.038, n = 3)
4 h
0.031 g/L (0.030 – 0.033, n = 3)
3.3.2 Antiproteinase K activity
Table 4 shows that we screened the synthesized 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives (4-h) against the proteinase K enzyme at concentrations of 0.1 to 0.8 mg/mL. The activity of the proteinase K inhibitor was demonstrated in a dose-dependent fashion. The compounds 4c and 4d showed greater activity than the other compounds (4a, 4b, 4e, 4f, 4 g, and 4 h) at all concentrations. These poor compounds had the least proteinase K inhibitory activity at 0.1 mg/mL, with only 13 % ± 2.53 % for compound 4f and 54 % ± 1.96 % for compound 4e at 0.7 mg/mL. Compounds 4c and 4d demonstrated the highest inhibition (63 % ± 4.66 %) and (63 % ± 5.12 %) at a concentration of 0.7 mg/mL, respectively.
Conc. mg/l
4a
4b
4c
4d
4e
4f
4 g
4 h
0.1
15.3463 % ± 4.26 %
14.863 % ± 3.78 %
15.8963 % ± 4.45 %
16.4563 % ± 5.09 %
15.57 % ± 4.11 %
13 % ± 2.53 %
16.12 % ± 4.77 %
16.56 % ± 5.02 %
0.2
18.34 % ± 4.65 %
17.45 % ± 5.16 %
18.98 % ± 4.18 %
19.46 % ± 4.15 %
18.56 % ± 3.99 %
16.40 % ± 5.08 %
18.76 % ± 5.61 %
18.76 % ± 5.13 %
0.3
22.66 % ± 4.46 %
21.76 % ± 3.88 %
23.46 % ± 4.72 %
23.76 % ± 5.26 %
22.56 % ± 4.89 %
23.46 % ± 5.23 %
22.26 % ± 3.68 %
23.46 % ± 4.98 %
0.4
32.46 % ± 5.02 %
31.76 % ± 3.90 %
33.16 % ± 4.49 %
33.46 % ± 5.06 %
32.86 % ± 4.83 %
33.36 % ± 5.25 %
33.46 % ± 4.62 %
33.36 % ± 4.36 %
0.5
42.76 % ± 4.33 %
40.45 % ± 4.87 %
41.34 % ± 4.29 %
43.56 % ± 4.79 %
42.46 % ± 5.11 %
43.36 % ± 5.04 %
42.46 % ± 4.34 %
42.56 % ± 5.19 %
0.6
52.56 % ± 3.18 %
51.96 % ± 5.01 %
50.35 % ± 5.21 %
55.46 % ± 4.38 %
52.36 % ± 5.12 %
52.66 % ± 4.61 %
50.46 % ± 4.09 %
52.86 % ± 4.69 %
0.7
61.86 % ± 4.47 %
60.56 % ± 4.08 %
63 % ± 4.66 %%
63 % ± 5.12 %
54 % ± 1.96 %
61.36 % ± 4.89 %
53.96 % ± 4.17 %
61.56 % ± 4.29 %
0.8
60.36 % ± 5.11 %
58.66 % ± 4.67 %
61.26 % ± 3.88 %
61.36 % ± 4.78 %
60.46 % ± 4.44 %
60.36 % ± 4.55 %
52.26 % ± 5.42 %
60.26 % ± 4.77 %
Compound 4d showed maximal proteinase K inhibition (%) at concentrations ranging from 0.2 to 0.8 mg/mL. Compounds 4 h and 4 g reached maximum action at concentrations of 0.1 and 0.4 mg/mL, similar to compound 4d. These results are in comparison to a few prior studies that achieved proteinase K inhibition at concentrations as high as 75 % (Bakht et al., 1944).
A proteinase K inhibitor cocktail (Sigma) was used as the positive control, and the inhibitory potential was found to be close to maximum inhibition (80 ± 2.48). IC50 results for compounds 4a-h for antiproteinase K activities demonstrated a range of 0.042–0.048 mg/L.
Compounds
IC50
4a
0.046 g/L (0.044 – 0.048, n = 3)
4b
0.047 g/L (0.045 – 0.049, n = 3)
4c
0.048 g/L (0.045 – 0.051, n = 3)
4d
0.046 g/L (0.044 – 0.047, n = 3)
4e
0.046 g/L (0.045 – 0.049, n = 3)
4f
0.044 g/L (0.042 – 0.046, n = 3)
4 g
0.042 g/L (0.040 – 0.043, n = 3)
4 h
0.046 g/L (0.045 – 0.048, n = 3)
The most promising candidates in terms of antiproteinase K activity were reported with IC50 values of 0.047 and 0.048 mg/L, respectively, for compounds 4b and 4c.
