

Translate this page into:
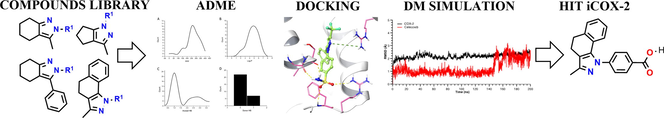
Molecular modeling and structural analysis of some tetrahydroindazole and cyclopentanepyrazole derivatives as COX-2 inhibitors
⁎Corresponding authors. epolo@utalca.cl (Efraín Polo-Cuadrado), mgutierrez@utalca.cl (Margarita Gutiérrez)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Abstract
A theoretical protocol based on ADME-Docking-Molecular Dynamics was used to predict anti-inflammatory activity based on the inhibition of the COX-2 enzyme. Most of the compounds presented a pi-cation type interaction with Arg106 residue, key in stabilization within the active site. Theoretically, the compounds have excellent pharmacodynamic and pharmacokinetic properties. Compound 3ab exhibited a similar mode of binding and stability in the active site of COX-2 compared to the reference drug celecoxib.
Abstract
In an attempt to rationalize the search for new potential anti-inflammatory compounds on the COX-2 enzyme, we carried out an in silico protocol that successfully combines the prediction of physicochemical and pharmacokinetic properties, molecular docking, molecular dynamic simulation, and free energy calculation. Starting from a small library of compounds synthesized previously, it was found that 70% of the compounds analyzed satisfy with the associated values to physicochemical principles as key evaluation parameters for the drug-likeness; all the compounds presented good gastrointestinal absorption and cerebral permeability and they showed an interaction with the Arg 106 residue of the COX-2 isoenzyme. Finally, it was obtained that compound 3ab has a binding mode, binding energy, and stability in the active site of COX-2 like the reference drug celecoxib, suggesting that this compound could become a powerful candidate in the inhibition of the COX-2 enzyme. In addition, we realized the crystallographic analysis of compounds 3j, 3r, and 3t defining the crystal parameters and the Packing interactions.
Keywords
Tetrahydroindazole
Pyrazole
COX-2 enzyme
Molecular Docking
Molecular dynamics simulation
Crystal structure

1 Introduction
Cyclooxygenase (COX) or prostaglandin-endoperoxide synthase, is an enzyme responsible for transforming arachidonic acid into inflammatory mediators such as prostacyclins, levuloglandins, thromboxane A2 and prostaglandins (PGs), which in response to the processes, usually generate unwanted effects such as pain and fever (Almansa et al., 2001; Penning et al., 1997). Williams et al. (1999), it was believed that the COX enzyme existed as a single compound, which caused that the processes related to inflammatory responses were treated with what is now known as non-selective non-steroidal anti-inflammatory drugs (NSAIDs). However, the COX enzyme was found to exist in two different isoenzymes called COX-1 and COX-2, and a splice variant of COX-1 called COX-3 (Stachowicz, 2021). COX-1 is responsible for producing the physiologically important PGs present in tissues such as the gastrointestinal tract and the kidney, while COX-2 is the main responsible for inflammatory responses and COX-3, according to the literature, could contain a capacity of remission phase in chronic inflammatory conditions and could be participating in the growth of cervical, ovarian, leukemia and colon cancer (Fitzpatrick, 2004; Kam and So, 2009; Penning et al., 1997; Sharma et al., 2019). These results suggested that COX-2 inhibitory compounds could be potent anti-inflammatory agents that would not produce the typical side effects (e.g., gastrointestinal ulcers, perforation, and bleeding) associated with non-selective anti-inflammatory drugs that inhibit both enzymes (Masferrer et al., 1994). For this reason, in recent years, new compounds have been developed as selective inhibitors for the COX-2 isoenzyme, which have shown significant efficacy in relieving conditions such as pain, inflammation, fever, pyretic diseases, and thrombotic.
The functioning of the COX-2 isoenzyme is related to different stages of tumor development, such as initiation, promotion, malignant conversion, metastasis, immune surveillance, and responses to therapy (Grivennikov et al., 2010). This relationship is believed to be a consequence of the enzyme’s ability to convert procarcinogens into carcinogens, inhibit apoptosis, promote angiogenesis, increase prostaglandin production, and therefore inflammatory responses (Xu, 2002). One of the strongest associations between inflammatory responses and cancer is the increased risk of inflammatory bowel diseases. However, inflammation also plays an essential role in developing other cancers, for example, cancers of the prostate, endometrium, bladder, and pancreas (Brasky et al., 2013; Neill et al., 2013; Wang et al., 2011).
In several pathological contexts has been shown that the brain is susceptible to inflammatory processes due to its low regenerative capacity and specific immune processes given by the blood–brain barrier (Elie et al., 2019). Therefore, increased COX-2 expression and PG production have been related to some neurodegenerative processes (Teismann et al., 2003), such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) and various neuropsychiatric disorders such as autism and schizophrenia, and depression (Perry et al., 2010; Prata et al., 2017; Singhal and Baune, 2017). The brain’s immune responses to various lesions are called neuroinflammation, which generates a series of biological events, the main one being the activation of microglial cells (Jacobs and Tavitian, 2012), which can mediate protective and regenerative mechanisms or, aggravate lesions, contributing to neurodegeneration (Czeh et al., 2011). For example, microglial cells attempt to degrade amyloid fibrils (associated with AD) by phagocytosis; even so, this degradation is incomplete, leading to a chronic inflammatory state (McGeer and McGeer, 1996, 1995). Some studies have reported more than 40 proteins characteristic of an inflammatory response, in which complement proteins, inflammatory cytokines, acute phase reactants, and many proteases and protease inhibitors are found in low concentrations in brain tissue autopsies in patients with AD (McGeer et al., 1996). These findings may suggest that AD involves a chronic inflammatory process and that anti-inflammatory agents may have a role in treating AD (Flynn and Theesen, 1999).
Celecoxib is COX-2 selective NSAIDs, usually used for pain relief in patients with osteoarthritis, dysmenorrhea, and rheumatoid arthritis (see Fig. 1). Within its structure is the pyrazole group an important pharmacophore center, which has demonstrated interesting pharmacological properties like antimicrobial, antifungal, anti-inflammatory, analgesic, antipyretic, cytotoxic, antitumor, anti-leukemic, among others (Alcaro et al., 2010; Curran et al., 2010; Kaping et al., 2016; Zask et al., 2009). Also, it has been reported that a large number of azole compounds, including those derived from pyrazole are very potent and selective COX-2 inhibitors (Atatreh et al., 2019; Dennis Bilavendran et al., 2020; Prasher and Sharma, 2020). For example, Paul Beswick et al. Synthesized a series of 2,3-diaryl-pyrazole [1,5-b]pyridazines (see Fig. 1) and evaluated their inhibitory capacity for the COX-2 enzyme, finding that the compounds obtained in the majority of the cases had good inhibitory profiles (Beswick et al., 2004). Likewise, Carmen Almansa et al., published the synthesis of a new series of pyrazole [1,5-a]pyrimidines (see Fig. 1) and evaluated their inhibitory capacity of the COX-2 isoenzyme, reporting that within the synthesized series, there were compounds with high efficacy and inhibitory selectivity (Almansa et al., 2001).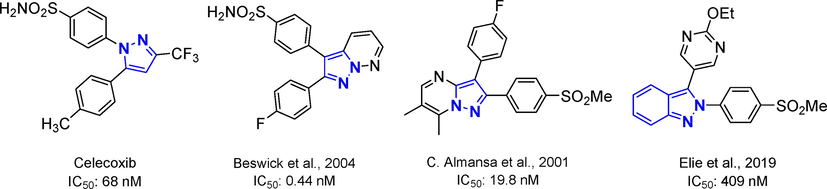
Structure of Celecoxib and some azole-derived compounds that have shown good inhibitory activities of the cox-2 enzyme.
