

In silico and docking studies on the binding activities of Keap1 of antioxidant compounds in non-oilseed legumes
⁎Corresponding author at: Department of Food Science and Biotechnology, Graduate School, Kyungpook National University, Daegu 41566, Korea. sang@knu.ac.kr (Sang-Han Lee)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We used in silico methods to predict the physiochemical and pharmacological characteristics, toxicity, and biological activities of the screened compounds. All compounds showed positive results while calculating their physiochemical and pharmacokinetic descriptors. Using the Prediction of Activity Spectra for Substances (PASS) software on compounds form non-oilseed legumes, we identified compounds (mainly 4 polyphenol compounds) with anti-infective, anti-eczematic, antimutagenic, muco-membranous protector, fibrinolytic, anticarcinogenic, hepato-protectant, cardio-protectant, antioxidant, and astringent effect. PASS predicted HO-1 expression enhancing and free radical scavenging properties for gallic acid, coumaric acid, catechin, and epicatechin. Data about validation protocols for molecular docking of ligand IVV to Keap1 was performed by root mean square deviation (RMSD) value is used to validate docking protocol and representation mainly for analyzing stability of protein and predicting conformational changes of protein. Molecular docking is a powerful technique for studies of receptor-ligand interaction and has led to the discovery of Keap1-Nrf2 small molecule inhibitors. Keap1 inhibits the degradation of Nrf2. Our results suggest that screened compounds from non-oilseed legumes can effectively interact with the Keap1 binding site and dissociate Keap1 and Nrf2. The screened compounds from non-oilseed legumes that displayed high binding affinities with Keap1 are promising new Nrf2 activators. We performed molecular docking to identify the molecular interactions of gallic acid, catechin, and epicatechin with Keap1. Non-oilseed legumes plant is a natural source of potent antioxidant that may prevent diseases and could be potentially used as functional food, nutraceuticals, and drugs.
Keywords
In silico
Keap1
Molecular docking
Nrf2
Non-oilseed legumes

1 Introduction
Phytochemicals are biologically active natural compounds with nutritional and therapeutic properties. Polyphenolic compounds are a group of phytochemicals with at least one hydroxylated benzene ring. The compounds in this wide and dissimilar group are generally categorized by their number of carbon atoms. Phenolic acids are a notable subgroup of phenolic compounds with either a C6–C1 (hydroxybenzoic acids) or a C6–C3 (hydroxycinnamic acids) skeleton, composed of a phenolic ring and a carboxyl substituent. Simple phenols, phenolic acids, coumarins, cinnamic acid derivatives, flavonoids, chalcones, anthocyanins, betacyanin, xanthones, benzophenones, lignin, lignans, quinone, and tannins are the main subgroups of natural phenolic compounds (Pengelly, 2004). Most healthy foodstuff contains phenolic compounds including legumes. Non-oilseed legumes compounds have identified such as gallic acid, coumaric acid, catechin and epicatechin (Diniyah et al., 2020a, 2020b) and they contribute to health benefits mainly through their antioxidative properties (Huang et al., 2019; Feng et al., 2018; Nakano et al., 2018; Niu et al., 2017).
Currently in silico methods attract attention because they can potentially replace some animal research, thus minimizing and ethical concerns. These methods can characterize and predict human and environmental toxicity (Raji et al., 2017; Hartung & Hoffmann, 2009). In silico tools also help to combine various up-to-date computational and experimental approaches more efficiently than a battery of laboratory experimental analyses (Devi et al., 2015). Medicinal chemists use in silico techniques such as virtual screening to quickly and efficiently evaluate the pharmacological behavior and receptor interactions of compounds (Goel et al., 2011). Besides structure–activity relationship analysis, in silico methods can also predict the properties of new structures or compounds based on databases of experimentally determined toxicity data. Progress in computer-supported modeling has resulted in a better understanding of the molecular mechanisms and toxicity of phytochemicals and their metabolites (Boobis et al., 2002; Nigsch et al., 2009).
