

Translate this page into:
Molecular mechanics and dynamics simulation of CD-47/SIRPα blockade study: A computational study on overcoming immunotherapeutic resistance in pancreatic ductal adenocarcinoma
⁎Corresponding author. saioluwatosin@gmail.com (Oluwatosin A. Saibu)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The sting of pancreatic ductal adenocarcinoma remains an irksome burden to the human populace. The focus of recent research has switched from finding the perfect medicinal medication to blocking immunological checkpoint proteins such as the signal regulating protein alpha-cluster of differentiation 47 (SIRPα-CD47). The search for CD47/SIRPα inhibitors with excellent oral bioavailability and permeability continues to elude researchers. This research aims to identify bioactive molecules with negligible side as inhibitor SIRPα-CD47 signaling cascade. Bioactive flavonoids from African medicinal plants were virtually screened against the SIRP-α binding site of CD47 using Schrodinger suite 2017-v1. The docking score was validated and complex stability performed using Gromacs. Among the bioactive flavonoids, five (5) compounds were predicted as potent inhibitors of CD47 with pelargonidin observed to have the best binding affinity of −6.715 kcal/mol. Validation using QSAR and pharmacophore modeling further confirm the interaction with predicted pIC50 range of 5.981 to 6.841 µM and fitness score of 1.109 to 1.530. Drug-likeness prediction revealed that all hit compounds obey Lipinski’s rule of five. The MD simulation result predicted the stability of pelargonidin and malvidin comparable to the standard drug NCGC00138783. The quantum mechanics estimation revealed that, the hit compounds have proton donating and accepting ability hence, they possess inhibitory potential. From the molecular docking, post-docking and MD simulation analyzes of this study, Pelargonidin, malvidin and Peonidin were proposed as suitable candidates that could be probed further for developing a new target-specific immunotherapeutic agent against PDAC.
Keywords
PDAC
CD47
SIRPα
ADME
In silico
Immunotherapeutic

1 Introduction
Over the past ten years, cancer has remained one of the top causes of death despite extensive study and rapid advancement (Sandeep and Sobhia, 2018). Pancreatic ductal adenocarcinoma (PDAC) was described by World Health Organization as one of the most prevalent and fatal types of cancer and one of the contributors to this unpleasant burden (Da-Costa et al., 2020). The increase in recently identified cases of pancreatic ductal adenocarcinoma is still a topic of discussion in cancer research. The cure remains elusive, and multiple attempts at developing the optimum treatment for PDAC, a disease with low survival, have been unsuccessful (Beatty et al., 2017). As a result, research has shifted toward immunotherapy. Immunotherapy is a cancer treatment strategy that takes advantage of the immune system's specificity and heterogeneity (Yang, 2015). Unfortunately, PDAC has resisted immunotherapy, varying from small molecule antibodies to viral modifications during treatment and various alterations in its signaling pathway could explain the reasons behind resistance mechanisms (Alausa et al., 2022). The limitation of cytotoxic T-cell responses to cancer cells is distinguishing trait cancers developed to evade host immunological responses. To treat and manage various malignancies, immunotherapeutic research has recently moved its focus to block checkpoint proteins such as PD-1, CTLA-4, LAG3, TIM3, TIGI T, and BTLA (Sarantis et al., 2020). In healthy conditions, immune checkpoint proteins regulate immune response by limiting autoimmunity. However, they hinder cytotoxic T-cell activity in cancer by inhibiting interactions between T-cells and antigen-presenting cells or malignant cells (Sharma and Allison, 2015).
Since its emergence in the late 1990s, the SIRPα-CD47 checkpoint has been demonstrated to be essential for cancer immune evasion (Jiang et al., 1999; Logtenberg et al., 2019). Fig. 1 showed the crystal structure of the CD47 with the red, green, grey and blue colours representing the N-terminal amino acids, C-terminal amino acids, α-helixes and β-fold sheets respectively.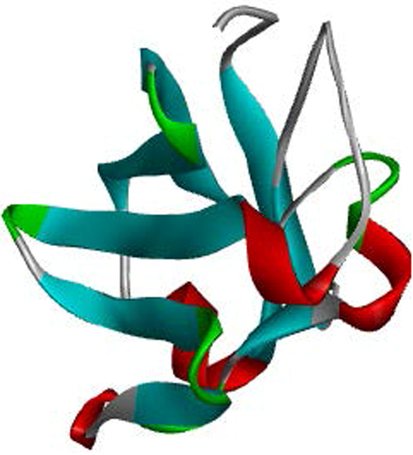
Crystal structure of CD47.
All human cells express the trans-membrane protein CD-47, but specific tumour cells have high levels of this protein (Zola et al., 2020). It is a cell surface glycoprotein related to the immunoglobulin superfamily that binds to several different proteins, including signal regulatory protein (SIRP), thrombospondin 1, and integrin (Zola et al., 2020). Numerous cancers rely on the tumour antigen CD47 for growth and spread. The focus of research has been on creating therapeutic drugs that significantly disrupt the SIRPα-CD47 signalling cascade. This cascade prevents dendritic cells from phagocytosing tumours, deactivating innate immunity, and ultimately leading to tumour regression (Alausa et al., 2022). Phagocytosis is inhibited by the interaction of CD47 with SIRPα, which sends the macrophages a “don't eat me” signal (Brown and Frazier, 2001). As a result, tumour cells might avoid immune monitoring by the inhibition of phagocytic processes when CD47 is overexpressed. It takes more than only CD47 inhibition to activate macrophage anti-tumour action. Recent studies indicated that the CD47-SIRPα axis, like the PD-1/PD-L1 in solid tumours is an essential immune checkpoint in various cancers.
Traditional medicine has a rich history of using natural substances to treat cancer. Additionally, about 60% of current anti-cancer therapeutics was derived from natural products and medicinal plants (Takahashi, 2018). Hence, this research focuses on discovering natural compounds capable of alleviating immunotherapeutic resistance in PDAC by targeting the CD47 using computational molecular mechanics, molecular dynamics, and quantum investigations.
2 Methods
The E-pharmacophore hypothesis development and screening, virtual screening, MM-GBSA calculation and QSAR modeling were performed using Maestro Schrodinger 2017v1 software. The ADME analysis was performed using web server, quantum calculation was carried out using Spartan 14 software and MD simulation by Gromacs. All computational studies were performed on a Dell computer with a Windows-10 OS, an Intel core i3 processor, and 8 GB RAM.
2.1 Protein preparation
The three-dimensional X-ray crystallographic structure of a complex of human signal regulatory protein SIRPα complex with CD47 (PDB ID: 2JJS) was chosen. The protein was prepared using the Schrodinger suite's protein preparation package. The preparation included the assignment of hydrogen bonds, bond orders, hydrogen addition, optimization, protein minimization, removal of all chains excluding chain C (CD47 chain), and deletion of waters (Newman et al., 2003).
