

Translate this page into:
New benzoxazole-based sulphonamide hybrids analogs as potent inhibitors of α-amylase and α-glucosidase: Synthesis and in vitro evaluation along with in silico study
⁎Corresponding authors. fazalstar@gmail.com (Fazal Rahim), Sono_waj@yahoo.com (Wajid Rehman)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The development of drugs resistance in diabetes mellitus is a growing clinical problem, creates many challenges for patient. To overcome these problems, there is a serious deficiency of anti-diabetic agents, may be synthesized that inhibit alpha amylase and alpha glucosidase activity. Here, we have design and synthesized benzoxazole based sulphonamide derivatives and evaluated for their anti-diabetic activity. Twenty-two benzoxazole based sulphonamide derivatives were synthesized by reacting 2-aminophenol with carbon disulphide in the presence of base (Et3N) to obtained 2-marcapto benzoxazole which was further dissolved in ethanol by slow addition of different substituted phenacyl bromide in the presence of triethylamine, afforded varied S-substituted benzoxazole products. These products were dissolved in ethanol and hydrazine hydrate was added an excess in the presence of acetic acid to gives Schiff base. This Schiff base products were further dissolved in THF along with different substituted benzene sulphonyl chloride followed by addition of few drops of Et3N, yielded benzoxazole based sulphonamide derivatives (1–22). Moreover, SAR was established for the synthesized compounds and molecular docking studies were conducted for the potent moieties in order to explore the binding modalities of analogs. Among the tested series few analogues were found few folds better potential than standard drug but analog 1 (IC50 = 1.10 ± 0.20 µM, 1.20 ± 0.30 µM), showed promising anti-diabetic activity against α-amylase and α-glucosidase (11.12 ± 0.15 µM and 11.29 ± 0.07 µM respectively).
Keywords
Benzoxazole
alpha-amylase and alpha-glucosidase
SAR
Molecular docking

1 Introduction
Diabetes mellitus (DM) is considered as serious health problem worldwide. Each year, the number of diabetic patient increases therefore it is also known as lifestyle related syndrome. Almost 366 million people suffered from diabetes when tested in 2011 by international Diabetes Federations and the number is therefore expected to be increase up to 522 million by 2030 (Whiting et al., 2011). In the developed countries, Type-II diabetes is more dominant is defined due to impaired insulin secretion as well as reduction in sensitivity of insulin (Taylor et al., 1994; Porter et al., 1991; Butler et al., 2003; Tang et al., 2010; Wu et al., 2007). α-Amylase secreted from pancreas is a hydrolyzing enzyme which convert glycogen and starch to maltose while it is further converted to glucose by α- Glucosidase enzyme which present in small intestine (Salar et al., 2017). Therefore, the general term post-prandial glycemia (PPHG) is used for high blood glucose level occurred by hydrolysis of starch to maltose and then to glucose. This PPHG indicate the primary information about type-II diabetes (Sun et al., 2017) and cause various complication including retinopathy, nephropathy, neuropathy and cardiovascular as well as it can damage organs which plays significant role in the body (Campas et al., 2012; Rask-Madsen et al., 2013). In this regard, the activity of both the enzyme can be prohibited by various drugs candidates to treat diabetes mellitus such as miglitol, voglibose and acarbose are well-known marketed drugs. But various investigations proved that these drugs produce some serious complications including diarrhea, and abdominal pain (Miller et al., 2014; Raghu et al., 2019).(See Graph 1 Graph 2 Scheme 1 Table 1 Table 2 Table 3).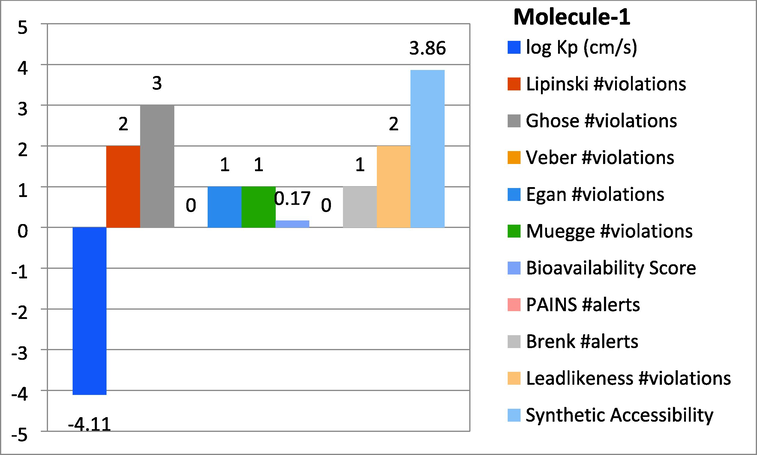
Represent the ADMET properties of compound-1.
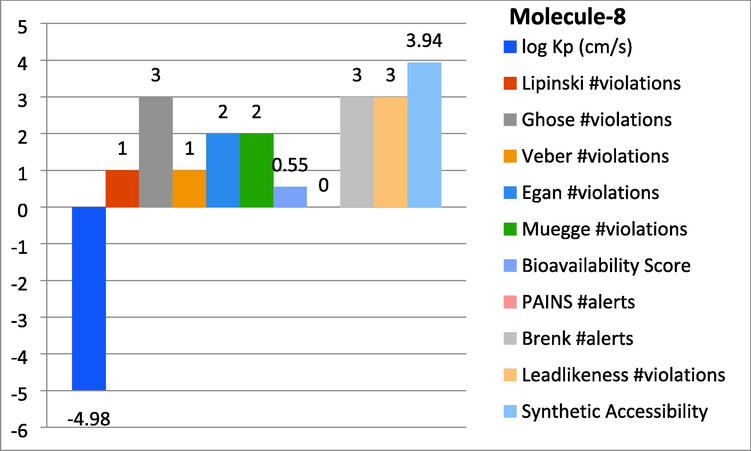
Represent the ADMET properties of compound-8.
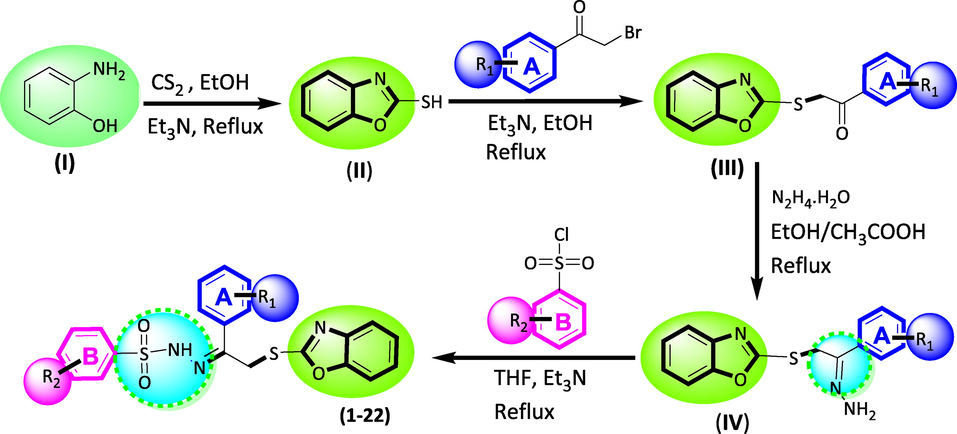
synthesis of benzoxazole based benzene sulphonamide derivatives.
S/N0
Alpha glucosidase IC50 = ±µM
A lpha amylase IC50 = ±µM
Ring A
Ring B
1
1.10 ± 0.20
1.20 ± 0.30
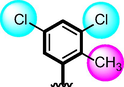
2
2.10 ± 0.10
2.40 ± 0.10
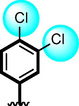
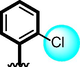
3
8.20 ± 0.20
9.10 ± 0.20
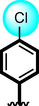
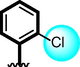
4
21.60 ± 0.30
22.20 ± 0.40
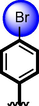
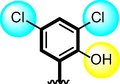
5
2.30 ± 0.10
2.70 ± 0.10
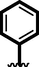
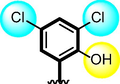
6
1.70 ± 0.10
1.60 ± 0.10

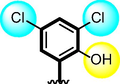
7
3.10 ± 0.10
2.90 ± 0.10
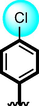
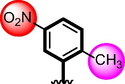
8
1.30 ± 0.10
1.50 ± 0.10
9
6.20 ± 0.20
7.30 ± 0.20
10
1.50 ± 0.10
1.70 ± 0.10
11
22.40 ± 0.40
23.20 ± 0.40
12
11.20 ± 0.20
11.50 ± 0.20
13
19.50 ± 0.30
21.10 ± 0.40
14
10.40 ± 0.20
11.20 ± 0.20
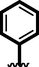
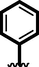
15
10.30 ± 0.20
11.10 ± 0.20

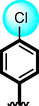
16
15.40 ± 0.30
14.30 ± 0.30
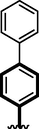
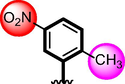
17
14.90 ± 0.30
15.90 ± 0.30
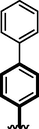
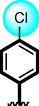
18
8.60 ± 0.20
9.30 ± 0.20
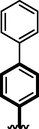

19
5.70 ± 0.20
6.30 ± 0.20
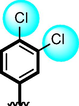
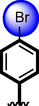
20
16.20 ± 0.20
17.10 ± 0.20
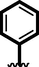
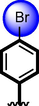
21
8.70 ± 0.20
9.20 ± 0.20
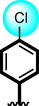
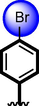
22
9.43 ± 0.20
10.10 ± 02
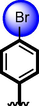
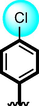
Acarbose
11.29 ± 0.07
11.12 ± 0.15
Analog-1 against Alpha Amylase
Mode
Affinity Kcal/mol
Dist from rmsd l.b
Best mode rmsd u.b
1
−9.5
0.000
0.000
2
−9.2
1.170
2.262
3
−9.2
1.946
7.397
4
−8.9
3.324
8.326
5
−8.6
3.689
8.264
6
−8.5
2.732
4.584
7
−8.5
2.618
3.585
8
−8.5
2.720
4.011
9
−8.5
2.607
7.822
Analog-1 against Alpha Glucosidase
1
−9.7
0.000
0.000
2
−9.7
0.767
1.741
3
−9.7
4.164
6.409
4
−9.6
1.460
2.102
5
−9.5
4.351
6.340
6
−9.5
4.742
7.553
7
−9.4
4.330
6.675
8
−9.2
3.563
5.712
9
−9.1
2.640
3.857
Analog-8 against Alpha Amylase
Mode
Affinity Kcal/mol
Dist from rmsd l.b
Best mode rmsd u.b
1
−9.0
0.000
0.000
2
−8.8
3.829
7.253
3
−8.7
3.662
8.688
4
−8.4
2.923
4.679
5
−8.4
3.213
4.342
6
−8.3
3.030
4.156
7
−8.2
3.471
6.806
8
−8.2
2.421
3.197
9
−8.2
2.258
7.296
Analog-8 against Alpha Glucosidase
1
−9.8
0.000
0.000
2
−9.8
0.764
1.641
3
−9.7
4.167
6.109
4
−9.3
1.450
2.002
5
−9.3
4.381
6.140
6
−9.3
4.722
7.353
7
−9.3
4.210
6.575
8
−9.2
3.523
5.812
9
−9.1
2.610
3.677
Benzoxazole is an important analog present in many biological active compounds showed diverse biological activities including antiviral, antimicrobial, analgesic and antidepressant activity. As well as used for the treatment of pathological diseases likes hepatitis, diabetes, alzheimer’s and coronary heart diseases. Number of marketed drugs (Fig. 2) shows promising biological activity having benzoxazole ring like Nakijinol-B (Anticancer) 1, Calcimycin (Antimicrobial) 2, Zinbo-5 (Antifungal) 3, Pseudopteroxazoles (Antitubercular) 4 (Chaney et al., 1974; McKee et al., 2008; McCulloch et al., 2011) as shown in Figure-1. Therefore, it would be essential to design and develop safe and effective therapeutic agent with less side effect in order to treat type-II diabetes mellitus. Moreover, rationale of the current study also contrasts the biological profile with previously reported derivatives as shown in Fig. 2. (See Fig. 3 Fig. 4 Fig. 5 Fig. 6 Fig. 7 Fig. 8 Fig. 9 Fig. 10 Fig. 11).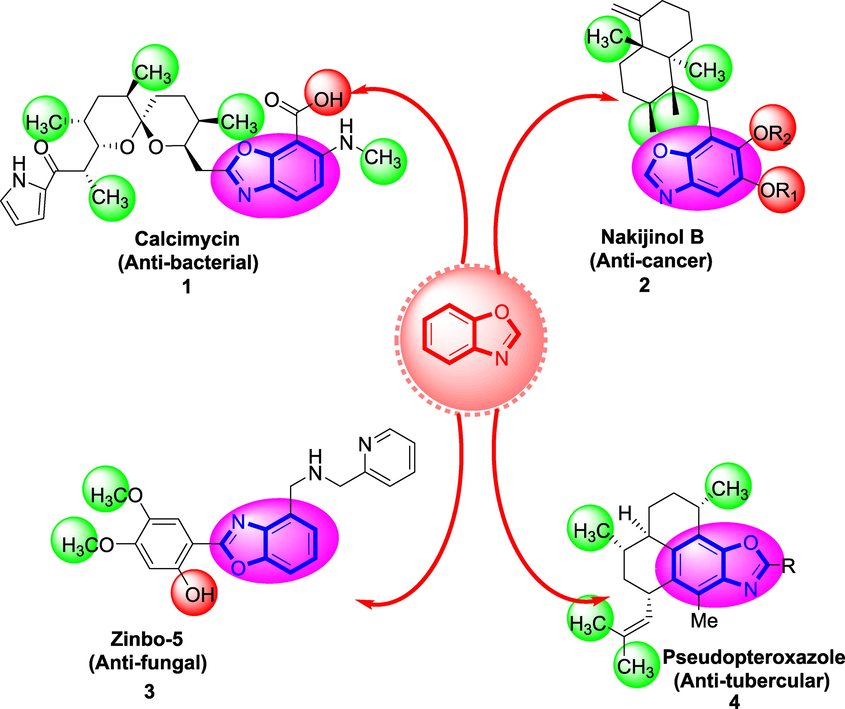
Various bioactive drugs of benzimidazole.