The literature shows that molecular docking has been crucial to study receptor-ligand interactions in the inhibition of enzymes related to antioxidant activity. This technique has clarified the possible active region of the receptor, which amino acid residues are involved in the interactions, and which atoms directly interact with the ligand (Gupta et al., 2018). Molecular docking has helped in the elucidation of the antioxidative mechanism of some compounds. Their antioxidant activity was determined using biological tests, and the AutoDock 4.0 program characterized the ligand-receptor interaction by calculating the binding free energy and inhibition constant (Ki) (Bandari et al., 2017). A molecular docking study with AutoDock 4.2.6 evaluated the antioxidant activity of a novel structural class of PPARα/γ receptor ligands (Niu et al., 2017).
The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) plays an essential role in regulating a series of phase II detoxification enzymes and non-enzymes antioxidants (Duan et al., 2016). Normally, the kelch-like ECH-associated protein 1 (Keap1) binds to Nrf2, retains it in the cytoplasm, and prevents its degradation. Keap1 acts as a substrate adaptor that negatively regulates Nrf2 activity under physiological conditions (Ji et al., 2015). Based on the scavenging ability of antioxidant compounds, we predicted the physiochemical, pharmacokinetic, and toxic properties of antioxidant compounds from non-oilseed legumes extract previously identified in our laboratory. Besides, we carried out chemometric analyses to establish a relationship between the molecular and electronic properties of these compounds and their antioxidant activity.
2 Materials and methods
2.1 Protein, ligand, and screened compounds selection and preparation
We obtained the protein (the crystal of Keap1), ligand (IVV), and screened compounds from the National Center for Biotechnology Information (Sayers et al., 2012) and PubMed (Shultz, 2007) databases, and the 3D structures from the Protein Data Bank (PDB code: 4L7B). We retrieved the 3D structures of the screened compounds of non-oil seed legumes (gallic acid, coumaric acid, catechin and epicatechin as shown in Fig. 1) from the PubChem database, in SDF (structure data file) format. (Kim et al., 2016). We then prepared the chemical compounds using the default parameters of the Ligprep module in the Schrödinger suite (LigPrep, 2018).
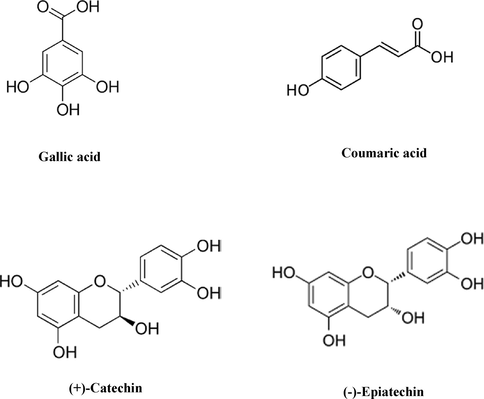
- Chemical structure of screened compounds of non-oilseed legumes.
2.2 In silico prediction of the physiochemical and pharmacokinetic parameters, and toxic risk
To estimate the physiochemical and pharmacokinetic parameters, possible metabolism, and toxicity risks (mutagenicity, carcinogenicity, cardiotoxicity, skin irritation, and hepatotoxicity) of the screen compounds, we conducted a computational simulation study on the SwissAdme (Daina et al., 2017), AdmetSAR and pkCSM (Pires et al., 2015) webservers. We used PASS-online for biological or pharmacological activities spectrum prediction. Prediction results were expressed in percentage of probable activity (Pa) and probable inactivity (Pi). Pa and Pi values vary from 0.000 to 1.000, thus, here, we considered only activities with Pa > Pi and Pa > 0.700. We also checked Lipinski’s rule of five, which are often used in rational drug design, to estimate the bioavailability of these ligands. We used simplified molecular data input format (SMILES) and SDF file formats to check the properties of the ligands.