Pyrazoles and their derivatives, have shown a wide variety of biological activities such as anti-inflammatory, antimicrobial, anticancer, anti-HIV, anti-hypertensive and have also been widely studied for their potential as selective inhibitors of the COX-2 enzyme (Elie et al., 2019; Minu et al., 2009; Plescia et al., 2010; Ranise et al., 1983; Thangadurai et al., 2012). Jhonatan Eli et al. Synthesized a series of 2-arylazaindazoles (see Fig. 1), which showed promising results as molecular scaffolds with the ability to inhibit the activity of COX-2 at the submicromolar level (Elie et al., 2019).
In this work, we present the synthesis of a novel group of azoles, which underwent a crystallographic study together with a theoretical-computational analysis of some of their structural properties and a molecular docking against the COX-2 isoenzyme.
2 Experimental
2.1 Data sets
We have recently reported an efficient solvent-free microwave-assisted synthesis and characterization (view supplementary information) of a series of azole-type derivatives from a condensation involving 1,3-diketones and arylhydrazines precursors with moderate to high yields (Polo et al., 2021). Due to the structural similarity with respect to some drugs and compounds previously reported with anti-inflammatory capacity, this series of compounds were chosen in the present theoretical study in order to detect a possible compound with inhibitory capacity on COX-2. Fig. 2 shows the structures of the selected compounds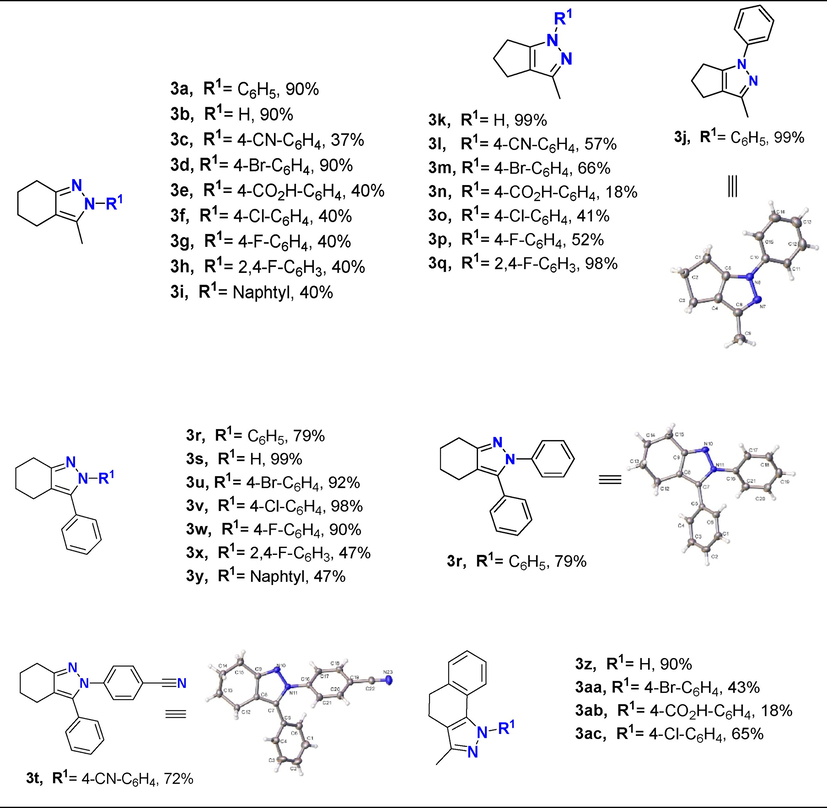
Synthesis of compound 3a-ac. ORTEP of compounds 3j, 3r, and 3t. Thermal ellipsoid was drawn with 30% of probability.
2.2 Molecular docking and ADME prediction
Computational calculations were performed using Schrödinger Small-Molecule Drug Discovery Suite (Schrödinger Release, 2021a). The atomic coordinates for protein were extracted from the X-ray crystal structures of COX-2 (PDB ID: 3LN1) (Wang et al., 2010). The initial setup of COX-2 enzyme for calculations was prepared using Protein Preparation Wizard of Schrödinger (Madhavi Sastry et al., 2013; Schrödinger Release, 2021b). The center of the grid box was situated using the location of co-crystallized original ligands and was refined and set up as 20 Å. To generate the minimized energy 3D structures LigPrep module (Schrödinger Release, 2021c) was used under the OPLS-AA force field (Jorgensen et al., 1996). Molecular docking studies were performed using Glide (Schrödinger Release, 2021d). Glide finds the best possible ligand binding posse in a protein grid space, evaluating the energy interactions between them (Friesner et al., 2004).
The absorption, distribution, metabolism, excretion properties of the compounds were predicted using QikProp software (Polo et al., 2021; Schrödinger Release, 2021e) through the calculations processed in a normal mode of different descriptors such as octanol/water partitioning coefficient (Log P), surface areas of polar nitrogen, and oxygen atoms (PSA), aqueous solubility (Log S), among others. The acceptability of the compounds based on Lipinski’s rule (Daina et al., 2017; Lipinski et al., 1997) was also evaluated. Likewise, the free access tool swissADME (http://www.swissadme.ch) was used to analyze the synthesized compounds’ pharmacokinetics, such as the blood–brain barrier and the ability to inhibit cytochrome P450 enzymes.
2.3 Molecular dynamics simulations
We performed two independent MD simulations for the lowest binding energy docking ligand and the celecoxib as reference compound using Desmond program in Schrödinger suite 2021–1(“SC ’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing,” 2006; Schrödinger Release, 2021f). Each protein–ligand complex was solvated in aqueous solutions using an explicit TIP3P water model. Each system was neutralized with 0.15 mol/L of NaCl to maintain an ionic concentration and parametrized with OPLSe force field. The MD simulations were carried out for a total of 200 ns with a recording interval of 100 ps with an isothermal isobaric assembly (NPT) and standard conditions of T = 310 K and P = 1 atm. The Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) approach was used as a prime module from Schrödinger Suite and default settings (Jacobson et al., 2002; Schrödinger Release, 2021).
