

Translate this page into:
Investigation of the microscopic mechanism of H2O adsorption by low-rank coal slit pores through GCMC and MD simulations
⁎Corresponding author at: College of Safety Science & Engineering, Liaoning Technical University, Fuxin 123000, China. LXQab161616@163.com (Xiaoqing Liu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Abstract
This study investigates the influence of pore structure on H2O adsorption in low-rank coal (LRC) models at a molecular level, employing variously sized LRC slit-pore models·H2O adsorption behavior is analyzed across different pore sizes ranging from 0.001 to 100 kPa fugacity, by employing the Grand canonical Monte Carlo (GCMC) method and molecular dynamics (MD) simulations to explore saturated adsorption kinetics. The findings indicate a positive correlation between H2O adsorption levels and both fugacity and pore diameter. Furthermore, at constant pore diameter, the adsorption heat increases exponentially with increasing adsorption levels. Notably, H2O adsorption in LRC slit pores exhibits multilayer adsorption characteristics. First, adsorption occurs as monomolecular layers on the LRC surface, transitioning to secondary adsorption sites formed by previously adsorbed H2O molecules with increasing fugacity, ultimately saturating the pores. This adsorption process exhibits a correlation with a shift in potential energy distribution from high to low potential energy directions. Moreover, the ordering of H2O molecule arrangements between hydrogen atoms, oxygen atoms, and H2O molecules decreases with increasing pore diameters. Larger pore diameters correspond to increased H2O molecule diffusion, the stronger interaction between the LRC slit pores and H2O molecules but reduced interaction with individual H2O molecules. Overall, this study establishes a theoretical foundation for further studies on coal pore wetting and offer valuable insights for studies aimed at reducing dust and preventing gas outbursts.
Keywords
LRC slit-pore model
Slit pore diameter
Adsorption active site
Energy distribution
Kinetic properties

1 Introduction
Coal serves as a pivotal fossil fuel across various domains and stands as one of the primary sources globally for electricity generation and a foundational material for numerous chemical products (Zhu et al., 2019; Xie, 2019; Xie, 2017). Despite its multifaceted utility, the utilization of coal entails environmental, climatic, and geological challenges, including notable risks such as coal and gas hazards arising from mining activities (Zhao and Liu, 2022). Within coal seams, water is inherently present in various forms: intrinsic, extrinsic, and chemosynthetic (Jin et al., 2017). Moisture assumes a critical role in both the extraction and application of coal. Its presence mitigates coal dust generation and dispersion, thereby reducing the risk of dust explosions in mines, as well as curbing greenhouse gas emissions (Qin et al., 2022; Zou et al., 2021; Shekarian et al., 2021). Injection of water into coal seams is a common practice aimed at averting disasters such as spontaneous combustion, coal and gas outbursts, explosions, and surface subsidence (Lun et al., 2022; Yue et al., 2022; Jiang et al., 2022; Gao et al., 2023). However, moisture compromises the calorific value of coal, elevates transportation expenses, and undermines coal storage stability. In coal flotation, the pronounced hydrophilicity of the LRC surfaces engenders poor floatability of fine coal particles and necessitates increased use of flotation agents (Zhao and Liu, 2022; Zhao and Liu, 2023; Bao et al., 2022; Li et al., 2021; Niu et al., 2022). Given the prevalence of water molecules in these processes, examining the interaction between coal and water molecules, as well as the wettability of coal surfaces, emerges as imperative for the safe extraction and efficient utilization of coal.
Coal surface wettability correlates with coal rank, oxygen-containing functional groups (OFGs), particle size, surface morphology, pore structure, and the degree of defects (You et al., 2018; Horikawa et al., 2021; Li et al., 2013; Wang et al., 2020). Molecular simulation is extensively applied across various domains such as materials science, biochemistry, drug design, and nanotechnology. This method effectively predicts the properties, behaviors, and interactions of substances, simulating their atomic and molecular dynamics across different temporal and spatial scales (Hao et al., 2022; Bai et al., 2021; Li et al., 2019; Wang et al., 2020; Long et al., 2021; Ge et al., 2023; Swenson and Stadie, 2019). Zhang et al. conducted a study on the wettability of coal seam surfaces in the Pingdingshan mining area, employing experiments such as contact angle measurements and scanning electron microscopy tests. Their findings indicated the significant influence of moisture content, ash composition, mass fraction of inorganic mineral constituents, and OFG content on coal seam wettability. Notably, Group E coal seams exhibited the highest content of inorganic mineral SiO2, characterized by rough and loose surface morphology, indicative of pronounced wettability (Zhang et al., 2021). Additionally, Zhang et al. investigated the relationship between coal rank and water wettability through experimental analysis and molecular simulation. They deduced that low-rank coals exhibited the highest hydrophilicity, while bituminous coals exhibited lower wettability. The number of hydrogen bonds emerged as a pivotal criterion for assessing wettability strength (Zhang et al., 2020). Furthermore, Zhao et al. analyzed the bonding mechanism between OFGs in coal molecules and H2O molecules using density functional theory (DFT). Their analysis revealed that H 1 s and O 2p orbitals interacted to form hydrogen bonds, with carboxylate groups exhibiting the strongest affinity for H2O molecules (Zhao and Liu, 2022). Zhao et al. employed electrostatic potentials and frontier orbital analyses in DFT to predict the relative strength of wettability across various OFG surfaces. Their findings were consistent with the calculated interaction energies between functional groups and H2O molecules (Zhao et al., 2023). In another study, Xu et al. utilized first-principle molecular dynamics to simulate the ordered arrangement of H2O molecules on lignite surfaces. They determined that the distance between the carboxyl group and H2O molecules required for hydrogen bond formation ranged from 0.151 to 0.186 nm. Additionally, a stable hydrogen bonding network formed among H2O molecules proximal to the carboxyl group. Conversely, ether bonds did not engage in hydrogen bonding with H2O molecules; instead, hydrogen bonding was exclusively observed between H2O molecules (Xu et al., 2022). Jiang et al. conducted macroscopic experiments and MD simulations to examine the interaction between different functional groups and water molecules. Both sedimentation experiments and contact angle tests indicated that an increase in OFGs promoted enhanced hydrophilicity on coal surfaces. Moreover, the interfacial hydrophilicity was notably stronger in polar group simulation systems compared to nonpolar ones, corroborating experimental findings (Jiang et al., 2023). In a related investigation, Jin et al. explored H2O adsorption behavior on Tashan bituminous coal by constructing a coal macromolecule model and employing GCMC simulations. Their analysis revealed that the quantity of adsorbed H2O molecules was contingent upon temperature and pressure. Notably, a significant increase in adsorption occurred, leading to the formation of water clusters at 298.15 K and 20 kPa, indicating possible capillary coalescence (Jxxin et al., 2017). Zhang et al. delved into the impact of pore structure on the wettability of LRC using the GCMC method and MD simulations. They observed that H2O adsorption in LRC pores conformed to Langmuir isothermal adsorption, with adsorption quantity escalating with porosity. Additionally, H2O predominantly existed in clustered form within larger pores (Zhang et al., 2022). While studies have predominantly investigated the influence of OFGs on the interaction between coal and H2O molecules, the significance of targeting coal pore structure on H2O adsorption has been scarcely explored. Analysis of multilayer adsorption of H2O molecules from various perspectives, particularly concerning the LRC slit-pore model and energy distribution during H2O adsorption, remains an underexplored area requiring further study.