2.3 Molecular docking study
To explore the possible binding mode of Keap1 ligand IVV and the ability of gallic acid, coumaric acid, catechin, and epicatechin to inhibit this interaction, we performed molecular docking simulations using AutoDock Vina (Trott and Olson, 2009) through the DockingApp’s interface (Di Muzio et al., 2017). All the water molecules and ligands were removed from the initial structure of Keap1 and polar hydrogen atoms were added before docking. The grid box was centered for each target on the native binding site. The grid box size was set to 16 Å × 16 Å × 16 Å with its center at position × = − 2.4, y = 2.8 and z = − 29.21. ΔG values, Receptor-ligand interaction data (binding affinity BA), and inhibition constant (Ki) values regarding the inhibition of these receptors were obtained using the AutoDock 4.2.6 and Vina programs (Morris et al., 1998), respectively, based on a standard protocol established by our research group for each analyzed receptor (Cruz et al., 2018; dos Santos et al., 2018; Silva et al., 2019). The figures were generated using Pymol and discovery studio.
3 Results
Based on previous data, we hypothesized that phenolic compounds such as flavonoids could be the primary constituents in non-oilseed legumes since polyphenols and flavonoids normally show a very strong antioxidant potential. Moreover, we used in silico simulation to predict the physiochemical and pharmacological characteristics, biological activities, and toxic risk of the selected compounds. Drug candidates need to meet certain criteria. We predicted the physiochemical, pharmacokinetic, toxic, and pharmacological properties of the compounds obtained from the docking simulation study. We then used the pkCSM, admetSAR and SwissAdme webservers to determine the toxicity profiles of the compounds by evaluating various physiochemical and pharmacokinetic (Table 1), and toxicity properties (Table 2). Furthermore, we evaluated the in-silico toxicity of constituents of non-oilseed legume and found a very low toxicity probability.
| Parameter | A1 | A2 | A3 | A4 |
|---|---|---|---|---|
| Molecular weight | 170.12 g/mol | 164.16 g/mol | 290.27 g/mol | 456.40 g/mol |
| Num. heavy atoms | 12 | 12 | 21 | 33 |
| Num. arom. heavy atoms | 6 | 6 | 12 | 18 |
| Num. rotatable bonds | 1 | 2 | 1 | 5 |
| Num. H-bond acceptors | 5 | 3 | 6 | 10 |
| Num. H-bond donors | 4 | 2 | 5 | 6 |
| Water solubility | −2.56 | −2.38 | −3.12 | −2.94 |
| Class solubility | Very soluble | Soluble | Soluble | Moderately |
| Bioavailability | 0.56 | 0.85 | 0.55 | 0.55 |
| Log Po/w (XLOGP3) | 0.70 | 1.46 | 0.36 | 1.85 |
| CaCO2 permeability | −0.081 | 1.21 | −0.283 | −0.277 |
| Total clearance | 0.518 | 0.662 | 0.183 | −0.132 |
| BBB permeability | −1.102 | −0.225 | −1.054 | −1.787 |
| CNS permeability | −3.74 | −2.418 | −3.298 | −3.721 |
| Intestinal absorption | 43.374 | 93.494 | 68.829 | 80.844 |
| CYP2D6 substrate | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No |
| Skin permeability | −2.735 | −2.715 | −2.735 | −2.735 |
A1: gallic acid; A2: coumaric acid; A3: catechin; A4: epicatechin.
Web server: pkCSM ( https://biosig.unimelb.edu.au/pkcsm .), admetSAR ( https://lmmd.ecust.edu.cn/admetsar2) and SwissAdme ( https://www.swissadme.ch/).
| Parameter | Gallic acid | Coumaric acid | Catechin | Epicatechin |
|---|---|---|---|---|
| Renal OCT2 substrate | No | No | No | No |
| Hepatotoxicity | No | No | No | No |
| Carcinogenicity (binary) | No | No | No | No |
| Mutagenecity | No | No | No | No |
| Cardiotoxicity | No | No | No | No |
| Skin toxicity | No | No | No | No |
A1: gallic acid; A2: coumaric acid; A3: catechin; A4: epicatechin.