2.4 Free energy calculation
The molecular MM/GBSA method was used to estimate the most active complexes binding free energy. For MM/GBSA calculations, only 100 ns were extracted for analysis from a 200 ns of MD simulation, and the explicit water molecules and ions were removed. The ΔGbind analysis was performed on three subsets of each system: the ligand alone, the protein alone, and the protein–ligand complex. The total free energy (ΔGbindTot) was calculated with the following equation:
-HMM is the bonded and Lennard–Jones energy terms
-Gsolv is the polar contribution of solvation energy and non-polar contribution to the solvation energy
-T is the temperature
-ΔSconf corresponds to the conformational entropy (Hayes and Archontis, 2012).
2.5 Crystallography
Some suitable crystals of all compounds were measured, and their diffraction data were collected at ca. 296 K of D8 Venture diffractometer equipped with a bidimensional CMOS Photon 100 detector, using graphite monochromated Mo-Kα (λ = 0.71073 Å). The diffraction frames were integrated using the APEX3 package (Bruker, 2016) and were corrected for absorptions with SADABS. The structure of the title compound was solved by intrinsic phasing (Sheldrick, 2015) using the OLEX 2 program (Dolomanov et al., 2009). All the structures were then refined with full-matrix least-squares methods based on F2 (SHELXL-2014) (Sheldrick, 2015). For all compounds, non-hydrogen atoms were refined with anisotropic displacement parameters. Compound 3j shows a positional disorder modeled using FVAR in a 57:43 ratio, over two positions, respectively, and twinned refinement with twin operation 2-fold rotation about u,v,w = [1 0 0], with twin fraction BASF = -3.00(9). All hydrogen atoms were included in their calculated positions, assigned fixed isotropic thermal parameters, and constrained to ride on their parent atoms. A summary of the details about crystal data, collection parameters, and refinement are documented in Table 2, and additional crystallographic details are in the CIF files. ORTEP views were drawn using OLEX2 software (Dolomanov et al., 2009). Additionally, In the crystal, the molecules are linked by Hydrogen bond, π-π and C – H ···π interactions. In case of compound 3j, this shows a π – π between pyrazole moiety with distance 4.002(4) Å and shift distance 1.794(2)°, and a C – H ···π interaction with a distance 3.112 Å and C – H ···π angle of 130.8(4)° (symmetry operation: 1/2+x; 1/2-y; +z). In compound 3r, this shows two C – H ···π interactions between cyclohexyl hydrogen and N-attached phenyl ring, and C-attached Phenyl ring and pyrazole moiety, with a distance of 2.957(10) Å and 2.964(10) Å, and angles of 147.1(1)° and 154.2(14)°, respectively (symmetry operations: +x; −1 + y; +z and + x; 1/2-y; -1/2+z, respectively). Finally, compound 3 t shows an intermolecular hydrogen bond interaction graph-set motif (Bernstein et al., 1995)
, along to c-axis [D – H: 0.93 Å; H···A: 2.59 Å; D···A: 3.432(2)Å; ∢D – H ···A: 151.0; symmetry operation: -1/2+x; 1/2 -y; +z] (see Figs. 9 and 10 for more details). C – H ···π interaction with a distance 3.286(6) Å and angle of 159.5(11)° (symmetry operations: 1/2+x; 1/2-y ; -1/2+z).
Compound
3j
3r
3 t
Empirical Formula
C13H14N2
C19H18N2
C20H17N3
Formula mass, g mol−1
198.26
274.35
299.36
Collection T, K
295.44
295.55
295.73
crystal system
orthorhombic
Monoclinic
monoclinic
space group
Pna21
P21/c
P21/n
a (Å)
7.999(9)
15.059(6)
9.0102(11)
b (Å)
14.234(17)
8.256(3)
16.3110(17)
c (Å)
9.871(12)
12.752(4)
10.9767(12)
β (°)
111.453(7)
94.671(3)
V (Å3)
1124(2)
1475.7(9)
1607.8(3)
Z
4
4
4
ρcalcd (gcm−3)
1.172
1.235
1.237
Crystal size (mm)
0.22 × 0.17 × 0.13
0.17 × 0.09 × 0.07
0.25 × 0.17 × 0.13
F(0 0 0)
424.0
584.0
632.0
abs coeff (mm−1)
0.070
0.073
0.074
2θ range (°)
5.724 to 48.976
5.726 to 55.5
5.63 to 52.838
range h,k,l
−9 / 9, −16/ 16, −11 / 11
−19 / 19, −10 /10, −16 / 16
−11 / 11, −20 / 19, −13 / 13
No. total refl.
8273
16,383
15,228
No. unique refl.
1867 [Rint = 0.1458, Rsigma = 0.1002]
3444 [Rint = 0.0823, Rsigma = 0.0591]
3303 [Rint = 0.0394, Rsigma = 0.0284]
Comp. θmax (%)
99.9
99.0
99.8
Max/min transmission
0.991/0.986
0.995/0.992
0.990/0.985
Data/Restraints/Parameters
1867/1/122
3444/0/191
3303/0/209
Final R [I > 2σ(I)]
R1 = 0.0622, wR2 = 0.1338
R1 = 0.0531, wR2 = 0.1041
R1 = 0.0418, wR2 = 0.0994
R indices (all data)
R1 = 0.1279, wR2 = 0.1616
R1 = 0.1034, wR2 = 0.1244
R1 = 0.0576, wR2 = 0.1146
Goodness of fit/F2
1.034
1.035
1.061
Largest diff. Peak/hole (eÅ−3)
0.23/-0.17
0.20/-0.19
0.20/-0.18
Flack Parameter
−3(9)
---
---
3 Results and discussion
3.1 Molecular docking and ADME prediction
3.1.1 Physicochemical and pharmacokinetic properties
In search of new drugs candidates, the new molecules should present a potential biological activity directed at one or more molecular targets involved in developing a disease, so it is essential to establish pharmacokinetic and toxicity parameters in this process. For this reason, a comparison of the physicochemical and pharmacokinetic properties for synthesized compounds and celecoxib was carried out through an in silico approach to identify its possible anti-inflammatory potential.
