

Translate this page into:
Molecular structure design and soft template synthesis of aza-, oxaaza- and thiaazamacrocyclic metal chelates in the gelatin matrix
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The data about of soft template synthesis proceeding in gelatin matrices in [3d-element M(II) ion – (N,S)- or (N,O,S)-ambidentate ligson – mono- or dicarbonyl ligson] systems, have been considered and discussed. The chemical nature of the final products of template synthesis formed under these specific conditions, has been compared with the chemical nature of the final products formed by template synthesis in solutions. It has been noted that in many cases, the nature and chemical composition of these products differ substantially. Specific features of the DFT calculated molecular structures of the macrocyclic compounds that can be formed due to the template synthesis in the systems indicated above, have been discussed, too. The review covers the period 1990–2015.
Keywords
Soft template synthesis
Molecular structure design
Metal chelate

1 Introduction
As known, processes of the so-called self-assembly of molecules are among the most important chemical reactions used presently in modern synthetic organic, organometallic, and coordination chemistry. In terms of each of such process, the target product is formed as a result of specific association of individual finer “building blocks”. These processes are now rather abundant in the synthesis of macrocyclic compounds, first of all, macrocyclic metal chelates containing atoms of various d-elements. One of the most significant varieties of “self-assembly” is template synthesis for which the association of molecules of the starting substances gains its specific directivity under the action of this or another “template”. As a rule, the role of such a “template” is played by any metal ion that further incorporates the macrocyclic metal chelate formed by self-assembly. It should specially be mentioned that such template present in the reaction system does not simply “conducts” the template process; in the absence of it, this process does not occur at all. Template reactions often turn out to be key in the synthesis of macroheterocyclic compounds; in this case, the metal chelates formed firstly, are subjected to removal of metal ion (the so-called demetallation). According to available data (Kasuda and Tsutsui, 1980), the first such a process was quite occasionally carried out as early as 1927 by Japanese chemists due to the reaction of 1,2-dibromobenzene and copper(I) cyanide in pyridine. Besides, instead of the expected simple target product, namely benzenedicarbonitrile-1,2 1,a, a dark blue substance, which many years later was identified as copper(II) phthalocyanine 1,b, was obtained.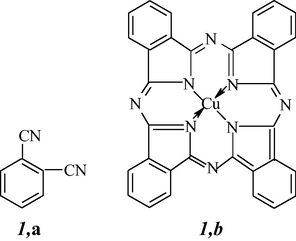
The priority of discovery of template synthesis is officially ascribed to Curtis (New Zealand), who at the beginning of 1960 synthesized the Ni(II) tetraazamacrocyclic chelate 1,c containing four nitrogen atoms, from such comparatively simple chemical agents as ethanediamine-1,2, acetone, and nickel(II) perchlorate (Curtis, 1960, 1968; Busch, 1963).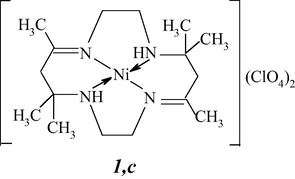
Further a huge number (now measured by five-unit number) of similar chelates were synthesized, so that this variant of complex formation has already been studied fairly well to the beginning of the XXI century; in particular, comprehensive monographs (Melson, 1979; Yatsimirskii et al., 1987; Lindoy, 1989; Davies et al., 1996; Skopenko et al., 1997; Gerbeleu et al., 1999; Garnovskii et al., 1999, 2000; Gloe, 2005) were devoted to this process.
At the present time, the number of published works on the given field of chemistry amounts to many thousands. In particular, such a synthetic method was applied to obtain complexes with organic ligands of a very complicated structure: porphyrins and phthalocyanines, preparation of which is very labor-consuming processes, and the so-called metal-encapsulated compounds (Voloshin, 1998). The related reactions are also leading in the synthesis of aza-, azaoxa-, and azathiamacrocycles, crown ethers, and other systems with closed contours containing various heteroatoms in the frameworks [see, for example, House and Curtis, 1961; Christensen et al., 1974; Nelson, 1980; Kodama et al., 1984; Hancock et al., 1987; Hancock and Martell, 1989; Westerby et al., 1991; Bertolo et al., 1999; Kumar and Alexander, 1999; Chandra and Gupta, 2002; Costisor and Linert, 2004; Khan et al., 2004; Cronin, 2005; Borisova et al., 2006; Niasari and Davar, 2006; Khandar et al., 2007; Niasari et al., 2007; Chandra et al., 2007; Ilhan et al., 2007; Llhan et al., 2007; Singh et al., 2007; Khanmohammad et al., 2007; Firdaus et al., 2008; Keypour et al., 2008; Mikhailov, 2008a; Mikhailov and Chachkov, 2009a, 2010; Nath et al., 2009; Tokarev et al., 2010; Papini et al., 2009; Singh and Kumar, 2010; Fabbrizzi et al., 2010; Rafat and Siddiqui, 2011; Gurumoorthy et al., 2012; Aquilanti et al., 2011; Walker et al., 2012; Singh and Sharma, 2014; Beynek et al., 2015]. This took place primarily because template synthesis leads, in the predominant majority of cases, to either the formation of additional metallocycles, or cross-linking of the metallocycles present earlier in the complex into a single closed contour and to the formation of macrocyclic compounds. (The latter presently imply compounds with a closed contour containing not less than nine atoms, at least three of which perform the function of donor centers (Gerbeleu et al., 1999)). In view of the above, as it seems to us, it is no exaggeration to say that the template synthesis occupies one of the most important positions in the hierarchy of synthetic methods in the modern coordination chemistry. Moreover, it is one of the most important synthetic approaches in supramolecular chemistry as well (Lehn, 1995a, 1995b). The final products of these reactions have a complex of nontrivial physicochemical properties and thus find exclusively diverse use: the list of branches where they are demanded includes metallurgy and medicine, industrial biotechnology and catalysis, microelectronics and agriculture, and many other areas of human activity. Also, the so-called molecular nanotechnology was added to this list in the recent years (Eremenko, 2008). It should be noted in this connection that many articles in the field of the template synthesis cited above, and namely (Chandra and Gupta, 2002; Costisor and Linert, 2004; Khan et al., 2004; Cronin, 2005; Borisova et al., 2006; Niasari and Davar, 2006; Khandar et al., 2007; Niasari et al., 2007; Chandra et al., 2007; Ilhan et al., 2007; Llhan et al., 2007; Singh et al., 2007; Khanmohammad et al., 2007; Firdaus et al., 2008; Keypour et al., 2008; Mikhailov, 2008b; Mikhailov and Chachkov, 2009a, 2010; Nath et al., 2009; Tokarev et al., 2010; Papini et al., 2009; Singh and Kumar, 2010; Fabbrizzi et al., 2010; Rafat and Siddiqui, 2011; Gurumoorthy et al., 2012; Aquilanti et al., 2011; Walker et al., 2012; Singh and Sharma, 2014; Beynek et al., 2015), were published in the 21st century, so that the interest in this method and its improvement remain high. Besides, among them are monographs and review articles, in particular (Gloe, 2005; Costisor and Linert, 2004; Cronin, 2005; Borisova et al., 2006; Mikhailov, 2008a).
At present time, template synthesis is carried out, as a rule, in liquid-phase homogeneous systems (solutions) or in such heterogeneous systems where at least one of the phases is liquid and another can be the solid or rarely gas phase. However, another variant is possible, when one of the parts of the system is a gel formed by any high-molecular-weight compound. Among these systems are, in particular, immobilized matrix implants based on natural high-molecular compounds in which the initial substances and complex formation reaction products with this or another degree of rigidity were detected in the bulk of the corresponding polymer support. The present review is devoted to such systems and processes of template synthesis of macrocyclic metal chelates occurring in these systems.
2 General regularities of template processes
In the general case, template synthesis presently implies a variety of complexing reactions where the metal ion with certain stoichiometry and electronic structure acts as a template for the formation from the corresponding initial substances the singly possible or prevailing (under the reaction conditions) products, the synthesis of which under other reaction conditions is either very difficult, or cannot be realized at all (Gerbeleu et al., 1999). Each of the compounds formed represents a metal complex with the chelate polydentate ligand (chelant), which is obtained according to the scheme [metal ion + building blocks of the future ligand (ligand synthones or ligsons) → complex] but not classical scheme for metal complexes (metal ion + ligand → complex). Template synthesis includes two aspects: the ligand system formation due to the organizing and directing role of complexing metal and reactions of organic compounds associated with this ion (reactions of coordinated ligands) (Davies et al., 1996; Gerbeleu et al., 1999; Garnovskii et al., 1999). This process usually occurs under fairly rigid conditions and requires elevated temperature in the reaction system for rather prolonged (several hours and more) time (Gerbeleu et al., 1999). This is partially related to a significant decrease in entropy upon the construction of macrocyclic compounds from individual structural fragments (ligand synthones), which is due to a decrease in the number of rotational and vibrational degrees of freedom followed by a decrease in the probability of formation of reaction products containing closed contours of atoms. This process could be facilitated to a significant extent when it is carried out in the so-called organizing systems favoring structural reorganization of chemical compounds, in particular, coordination compounds, formed in the reaction. In principle, any process of self-assembly is accompanied by the formation of chemical compounds with a more complicated composition than the initial substances in general and ligsons in particular. The general concept of chemical thermodynamics assumes that this process should result in a noticeable decrease in the entropy of the reaction system. According to the classical Gibbs–Helmholtz equation for the isobaric chemical process (2.1)
Since both (ΔSo)os and ΔS0 are negative values, |(ΔS0) − (ΔSo)os| < |(ΔS0)|. As can easily be noticed, similar forced decrease in entropy could result in a decrease in the slope ratio of the linear dependence ΔG0(T) to the abscissa, due to which the temperature range of this template process increases. This fact is illustrated by means of Fig. 2.1.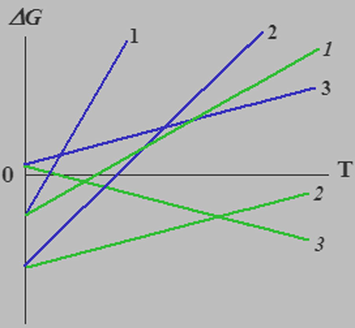
Schematic view of the ΔG0(T) (1, 2, 3) and
(1, 2, 3) dependences for three variants of the template process. The lines 1, 1 and 2, 2 are related to the template processes with ΔH < 0 which, in principle, can be implemented in the systems without a compulsory decrease in the entropy; the lines 3, 3, for the template processes with ΔH > 0, which can be implemented only in the systems with a compulsory decrease in the entropy. The lower slope of the 1, 2, and 3 lines is clearly seen, unlike that of the 1, 2 and 3 lines.