Web server: pkCSM ( https://biosig.unimelb.edu.au/pkcsm .), admetSAR ( https://lmmd.ecust.edu.cn/admetsar2) and SwissAdme ( https://www.swissadme.ch/).
The physiochemical and pharmacokinetic descriptors of all the compounds were positive (Table 1). As described in Table 1, all calculated descriptors were within the satisfactory range, with properties including, QPlogPo/w (octanol/water partition coefficient), QPlogS (aqueous solubility) (Jorgensen and Duffy, 2002, 2000), molecular weight, and number of H bond acceptors and donors (Cavalli et al., 2002). All four screened compounds showed (Table 2) no inhibitory activity and positive results toward Renal OCT2 substrate, mutagenicity, hepatotoxicity, carcinogenicity, skin toxicity (Martin, 2005), percentage of human intestinal absorption (Pires et al., 2015) and possible metabolism through the CYP family (Hollenberg, 2002). A1, A3 and A4 were unable to penetrate the CNS (log PS ← 3 < − 3) (Pires et al., 2015).
In our study, PubChem CID: 370 [IUPAC name: 3,435-trihydroxybenzoic acid], CID: 637,542 [IUPAC name: (E)-3(4-hydroxyphenyl) prop-2-enoic acid], CID: 72,276 [IUPAC name: (2R,3R)-2-(3,4-dihydroxyphenyl)-3,4-dihydro-2H-chromene-3,5,7-triol], CID: 65,064 [IUPAC name: (2r,3r)-5,7-dihydroxy-2(3,4,5-trihydroxyphenil)-3,4-dihydro-2H-chromen-3-yl]3,4,5-trihydroxybenzoate] were denoted as A1, A2, A3, and A4, respectively.
Moreover, we performed the in-silico simulation to predict the pharmacological properties of the screened compounds (Table 3). PASS identified compounds that could have antiseptic, anti-infective, anti-eczematic, antimutagenic, muco-membranous protective, fibrinolytic, anticarcinogenic, hepato-protective, cardio-protective, antioxidant, and astringent effects. This supports the potential use of non-oilseed legumes for diabetes and metabolic syndromes. Additionally, PASS predicted HO-1 expression enhancing and free radical scavenging properties for gallic acid, coumaric acid, catechin, and epicatechin (probable activity, Pa > 0.7).
| Phytoconstituents | Main Predicted Properties by PASS-Online | Probable activity (Pa#) | Probable inactivity (Pi#) |
|---|---|---|---|
| Gallic acid | Antiseptic Superoxide dismutase inhibitor Antieczematic Antiinfective Astringent HO-1 expression enhancer Oxidoreductase inhibitor |
0.910 0.898 0.855 0.828 0.812 0.732 0.717 |
0.003 0.004 0.009 0.001 0,005 0.005 0.013 |
| Coumaric acid | Antimutagenic HO-1 expression enhancer Fibrinolytic Antiseptic Anti-eczematic |
0.886 0.783 0.749 0.729 0.707 |
0.002 0.004 0.009 0.005 0.043 |
| Catechin | Muco-membranous protector HO-1 expression enhancer Lipid peroxidase inhibitor Free radical scavenger Antioxidant Anticarcinogenic |
0.962 0.939 0.888 0.842 0.810 0.795 |
0.003 0.002 0.002 0.002 0.003 0.005 |
| Epicatechin | HO-1 expression enhancer Free radical scavenger Fibrinolytic Antimutagenic Anticarcinogenic Hepato-protectant Astringent Cardio-protectant Anti-eczematic Antioxidant |
0.982 0.960 0.947 0.934 0.861 0.840 0.838 0.837 0.750 0.739 |
0.001 0.001 0.002 0.002 0.004 0.003 0.001 0.003 0.030 0.004 |
Using PASS-online web server: https://www.pharmaexpert.ru/passonline/.