LRC is distinguished by its abundant reserves and cost-effective extraction processes worldwide, making it a viable energy resource. The implementation of coal bed water injection serves a multitude of purposes, including enhancing coalbed methane recovery rates, mitigating gas outbursts, reducing coal dust suspension concentrations to enhance mine safety, and modifying coal bed mechanical properties to improve stability. Given the inherent connection between LRC and H2O molecules in practical applications, employing molecular simulation to scrutinize H2O adsorption characteristics in LRC assumes paramount importance. In this study, we investigate the impact of coal pore structure on H2O adsorption and diffusion at a molecular level. To accomplish this, we developed LRC slit-pore models with varying pore diameters. Utilizing GCMC simulation, we examined the adsorption quantity, isosteric heat of adsorption, energy distribution, and probability density field distribution of H2O molecules across nanoporous, microporous, and mesoporous LRC models to elucidate the adsorption mechanism. Additionally, MD simulations were employed to analyze the kinetic properties of H2O in distinct LRC slit-pore models. The findings of this research provide theoretical foundations for further exploration into coal pore wetting and offer valuable insights for studies aimed at reducing dust and preventing gas outbursts.
2 Methods
2.1 Model building and optimization
The coal molecule comprises a continuous polycyclic aromatic hydrocarbon structure composed of carbon atoms and containing various heteroatoms. In this study, the Wender coal model, illustrated in Fig. 1(a), was selected to represent the LRC molecule (Xia et al., 2019). The molecular formula of the Wender coal model is C43H46O12, with a molecular weight of 754 and elemental content of 25.46 % O, 6.10 % H, and 68.44 % C. The abundant OFGs and fewer branched chains of the Wender coal model are consistent with the structural features of the LRC. Extensive research has demonstrated the congruence of the Wender coal model with LRC properties (Zhao and Liu, 2023; Zhang et al., 2020), confirming its suitability for investigating the adsorption-diffusion characteristics of H2O in LRC slit pores. Geometry Optimization tasks were conducted using the Materials Studio software to optimize the structural energy of individual coal molecules. Periodic boundary conditions were applied to these molecules to simulate realistic coal behavior. Subsequently, the single coal molecules were arranged within a crystal cell measuring 10.15 × 10.15 × 10.15 Å using the Amorphous Cell module. The cell model was simulated with geometry optimization and annealing kinetics at temperatures ranging from 298 K to 1498 K. Upon simulation, the density stabilized at 1.12 g/cm3, the energy was minimized and stabilized, and the optimal configuration was achieved, as depicted in Fig. 1(b). Geometry optimization and annealing were performed with medium accuracy, employing the COMPASS II force field to assign corresponding force field parameters to the atoms in the model. Details of the force field parameters are provided in the Appendix, along with the use of Forcefield assigned to assign charges (Meng et al., 2021; Xia et al., 2019). The COMPASS II force field was selected for its broader applicability and higher accuracy compared to the Dreiding force field. Numerous scholars have employed the COMPASS II force field to investigate coal adsorption characteristics with various gas molecules, yielding results consistent with experimental findings (Tan et al., 2022; Wang et al., 2020). Long-range electrostatic interactions were analyzed using Ewald summation with an accuracy of 1.0 × 10-3 kcal/mol, while van der Waals interactions were examined via Atom-based analysis with a truncation radius of 12.5 Å (You et al., 2019). Detailed parameter settings are outlined in Table 1. Finally, the single-cell model underwent cell expansion to construct the slit-pore model, as illustrated in Fig. 1(c). The structure of the H2O molecule is presented in Fig. 1(d), with an optimized bond length of 0.93 Å and a bond angle of 104.55°.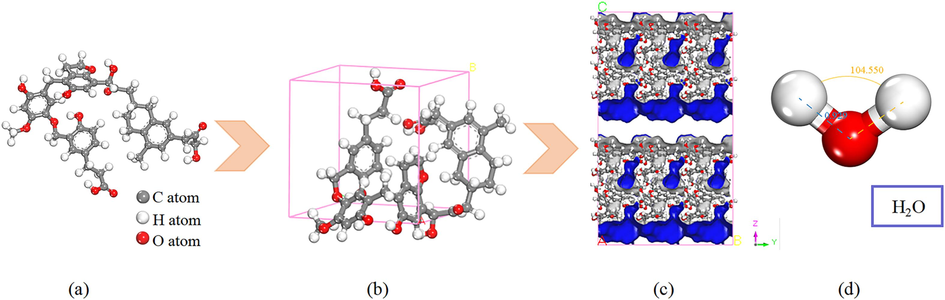
Structure of coal and H2O molecules: (a) Wender coal molecule, (b) single-coal cell model, (c) slit pore model, and (d) H2O molecule.