Therefore, it can be expected that for template synthesis in organizing systems complex formation reactions that are thermodynamically inadmissible under traditional conditions, i.e., without the above indicated ordering, and this is possible without changing the assortment and composition of formed metal chelates.
3 Gelatin as a polymer massif for template processes
Immobilized matrices (in particular, thin-film implants) on the basis of high-molecular compounds with immobilized metal complexes represent a typical example of the reaction medium in which the reaction system is preordered and, hence, the entropy undergoes forced decreasing. They can be divided into two categories (Pomogailo, 1988). In the first one, a chemical bond is observed between molecules of the immobilized substance and high-molecular-weight compound molecules, whereas the second category is characterized by the interaction due to intermolecular (in particular, van der Waals) bonds only. Such known natural compounds as gelatin, agar, and pectin belong to high-molecular compounds that, in principle, can be used for the immobilization of metal complexes. Each of these compounds is characterized by isotropic physicochemical properties, transparency, hydrophilicity, and plasticity. All these high-molecular compounds easily form gels, viz. disperse systems based on low-molecular liquids (including water) possessing some properties of solids, in particular, the absence of fluidity at low shear stress, capability of shape retaining, and noticeable strength and elasticity. Three substances indicated above are appropriate, in principle, for the construction of immobilized matrix systems. However, gelatin that can form, first, comparatively stable gels even at a high (up to 99.9 wt.%) water content and, second, thin-film implants on various substrates (glass, polymeric, and metallic) should be acknowledged to be the most suitable for complexing and template synthesis of macrocyclic metal chelates. In addition, massifs of the indicated high-molecular compound can be considered as quasi-elastic, resin-like polymers, since they contain infinite networks formed by low rows of molecules connected to each other through a restricted number of transverse bonds. Therefore, the gelatin structure seems to be rather convenient for the formation of immobilized matrix systems, because this structure, on the one hand, does not allow any rigid crystalline blocks to take place (unlike agar-agar) and, on the other hand, possesses fairly high number of cells for accepting and consequent fixing molecules of the immobilized chemical compound. In addition, these cells, even being-filled by such molecules, retain certain freedom of migration in space. By having the polymer with this structure, one can obtain immobilized systems with a fairly uniform distribution of the immobilized substance in this or another part of the polymer massif and with good steric accessibility of molecules of this substance for diverse chemical processes. Owing to this, almost all works on studying template synthesis under specific conditions of metal complex immobilized matrix systems are connected with the so-called gelatin-immobilized matrices (GIM).
In its chemical nature, gelatin is known to be a poly-disperse mixture of low-molecular polypeptides of a general formula 3,a (R1, Ri,– various radicals)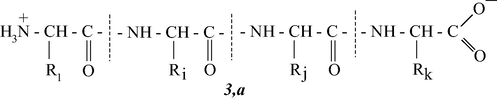
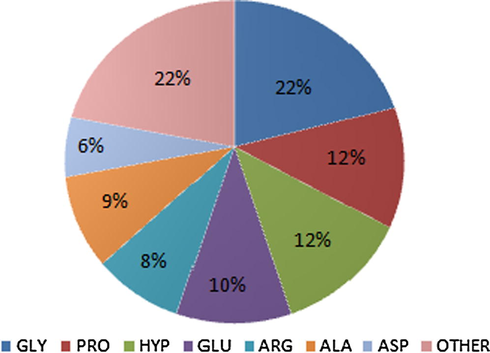
General amino acid composition of gelatin (GLY– glycine, PRO– proline, HYP– hydroxyproline, GLU– glutamine, ARG– arginine, ALA– alanine, ASP– asparagine).
Gelatin molecules consist of three polypeptide chains with almost the same molecular weight, two of which are usually nearly identical to each other by the set and sequence of amino acids (the so-called α1-chains), whereas the third one (α2-chain) differs from them in this respect (Ramachadran, 1967; Boedtker and Doty, 1954; Chen et al., 1991). Model of such a structure is shown in Fig. 3.2. The typical stoichiometric composition of this high-molecular compound expressed in the amount and assortment of α–chains in the macromolecule is, as a rule, (α1)2α2 and, rarely, (α1)3 (Groome and Clegg, 1965; Stravich and Nimni, 1971). In the publications (Rich and Crick, 1955; Cowan et al., 1955; Chen et al., 1991), the structure for gelatin molecules in which a helical structure with the left travel is ascribed to each α–chain, was proposed. In terms of this structure, all the three polypeptide helices are interlaced into a single right-handed helix, the decisive role in the stabilization of which belongs to hydrogen bonds (Rich and Crick, 1955). Besides, α2-chains are generally characterized by the same set of polypeptide fragments as α1-chains, but their aminoacid sequence contains less such amino acids as proline, hydroxyproline, and lysine, while tyrosine, valine, histidine, and hydroxylysine predominate (James, 1977). It was found (Ramachadran, 1967) by electron microscopy that the diameter of a gelatin macromolecule was only 1.4 nm, whereas its length was 285 nm. The macromolecule is sharply asymmetric and anisometric (Ramachadran, 1967; Boedtker and Doty, 1954). These data are well consistent with similar results (Boedtker and Doty, 1954) of measurements of light scattering and viscosity of gelatin solutions. Based on these data, we attempted to estimate the average size of an individual intermolecular cavity in the gelatin massif. Among their number, the volume of the polymer gelatin layer (Ve) with a surface area of 1 cm2 and a thickness of 10 μm are equal to (1.0·1.0·10·10−4) cm3 = 1.0·10−3 cm3, and, hence, the gelatin mass in this massif is (0.5·1.0·10−3) g = 0.5·10−3 g (at an average density of 0.5 g·cm–3). As mentioned above, the molecular mass of gelatin (MGel) is ∼(2.0–3.0)·105; therefore, the number of its molecules in this massif is (0.5·10−3/MGel)·6.02·1023 = (1.0–1.5)·1015. Taking the length and diameter of the gelatin molecule indicated above and assuming that the molecule can be identified as a narrow-band cylinder, its volume VM will be equal to (1/4πDh2) = (1/4)·3.14·(2850·10−8 cm)·(14·10−8 cm)2 = 4.38·10−19 cm3. For the maximally dense packing, these molecules will occupy the total volume equal to (4.38·10–19·(1.0–1.5)·1015) cm3 = (4.38–6.57)·10–4 cm3. It can be postulated that the volume of the cell-cavities we are interested in is equal to the total volume of the polymer massif minus the volume occupied by gelatin molecules just calculated by us: [1.0·10–3 − (4.38–6.57)·10–4] cm3, which is (3.43–5.62)·10–4 cm3. In the first (although and rather rough) approximation, we can accept that each gelatin molecule participates in the formation of only one cell. Then the average volume of the cell can be determined as a quotient of the division of their total volume into the number of gelatin molecules and, as it is easily seen, is equal to (2.29–5.62)·10–19 cm3. The linear size of this averaged cell (if assuming its spherical shape) is D = (6V/π)1/3 = [6·(2.29–5.62)·10–19 cm3/3.14]1/3 = 7.60–10.22 nm, and if assuming the cubic shape, a = V1/3 = [(2.29–5.62)·10–19 cm3]1/3 = 6.13–8.24 nm. The fragment of gelatin structure containing intermolecular cavities is shown in the Fig. 3.3. As can be seen from these data, rather large molecules of the immobilized substance may be incorporated into the cell of similar sizes. Rather large gaps between the chains of the spatial network in the molecular structure of gelatin allow molecules and ions of low-molecular substance, unlike large colloidal particles or macromolecules, to diffuse into intermolecular cavities of GIM nearly as easy as into liquid-phase solvents.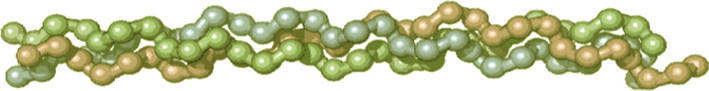
Schematic image of three polypeptide chains form a triple-helix of gelatin. This molecule is stabilized by the formation of covalent cross-links, both within the gelatin triple helix and between gelatin helices.
The fragment of gelatin structure containing intermolecular cavities (left); the general plane of intermolecular cavity in which can be nanoparticles of various chemical compounds formed in chemical processes in the gelatin matrix (right).
A very important feature of gelatin that distinguishes it from other aforementioned natural high-molecular (agar and pectin) is that gelatin is ampholyte (James and Mees, 1972; James, 1977) and its macromolecule gains a negative charge in an alkaline medium, due to which the proton_donor activity of the immobilized compounds becomes more significant than that in an aqueous solution. The most part of the presently known processes of template synthesis is associated with the intramolecular formation of water due to mobile hydrogen atoms of one ligson (A) and oxygen atoms of another ligson (B) (Gerbeleu et al., 1999). The more mobile the hydrogen atoms in the corresponding ligson, the higher the probability of this process. In turn, this is directly related to its proton-donor ability. Since deprotonated forms of ligsons are involved, in many cases, at least indirectly in the formation of macrocyclic metal chelates, theoretically it can be expected that the self-assembly of this type in GIM should be more efficient than under traditional conditions (in solution or in the solid phase). It was mentioned as far as at the end of the last century (Mikhailov, 1995, 1997) that the character of template synthesis in GIM in the whole series of ternary systems metal ion—ligson A—ligson B differs significantly from that of the template synthesis in solutions and in the solid phase. Similar differences are most pronounced in the case of ambidentate ligsons A containing three or more donor sites and capable, depending on the complex formation conditions, of coordinating to this or another metal ion by different ways; these ligsons are considered in almost all works devoted to template synthesis in GIM.