The oral availability of the synthesized compounds was studied based on the Lipinski criteria (Lipinski et al., 1997). Fig. 3 shows the drug-likeness distribution graphs of synthesized compounds. The MW distribution ubicates all compounds in the established range for Lipinski’s rule (MW < 500 uma), where most compounds present a molecular weight between 250 and 300 g/mol. the Log P distribution shows a maximum frequency at values of 4 and 5 units, but there is also a considerable frequency over the five units. Lipinski’s rule of five where donor hydrogen bond must be less than five and acceptor must be less than 10, none of the compounds exceed this range, and there is no violation of the rule. Concerning donor component (donor HB), maximum frequency (23) was found with a value of 1, while for acceptor (HBA) was of 0 and 1 unit, with a higher frequency at 0. Finally, rotatable bonds were determined for each of these compounds, and analyzed their conformational flexibility. All yielded a lower value than the reference compound.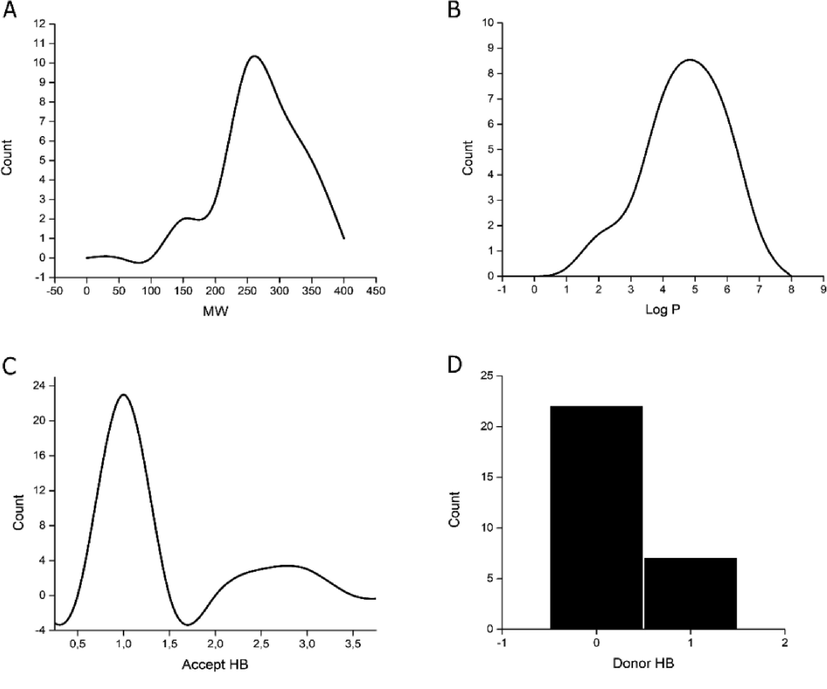
Distribution graphs of synthesized compounds molecular properties.
In summary, 70% of compounds did not violate the Lipinski rule, unlike the reference drug. The rest compound presented only one violation corresponding to the partition coefficient exceeding the established range. (Tables S1 and S2).
Gastrointestinal absorption and brain permeability are two essential criteria in determining the bioavailability of a drug. These two parameters were determined for the synthesized compounds from the construction of a boiled egg diagram based on the descriptors LogP and the topological polar surface area (TPSA) (Daina and Zoete, 2016), using the open-access tool swissADME (http://www.swissadme.ch) (Daina et al., 2017) (Fig. 4). All compounds showed good gastrointestinal absorption and permeability of the blood–brain barrier, unlike celecoxib, which shows low permeability. Compounds 3ab, 3e, and 3n are not substrates for P-glycoprotein (P-gp), which could indicate that their bioavailability would not be reduced. These three compounds have carboxylate groups in their structure.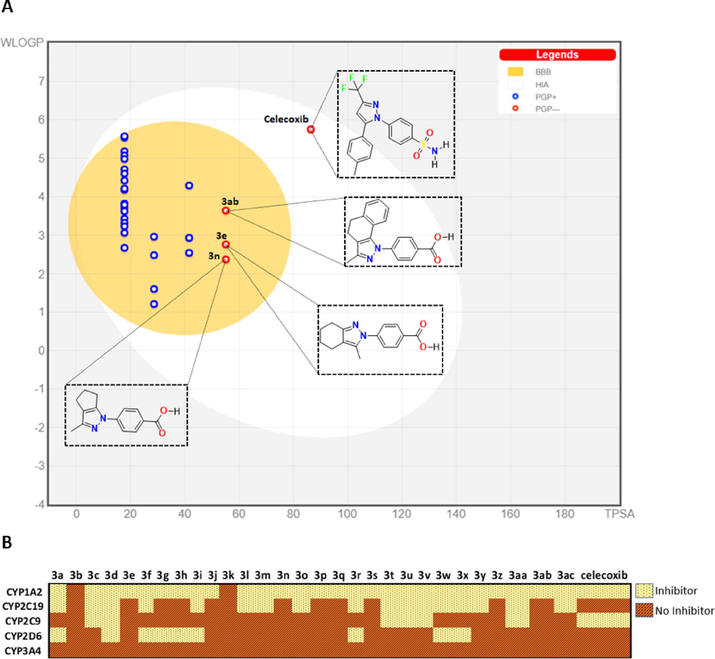
Pharmacokinetic profile. (A) BOILED-Egg diagram of synthesized compounds. White region: high probability of passive absorption by the gastrointestinal tract; yellow region (yolk): high probability of brain penetration. Yolk and the white region are not mutually exclusive. Bluepoint: P-gp substrate. Redpoint: No P-gp substrate. (B) Inhibitory capacity on the set of CYP450 enzymes.
Drug metabolism occurs mainly within the liver through enzymes that make up cytochrome P450 (CYP450). The inhibition of these enzymes can generate an accumulation of xenobiotics that need to be metabolized and thus, not reach toxic levels within the body (Ghalehshahi et al., 2019). Through the swissADME, it was possible to determine the pharmacokinetic aspect for the synthesized compounds. It was also determined whether these compounds could be inhibitors of the five most important isozymes of CYP450 involved in metabolizing drugs such as CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. The predictive results show that none of the compounds inhibits CYP3A4 enzyme, the most abundant in the liver and an active participant in the metabolism of approximately 60% of known drugs (Zhou et al., 2005). Compounds 3b and 3k do not show any inhibition on the five subfamilies analyzed. Likewise, 28% of the compounds showed inhibitory capacity on a single CYP subfamily, among which the compounds 3ab, 3n, and 3e mentioned above stand out. These three compounds inhibit the CYP1A2 subfamily, involved in caffeine metabolism (Cornelis et al., 2006). This calculation allows us to have an overview of the pharmacokinetics and the effects that the synthesized compounds could have on CYP450 enzymes since possible risks due to accumulation of substances at toxic levels due to the inhibition of these enzymes must be analyzed.
3.1.2 Docking studies
We carried out molecular docking calculations to examine to a molecular level, the potential binding modes of most promissory ligands to the COX-2 as potential inhibitors. Based on structural information from the COX-2 crystal structures in complex with celecoxib, we established the binding site for the synthesized molecules for COX-2 enzyme. We used Glide for Carry out molecular docking calculations (Schrödinger Release, 2021d). Glide software uses a series of hierarchical filters that include a systematic searching approach, sampling the ligands positional, conformational, and orientation space (Friesner et al., 2004). Results determined that 86% of synthesized compounds show a cation-π interaction with Arg106 residue. In converting arachidonic acid into prostaglandin G2, Arg106 interacts with the carboxylate group from the substrate (Greig et al., 1997). The specific protein–ligand interactions observed in the representative docking poses of lead compounds are summarized in Table 1 and Table S3.