Geometry Optimization
Algorithm
Smart
Max.iterations
5 × 103 (Zhao and Liu, 2022)
Energy
1 × 10-3 kcal/mol
Force
0.5 kcal/mol/Å (Zhao and Liu, 2023)
Anneal
Annealing cycles
5
Total number of steps
6 × 105
Ensemble
NPT (Zhao and Liu, 2022)
Time step
1.0 fs
Thermostat
Andersen
Barostat
Berendsen
Coal possesses a natural porous structure characterized by complexity. According to the International Union of Pure and Applied Chemistry (IUPAC), pores are categorized into macropores, mesopores, and micropores, with size boundaries set at 50 nm and 2 nm, while micropores smaller than 1 nm are referred to as nanopores (Sharon et al., 2015). Microporous pores constitute the majority of coal pores, whereas macroporous pores are less prevalent. In this study, we investigate the H2O adsorption mechanism in coal by establishing models of LRC with nanopores of 0.5 and 1 nm, micropores of 1.5 and 2 nm, and mesopores of 2.5 and 3 nm. First, the single-cell model was expanded to 3 × 3 × 2 cells. Subsequently, slit-shaped pore structures with varying pore diameters were constructed by introducing vacuum layers with different thicknesses between the two layers of expanded cell models using the Build Layers tool. Fig. 2 illustrates the different slit hole models and cell parameters. The model conforms to the orthorhombic crystal system with interprong angles α, β, and γ set at 90°, prong lengths a and b measuring 3.12 nm, and prong lengths c measuring 5.39/5.89/6.39/6.89/7.39/7.89 nm, respectively. To ensure comparability with the LRC slit-pore model (b)(c)(d)(e)(f) depicted in Fig. 2, model (a) does not impose restrictions such as rotations on the H2O molecular configuration. Additionally, the empty space above the LRC model contains H2O molecules during the simulation. However, the impact of these molecules on H2O adsorption in the slit pores is minimal and can be disregarded.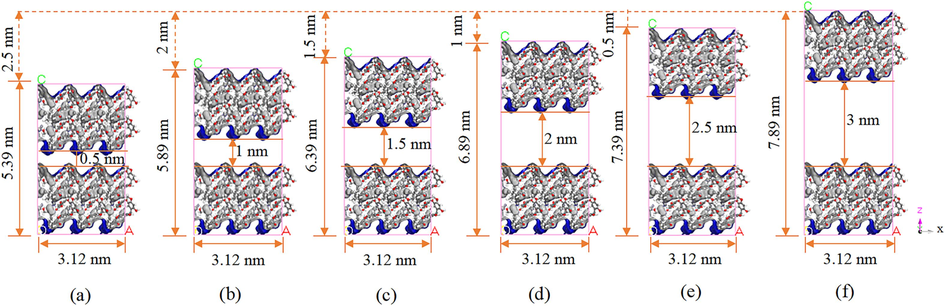
LRC molecular slit pore model illustrating free volume (blue areas) and volume occupied by the atomic skeleton (gray areas) at various pore sizes: (a) 0.5 nm, (b) 1 nm, (c)1.5 nm, (d) 2 nm, (e) 2.5 nm, and (f) 3 nm.
2.2 Simulation and calculation methods
2.2.1 GCMC simulations
Before adsorption simulations, the structural optimization of the LRC slit-pore model was performed. The Materials Studio software facilitated the calculation of isothermal adsorption and kinetic properties of H2O molecules within various LRC slit pores. Isothermal adsorption of LRC slit-pore model was performed using the Sorption module based on the GCMC method. The Adsorption isotherm task term was employed, with the computational method selected as Metropolis (Mosher et al., 2013), and the adsorbate designated as the geometry-optimized H2O molecule. The simulation encompassed a fugacity range of 0.001–100 kPa, with 10 fugacity steps, conducted at a temperature of 298 K. A total of 2 × 106 Monte Carlo steps were executed during the isothermal adsorption simulation, with 106 equilibration steps followed by 106 production steps for statistical averaging. The parameter settings for the force field and non-covalent interaction forces mirrored those used in the geometry optimization task item. Upon completion of the isothermal adsorption, data on isothermal adsorption quantity, isosteric heat of adsorption, probability density field distribution, and energy distribution were obtained.
2.2.2 MD simulations
The kinetic properties of H2O within LRC slit pores of varying diameters were investigated through simulations conducted using the Locate task item within the Sorption module, with the loading amount set to the saturated adsorption level achieved during isothermal adsorption. The maximum loading steps were set at 1 × 106, with 1 × 106 production steps and temperature controlled at 298 K over four temperature cycles. Subsequently, geometry optimization and MD simulations were executed using the Forcite module for different LRC slit-pore models/H2O molecular systems. Canonical ensemble NVT was employed for MD simulations (Nie et al., 2023), with the Nose thermostat selected to maintain a constant temperature of 298 K (Song et al., 2017). The initial rate followed a Boltzmann random distribution. Each simulation comprised 1 × 106 steps, with a total simulation time of 1000 ps and a time step of 1 fs. Equilibration of the system was allowed for the first 500 ps, with data from the subsequent 500 ps utilized for characterizing the kinetic behavior of H2O molecules within the LRC slit-pore model, including H2O diffusion and the interaction strength between H2O and the LRC slit-pore model.
3 Results and discussion
3.1 Adsorption isotherms
The adsorption isotherms of H2O molecules within the LRC slit-pore model, featuring various pore sizes, are illustrated in Fig. 3(a). Additionally, the Freundlich isothermal adsorption model was employed for fitting, yielding satisfactory results as depicted in Fig. 3(b). Analysis of Fig. 3 reveals that, at the same fugacity, larger apertures of the slit pores corresponded to higher H2O adsorption amounts. For instance, at a fugacity of 100 kPa, the adsorption amounts of H2O by 0.5, 1, 1.5, 2, 2.5, and 3 nm LRC slit pores were 16.55, 22.05, 27.74, 33.51, 38.43, and 44.23 mmol/g, respectively. Compared with the 0.5 nm nanopore slit model, the other five LRC slit-pore models exhibited increased adsorption by 33.19 %, 67.56 %, 102.44 %, 132.15 %, and 167.20 %, respectively. This phenomenon can be attributed to the dependency of H2O adsorption in the LRC model on both the kinetic diameter of the H2O molecule itself and the aperture of the slit pore. The kinetic diameter of the H2O molecule was 0.28 nm, with pore diameters greater than 0.28 nm serving as effective adsorption pore diameters within the determined LRC model. Hence, the pore diameters of the slit pores primarily dictate the adsorption amount. Below a fugacity of 10 kPa, the adsorption of H2O in the LRC slit pores of varying sizes exhibited gradual increases, indicating monomolecular layer adsorption. Beyond 