4 Metal hexacyanoferrates(II) gelatin-immobilized matrices (MHF-GIM)
The so-called metal hexacyanoferrate(II) GIM containing hexacyanoferrates(II) of M(II), M(III), and M(IV) where M are p-, d-, or f-elements (in further – MHF-GIM), as immobilized metal complexes are very promising for template synthesis in gelatin-immobilized matrices. The indicated coordination compounds are characterized by rigid fixation in the polymer massif, optimum kinetic lability, and very low solubility in water. Template synthesis MHF-GIM could occur on their contact with aqueous solutions containing a combination of the corresponding ligand synthones. The technology of manufacturing gelatin-immobilized matrix implants containing hexacyanoferrates(II) of d-elements is described in a whole series of works (see, for example, Mikhailov and Budnikov, 1989; Mikhailov and Polovnyak, 1990a, 1990b, 1990c, 1991; Mikhailov, 1991, 1992, 1996, 1998; Mikhailov and Rozhentsov, 2001). In all cases, gelatin-immobilized matrices containing elemental silver (Ag-GIM), which, in turn, were obtained from halogen-silver GIM using the technology known in the halogen-silver technology (James and Mees, 1972; James, 1977), served as the starting raw materials. The most general approach to the immobilization of hexacyanoferrates(II) of d-elements in the gelatin matrix was described (Mikhailov, 1998). The essence of the approach is the transformation of Ag-GIM into a silver(I)hexacyanoferrate(II) GIM due to the treatment of the Ag-GIM with an aqueous solution of potassium hexacyanoferrate(III) K3[Fe(CN)6] followed by the electrophilic substitution of Ag(I) by the corresponding ion M(II), M(III), and M(IV). This substitution occurs under the action of chlorides of these ions according to general Eqs. (4.1–4.4)
(immobilized chemical compounds in braces are bold faced). The by-product, namely silver(I) chloride, is removed from GIM by the treatment with an aqueous solution of sodium thiosulfate Na2S2O3. As a result of this process, soluble Ag(I) complex diffusing from the GIM to the contacting solution, takes place according to the general Eq. (4.5)
In some cases, gelatin-immobilized hexacyanoferrates(II) of d-elements can be obtained by a simpler way using reactions (4.6–4.9), whose participants are coordination compounds of M(II) and M(III) with various oxy acids, in particular, oxalic (H2Ox), citric (H3Ct), and tartaric (H4Tart) acids
The formed poorly soluble compound of Ag(I), namely, Ag4[Fe(CN)6], as AgCl, is removed from GIM by the action of Na2S2O3 using the equation
Heterobinuclear hexacyanoferrates(II) containing two different d-elements (see the respective information in publications (Mikhailov et al., 2003a, 2003b, 2003c, 2003d, 2004a, 2004b; Mikhailov, 2008b) can be obtained along with mononuclear hexacyanoferrates(II) of d-elements using electrophilic substitution reactions in MHF-GIM. In principle, these coordination compounds can be used for realization of template synthesis in GIM, too (although template reactions with these participations were not carried out up to now).
5 Template synthesis of aza-, oxaaza- and thiaazamacrocyclic metal chelates in the MHF-GIM
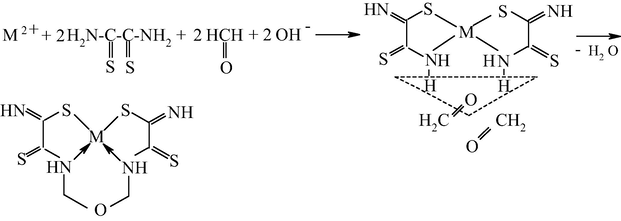
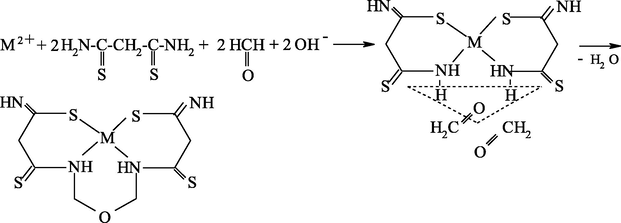
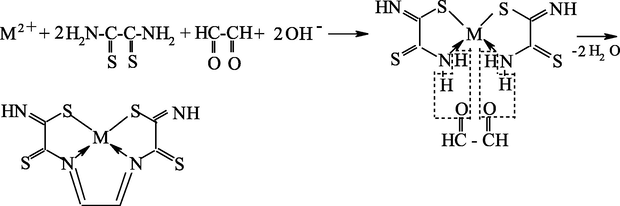
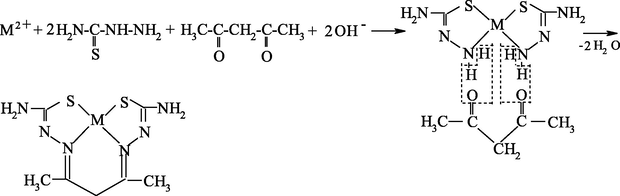
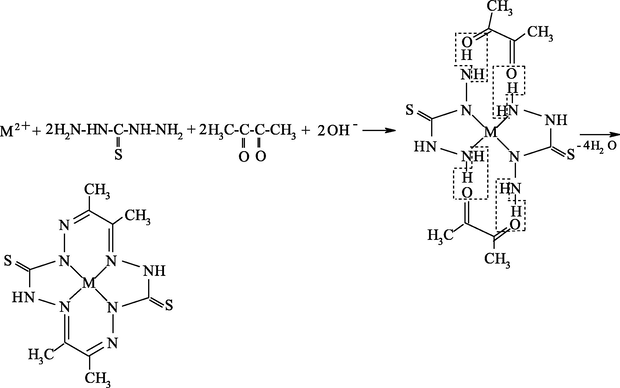
As already mentioned, it seems most reasonable to use ambidentate ligsons in GIM in template processes. The (N,S)- and (N,O,S)-donor atomic organic compounds capable to coordination to the metal ion through the nitrogen, sulfur or oxygen atoms, and namely, ethanedithioamide H2NC(⚌S)C(⚌S)NH2, thiocarbamoylmethaneamide H2NC(⚌S)C(⚌O)NH2, hydrazinomethanethioamide H2NHNC(⚌S)NH2, hydrazinomethanethiohydrazide H2NHNC(⚌S)NHNH2, and propanedithioamide H2NC(⚌S)CH2C(⚌S)NH2, were used as ambidentate ligsons (Mikhailov and Chachkov, 2009a, 2009b, 2010; Mikhailov et al., 1999, 2000a, 1998, 2008a, 2000b, 2003e, 2003f, 2003g, 2008b, 2009, 2001a, 2001b, 2007, 1997, 2004c, 2013a; Mikhailov and Khamitova, 2000, 1998c, 1998d, 1999; Mikhailov, 2001a, 2000a, 2001b, 2014, 2001c, 2002a, 2002b, 2002c, 2000b; Mikhailov and Khamitova, 1998a, 1998b; Chachkov and Mikhailov, 2010a, 2011a, 2012a; Kazymova et al., 2010). In these researches cited, organic compounds containing one or two carbonyl groups (C⚌O), were used as the second ligsons: methanal H2C⚌O, ethanal H3CCH(⚌O), propanone H3CC(⚌O)CH3, ethanedial CH(⚌O)CH(⚌O), butanedione-2,3 H3CC(⚌O)C(⚌O)CH3, and pentanedione-2.4 H3CC(⚌O)CH2C(⚌O)CH3. The data on the nature of metal chelate complexes formed in ternary systems [M(II) ion – (N,S)-, (N,O,S)-donoratomic ambidentate ligson A—mono-, dicarbonyl-containing ligson B] involving various metal ions and the above indicated organic compounds are presented in Tables 51 and 52. As it can be seen from the data presented in them, the assortment of metal chelates formed by template synthesis in metal hexacyanoferrate(II) GIM, in almost of indicated systems is more significant and diverse than that in the case of the same process in an aqueous solution. Moreover, in some cases, template synthesis occurs only in these GIM, whereas simple (non-macrocyclic) chelate complexes are formed in an aqueous solution or in solid phase. The internal coordination sphere of these simple complexes contains only molecules of corresponding nitrogen-, oxygen-, and sulfur-containing ligson A. In the case of monocarbonyl ligsons B, as a rule, macrotricyclic complexes in which two donor nitrogen atoms are in the composition of the 6-membered ring, are formed, whereas macrotricyclic complexes, where two donor N atoms are in the 5-membered rings, are formed in the case of the dicarbonyl ligsons. Therefore, note that the composition and structure of chelants formed in the considered systems depend more considerably on the nature of ligsons involved in template synthesis, than on the nature of the M(II). Anyway, this phenomenon is observed for the few M(II), whose hexacyanoferrate(II) GIM were used for the moment in experiment [Co(II), Ni(II), and Cu(II)]. Chemism of all template processes proceeding in the MHF-GIM, and studied by us, has been connected with sort Schiff condensation with the intramolecular formation of water, in particular according to Schemes 5.1–5.3 in the case monocarbonyl ligsons and according to Schemes 5.4–5.6 in the case of dicarbonyl ligsons.