Compa
glide score
glide lipob
Interaction type
HB
π-cat
π-π stacking
3y
−9.67
−6.00
–
Arg106
3x
−9.64
−5.12
–
Arg106
3w
−9.49
−5.12
–
Arg106
3ab
−9.37
−4.99
Phe504, Arg499
Arg106
3ac
−9.32
−5.19
–
Arg106
3t
−9.26
−5.13
–
Arg106
3r
−9.24
−5.15
–
Arg106
3aa
−9.21
−5.28
Arg499
Arg106
3v
−9.18
−5.42
–
Arg106
Tyr341
3u
−9.07
−5.22
–
Arg106
celecoxibc
−10.23
−4.79
Phe504, Arg499,Leu338, Ser339
Arg106
The pyrazole ring of compound 3ab exhibits a cation-π interaction with the Arg106 residue, located in the hydrophobic zone of the COX-2 active site. Likewise, the carboxylate function of this ligand shows hydrogen bond interactions with residues Phe504 and Arg499, which are like those presented by the sulfonamide group from Celecoxib (Fig. 5). Arg499 increases the polarity of COX-2 active site, and this interaction is critical in the selectivity of COX-2 for inhibitor compounds since, in the COX-1 active site, Arg499 is replaced by His (Rouzer and Marnett, 2020). Similarly, the cyclohexane ring of 3ab presents hydrophobic interactions with residues such as Ser516, Leu517, Ala513 and Val335 in the range of 3.36–4.34 A. (see Figure S85).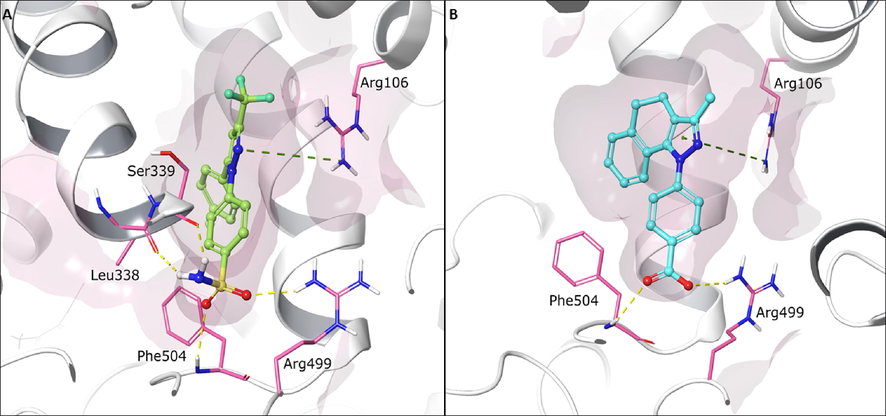
Predicted binding conformations into COX-2 active site of (A) reference ligand celecoxib (green) and (B) compounds 3ab (cyan). Green dotted lines represent cation - π interaction. Yellow dotted lines represent hydrogen bond interaction. Relevant amino acids are shown in thin tube. Compounds are shown in ball-and-stick representation. The secondary protein structure is depicted as white ribbons.
On the other hand, compounds 3y, 3x, and 3w also yielded the best binding energy within COX-2, showing cation-π interaction of the pyrazole ring with the Arg106 residue. Despite not forming hydrogen bonds with crucial amino acids in the catalytic pocket, these compounds present a considerable value in the lipophilic component released by Glide, which indicates a high hydrophobic affinity for the residues found in the hydrophobic channel of the active site.
Finally, 2H-indazole derivatives have been synthesized as COX-2 inhibitors (Pérez-Villanueva et al., 2017) and due to the structural similarity to our compounds, the most active compound against COX-2 in the series was compared with compound 3ab and it was determined that they possess similar orientation and mode of binding in the active site of the protein (see Figure S86).
Because compound 3ab exhibited the conventional binding mode usually adopted by the reference inhibitor, a molecular dynamics study was performed. To examine the overall structure stability of protein – ligand complex.
3.2 Molecular dynamics simulations
We used molecular dynamics (MD) simulations for studying and analyzing from atomistic insight the enzyme-ligand interactions and evaluate the differences in the binding mode of celecoxib and molecule 3ab in complex with COX-2 enzyme during 200 ns of simulation. The MD analysis revealed small but significant differences between 3ab-COX-2 complex compared with celecoxib-complex. The Interaction fraction plots show the main contacts and contributions over the molecular simulation trajectory of 200 ns. In the 3ab-COX-2 complex, the residues Arg106 and Arg499 engaged mainly in hydrogen bonds interactions direct or water-mediated, and ionic interactions with both oxygens of the carboxylate group with occupancy of over 95% during the time of the trajectory. The Phe504, Tyr371 and Leu370 residues also played an essential role in the protein–ligand interactions through hydrophobic contacts whit the aromatic ring of pyrazole moiety. Phe504 residue was engaged over 50% of the trajectory time through π-π stacking interaction.
As shown in Fig. 6, molecule 3ab displayed a considerable number of water-mediated interactions contributing to system stabilization. Notwithstanding, RMSD trajectories of the 3ab-COX-2 complex during the MD showed high deviations (∼3.5 Å) staying stable during the simulation time, with fluctuations attributed to carboxylate moiety, which is comparatively higher than other groups of the compound 3ab. Similarly, we characterized and compared the intermolecular interactions of celecoxib with the COX-2 enzyme. The celecoxib-COX-2 interaction study revealed that the celecoxib interaction is conserved, showing similar contacts to molecule 3ab. However, the interactions are mainly governed by hydrophobic and hydrogen bonding interactions with aromatic residues such as Phe504 and hydrophobic ones such as Ile503 and Leu338 with an occupancy over 95%. We also observed less water-mediated H-bonding in the MD in the complex with celecoxib compared with the 3ab molecule complex.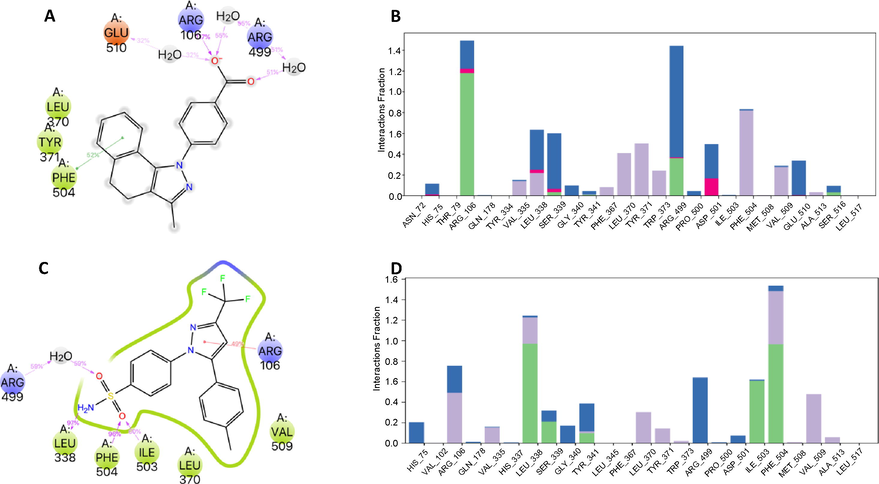
Interactions of compound 3ab (A) and Celecoxib (C) with COX-2 enzyme were monitored throughout the simulation. We show interactions that occur more than 30.0% of the simulation time in the MD trajectory. B). Interaction fraction plots depicting enzyme COX-2 contacts with 3ab compound during 200 ns MD simulation Hydrophobic interactions, H-bonds, ionic interactions, and water bridges are shown in purple, green, fuchsia, and blue, respectively. The stacked bar charts are normalized throughout the trajectory: for example, a value of 0.7 suggests that 70% of the simulation time, the specific interaction is maintained. Values over 1.0 are possible as some protein residue may make multiple contacts of the same subtype with the ligand. D). Interaction fraction plots depicting enzyme COX-2 contacts with celecoxib.
