

Insilico exploration of the potential inhibitory activity of DrugBank compounds against CDK7 kinase using structure-based virtual screening, molecular docking, and dynamics simulation approach
⁎Corresponding author. ahussain591@gmail.com (Afzal Hussain) afzalhussain@manit.ac.in (Afzal Hussain)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The CDK-activating complex (CAK), which includes CDK7, cyclin H, and the RING-finger protein (MAT1), drives cell cycle advancement via T-loop phosphorylation of cell cycle CDKs.
The heterotrimeric CAK complex is a component of TFIIH, a generic transcription factor with dual functions in transcription and cell cycle control. CDK7 facilitates transcription by phosphorylating RNA polymerase II (Pol II) at active gene promoters. The “hallmark of cancer” has been attributed to cell cycle dysregulation, as well as aberrant transcriptions mediated by various pathways found in a variety of malignancies. Furthermore, clinical outcomes show that CDK7 levels are abundantly produced in many types of malignancies, implying that it may play a role in tissue maintenance. As a result, CDK7 is regarded as a malignant therapeutic target. Selective CDK7 inhibitors (CDK7i) have been found to work as anti-cancer medications. Drugs being repurposed for CDK7 kinase treatments is a viable strategy to swiftly uncover powerful therapeutic options for some of the most challenging forms of cancer. All of the DrugBank database chemicals, as well as the CDK7 kinase protein, were prepared, and Maestro (Schrödinger Suite) and GROMACS software suite were used to perform Docking, ADMET, MMGBSA, and MD simulation analyses. After screening the DrugBank molecules against CDK7 kinase, compounds including DB07075, DB07163, DB07025, DB01204, DB03916, DB02943, DB07812, and DB07959 were discovered to fit in the active site of the CDK7 kinase and demonstrate tight interactions. The top three docked compounds were tested, and the MD simulation revealed that they were stable with the target protein at 200 ns. As a result, these chemicals have the potential to be effective CDK7 Kinase inhibitors. As a final result, we present DB07075 (3-(5-[4-(aminomethyl)piperidin-1-yl]methyl-1H-indol-2-yl)-1H-indazole-6-carbonitrile) is a reversible inhibitor because it inactivates an enzyme through non-covalent, reversible interactions that could be a more promising inhibitor of CDK7 kinase by interacting with CDK7 kinase. This novel molecule, DB07075 has met all in silico criteria, necessitating further in vitro and in vivo research, especially in clinical trials.
Keywords
CDK7 kinase
Inhibitor
Drug repurposing
Virtual Screening
Molecular Docking
Molecular Simulation

- CDK7
-
Cyclin-dependent kinase 7
- RMSD
-
Root-mean-square deviation
- HTVS
-
High-throughput virtual screening
- MD
-
Molecular dynamics
- RMSF
-
root mean square fluctuation
Abbreviations
1 Introduction
The T-loop phosphorylation required for activation of CDKs 1, 2, 4, and 6, which regulate cell cycle progression, is provided by the CDK-activating kinase (CAK), which contains Cyclin-dependent kinase 7 (CDK7), cyclin H, and MAT1 (Larochelle et al., 1998). CAK serves a role in transcription regulation as a component of the universal transcription factor, TFIIH. CDK7 aids transcription initiation by phosphorylating the C-terminal domain (CTD) of RNA polymerase II (Pol II) at serine 5 (Ser5) at active gene promoters (Sava et al., 2020). CDK7 phosphorylates CDK9, which then phosphorylates the Pol II CTD at Ser2 to trigger transcription elongation. (Bowman and Kelly 2014). The activity of a number of transcription factors, including p53, oestrogen receptor (ER), retinoic acid receptors (RARs), and androgen receptor (AR), is regulated by CDK7-mediated phosphorylation (Patel et al., 2016). Due to its dual role in regulating the cell cycle and transcription, CDK7 has been investigated as an anticancer therapeutic target, and a number of selective inhibitors of CDK7 have been developed and tried as cancer therapy (Morgan 1995). Cancer cells are more reliant than normal cells on high amounts of super-enhancer (SE)-driven transcription mediated by specific oncogenic drivers, such as RUNX1 in acute lymphoblastic lymphoma (ALL) and N-MYC in neuroblastoma, as evidenced by preclinical investigations (Bradner et al., 2017, Sengupta and George 2017). Only four selective CDK7 inhibitors have progressed to Phase I/II clinical studies for the treatment of advanced solid tumours: ICEC0942, SY-1365, SY-5609, and LY340515 (Kolloch et al., 2022). It's critical to develop selective CDK7 inhibitors and understand their mechanism of action in cancer in order to take advantage of their potential for usage in combination therapies (Sava et al., 2020). As a result, understanding the role of CDK7 in normal vs tumour cells, as well as the underlying causes of CDK7 suppression in cancer, are crucial areas of medical and/or pharmaceutical science study.
Altogether new cancer drugs discovery is not a trivial task. Surprisingly, existing successful cancer treatments could be leveraged to uncover specific promising therapeutic drug candidates by using pre-existing refined pharmacokinetics and delivering safe molecules with no side effects (Link 2019). Furthermore, computer-aided drug design (CADD) procedures have been identified as a viable option. For anticancer drug development, structure-based virtual screening (SBVS) and molecular docking have been frequently used (Baig et al., 2016). It is regarded as the beginning point for the discovery of novel lead compounds, which will pave the way for the development of many therapeutics for a variety of ailments. After a thorough search of the DrugBank database, the selective SBVS approach and molecular dynamics simulation was used in this study. The Schrödinger software suite was used to create the docking framework against the target protein and also GRMACS 2021 was applied for the simulation study of the target with drug molecules (Bhachoo and Beuming 2017).