In aqueous solution
In metal hexacyanoferrate(II) GIM
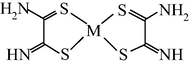
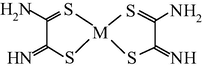
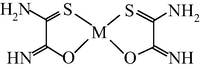
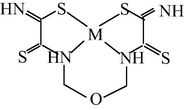
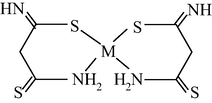
M(II) – ethanedithioamide – ethanedial (M = Ni, Cu) (Mikhailov et al., 1999, 2001a, 2001b, 2007; Mikhailov and Khamitova, 2000, 1998b; Mikhailov, 2001a, 2000b)
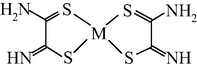
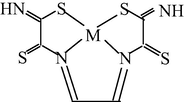
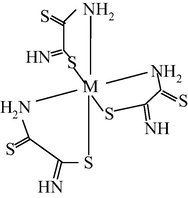
M(II) – ethanedithioamide – ethanedial (M = Co) (Mikhailov and Khamitova, 2000, 1999; Mikhailov et al., 2000a, 1997)
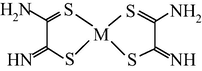
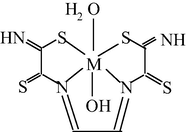
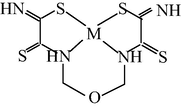
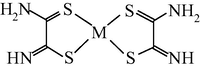
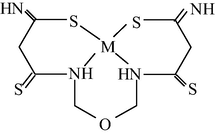
M(II) – propanedithioamide – ethanedial (M = Ni, Cu) (Mikhailov et al., 2003e)
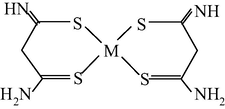
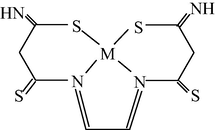
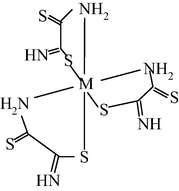
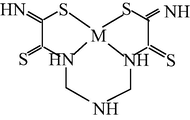
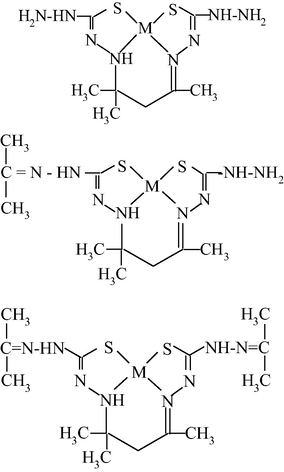
M(II) – hydrazinomethanethiohydrazide – butanedione-2,3 (M = Cu) (Mikhailov et al., 2004c)
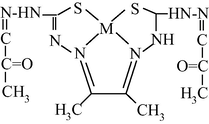
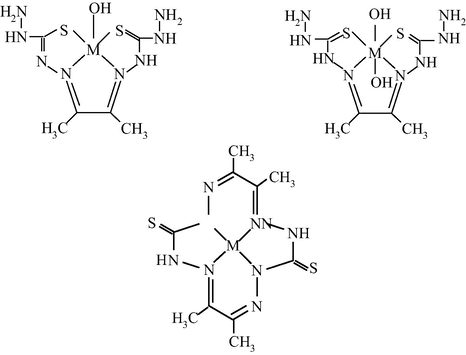
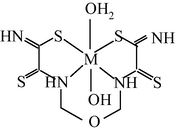
M(II) – hydrazinomethanethioamide – butanedione-2,3 (M = Co, Ni, Cu) (Mikhailov et al., 2013a)
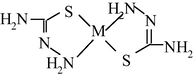
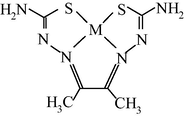
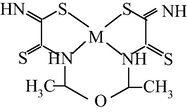
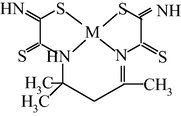
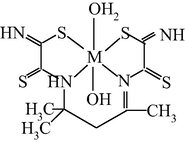
M(II) – hydrazinomethanethioamide – pentanedione-2,4 (M = Co, Ni, Cu) (Kazymova et al., 2010)
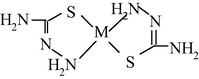
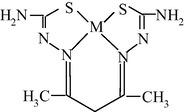


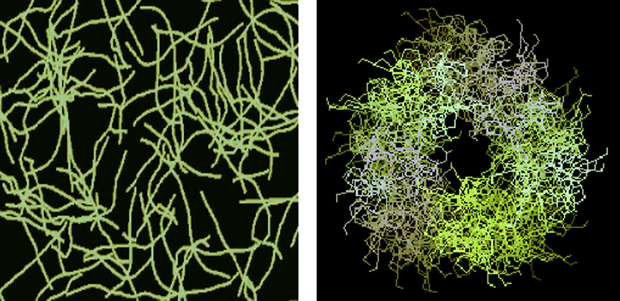
By finishing the Section 5, note that, based on the indicated above sizes of intermolecular cavities, it can be expected that immobilized macrocyclic chelates formed in them due to self-assembly will be composed of nanoparticles. The electronic microscopic studies of some metal chelate GIM confirm this assumption. The photographs of nanoparticles of some of these metal chelates immobilized in the GIM, are shown in Figs. 5.1–5.3. In this connection, it is not surprising that attempts to isolate chemical substances formed in these cavities from the GIM resulted in the isolation of the substances in the amorphous state or as very small crystals unsuitable for single-crystal X-ray diffraction analysis. Partially for this reason, crystal structures of the macrocyclic complexes formed under these specific conditions of complexing, have not been experimentally determined so far.
The SEM photographs of nanoparticles of Co(II) macrotricyclic metal chelate formed in a template synthesis in the MHF-GIM in the Co(II) – ethanedithioamide – methanal triple system.

The SEM photographs of nanoparticles of Ni(II) macrotricyclic metal chelate formed in a template synthesis in the MHF-GIM in the Ni(II)– hydrazinomethanethioamide– pentanedione-2,4 triple system.

The SEM photographs of nanoparticles of Cu(II) macrotricyclic metal chelate formed in a template synthesis in the MHF-GIM in the Cu(II)– ethanedithioamide– ethanedial triple system.
6 Quantum chemical design of molecular structures of macrocyclic metal chelates
Recently we published a series of works where the quantum chemical simulation by the density functional theory (DFT) was performed for molecular structures of diverse macrocyclic metal chelates that are really formed and can be formed in principle in the course of template synthesis in MHF-GIM involving various (N,O,S)-ambidentate ligsons and compounds containing C⚌O groups. The main attention was given to the macrocyclic chelates of 3d-elements, viz. Co(II), Ni(II), and Cu(II), and their neighbors in the D.I. Mendeleev Periodic System, namely Mn(II), Fe(II) and Zn(II), which contain three conjugated metal chelate rings (macrotricyclic complexes) (Chachkov and Mikhailov, 2010a, 2008, 2009a, 2009b, 2010b, 2010c, 2010d, 2011b, 2011c, 2012b, 2012, 2012c, 2013a, 2014a, 2014b; Chachkov et al., 2009; Mikhailov et al., 2012; Chachkov et al., 2013; Mikhailov and Chachkov, 2014a, 2014b, 2014c, 2014d, 2015a, 2015b, 2015c, 2015d, 2016, 2015e). Somewhat less attention was given to macrocyclic complexes with four conjugated metal chelate rings (macrotetracyclic complexes) (Chachkov and Mikhailov, 2009c, 2013b, 2013c, 2014c, 2014d, 2014e, 2014f, 2013d, 2013e; Mikhailov and Chachkov, 2013a, 2013b, 2013c, 2015f, 2015g; Mikhailov et al., 2013b). (565) Macrotricyclic complexes are most abundant among the former, and many publications (Chachkov and Mikhailov, 2010a, 2011a, 2012a, 2008, 2009a, 2009b, 2010b, 2010c, 2011b, 2012b, 2013a; Chachkov et al., 2009, 2013; Mikhailov and Chachkov, 2014d, 2015a, 2015b, 2015c, 2015d) are devoted to them. (555) Macrotricyclic complexes are considerably poorly presented (Chachkov and Mikhailov, 2011c, 2012b, 2014a, 2014b; Mikhailov and Chachkov, 2012, 2014b, 2015a, 2016); two papers (Chachkov and Mikhailov, 2010d; Mikhailov and Chachkov, 2014c) are devoted to (666) macrotricyclic complexes, and only one publication (Mikhailov and Chachkov, 2014a) considers (545) macrotricyclic complexes. Among the second (macrotetracyclic) complexes, (5656) macrotetracyclic (Chachkov and Mikhailov, 2009c, 2013c, 2013d, 2014c, 2014f; Mikhailov and Chachkov, 2013a), (5456) macrotetracyclic (Chachkov and Mikhailov, 2013b, 2013c, 2014d; Mikhailov et al., 2013b), (5555) macrotetracyclic (Mikhailov and Chachkov, 2013b; Chachkov and Mikhailov, 2013c) and (5454) macrotetracyclic (Chachkov and Mikhailov, 2014e) complexes are described (Hereinafter, figures in parentheses designate the number of atoms in each metal chelate ring in the composition of the macrocycle; the sequence of figures corresponds to the order of arrangement of cycles with the corresponding memberness in the macrocycle for clockwise movement along the perimeter). When characterizing structural features of these metal complexes, it is worth mentioning that the most of them are non-coplanar against expectations based on rigidity of their structures. The degree of deviation from co-planarity, which in the case of chelate modes can quantitatively be characterized by the difference (BAS – 360°), where BAS