4 Discussion
Fig. 2A–C shows the validation protocols data for the molecular docking of ligand IVV to Keap1 in a three-dimensional ribbon binding map. Generally, the root mean square deviation (RMSD) value is used to validate docking protocols and when analyzing protein stability and predicting conformational changes. The RMSD value depends on the binding interaction and energy between the protein and its ligand (Durairaj, 2015). According to the literature, the RMSD values expressing the relationship between the calculated X-ray crystallographic data of the complexed ligand and the simulated result must be less than 2.0 Å (da Silva et al., 2018; Hevener et al., 2009). Low RMSD values reflect low variation and would be acceptable. Our RMSD value is low (1.85 Å) and attests that the protocol we used can be applied in molecular docking analyses.
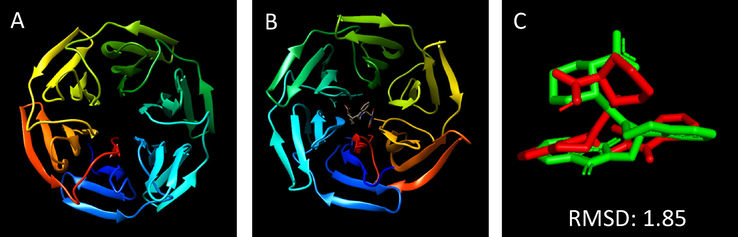
-
Comparison of molecular docking by Keap1 and its ligand IVV. (A) Crystal structure of Keap1 (PDB: 4L7B). (B) Crystal structure of Keap1 (PDB: 4L7B) with the original ligand IVV; (C) IVV superimposed structure.
Fig. 3 shows the binding site of IVV within Keap1. The degree of binding of the ligand with the protein refers to the binding affinity. The energy released due to the bond formation, or rather, interaction of the ligand and protein is termed in form of binding energy. The free energy of the favorable reaction is negative (Asthana, 2014). The affinity binding energy of small molecular ligand IVV to Keap1 was predicted to be − 9.2 kcal/mol and we used this value to classify the best poses obtained in the molecular docking analyses. Only the smallest affinity binding energy values for the best poses are shown (Fig. 3). The lower the affinity binding energy, the more significant the interaction between the receptor and the ligand (da Silva et al., 2018). Interestingly, the calculated affinity binding energy between ligand IVV, Keap1 and gallic acid, coumaric acid, catechin, and epicatechin were − 6.2, −5.8, −8.4, and − 8.5 kcal/mol, respectively, which is higher than the binding affinity of IVV and Keap1 (−9.2 kcal/mol). It is possible to verify the tendency of the binding affinity value to decrease by increasing the number of interactions (Da Silva et al., 2018). The molecular docking results of these interactions are shown in Fig. 4.
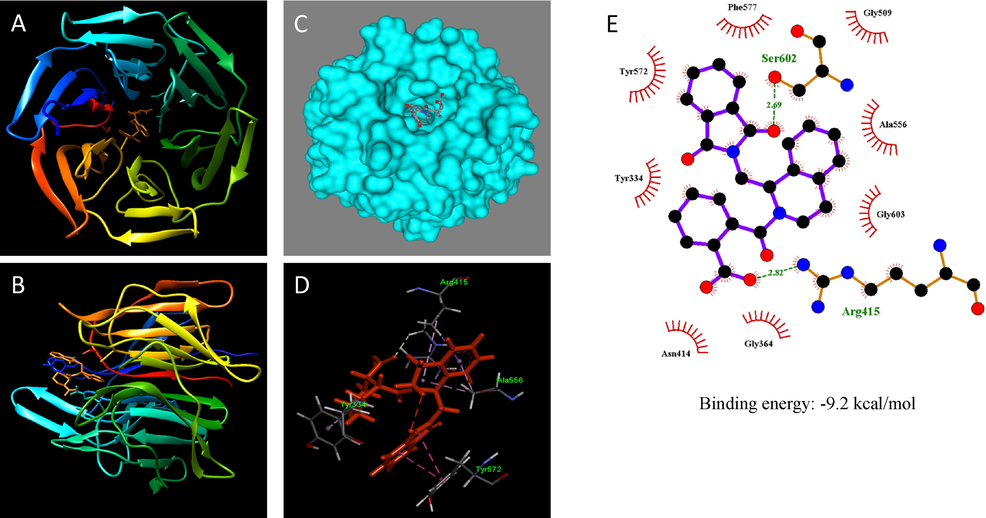
-
Molecular docking results of IVV with Keap1. Crystal structure of Keap1 with the original ligand IVV (orange) (PDB: 4L7B). (A) Front view; (B) Side view; (C) Surface view; (D) 3D view (E) 2D representation of the interactions.