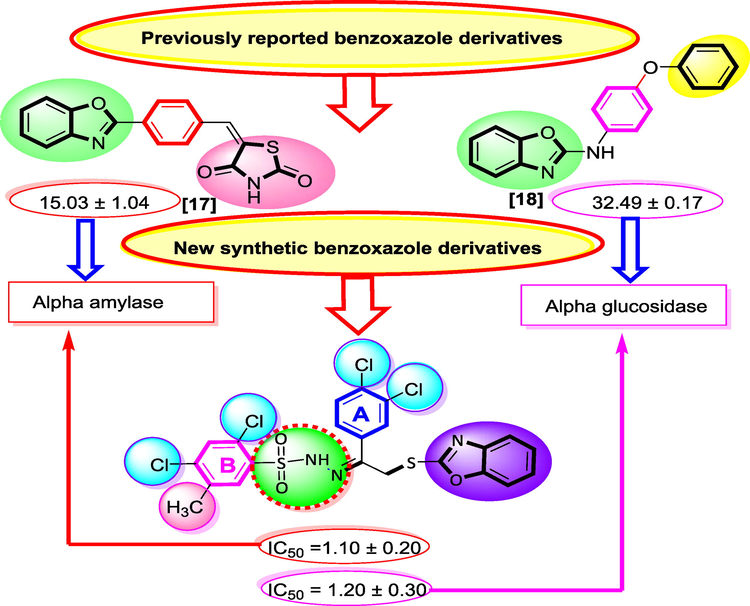
Rational study of current analog.
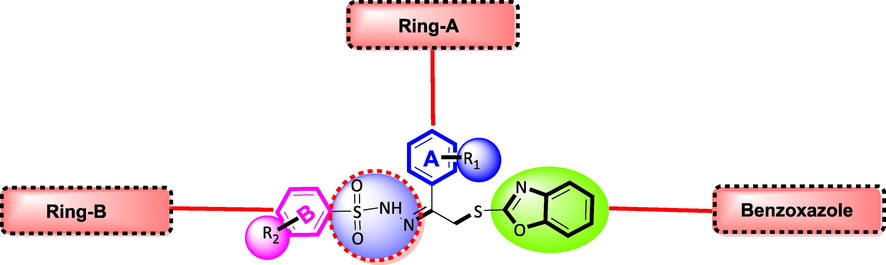
general representation of molecule.
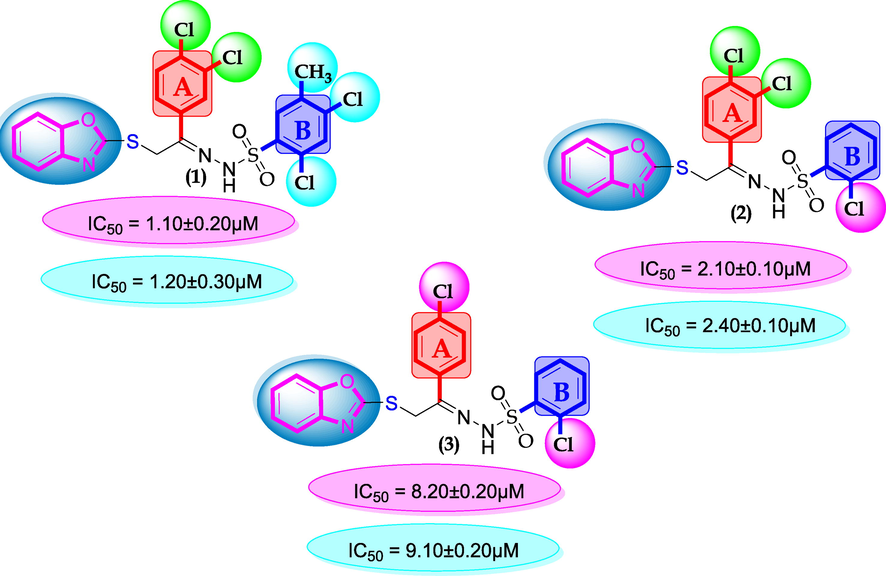
SAR for compounds 1, 2 and 3.
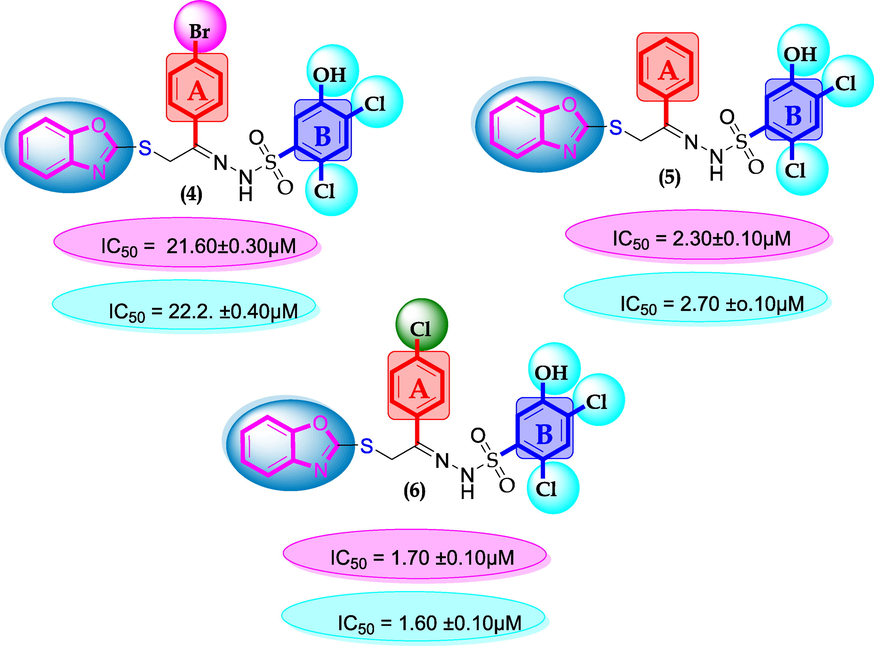
SAR for compounds 4, 5 and 6.
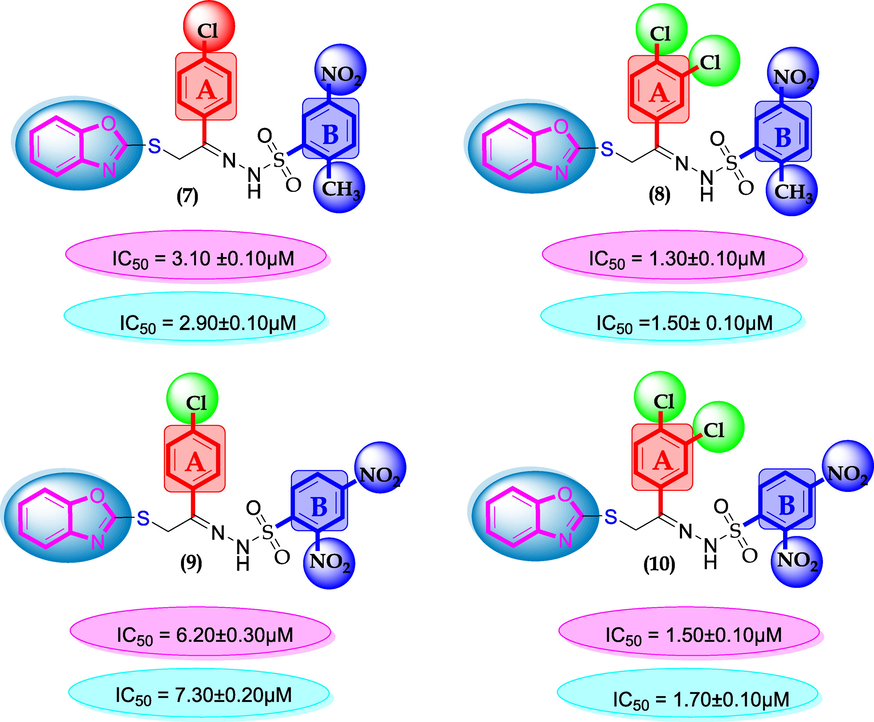
SAR for compounds 7, 8, 9 and 10.
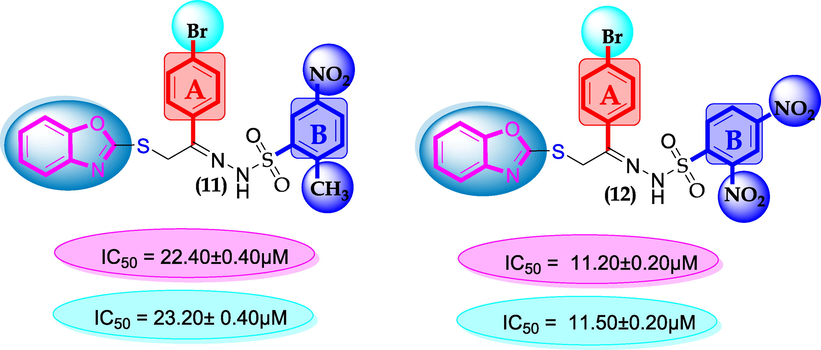
SAR for compounds 11 and 12.
