

Translate this page into:
NMR investigation of substituent effects on strength of π-π stacking and hydrogen bonding interactions to supports the formation of [2 + 2] photodimerization in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes
⁎Corresponding authors. biglari.z@lu.ac.ir (Zeinab Biglari), biglariz@gmail.com (Zeinab Biglari), Gholipour.a@lu.ac.ir (Alireza Gholipour) Alir.gholipour@gmail.com (Alireza Gholipour)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
We have investigated the ability of para-X-phenylboronic acid (para-X-ba) to enable reactivity of trans-1,2-bis(4-pyridyl)ethylene (bpe) to direct intermolecular [2 + 2] photodimerization via computational chemistry. Para-X-ba would support the formation of discrete four component hydrogen bonded molecular assemblies wherein π-π stacking of a pyridyl-functionalized alkene would conform to undergo [2 + 2] photodimerization.
We have demonstrated by computational 1H NMR data the effect of electron-withdrawing and donating substituents in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes to assemble bpe into π-π stacking via –(B)O − H···N– hydrogen bonds to react to afford (para-X-ba):::tpcb:::(para-X-ba) complexes (X = NO2, CN, F, Cl, Br, C(O)CH3, OCH3, OH, NH2 and H where || and ∙∙∙ denote π-π stacking and hydrogen bonds).
Also, these interactions have been investigated at M05-2X/6-311++G** level of theory in detail in terms of the energetic, geometrical parameters and electron density properties to characterize and to examine the strengthening of the interactions. There are good relationships between the NMR, AIM, energy data and Hammett constants.
Keywords
Photodimerization
Computational chemistry
Hammett constants
π-π Stacking
Hydrogen bonding

1 Introduction
Boronic acids have been widely used as reagents for the targeted synthesis of compounds having applications in the areas of pharmaceuticals, agrochemicals etc. It should be noted that there are potential drawbacks to using boronic acids as supramolecular building blocks, especially in the presence of heterocyclic compounds (Zheng et al., 2010, 2012; Das et al., 2017; Cuenca et al., 2016; Hall, 2006; Li et al., 2017; Diemoz and Franz, 2019). Boronic acids can be regarded as “green” compounds due to their low toxicity and their ultimate degradation into the environmentally friendly boric acid (Hall, 2005). The reversible hydrogen bonding interactions that boronic acids can take part in has seen a significant increase in the applications of boronic acid based systems in self-assembly sensing, and separation science (James, 2016; Nishiyabu et al., 2011; Kubo et al., 2015; Fujita et al., 2008; Corbett et al., 2006). While there has been considerable work on the construction of supramolecular frameworks and architectures based on organoboronic acids; (Saleem et al., 2019; Chen et al., 2020) there has been significantly less work on the ability of boronic acids to form hydrogen-bonded complexes.
The evidence for the formation of hydrogen bonds and π-π stacking in boronic acids is substantial and its relevance at the solid state has been essentially limited to the self-associated architectures and data regarding the thermodynamics of the interactions with complementary recognition motifs in solution are essentially unknown (Shang et al., 2015; Zhao et al., 2016; Iribarren et al., 2019; Campos-Gaxiola et al., 2010; Rettig and Trotter, 1977; Cyrański et al., 2008). These interactions are among the most prevalent non-covalent interactions present in both natural systems and synthetic (Meyer et al., 2003; Burley and Petsko, 1985). These interactions play an important role in determining the conformation of organic molecules, (Hughes and Waters, 2006) the structure and stability of proteins, and directing the stereoselectivity of organic transformations (Pecsi et al., 2010).