RMSD trajectories of COX-2-celecoxib complex during the MD showed initially slightest deviations (∼1.0 Å), showing high stability during this simulation time. Still, in the last simulation time, the celecoxib RMSD increases significantly and reaches a deviation around ∼2.0 Å (Fig. 7). The last fluctuations were attributed to sulfamoylphenyl moiety.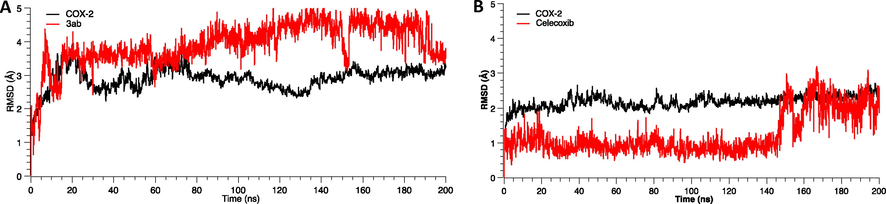
Root Mean Square Deviation (RMSD) as a function of simulated times for the complexes formed between COX-2 enzyme, compound 3ab and celecoxib.
3.3 Free energy calculation
As MD simulation-derived molecular mechanics generalized Born surface area (MM/GBSA) calculations incorporate receptor/ligands complexes motions and treat solvation effects better than the docking, we used this method to calculate the free energy of each complex. The binding free energy was estimated after the MD simulation; the last 100 ns for both complexes gave as results energy values of −67.3 ± 4.3 kcal/mol and −57.4 ± 5.4 kcal/mol celecoxib and molecule 3ab, respectively (Fig. 8). The MM/GBSA results show that celecoxib has a lesser binding affinity being the coulombs energy (-18,5 kcal/mol), the component of high contribution. However, the compound 3ab depicted the lowest lipophilic energy (dG_bind_Lipo) (−32.6 kcal/mol) with COX-2 enzyme, while celecoxib showed relatively higher dG_bind_Lipo (−23.0 kcal/mol) being this energetic component the one that contributes the most to the total binding affinity.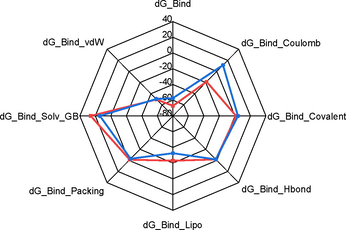
The radar graph shows the MM/GBSA binding energy values of molecule 3ab (blue) and celecoxib (red) and the energy decomposition values in the axes.
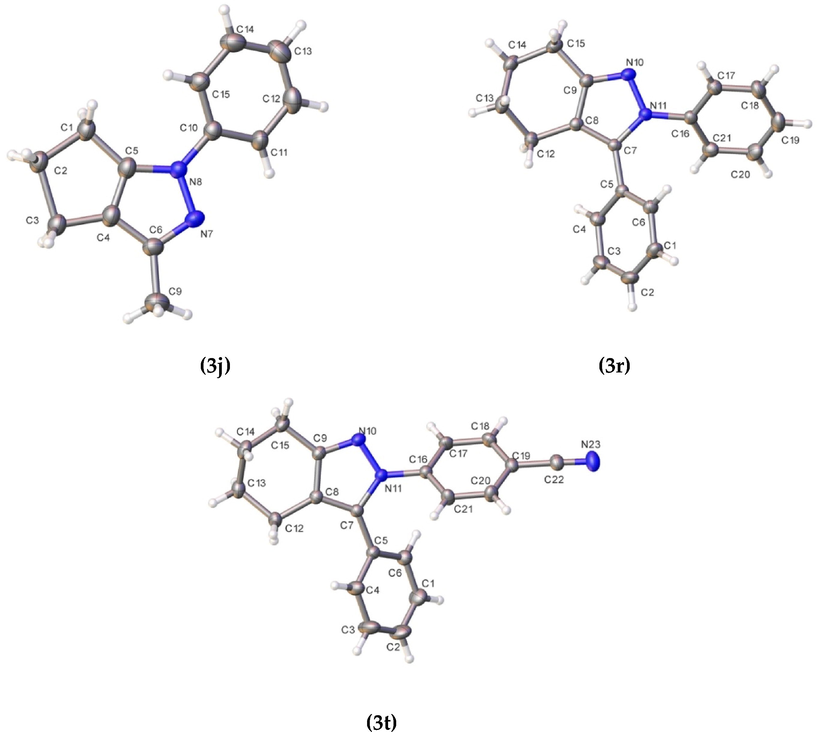
ORTEP of compounds 3j, 3r, and 3t. Thermal ellipsoid was drawn with 30% of probability.
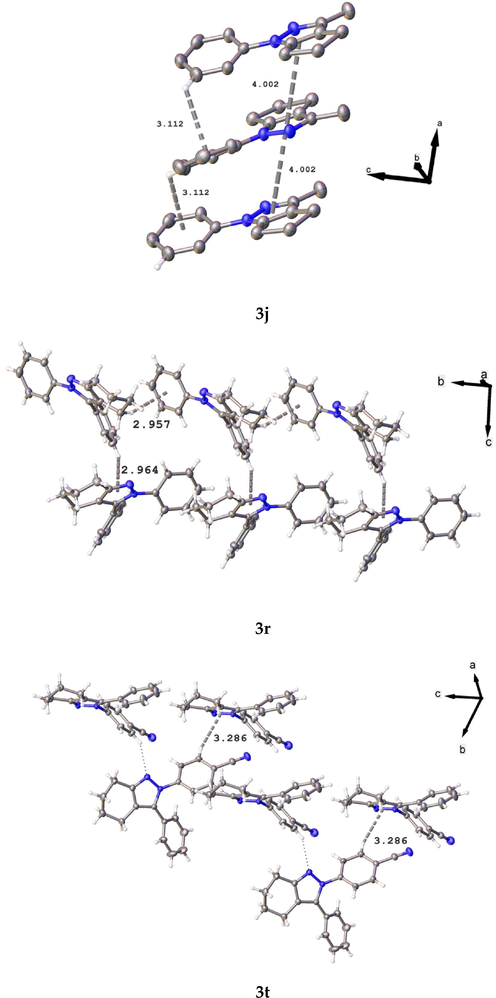
Packing interactions in compounds 3j, 3r, and 3t.
3.4 Crystallography
The molecular structure of compound 3j corresponds to a 3,1-disubstituted cyclopenta[d]pyrazole; meanwhile, 3r and 3 t correspond to 3,2-disubstituted tetrahydro-2H-indazole. In compound 3j, this shows a non-centrosymmetric setting, and the other two show a centrosymmetric setting. All compounds have normal bond distances and angles (Allen et al., 1987). Dihedral angles between the least-square mean plane of the phenyl ring and heterocycle for 3j is 19.2(2)°. In the compounds 3r and 3 t, the dihedral angles are 33.32(7)°/42.41(7), and 131.03(6)/ 37.60(7) for the rings attached to N11 and C7, respectively.