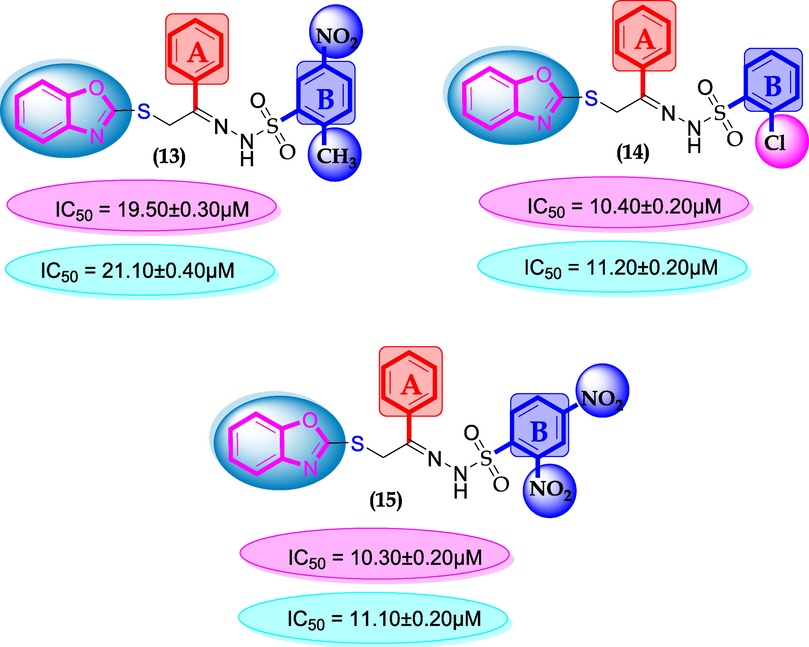
SAR for compounds 13, 14 and 15.
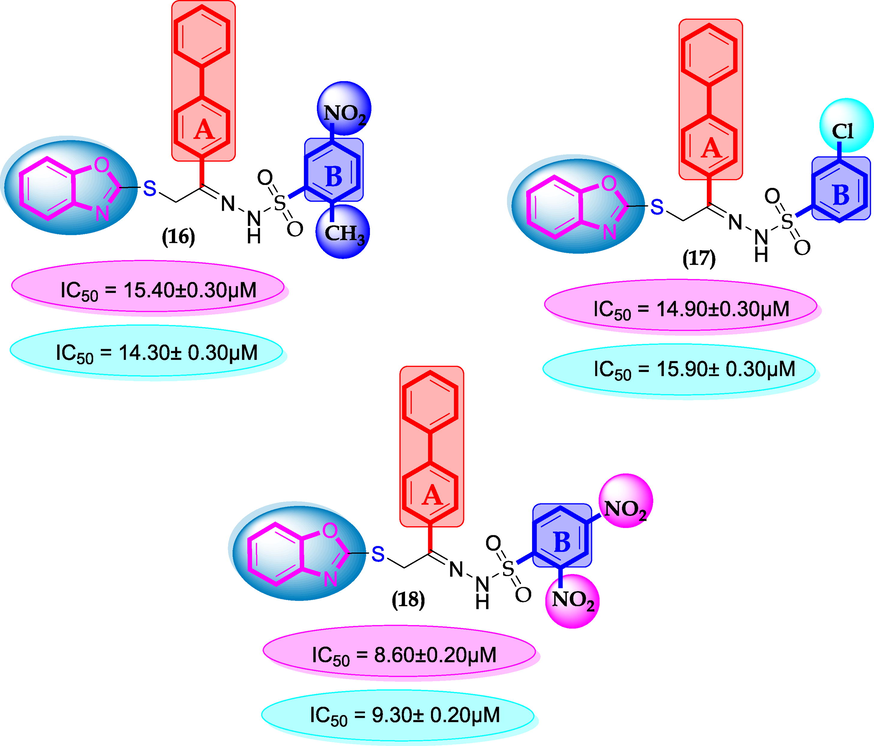
SAR for compounds 16, 17 and 18.
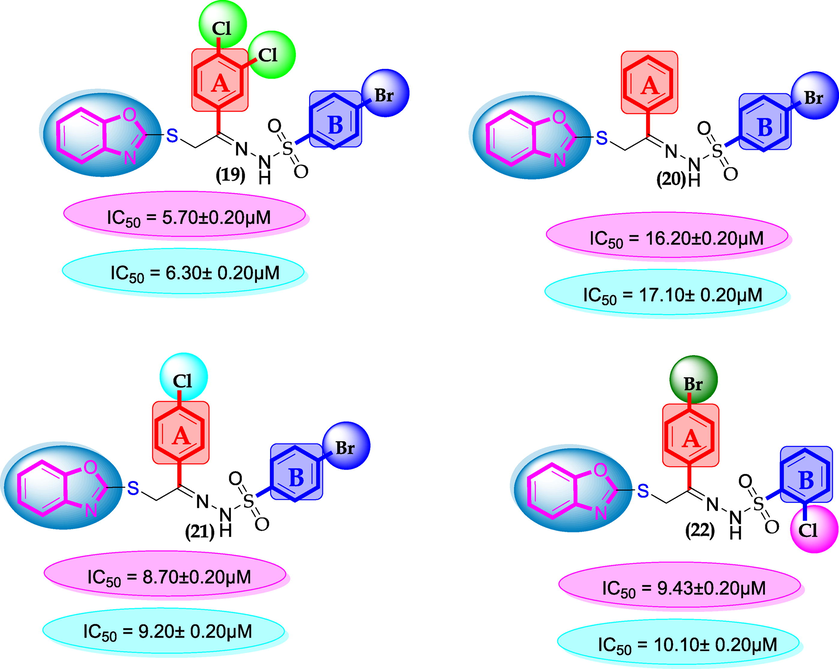
SAR for compounds 19, 20, 21 and 22.
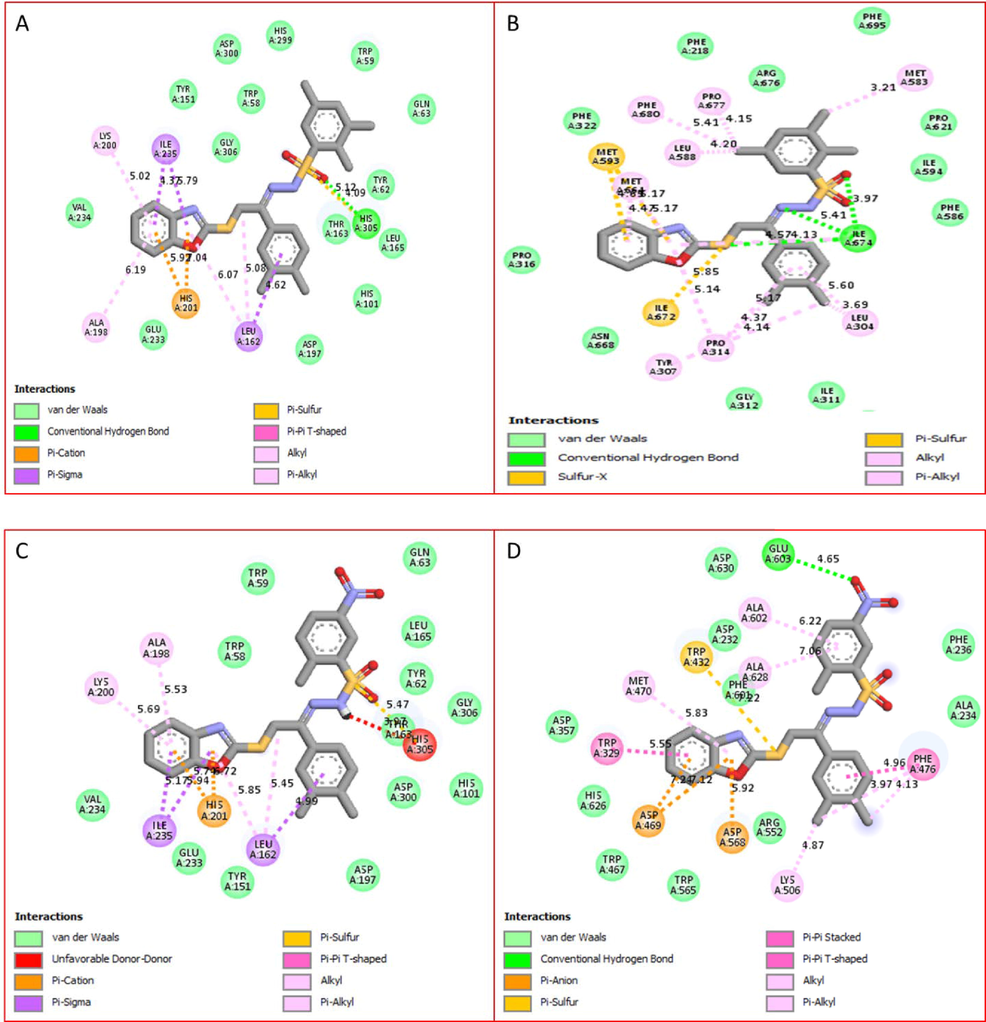
(A–D). The PLI profile for potent compounds against the Alpha amylase and Alpha glucosidase indicate the surface of the corresponding enzyme. (A-D) represent the PLI profile for compounds 1(A and B) and 8 (C and D) by 2D, respectively.
2 Experimental
2.1 Assay protocol
2.1.1 α-Amylase inhibitory activity
Inhibition criteria were established for α-amylase (0.5 mg/mL in phosphate buffer; 0.2 mM maintained at pH 6.9) by the method was described earlier (Salar et al., 2019; Taha et al., 2018). 500 µL of α-amylase along with incubation of 500 µL of test sample (1–100 µg/mL) was stirred almost 10 min at 100 °C. 1 % starch solution was mixed after pre-incubations (500 μL, in 0.2 mM phosphate buffer maintained at pH 6.9) for another 10 min at 25 °C. After completions, cool the mixture and diluted with distilled water (10 mL) were analyzed at 540 nm by recording absorbance (Imran et al., 2018). Standard drug acarbose was used.
The percentage of inhibition was estimated by employing the formula;
2.1.2 α-Glucosidase inhibitory activity
Inhibition criteria for α-glucosidase were established by using modified version published technique (Imran et al., 2018 Gollapali et al., 2018, Zhang, X. et al, 2022) 10 μL of sample at variable concentrations (1 to 100 μg/mL) was pre-mixed with 490 μL phosphate buffer (pH 6.8) along with α-glucosidase solution was prepared by mixing 1 mg in 100 mL phosphate buffer (pH 6.8) comprising of 200 mg bovine serum albumin. 10 μL of sample at variable concentrations and 250 μL of 5 mM p-nitrophenyl α-d-glucopyranoside and pre incubated were added at 37 °C. Moreover, 250 μL α-glucosidase (0.15 unit/mL) was added after 5 min for further incubation for 15 min at 37 °C. Added 2000 μL of Na2CO3 (200 mM). α-Glucosidase activity was computed at 400 nm on Shimadzu 265 UV–Vis spectrophotometer (Japan) and the amount of released was computed under the positive control of standard drug acarbose. The optimum concentration needed to hydrolyze 50 % of α-glucosidase was defined as the IC50 value.
The percentage of inhibition was estimated by employing the formula;
2.1.3 Molecular docking protocol
According to Crippen's fragmentation method, the partition coefficient (log P) values were predicted using Crippen's fragmentation by the CS ChemProp module of ChemDraw Ultra 12 (CambridgeSoft, Cambridge, MA, USA). (Ghose, A.K. et al.,1987).The polar surface area (tPSA) was calculated by the atom-based method (Ertl, P., et al. 2000). Chemdraw (14.0) was used to generate the structures of compounds 8 and 9, which were then imported into the auto dock tool. Following a final check of all atom and bond types and the addition of any required hydrogen atoms, the Gasteiger-Marsili charges were assigned. Marvin determined the substances' pKa values and the ionisation states that correspond to physiological conditions (pH 7.4). (ChemAxon). The molecules were docked to alpha glucosidase and alpha amylase. It was necessary to build the enzyme structure before docking with GoldSuite (CCDC). The hydrogen atoms were added, certain water molecules (616, 634, and 643) were kept, and all of the histidine residues were protonated at Nε. All amino acid residues within a radius of around 12 from the known compound, acarbose, were considered to be within the binding site. There were used a common set of genetic algorithms. The population size was 100, and there were 100,000 operations. Nine conformations of each chemical were as a result obtained. DSV provided results visualization (2020).