is the sum of bond angles X—M—Y (X and Y are donor atoms, and M is the metal atom in the chelate node). In the case of chelate cycles, the degree of deviation is characterized by the difference between the sums of bond angles between the atoms in the 4-, 5- and 6-membered chelate rings (BAS4, BAS5, and BAS6, respectively) and the sum of angles in the corresponding planar polygon (360, 540, and 720°, respectively). In several cases, this value is rather significant (several tens of degrees). For all (545) macrotricyclic metal chelates, chelate nodes are formed with the tetragonal pyramidal orientation of the donor oxygen or sulfur atoms relative to the complexing atom M(II), for which the values of (BAS – 360°) are negative (Mikhailov and Chachkov, 2014a). On the contrary, the (555) macrotetracyclic metal chelates are characterized by either rigidly planar structure of the chelate node, or only slight deviation from co-planarity (Chachkov and Mikhailov, 2011c, 2012b, 2014a, 2014b; Mikhailov and Chachkov, 2012, 2014b, 2014b). For both (565) macrotricyclic and (545) macrotricyclic metal chelates, the chelate node almost always deviate to this or another extent from co-planarity. However, the metal chelates in which (BAS – 360°) < 0 and, hence, the structure of their chelate nodes is pyramidal, and the metal chelates, where (BAS – 360°) > 0 and the pseudo-tetrahedral or even quasi-tetrahedral structure of these node is observed, were mentioned. It should be emphasized that the complexes with the former ratio are met much more frequently than the complexes with the second ratio (Chachkov and Mikhailov, 2010a, 2008, 2009a, 2009b, 2010b, 2010c, 2011b, 2012c, 2013a, 2014d; Chachkov et al., 2009, 2014d; Chachkov et al., 2013); all the data show that the tendency to “tetrahedrization” of the chelate mode increases with an increase in the overall memberness of the rings and becomes distinctly pronounced for the (666) macrotricyclic metal chelates (Chachkov and Mikhailov, 2010d; Mikhailov and Chachkov, 2014c). The typical molecular structures of these complexes are shown in Figs. 6.1–6.6. As for co-planarity of metal chelate rings, both complexes with rigidly planar rings and with rings having very significant deviations from co-planarity are observed, and no interrelation between the degree of co-planarity of the chelate node and metal chelate rings in specific complex was revealed so far. Therefore, it should be mentioned that in the (545)- and (565) macrotricyclic metal chelates both the 4-membered and 5- and 6-membered metal chelate rings are non-coplanar, as a rule. The only exceptions are the iron(II) and nickel(II) (565) macrotricyclic complexes with 4,6-dimethyl-2,3,7,8-tetraazanonediene-3,6-dithioamide-1,9 formed in the template synthesis in the systems M(II) ion – hydrazinomethanethioamide – pentanedione-2,4 (Chachkov et al., 2013). The complexes of other M(II) considered in this work, namely, Mn(II), Co(II), Cu(II), and Zn(II), are also quasi-planar and the values of (BAS5 – 540°) and (BAS6 – 720°) for their 5- and 6-membered rings, respectively, very insignificantly (not more than by 10°) differ from zero. Remarkably in these metal chelates, unlike other calculated (565) macrotricyclic complexes, the 6-membered ring contains two double bonds. It is not excluded that this fact is the main factor providing co-planarity of the metal chelates listed above. The (555) macrotricyclic metal chelates, on the contrary, are characterized by planar molecular structures (Chachkov and Mikhailov, 2011c, 2012b, 2014a, 2014b; Mikhailov and Chachkov, 2012, 2014b). The Mn(II), Fe(II), Co(II), Ni(II), Cu(II), and Zn(II) complexes with such tetradentate ligands as 2,7-dithio-3,6-diazaoctadiene-3,5-dithioamide-1,8 and 4,5-dimethyl-1,8-dimercapto-2,3, 6,7-tetraazaoctatetraene-1,3,5,7-diamine-1,8 (Mikhailov and Chachkov, 2012) are typical examples. The molecular structures of some of these complexes are shown in Figs. 6.2 and 6.3. Note that qualitatively the structures of the macrotricyclic complexes depend rather poorly on the nature of M(II) and its electronic configuration. In all these complexes, the M—X and M—Y bonds (X, Y – N, O or S donor atoms) in both the chelate nodes with the same set of donor atoms (of the MX4 or MY4 type) and the chelate nodes with different sets (of the MX2Y2 type) become usually shorter on going from Mn to Ni and elongate on going from Ni to Zn (Chachkov and Mikhailov, 2010a, 2008, 2009a, 2009b, 2010b, 2010c, 2010d, 2011b, 2011c, 2012b, 2012c, 2013a, 2014a, 2014b; Chachkov et al., 2009, 2013; Mikhailov and Chachkov, 2012, 2014a, 2014b, 2014c, 2014d; Mikhailov et al., 2012). As should be expected from the general theoretical concepts, the M—S bonds are substantially longer than the M—N and M—O bonds. It is noteworthy that in several cases, the metal chelate cycles with the same number of atoms and their set in the same complex are not identical, nevertheless, by the values of bond angles between the composing atoms, but the sums of angles in these metal chelate rings are exactly or nearly equal to each other. In all of the above complexes with (NSSN)-coordination template tetradentate ligand to M(II), donor nitrogen (and sulfur) atoms are in cis-position relative to each other. Nevertheless, in two recently published papers (Mikhailov and Chachkov, 2015a, 2015b) it was found the possibility of the existence also of template (565) macrotricyclic complexes with trans-coordinated donor atoms N and S of tetradentate ligand to a complexator M(II).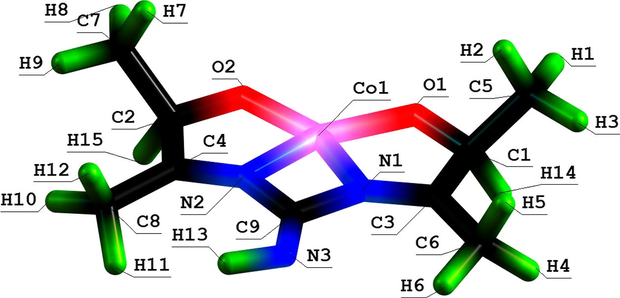
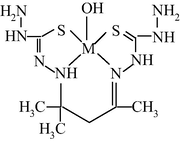
Molecular structure of (545) oxaazamacrotricyclic Co(II) chelate with 3,7-dimethyl-4,6-diazanonen-3,6-diol-2,8.
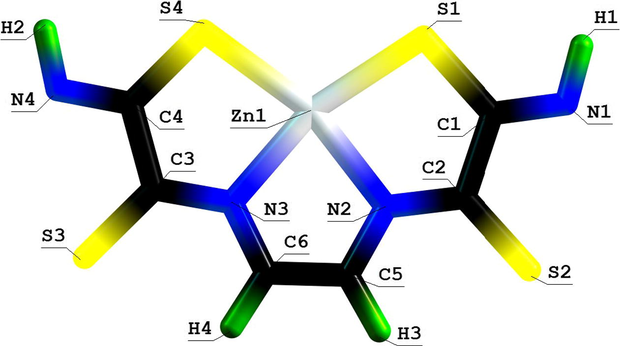
Molecular structure of (555) thia-azamacrotricyclic Zn(II) chelate with 2,7-dithio-3,6-diazaoctadien-3,5-dithioamide-1,8.
Molecular structure of (555) thia-azamacrotricyclic Ni(II) chelate with 4,5-dimethyl-1,8-dimercapto-2,3,6,7-tetraazaocta-tetraen-1,3,5,7-diamine-1,8.
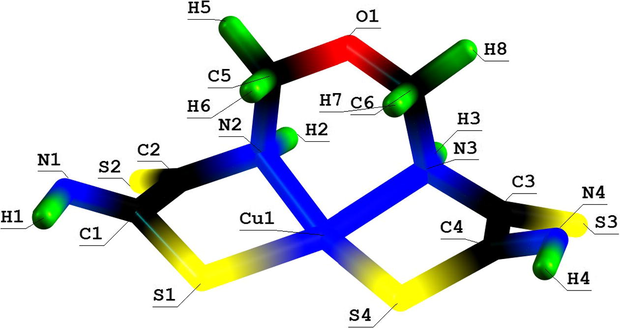
Molecular structure of (565) oxathiaazamacrotricyclic Cu(II) chelate with 2,8-dioxa-3,7-diaza-5-oxanonandithioamide-1,9.
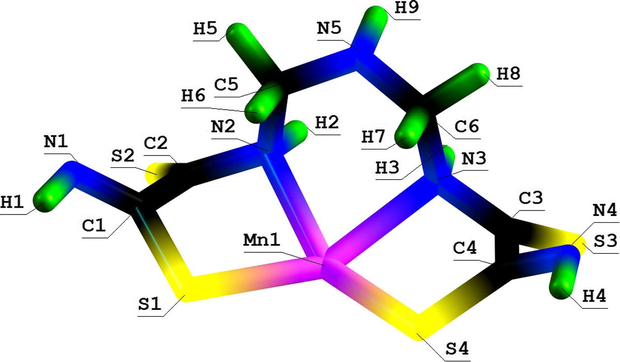
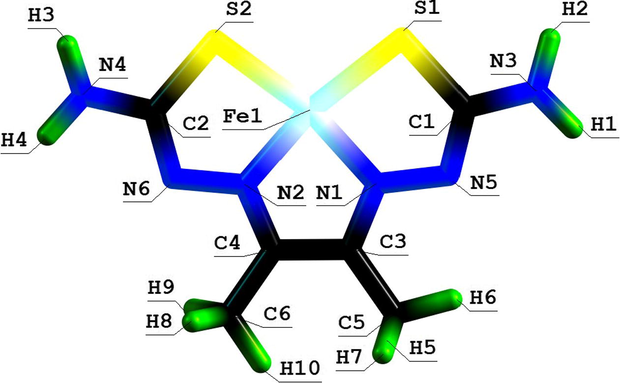
Molecular structure of (565) thia azamacrotricyclic Mn(II) chelate with 2,8-dithio-3,5,7-triazanonandithioamide-1,9.