10 kPa, a sharp increase in H2O molecule adsorption was observed, signifying the formation of multilayer adsorption and the appearance of numerous water clusters until the slit pores of low-rank coal reached saturation. An increase in fugacity leads to adsorption saturation, reaching adsorption equilibrium at a specific fugacity point where the adsorption rate stabilizes. Notably, the fugacity point of adsorption equilibrium varied across the LRC slit-pore models of different pore sizes. For instance, adsorption equilibrium occurred at approximately 50 kPa for a pore diameter of 0.5 nm, at 60 kPa for pore diameters of 1 and 1.5 nm, and at approximately 80 kPa for a pore diameter of 2 nm. Moreover, no equilibrium was observed even at fugacities exceeding 80 kPa for pore diameters of 2.5 and 3 nm. The greater the pore size of the LRC slit-pore model, the higher the fugacity point of adsorption equilibrium, making adsorption saturation more challenging. The existence of multilayer adsorption of H2O molecules on the LRC slit-pore model was indicated by the energy distribution (Fig. 6) and the interaction energy of H2O molecules with the LRC slit-pore model in Fig. 10. Notably, the formation of potential energy bimodal peaks at low fugacities and the increasing interaction energy of individual H2O molecules with the LRC pore model with increasing pore diameter confirmed the occurrence of multilayer adsorption of H2O molecules. Yang et al. analyzed the adsorption behavior of low-rank coal and high-rank coal using water vapor isothermal adsorption tests and analyzed them using the Freundlich model (Yang et al., 2021). Their findings revealed a transition from monolayer adsorption of H2O molecules to multilayer adsorption with increasing water content, eventually leading to capillary condensation. They concluded that the difference in coal water absorption was primarily related to the degree of pore development, aligning with the conclusions drawn from the adsorption of H2O molecules in different LRC slit-pore models in this paper.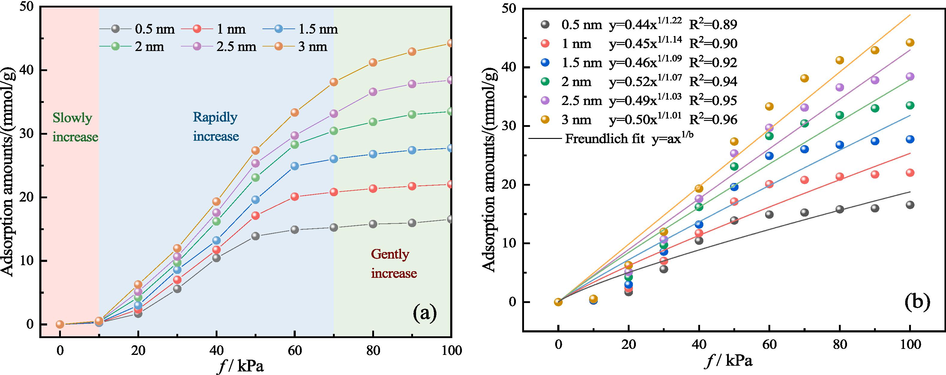
Adsorption amounts of H2O molecules at various pore diameters. (a) adsorption isotherm (b) Freundlich fit.
3.2 Isosteric heat of adsorption
H2O adsorption by the LRC slit-pore model was assessed from an energy perspective by analyzing the isosteric adsorption heat. Fig. 4 illustrates the relationship between the adsorbed H2O amount and the corresponding isosteric adsorption heat across different pore diameters. As depicted in Fig. 4, within the same LRC slit-pore model, the adsorption heat exhibited an exponential increase with rising H2O molecular adsorption capacity, exhibiting an average growth rate of approximately 160.56 %. When the adsorption amount remained below 0.56 mmol/g, the isosteric adsorption heat measured less than 42 kJ/mol, indicating physical adsorption where H2O primarily adsorbed onto LRC molecules at the main adsorption site (Zhao and Liu, 2022; Wang et al., 2020). However, upon surpassing the 0.56 mmol/g threshold, the isosteric adsorption heat increased sharply before stabilizing, consistently exceeding 42 kJ/mol. For example, a 3 nm slit pore adsorption of 44.23 mmol/g, the isosteric adsorption heat peaked at 70.40 kJ/mol, surpassing the range of physical adsorption. This notable increase suggests a shift in the adsorption site, where the H2O adsorbed on the LRC acts as the active site for adsorption (the second adsorption site) and thus adsorbs the H2O molecules. As the isosteric adsorption heat stabilized, it indicated saturation of the second adsorption site, with an accumulation of H2O molecules forming water clusters within the LRC slit pores. At this stage, the H2O-H2O interaction outweighed the interaction between LRC and H2O. Notably, there was no direct correlation between adsorption heat and LRC slit pore diameter. In summary, the adsorption mechanism of H2O by the LRC slit-pore model differs from that of other gases, characterized by multilayer adsorption, changing active adsorption sites, and a sharp increase in isosteric adsorption heat.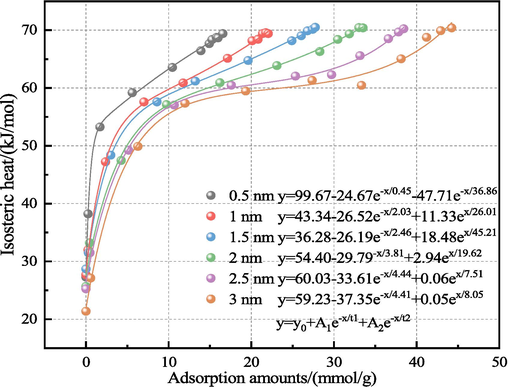
Correlation between adsorption amount and isosteric adsorption heat.
3.3 Probability density field distribution
Visualizing the adsorption of H2O within the LRC slit pores is facilitated by the probability density field distribution (Wu et al., 2019). Fig. 5 illustrates the fluctuation in H2O adsorption density within 0.5 nm LRC slit pores across eight fugacities: 1, 5, 10, 20, 30, 50, 70, and 100 kPa. The closer to the blue area in the figure, the higher the density of H2O molecules. At a fugacity of 1 kPa, H2O predominantly occupied the adsorption sites on the LRC surface, particularly the OFGs, which are inclined to form hydrogen bonds with H2O, while the presence of H2O within the slit pores was minimal. With an increase in fugacity to 5 kPa, albeit with the adsorbed H2O on the LRC escalated, a lower density within the slit pores, where the interaction between LRC and H2O prevailed. Subsequently, at 10 kPa fugacity, the density of H2O within both the LRC and the slit pores experienced a moderate increase, accompanied by a slight aggregation of H2O within the pores. By the time the fugacity reached 20 kPa, H2O concentration within the slit pores increased substantially, forming denser water clusters. When the fugacity increased to 50 kPa, the LRC slit pores approached saturation with H2O, exhibiting a very high density, signifying saturation of adsorption where H2O molecules occupied the second adsorption site. However, due to the aperture limitation of the slit pores, there was no substantial increase in H2O density when the fugacity was further increased to 100 kPa, akin to the degree of H2O aggregation observed at 50 kPa. This study comprehensively analyzed the probability density field of H2O within 0.5 nm slit pores across different fugacities, with the distribution of the probability density field of H2O in slit pores of other aperture sizes demonstrating similar patterns.