2.2 General method for the synthesis of benzoxazole based sulphonamide derivatives
2-Marcapto benzoxazole was obtained by reacting 2-aminophenol and carbon di-sulphide was added to the pre-heated solution of ethanol in the presence of Et3N, afforded 2-Marcapto benzoxazole was then refluxed along with different substituted phenacyl bromide (R1) in ethanol triethylamine were added as catalyst, yielded 2-(benzo[d]oxazol-2-ylthio)-1-phenylethan-1-one was then dissolved in ethanol and hydrazine hydrate was added slowly an excess, followed by addition of few drops of acetic acid, obtained Schiff base upon further treating with different substituted benzene sulphonyl chloride (R2) in THF in the presence of ET3N under refluxed conditions, obtained benzoxazole based sulphonamide derivatives (1–22).
2.2.1 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(3,4-dichlorophenyl)ethylidene)-3,5-dichloro-2-hydroxybenzenesulfonohydrazide
Yield 82 %, m.p. 228–30 °C, Light green. FT-IR v, (cm−1): OH (3584), NH (3475), Sp3CH (2990), C⚌N (1655), C⚌C (1580), C—O (1230), S⚌O (1170), C-Cl (7 5 7). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.68 (s, 1H, OH), 8.31 (d, J = 2.0 Hz, 1H, Aromatic-H), 8.03 (dd, J = 6.2, 2.1 Hz, 1H, Aromatic-H), 7.89 (d, 7.6 Hz, 1H, Aromatic-H), 7.55 (dd, J = 9.7, 2.5 Hz, 1H, Aromatic-H), 7.50 (dd, J = 8.5, 2.0 Hz, 1H, Aromatic-H), 7.38 (d, J = 2.5 Hz, 1H, Aromatic-H), 7.30–7.21 (m, 1H, Aromatic-H), 7.15–7.11 (m, 1H, Aromatic-H), 5.24 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ168.0, 149.1, 132.1, 130.3, 128.0, 128.2 126.7, 125.3, 125.2, 124.2, 122.3, 122.2, 121.6, 120.3, 120.1, 119.3, 109.4, 39.9, 20.4, 20.5, 14.7. HR-EI-MS: m/z calcd for C21H13Cl4N3O4S2 [M]+ 577.27200; Found: 577.27189.
2.2.2 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(3,4-dichlorophenyl)ethylidene)-2-chlorobenzenesulfonohydrazide
Yield 89 %, m.p. 231–33 °C, Light green. FT-IR v, (cm−1): NH (3473), Sp3CH (2987), C⚌N (1658), C⚌C (1584), C—O (1225), S⚌O (1172), C-Cl (7 5 9). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 8.30 (d, J = 1.9 Hz, 1H, Aromatic-H), 8.02 (dd, J = 6.8, 2.4 Hz, 1H, Aromatic-H), 7.90 (d, J = 8.3 Hz, 1H, Aromatic-H), 7.87 (dd, J = 7.6, 1.9 Hz, 1H, Aromatic-H), 7.60 (dd, J = 6.1, 3.0 Hz, 1H, Aromatic-H), 7.37–7.30 (m, 1H, Aromatic-H), 7.29–7.20 (m, 1H, Aromatic-H), 7.13 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.07 (d, J = 8.1 Hz, 2H, Aromatic-H), 5.28 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ165.0, 155.6, 151.9, 141.5, 139.7, 135.7, 133.5, 133.5, 133.3, 131.5, 130.6, 130.3, 130.2, 128.7, 127.1, 126.3, 124.8, 123.8, 119.1, 110.6, 37.2. HR-EI-MS: m/z calcd for C21H14Cl3N3O3S2 [M]+ 526.83100; Found: 526.83073.
2.2.3 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-chlorophenyl)ethylidene)-2-chlorobenzenesulfonohydrazide
Yield 83 %, m.p. 234–35 °C, Light green. FT-IR v, (cm−1): NH (3471), Sp3CH (2985), C⚌N (1654), C⚌C (1582), C—O (1222), S⚌O (1170), C-Cl (7 5 7). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 8.09 (d, J = 8.1 Hz, 2H, Aromatic-H), 7.87 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.69 (d, J = 8.1 Hz, 2H, Aromatic-H), 7.61 (dd, J = 9.6, 3.6 Hz, 1H, Aromatic-H), 7.32 (d, J = 6.4 Hz, 2H, Aromatic-H), 7.37–7.31 (m, 1H, Aromatic-H), 7.19–7.14 (m, 1H, Aromatic-H), 7.13 (dd, J = 8.4, 4.3 1H, Aromatic-H), 5.21 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ 168.0, 167.9, 151.9, 141.5, 139.7, 136.6, 133.3, 132.1, 130.3, 130.2, 129.0, 128.9, 126.1, 127.1, 122.2, 119.1, 114.4, 109.4, 39.9, 20.4, 8.58. HR-EI-MS: m/z calcd for C21H15Cl2N3O3S2 [M]+ 492.38900; Found: 492.38888.
2.2.4 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-bromophenyl)ethylidene)-3,5-dichloro-2-hydroxybenzenesulfonohydrazide
Yield 86 %, m.p. 238–40 °C, Light brown. FT-IR v, (cm−1): OH (3588), NH (3479), Sp3CH (2993), C⚌N (1659), C⚌C (1586), C—O (1227), S⚌O (1174), C-Cl (7 5 5), C-Br (6 1 4). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 11.1 (s, 1H, OH), 8.09 (d, J = 7.6 Hz, 2H, Aromatic-H), 7.99 (d, J = 7.5 Hz, 2H, Aromatic-H), 7.88–7.85 (m, 1H, Aromatic-H) 7.73–7.71 (m, 1H, Aromatic-H), 7.61 (t, J = 7.4 Hz, 1H, Aromatic-H), 7.50 (d, J = 2.7 Hz, 2H, Aromatic-H), 7.49 9d, J = 2.4 Hz, 1H, Aromatic-H), 7.35 (dd, J = 6.7, 2.4 Hz, 1H, Aromatic-H), 7.15 (dd, J = 6.0, 3.2, 1H, Aromatic-H), 5.30 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ 168.0, 155.6, 152.8, 151.9, 149.1, 132.1, 131.7, 130.4, 130.2, 130.1, 128.0, 126.7, 125.3, 125.2, 122.2, 122.0, 115.8, 109.4, 109.1, 39.9, 26.6.HR-EI-MS: m/z calcd for C21H14BrCl2N3O4S2 [M]+ 587.28400; Found: 587.28382.
2.2.5 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-phenylethylidene)-3,5-dichloro-2-hydroxybenzenesulfonohydrazide
Yield 72 %, m.p. 233–35 °C, Light green. FT-IR v, (cm−1): OH (3587), NH (3477), Sp3CH (2997), C⚌N (1664), C⚌C (1586), C—O (1233), S⚌O (1175), C-Cl (7 5 9). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.91 (s, 1H, OH), 7.86 (dd, J = 7.2, 2.5 Hz, Aromatic-H), 7.73 (t, J = 7.3 Hz, 1H, Aromatic-H), 7.58 (d, J = 3.4 Hz, 1H, Aromatic-H) 7.50 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.49 (d, J = 2.5 Hz, 1H, Aromatic-H) 7.35 (d, J = 6.0 Hz, 1H, Aromatic-H), 7.19–7.19 (m, 1H, Aromatic-H), 5.00 (s, 2H,CH2). 13C NMR (125 MHz, DMSO‑d6): δ165.0, 155.6, 151.9, 145.2, 142.7, 141.5, 139.8, 134.0, 131.0, 130.2, 128.8, 128.8, 128.2, 128.2, 127.0, 124.8, 123.8, 123.0, 119.1, 110.6, 37.2.HR-EI-MS: m/z calcd for C21H15Cl2N3O4S2 [M]+ 508.38800; Found: 508.38775.
2.2.6 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-chlorophenyl)ethylidene)-3,5-dichloro-2-hydroxybenzenesulfonohydrazide
Yield 76 %, m.p. 237–38 °C, Light yellow. FT-IR v, (cm−1): OH (3590), NH (3480), Sp3CH (2995), C⚌N (1672), C⚌C (1586), C—O (1235), S⚌O (1172), C-Cl (7 5 9). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.91 (s, 1H, OH), 7.86 (dd, J = m7.2, 2.5 Hz, 2H, Aromatic-H), 7.73 (t, J = 7.3 Hz, 2H, Aromatic-H), 7.58 (d, J = 3.4 Hz, 1H, Aromatic-H) 7.50 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.49 (d, J = 2.5 Hz, 1H, Aromatic-H), 7.35 (d, J = 6.0 Hz, 2H, Aromatic-H), 7.19–7.10 (m, 1H, Aromatic-H) 3.10 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ167.0, 154.6, 152.8, 151.9, 141.5, 136.6, 135.0, 132.1, 128.9, 128.9, 128.6, 128.2, 128.2, 126.1, 125.9, 124.8, 124.0, 123.8, 119.1, 110.6, 37.2.HR-EI-MS: m/z calcd for C21H14Cl3N3O4S2 [M]+ 542.83000; Found: 542.82968.
2.2.7 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-chlorophenyl)ethylidene)-2-methyl-5-nitrobenzenesulfonohydrazide
Yield 74 %, m.p. 231–32 °C, Light gray. FT-IR v, (cm−1): NH (3480), Sp3CH (2996), C⚌N (1657), C⚌C (1588), C⚌N (1357), C—O (1232), S⚌O (1175), C-Cl (7 5 9). 1H NMR (600 MHz, DMSO‑d6): δ12.53 (s, 1H, NH), 8.28 (s, 1H, Aromatic-H), 8.09 (d, J = 8.3 Hz, 1H, Aromatic-H), 7.86 (d, J = 7.2 Hz, 2H, Aromatic-H), 7.62 (d, J = 8.22 Hz, 1H, Aromatic-H), 7.59 (d, J = 7.02 Hz, 2H, Aromatic-H), 7.46 (d, J = 8.9 Hz, 1H, Aromatic-H), 7.13 (d, J = 8.4 Hz, 1H, aromatic-H), 2.62 (s, 2H, CH2), 2.56 (s, 3H, CH3), 13C NMR (125 MHz, DMSO‑d6): δ 165.0, 155.6, 151.9, 145.2, 142.7, 141.5, 139.8, 136.6, 132.1, 130.2, 128.9, 128.9, 128.2, 128.2, 127.0, 124.8, 123.8, 123.0, 119.1, 110.6, 37.2, 22.0.HR-EI-MS: m/z calcd for C22H17ClN4O5S2 [M]+ 516.97100; Found: 516.97087.