Additionally, according to Cremer & Pople’s parameters (Cremer, 1984), for 5-membered rings, in compound 3j, it was not easy to determined readily. However, according to the calculation, this shows the closest envelope conformation on C2 atom (Q(2) = 0.289/0.094 Å; ϕ = 217(3)/139(11)°). In the case of compounds 3r and 3 t these show a half-chair conformation (QT = 0.509 Å; θ = 130.1(2)°; ϕ = 35.4(3)° and QT = 0.492 Å; θ = 49.5.1(2)°; ϕ = 202.9(3)°, respectively).
4 Conclusions
Crystallographic analysis of 3j, 3r, and 3t, showed standard bond angles and distances. 3j presented asymmetric center adjustment while the last two showed a non-centered symmetric fit; additionally, it was evidenced that compounds 3r and 3t presented a half-chair conformation in the pyrazole ring.
Theoretical study of oral availability of the reported compounds (3a-3ac) found that around 70% of these complied with Lipinski’s rules. All compounds showed good gastrointestinal absorption and blood–brain barrier, and in general, they do not interrupt the enzymatic processes of CYP450 mediated in the liver.
Equally important, in the molecular coupling study, it was found that 86% of the compounds analyzed showed a cation-π interaction with the Arg106 residue of the COX-2 enzyme. In addition, it is essential to mention that compound 3ab exhibited the conventional binding mode usually adopted by the reference inhibitor, exhibiting interactions with residues Arg106, Arg499, and Phe504. Interactions of the 3ab-COX-2 complex with Arg106 residue were mainly through hydrogen bonds or water-mediated H-bonding and ionic interactions of the oxygens of the carboxylate group. Trajectory analysis of Molecular dynamics showed that the studied complexes COX-2-celecoxib and COX-2-3ab displayed structural stability during the MD simulations. The estimated free energies of binding of these two compounds are −67.3 and −57.4 kcal/mol for celecoxib and compound 3ab, respectively. The contribution of lipophilic energy was energetically favorable for the compound 3ab, while for celecoxib, the contribution of Coulomb energy was predominant.
In conclusion, many of the analyzed compounds presented good ADME properties, which is a good indication that these molecules could potentially apply to the pharmacological field. Is it possible to infer that compound 3ab is a candidate with the potential to inhibit the COX-2 isoenzyme. Our next step in this search for COX-2 inhibitors will be the experimental evaluation of compound 3ab, hoping to obtain remarkable results and have a hit compound in developing COX-2 inhibitors.
Acknowledgments
The authors acknowledge to FONDECYT program (project number 1200531) to support this research, and also to FONDEQUIP program (EQM 130021 and 180024). Y.A.R.N. thanks FONDECYT Post-Doctoral Fellowship No. 3190557. Y.D thanks FONDECYT 11201113.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Hit identification and biological evaluation of anticancer pyrazolopyrimidines endowed with anti-inflammatory activity. ChemMedChem. 2010;5:1242-1246.
- [CrossRef] [Google Scholar]
- Allen, F.H., Kennard, O., Watson, D.G., Brammer, L., Orpen, A.G., Taylor, R., 1987. Tables of Bond Lengths Determined by X-Ray and Neutron-Diffraction. 1. Bond Lengths in Organic-Compounds. J. Chem. Soc., Perkin Trans. 2 S1–S19. https://doi.org/Doi 10.1039/P298700000s1
- Synthesis and SAR of a new series of COX-2-selective inhibitors: Pyrazolo[1,5-α]pyrimidines. J. Med. Chem.. 2001;44:350-361.
- [CrossRef] [Google Scholar]
- Anti-inflammatory drug approach: Synthesis and biological evaluation of novel pyrazolo[3,4-d]pyrimidine compounds. Bioorg. Chem.. 2019;86:393-400.
- [CrossRef] [Google Scholar]
- Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chemie Int. Ed. English. 1995;34:1555-1573.
- [CrossRef] [Google Scholar]
- Identification of 2,3-diaryl-pyrazolo[1,5-b]pyridazines as potent and selective cyclooxygenase-2 inhibitors. Bioorg. Med. Chem. Lett.. 2004;14:5445-5448.
- [CrossRef] [Google Scholar]
- Non-steroidal anti-inflammatory drugs and endometrial cancer risk in the VITamins and Lifestyle (VITAL) cohort. Gynecol. Oncol.. 2013;128:113-119.
- [CrossRef] [Google Scholar]
- Bruker, APEX3 Software, 2016. Bruker AXS Inc. V2016.1, Madison, Wisconsin, USA.
- Coffee, CYP1A2 Genotype, and Risk of Myocardial Infarction. JAMA. 2006;295:1135.
- [CrossRef] [Google Scholar]
- On the correct usage of the Cremer-Pople puckering parameters as quantitative descriptors of ring shapes – a reply to recent criticism by Petit, Dillen and Geise. Acta Crystallogr. Sect. B Struct. Sci.. 1984;40:498-500.
- [CrossRef] [Google Scholar]
- Pyrazolopyrimidines as highly potent and selective, ATP-competitive inhibitors of the mammalian target of rapamycin (mTOR): Optimization of the 1-substituent. Bioorganic Med. Chem. Lett.. 2010;20:1440-1444.
- [CrossRef] [Google Scholar]
- SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep.. 2017;7:42717.
- [CrossRef] [Google Scholar]
- A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem. 2016;11:1117-1121.
- [CrossRef] [Google Scholar]
- Synthesis and discovery of pyrazolo-pyridine analogs as inflammation medications through pro- and anti-inflammatory cytokine and COX-2 inhibition assessments. Bioorg. Chem.. 2020;94:103484
- [CrossRef] [Google Scholar]
- OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr.. 2009;42:339-341.
- [CrossRef] [Google Scholar]
- Design of selective COX-2 inhibitors in the (aza)indazole series. Chemistry, in vitro studies, radiochemistry and evaluations in rats of a [18F] PET tracer. J. Enzyme Inhib. Med. Chem.. 2019;34:1-7.
- [CrossRef] [Google Scholar]
- Cyclooxygenase Enzymes: Regulation and Function. Curr. Pharm. Des.. 2004;10:577-588.
- [Google Scholar]
- Pharmacologic management of Alzheimer disease: Part III: Nonsteroidal antiinflammatory drugs - Emerging protective evidence? Ann. Pharmacother.. 1999;33:840-849.
- [CrossRef] [Google Scholar]
- Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem.. 2004;47:1739-1749.
- [CrossRef] [Google Scholar]
- Synthesis, CYP 450 evaluation, and docking simulation of novel 4-aminopyridine and coumarin derivatives. Arch. Pharm.. 2019;Weinheim). 352:1800247.
- [CrossRef] [Google Scholar]
- The Interaction of Arginine 106 of Human Prostaglandin G/H Synthase-2 with Inhibitors Is Not a Universal Component of Inhibition Mediated by Nonsteroidal Anti-inflammatory Drugs. Mol. Pharmacol.. 1997;52:829-838.