Probability density field distribution of H2O at different fugacities: (a) 1 kPa, (b) 5 kPa, (c) 10 kPa, (d) 20 kPa, (e) 30 kPa, (f) 50 kPa, (g) 70 kPa, and (h) 100 kPa.
3.4 Energy distribution
The number of adsorption sites and the strength of adsorption were determined based on the energy distribution of H2O during adsorption within the LRC slit-pore model. Fig. 6 depicts the energy distribution of H2O adsorption within 0.5 nm LRC slit pores at varying fugacities. The number of peaks indicated the number of adsorption sites, with lower energies suggesting stronger adsorption. At a fugacity of 1 kPa, a distinct potential energy peak around −27 kJ/mol, corresponding to the preferred adsorption site of H2O, was observed, indicating that all H2O molecules were adsorbed on the LRC. With increasing fugacity, the energy of H2O adsorption within the LRC slit pores exhibited a bimodal distribution, signifying the presence of two adsorption sites for H2O, each with different interaction regions. When the fugacity increased to 5 kPa, the potential energy peak at −27 kJ/mol gradually diminished, while the peak at −45 kJ/mol increased, indicating the emergence of a second adsorption site alongside the preferred one, although most H2O molecules were still adsorbed at the preferred site. The formation of the second adsorption site evolved gradually with increasing adsorption, with adsorbed H2O molecules serving as new adsorption sites. When the fugacity reached to 10 kPa, the potential energy peaks at −27 kJ/mol and −45 kJ/mol were close to each other, and the preferred and second adsorption sites occupied the same ratio. The emergence of potential energy bimodal peaks at low fugacity indicated the multilayer adsorption of H2O within the LRC slit-pore model and the existence of two adsorption sites for H2O molecules. Given that H2O molecules exhibit a Boltzmann distribution, the ratio of H2O molecules between the potential bimodal peaks at low fugacity was approximately 1:5, as revealed by the simulation of the RDF between H2O and H2O, with the peak area of the RDFH2O-H2O bimodal peaks being roughly 1:5. When the fugacity increased to 20 kPa, a peak in the −27 kJ/mol energy region was absent, and the only peak existed in the −45 kJ/mol potential energy region, shifting to a lower energy region (-50 kJ/mol). Thus, H2O saturated the preferred adsorption site, and all adsorption shifted to the second adsorption site, leading to enhanced H2O-H2O interactions and a substantial increase in the amount of H2O adsorbed within the LRC slit pores. When the fugacity increased to the range of 50 kPa to 100 kPa, the peak potential energy ranged from approximately −66 kJ/mol to −68 kJ/mol in the low-energy region, indicating a higher concentration of H2O adsorption in this region where the H2O-H2O interaction was the strongest. Specifically, at 50 kPa, the adsorption of H2O at the second adsorption site reached saturation, leading to the filling of the LRC slit pores with H2O. Svabova et al. conducted isothermal water vapor adsorption tests using three coal samples and concluded that adsorption primarily occurred at the primary adsorption site at lower pressures. However, with increasing pressure, H2O molecules gradually shifted to the secondary adsorption site, wherein secondary adsorption became more prominent (Svabova et al., 2011). These findings aligned with the simulation results.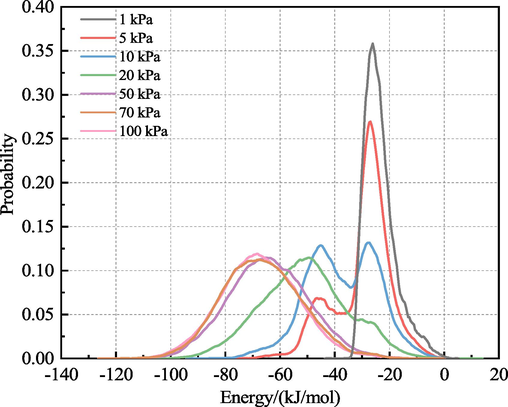
Energy distribution of H2O in the LRC slit-pore model.
A comparison of the energy distribution of H2O adsorbed in the LRC slit pores at various fugacities revealed a shift in adsorption sites for H2O molecules. Increasing fugacity resulted in a shift of the potential energy distribution of H2O toward lower potential energy, with the high energy region representing the preferred adsorption site. Conversely, in the case of CO2 gas adsorption in coal, the potential energy distribution occurred in the region with the lowest interaction energy, indicating the preferential adsorption site for CO2. The position of the potential energy peak shifted to the right with increasing fugacity. These differences in the direction of potential energy movement and the preferential adsorption sites of H2O molecules and CO2 gas indicated notable variations in their adsorption properties. The adsorption mechanism of H2O in coal was assessed based on the energy distribution of H2O in the LRC slit-pore model, confirming the conclusions drawn from the adsorption amount of H2O molecules, the distribution of probability density field, and the adsorption heat. These findings provided a deeper understanding of the adsorption behavior of H2O molecules in coal.
3.5 Kinetic properties of H2O molecules
3.5.1 Diffusion properties of H2O
The size of the LRC pore diameter influenced the migratory diffusion characteristics of H2O. The mean square displacement (MSD) and the self-diffusion coefficient (D) helped elucidate the diffusion rate of H2O within the slit pores of varying pore diameters. Stable data from 0 to 250 ps were selected to analyze the MSD and calculate the self-diffusion coefficient using the following expression (Hao et al., 2022):
The self-diffusion coefficient was derived from the Einstein equation with the following expression (Hao et al., 2022):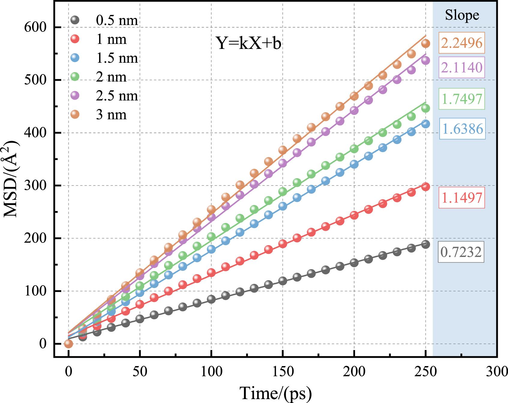
Self-diffusion coefficients of H2O in the LRC slit-pore model.