2.2 Grid generation
The receptor grid was created using the amino acids found on the surface of CD47 that are important for binding to SIRPα (Sandeep and Sobhia, 2018). GLU 35, THR 99, ARG 103, GLU 97, THR 102, GLU 100, GLU 104, LEU 101 and GLU 106 are among the amino residues. The coordinates for the X, Y, and Z axis are 28.98, −14.73, and 35.32 respectively.
2.3 Ligand preparation
Flavonoids with cancer-inhibitory properties from medicinal plants were retrived from literature. The flavonoids' three-dimensional structure was downloaded in Sdf format from Pubchem database. The compounds were prepared with the Maestro Schrodinger software's LigPrep module . Fig. 2 shows the initially screened top ranking ligand structures as well as their chemical names.
2D Structure and name of the selected flavonoids.
2.4 Structure-based screening
GLIDE structure-based screening includes three precision methods: HTVS, standard precision (SP) docking, and XP docking. This study used two docking precisions to obtain a potential lead molecule quickly. HTVS can swiftly screen a vast number of molecules; however, the sampling techniques were constrained, making it difficult to interpret the results. As a result, SP was used to dock all the ligands (16) with high glide score from the HTVS precision analysis, which chooses an appropriate binding pose from a broad pool of ligands.
2.5 Generation of pharmacophore hypothesis
The standard drug (NCGC00138783) was used to generate an energy optimized pharmacophore model for the crystal structure of CD47. The model was generated from protein-ligand option of the phase develop pharmacophore tool of schrodinger suite (2017–1) (Omoboyowa et al., 2023). The pharmacophore model is shown in Fig. 3.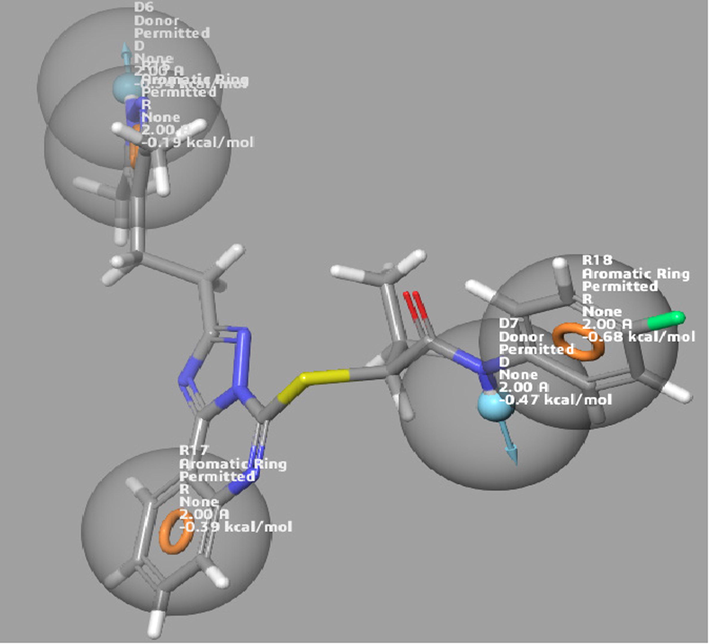
The pharmacophore hypothesis generated with the standard drug.
The virtual screening base on E-pharmacophore was performed with the five (5) flavonoids with top docking scores after preparation using macro model minimization. The pharmacophore-based analysis was carried out with phase module to generate a subset of molecules having chemical features for binding to CD47 according to the generated model. The fitness scores were used to justify the best hits.
2.6 Development of automated QSAR model
By blasting the FASTA sequence of the protein received from PDB, the protein inhibitors with their corresponding IC50 were extracted from the CHEMBL database (https://www.ebi.ac.uk/chembl/) with the chain C (CD47 chain) sequence (shown below) used for the blasting. The inhibitory chemicals were translated to sdf format using the Data-Warrior software (v.2) (Omoboyowa et al., 2022). The sdf format of the inhibitors was imported into the Schrodinger suite workspace and prepared using the macro model minimization tool. The QSAR model for the protein was developed. The best-projected rank was kpls molprint 24, with the model's prediction precision measured by the ranking score, RMSE, SD, Q2, and R2. The pIC50 of a lead compound was predicted using this approach.
2.7 CD47 FASTA sequence for CHEMBL blast
>2JJS_2|Chains C|LEUKOCYTE SURFACE ANTIGEN CD47|HOMO SAPIENS (9606).
QLLFNKTKSVEFTFGNDTVVIPCFVTNMEAQNTTEVYVKWKFKGRDIYTFDGALNKSTVPTDFSSAKIEVSQLLKGDASLKMDKSDAVSHTGNYTCEVTELTREGETIIELKYRVVSWSTRHHHHHH.
2.8 Free binding energy calculation (MM-GBSA)
The docking results showed that the selected ligands bind to the protein's active site via the receptor grid. However, it was unclear if this binding would be sufficient to cause a biological response as this depends primarily on the free binding energies of the protein–ligand complex. The binding free strengths of the top-ranking compounds and the reference drug were determined using the MM-GBSA module of the Schrodinger software.
2.9 Quantum chemical methods
Theoretical calculations have been widely studied through a powerful tool denoted as the Density Functional Theory method (DFT) due to its structural and spectral explanation of organic molecules. All calculations were performed and computed by spartan 14 software by wavefunction Inc on the top five hit compounds. The study was carried out with complete optimization of all geometrical variables via 6-31G* basis set, and this was accomplished with B3LYP density functionals (Huang et al., 2021). Frontier molecular orbitals (FMOs) energy was calculated, i.e., for the top five hit compounds identified, which by calculation deduce the energy band gap (Eq (1), which according to Koopman's theorem, predicts the interacting center.
2.10 Insilco ADMET prediction
We were interested in the drug-likeness properties of the top five hits identified through molecular docking. This was accomplished with the help of the QikProp module. Then, additional ADME properties were obtained from the online web server AdmetSar (https://lmmd.ecust.edu.cn/). The ligands' chemical structures were provided. The structures were translated to their canonical simplified molecular-input line-entry system (SMILE) to determine the ligand's physicochemical properties and pharmacokinetic models (Jensen, 2001). The drug-likeness of can be used to establish if a ligand is suitable for oral administration. The in silico prediction is based on Lipinski's rules (molecular weight, hydrogen bond donor, and hydrogen bond acceptor) (Cheng et al., 2012).
2.11 Molecular dynamic (MD) simulation
After the docking screening, the top ligand–protein complexes were further submitted to molecular dynamics simulation. Other approaches are needed for validation because the dynamics of the complexes were not considered during the molecular docking procedure. Using molecular dynamic simulations, the stability of docked Protein-Ligand complexes was evaluated. LiGRO (Yao et al., 2017) was used to set up the simulation system, and a GUI-based tool produced the system file needed to execute the simulation. The target was dissolved in a cubic box filled with TIP3P molecules of water with 150 mM NaCl ion concentration. The target was parameterized using the Amber99sb force field, and the ligand molecules were parameterized using LiGRO's ACPYPE module. The study by Omoboyowa et al. served as the guide for preparing all other simulation systems and parameters. GROMACS 5.1.5 was used to generate a run for each complex system that lasted 100 ns (Omoboyowa et al., 2022).