The objective of the research was to develop novel noncovalent ATP-competitive CDK7 Kinase inhibitors with high binding interaction in the binding region. Most protein kinase-approved drugs with clinical approval belong to the ATP-competitive inhibitor class. By concentrating on the ATP-binding pocket, these substances block the catalytic activity and prevent the phosphorylation of the substrate. We, therefore, also focus on such ATP-competitive inhibitors in our research. This study used a drug repurposing strategy to find potential CDK7 Kinase inhibitors, which were then evaluated for absorption, distribution, metabolism, excretion, and toxicity (ADMET) and Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) and MD simulation analysis to find novel non covalent inhibitors that could be used as potential leads for treating cancer disease.
2 Materials and methods
2.1 Selection and preparation of the target structure
The 3D (three-dimensional) structure of CDK7 kinase target protein (PDB ID: 1UA2) was downloaded from the Protein Data Bank (https://www.rcsb.org). Protein Preparation Wizard of Schrodinger Suite was used to prepared the protein and used for further analysis. Hydrogen atoms were added to the protein structure to optimize hydrogen bonding. Finally, using the OPLS-2005 (Optimized Potentials for Liquid Simulations) force field and a 0.30 Å RMSD (root-mean-square deviation) cutoff, minimization was performed (Lolli et al., 2004).
2.2 Preparation of DrugBank database compounds
Compounds were first extracted from the DrugBank database. Ligprep, a ligand preparation procedure of Schrodinger software, was utilized to prepare high-quality structures with all atoms. The bond angles and bond ordering were assigned, and the minimization methods, which included the OPLS-2005 force field and the Epik feature to retain the ligand in the proper protonation position, were run (Wishart et al., 2018).
2.3 Preparation of the grid
The Glide framework was used to generate the Grid of the prepared target protein. The centroid of the CDK7 kinase structure was chosen to be the adenosine triphosphate (ATP) molecule. The partial charge cutoff was set to 0.25 and the scaling factor was set to 1.0 (Hussain and Hussain 2022).
2.4 Molecular screening and docking
All the prepared compounds from DrugBank database were chosen for the analysis of Lipinski properties and bioactivity. To get the best score, HTVS (High-throughput virtual screening), SP (standard precision), XP (extra precision) screening, and Docking were employed followed by selection of the XP description information for pose viewing (Hussain and Verma 2017, Hussain et al., 2017).
2.5 Drug-like properties analysis
The selected DrugBank compounds were analyzed to understand their “Drug-Like” properties. The ADME (absorption, distribution, metabolism and excretion) properties of selected ligands were investigated using the Schrodinger suite's QikProp tool. The tool predicted the physiochemical properties with a detailed analysis of molecular weight (MW), hydrogen bond acceptor (HBA), total solvent accessible surface area (SASA), human oral absorption, hydrogen bond donor (HBD), predicted aqueous solubility (QP log S), etc (QikProp 2010).
2.6 Drug-target binding energy analysis using the MM-GBSA method
The Drug-Target Binding Energy was calculated using Schrodinger Software and subsequently the calculated binding energy was used to evaluate the stability of the target protein and ligand complexes (Hussain and Verma 2016).
2.7 Molecular dynamics (MD) simulation
MD simulation was done for 200 ns time frame on GROMACS 2021.3 software using the CHARMM27 all-atom force field on the docked complexes of the topmost screened three ligands (best XP docking score), as well as the protein structure of CDK7 Kinase (PDB code: 1UA2). The SwissParam was used to acquire the ligand coordinates and topological parameters. The neutralization system included the addition of Na + and Cl- ions, as well as solvation utilizing the TIP3P water cube model. The steepest descent algorithm was used to accomplish energy minimization for a maximum of 50,000 steps. After that the structures stabilized to a maximum force of 1000 kJ mol−1 nm−1, then two-step equilibrium was completed at 300 K, 1.0 atm, as well as 50,000 steps, 300 K using the NVT ensemble and Berendsen thermostat, correspondingly. The LINCS (linear constraint solver) approach was utilized to constrain covalent bonds, whereas the particle mesh Ewald approach was employed to tackle long-range coulombic contacts. Production Molecular Dynamics (MD) experiments were run for 200 ns at a fixed temperature of 300 K, and Every 2.0 ps, MD trajectories were updated. GROMACS in-built tools including H-bond, rmsf, gmx rmsd, gyrate, and the H-bonds module included in the VMD (visual molecular dynamics) program were used to do the post-MD analysis of trajectories. During the MD run, the H-bonds java applet of VMD was used to look for intermolecular H-bonds established by the ligand with the entire protein and also particular contacts with the crucial binding site residues of the CDK7 Kinase at a donor–acceptor range cutoff of 3.5 and a bond angle cutoff of 20Ao in each of the three complexes (Abraham et al., 2015, Kumar et al., 2021).
3 Results
3.1 Docking with the target protein
The docking was aimed to screen the best novel drugs that target CDK7 kinase. Thus, the DrugBank database was scored extensively for molecules that bind to the target protein with the highest affinity and interact with it efficiently. The docking scores were documented such as DB07075 (-13.706), DB07163 (-11.238), DB07025 (-11.21), DB01204 (-10.591), DB03916 (-10.574), DB02943 (-10.5), DB07812 (-10.48), DB07959 (-10.461). All of these compounds were found to bind to residues in the target protein's active site, where the ATP-binding site is located. It proved that the drug molecules were tightly bound to the active sites of the target proteins and had improved inhibitory activities. All the selected screened compounds didn't contain the Cysteine residue but showed a tight interaction with the target protein; therefore, the noncovalent inhibitors are also having the potential against CDK7 Kinase as the recent study published for SY-5609 (noncovalent inhibitor) (Kolloch et al., 2022). Glide's enhanced precision (XP) mode was used to perform the ligand docking calculations. Docking results of top eight compounds are listed in Table 1 and Table 2. The binding mode of interaction between the target and the DrugBank compounds is depicted in the Docking and LigPlot interaction diagrams (Fig. 1). To compare the docking scores, a thorough plot chart was created (Fig. 2).