3.3.3 Structure activity relationship (SAR) of studied compounds (4a-h)
We synthesized a series of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives (4-h) through a one-pot multicomponent reaction, which involved condensation of substituted aldehydes (1a–d), cynoacetate (2), and urea/thiourea (3), using TiO2 as a photocatalyst in ethanolic medium. All the compounds demonstrated better PLA2 (%) activity than proteinase K (%) activity. After establishing the structure–activity relationship (SAR) of all studied compounds, we found that compounds (4b, 4f) with electron-donating substituents exhibit good PLA2 Inhibitory (%) activity compared to those with electron withdrawing substituents (4a, 4e) or those with heterocyclic moieties (4c, 4d, 4 g, 4 h). On the other hand, compounds (4c, 4d) showed comparatively better Proteinase K inhibitory (%) activity with heterocyclic rings as compared to other electron-donating or substituent-withdrawing compounds. In general, all the compounds tested against PLA2 (%) and proteinase K (%) act in a dose-dependent manner.
4 Conclusion
To summarize, we synthesized and tested a range of 1,2,3,4-tetrahydropyrimidine-5-carbonitrile derivatives for antiphospholipase (PLA2) and proteinase K inhibitory properties. Titanium dioxide acts as an as an excellent photocatalyst and produces a high yield in least reaction times. Significant biological activity against enzymes was discovered in some of these compounds, particularly against PLA2. In the optimization of these compounds using DFT, negative values were found for the HOMO and LUMO. This suggests that the molecules possess energetic stability, a crucial factor for the formation of a robust ligand–protein complex. MD simulations confirmed the docking results and confirmed that the compounds are stable when they are combined with PLA2 and proteinase K. Additionally, the pharmacokinetic investigation indicated that the potential compounds did not exhibit any issues that would impede further studies.
Furthermore, compounds featuring electron-donating substituents demonstrated enhanced proteinase inhibitory activity (%) on PLA2, whereas those featuring heterocyclic rings demonstrated superior activity (%) on proteinase. In a futuristic study, more similar types of compounds will be tried by other photocatalysts and evaluated for antiphospholipase (PLA2) and proteinase K inhibitory effects.
Statistical Analysis: Data are expressed as mean ± standard error of the mean (n = number of experiment) and the median effective concentrations (IC50) with 95 % confidence intervals (CI). P < 0.05 is considered to be statistically significant. IC50 values were calculated using GraphPad prism4 program (GraphPad, San Diego, CA, USA).
CRediT authorship contribution statement
Md. Afroz Bakht: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. Imtiaz Ali: Data curation, Formal analysis, Investigation, Resources, Writing – review & editing. Gagandeep Singh: Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Acknowledgments
The author extends his appreciation to the Deputyship for Research & Innovation, Ministry of Education in Saudi Arabia for funding this research work through project number (2022/02/21704).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1–2:19-25.
- [CrossRef] [Google Scholar]
- I. Ahmad, V. Jagatap, H. Patel, Application of density functional theory (DFT) and response surface methodology (RSM) in drug discovery, in: C. Egbuna, M. Rudrapal, H.B.T.-P. Tijjani Computational Tools and Databases in Drug Discovery (Eds.), Phytochem. Comput. Tools Databases Drug Discov., Elsevier, 2023: pp. 371–392. https://doi.org/10.1016/B978-0-323-90593-0.00004-6.
- Synthesis and biological evaluation of certain 3-substituted benzylideneamino-2-(4-nitrophenyl) quinazolin-4 (3 H)-one derivatives. J. Enzyme Inhib. Med. Chem.. 2015;30:270-276.
- [Google Scholar]
- Photocatalytic-driven three-component synthesis of 1, 2, 3, 4-tetrahydropyrimidine-5-carbonitrile derivatives: A comparative study of organocatalysts and photocatalysts. J. Photochem. Photobiol. A Chem.. 2023;436:114358
- [Google Scholar]
- Synthesis, antimicrobial, anticancer, PASS, molecular docking, molecular dynamic simulations & pharmacokinetic predictions of some methyl β-D-galactopyranoside analogs. Molecules. 2021;26:7016.
- [Google Scholar]
- Methyl β–D–galactopyranoside esters as potential inhibitors for SARS–CoV–2 proteinase K enzyme: synthesis, antimicrobial, PASS, molecular docking, molecular dynamics simulations and quantum computations. Glycoconj. J.. 2022;39:261-290.
- [Google Scholar]
- NSAIDs as potential treatment option for preventing amyloid β toxicity in Alzheimer’s disease: An investigation by docking, molecular dynamics, and DFT studies. J. Biomol. Struct. Dyn.. 2018;36:2099-2117.