2.12 MD simulation trajectories analysis
Using conventional GROMACS tools, the trajectories of the MD simulation were evaluated for the root mean square fluctuation, radius of gyration (RG), H-bond mapping and root mean square deviation. The generated trajectories were shown in the PyMOL visualization graphics system (Version 2.0 Schrödinger, LLC.). Using the Molecular-dynamics-Interaction-plot tool, the interaction proportions of the target residues interacting with the compounds were computed (Jiang et al., 2021).
3 Results and discussion
3.1 Molecular docking, MMGBSA and interaction profiling
The protein CD47 was docked by the selected flavonoid derivatives to predict binding energy and interaction with the protein. Before docking was carried out, the protein was analyzed for its residues, which were vital for interacting with its partner protein CD-47.
Pelargonidin and (+)-Gallocatechin exhibited the highest binding energies of −6.715 kcal/mol and −6.353 kcal/mol respectively compared to the standard drug, which has a binding energy of −3.445 kcal/mol (Fig. 4). Thus, the docking results were analyzed, and it was finally reported that among the top flavonoid compounds, Pelargonidin and (+)-Gallocatechin exhibit the best binding interaction, essential in identifying and developing new therapeutics targeting CD47. This will block its interaction with CD47 as a strategy to prevent cancer. The 2D interaction diagram shows the residues involved in the ligand's binding (Fig. 5) and the 3D binding complexes were presented in Fig. 6. Pelargonidin, the top-ranked ligand, formed H-bonds with THR 34, GLU 97, LEU 101, and GLU 104. While the other ligands including the standard drug, interacted via H-Bond with GLU 104, a critical amino residue in the CD47 binding site. Malvidin and Peonidin had another unique interaction with TYR 37, called Pi-Cation. VAL 36, TYR 37, ALA 53, and LEU 101 were the same interacting hydrophobic amino acids in the top-ranked ligand and the standard drug. Additionally, all reported ligands, including the standard drug, have similar interactions with the following key hydrophobic amino acids: TYR 37, ALA 53, and LEU 101 as presented in Table 1. The formation of H-bond and other hydrophobic interactions between small molecules and the amino acid residues at the binding domain of the target is vital for their inhibitory potential (Omoboyowa, 2022). The Schrodinger suite's Prime module's MM-GBSA approach was used to calculate the binding energies of the top compounds with the highest docking scores. The lower the score, the higher the binding energy, this method provide a reliable statistical post-docking examination of docked complexes. The relative free binding energies of pelargonidin, (+)-gallocatechin, malvidin, peonidin, and catechin are −29.25, −15.87, −29.59, −24.92, and −16.00, respectively. Pelargonidin, Malvidin, and Peonidin exhibit higher binding energies than the reference drug, per the results of the MM-GBSA (−22.31).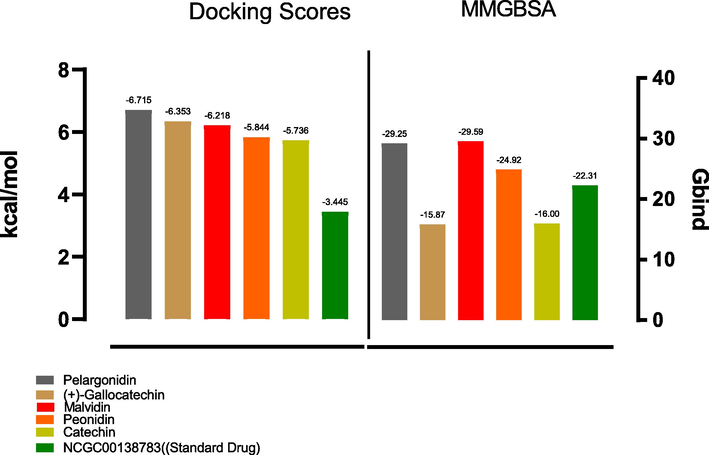
Graph depicting the docking and MM-GBSA scores for the top five hits including the standard drug.
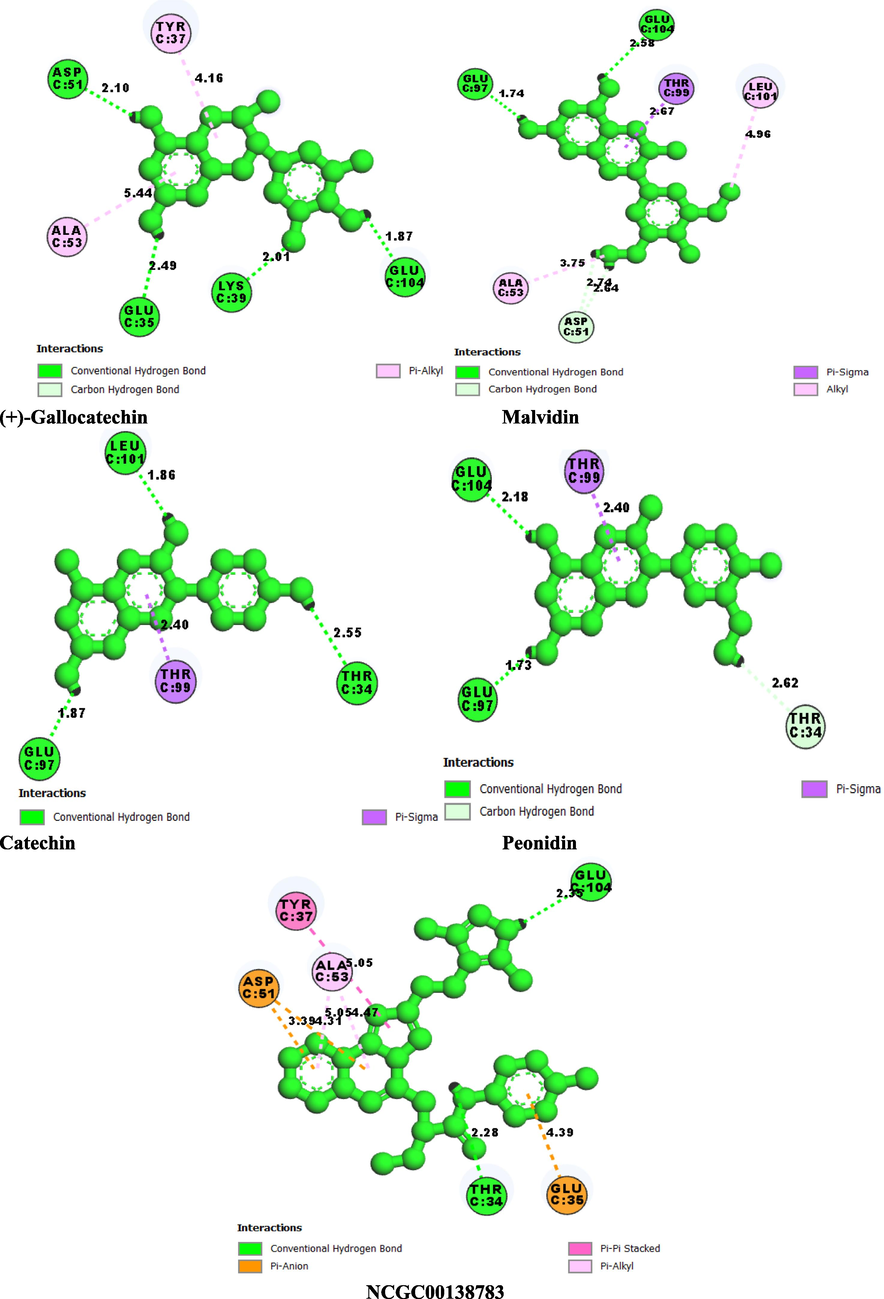
2D molecular interaction SIRPα binding pocket with hit compounds. (a) Pelargonidin (b) (+)-Gallocatechin (c) Malvidin (d) Peonidin (e) Catechin (f) NCGC00138783(Standard Drug).