| Sl. No. | Compound | IUPAC name | Mol. formula | Mol. Wt. | Docking Score | 2d Structure |
|---|---|---|---|---|---|---|
| 1 | DB07075 | 3-(5-{[4-(aminomethyl)piperidin-1-yl]methyl}-1H-indol-2-yl)-1H-indazole-6-carbonitrile | C23H24N6 | 384.477 | −13.706 |

|
| 2 | DB07163 | 7-[(2-aminoethyl)amino]-9-fluoro-5-(1H-pyrrol-2-yl)-2-azatricyclo[6.3.1.0â□́,Â1Â2]dodeca-1(11),4,6,8(12),9-pentaen-3-one | C17H15FN4O | 310.326 | −11.238 |
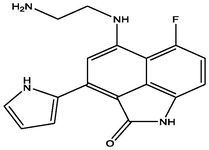
|
| 3 | DB07025 | 3-(5-{[4-(aminomethyl)piperidin-1-yl]methyl}-1H-indol-2-yl)-1H-quinolin-2-one | C24H26N4O | 386.489 | −11.21 |
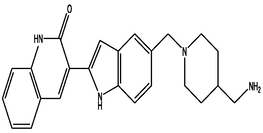
|
| 4 | DB01204 | 1,4-dihydroxy-5,8-bis({2-[(2-hydroxyethyl)amino]ethyl}amino)-9,10-dihydroanthracene-9,10-dione | C22H28N4O6 | 444.481 | −10.591 |
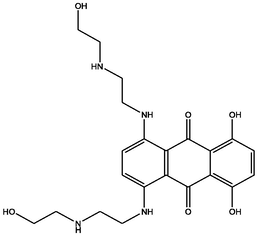
|
| 5 | DB03916 | 4-{2-[4-(2-aminoethyl)piperazin-1-yl]pyridin-4-yl}-N-(3-chloro-4-methylphenyl)pyrimidin-2-amine | C22H26ClN7 | 423.942 | −10.574 |
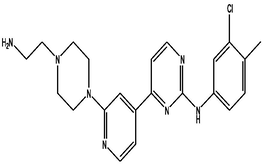
|
| 6 | DB02943 | 2-[(2R)-2-(4-aminobutanamido)-3-{[(4-methoxyphenyl)methyl]sulfanyl}propanamido]acetic acid | C17H25N3O5S | 383.463 | −10.5 |
|
| 7 | DB07812 | N-[(1S)-2-amino-1-phenylethyl]-5-{1H-pyrrolo[2,3-b]pyridin-4-yl}thiophene-2-carboxamide | C20H18N4OS | 362.448 | −10.48 |
|
| 8 | DB07959 | 3-(1H-1,3-benzodiazol-2-yl)-1H-indazole | C14H10N4 | 234.256 | −10.461 |
|
| Interactions | ||||||
|---|---|---|---|---|---|---|
| Compounds H-Bonds Hydrophobic and Pi-Pi stacking (cutoff 4.00Ao) | ||||||
| Free Binding Energy | Number | Residue | Distance (Ao) | Number | Residue | |