- [Google Scholar]
- In silico and in vitro studies on the inhibition of laccase activity by Ellagic acid: Implications in drug designing for the treatment of Cryptococcal infections. Int. J. Biol. Macromol.. 2022;209:642-654.
- [Google Scholar]
- Synthesis and biological evaluation of octahydroquinazolinones as phospholipase A2, and proteinase K inhibitors: Experimental and theoretical exploration. Molecules. 1944;2023:28.
- [Google Scholar]
- Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098-3100.
- [CrossRef] [Google Scholar]
- Vanillic acid as phospholipase A2 and proteinase Ks inhibitor: In vitro and computational analyses. Biotech. Appl. Biochem.. 2021;68:486-496.
- [Google Scholar]
- Particle mesh Ewald: An N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys.. 1993;98:10089-10092.
- [Google Scholar]
- Determination of phospholipase A2 activity by a colorimetric assay using a pH indicator. Toxicon. 1987;25:1181-1188.
- [Google Scholar]
- Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev.. 2011;111:6130-6185.
- [Google Scholar]
- Synthesis, antiphospholipase A2, antiproteinase K, antibacterial evaluation and molecular docking analysis of certain novel hydrazones. Molecules. 2016;1664:1-17.
- [Google Scholar]
- Exploring ligand stability in protein crystal structures using binding pose metadynamics. J. Chem. Inf. Model.. 2020;60:1528-1539.
- [CrossRef] [Google Scholar]
- Aladahalli S. Giresha , Deepadarshan Urs , J.G. Manjunatha , P. Sophiya , B. H. Supreetha , Shankar Jayarama & K. K. Dharmappa, Group IIA secreted phospholipase A2 inhibition by elemolic acid as a function of anti–infammatory activity, Sci.Rep, 12(2022) 7649.
- Synthetic approaches and functionalizations of imidazo [1, 2-a] pyrimidines: an overview of the decade. RSC Adv.. 2015;5:81608-81637.
- [Google Scholar]
- Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform.. 2012;4:17.
- [CrossRef] [Google Scholar]
- LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem.. 1997;18:1463-1472.
- [CrossRef] [Google Scholar]
- Synthesis, antimicrobial, molecular docking and molecular dynamics studies of lauroyl thymidine analogs against SARS-CoV-2: POM study and identification of the pharmacophore sites. Bio. Chem.. 2022;125:105850
- [Google Scholar]
- Molecular dynamics simulations: advances and applications. Adv. Appl. Bioinform. Chem.. 2015;8:37-47.
- [CrossRef] [Google Scholar]
- Efficient rapid access to Biginelli for the multicomponent synthesis of 1, 2, 3, 4-tetrahydropyrimidines in room-temperature diisopropyl ethyl ammonium acetate. ACS Omega. 2019;4:22313-22324.
- [Google Scholar]
- A comprehensive review on pyrimidine analogs-versatile scaffold with medicinal and biological potential. J. Mol. Struct.. 2021;1246:131168
- [CrossRef] [Google Scholar]
- CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem.. 2008;29:1859-1865.
- [CrossRef] [Google Scholar]
- L.P. Kagami, G.M. das Neves, A.W.S. da Silva, R.A. Caceres, D.F. Kawano, V.L. Eifler-Lima, LiGRO: a graphical user interface for protein-ligand molecular dynamics., J. Mol. Model. 23 (2017) 304. https://doi.org/10.1007/s00894-017-3475-9.
- L.P. Kagami, G.M. das Neves, L.F.S.M. Timmers, R.A. Caceres, V.L. Eifler-Lima, Geo-Measures: A PyMOL plugin for protein structure ensembles analysis., Comput. Biol. Chem. 87 (2020) 107322. https://doi.org/10.1016/j.compbiolchem.2020.107322.
- An efficient and rapid dehydrogenation of 4-aryl-3, 4-dihydropyrimidin-2 (1H)-ones (DHPMs) using CAN/HCl. Tetrahedron Lett.. 2012;53:5541-5543.
- [Google Scholar]
- Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol.. 2002;9:646.
- [Google Scholar]
- Chemical descriptors, PASS, molecular docking, molecular dynamics and ADMET predictions of glucopyranoside derivatives as inhibitors to bacteria and fungi growth. Org. Commun.. 2022;15:184-203.
- [Google Scholar]
- 2-Oxoesters: A novel class of potent and selective inhibitors of cytosolic group IVA phospholipase A2. Sci. Rep.. 2017;7:7025.
- [Google Scholar]
- CHARMM-GUI supports the Amber force fields. J. Chem. Phys.. 2020;153:35103.
- [CrossRef] [Google Scholar]
- Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785-789.
- [CrossRef] [Google Scholar]
- Radius of gyration as an indicator of protein structure compactness. Mol. Biol.. 2008;42:623-628.