Slit pore aperture
MSD fitting curves
R2
Self-diffusion coefficient (D, cm2/s)
0.5 nm
Y = 0.7232 X + 9.7789
0.9981
1.21 × 10-5
1 nm
Y = 1.1497 X + 15.2632
0.9975
1.92 × 10-5
1.5 nm
Y = 1.6386 X + 12.7939
0.9992
2.73 × 10-5
2 nm
Y = 1.7497 X + 20.6512
0.9969
2.92 × 10-5
2.5 nm
Y = 2.1140 X + 20.7099
0.9978
3.52 × 10-5
3 nm
Y = 2.2496 X + 21.5591
0.9974
3.75 × 10-5
3.5.2 Radial distribution function (RDF)
The RDF characterizes the distribution of one class of atoms or molecules in the system relative to another class of atoms or molecules within a specific distance r + dr (Zhao and Liu, 2022; Xiang et al., 2014). It responds to the level of ordering of H2O within the LRC slit pores of varying pore sizes using the following expression (Zhou et al., 2022):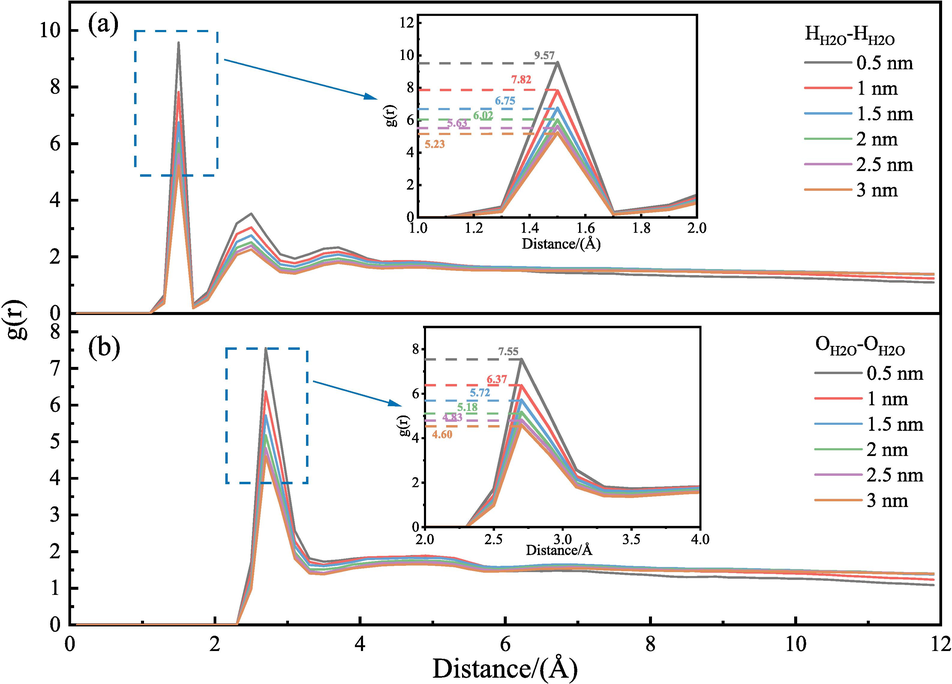
RDF between atoms in H2O molecule.
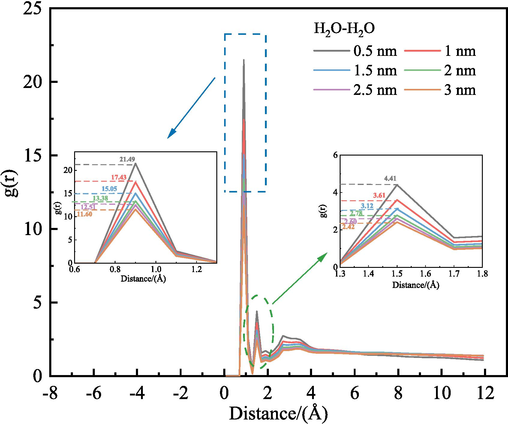
RDF between H2O molecule and H2O molecule.
3.5.3 Effect of pore size of slit pores on the adsorption of LRC/H2O
The strength of H2O adsorption was evaluated by calculating the interaction energies between different LRC slit-pore models and H2O. The script was utilized to analyze 101 frames of interaction energy post-model adsorption equilibrium, focusing on the last 80 frames to calculate the average interaction energy. The expression for each frame of interaction energy/adsorption energy is as follows (Ding et al., 2023):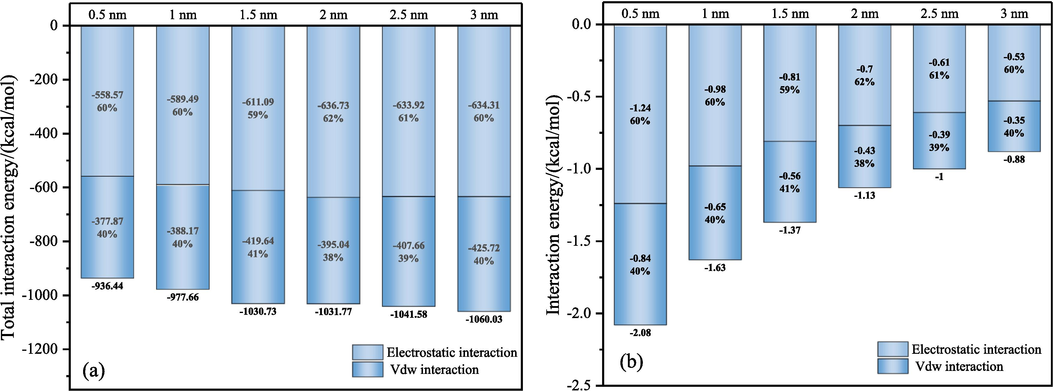
Interaction energy between H2O (individual H2O molecules) and LRC slit-pore models.