| DB07075 | −58.32 | 5 | GLU99, ASP97, MET94, ASP92, ASP155 | (1.94,, 2.58, 1.67, 2.30, 2.43) | 11 | VAL100, MET94, PHE93, PHE91, ILE75, ALA154, ALA39, VAL26, LEU18 Salt Bridge GLU99, ASP97 |
| DB07163 | −50.19 | 3 | ASN141, MET94, ASP92, | (2.81, 2.00, 2.39) | 8 | VAL26, ALA154, PHE91, ALA39, PHE93, MET94, LEU144, LEU18 |
| DB07025 | −68.53 | 3 | MET94, ASP97, GLU99 | (2.06, 2.39, 2.05) | 11 | VAL100, MET94, PHE93, LEU144, PHE91, ALA154, ILE75, ALA39, VAL26, LEU18 Salt Bridge Asp97 |
| DB01204 | −60.24 | 5 | ASP155, ASP92, MET94, ASP97, TPO170 | (2.25, 2.05, 2.78, 1.74, 1.54) | 10 | LEU144, ALA154, PHE91, PHE93, MET94, VAL100, LEU18, VAL26, ALA39, ILE75 |
| DB03916 | −47.63 | 3 | TPO170, GLU20, MET94 | (2.95, 2.04, 2.06) | 10 | VAL26, LEU18, ILE75, LEU144, ALA154, ALA39, MET94, PHE93, PHE91 Salt Bridge TPO170 |
| DB02943 | −58.09 | 6 | ASN142, ASN141, SER161, LYS41, MET94, GLU99 | (1.91, 1.82, 1.79, 2.15, 2.08, 1.72) | 13 | VAL100, LEU144, VAL26, PHE23, ALA39, LEU18, ILE75, ALA154, MET94, PHE93, PHE91 Salt Bridge GLU99, ASP97 |
| DB07812 | −45.10 | 4 | GLU99, ASP97, MET94, ASP92 | (1.76, 2.60, 2.07, 2.07) | 9 | VAL100, VAL26, MET94, PHE93, ILE75, PHE91, ALA39, LEU18, LEU144 |
| DB07959 | −45.88 | 2 | MET94, ASP92 | (1.82, 1.99) | 9 | LEU18, VAL26, ALA154, ALA39, ILE75, LEU144, PHE91, PHE93, MET94 |
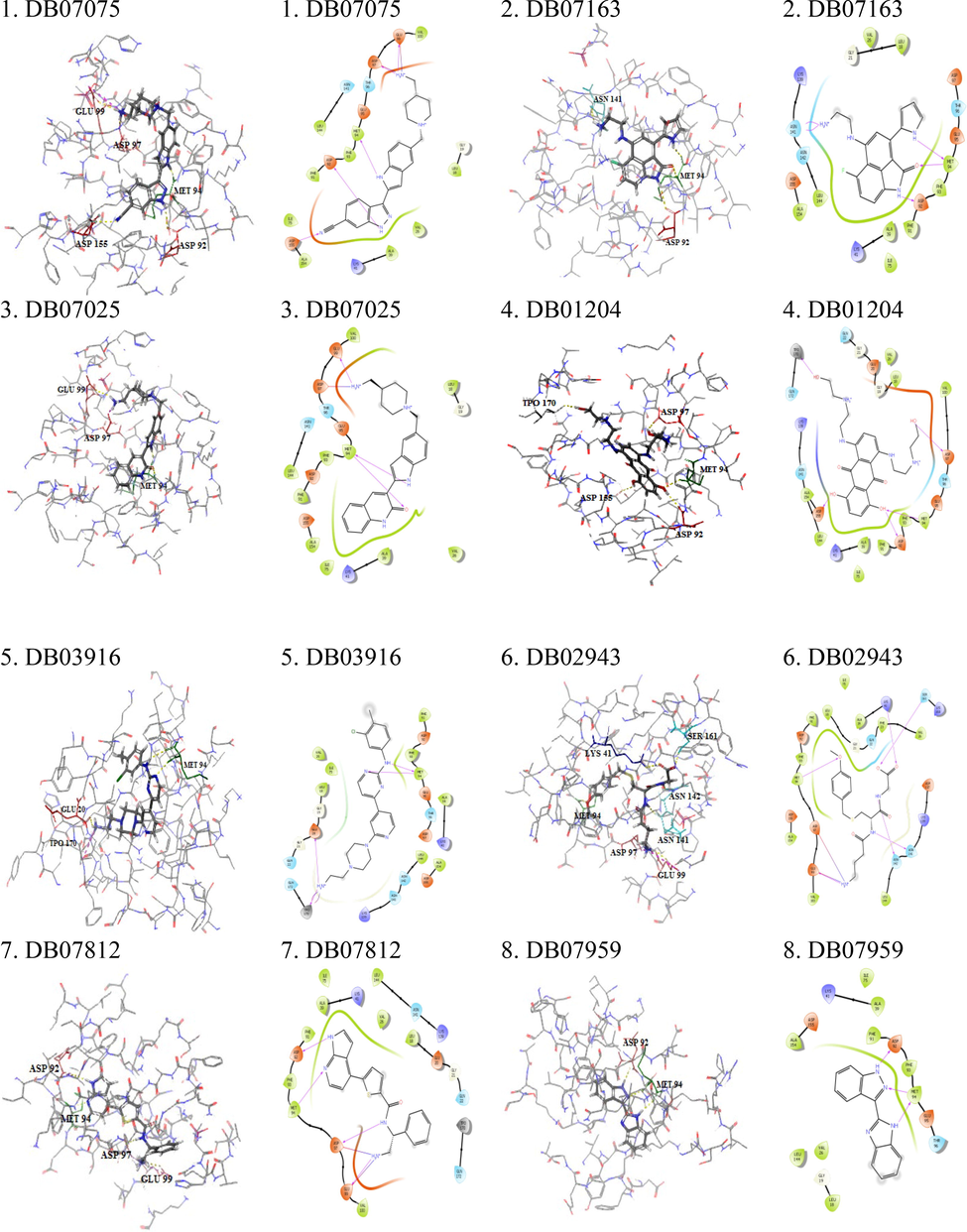
- Docked complex of top eight DrugBank screened compounds with the CDK7 kinase active siteand corresponding ligPlot interaction diagram.
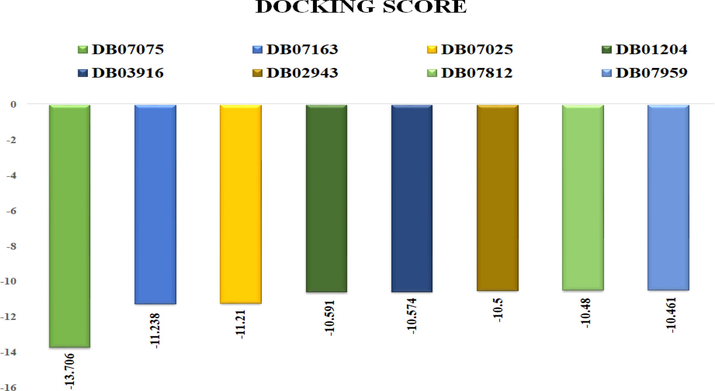
- The docking score of the top eight lead compounds from Drugbank which are showing the highest binding affinity towards CDK7.