2.2.8 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(3,4-dichlorophenyl)ethylidene)-2-methyl-5-nitrobenzenesulfonohydrazide
Yield 78 %, m.p. 238–40 °C, Light gray. FT-IR v, (cm−1): NH (3482), Sp3CH (2998), C⚌N (1659), C⚌C (1590), C⚌N (1380), C—O (1234), S⚌O (1177), C-Cl (7 6 2). 1H NMR (600 MHz, DMSO‑d6): δ12.51 (s, 1H, NH), 8.53 (s,1H, Aromatic-H), 8.28 (d, J = 2.3 Hz, 1H, Aromatic-H), 8.0 (d, J = 8.2 Hz, 1H, Aromatic-H), 7.87 (d, J = 8.2 Hz, 1H, Aromatic-H), 7.62 (d, J = 8.5 Hz, 1H, Aromatic-H), 7.46 (d, J = 8.4 Hz, 1H, Aromatic-H), 7.26 (d, J = 6.0 Hz, 2H, Aromatic-H), 7.13 (d, J = 8.0 Hz, 2H, Aromatic-H), 5.15 (s, 2H,CH2) 2.64 (s, 3H, CH3). 13C NMR (125 MHz, DMSO‑d6): δ167.9, 144.8, 132.1, 132.0, 131.4 131.2, 130.4, 128.4, 122.6, 122.2, 121.9, 109.4, 106.3, 102.2, 102.0, 101.7, 101.4, 101.1, 98.7, 98.4, 20.4, 19.5. HR-EI-MS: m/z calcd for C22H16Cl2N4O5S2 [M]+ 551.41300; Found: 551.41277.
2.2.9 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-bromophenyl)ethylidene)-2,4-dinitrobenzenesulfonohydrazide
Yield 80 %, m.p. 237–39 °C, Light yellow. FT-IR v, (cm−1): NH (3486), Sp3CH (3009), C⚌N (1660), C⚌C (1592), C⚌N (1382), C—O (1236), S⚌O (1178), C-Br (6 2 0). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 9.68 (s, 1H, Aromatic-H), 8.42 (dd, J = 9.0, 2.5 Hz, 1H, Aromatic-H), 8.41 (d, J = 6.4 Hz, 1H, Aromatic-H), 8.16 (d, J = 9.0 Hz, 2H, Aromatic-H), 8.09 (d, J = 8.3 Hz, 2H, Aromatic-H), 7.70 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.26 (d, J = 8.2 Hz, 2H, Aromatic-H), 5.18 (s, 2H, CH2). 13C NMR (125 MHz DMSO‑d6): δ168.1, 168.0, 152.0, 132.1, 130.3, 129.0, 128.7, 128.5, 122.2, 121.5, 120.3, 119.1, 116.6, 113.6, 113.4, 110.8, 109.4, 45.6, 39.9, 20.3, 8.52. HR-EI-MS: m/z calcd for C21H14BrN5O7S2 [M]+ 592.39500; Found: 592.39472.
2.2.10 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(3,4-dichlorophenyl)ethylidene)-2,4-dinitrobenzenesulfonohydrazide
Yield 86 %, m.p. 232–33 °C, Light yellow. FT-IR v, (cm−1): NH (3486), Sp3CH (3040), C⚌N (1666), C⚌C (1590), C⚌N (1384), C—O (1237), S⚌O (1178), C-Cl (7 6 9). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.68 (s, 1H, Aromatic-H), 8.41 (d, J = 6.5 Hz, 1H, Aromatic-H), 8.30 (d, J = 2.0 Hz, 1H, Aromatic-H), 8.02 (d, J = 6.3 Hz, 1H, Aromatic-H), 9.91 (d, J = 8.4, 1H, Aromatic-H), 7.63 (d, J = 7.2 Hz, 1H, Aromatic-H), 7.39 (d, J = 7.3 Hz, 2H, Aromatic-H), 7.15 (d, J = 6.5 Hz, 2H, Aromatic-H), 5.29 (s, 2H, CH2).13C NMR (125 MHz, DMSO‑d6): δ190.7, 167.9, 149.9, 136.8, 135.0, 132.2, 132.1, 131.9, 131.2, 130.4, 128.5, 128.4, 124.4 122.2, 120.3, 113.2, 119.4, 109.4, 40.8, 39.9, 20.4. HR-EI-MS: m/z calcd for C21H13Cl2N5O7S2 [M]+ 582.38300; Found: 582.38281.
2.2.11 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-bromophenyl)ethylidene)-2-methyl-5-nitrobenzenesulfonohydrazide
Yield 89 %, m.p. 239–40 °C, Light yellow. FT-IR v, (cm−1): NH (3481), Sp3CH (2997), C⚌N (1658), C⚌C (1585), C⚌N (1340), C—O (1220), S⚌O (1165), C-Br (6 1 0). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 8.50 (s, 1H, Aromatic-H), 8.09 (d, J = 8.4 Hz, 1H, Aromatic-H), 7.68 (d, J = 7.9 Hz, 2H, Aromatic-H), 7.62 (d, J = 8.8 Hz, 1H, Aromatic-H) 7.61 (d, J = 8.8 Hz, 2H, Aromatic-H), 7.46 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.36 (d, J = 6.1 Hz, 2H, Aromatic-H), 5.31 (s, 2H,CH2), 2.65 (s, 3H, CH3). 13C NMR (125 MHz, DMSO‑d6): δ 165.0, 155.6, 151.9, 145.2, 142.7, 141.5, 139.8, 136.6, 132.1, 130.2, 128.9, 128.9, 128.2, 128.2, 127.0, 124.8, 123.8, 123.0, 119.1, 110.6, 37.2, 22.0. HR-EI-MS: m/z calcd for C22H17BrN4O5S2 [M]+ 561.42500; Found: 561.42474.
2.2.12 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-bromophenyl)ethylidene)-2,4-dinitrobenzenesulfonohydrazide
Yield 83 %, m.p. 230–31 °C, Light yellow. FT-IR v, (cm−1): NH (3482), Sp3CH (3030), C⚌N (1660), C⚌C (1566), C⚌N (1320), C—O (1210), S⚌O (1155), C-Cl (7 4 2), C-Br (6 1 5). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.68 (s, 1H, Aromatic-H), 8.42 (d, J = 9.0, 2.5 Hz, 1H, Aromatic-H), 8.16 (d, J = 8.9 Hz, 1H, Aromatic-H), 8.01 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.84 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.43 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.02 (d, J = 8.9 Hz, 2H, Aromatic-H), 5.28 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ191.6, 167.9, 150.1, 146.0, 133.8, 132.1, 132.0, 131.7, 130.4, 130.1, 128.7, 128.5, 128.2, 124.4, 122.2, 121.5, 113.2, 109.4, 40.9, 39.9.6, 20.4. HR-EI-MS: m/z calcd for C21H14BrN5O7S2, [M]+ 592.39500; Found: 592.39489.
2.2.13 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-phenylethylidene)-2-methyl-5-nitrobenzenesulfonohydrazide
Yield 84 %, m.p. 233–35 °C, Light yellow. FT-IR v, (cm−1): NH (3465), Sp3CH (2950), C⚌N (1620), C⚌C (1550), C⚌N (1310), C—O (1210), S⚌O (1140). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 8.50 (d, J = 2.6 Hz, 1H, Aromatic-H), 8.09 (d, J = 8.3 Hz, 1H, Aromatic-H), 7.88 (d, J = 8.1 Hz, 1H, Aromatic-H), 7.59 (d, J = 6.2 Hz, 2H, Aromatic-H) 7.50 (m, 1H, Aromatic-H), 7.46 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.15 (dd, J = 8.9 Hz, 1H, Aromatic-H), 7.12 (d, J = 6.0, 3.4 Hz, 1H, Aromatic-H), 5.32 (s, 2H,CH2), 2.77 (s, 3H, CH3). 13C NMR (125 MHz, DMSO‑d6): δ 165.0, 155.6, 151.9, 145.2, 142.7, 141.5, 139.8, 134.0, 131.0, 130.2, 128.8, 128.8, 128.2, 128.2, 127.0, 124.8, 123.8, 123.0, 119.1, 110.6, 37.2, 22.0.HR-EI-MS: m/z calcd for C22H18N4O5S2 [M]+ 482.52900; Found: 482.52866.
2.2.14 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-phenylethylidene)-2-chlorobenzenesulfonohydrazide
Yield 89 %, m.p. 236–37 °C, Light green. FT-IR v, (cm−1): NH (3430), Sp3CH (2953), C⚌N (1623), C⚌C (1554), C⚌N (1317), C—O (1219), S⚌O (1142), C-Cl (7 5 0). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 8.08–8.05 (m, 1H, Aromatic-H), 7.87 (t, 1H, Aromatic-H), 7.84 (d, J = 7.7 Hz, 2H, Aromatic-H), 7.72 (d, J = 7.3 Hz, 2H, Aromatic-H), 7.66 (dd, J = 6.2, 2.5 Hz, 1H, Aromatic-H), 7.60 (dd, J = 8.5, 3.1 Hz, 1H, Aromatic-H), 7.47–7.41(m, 1H, Aromatic-H), 7.39–7.30 (m, 1H, Aromatic-H), 7.13 (dd, J = 7.8, 2.3 Hz, 1H, Aromatic-H), 5.28 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ165.0, 155.6, 151.9, 141.5, 139.7, 134.0, 133.3, 131.5, 131.0, 130.2, 128.8, 128.8, 128.7, 128.2, 128.2, 124.8, 123.8, 127.1, 119.1, 110.6, 37.2. HR-EI-MS: m/z calcd for C21H16ClN3O3S2 [M]+ 457.94700; Found: 457.94685.
2.2.15 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-phenylethylidene)-2,4-dinitrobenzenesulfonohydrazide
Yield 80 %, m.p. 240–41 °C, Light green. FT-IR v, (cm−1): NH (3434), Sp3CH (2957), C⚌N (1629), C⚌C (1544), C⚌N (1321), C—O (1223), S⚌O (1145). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 9.67 (s, 1H, Aromatic-H), 8.42 (d, J = 8.9, 2.5 Hz, 1H, Aromatic-H), 8.35 (d, J = 9.0 Hz, 1H, Aromatic-H), 8.16 (d, J = 9.0 Hz, 2H, Aromatic-H), 8.00 (dd, J = 7.5 Hz, 2H, Aromatic-H), 7.56 (dd, J = 9.1, 3.1 Hz, 1H, Aromatic-H) 7.49 (t, J = 6.9 Hz, 2H, Ar), 7.15–7.10 (m, 1H, Aromatic-H), 5.25 (s, 2H, CH2). 13C NMR (125 MHz DMSO‑d6): δ 165.0, 155.6, 152.0, 151.9, 148.1, 141.5, 140.5, 134.0, 131.0, 130.3, 129.1, 128.8, 128.8, 128.2, 124.8, 123.8, 119.1, 114.5, 110.6, 37.2, 28.2. HR-EI-MS: m/z calcd for C21H15N5O7S2 [M]+ 513.49900; Found: 513.49886.
2.2.16 (E)-N'-(1-([1,1′-biphenyl]-4-yl)-2-(benzo[d]oxazol-2-ylthio)ethylidene)-2-methyl-5-nitrobenzenesulfonohydrazide
Yield 71 %, m.p. 242–44 °C, Light green. FT-IR v, (cm−1): NH (3437), Sp3CH (2959), C⚌N (1640), C⚌C (1546), C⚌N (1326), C—O (1229), S⚌O (1144). 1H NMR (600 MHz, DMSO‑d6): δ12.53 (s, 1H, NH), 8.28 (s, 1H, Aromatic-H), 8.18 (d, J = 7.9 Hz, 1H, Aromatic-H), 8.08 (dd, J = 6.7, 2.5 Hz, 2H, aromatic-H), 7.92 (d, J = 7.9 Hz, 1H, Aromatic-H), 7.80 (d, J = 7.5 Hz, 2H, Aromatic-H), 7.64 (d, J = 8.8 Hz, 2H, Aromatic-H),7.62 (t, J = 7.9 Hz, 2H, Aromatic-H), 7.54 (t, J = 7.6 Hz, 2H, aromatic-H), 7.47 (d, J = 6.2 Hz, 2H, Aromatic-H), 7.46 (d, J = 7.3 Hz, 2H, Aromatic-H), 5.31 (s, 2H, CH2), 2.61 (s, 3H, CH3). 13C NMR (191.8, 150.3, 147.5, 145.4, 144.8, 144.2, 138.5, 133.5, 132.1, 129.2, 129.0, 128.8, 128.6, 127.0, 127.0, 126.8, 124.7, 123.3, 122.2, 121.1, 113.1, 119.1, 110.6, 109.4, 102.6, 37.2, 20.1, 8.48. HR-EI-MS: m/z calcd for C28H22N4O5S2 [M]+ 558.62700; Found: 558.62671.