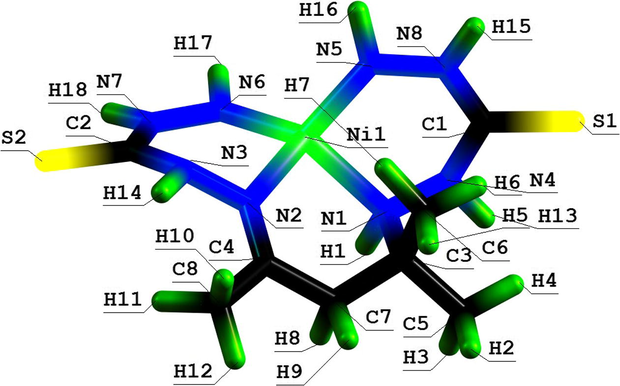
Molecular structure of (666) aza-macrotricyclic Ni(II) chelate with 3,9-dithio-4,8-diaza-6-oxaundecanedithioamide-1,11.
As can be seen from Tables 5.1 and 5.2, in the all triple systems indicated in them, as a result of template synthesis in GIM, only macrotricyclic metal chelates are formed (the singular exception is only the Cu(II)– hydrazinomethanethiohydrazide– butanedione-2,3 system considered in Mikhailov et al. (2004)). However, quantum-chemical calculations by the DFT method clearly evidence about the possibility of formation in these same ternary systems, also, conforming macrotetracyclic metal chelates in which the chelate ligand is a macrocyclic compound. As a whole, the molecular structures of the such metal chelates are characterized by the same features as for the molecular structures of the macrotricyclic complexes. The most part of them is not planar, and the degree of deviation from co-planarity sometimes is very significant (for the 6-membered chelate cycles, it can reach almost 90°). The pyramidal coordination with the BAS smaller than 360° is more typical of their metal chelate nodes of the general formula MN4 (M = Mn, Fe, Co, Ni, Cu, Zn) than the planar or tetrahedral coordination with BAS equal to or larger than 360° (Chachkov and Mikhailov, 2009c, 2013b, 2013c, 2013d, 2013e, 2014c, 2014d, 2014e, 2014f; Mikhailov and Chachkov, 2013a, 2013b, 2013c, 2015f, 2015g; Mikhailov et al., 2013b). The rigidly planar or close to planar group of the nitrogen atoms in the chelate node is explicitly predominant for both the pyramidal and planar geometry of the metal chelate ring. A characteristic example is the (5656) macrotetracyclic chelates of the listed above M(II) ions with 1,8-dioxa-3,6,10,13-tetraazacyclotetradecanetetrathione-4,5,11,12 (Chachkov and Mikhailov, 2009c). For these coordination compounds, the value of BAS in the MN4 chelate nodes is 335.7, 354.9, 356.7, 358.5, 354.4, and 348.0° in the Mn(II), Fe(II), Co(II), Ni(II), Cu(II), and Zn(II) complexes, respectively, whereas the sum of non-bond angles between the nitrogen atoms in the N4 group in each complex is 360.0°. It is remarkably that, in spite of equality of the latter sums, the sum for different M(II) is composed of angles that differ in value and are equal in pairs to each other. For example, in the Mn(II) complex with this macrocyclic ligand, these angles are 88.6 and 91.4°; those in the Ni(II) complex are 89.9 and 90.1°; and in the Zn(II) complex, they are 89.3 and 90.7°. The 5-membered metal chelate rings in each complex are quite identically to each other by both the sum of bond angles between the atoms composing them and the separately taken similar angles. Nevertheless, the both values substantially depend on the nature of M(II) (515.5, 530.4, 531.8, 534.9, 528.7, and 522.3° in the Mn(II), Fe(II), Co(II), Ni(II), Cu(II), and Zn(II) chelates, respectively). At the same time, the 6-membered metal chelate rings differ strongly, in spite of the complete identity by the nature of composing atoms and the order of their arrangement in the cycle. Therefore, in all cases, the 6-membered metal chelate rings containing the N–H bonds with the hydrogen atoms located inside the rings have a noticeably higher degree of distortion from co-planarity than the cycles containing no similar bonds.
As it was already mentioned, almost all described by us (5656) macrotetracyclic metal chelates are non-coplanar. Among the few exceptions from this rule are the Fe(II), Co(II), Ni(II), and Cu(II) chelates with 3,10-dithio-6,7,13,14-tetramethyl-1,2,4,5,8,9,11,12-octaazacyclodecanetetraene-1,5,7,12 (the Mn(II) and Zn(II) chelates with this ligand have a quasi-pyramidal structure of the chelate node). The formation of these complexes in the template synthesis in M(II)– hydrazinomethanethiohydrazide– butanedione-2,3 ternary systems was considered before (Mikhailov et al., 2004c; Mikhailov and Chachkov, 2013a). These chelates are distinguished by one more unique feature, which was not observed for other planar complexes: the electrical moments of the dipole calculated form them are 0.00 (in the Fe(II) complex), 0.01 (in the Co(II) complex), and 0.02 (in the Ni(II) and Cu(II) complexes), i.e., are almost zero. It should be mentioned for the molecular structures of the macrotetracyclic metal chelates with other sets of four metal chelates that the (5555) macrotetracyclic complexes, as the (555) macrotricyclic complexes, are characterized by the co-planar or very close to co-planar configuration of the both metal chelate node MN4 and the 5-membered heterocycles in these complexes (Mikhailov and Chachkov, 2013b; Chachkov and Mikhailov, 2013c). For the (5454) macrotetracyclic compounds, the tetragonal pyramidal structure of the metal chelate node is predominant (Chachkov and Mikhailov, 2014e). These facts are clearly illustrated by the molecular structure of the Ni(II) complex with 4,5,9,10-tetramethyl-1,3,6,8-tetraazacyclodecadiene-5,8-diimine-2,7 presented in Fig. 6.7, and with the molecular structure of the Co(II) complex with 1,4,7,10-tetraazadodecatriene-1,3,8-tetrathione-5,6,11,12 presented in Fig. 6.8. It is characteristic that the deviation of the first of indicated chelates from co-planarity is very significant: 35.4° (although the group of atoms N4 itself is planar). It is also interesting that the 4- and 5-membered rings are pairwise the same by the sum of bond angles in them and by these angles themselves. The both 4-membered metal chelate rings are rigidly planar (each sum of bond angles is 360.0°) and the both 5-membered metallocycles are nearly planar (each sum of bond angles is 538.9°, which is only 1.1° smaller than the sum of internal angles in the planar pentagon). Chelates of other aforementioned ions of 3d-elements M(II) with this macrocyclic ligand have similar specific features. Only the Zn(II) chelate is kept aside for which the deviation from co-planarity by all listed parameters is noticeably higher than that for the complexes of other M(II) (Chachkov and Mikhailov, 2014e). The tetragonal pyramidal structure of the metal chelate node is also predominant for the (5656) macrotetracyclic compounds, although, as it was already mentioned above, among them there are also metal chelates with a strong plane structure, such as the M(II) complexes with 3,10-dithio-6,7,13,14-tetramethyl-1,2,4,5,8,9,11,12-octaaza-cyclodecanetetraene-1,5,7,12 (M = Fe, Co, Ni, Cu) (see Figs. 6.9 and 6.10).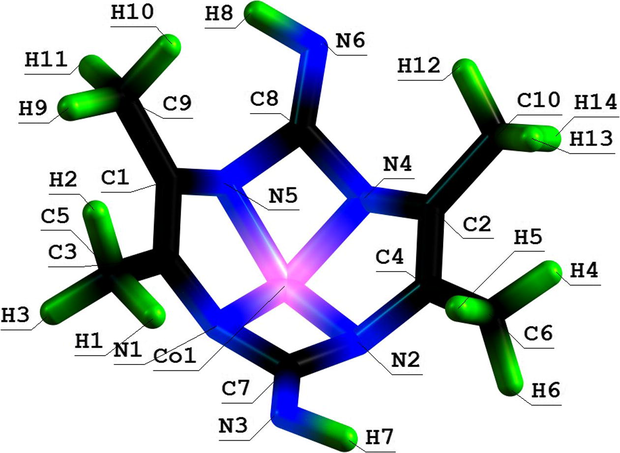
Molecular structure of (5454) aza-macrotetracyclic Co(II) chelate with 4,5,9,10-tetramethyl-1,3,6,8-tetrazacyclodecadien-5,8-diimine-2,7.
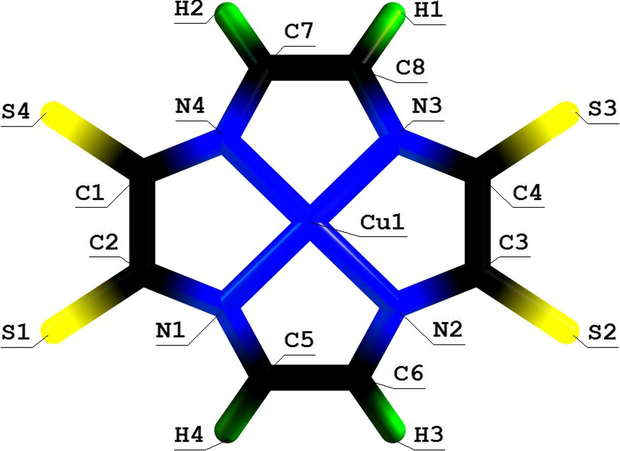
Molecular structure of (5555) aza-macrotetracyclic Cu(II) chelate with 1,4,7,10-tetraazadodecatriene-1,3,8-tetrathione-5,6,11, 12.
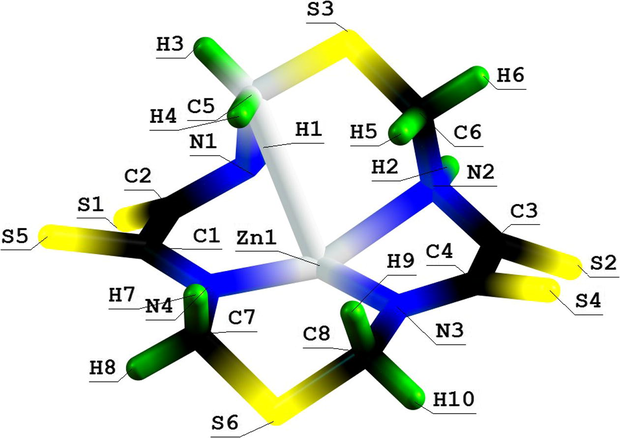
Molecular structure of (5656) thia azamacrotetracyclic Zn(II) chelate with 1,8-dithia-3,6,10,13-tetraazacyclotetradecanetet-rathione-4,5,11,12.