-
Comparison of binding residues of selected polyphenol compounds in non-oilseed legumes with Keap1. Gallic acid (A), coumaric acid (B), catechin (C), and epicatechin (D) with Keap1 are shown on their binding residues. Crystal structure of Keap1 (PDB: 4L7B) and binding interaction with gallic acid (pink), coumaric acid (yellow), catechin (red), and epicatechin (green). (i) Front view; (ii) Side view; (iii) Surface interaction; (iv) 3D view (v) 2D representation of the interactions.
Molecular docking is a very powerful technique for research of receptor-ligand interaction inhibition of enzymes related to antioxidant activity of compounds and has allowed the discovery of Keap1-Nrf2 small molecule inhibitors (Li et al., 2020), ERK-Nrf2-Keap1-mediated antioxidative response inhibition (Feng et al., 2018). To investigate whether our screened compounds could directly inhibit the protein–protein interaction between Keap1 and Nrf2, we assessed their ability to bind to the Keap1 kelch domain using AutoDock Vina molecular docking software. The Kelch domain of Keap1 is considered as the Nrf2 peptide binding site, and several reported inhibitors that directly bind to this site interrupt Nrf2-Keap1 interaction, promoting Nrf2 nuclear translocation (Pang et al., 2016). Thus, we assumed that our screened compounds-induced Nrf2 activation might be also associated with direct binding to the Kelch domain of Keap1. Keap1 is considered as an inhibitor for degrading Nrf2. Our results showed that four phytochemicals can effectively interact with the Keap1 binding site and dissociate Keap1 and Nrf2.
The Keap1-Nrf2/ARE signaling pathway is an important defense system against exogenous and endogenous oxidative stress injury. It is a powerful oxidation–reduction system and is also an isobiotic sensitive signaling pathway which protects cells from injury and death (Chirumbolo and Bjørklund, 2018). In absence of stress, Keap1 is an E3 ubiquitin ligase substrate adaptor that targets Nrf2 for rapid proteasomal degradation, which limits the cytoplasmic concentration of Nrf2. Gallic acid, coumaric acid, catechin, and epicatechin bind to Keap1, disturbing its interaction with IVV (Fig. 4). Competing with the Nrf2 binding site of Keap1 is an alternative pathway for the regulation of Nrf2 activation. Our molecular docking results indicated that gallic acid, coumaric acid, catechin, and epicatechin could stabilize Keap1. Our results suggest that nuclear translocation of Nrf2 promoted by gallic acid, coumaric acid, catechin, and epicatechin might be caused by the fact that they bind to Keap1 and disturb protein–protein interaction between Keap1 and Nrf2. Besides, gallic acid disturbs protein–protein interaction between Keap1 and Nrf2 which might also contribute to the translocation of Nrf2 (Feng et al., 2018). Li et al., (2020) found that phytochemicals that had high binding affinity with Keap1 are promising new Nrf2 activators.
Funding
This study is supported by the National Research Foundation of Korea (NRF) grant, funded by the Ministry of Science and ICT (2020R1A2C2011495 and 2021R1I1A1A058062).