2.2.17 (E)-N'-(1-([1,1′-biphenyl]-4-yl)-2-(benzo[d]oxazol-2-ylthio)ethylidene)-2-chlorobenzenesulfonohydrazide
Yield 82 %, m.p. 238–40 °C, Light green. FT-IR v, (cm−1): NH (3438), Sp3CH (2964), C⚌N (1647), C⚌C (1548), C⚌N (1327), C—O (1230), S⚌O (1146), C-Cl (7 5 1). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 8.18 (d, J = 7.9 Hz, 2H, Aromatic), 7.92 (d, J = 7.9 Hz, 2H, Aromatic-H), 7.80 (d, J = 7.7, Hz, 2H, Aromatic), 7.88 (d, J = 7.4 Hz, 2H, Aromatic-H), 7.80 (d, J = 7.5 Hz, 2H, Aromatic-H), 7.78–7.73 (m, 1H, Aromatic-H), 7.63 (dd, J = 9.0, 3.0 Hz, 1H, Aromatic-H), 7.56 (dd, J = 8.1, 3.0 Hz, 1H, Aromatic-H), 7.54 (t, J = 7.4 Hz, 2H, Aromatic-H), 7.47–7.41 (m, 1H, Aromatic-H), 7.33–7.21 (m, 1H, Aromatic-H), 7.30 (dd, J = 8.8, 3.5 Hz, 1H, Aromatic-H), 5.40 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ 197.5, 168.0, 151.9, 144.9, 144.5, 135.5, 132.1, 130.6, 130.3, 130.0, 129.3, 129.1, 129.0, 128.9, 128.8, 128.3, 127.4, 127.1, 127.0, 126.9, 126.8, 126.1, 125.1, 122.2, 114.4, 109.4.6, 26.7. HR-EI-MS: m/z calcd for C27H20ClN3O3S2 [M]+ 534.04500; Found: 534.04482.
2.2.18 (E)-N'-(1-([1,1′-biphenyl]-4-yl)-2-(benzo[d]oxazol-2-ylthio)ethylidene)-2,4-dinitrobenzenesulfonohydrazide
Yield 77 %, m.p. 238–39 °C, Light yellow. Light green. FT-IR v, (cm−1): NH (3410), Sp3CH (2930), C⚌N (1610), C⚌C (1520), C⚌N (1311), C—O (1208), S⚌O (1122). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 9.67 (s, 1H, Aromatic-H), 8.42 (dd, J = 8.9, 2.4 Hz, 1H, Aromatic-H), 8.16 (d, J = 8.2 Hz, 1H, Aromatic-H), 8.16 (d, J = 8.2 Hz, 1H, Aromatic-H), 7.91 (d, J = 8.1 Hz, 2H, Aromatic), 7.87 (dd, J = 6.0, 3.1 Hz, 2H, Aromatic-H), 7.79 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.66–7.59 (m, 1H, Aromatic-H), 7.54 (dd, J = 8.0, 2.4 Hz, 1H, Aromatic-H), 7.49 (m, 1H, Aromatic-H), 7.28 (dd, J = 9.3, 3.1 Hz, 1H, Aromatic-H) 7.15–7.10 (m, 1H, Aromatic-H), 7.02 (t, J = 8.9 Hz, 1H, Aromatic-H), 5.25 (s, 2H,CH2). 13C NMR (125 MHz, DMSO‑d6): δ 198.0, 171.6, 165.0, 156.9, 149.1, 146.1, 143.5, 140.8, 140.5, 132.9, 130.3, 129.7, 129.7, 129.2, 129.2, 129.1, 128.0,128.0, 127.9, 127.9, 127.6, 124.8, 123.8, 119.1, 114.5, 110.6, 37.2. HR-EI-MS: m/z calcd for C27H19N5O7S2 [M]+ 589.59700; Found: 589.59674.
2.2.19 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(3,4-dichlorophenyl)ethylidene)-4-bromobenzenesulfonohydrazide
Yield 79 %, m.p. 233–35 °C, Light yellow. FT-IR v, (cm−1): NH (3434), Sp3CH (2977), C⚌N (1630), C⚌C (1549), C—O (1226), S⚌O (1176), C-Cl (7 5 3), C-Br (6 1 2). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 8.04 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.89 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.75 (d, J = 8.1 Hz, 1H, Aromatic-H), 7.60 (d, J = 6.1, 1.9 Hz, 1H, Aromatic-H), 7.56–7.50 (m, 1H, Aromatic-H), 7.48 (dd, J = 6.3, 2.1 Hz, 1H, Aromatic-H), 7.41 (d, J = 6.1, 2.1 Hz, 1H, Aromatic-H), 7.14–7.10 (m, 1H, Aromatic-H), 7.06 (s, 1H, Aromatic-H), 5.16 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ 169.0, 157.6, 152.9, 146.0, 141.5, 135.7, 133.5, 133.5, 131.9, 131.9, 130.6, 130.3, 129.5, 129.5, 126.3, 126.3, 124.8, 123.8, 111.1, 109.6, 37.2.HR-EI-MS: m/z calcd for C21H14BrCl2N3O3S2 [M]+ 571.28500; Found: 571.28483.
2.2.20 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-phenylethylidene)-4-bromobenzenesulfonohydrazid
Yield 74 %, m.p. 236–38 °C, Light gray. FT-IR v, (cm−1): NH (3436), Sp3CH (2978), C⚌N (1632), C⚌C (1550), C—O (1228), S⚌O (1177), C-Br (6 1 1). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 7.87 (dd, J = 6.1, 3.2 Hz, 1H, Aromatic-H), 7.78 (d, J = 8.4 Hz, 1H, Aromatic-H), 7.73 (d, J = 8.7 Hz, 2H, Aromatic-H), 7.65 (d, J = 8.5 Hz, 2H, Aromatic-H), 7.60 (d, J = 7.7 Hz, 1H, Aromatic-H), 7.48 (m, 1H, Aromatic-H), 7.15 (t, J = 6.1 Hz, 1H, Aromatic-H), 5.20 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ168.0, 155.6, 151.9, 147.5, 146.0, 142.5, 134.0, 132.1, 131.9, 131.8, 131.0, 130.6, 129.5, 127.7, 126.0, 122.2, 114.4, 109.4, 39.9, 20.4, 20.2.HR-EI-MS: m/z calcd for C21H16BrN3O3S2 [M]+ 502.40100; Found: 502.40073.
2.2.21 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-chlorophenyl)ethylidene)-4-bromobenzenesulfonohydrazide
Yield 85 %, m.p. 231–33 °C, Light green. FT-IR v, (cm−1): NH (3437), Sp3CH (2979), C⚌N (1633), C⚌C (1551), C—O (1229), S⚌O (1178), C-Cl (7 4 9), C-Br (6 1 0). 1H NMR (600 MHz, DMSO‑d6): δ 12.5 (s, 1H, NH), 8.10 (d, J = 8.1 Hz, 2H, Aromatic-H), 7.78 (d, J = 8.4 Hz, 2H, Aromatic-H), 7.76 (d, J = 6.9 Hz, 2H, Aromatic-H), 7.75 (d, J = 7.4 Hz, 2H, Aromatic-H), 7.60 (d, J = 8.8 Hz, 2H, Aromatic-H), 7.16 (d, J = 7.4 Hz, 2H, Aromatic-H), 3.07 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ 166.0, 156.6, 152.9, 147.0, 143.5, 137.6, 132.1, 131.9, 131.9, 129.5, 129.5, 128.9, 128.9, 128.2, 128.2, 126.3, 124.8, 123.8, 119.1, 110.6, 37.2. HR-EI-MS: m/z calcd for C21H15BrClN3O3S2 [M]+ 536.84300; Found: 536.84280.
2.2.22 (E)-N'-(2-(benzo[d]oxazol-2-ylthio)-1-(4-bromophenyl)ethylidene)-2-chlorobenzenesulfonohydrazide
Yield 78 %, m.p. 229–31 °C, Light gray. FT-IR v, (cm−1): NH (3438), Sp3CH (2980), C⚌N (1634), C⚌C (1552), C—O (1230), S⚌O (1179), C-Cl (7 4 7), C-Br (6 0 8). 1H NMR (600 MHz, DMSO‑d6): δ12.5 (s, 1H, NH), 8.07 (d, J = 8.1 Hz, 2H, Aromatic-H), 8.00 (d, J = 8.2 Hz, 2H, Aromatic-H), 7.87 (d, J = 7.2 Hz, 2H, Aromatic-H), 7.83 (d, J = 8.1 Hz, 2H, Aromatic-H), 7.78–7.74 (m, 1H, Aromatic-H), 7.56 (dd, J = 8.4, 4.5 Hz, 1H, Aromatic-H), 7.46–7.44 (m, 1H, Aromatic-H), 7.36 (dd, J = 8.9, 3.8 Hz, 1H, Aromatic-H), 5.21 (s, 2H, CH2). 13C NMR (125 MHz, DMSO‑d6): δ167.0, 157.6, 153.9, 143.5, 139.7, 134.3, 132.0, 131.9, 131.7, 131.5, 130.2, 128.7, 128.6, 128.6, 125.4, 127.1, 124.8, 123.8, 119.1, 110.6, 37.2. HR-EI-MS: m/z calcd for C21H15BrClN3O3S2 [M]+ 536.84300; Found: 536.84277.
3 Results and discussion
3.1 Chemistry
The strategy adopted for the synthesis of 2-Marcapto benzoxazole as according to reported method already present in the literature for compound (III) but the present study were conducted with minor modification to obtain targeted compounds (1–22). 2-aminophenol (I) (1-mmol) was dissolved in ethanol and carbon di-sulphide was added to the pre-heated solution in the presence of Et3N. After being refluxed the target compounds were achieved (II). 2-Marcapto benzoxazole (II) was then refluxed along with different substituted phenacyl bromide in ethanol solvent while few drops of triethylamine, added as catalyst, yielded 2-(benzo[d]oxazol-2-ylthio)-1-phenylethan-1-one (III). Compound (III) was further dissolved in ethanol and hydrazine hydrate was added slowly an excess followed by addition of few drops of acetic acid, obtained Schiff base (IV). This Schiff base compound (IV) and different substituted benzene sulphonyl chloride were dissolved in THF in the presence of triethylamine under refluxed conditions. After completion of reaction, benzoxazole based sulphonamide derivatives (1–22) were obtained as shown in scheme-1. These products were dried, washed with n-hexane and fine powder were collected. Furthermore, Thin layer chromatography (TLC) were used at every step to confirm the products formation as well as varied spectroscopic techniques such as H NMR, C NMR and HREI-MS were used in order to confirm the basic skeleton of the synthesized compounds.
3.2 General representation of molecule
In order to explain biological activity of synthesized compounds, by this consideration we divided synthesized molecule into three parts; a benzoxazole moiety, ring-A and ring-B. These aromatic rings showed different activity profile due to varied substitutions and their position as shown in Figure-3.
4 Biological profile
Biological profiles of the tested scaffolds were found with varied range of inhibitions due to the substitution pattern. Nature of substituents, number and position are the factors responsible for increase or decrease the biological potentials of compounds. In this regard we compared the following same substituted analogues and their inhibitory profile in SAR study against standard drug acarbose with α-amylase and α-glucosidase having IC50 = 9.20 ± 0.10 µM and 10.10 ± 10 µM respectively.