- [CrossRef] [Google Scholar]
- Hayes, J.M., Archontis, G., 2012. MM-GB(PB)SA Calculations of Protein-Ligand Binding Free Energies, in: Molecular Dynamics - Studies of Synthetic and Biological Macromolecules. InTech. https://doi.org/10.5772/37107
- Noninvasive molecular imaging of neuroinflammation. J. Cereb. Blood Flow Metab.. 2012;32:1393-1415.
- [CrossRef] [Google Scholar]
- On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol.. 2002;320:597-608.
- [CrossRef] [Google Scholar]
- Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc.. 1996;118:11225-11236.
- [CrossRef] [Google Scholar]
- COX-3: Uncertainties and controversies. Curr. Anaesth. Crit. Care. 2009;20:50-53.
- [CrossRef] [Google Scholar]
- A facile, regioselective synthesis of novel 3-(N-phenylcarboxamide)pyrazolo[1,5-a]pyrimidine analogs in the presence of KHSO 4 in aqueous media assisted by ultrasound and their antibacterial activities. Mol. Divers.. 2016;20:379-390.
- [CrossRef] [Google Scholar]
- Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev.. 1997;23:3-25.
- [CrossRef] [Google Scholar]
- Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des.. 2013;27:221-234.
- [CrossRef] [Google Scholar]
- Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. U. S. A.. 1994;91:3228-3232.
- [CrossRef] [Google Scholar]
- Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann. N. Y. Acad. Sci.. 1996;777:213-220.
- [CrossRef] [Google Scholar]
- The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res. Rev.. 1995;21:195-218.
- [CrossRef] [Google Scholar]
- Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: A review of 17 epidemiologic studies. Neurology. 1996;47:425-432.
- [CrossRef] [Google Scholar]
- Synthesis, antimicrobial activity and QSAR studies of new 2,3-disubstituted-3,3a,4,5,6,7-hexahydro-2H-indazoles. Bioorganic Med. Chem. Lett.. 2009;19:2960-2964.
- [CrossRef] [Google Scholar]
- Aspirin, nonsteroidal anti-inflammatory drugs, paracetamol and risk of endometrial cancer: A case-control study, systematic review and meta-analysis. Int. J. Cancer. 2013;132:1146-1155.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of the 1,5-Diarylpyrazole Class of Cyclooxygenase-2 Inhibitors: Identification of 4-[5-(4-Methylphenyl)-3- (trifluoromethyl)-1 H -pyrazol-1-yl]benzenesulfonamide (SC-58635, Celecoxib) J. Med. Chem.. 1997;40:1347-1365.
- [CrossRef] [Google Scholar]
- Synthesis and Biological Evaluation of 2H-Indazole Derivatives: Towards Antimicrobial and Anti-Inflammatory Dual Agents. Molecules. 2017;22:1864.
- [CrossRef] [Google Scholar]
- Microglia in neurodegenerative disease. Nat. Rev. Neurol.. 2010;6:193-201.
- [CrossRef] [Google Scholar]
- Plescia, S., Raffa, D., Plescia, F., Casula, G., Maggio, B., Daidone, G., Raimondi, M.V., Cusimano, M.G., Bombieri, G., Meneghetti, F., n.d. Synthesis and biological evaluation of new indazole derivatives. Gen. Pap. Ark. 2010, 163–177. https://doi.org/10.3998/ark.5550190.0011.a14
- Microwave-assisted synthesis, biological assessment, and molecular modeling of aza-heterocycles: Potential inhibitory capacity of cholinergic enzymes to Alzheimer’s disease. J. Mol. Struct.. 2021;1224:129307
- [CrossRef] [Google Scholar]
- “Azole” as privileged heterocycle for targeting the inducible cyclooxygenase enzyme. Drug Dev. Res.. 2020;82:167-197.
- [CrossRef] [Google Scholar]
- Bridging Autism Spectrum Disorders and Schizophrenia through inflammation and biomarkers - pre-clinical and clinical investigations. J. Neuroinflammation. 2017;14:1-33.
- [CrossRef] [Google Scholar]
- Derivatives of 4,5,6,7-tetrahydro-7,8,8-trimethyl-3-phenylamino-4,7-methano-2H-indazole with hypoglycemic and hypotensive activities. Farm. Ed. Sci.. 1983;38:101-111.
- [CrossRef] [Google Scholar]
- Structural and Chemical Biology of the Interaction of Cyclooxygenase with Substrates and Non-Steroidal Anti-Inflammatory Drugs. Chem. Rev.. 2020;120:7592-7641.
- [CrossRef] [Google Scholar]
- ACM/IEEE Conference on Supercomputing, 2006. New York, NY, USA: Association for Computing Machinery; 2006.
- Schrödinger Release 2021-1:, 2021. Prime. Schrödinger, LLC New York, NY.
- Schrödinger Release 2021-2:, 2021a. Maestro. Schrödinger, LLC New York, NY.
- Schrödinger Release 2021-2:, 2021b. Protein Preparation Wizard. Epik, Schrödinger, LLC, New York, NY, 2021; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY.
- Schrödinger Release 2021-2:, 2021c. LigPrep. Schrödinger, LLC New York, NY.
- Schrödinger Release 2021-2:, 2021d. Glide. Schrödinger, LLC New York, NY.
- Schrödinger Release 2021-2:, 2021e. QikProp. Schrödinger, LLC New York, NY.
- Schrödinger Release 2021-2: 2021f. Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools. Schrödinger, New York, NY.
- Study of Cyclooxygenase-3 on the Bases of Its Facts and Controversies. Int. J. Pharmarceutical Sci. Res.. 2019;10:387-392.
- [Google Scholar]
- Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem.. 2015;71:3-8.
- [CrossRef] [Google Scholar]
- Microglia: An interface between the loss of neuroplasticity and depression. Front. Cell. Neurosci.. 2017;11
- [CrossRef] [Google Scholar]
- Deciphering the mechanisms of regulation of an excitatory synapse via cyclooxygenase-2. A review. Biochem. Pharmacol.. 2021;192:114729
- [CrossRef] [Google Scholar]
- Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. U. S. A.. 2003;100:5473-5478.
- [CrossRef] [Google Scholar]
- Indazole: A medicinally important heterocyclic moiety. Med. Chem. Res.. 2012;21:1509-1523.
- [CrossRef] [Google Scholar]
- The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett.. 2010;20:7159-7163.
- [CrossRef] [Google Scholar]
- COX inhibitors directly alter gene expression: Role in cancer prevention? Cancer Metastasis Rev.. 2011;30:641-657.
- [CrossRef] [Google Scholar]
- The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999;18:7908-7916.
- [CrossRef] [Google Scholar]
- COX-2 inhibitors in cancer treatment and prevention, a recent development. Anticancer. Drugs. 2002;13:127-137.
- [CrossRef] [Google Scholar]
- ATP-competitive inhibitors of the mammalian target of rapamycin: Design and synthesis of highly potent and selective pyrazolopyrimidines. J. Med. Chem.. 2009;52:5013-5016.
- [CrossRef] [Google Scholar]
- Mechanism-Based Inhibition of Cytochrome P450 3A4 by Therapeutic Drugs. Clin. Pharmacokinet.. 2005;44:279-304.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2021.103540.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