3.2 Analyzing ADMET and drug-likeliness characteristics of docked compounds
QikProp tool is used to read the molecular properties of the selected compounds in order to satisfy Lipinski's rule of five, which is critical for rational drug design. The selected drug compounds acquired from DrugBank were evaluated for drug-likeness as well as physiochemical properties. The best hits were then evaluated for drug-likeliness using Lipinski's rule, bioactivity rating, and ADME characteristics such as Total Solvent Accessible Surface, Predicted Aqueous Solubility, Human Oral Absorption, Molecular Weight (MW), Hydrogen Bond Donor (HBD), Hydrogen Bond Acceptor (HBA), Total Solvent Accessible Surface (QP log S).Other drug-like properties including Predicted Polarizability (QPpolrz), Total Solvent Accessible Surface Area (SASA), Predicted octanol/water partition coefficient (QPlogPo/w), Predicted hexadecane/gas partition coefficient (QPlogPC16), Predicted octanol/gas partition coefficient (QPlogPC16), Predicted octanol/gas partition coefficient (QPlogPC16), and Predic (QPlogKp) were evaluated as well (Table 3 and Table 4).These ligands' physiochemical characteristics and ADME metrics indicate that they could be used as drug candidates in the future.
| Sl. No. | Compound Ida | Mol.Wt.b | QPlog Sc | SASAd | H-Bond Acceptore | H-Bond Donorf | HumanOral Absorptiong |
|---|---|---|---|---|---|---|---|
| 1 | DB07075 | 384.477 | −4.439 | 707.504 | 6 | 4 | 54.295 |
| 2 | DB07163 | 310.326 | −2.301 | 545.191 | 5 | 5 | 67.743 |
| 3 | DB07025 | 386.489 | −4.179 | 713.494 | 5 | 3 | 72.065 |
| 4 | DB01204 | 444.481 | −1.039 | 737.283 | 10 | 4 | 27.155 |
| 5 | DB03916 | 423.942 | −4.465 | 775.995 | 7 | 3 | 80.124 |
| 6 | DB02943 | 383.463 | −1.559 | 696.324 | 8 | 4 | 20.734 |
| 7 | DB07812 | 362.448 | −3.893 | 642.721 | 5 | 4 | 79.131 |
| 8 | DB07959 | 234.256 | −3.645 | 463.16 | 3 | 2 | 100 |
| Sl. No. | Compound | QPpolrz | QPlogPoct | QPlogPo/w | QPlogPC16 | QPlogPw | QPlogKp | CIQPlogS | QPlogKhsa |
|---|---|---|---|---|---|---|---|---|---|
| 1 | DB07075 | 43.3 | 23.629 | 2.107 | 14.197 | 14.883 | −7.978 | −4.583 | 0.497 |
| 2 | DB07163 | 31.301 | 20.61 | 1.316 | 10.822 | 14.817 | −5.27 | −3.312 | −0.159 |
| 3 | DB07025 | 45.095 | 22.532 | 3.292 | 14.131 | 12.707 | −6.653 | −4.148 | 0.827 |
| 4 | DB01204 | 38.786 | 24.367 | 0.689 | 14.431 | 16.253 | −7.998 | −2.932 | −0.385 |
| 5 | DB03916 | 47.279 | 23.903 | 3.399 | 15.026 | 14.347 | −5.786 | −3.996 | 0.555 |
| 6 | DB02943 | 36.233 | 21.573 | −1.119 | 13.266 | 17.993 | −6.439 | −1.858 | −0.954 |
| 7 | DB07812 | 39.914 | 21.946 | 2.907 | 13.803 | 14.356 | −4.391 | −4.618 | 0.352 |
| 8 | DB07959 | 27.933 | 13.742 | 2.608 | 8.97 | 9.376 | −1.858 | −3.925 | 0.125 |
QPplorz Predicted polarizability in cubic angstroms (permissible scale is: 13.0 – 70.0).
QPlogPoct Predicted octanol/gas partition coefficient (permissible scale is: 8.0 – 35.0).
QplogPo/w Predicted octanol/water partition coefficient (permissible scale is: –2.0 – 6.5).
QPlogPC16 Predicted hexadecane/gas partition coefficient (permissible scale is: 4.0 – 18.0).
QPlogPw Predicted water/gas partition coefficient (permissible scale is: 4.0 – 45.0).
QPlogKp Predicted skin permeability, log Kp (permissible scale is: –8.0 –1.0).
CIQPlogS Conformation-independent predicted aqueous solubility (permissible scale is: –6.5 – 0.5).
QPlogKhsa Prediction of binding to human serum albumin (permissible scale is: –1.5 – 1.5).
3.3 Evaluation of drug-target binding energy of the screened compounds
The Prime MM-GBSA module is very useful for determining protein–ligand binding affinity. It uses a mix of the Surface Generalized Born Solvation Model for Polar Salvation (GSGB), non-polar solvation (GNP), and OPLS Molecular Mechanics Energies (EMM) to calculate nonpolar solvent accessible surface area and van der Waals interactions.This module was applied to the protein–ligand complexes that were chosen. The following equations were used to compute the changes in total free energy caused by ligand binding (Fig. 3).

- MM-GBSA energy calculation.
Finally, the binding free energy of selected DrugBank compounds as complexes were anticipated and calculated (Table 5).
| Sl. No. | Compounda | Gevdwb | GsolLipoc | Gcould | Gcovalente | GsolGBf | ΔG bindg |
|---|---|---|---|---|---|---|---|
| 1 | DB07075 | −39.7615 | −19.27918446 | −113.2081254 | 6.56478033 | 112.9901 | −58.32409972 |
| 2 | DB07163 | −38.5721 | −16.62401513 | −49.37464318 | 1.036189811 | 57.06864 | −50.19531003 |
| 3 | DB07025 | −43.1235 | –22.74675736 | −95.32570219 | 1.577161145 | 95.17995 | −68.53077515 |
| 4 | DB01204 | −40.1834 | –23.86333743 | −118.4126977 | 5.854120515 | 121.4921 | −60.24244552 |
| 5 | DB03916 | −47.7613 | −15.96167093 | −67.70271742 | 1.4706851 | 85.1289 | −47.6302016 |
| 6 | DB02943 | −45.1735 | −17.44727424 | −43.06485389 | 9.674905475 | 43.75793 | −58.09810364 |
| 7 | DB07812 | −42.098 | −13.13177461 | −70.92995582 | 2.522891931 | 84.98804 | −45.1075747 |
| 8 | DB07959 | −31.6081 | −13.89082187 | −25.56752352 | 2.185984024 | 26.33351 | −45.88439556 |
3.4 Superimposed structure
Superimpose is a technique for determining whether or not all of the selected hits occupied the same active binding sites. For this investigation, all of the DrugBank hits were precisely superimposed on the crystal structure (PDB ID: 1UA2) that inhabited in the target's active site pocket. Surprisingly, the binding pattern of the selected DrugBank compounds resembled that of the reference chemical, ATP. The superimposed structures are critical for further research. Indeed, the docked drug compounds were shown to be entirely occupied within the active site region of CDK7 Kinase, as evidenced by the binding patterns displayed by the selected drug compounds. CDK7 Kinase and drug molecules were superimposed and displayed (Fig. 4).