The comparison criteria were set for chloro-substituted analogues (1, 2 and 3). These molecules possess chloro-moities at both ring A and B and their inhibitory profile were found different from one another due to different position of chloro groups. In compounds (1 and 2) ring-A contain two chlorine groups at meta and para-position while ring-B (compound 1) contain chloro group at ortho and para-position with one methyl moiety at meta-position and compound 2, contain only one chlorine group at ortho-position, showed different inhibitory concentrations in both α-amylase and α-glucosidase with IC50 = 1.10 ± 0.20, 1.20 ± 0.30 µM and 2.10 ± 0.10, 2.40 ± 0.10 µM respectively. When both these compounds weere compared to analog (3) having one chloro-group at para-position of ring-A and ring-B contain chloro-group at ortho-position, showed few fold less activity profile with IC50 = 8.20 ± 0.20 µM and 9.10 ± 0.20 µM. Thus, it was clearly observed that activity profile was dominant in case of compound 1 might be due to the presence of methyl moiety and one chloro group attach to para-position at ring-B while compound 2, was ranked second shows somewhat less activity on both the cases. Analog 3, was found with comparable activity to standard drug acarbose as shown in Figure-4.
Similarly, chloro-substituted compounds bearing hydroxyl moiety at meta-position on aromatic ring-B of compounds 4, 5 and 6 displayed varied range of inhibitiory profile. All these compounds having two chloro-moieties at ortho and para-position of ring-B while ring-A contain varied substituents. In case of compound 4, bromo moiety being bulky group itself reduce the chances of interactions thus molecule was found with somewhat lower activity in both α-amylase and α-glucosidase (IC50 = 21.60 ± 0.30 µM and 22.2 ± 0.40 µM) than standard acarbose (IC50 9.20 ± 0.10 µM and 10.10 ± 10 µM respectively). While scaffolds 5 and 6, were found with eight and nine folds better activity (IC50 = 2.30 ± 0.10 µM, 2.70 ± 0.10 µM and 1.70 ± 0.10 µM, 1.60 ± 0.10 µM respectively) than standard drug. This might be the absence of bromo-group at ring-A in compound 5, increases the chances of interactions as well as compounds 6, was found an active moiety among hydroxyl-substituted compounds it may be the presence of chloro-group at para-position of ring-A showed better interactions than others as shown in Figure-5.
Compounds 7, 8 9 and 10 were compared due to the presence of chloro and nitro moieties, attached to aromatic ring-A and B. The positions of substitution pattern are the main purpose of interaction and their potency. Compound 7 (IC50 = 3.10 ± 0.10 µM and 2.90 ± 0.10 µM) and compound 9 (IC50 = 6.20 ± 0.30 µM, 7.30 ± 0.20 µM) bearing chloro-group at para-position of ring-A while nitro group at ortho, meta and para-position as well as methyl moiety at ortho-group on ring-B. The change in their activity in case of analog 7, showed significant profile it might be the presence of methyl-group. While in case of analog 9, the activity is somewhat lower than analog 7, clearly indicate the presence of electron with drawing group a ortho-position reduce the activity profile of compound. Compounds 8 and 10 were found better activity due to the presence of two chloro-groups at ring-A while ring-B contain varied substituents. The presence of methyl moiety in compound 8 (IC50 = 1.30 ± 0.10 µM and 1.50 ± 0.10 µM) increases the chances of interactions as in case of analog 7. When nitro group replace methyl moiety the interaction of compound 10 (1.50 ± 0.10 µM and 1.70 ± 0.10 µM) reduces due to deactivation of the ring take place. In such comparison criteria analogues bearing methyl-groups on ring-B, displayed significant activity than nitro-substituted analogues as shown in Figure-6.
Scaffolds 11 and 12 were found somewhat poor in inhibitory profile when compared with standard drug acarbose it might be the presence of bulky group bromine at ring-A which reduce the interactions of molecules as shown in Figure-7.
Substituents are the important factor for increasing or decreasing the activity profile in this regard compounds 13, 14 and 15 having no-substituents at ring-A; displayed comparable activity with standard drug acarbose. This comparative might be the presence of attached substituents at ring-B as shown in Figure-8.
Compounds 16, 17 and 18 were found with comparable inhibitory activity due to the presence of biphenyl moiety at ring-A and varied attached substituents at ring-B. Among compound 18 (IC50 = 8.60 ± 0.20 µM and 9.930 ± 0.20 µM) was regarded as the active one in both α-amylase and α-glucosidase. It might be the presence of two nitro-group at ortho and para-position of ring-B while the remaining two compounds 16 (IC50 = 15.40 ± 0.30 µM and 14.30 ± 0.30 µM) and 17 (IC50 = 14.90 ± 0.30 µM and 15.90 ± 0.30 µM) were found with comparable activity with standard drug acarbose as shown in Figure-9.
Group of chloro-substituted compounds bearing bromine moiety at ring-B were compared with each other as well as with standard drug acarbose. Among analog 19 (IC50 = 5.70 ± 0.20 µM and 6.30 ± 0.20 µM, was found active, it might be the presence of two chloro-groups while analog 21 (IC50 = 8.70 ± 0.20 µM and 9.20 ± 0.20 µM) and 22 (IC50 = 9.43 ± 0.20 µM and 10.10 ± 0.20 µM) bearing one chloro groups at ring-A and ring-B respectively were found with comparable activity to standard drug acarbose. In this group only member analog 20 (IC50 = 16.20 ± 0.20 µM and 17.10 ± 0.20 µM), was found not active as shown in Figure-10.
In the above mention SAR study the substitution pattern and nature of substituents is only responsible for potent behavior. Thus, by such analysis compounds 1 (IC50 = 1.10 ± 0.20 µM, 1.20 ± 0.30 µM), 2 (IC50 = 2.10 ± 0.10 µM, 2.40 ± 0.10 µM), 6 (IC50 = 1.70 ± 0.10 µM, 1.60 ± 0.10 µM), 8 (IC50 = 1.30 ± 0.10 µM, 1.50 ± 0.10 µM), 10 (IC50 = 150 ± 0.10 µM, 1.70 ± 0.10 µM) were found few folds better potentials when compared to standard drug acarbose (α-amylase with IC50 = 9.20 ± 0.10 µM and α-glucosidase IC50 = 10.10 ± 0.10 µM). The changed observed in the activity of analogs might be the presence substituents. The biological system depends on the nature, position and number of substituents attached to aromatic ring which increase or decrease activity profile of analogs. The inhibitory profiles were strong when an electron donating group (EDG) attached to the ring whereas found weak when electron withdrawing group (EWG) attached to the ring. Increase the negative charge by EDG might increase the inhibitory potential of analogs. When compare the inhibitory profile of above mention analogs difference found due to presence of varied substituents on different position of aromatic ring which clearly indicate the effects of attached moieties.
5 Molecular docking studies
Molecular docking study was performed by using discovery studio visualizer (DSV) and auto Dock vina 1.5.6. (Kharb, M. et al., 2012, Li, Z.et al., 2015, Rao, C.et al., 2021 & Khan, S. et al., 2022) The synthesized compounds were docked against alpha amylase and alpha glucosidase and their structure were obtained from protein data bank (PDB) by searching codes such as 1b2y (Chain-A, Resolution = 3.20 Å, Native ligand Pyroglutamic acid and Residue == 496 amino acid) & 3w37 (Chain-A, Resolution = 1.70 Å, Native ligand Acarbose and Residue == 913 amino acid).
Initially, preparation of protein was done by using DSV in order to remove water molecules both protein and selected compounds were saved in PDB format. These PDB format were then open in auto dock tools by adding polar hydrogen to protein as well as kollman and gasteiger charges. While ligand was prepared by torsion tree in which choose root and detect root were identified. These prepared protein and ligand were saved as PDBQT format. Furthermore, configuration file was generated along with X,Y and Z axis (the dimension of these coordinates were 80 and grid box center 25.555 Å, 61.538 Å and 51.515 Å) then followed docking procedure by using command prompt which generated 9 poses for the selected molecule. The top ranked molecules was docked with protein PDBQT format in DSV. The protein ligand interaction (PLI) of molecules with active site of enzymes was summarized in Figure-11.
5.1 Docking results
Molecular docking was performed in order to explore binding mode of synthesized compounds against targeted enzyme, i.e., Alpha amylase and Alpha glucosidase. Both the enzyme crystallographic coordinates were retrieved from www.rcsb.org. Docking procedure revealed that the selected compounds had numerous potentials when tested against targeted enzymes. Few compounds possess electron with drawing group while some have electron donating group but the potent moieties have chloro and nitro-group illustrated in Figure-11, showed the superposed surface complex structure. Moreover, varied substituted compounds were docked then observed different binding modalities against both the targeted enzyme. In this regard chloro and nitro-substituted compounds exhibited better potential against these enzymes.
Specifically, In the case of efficacious compound 1 and 8, the protein–ligand interaction (PLI) profile not only listed the best potential (in silico), but also demonstrated better potency in vitro study for both alpha amylase and alpha glucosidase and their lowest binding affinity as shown in Table-2 and 3 respectively.
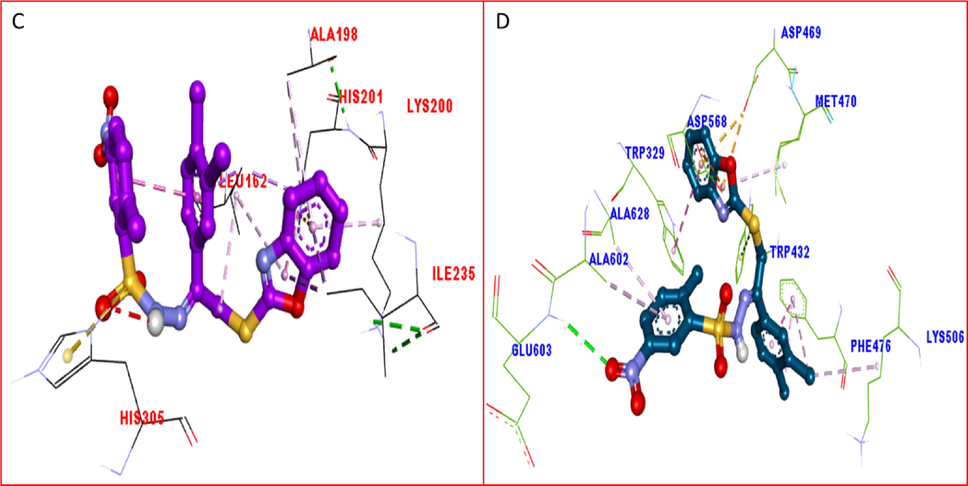
The protein ligand interaction profile for compound 1 having chloro groups, showed different interactive residues for alpha amylase as shown in Figure-11-A having distance between ligand and protein ranging from 4.09 to 619A ad their binding interactions such as His305(HB), LEU162 (π-σ), ILE-235(π-σ), HIS201(π-Cation), lys200(π-alkyl), Ala198(alkyl), Similarly, for alpha glucosidase having distance between ligand and protein ranging from 3.21 to 6.74A ad their binding interactions the interactive residues are MET583(π-Alkyl), ILE674(HB), MET593(π-X), ILE672(π-S), PRO314(π-Alkyl), PHE680(Alkyl), LEU304(Alkyl) and MET664(π-Alkyl) as shown in Figure-11-B.
The most potent compound 8 displayed remarkable behavior due to various PLI profile. This compound bearing nitro-moiety at aromatic ring have significant interactive residue for alpha amylase having distance between ligand and protein ranging from 3.79 to 597A ad their binding interactions such as LEU162(π-σ), ILE235(π-σ), HIS201(π-Cation), THR163(VW-ineractions), ALA198(π-Alkyl), TYS200(Alkyl) as shown in Figure-11-C. While against alpha glucosidase having distance between ligand and protein ranging from 3.97 to 7.06A ad their binding interactions such as PHE476(π-π Stacked), TRP432(π-S), ASP568(π-Anion), MET470(Alkyl),LYS506(π-Alkyl), TRP329(π-π Stacked), ALA602(Alkyl) etc as shown in Figure-11-D.