Fig. 11 illustrates the relative concentration distribution of H2O molecules along the Z-axis in the LRC slit-pore/H2O system. The adsorption rate of H2O in the LRC slit pores was 33.36 % when the pore diameter was 0.5 nm. With an increase in pore diameter, the adsorption rate of H2O gradually rises, exceeding 50 % for all pore diameters exceeding 1 nm. When the pore diameter reaches 3 nm, the adsorption rate peaks at 74.75 %. A substantial amount of H2O is adsorbed in the LRC slit with larger pore sizes, weakening the interaction of individual H2O molecules with the LRC. Conversely, fewer H2O molecules are adsorbed in small aperture slit pores, resulting in greater force exerted by low-rank coal in micropores compared to mesopores on individual H2O molecules. The pore diameter magnitude significantly influences H2O adsorption in the LRC slit-pore model, which is pivotal for the investigation of coal pore wetting.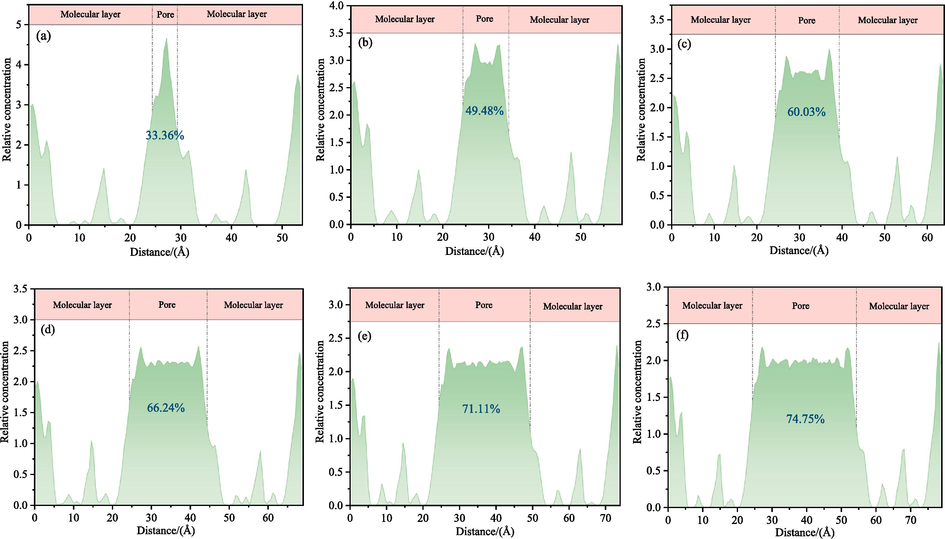
Relative concentration distribution of H2O molecules along the Z-axis: (a) 0.5 nm, (b) 1 nm, (c) 1.5 nm, (d) 2 nm, (e) 2.5 nm, and (f) 3 nm.
4 Conclusions
This study investigated the influence of LRC slit pore size on the adsorption-diffusion properties of H2O through GCMC and MD simulations, enhancing the understanding of H2O adsorption mechanisms in LRC and providing new insights at the molecular scale.
The adsorption of H2O in LRC slit pores exhibited a positive correlation with fugacity, and adsorption was enhanced with increasing slit pore diameter. This adsorption followed a multilayer pattern. Adsorption primarily occurred on the LRC surface and within the slit pores.
For adsorption of H2O molecules in the LRC slit pores, the potential energy distribution occurred in the region with the highest interaction energy, indicating the preferential adsorption site for H2O molecules. The position of the potential energy peak shifted to the left with increasing fugacity. Larger pore diameters corresponded to stronger interactions between H2O and the LRC slit-pore model, while interactions between individual H2O molecules and the model decreased. The bimodal distribution of energy upon adsorption of H2O molecules and the decreasing interaction of individual H2O molecules with the LRC slit-pore model with increasing pore diameter confirmed the occurrence of multilayer adsorption of H2O molecules.
The peak RDF between hydrogen atoms, oxygen atoms, and H2O molecules in the H2O decreased with increasing pore diameter. Conversely, the diffusion rate gradually enhanced. The increase in LRC slit pore diameter facilitated H2O molecule self-diffusion. The analysis of the RDF matched the results of the calculated self-diffusion coefficients.
The microscopic mechanisms governing H2O molecules adsorbed by the LRC slit-pore model were elucidated through molecular simulation, which may provide valuable insights for future studies aimed at reducing dust and preventing gas outbursts.
CRediT authorship contribution statement
Dan Zhao: Conceptualization, Funding acquisition, Methodology, Supervision. Xiaoqing Liu: Data curation, Writing – original draft, Writing – review & editing, Visualization, Software, Investigation, Validation.
Acknowledgments
This work was supported by the National Natural Science Foundation of China(52374202) and the Liaoning Provincial Department of Education Project(LJ2019JL025). The authors would like to thank all the reviewers who participated in the review, as well as MJEditor (www.mjeditor.com) for providing English editing services during the preparation of this manuscript.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular simulation of N2 and CO2 injection into a coal model containing adsorbed methane at different temperatures. Energy. 2021;219:119686
- [Google Scholar]
- Investigation on mechanism of the oleic acid/methyl oleate/diesel ternary compound collector in low-rank coal flotation. Fuel. 2022;320:123894
- [Google Scholar]
- Study on coal wettability under different gas environments based on the adsorption energy. ACS Omega. 2023;8:22211-22222.
- [Google Scholar]
- Mechanism of micro-wetting of highly hydrophobic coal dust in underground mining and new wetting agent development. Int J Min Sci Techno. 2023;33:31-46.
- [Google Scholar]
- Microkinetic models with interacting adsorbates and langmuir-hinshelwood steps: solving the master equation on small periodic tiles. J. Phys.Chem C. 2023;127(49):23687-23695.
- [Google Scholar]
- Adsorption and diffusion of methane in coal slit pores: insights into the molecular level. Energy Fuels. 2022;36:880-886.
- [Google Scholar]
- Temperature dependence of water cluster on functionalized graphite. Carbon. 2021;183:380-389.
- [Google Scholar]
- Molecular dynamics simulations and experimental characterization of the effect of different concentrations of [Bmim][Cl] in aqueous solutions on the wettability of anthracite. Fuel. 2022;324:124618
- [Google Scholar]
- Investigation on the effect of functional groups on the wettability of coal dust: experiments and theoretical validation. Fuel. 2023;351:128987
- [Google Scholar]
- H2O adsorption mechanism in coal basing on Monte Carlo method. J. China Coal Soc.. 2017;42(11):2968-2974.
- [Google Scholar]
- H2O adsorption mechanism in coal basing on Monte Carlo method. J. China Coal Soc.. 2017;42(11):2968-2974.
- [Google Scholar]
- Molecular simulation of adsorption of gas in coal slit model under the action of liquid nitrogen. Fuel. 2019;255:115775
- [Google Scholar]
- Wetting and interfacial properties of water on the defective graphene. J. Phys. Chem. C. 2013;117:14106-14112.