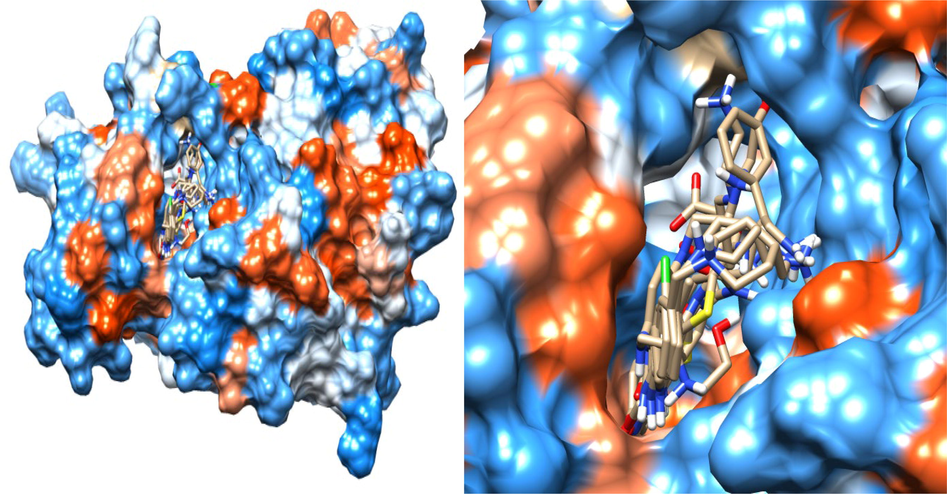
- Superimposed model of the eight selected DrugBank Hit in the active site of the target.
3.5 Structural analysis of systems using molecular dynamic simulation (MD)
In numerous biological contexts, MD simulation is used to represent structural disturbances and actual mobility of a receptor with ligand complex. RMSD, a well-developed measure for protein–ligand stability and equilibration, was used to validate the docking orientations, and the overall changing patterns of behavior for the entire protein structure were studied during the MD simulation study.
3.6 Root-mean-square deviation (RMSD)
The RMSD eventually determines how much a protein changes when its primary coordinates are modified. The RMSD measurements for the protein backbone and potential inhibitors were assessed using the basic structure as a starting point (0 to 200 ns). The backbone is an essential part of the target structure. Hence, we have shown the overall backbone simulation concerning the time. The RMSD values were determined for the free CDK7 receptor, CDK7 + DB07075, CDK7 + DB07163, and CDK7 + DB07025 complexes as well (Fig. 5). Surprisingly, among all the parameters examined thus far, the RMSD values grew rapidly from 0 to 50 ns and remained stable after establishing equilibration for the length of the simulation. The complexes CDK7 + DB07075, CDK7 + DB07163, and CDK7 + DB07025 had lower RMSD values and were nearly equivalent to the receptor (CDK7). These three drug candidates were shown to be stable when bound to CDK7, implying that they could serve as a good foundation for further research.
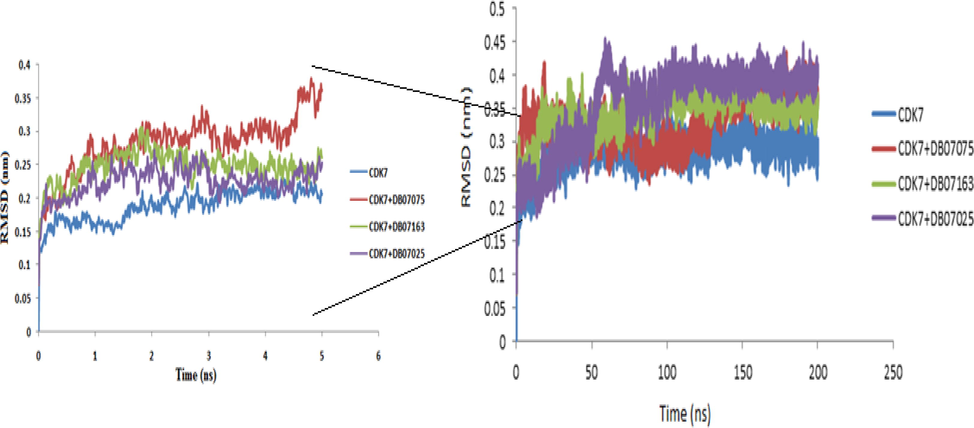
- The plot of the root - mean - square deviation (RMSD) of the receptor backbone vs simulation duration for a solvated CDK7 receptor complexes with DB07075, DB07163 and DB07025 during a 200 ns molecular dynamics simulation. The 5 ns simulation graph is shown in the zoom panel on the left side.
3.7 Root-mean-square fluctuations (RMSF)
Backbone RMSF values were calculated, and RMSF charts were created and analyzed. The findings revealed differences at the atomic level. The RMSF values of CDK7 receptor, as well as CDK7 + DB07075, CDK7 + DB07163, and CDK7 + DB07025 complexes, are displayed (Fig. 6).