In alpha amylase the hydrogen bond interaction is weak the bond length was found with varied ranges while in case of alpha glucosidase hydrogen bond interaction is strong in the range of 3A.
The only differences found in both compound 1 and 8 are the attached substituents and PLI profile, in both the case such as chloro and nitro are attached to para position of aromatic ring in which chloro group increase the nucleophilic character of the ring while nitro being electron with drawing moiety decrease the nucleophilic character therefore the moiety having very weak interaction but the presence of benzoxazole and sulphonamide moieties they shows strong interaction with enzyme active site. While in case of chloro-substituted analog the presence of more nucleophilic π-system the interaction of the compounds were observed maximum as shown in Figs. 12 and 13.(See Fig. 14).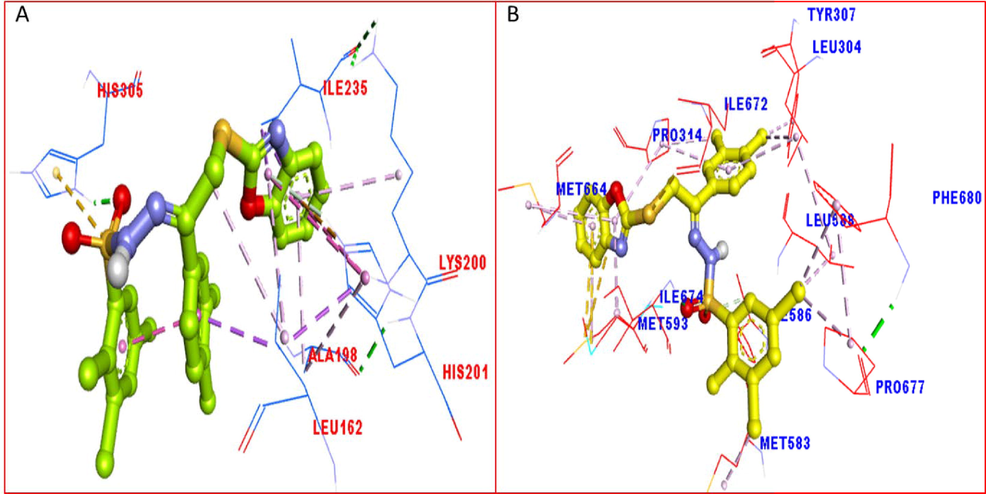
The PLI profile for potent compounds-1 against Alpha amylase and Alpha glucosidase enzyme indicate the surface of the corresponding enzyme by 3D.
The PLI profile for potent compounds-8 against Alpha amylase and Alpha glucosidase enzyme indicate the surface of the corresponding enzyme by 3D.
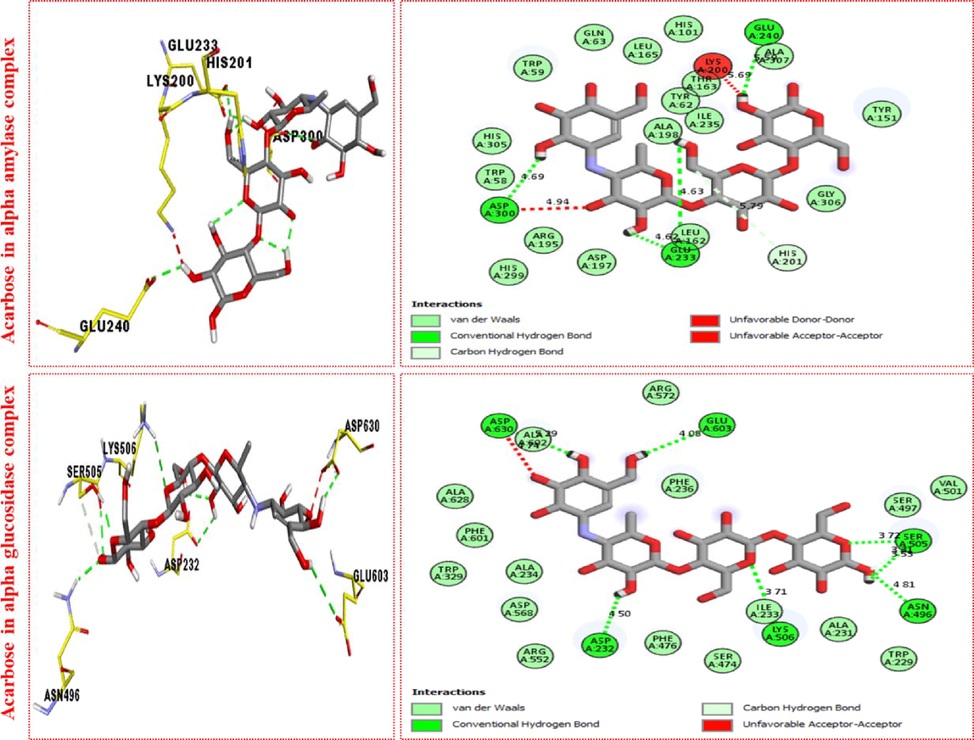
The PLI profile for potent acarbose against Alpha amylase and Alpha glucosidase enzyme indicate the surface of the corresponding enzyme.
Overall docking studies represent that scaffolds 1 and 8 having electron donating and with drawing moiety showed better result in both the targeted enzymes. The interaction of scaffolds was observed by hydrogen bonding, pi-pi interaction, pi-sulphur and pi-cation. Scaffolds showed all the mention interactions due to various rings moieties and heteroatoms. The better interaction of analogs 1 and 8 were found excellent against alpha amylase and alpha glucosidase due to attached substituents which dominantly shows greater potential as compared to other substituted analogs of the series. Analogs 1 and 8 both having chloro moieties a varied position of aromatic ring such increase the nucleophilic character of the ring thus, increases the interactions. The nucleophilic character of analogs 1 and 8 were found maximum due to greater number of chlorine attached to aromatic ring which cover the protein from different directions, showed better potential as compared to others (Figure-11–13). When compared with acarbose a standard drug (Figure-14) these moieties were found with much potent behavior.
The comparison study of acarbose standard drug exhibited the following interactions against α‐amylase enzyme with distance from 4.62 to 5.79A and their interactive residues such as ASP-A-300(H-B), ASP-A-300(Donor-donor), GLU-A-233(H-B). LEU-A-162(VW), HIS-A-201(C—H), ALA-A307(VW), GLU-A-240(H-B) and LYS-A-200 (Acceptor-acceptor), etc. Similarly, α‐glucosidase showed a varied range of distance from 3.71 to 5.29A and their interactive residues such as ASP-A-630(H-B), ALA-A-492(VW), GLU-A-603(H-B), SER-A-505(H-B), ASN-A-496(H-B), ILE-A-233(VW) and ASP-A-232(H-B),etc as illustrated in figure-13. Most of the synthesized ligand showed better interaction when compared with standard drugs acarbose due to attached functional group. RMSD value for acarbose was −8.73A.
6 ADMET prediction
In this study the molecules were found with different properties to ensure the Absorption, Distribution, Metabolism, Excretion, and Toxicity through online software SwissADME. By this study log Kp, Lipinski, Ghose, Veber, gan, Muegge violations, Bioavailability score, PAINS, Brenk alerts as well as Leadlikeness violations were monitored. The subjected candidates 1 and 8 were found with significant results as shown in graph-1 and 2.
7 Conclusion
Benzoxazole based sulphonamide derivatives (1–22) were synthesized successfully by simple and efficient way. All the synthesized derivatives were characterized thorugh different spectroscopic techniques such as H NMR, C NMR and HREI-MS. Moreover, these derivatives were screened against α-amylase and α-glucosidase. Among scaffolds 1 and 8 were found with significant activities for α-amylase and α-glucosidase inhibition. In addition, molecular docking studies also revealed the better binding interaction with active sites of enzymes. This study identifies new class of compounds as α-amylase and α-glucosidase inhibitors.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102-110.
- [Google Scholar]
- Chronic hyperglycemia and glucose toxicity: pathology and clinical sequelae. Postgrad. Med.. 2012;124:90-97.
- [Google Scholar]
- Structure of A23187, a divalent cation ionophore. J. Am. Chem. Soc.. 1974;96:1932-1933.
- [Google Scholar]
- Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem.. 2000;43:3714-3717.
- [Google Scholar]
- Atomic physicochemical parameters for three-dimensional structure-directed quantitative structure-activity relationships. 2. modeling dispersive and hydrophobic interactions. J. Chem. Inf. Comput. Sci.. 1987;27:21-35.
- [Google Scholar]
- Synthesis of Bis-indolylmethane sulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg. Chem.. 2018;80:112-120.
- [Google Scholar]
- Bioorg. Chem.. 2018;73:121-127.
- Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. Str: J. Molecul; 2022. p. :133812.
- Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Tech.. 2012;2:189-197.
- [Google Scholar]
- Adaptive molecular docking method based on information entropy genetic algorithm. App. Soft Comput.. 2015;26:299-302.
- [Google Scholar]
- One-pot syntheses of pseudopteroxazoles from pseudopterosins: a rapid route to non-natural congeners with improved antimicrobial activity. J. Nat. Prod.. 2011;74:2250-2256.
- [Google Scholar]
- Synthesis, metal ion binding, and biological evaluation of new anticancer 2-(2′-hydroxyphenyl) benzoxazole analogs of UK-1. Bioorg. Med. Chem.. 2008;16:1775-1783.
- [Google Scholar]
- New and emerging drugs and targets for type 2 diabetes: reviewing the evidence. Am. Health Drug Benefits.. 2014;7:452.
- [Google Scholar]
- In vitro and in silico inhibition properties of fucoidan against α-amylase and α-D-glucosidase with relevance to type 2 diabetes mellitus. Carbohydr. Polym.. 2019;209:350-355.
- [Google Scholar]
- Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation. 2021;17(3):404.
- [Google Scholar]
- Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metab.. 2013;17:20-33.
- [Google Scholar]
- New hybrid hydrazinyl thiazole substituted chromones: as potential α-amylase inhibitors and radical (DPPH & ABTS) scavengers. Sci. Rep.. 2017;7:1-17.
- [Google Scholar]
- New hybrid scaffolds based on hydrazinyl thiazole substituted coumarin; as novel leads of dual potential; in vitro α-amylase inhibitory and antioxidant (DPPH and ABTS radical scavenging) activities. Med. Chem.. 2019;15:87-101.
- [Google Scholar]
- Natural prenylchalconaringenins and prenylnaringenins as antidiabetic agents: α-glucosidase and α-amylase inhibition and in vivo antihyperglycemic and antihyperlipidemic effects. J. Agric. Food Chem.. 2017;65:1574-1581.
- [Google Scholar]
- Synthesis, α-amylase inhibitory potential and molecular docking study of indole derivatives. Bioorg. Chem.. 2018;80:36-42.
- [Google Scholar]
- Chin. Chem. Lett.. 2010;21:253-256.
- Insulin resistance or insulin deficiency: which is the primary cause of NIDDM? Diabetes. 1994;43:735-740.
- [Google Scholar]
- IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. J. Shaw, Diabetes Res. Clin. Pract.. 2011;94:311-321.
- [Google Scholar]
- Synthesis of (S)-2-ethoxy-3-phenylpropanoic acid derivatives and their insulin-sensitizing activity. Chin. J. Chem.. 2007;25:265-267.
- [Google Scholar]
- Synthesis and biological evaluation of coumarin derivatives containing oxime ester as α-glucosidase inhibitors. Arab. J. Chem.. 2022;15(9):104072
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104341.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