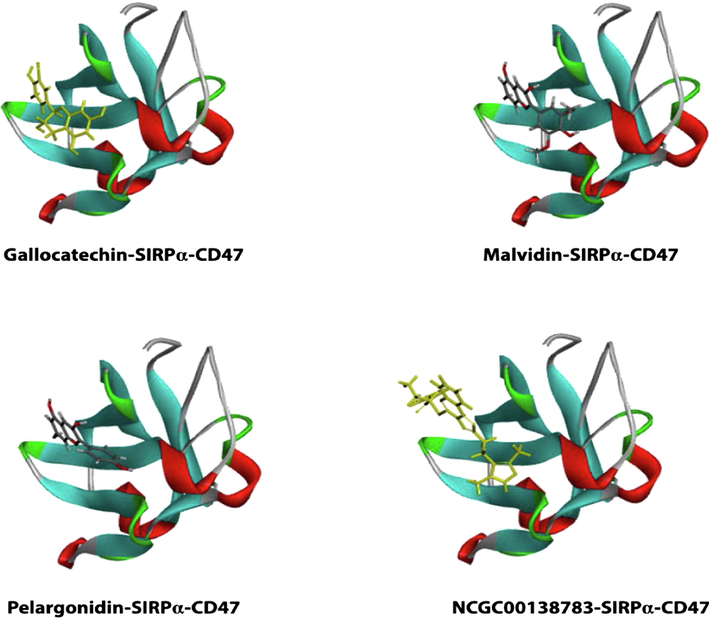
3D representation of hits with binding pocket of SIRPα.
Entry Name
H-Bond Residues
Hydrophobic Interacting amino acids
Other Interactions
Pelargonidin
LEU 101, GLU 104, GLU 97, THR 34
VAL 36, TYR 37, ALA 53, LEU 101
None
(+)-Gallocatechin
GLU 97, LYS 39, ASP 51, GLU 35, GLU 104
LEU 101, TYR 37, VAL 36, ALA 53
None
Malvidin
THR 34, LYS 39, GLU 97, LEU 101, GLU 104
ALA 53, TYR 37, LEU 101
Pi-Cation: TYR 37
Peonidin
ASP 51, LYS 39, GLU 104
ALA 53, VAL 36, TYR 37, LEU 101
Pi-Cation: TYR 37
Catechin
GLU 35, LYS 39, ASP 51, GLU 97, GLU 104
VAL 36, TYR 37, ALA 53, LEU 101
None
NCGC00138783(Standard Drug)
ASP 51, GLU 104
VAL 36, TYR 37, ALA 53, LEU 101
PI-PI STACKING: TYR 37
3.2 Virtual screening using E-pharmacophore model
Ligand-based pharmacophore approach is a vital computational model for drug design without macromolecular protein structure. This hypothesis is an ensemble of electronic and steric characters which are vital in ensuring molecular interactions with specific biological molecules and to stimulate or inhibit signaling pathways of such protein (Yang, 2010; Omoboyowa et al., 2023). In this study, Herein, the e-pharmacophore model was developed based on the standard drug-CD47 complex using four partitioning features. Fig. 3 showed the generated hypothesis with the standard drug. The model with the best fitness score has one hydrogen bond acceptor, two hydrogen bond donors, one aromatic ring and hydrophobic interaction.
The fitness scores of the five (5) hits and standard drug are shown in Table 2. All the hit compounds showed fitness score greater than 1.0 with (+)-gallocatechin and catechin having the highest score of 1.191 followed by Catechin (1.531). Although the standard drug showed higher fitness scores higher (1.530).
Entry Name
Fitness
Pelargonidin
1.185
(+)-Gallocatechin
1.109
Malvidin
1.191
Peonidin
1.143
Catechin
1.191
NCGC00138783(Standard Drug)
1.530
3.3 Auto QSAR analysis
As it uncovers the correlations between the structural characteristics of chemical compounds and their biological activities, the quantitative structure–activity relationship (QSAR) is a computational model significant in drug development (Omoboyowa et al., 2023). AutoQSAR is a machine-learning approach that generates streams of independent variable-building models with various topological and physiochemical descriptors (Dixon et al., 2016). The autoQSAR model divides the test compounds into a 25% test and a 75% train set, as shown in Table 5. The best predictive model from the experimental data was determined using the based partial least-squares regression (kpls) analysis: kpls molprint2D 29. The model parameters resulted in a standard deviation (S.D.) of 0.4354, R2 of 0.8774, RMSE of 0.4220, and Q2 of 0.8706 (Table 3). The experimental compounds' model showed more training sets (blue colour) than the test set (red colour) as observed in the scatter plot in Fig. 7. The distribution of the test and train set in Fig. 7 was consistent with the distribution observed in Table 5. The QSAR model was use to obtain the pIC50 of the hit compounds shown in Table 5, All the hit compounds and standard drug showed high pIC50 above 5.00 μM with pelargonidin and peonidin (6.001 μM) showing better pIC50 among the hit compounds comparable with the standard drug (6.841 μM).(See Table 4)
Model code
S.D
R2
RMSE
Q2
kpls_molprint2D_24
0.4354
0.8774
0.4220
0.8706
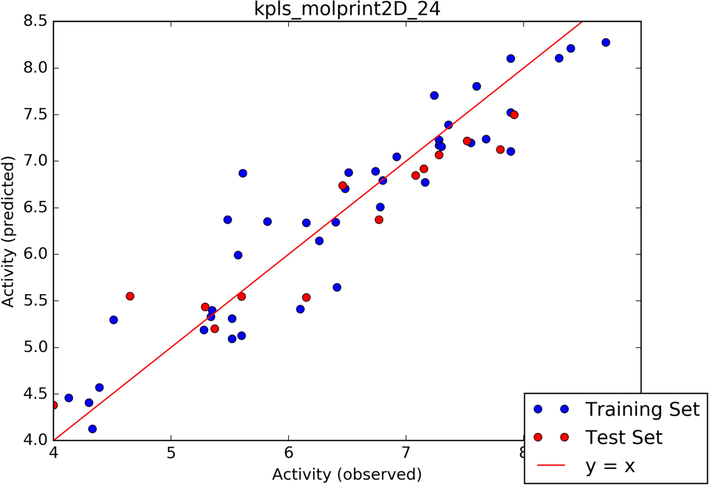
Scatter plot of the best model.