- [Google Scholar]
- Effect of mixed cationic/anionic surfactants on the low-rank coal wettability by an experimental and molecular dynamics simulation. Fuel. 2021;289:119886
- [Google Scholar]
- Adsorption and diffusion characteristics of CH4, CO2, and N2 in micropores and mesopores of bituminous coal: molecular dynamics. Fuel. 2021;292:120268
- [Google Scholar]
- Studying the micromechanism of water injection to suppress coal and gas protrusion. AIP Adv... 2022;12:125014
- [Google Scholar]
- Study of the wetting mechanism of SDBS solution on zhaozhuang coal surface. J. China Univ. Min. Technol.. 2021;50(2):381-388.
- [Google Scholar]
- Molecular simulation of methane adsorption in micro-and mesoporous carbons with applications to coal and gas shale systems. Int. J. Coal Geol.. 2013;109–110:36-44.
- [Google Scholar]
- Study on the effect of different surfactants on the wettability of coal dust based on Dmol3 module. J. China Coal Soc.. 2023;48(3):1255-1266.
- [Google Scholar]
- Insight into the low-rank coal flotation using amino acid surfactant as a promoter. Fuel. 2022;307:121810
- [Google Scholar]
- Liquid nitrogen’s effect on the mechanical properties of dried and water-saturated frozen coal. Energy Fuels. 2022;36:1894-1903.
- [Google Scholar]
- Pore characteristics of wilcox group coal, U.S. Gulf Coast region: implications for the occurrence of coalbed gas. Int. J. Coal Geol.. 2015;139:80-94.
- [Google Scholar]
- An analysis of contributing mining factors in coal workers’ pneumoconiosis prevalence in the United States coal mines. Int. J. Coal Sci. Techn.. 2021;8(6):1227-1237.
- [Google Scholar]
- Molecular simulation of CH4/CO2/H2O competitive adsorption on low rank coal vitrinite. Phys. Chem. Chem. Phys.. 2017;19:17773-17788.
- [Google Scholar]
- Langmuir’s theory of adsorption: a centennial review. Langmuir. 2019;35(16):5409-5426.
- [Google Scholar]
- Molecular simulation for physisorption characteristics of O2 in low rank coals. Energy. 2022;242:122538
- [Google Scholar]
- Research on adsorption characteristics of H2S, CH4, N2 in coal based on Monte Carlo method. Sci. Rep.. 2020;10:21882.
- [Google Scholar]
- Examination of adsorption behaviors of carbon dioxide and methane in oxidized coal seams. Fuel. 2020;273:117599
- [Google Scholar]
- Potential impact of CO2 injection into coal matrix in molecular terms. Chem. Eng. J.. 2020;401:126071
- [Google Scholar]
- Effect of vacancy defects on electronic properties and wettability of coal surface. Appl Surf Sci. 2020;511:145546
- [Google Scholar]
- Molecular simulation of coal-fired plant flue gas competitive adsorption and diffusion on coal. Fuel. 2019;239:87-96.
- [Google Scholar]
- Enhancement of the surface hydrophobicity of low-rank coal by adsorbing DTAB: an experimental and molecular dynamics simulation study. Fuel. 2019;239:145-152.
- [Google Scholar]
- Improving the adsorption of oily collector on the surface of low-rank coal during flotation using a cationic surfactant: an experimental and molecular dynamics simulation study. Fuel. 2019;235:687-695.
- [Google Scholar]
- Molecular simulation of the CH4/CO2/H2O adsorption onto the molecular structure of coal. Sci. China Earth Sci.. 2014;44(7):1418-1428.
- [Google Scholar]
- Research framework and anticipated results of deep rock mechanics and mining theory. Adv. Eng. Sci.. 2017;49(2):1-16.
- [Google Scholar]
- Research review of the state key research development program of China: deep rock mechanics and mining theory. J. China Coal Soc.. 2019;44(5):1283-1305.
- [Google Scholar]
- Quantum chemical caculation of the interaction between lignite and water molecules. J. China Univ. Min. Technol.. 2022;51(3):554-561.
- [Google Scholar]
- Research on differences of adsorbed water characteristics of low rank and high rank coals based on water vapor adsorption model. Safety in Coal Mines. 2021;52(4):7-12.
- [Google Scholar]
- Role of oxygen functional groups for structure and dynamics of interfacial water on low rank coal surface: a molecular dynamics simulation. Mol. Phys.. 2018;116(13):1670-1676.
- [Google Scholar]
- Influence of surfactant for improving dewatering of brown coal: a comparative experimental and MD simulation study. Sep. Purif. Technol.. 2019;210:473-478.
- [Google Scholar]
- Interaction mechanism of water movement and gas desorption during spontaneous imbibition in gas-bearing coal. Fuel. 2022;318:123669
- [Google Scholar]
- Molecular simulation investigation on the effect of pore structure on the wettability of low-rank coal. J. China Univ. Min. Technol.. 2022;51(6):1117-1127.
- [Google Scholar]
- Micro-wetting characteristics of coal dust and preliminary study on the development of dust suppressant in Pingdingshan mining area. J. China Coal Soc.. 2021;46(3):812-825.
- [Google Scholar]
- New insight into surface wetting of coal with varying coalification degree: an experimental and molecular dynamics simulation study. Appl. Surf. Sci.. 2020;511:145610
- [Google Scholar]
- Density functional calculation of H2O/CO2/CH4 for oxygen containing functional groups in coal molecules. ACS Omega. 2022;7:17330-17338.
- [Google Scholar]
- Effect of ionic surfactants on flotation of low-rank coal: a DFT calculation and MD simulation study. Mol. Phys.. 2022;120:e2140719.
- [Google Scholar]
- Monte Carlo and molecular dynamics simulations of CH4 molecules adsorption behavior in bituminous coal. Int. J. Low-Carbon Tec.. 2022;17:879-887.
- [Google Scholar]
- Study on the microscopic mechanism of adsorption and diffusion of hydrocarbon oil drops on coal surface using molecular dynamics simulations. Int. J. Quantum Chem.. 2023;123(23):e27229.
- [Google Scholar]
- Effect of oxygen-containing functional groups on the wettability of coal through DFT and MD simulation. Arab. J. Chem.. 2023;16(4):104606
- [Google Scholar]
- Reactive adsorption mechanism of O2 onto coal vitrinite during the low temperature oxidation process. Fuel. 2022;308:121802
- [Google Scholar]
- Upgrading low-quality oil shale using high-density gas-solid fluidized bed. Fuel. 2019;252:666-674.
- [Google Scholar]
- Gas flow laws in coal subjected to hydraulic slotting and a prediction model for its permeability-enhancing effect. Energ Source Part A 2021
- [Google Scholar]
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2024.105697.
Appendix A
Supplementary data
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