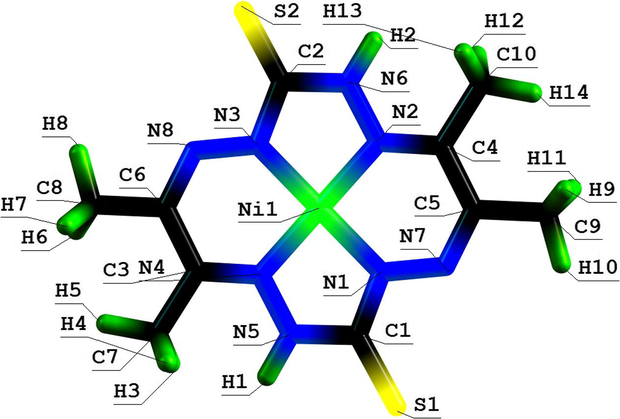
Molecular structure of (5656) azamacrotetracyclic Ni(II) chelate with 3,10-dithio-6,7,13,14-tetramethyl-1,2,4,5,8,9,11, 12-octaazacyclotetradecanetetraene-1,5,7,12.
Similar tetrahedral pyramidal structures of the metal chelate nodes MN4 are also typical of the (5456) macrotetracyclic metal chelates containing not two, as in the considered macrotricyclic and macrotetracyclic complexes, but already three types of chelate rings differed by the number of atoms (Chachkov and Mikhailov, 2013c, 2014d; Mikhailov and Chachkov, 2013c; Mikhailov et al., 2013b). An example for this structure is shown in Fig. 6.11. In this structure, as in the structures of the (5454) macrotetracyclic metal chelates, the values of BAS in the MN4 chelate nodes are also substantially lower than 360°, and the N4 group and the 4-membered metal chelate rings are nearly coplanar. However, the 5- and especially 6-membered rings exhibit s significant deviation from planarity (up to 25 and 80°, respectively). As a rule, in all these macrotetracyclic metal complexes, the M—N bond lengths in the MN4 chelate nodes are pairwise equal to each other (in the best case). However, in the Mn—Zn series they exhibit the same main regularity as in the case of the macrotricyclic complexes: on the whole, these bonds shorten on going from Mn to Ni and become longer on going from Ni to Zn (Chachkov and Mikhailov, 2009c, 2013c, 2013d, 2013e, 2013f, 2014c, 2014d, 2014e, 2014f; Mikhailov and Chachkov, 2013a; 2013c, 2013b, 2013c, 2015f, 2015g; Mikhailov et al., 2013b).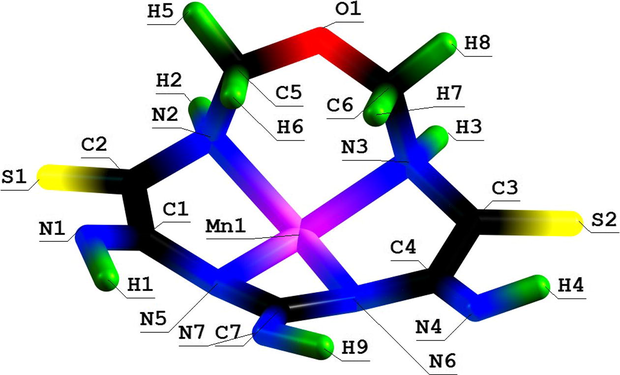
Molecular structure of (5456) oxaazamacrotetracyclic Mn(II) chelate with 5,7,9-triimino-1-oxa-3,6,8,11-tetraazacyclododecanedithione-4,10.
Recently we were also shown the possibility of existence (5676) macrotetracyclic M(II) chelates of 3d-elements indicated above, with 2,2,7,8,13,13-hexamethyl-1,5,6,9,10,14-hexaazacyclohexadecatetraene-4,6,8,10 containing 16-membered macrocycles and 7-membered metal chelate rings, but results of calculation of their molecular structures have not been published till.
7 Conclusion
As can be seen from aforesaid, template synthesis in metal hexacyanoferrate(II) GIM represent a very specific phenomenon in the modern coordination chemistry of d-elements, because they can provide gelatin-immobilized coordination (in fact, supramolecular) compounds that have not yet been obtained in a similar process in solution or in the solid phase. This difference is especially pronounced in the case when one of participants of the template reaction has a property of the ambidentate ligand capable of coordinating, depending on the nature of the “template” complexing agent, through different donor atoms (e.g., (N,O,S)-donoratomic organic compounds mentioned in this article). It can be assumed that potential possibilities of template synthesis in MHF-GIM are rather high and the above list of ternary systems metal ion—ligson A—ligson B, in which the synthesis occurs, can significantly be extended. In this connection, such a synthesis is worth of serious attention of chemists working in this specific area of chemical science, in particular, in coordination and supramolecular chemistry. Noteworthy is the possibility of its application in nanotechnology and close to its branches; related materials have been reported from different groups in the last years, in particular (Li et al., 2014, 2015, 2016; Tang et al., 2015, 2016). As for the results of quantum chemical calculations of coordination compounds formed as a result of template reactions discussed in this review, it should be noted that they allow not only to confirm at the whole their molecular structures, but, also, to identify a number of specific features. (In particular, the specifics of coordination of donor centers corresponding polydentate ligands to complexator M(II), the degree of co-planarity of metal chelate cycles and of chelate nodes, macrocycles and so forth). These data are particular importance due to the fact that the technique of the X-ray analysis of nanoparticles of metal complexes formed in gelatin matrix, and up to now is not elaborated at all, and, therefore, to obtain data on the structure of such complexes by experimental way is not possible at this moment. In perspective, the results of these calculations can be used for prognostication of the mechanism of formation of these same metal complexes, that, in its turn, will be to promote to further improve the processes of template synthesis of various metal macrocyclic compounds.
Acknowledgments
The present study was carried out with financial support in the framework of draft № 4.1584.2014/K to the competitive part of the state task of the Russian Federation on the 2014–2016 years.
References
- Arch. Biochem. Biophys.. 1961;94:20.
- Photographic Gelatin. Vol vol. 1. London: Academic Press; 1972.
- Photographic Gelatin. Vol vol. 1. London: Academic Press; 1976.
- Dalton Trans.. 2011;40:2764.
- J. Biol. Chem.. 1996;271:12003.
- Inclus. Phenom. Macrocycl. Chem.. 1999;5:191.
- Asian J. Chem.. 2015;27:4141.
- Life. 2005;57:161.
- Biomaterials. 2004;25:5675.
- J. Phys. Chem.. 1954;58:968.
- Chem. Rev.. 2006;107:46.
- Adv. Chem. Ser.. 1963;1:1.
- Phys. Chem. Chem. Phys.. 1999;1:5689.
- Russ. J. Gen. Chem.. 2008;78:1849.
- Russ. J. Gen. Chem.. 2009;79:1122.
- Russ. J. Inorg. Chem.. 2009;54:1952.
- Macroheterocycles. 2009;2(3–4):271.
- Russ. J. Inorg. Chem.. 2010;55:1243.
- Russ. J. Phys. Chem.. 2010;85:152.
- Macroheterocycles. 2010;3:161.
- Macroheterocycles. 2010;3:171.
- Russ. J. Inorg. Chem.. 2011;56:1935.
- Russ. J. Inorg. Chem.. 2011;56:223.
- Russ. J. Phys. Chem.. 2011;85:1475.
- Russ. J. Inorg. Chem.. 2012;57:981.
- Russ. J. Inorg. Chem.. 2012;57:205.
- Russ. J. Inorg. Chem.. 2012;57:1100.
- Russ. J. Gen. Chem.. 2013;83:1123.
- Russ. J. Inorg. Chem.. 2013;58:1073.
- Russ. J. Inorg. Chem.. 2013;2013(58):1203.
- Russ. J. Gen. Chem.. 2013;83:1937.
- Russ. J. Inorg. Chem.. 2013;58:1315.
- Russ. J. Inorg. Chem.. 2014;59:489.
- Russ. J. Gen. Chem.. 2014;84:1962.
- Russ. J. Inorg. Chem.. 2014;59:218.
- Russ. J. Gen. Chem.. 2014;84:315.
- Russ. J. Inorg. Chem.. 2014;59:349.
- Russ. J. Inorg. Chem.. 2014;59:1276.
- J. Struct. Chem.. 2009;50:613.
- Russ. J. Inorg. Chem.. 2013;58:548.
- Trans. Met. Chem.. 2002;27:329.
- Trans. Met. Chem.. 2007;32:240.
- J. Protein Chem.. 1991;10:535.
- Chem. Rev.. 1974;74:351.
- Metal Mediated Template Synthesis of Ligands. World Scientific Publishing; 2004. 308 p
- Nature. 1955;176:1062.
- Ann. Rep. Prog. Chem., Sect. A. 2005;101:319.
- J. Chem. Soc.. 1960;11:4409.
- Coord. Chem. Rev.. 1968;3:3.
- Synthetic Coordination Chemistry: Principles and Practice. Singapore—London: World Scientific; 1996. 452 p
- Draget K.I., Hattrem M.N., eds. Physical Properties of Gelatin Based Solid Emulsions. Trondheim: NTMU; 2013.
- Nanotechnology. 2008;3:2. (in Russian)
- Coord. Chem. Rev.. 2010;254:1628.
- Advances in Protein Chemistry. Vol Vol. 4. New York: Academic Press; 1948. 650 p
- Trans. Met. Chem.. 2008;33:467-473.