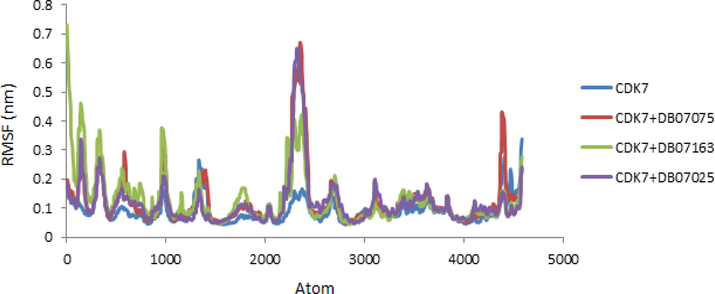
- RMSF values of CDK7 Kinase alone and in combination with DB07075, DB07163 and DB07025 were displayed versus atom number.
For the free receptor (CDK7) and complexes with low RMSF values, the data obtained from atomic fluctuations was nearly identical. Thus, the RMSF plot demonstrated that binding is stable within the receptors for all of the evaluated drug candidates, and there was no significant effect on the receptor's flexibility during the simulation study.
3.8 The radius of gyration (Rg)
The radius of gyration (Rg) data is utilized in the simulation to determine if the complex is stable in folded or unfolded forms. CDK7 receptor, CDK7 + DB07075, CDK7 + DB07163, and CDK7 + DB07025 complexes all share similar properties, according to the Rg plot. The Rg values of protein and protein-drug candidate complexes showed a substantial similarity and stable folded structure when compared to the protein complex. The graph of radius of the gyration is displayed (Fig. 7).
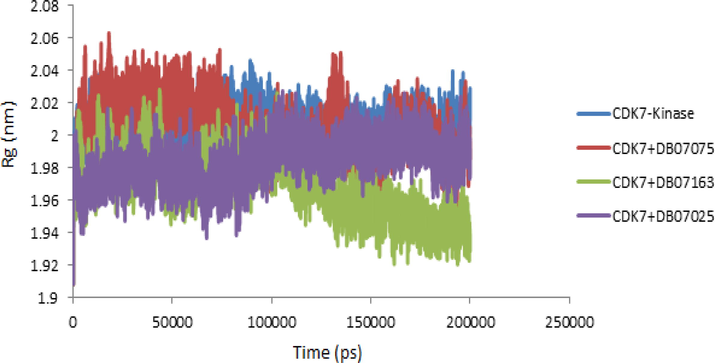
- The radius of gyration (Rg) values of CDK7 Kinase alone and in complex with the DB07075, DB07163 and DB07025 were plotted against Time (ps) to 200 ns.
3.9 Hydrogen bond analysis
Hydrogen bond analysis is used to learn more about drug molecular interactions, molecular recognition, and selectivity within receptors. The analysis of hydrogen bonds was used to adjust the protein–ligand interactions obtained from changes in secondary structures during MD simulation. MD simulation determines the model for interacting with the ligand for each receptor orientation. The number of hydrogen bonds produced in MD simulations of all complicated trajectories was investigated (Fig. 8). During MD simulations, the CDK7 + DB07075 complex generated more hydrogen bonds than the CDK7 + DB07163 or CDK7 + DB07025 complexes. DB07075, a molecule having a number of hydrogen bonds linked to CDK7, was discovered in the simulation data. CDK7 + DB07075, CDK7 + DB07163, and CDK7 + DB07025 were able to maintain a stable contact with CDK7′s binding pocket throughout the simulation time.
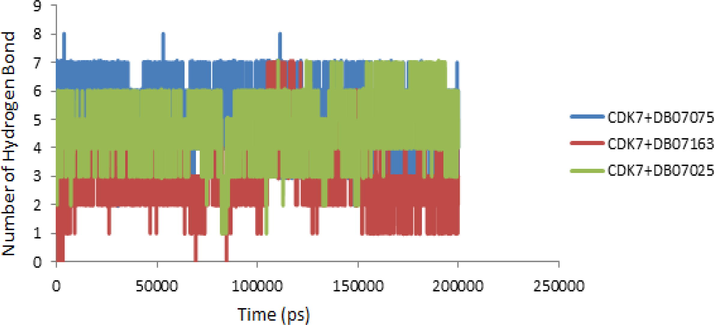
- Hydrogen bond patterns of CDK7 + DB07075, CDK7 + DB07163 and CDK7 + DB07025 complex during molecular dynamics simulation at 200 ns.
4 Discussion
According to the World Health Organization, cancer is the greatest cause of death worldwide, accounting for roughly ten million deaths in 2020. As a result, scientists and researchers are working tirelessly to develop effective therapeutic medications because there is an urgent need to find new therapies with fewer side effects to combat this terrible cancer disease. Before moving into the wet lab, in silico techniques like molecular docking, virtual screening, and Molecular Dynamic Simulations can help uncover new drug candidates. The reason for this is due to the multifarious features of these in silico technologies, which provide a clear exposition on which subsequent clinical research-related trials can be created and carried out. As a result, we used Schrodinger software to perform HTVS, SP, and XP virtual screening and docking of DrugBank database compounds to target the important enzyme CDK7 Kinase.
We identified a total of eight DrugBank compounds after screening and docking, but only three of them, DB07075 (-13.706), DB07163 (-11.238), and DB07025 (-11.21), were judged to be the best drug candidates with good score values and essential features. The results of the docking and LigPlot interaction investigations have also indicated that the identified drug candidates interact with the CDK7 Kinase molecule's active site residues.