Author contributions
N.D., M.B.A., and S.-H.L. performed the experiments. N.D., A.J., F.H.A., and H.-J.C., literature search, designed the research and analyzed the data. N.D. and S.-H.L. wrote the paper. N.D., M.B.A. and S.-H.L. revised the paper.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Synthesis of new chromeno-carbamodithioate derivatives and preliminary evaluation of their antioxidant activity and molecular docking studies. Bioorganic Med. Chem. Lett.. 2017;27:1256-1260.
- [CrossRef] [Google Scholar]
- In silico prediction of ADME and pharmacokinetics: Report of an expert meeting organised by COST B15. Eur. J. Pharm. Sci.. 2002;17:183-193.
- [CrossRef] [Google Scholar]
- Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K+ channel blockers. J. Med. Chem.. 2002;45:3844-3853.
- [CrossRef] [Google Scholar]
- Sulforaphane and 5-fluorouracil synergistically inducing autophagy in breast cancer: A possible role for the Nrf2-Keap1-ARE signaling? Food Chem. Toxicol.. 2018;112:414-415.
- [CrossRef] [Google Scholar]
- Computational design of new protein kinase 2 inhibitors for the treatment of inflammatory diseases using QSAR, pharmacophore-structure-based virtual screening, and molecular dynamics. J. Mol. Model.. 2018;24:225.
- [Google Scholar]
- da Silva Costa, J., da Silva Lopes Costa, K., Cruz, J.V., da Silva Ramos, R., Silva, L.B., Do Socorro Barros Brasil, D., de Paula da Silva, C.H.T., dos Santos, C.B.R., da Cruz Macedo, W.J., 2018. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 24, 576–594. https://doi.org/10.2174/1381612823666170711112510
- Da Silva Costa, J., Da Silva Ramos, R., Da Silva Lopes Costa, K., Do Socorro Barros Brasil, D., De Paula Da Silva, C.H.T., Ferreira, E.F.B., Dos Santos Borges, R., Campos, J.M., Da Cruz Macêdo, W.J., Dos Santos, C.B.R., 2018. An in silico study of the antioxidant ability for two caffeine analogs using molecular docking and quantum chemical methods. Molecules 23. https://doi.org/10.3390/molecules23112801.
- SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:1-13.
- [CrossRef] [Google Scholar]
- In silico approach of antibacterial compounds from mangrove - Avicennia marina through docking analysis. Biomed. Res. An Int. J. Med. Sci.. 2015;25:S52-S54.
- [Google Scholar]
- DockingAPP: a user friendly interface for facilitated docking simulations with AutoDock Vina. J. Comput. Aided. Mol. Des.. 2017;31:213-218.
- [Google Scholar]
- Lablab purpureus protects hacat cells from oxidative stress-induced cell death through nrf2-mediated heme oxygenase-1 expression via the activation of p38 and erk1/2. Int. J. Mol. Sci.. 2020;21:1-16.
- [CrossRef] [Google Scholar]
- Antioxidant potential of non-oil seed legumes of Indonesian’s ethnobotanical extracts. Arab. J. Chem. 2020
- [CrossRef] [Google Scholar]
- Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol.. 2018;222:107-120.
- [CrossRef] [Google Scholar]
- Antioxidant tert-butylhydroquinone ameliorates arsenic-induced intracellular damages and apoptosis through induction of Nrf2-dependent antioxidant responses as well as stabilization of anti-apoptotic factor Bcl-2 in human keratinocytes. Free Radic. Biol. Med.. 2016;94:74-87.
- [CrossRef] [Google Scholar]
- Gallic acid, a natural polyphenol, protects against tert-butyl hydroperoxide- induced hepatotoxicity by activating ERK-Nrf2-Keap1-mediated antioxidative response. Food Chem. Toxicol.. 2018;119:479-488.
- [CrossRef] [Google Scholar]
- PASS-assisted exploration of new therapeutic potential of natural products. Med. Chem. Res.. 2011;20:1509-1514.