ID
Set
pIC50 (observed)
pIC50 (predicted)
Residue error
1
Train
6.800
6.7053
0.2253
2
Train
7.2800
7.2276
−0.0524
3
Train
7.3600
7.3898
0.0298
4
Train
6.4100
5.6460
−0.7640
5
Train
6.7800
6.5088
−0.2712
6
Train
6.8000
6.7940
−0.0060
7
Train
4.3300
4.1273
−0.2027
8
Train
5.2800
5.1881
−0.0919
9
Train
7.8900
8.1051
0.2151
10
Train
5.5200
5.3111
−0.2089
11
Train
4.5100
5.2974
0.7874
12
Train
4.3900
4.5702
0.1802
13
Test
4.6500
5.5514
0.9014
14
Train
6.1500
6.3404
0.1904
15
Train
5.6100
6.8730
1.2630
16
Train
7.2800
7.1699
−0.1101
17
Train
6.5100
6.8792
0.3692
18
Train
7.2400
7.7064
0.4664
19
Train
6.7400
6.8927
0.1527
20
Train
4.3000
4.4081
0.1081
21
Train
6.4000
6.3470
−0.0530
22
Test
7.1500
6.9184
−0.2316
23
Test
4.0000
4.3802
0.3803
24
Train
7.6000
7.8038
0.2038
25
Train
7.5500
7.1972
−0.3528
26
Train
5.4800
6.3736
0.8926
27
Test
7.0800
6.8466
−0.2334
28
Test
6.4600
6.7386
0.2786
29
Train
5.5700
5.9920
0.4220
30
Train
7.8900
7.1059
−0.7841
31
Test
7.2800
7.0674
−0.2126
32
Train
7.3000
7.1560
−0.1440
33
Train
7.1600
6.7734
−0.3866
34
Test
5.2900
5.4361
0.1461
35
Train
7.8900
7.5223
−0.3677
36
Test
5.6000
5.5471
−0.0529
37
Test
7.8000
7.1266
−0.6734
38
Train
5.6000
5.1285
−0.4715
39
Train
6.1000
5.4129
−0.6871
40
Train
5.3400
5.3328
−0.0072
41
Train
5.3500
5.3987
0.0487
42
Test
7.5200
7.2172
−0.3028
43
Train
7.6800
7.2379
−0.4421
44
Train
8.7000
8.2774
−0.4226
45
Train
8.4000
8.2109
−0.1891
46
Train
7.6000
7.8038
0.2038
47
Train
6.9200
7.04796
0.1276
48
Train
5.8200
6.3538
0.5338
49
Test
5.3700
5.2028
−0.1672
50
Train
4.1300
4.4591
0.3291
51
Test
6.7700
6.3736
−0.3964
52
Train
8.3000
8.1063
−0.1937
53
Test
6.1500
5.5388
−0.6112
54
Test
7.9200
7.4993
−0.4207
55
Train
5.5200
5.0927
−0.4273
56
Train
6.2600
6.1460
−0.1140
Pubchem ID
Compound Name
Predicted pIC50 (µM)
65,084
(+)-Gallocatechin
5.981
159,287
Malvidin
5.981
440,832
Pelargonidin
6.001
441,773
Peonidin
6.001
NCGC00138783
Reference Drug
6.841
3.4 Quantum calculations
The development of density functional theory (DFT) of small molecules is an important tool use to describe the molecular interactions between molecules and gives information concerning electron transfer within the molecule which is required to predict the chemical stability and reactivity of a molecule (Balogun et al., 2021). The energy of high occupied molecular orbital (EHOMO) and low unoccupied molecular orbital (ELUMO) and energy gap (Eg) estimated from the quantum mechanics calculations are vital in predicting molecular reactivity of bioactive compounds (Omoboyowa et al., 2023). From the results presented in table Fig. 8, the complete geometry optimization of the selected flavonoid derivatives at their low energy level, in which all analysis were computed on these least optimized flavonoid molecules.
HOMO and LUMO density for the five flavonoid derivatives.
From the results presented in Table 6 and Fig. 8, the electron donation potential of the hit compounds was suggested by the EHOMO values which range from −5.63 to −9.25 eV and the electron accepting potential was predicted by the ELUMO values which range from −6.12 –n0.08 eV. Hence, the high EHOMO value and lower ELUMO value are necessary for the predicted reactivity of the compounds. Energy gap (Eg) is the difference between the HOMO and LUMO energies and has been reported by Uzzaman and Mahmud (2020), as a predictor of the molecule stability and reactivity, higher value of Eg denote greater stability with less bioavailability and low reactivity (Omoboyowa et al., 2023). The Eg of the hits were estimated between the range of 2.56 to 5.71 eV suggesting that the hit compounds are reactive and stable molecules.(See Table 7 and Table 8.
Compounds
Pelargonidin
−9.25
−6.34
2.91
(+)-Gallocatechin
−5.65
0.02
5.67
Malvidin
−8.68
−6.12
2.56
Peonidin
−8.83
−6.19
2.64
Catechin
−5.63
0.08
5.71
Entry Name
mol MW
Hbond Acceptors
Hbond Donors
iLogP
Polar Surface Area
Rule of Five
Pelargonidin
271.24
5
4
−2.44
94.06
Suitable
(+)-Gallocatechin
306.27
7
6
1.47
130.61
Suitable
Malvidin
331.30
7
4
−1.96
112.52
Suitable
Peonidin
301.27
6
4
−1.94
103.29
Suitable
Catechin
290.27
6
5
1.33
110.38
Suitable
NCGC00138783(Standard Drug)
503.59
6
2
3.13
126.16
Suitable
Model
Pelargonidin
(+)-Gallocatechin
Malvidin
Peonidin
Catechin
Ames mutagenesis
–
–
–
–
+
Acute Oral Toxicity (c)
III
IV
III
III
IV
Blood Brain Barrier
+
–
+
+
–
Caco-2
–
–
+
+
–
Carcinogenicity (binary)
–
–
–
–
–
CYP1A2 inhibition
–
–
+
+
–
CYP2C19 inhibition
–
–
+
+
–
CYP2C9 inhibition
–
–
+
+
–
CYP2C9 substrate
–
–
–
–
–
CYP2D6 inhibition
–
–
–
–
–
CYP2D6 substrate
–
+
–
–
+
CYP3A4 inhibition
–
–
+
+
–
CYP3A4 substrate
+
–
–
–
–
Hepatotoxicity
+
+
+
+
–
Human Ether-a-go-go-Related Gene inhibition
–
–
–
–
–
Human Intestinal Absorption
–
+
+
+
+
Human oral bioavailability
–
–
–
–
–
Acute Oral Toxicity
1.435115576
1.636945605
1.2353605
0.9769318
1.418269
P-glycoprotein inhibitor
–
–
+
–
–
P-glycoprotein substrate
–
–
–
–
–
PPAR gamma
+
+
+
+
+
Plasma protein binding
0.852887869
0.877940595
0.881655
0.9295143
0.987759
Subcellular localization
Nucleus
Mitochondria
Nucleus
Nucleus
Mitochondria
UGT catelyzed
–
+
+
+
+
Water solubility
−3.09788922
−3.101451725
−3.4489542
−3.343575
−3.101452
3.5 Prediction of physicochemical and ADMET-TOX properties
The in silico drug-likeness predictions are founded on Lipinski's rule of five; hydrogen bond donor, hydrogen bond acceptor and molecular weight (Cheng et al., 2012). The draggability of a molecule is generally based on this rule. To ascertain if substances could penetrate the central nervous system, the blood–brain barrier penetration was evaluated (Table 5) (Karelson et al., 1996). Out of the five predicted molecules, only (+)-Gallocatechin and Catechin are likely to penetrate the blood–brain barrier. In humans, many cytochrome P450s catalyze the metabolism of various substances, including xenobiotics and medicines.