Furthermore, using the selected drug candidates, ADMET and MMGBSA analyses have been undertaken, with the findings indicating that the values are in the favorable range. Finally, the RMSD values of the receptor and receptor-ligand complexes were computed, revealing that the interactions were stable throughout the simulation phase. The unbound receptor and the complex's backbone atoms have similar RMSF values, indicating that the drug candidates inside the CDK7 receptor are stable. The gyration radius bore a striking resemblance as well. Despite the fact that several studies are being conducted around the world to find strong inhibitors of cancer targets, no such studies have been conducted using DrugBank medicines targeting the CDK7 Kinase. Our unique methodologies have enabled us to exhibit the interaction of the selected drug candidates with the target CDK7 Kinase enzymes and highlighted their prospective therapeutic values to aid in the selection of the best pharmaceuticals for cancer therapies. Despite the limitations of in silico analyses such as virtual screening, docking methods, and MD simulations, the good news is that computer-aided drug design (CADD), drug discoveries, and interactions can be achieved. Hence, these in silico drug design, drug discovery and development can be done precisely to find the needle or rather “the novel molecules of life” from a haystack of complex cellular factories. However, more experimental and clinical research is needed to establish the compounds' inhibitory actions against CDK7 Kinase in order to employ them as prospective cancer treatments. Hence, several published literature shows that drug databases are reliable sources for identifying numerous novel compounds with excellent inhibition properties. These methods of screening novel compounds are highly effective in terms of both time and cost. Our research group focuses on finding a potent noncovalent inhibitor against CDK7 Kinase like SY-5609. Soon, our research will discover the drug acting with multiple molecular targets (CDK Kinase).
5 Conclusion
Over the last few decades, tremendous progress has been made in the treatment of life-threatening disorders. CDK7 inhibitors are thought to be potential anticancer drugs, however they all have substantial drawbacks that hinder their widespread usage. Repurposing existing medications could be a good way to find therapeutic interventions that have been demonstrated to be safe and effective in clinical trials. Based on virtual screening, molecular docking, and MD simulation, we found a few DrugBank compounds that can be employed as potent inhibitors of the CDK7 Kinase. Intriguingly, DB07075 was found to be tightly bound to the active site of CDK7 Kinase. Both the docking studies on DB07075, which scored −13.706 and the RMSD, yielded high results. In the case of DB07075, the RMSF, Rg, and the quantity of H-bonds were likewise determined to be in the favourable ranges. Overall, we believe that DB07075, also known as 3-(5-[4-(aminomethyl)piperidin-1-yl]methyl-1H-indol-2-yl)-1H-indazole-6-carbonitrile, is one of the most promising Anti CDK7 Kinase medicines. These chemicals, however, must be tested in a wet lab setting.
Acknowledgment
The authors want to acknowledge the Department of Bioinformatics, MANIT, for providing the research facility to complete the research workand also would like to thank College of Applied Medical Sciences, Buraydah Private Colleges, Al-Qassim 31717, Kingdom of Saudi Arabia for their support.We are also thankful to the Schrodinger team for providing the software facility.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1:19-25.
- [Google Scholar]
- Computer aided drug design: success and limitations. Curr. Pharm. Des.. 2016;22:572-581.
- [CrossRef] [Google Scholar]
- Investigating protein-peptide interactions using the Schrödinger computational suite. Methods Mol. Biol. (Clifton N.J.). 2017;1561:235-254.
- [CrossRef] [Google Scholar]
- RNA polymerase II transcription elongation and Pol II CTD Ser2 phosphorylation: a tail of two kinases. Nucleus (Austin, Tex.). 2014;5:224-236.
- [CrossRef] [Google Scholar]
- Computational repurposing of asthma drugs as potential inhibitors of SARS-CoV-2 M(pro) New Microb. New Infect.. 2022;47:100979
- [CrossRef] [Google Scholar]
- Recognition of new inhibitor of CDK9/Cyclin T1 complex as persuasive anticancer agent. Int. J. Adv. Biotechnol. Res.. 2016;7:654-668.
- [Google Scholar]
- Molecular docking and in silico ADMET study reveals 3- (5-{[4-(aminomethyl) piperidin-1-yl] methyl}-1h-indol-2-yl)-1h-indazole-6-carbonitrile as a potential inhibitor of cancer Osaka thyroid kinase. Biomed. Res.. 2017;28:5805-5815.
- [Google Scholar]
- Identification of novel inhibitors against Cyclin dependent kinase 9/Cyclin T1 complex as: anti cancer agent. Saudi J. Biol. Sci.. 2017;24:1229-1242.
- [CrossRef] [Google Scholar]
- Control of expression of key cell cycle enzymes drives cell line-specific functions of CDK7 in human PDAC cells. Int. J. Mol. Sci.. 2022;23
- [CrossRef] [Google Scholar]
- Identification of CDK7 inhibitors from natural sources using pharmacoinformatics and molecular dynamics simulations. Biomedicines. 2021;9:1197.
- [Google Scholar]
- Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev.. 1998;12:370-381.
- [CrossRef] [Google Scholar]
- Link, W., 2019. Anti-cancer Drugs–Discovery, Development and Therapy. International Manual of Oncology Practice, Springer: 95-111.
- Lolli, G., E. D. Lowe, N. R. Brown, et al., 2004. The crystal structure of human CDK7 and its protein recognition properties. Structure (London, England : 1993). 12, 2067-2079. https://doi.org/10.1016/j.str.2004.08.013.
- Expression of CDK7, Cyclin H, and MAT1 is elevated in breast cancer and is prognostic in estrogen receptor-positive breast cancer. Clin. Cancer Res. : Off. J. Am. Assoc. Cancer Res.. 2016;22:5929-5938.
- [CrossRef] [Google Scholar]
- QikProp, S. d., 2010. version 3.3, Schrödinger, LLC: New York, NY.
- CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev.. 2020;39:805-823.
- [CrossRef] [Google Scholar]
- Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer. 2017;3:269-281.
- [CrossRef] [Google Scholar]
- DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res.. 2018;46:D1074-D1082.
- [CrossRef] [Google Scholar]