- [Google Scholar]
- Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J. Mol. Biol.. 1994;235:625-634.
- [CrossRef] [Google Scholar]
- An efficient synthesis of 3, 4-dihydropyrimidin-2 (1H)-ones catalyzed by thiamine hydrochloride in water under ultrasound irradiation. Tetrahedron Lett.. 2010;51:3138-3140.
- [Google Scholar]
- Synthesis, characterization, synergistic antimicrobial properties and molecular docking of sugar modified uridine derivatives. Ovidius Univ. Ann. Chem.. 2021;32:6-21.
- [Google Scholar]
- Synthesis of dihydropyrimidinones via Biginelli multi-component reaction. Tetrahedron Lett.. 2012;53:1543-1545.
- [Google Scholar]
- Software update: the ORCA program system, version 4.0. WIREs Comput. Mol. Sci.. 2018;8:e1327.
- [CrossRef] [Google Scholar]
- M. Oh, S. Y. Jang, Ji-Yoon. Lee, J. W. Kim, Y. Jung, J. Kim, J. Seo, Tae-Su. Han, E. Jang , H. Y. Son, D. Kim, M. W. Kim , Jin-Sung. Park, Kwon-Ho. Song, Kyoung-Jin. Oh, W. K. Kim, Kwang-Hee. Bae, Yong-Min. Huh, S. H. Kim, D. Kim, Baek-Soo. Han, S. C. Lee, Geum-Sook. Hwang, Eun-Woo. Lee, The lipoprotein-associated phospholipase A2 inhibitor Darapladib sensitises cancer cells to ferroptosis by remodelling lipid metabolism, Nat. Commun, 14(2023) 5728.
- Effectiveness of estrogen and its derivatives over dexamethasone in the treatment of COVID-19. J. Biomol. Struct. Dyn. 2023:1-17.
- [CrossRef] [Google Scholar]
- Recent medicinal approaches of novel pyrimidine analogs: A review. Heliyon. 2023;9:e16773.
- [Google Scholar]
- Highly potent 2-oxoester inhibitors of cytosolic phospholipase A2 (GIVA cPLA2) ACS Omega. 2018;3:8843-8853.
- [Google Scholar]
- The use of global and local reactivity descriptors of conceptual DFT to describe toxicity of benzoic acid derivatives. Comput. Theor. Chem.. 2023;1226:114211
- [CrossRef] [Google Scholar]
- Green on-water multicomponent approach for the synthesis of pyrrolo [2, 3-d] pyrimidines. Tetrahedron Lett.. 2021;81:153336
- [Google Scholar]
- Discovery of putative natural compounds inhibitor of the germinant spore receptor CspC in Clostridioides difficile infection: Gaining insights via In silico and bioinformatics approach. Informatics Med. Unlocked.. 2023;42:101339
- [CrossRef] [Google Scholar]
- Identification of potential inhibitors of HER2 targeting breast cancer—A structure-based drug design approach. J. Biomol. Struct. Dyn. 2023:1-18.
- [CrossRef] [Google Scholar]
- Recent advances of visible-light photocatalysis in the functionalization of organic compounds. J. Photochem. Photobiol C: Photochem. Rev. 2022;50:100488
- [Google Scholar]
- Identification of a dual inhibitor of secreted phospholipase A2 (GIIA sPLA2) and SARS-CoV-2 main proteinase K. Pharmaceuticals. 2022;15:961.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- An approach to developing a force field for molecular simulation of martensitic phase transitions between phases with subtle differences in energy and structure. J. Am. Chem. Soc.. 2004;126:396-405.
- [Google Scholar]
- Development of a potent 2-oxoamide inhibitor of secreted phospholipase A2 guided by molecular docking calculations and molecular dynamics simulations. Bioorg. Med. Chem.. 2016;24:1683-1695.
- [Google Scholar]
- Synthesis and mechanism of novel fluorescent coumarin–dihydropyrimidinone dyads obtained by the Biginelli multicomponent reaction. New J. Chem.. 2015;39:2323-2332.
- [Google Scholar]
- Synthesis of pyrimidine derivatives via multicomponent reaction catalyzed by ferric chloride. Appl. Organomet. Chem.. 2020;34:e5921.
- [Google Scholar]
- monosaccharide derivatives: synthesis, antimicrobial, pass, antiviral and molecular docking studies against sars-cov-2 mpro inhibitors, Cellulose. Chem. Technol.. 2021;55:477-499.
- [Google Scholar]
- Applications of TiO2 nanotube arrays in environmental and energy fields: A review. Micropor. Mesopor. Mater.. 2015;202:22-35.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105798.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary Data 1
Supplementary Data 1