Thus, inhibition of cytochrome P450 isoforms may result in drug-drug interactions in which co-administered drugs fail to be metabolized and accumulate to toxic levels. The predicted compounds are CYP2D6 inhibitors and CYP2C9 substrates. All are predicted to be CYP3A4 substrates except for pelargonidin. Adverse drug administration-related effects are referred to as acute oral toxicity (Nyandoro, et al., 2018). Fortunately, all substances proved negative for acute toxicity and mutagenicity in the AMES test. Based on their acute oral toxicity, compounds are categorized into four classes. LD50 values for substances in Category I are less than or equal to 50 mg/kg. LD50 values for substances in Category II are higher than 50 mg/kg but lower than 500 mg/kg. LD50 values for substances in Category III are higher than 500 mg/kg but lower than 5000 mg/kg. The LD50 values of the substances in Category IV were higher than 5000 mg/kg. It is predicted that all the compounds will fall into Category III or IV. All of the compounds are thought to be non-carcinogenic. Drug solubility has previously been defined as a critical property of evaluating pharmaceuticals in the drug development cycle. This is because it helps to determine the concentration of the drug in the systemic circulation, resulting in a maximal optimal response (Walum, 1998). As a result of the presence of hydroxyl groups in the compounds, they were water-soluble. None of the substances is P-glycoprotein substrates (P-GB). The potassium channel, the human Ether-a-go-go Related Gene (hERG), is required for cardiac excitability control and regular heartbeat (Savjani et al., 2012). Since none of the chemicals inhibits the hERG gene, they cannot induce proarrhythmia. Pelargonidin, the lead compound, has been shown to have antioxidant properties after reducing oxidative markers. A research found the antioxidant efficacy of Pelargonidin via an increase in the level of the natural antioxidant enzymes catalase and superoxide dismutase (Lamothe et al., 2016; Mirshekar et al., 2010). The therapeutic potential of the screened compounds was determined using Christopher Lipinski's proposed rules of five (ROF) with a molecular weight of 500, HB donors of 5, HB acceptors of 10, and an octanol/water partition coefficient (log p 5) (Table 3) (Cheng et al., 2012). Except for gallocatechin, which possesses more than five HB donors, all the compounds met all the criteria.
3.6 Molecular dynamics simulation
Following the results generated from the molecular docking campaign, we employed the use of the following molecular dynamics parameters; RMSD, RMSF, HBOND and ROG in a 100 ns simulation to evaluate the dynamical stability of the three selected hit druglike candidates (Pelargonidin, (+)-Gallocatechin and Malvidin) and the reference drug (NCGC00138783). Their behavioral kinetics was also evaluated in both bound and unbound states with the protein structure (2JJS). Each complex was confined to an environment closely related to a normal physiological condition of a natural cell in terms of temperature, solvent, pressure, and ions throughout the simulation. To this end, we evaluated and compared the 100 ns spectrums of each prospective drug candidate complex against the reference drug (NCGC00138783).
3.7 Root mean square deviation
Using RMSD analysis, it is possible to determine the conformational and structural alterations of the backbone atoms of the protein–ligand entities (2JJS-NCGC00138783, 2JJS-GALLOCATECHIN, 2JJS-MALVIDIN, 2JJS-PELARGONIDIN and 2JJS). By comparison of RMSD spectrum of the referenced inhibitor and the unbound protein with the proposed hit compounds, one could speculate if the binding of the chemical entities is stable and capable of binding stably at the binding pocket of the receptor (Roy et al., 2017). Hence, we calculated and plotted the RMSD of each simulated complex for the entire 100 ns MD production run (Fig. 9). Looking at the RMSD spectrum, Pelargonidin and Malvidin depicted almost the same graphical pattern with the standard compound throughout the entire simulation suggesting their potential to act as a putative binder of the protein target. This is evidenced with their close average RMSD of 0.1899 nm and 0.186 nm respectively, compared with the 0.175 nm of the referenced compound. In contrast, Gallocatechin showed a different RMSD pattern compared to the standard drug and the apoprotein (2JJS). However, with mean RMSD value of 0.30 nm, the ligand average RMSD value falls below 0.5 nm value which is acceptable for a considerable stable system (Adelusi et al., 2020). Overall, the RMSD result showed that the binding of Malvidin, Pelargonidin, and Gallocatechin with the protein target are stable and suggest their ability to disrupt the SIRPα-CD47 signaling cascade.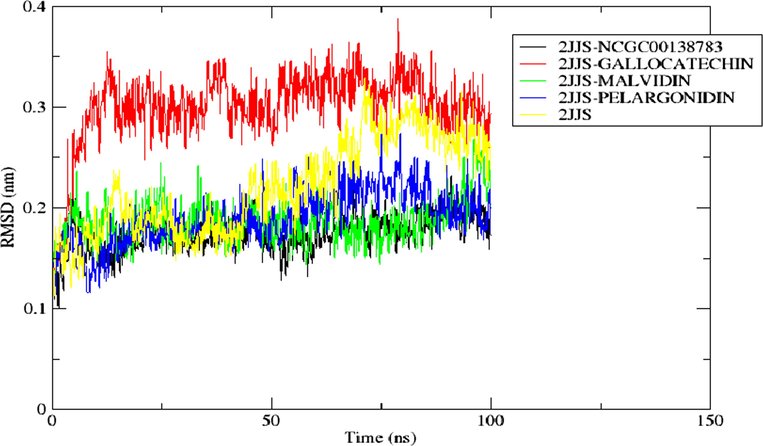
The RMSD of the backbone atom of the protein in top docking scored hit complexes throughout 100 ns simulation.