- [CrossRef] [Google Scholar]
- Docking techniques in pharmacology: How much promising? Comput. Biol. Chem.. 2018;76:210-217.
- [CrossRef] [Google Scholar]
- Validation of molecular docking programs for virtual screening against dihydropteroate synthase. J. Chem. Inf. Model.. 2009;49:444-460.
- [CrossRef] [Google Scholar]
- Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev.. 2002;34:17-35.
- [CrossRef] [Google Scholar]
- (-)-Epicatechin attenuates hepatic sinusoidal obstruction syndrome by inhibiting liver oxidative and inflammatory injury. Redox Biol.. 2019;22:101117
- [CrossRef] [Google Scholar]
- The involvement of p62-Keap1-Nrf2 antioxidative signaling pathway and JNK in the protection of natural flavonoid quercetin against hepatotoxicity. Free Radic. Biol. Med.. 2015;85:12-23.
- [CrossRef] [Google Scholar]
- Prediction of Drug Solubility from Monte Carlo Simulations. Bioorg. Med. Chem. Lett.. 2000;10:1155-1158.
- [CrossRef] [Google Scholar]
- Prediction of drug solubility from structure. Adv. Drug Deliv. Rev.. 2002;54:355-366.
- [CrossRef] [Google Scholar]
- PubChem substance and compound databases. Nucleic Acids Res.. 2016;44:D1202-D1213.
- [CrossRef] [Google Scholar]
- Li, M., Huang, W., Jie, F., Wang, M., Zhong, Y., Chen, Q., Lu, B., 2020. Since January 2020 Elsevier has created a COVID-19 resource centre with free information in English and Mandarin on the novel coronavirus COVID- 19 . The COVID-19 resource centre is hosted on Elsevier Connect , the company ’ s public news and information .
- LigPrep, 2018. Schrodinger, LLC. Retrieved from citeu- € like-article-id: 11059985 [WWW Document].
- Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem.. 1998;19:1639-1662.
- [CrossRef] [Google Scholar]
- Computational molecular docking and X-ray crystallographic studies of catechins in new drug design strategies. Molecules. 2018;23
- [CrossRef] [Google Scholar]
- Computational toxicology: an overview of the sources of data and of modelling methods. Expert Opin. Drug Metab. Toxicol.. 2009;5:1-14.
- [Google Scholar]
- A novel structural class of coumarin-chalcone fibrates as PPARα/γ agonists with potent antioxidant activities: Design, synthesis, biological evaluation and molecular docking studies. Eur. J. Med. Chem.. 2017;138:212-220.
- [CrossRef] [Google Scholar]
- Caffeic acid prevents acetaminophen-induced liver injury by activating the Keap1-Nrf2 antioxidative defense system. Free Radic. Biol. Med.. 2016;91:236-246.
- [CrossRef] [Google Scholar]
- Pengelly, A., 2004. The Constituents of Medicinal Plants: An Introduction to the Chemistry and Therapeutics of Herbal Medicine, 2nd ed. CABI.
- pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem.. 2015;58:4066-4072.
- [CrossRef] [Google Scholar]
- In vitro and in silico approaches to study the bioactivity of Citrus limon leaf extracts. J. Young Pharm.. 2017;9:290-295.
- [Google Scholar]
- Database resources of the National Center for Biotechnology Information. Nucleic Acids Res.. 2012;40:13-25.
- [CrossRef] [Google Scholar]
- Comparing test searches in PubMed and Google Scholar. J. Med. Libr. Assoc.. 2007;95:442-445.
- [CrossRef] [Google Scholar]
- Ligand- And structure-based virtual screening of 16-((diiso-butylamino)methyl)-6α-hydroxyvouacapane-7β,17β-lactone, a compound with potential anti-prostate cancer activity. J. Serbian Chem. Soc.. 2019;84:153-174.
- [CrossRef] [Google Scholar]
- Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2009;31:455-461.
- [Google Scholar]