- J. Biol. Chem.. 2005;280:4005.
- J. Biol. Chem.. 1992;267:15398.
- Garnovskii A.D., Kharisov B.I., eds. Direct Synthesis of Coordination and Organometallic Compounds. Lausanne—Amsterdam—London—New York: Elsevier; 1999. 244 p
- Modern Aspects of Synthesis of Metal Complexes (Mainn Ligands and Methods). Rostov-Don; 2000. 355 p (in Russian)
- J. Biol. Chem.. 2004;279:46921.
- Template Synthesis of Macrocyclic Compounds. Weinheim—New York—Chichester—Brisbane—Singapore—Toronto: Wiley-VCH; 1999. 565 p
- Gloe K., ed. Macrocyclic Chemistry: Current Trends and Future Perspectives. Dordrecht: Springer; 2000. 450 p
- Photographic Gelatin. London: Focal Press; 1965. p. 35
- Bull. Korean Chem. Soc.. 2012;33:2279.
- Chem. Rev.. 1989;89:1875.
- Inorg. Chim. Acta. 1987;133:221.
- Gelatin. In: Phillips G.O., Williams P.A., eds. Handbook of Hydrocolloids. Cambridge: Woodhead Publishing Ltd.; 2009. p. :67-86.
- [Google Scholar]
- Chem. Ind.. 1961;42:1708.
- J. Mol. Biol.. 1973;79:137.
- Trans. Met. Chem.. 2007;32:584.
- The Theory of the Photographic Process. New York: Macmillan; 1977.
- The Theory of the Photographic Process. New York: Macmillan; 1972.
- Coord. Chem. Rev.. 1980;32:67.
- Int. Symp. Advances Sciences in Organic Chemistry (Miskhor, Crimea, June 21–25, 2010). 2010. p. :C83.
- Inorg. Chim. Acta. 2008;361:1415.
- Synth. React. Inorg. Met. Org. Chem.. 2004;34:1305.
- Polyhedron. 2007;26:33.
- Inorg. Chim. Acta. 2007;360:579.
- J. Chem. Soc., Dalton Trans.. 1984;4:673.
- Polyhedron. 1999;18:1561.
- Supramolecular Chemistry. Weinheim: Verlag Chemistry; 1995. 356 p
- Supramolecular Chemistry. Concepts and Perspectives. Weinheim: VCH; 1995.
- Chem. Eur. J.. 2014;20:6027.
- Angew. Chem. (Int. Ed.). 2015;54:11073.
- APL Mater.. 2016;4:040703.
- [CrossRef]
- Polym. Int.. 2002;51:233.
- The Chemistry of Macrocyclic Ligand Complexes. Cambridge: Cambridge Univ. Press; 1989.
- Trans. Met. Chem.. 2007;32:1012.
- Melson G.A., ed. Coordination Chemistry of Macrocyclic Compounds. New York, London: Plenum Press; 1979. 664 p
- Indian J. Chem.. 1991;30A:252.
- J. Inorg. Chem. USSR. 1992;1992(37):172.
- Russ. Chem. Rev.. 1995;64:657.
- Trans. Met. Chem.. 1996;21:363.
- Rev. Inorg. Chem.. 1997;18:287.
- Russ. J. Gen. Chem.. 1998;68:827.
- Trans. Met. Chem.. 2000;25:552.
- Russ. J. Coord. Chem.. 2000;26:804.
- Int. J. Inorg. Mater.. 2001;3:1053.
- Heterocycl. Commun.. 2001;7:79.
- Russ. J. Gen. Chem.. 2001;2001(71):1676.
- Russ. J. Gen. Chem.. 2002;72:1525.
- Russ. J. Coord. Chem.. 2002;28:363.
- Russ. J. Coord. Chem.. 2002;28:32.
- J. Coord. Chem.. 2008;61:1333.
- Russ. Chem. Bull.. 2008;57:8.
- Eur. Chem. Bull.. 2014;3:935.
- Bull. Chem. Soc. Jpn.. 1989;62:4016.
- J. Coord. Chem.. 2009;62:1058.
- Macroheterocycles. 2009;2:271.
- J. Coord. Chem.. 2010;63:4309.
- Russ. Chem. Bull.. 2012;61:1531.
- Russ. J. Inorg. Chem.. 2013;58:174.
- Inorg. Chim. Acta. 2013;408:199.
- Inorg. Chim. Acta. 2013;408:246.
- Russ. J. Inorg. Chem.. 2014;59:101.
- Eur. Chem. Bull.. 2014;3:367.
- Russ. J. Inorg. Chem.. 2014;59:1283.
- Russ. J. Inorg. Chem.. 2014;59:1472.
- Russ. J. Inorg. Chem.. 2015;60:187.
- Russ. J. Gen. Chem.. 2015;85:628.
- Russ. J. Inorg. Chem.. 2015;60:889.
- Russ. J. Inorg. Chem.. 2015;60:964.
- Russ. J. Inorg. Chem.. 2015;60:1253.
- Russ. J. Inorg. Chem.. 2015;60:1117.
- Russ. J. Inorg. Chem.. 2015;60:1354.
- Russ. J. Inorg. Chem.. 2016;61:208.
- Mendeleev Commun.. 1998;3:96.
- Russ. J. Phys. Chem.. 1998;72:921.
- Russ. J. Gen. Chem.. 1998;68:1187.
- Russ. J. Coord. Chem.. 1998;24:629.
- Russ. Chem. Bull.. 1999;48:1975.
- Trans. Met. Chem.. 2000;2000(25):26.
- J. Inorg. Chem. USSR. 1990;35:1169.
- J. Inf. Record. Mater.. 1990;18:21.
- J. Inf. Record. Mater.. 1990;18:225.
- J. Imaging Sci.. 1991;35:258.
- Russ. J. Gen. Chem.. 2001;71:809.
- Russ. J. Gen. Chem.. 1997;67:1935.
- J. Soc. Photogr. Sci. Technol. Jpn.. 1998;61:387.
- Trans. Met. Chem.. 1999;24:503.
- Heterocycl. Commun.. 2000;6:137.
- Heterocycl. Commun.. 2000;6:357.
- Int. J. Inorg. Mater.. 2001;3:161.
- Heterocycl. Commun.. 2001;7:359.
- Russ. J. Coord. Chem.. 2003;29:115.
- Russ. J. Coord. Chem.. 2003;29:327.
- Russ. J. Gen. Chem.. 2003;73:847.
- Russ. J. Coord. Chem.. 2003;29:630.
- Trans. Met. Chem.. 2003;28:592.
- Trans. Met. Chem.. 2003;2003(28):665.
- Heterocycl. Commun.. 2003;9:61.
- Russ. J. Gen. Chem.. 2004;74:7.
- Russ. J. Coord. Chem.. 2004;30:639.
- Trans. Met. Chem.. 2004;29:732.
- Trans. Met. Chem.. 2007;32:56.
- Macroheterocycles. 2008;1:90.
- Russ. J. Gen. Chem.. 2008;78:258.
- Russ. J. Gen. Chem.. 2009;79:24.
- Russ. J. Inorg. Chem.. 2012;57:1570.
- Russ. J. Inorg. Chem.. 2013;58:1518.
- Centr. Eur. J. Chem.. 2013;11:1822.
- Biomacromology. 2009;10:2565.
- J. Coord. Chem.. 2009;62:3629.
- Pure Appl. Chem.. 1980;52:2461.
- Inorg. Chem. Commun.. 2006;9:175.
- Trans. Met. Chem.. 2007;32:9.
- Biopolymers. 2006;84:181.
- Biopolymers. 2006;84:421.
- Proc. Nat. Acad. Sci. USA. 2006;103:9001.
- Dalton Trans.. 2009;38:177.
- Proc. Nat. Acad. Sci. USA. 2008;105:2824.
- Phillips G.O., Williams P.A., eds. Handbook of Hydrocolloids. London: Woodhead Publishing Ltd.; 2000. 450 p
- Structure. 1997;5:359.
- Pomogailo A.D., ed. Polymer_Immobilized Metallocomplex Catalysts. Moscow: Nauka; 1998. 303 p (in Russian)
- J. Korean Chem. Soc.. 2011;55:912.
- Treatise on Collagen. Vol vol. I. New York: Academic Press; 1967. p. 187
- Nature. 1955;176:915.
- Gelatine Handbook: Theory and Industrial Practice. Weinheim: Wiley-VCH Verlag; 2007.
- J. Coord. Chem.. 2010;63:4007.
- Asian J. Chem.. 2014;26:376.
- Trans. Met. Chem.. 2007;32:1051.
- Skopenko, V.V., Garnovskii, A.D., Kokozei, V.N., Kuzharov, A.S., Gokhon_Zorilla, G., Burlov, A.S., Vasil’eva, O. Yu, Pavlenko, V.A., Kharisov, B.I., Kherets, B.M., Blanko, L.M., Garnovskii, D.A., 1997. In: Skopenko, V.V. (Ed.), Direct Synthesis of Coordination Compounds, Venturi, Kiev, 172 p (in Russian).
- Biochemistry. 1971;10:3005.
- Angew. Chem. (Int. Ed.). 2015;54:588.
- Chem. Commun.. 2016;52:505.
- Polyhedron. 2009;28:2010.
- Eur. J. Biochem.. 1999;259:513.
- J. Biol. Chem.. 2003;278:12241.
- Pharm. Des.. 2007;13:3608.
- J. Biol. Chem.. 1963;238:2003.
- Mendeleev Chem. J. (Engl. Transl.). 1998;42:5.
- Tetrahedron Lett.. 2012;53:6548.
- Inorg. Chem.. 1991;30:2109.
- Wird A.G., Courts A., eds. The Science and Technology of Gelatin. New York: Academic Press; 1977.
- Synthesis of Macrotricyclic Compounds. Kiev: Naukova Dumka; 1987. 279 p (in Russian)