3.8 Root mean square fluctuation of the top docking scored hit complexes
For examining the structural fluctuation of the amino acid residues of the bound and unbound complexes, as well as changes in the position of the ligand, the Root Mean Square Fluctuation (RMSF) is a commonly used metric. A higher average RMSF value denotes a system with more fluctuating residues while a lower average value corresponds to a system with less fluctuating residues (Roy et al., 2017). According to the result represented in Fig. 12, all the bounded system including the standard (2JJS-GALLOCATECHIN, 2JJS-MALVIDIN, 2JJS-PELARGONIDIN, and 2JJS-NCGC00138783) demonstrate similar RMSF pattern. In comparison with the mean RMSF of the unbound receptor (0.199 nm), 2JJS-GALLOCATECHIN, 2JJS-MALVIDIN, 2JJS-PELARGONIDIN, and 2JJS-NCGC00138783, have lower average RMSF values of 0.146 nm, 0.135 nm, 0.152 nm, and 0.138 nm respectively. Interestingly, the RMSF result is consistent with the RMSD analysis which revealed that the binding of the ligands do not disrupt the conformational dynamics of the protein by depicting a lower average RMSD (with the exception of Gallocatechin) and RMSF values compared with the unbound form of the receptor (2JJS) (Fig. 10).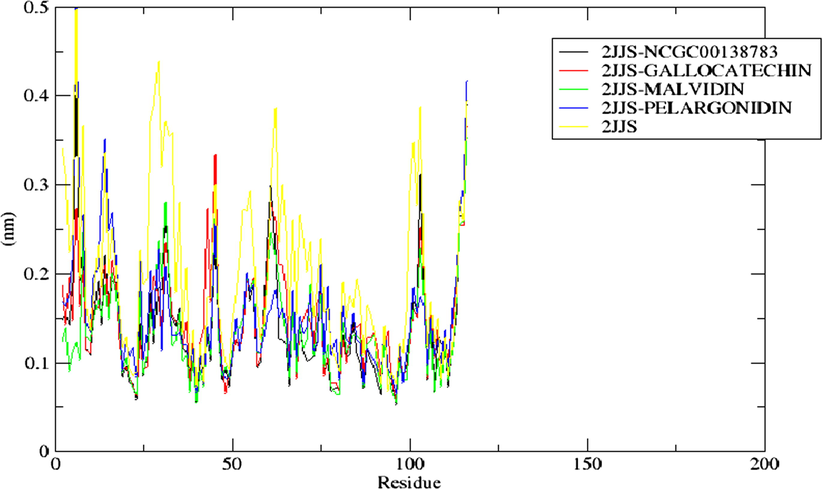
The RMSF of the residues present in the protein (2JJS) of top docking scored hit complexes throughout the simulation period.
3.9 Hydrogen bond mapping of top docking scored hit complexes
Intermolecular H-bond is an important type of interaction that exists between protein–ligand complexes. Higher H-bond could be responsible for the greater stability of a compound at the binding pocket of the receptor while lower H-bond may indicate a lower stable system (Lee et al., 2012). In the H-bond spectrum depicted in Fig. 11, Pelargonidin-one of the promising drug candidates showed thicker H-bond spectrum compared to all other binary complexes including the referenced inhibitor. Statistically, Pelargonidin, Malvidin, Gallocatechin, and the standard drug averaged 1.85, 0.44, 0.46, and 0.53 H-bonds respectively with their targeted receptor. Although Malvidin, and Gallocatechin have lower intermolecular H-bond, however, the difference between their values with the standard drug is not statistically significant and hence they could also be a good inhibitor of the protein using the H-bond metric.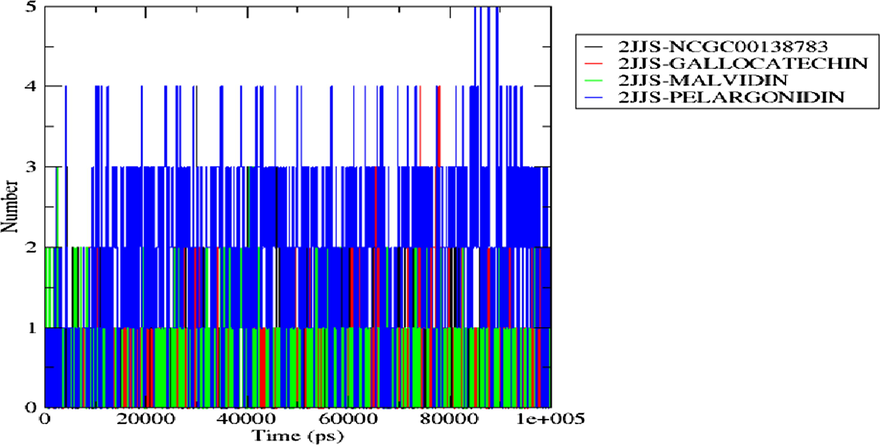
H-bond distribution of the top docking scored hit complexes during 100 ns simulation run in the active site of the protein target.
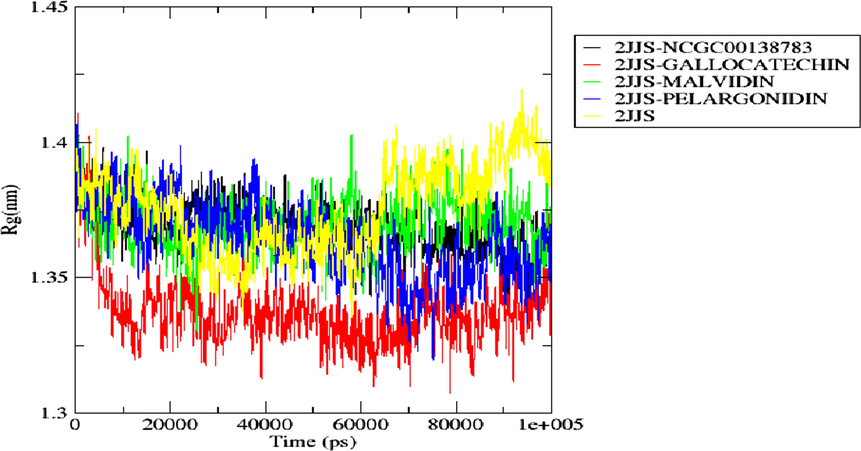
Radius of gyration spectrum of the top docking scored hit complexes during 100 ns simulation run.
3.10 Radius of gyration
The compactness of the secondary structures of the bound and unbound form of a simulated receptor is measured by radius of gyration (ROG). From this, one could determine if a system is stably folded or not. Fig. 12 shows the ROG plots of the apoprotein (2JJS) and the complexes (2JJS-NCGC00138783, 2JJS-Gallocatechin, 2JJS-Malvidin, 2JJS-Pelargonidin) as a function of time. Like the ROG result, Pelargonidin and Malvidin depicted similar ROG pattern with the standard drug throughout the entire simulation time. Their average ROG values are 1.364 nm, 1.370 nm, and 1.369 nm respectively. In contrast, Gallocatechin demonstrated different ROG pattern with the standard drug, but it has the lowest ROG value (1.337 nm) when compared with all other bounded systems. Our overall analysis of ROG indicates that the binding of the ligands does not distort the structural compactness of the receptor as they all averaged lower ROG vales when compared with the apoprotein (1.376 nm) as shown in Fig. 12.
4 Conclusion
The interaction of CD47-SIRPα signaling allows cancer cells to elude immune detection and clearance by suppressing phagocytic action. As a result, this research aimed to predict a therapeutic drug that may circumvent immunotherapeutic resistance in PDAC by suppressing SIRPα-CD47 signaling with minimal side effects in humans.
After computational analysis using molecular docking, molecular dynamics, and MM/GBSA quantum chemical calculations, the screened compounds (pelargonidin, malvidin, and peonidin) have higher binding energies than the reference medication. They also demonstrated better ideal stability and desirable intermolecular interactions in the selective pockets of CD47 compared to the reference medication (NCGC00138783). This study contributes to a better knowledge of the stability and interaction profile of protein–ligand complexes and the mechanism of inhibition involved in CD47-ligand complexes. According to the pharmacokinetic study, pelargonidin, malvidin and peonidin possess novel physicochemical characteristics and drug-like features. Additional in-vivo or in-vitro research is required to evaluate these ligands' pharmacological and biological activities in overcoming immunotherapeutic resistance in PDAC via CD47-SIRPα inhibition.
Author contributions
OAS conceived and designed the study. TMA, DSB, OOO and SOH contributed to the literature search; ATO, AJA and GOA contributed to data collection; OAS, TMA and DAO wrote the manuscript draft; OAS and TMA interpreted the results of the MD simulation; TMA OOO and ATO contributed to the figures. TAB, OAS, TMA, TTO, and AJA contributed to editing and proofreading of the manuscript. DAO supervised the research. All authors agreed to the final manuscript.
Acknowledgement
The authors wish to appreciate Olamide Olaoba (University of Missouri Columbia) for his insight during this study.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Molecular modeling in drug discovery. Inform. Med. Unlocked.. 2020;24:100880
- [CrossRef] [Google Scholar]
- Overcoming immunotherapeutic resistance in PDAC: SIRPα-CD47 blockade. Pharmacol. Res.. 2022;106264
- [Google Scholar]
- Computational evaluation of bioactive compounds from Colocasia affinis schott as a novel EGFR inhibitor for cancer treatment. Cancer Inf.. 2021;20:1-12.
- [CrossRef] [Google Scholar]
- Deploying immunotherapy in pancreatic cancer: defining mechanisms of response and resistance. Am. Soc. Clin. Oncol. Educ. Book. 2017;37:267-278.
- [CrossRef] [Google Scholar]
- Integrin-associated protein (CD47) and its ligands. Trends Cell Biol.. 2001;11:130-135.
- [Google Scholar]
- AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Information Model.. 2012;52:3099.
- [Google Scholar]
- Trends in the incidence of pancreatic adenocarcinoma in all 50united states examined through an age period Cohort analysis. JNCI Cancer Spectr.. 2020;4(4):33.
- [Google Scholar]
- AutoQSAR: an automated machine learning tool for best-practice quantitative structural-activity relationship modeling. Future Med. Chem.. 2016;8(15):1825-1839.
- [Google Scholar]
- Structural analysis and binding sites of inhibitors targeting the CD47/SIRPα interaction in anticancer therapy. Comput. Struct. Biotechnol. J.. 2021;19:5494-5503.
- [CrossRef] [Google Scholar]
- Polarization consistent basis sets: Principles. J. Chem. Phys.. 2001;115:9113-9125.
- [CrossRef] [Google Scholar]
- Integrin-associated protein is a ligand for the p84 neural adhesion molecule. J. Biol. Chem.. 1999;274:559-562.
- [Google Scholar]
- Targeting CD47 for cancer immunotherapy. J. Hematol. Oncol.. 2021;14:180.
- [CrossRef] [Google Scholar]
- Quantum-chemical descriptors in QSAR/QSPR studies. Chem. Rev.. 1996;96:1027-1043.
- [CrossRef] [Google Scholar]
- The Human Ether-a-go-go-related Gene (hERG) potassium channel represents an unusual target for protease-mediated damage. J. Biol. Chem.. 2016;291:20387-20401.
- [CrossRef] [Google Scholar]
- Conformational sampling of flexible ligand-binding protein loops. Bull. Kor. Chem. Soc.. 2012;33:770-774.
- [Google Scholar]
- Glutaminyl cyclase is an enzymatic modifier of the CD47-SIRP alpha axis and a target for cancer immunotherapy. Nat. Med.. 2019;61:619.
- [Google Scholar]
- Chronic oral pelargonidin alleviates streptozotocin-induced diabetic neuropathic hyperalgesia in rat: involvement of oxidative stress. Iran. Biomed. J.. 2010;14:33-39.
- [Google Scholar]
- Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod.. 2003;66:1022-1037.
- [Google Scholar]
- In silico pharmacokinetic and molecular docking studies of N-cinnamoyltetraketide derivatives as inhibitors of cyclooxygenase-2 enzyme. Tanzania J. Sci.. 2018;44:1-15.
- [Google Scholar]
- Exploring molecular docking with E-pharmacophore and QSAR models to predict potent inhibitors of 14-α-demethylase protease from Moringa spp. Pharmacol. Res.- Mod. Chin. Med.. 2022;4:100147
- [Google Scholar]
- Inhibitory potential of phytochemicals from Chromolaena odorata L. against apoptosis signal-regulatory kinase 1: a computational model against colorectal cancer. Comput. Toxicol.. 2022;23:100235
- [CrossRef] [Google Scholar]
- Structure-based in silico investigation of antagonists of human ribonucleotide reductase from Annona muricata. Inf. Med. Unlocked. 2023;38:101225
- [Google Scholar]
- Pelargonidin-PLGA nanoparticles: fabrication, characterization, and their effect on streptozotocin induced diabetic rats1. Indian J. Exp. Biol.. 2017;55:819-830.
- [Google Scholar]
- Design of anti-CD47 molecules using 1n silico approaches. Br. J. Pharm. Med. Res.. 2018;3:1270-1295.
- [Google Scholar]
- Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest Oncol.. 2020;12:173-181.
- [CrossRef] [Google Scholar]
- Drug solubility: importance and enhancement techniques. ISRN Pharm.. 2012;195727
- [CrossRef] [Google Scholar]
- Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;16:205-214.
- [Google Scholar]
- Molecular functions of SIRPα and its role in cancer. Biomed. Rep.. 2018;9:3-7.
- [CrossRef] [Google Scholar]
- Structural modification of aspirin to design a new potential cyclooxygenase (COX-2) inhibitors. Silico Pharmacol.. 2020;8:1.
- [Google Scholar]
- Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov. Today. 2010;15:444-451.
- [Google Scholar]
- Cancer immunotherapy: harnessing the immune system to battle cancer. J. Clin. Invest.. 2015;125(9):3335-3337.
- [Google Scholar]
- Prostate cancer down-regulated SIRP-α modulates apoptosis and proliferation through p38-MAPK/NF-κB/COX-2 signaling. Oncol. Lett.. 2017;13:4995-5001.
- [Google Scholar]
- Leukocyte and Stromal Cell Molecules: The CD Markers. New Jersey: Wiley; 2020. p. :591.
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2023.105265.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