To the best of our knowledge, we have investigated the ability of para-X-ba where: X = NO2, CN, F, Cl, Br, C(O)CH3, OCH3, OH, NH2 and H to enable reactivity of trans-1,2-bis(4-pyridyl)ethylene (bpe) to direct intermolecular [2 + 2] photodimerization via computational chemistry (Scheme 1). Para-X-ba would support the formation of discrete four-component hydrogen-bonded molecular assemblies wherein π-π stacking of a pyridyl-functionalized alkene would conform to undergo [2 + 2] photodimerization.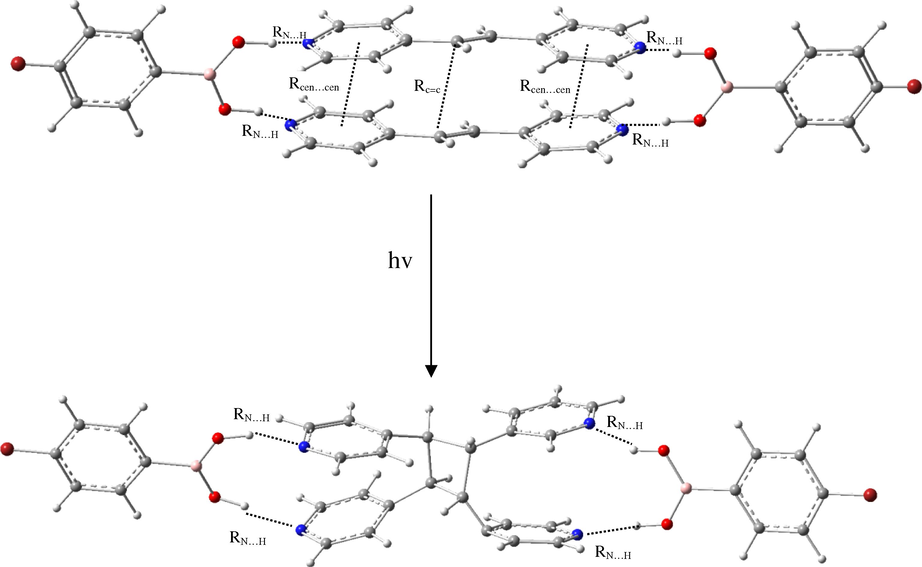
Molecular models used in the present study. (Red spherical = NO2, CN, C(O)OCH3, F, Cl, Br, H, OCH3 OH and NH2 substituents).
Our efforts to utilize the complexes based on para-X-ba to enable photoreactivity of bpe are inspired by work of MacGillivray et Al. (Alvarado et al., 2018) who experimentally demonstrated a propensity of related para-X-ba and bpe to assemble by a combination of π-π stacking contacts and hydrogen bond interactions. The work of MacGillivray et al. (Alvarado et al., 2018) prompted us to explore potential to employ para-X-ba to direct an intermolecular [2 + 2] photodimerization of bpe by computational chemistry.
2 Computational chemistry
To shed further light on the electronic and geometries properties ruling the formation and the stability of the (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes (X = NO2, CN, F, Cl, Br, C(O)CH3 , OCH3, OH, NH2 and H where || and ∙∙∙ denote π-π stacking and hydrogen bonds), DFT geometry optimization and frequency calculations were carried out at the M05-2X/6–311++G** level (Zhao and Truhlar, 2008) using the GAMESS program (Schmidt et al., 1993). The geometries have been optimized using the the gradient CP-corrected methods (Boys and Bernardi, 1970). Calculations were followed by harmonic vibrational frequencies calculations at the same level of theory and it confirms that the optimized structures have energetic minima on their potential energy surfaces.
In following, the 1HNMR calculations of the chemical shielding were performed for structure of the complexes. The chemical shielding of protons were computed at M05-2X/6–311++G** levels using SPINSPIN keyword. The shielding values were used to calculate the chemical shifts of hydrogen with respect to TMS (δH = σTMS − σH). Also, we employed the nucleus independent chemical shift (NICS) index that is one of the most widely employed indicators of aromaticity, which was proposed by Schleyer and co-workers (Ebrahimi et al., 2009a, 2009b; Ghafari and Gholipour, 2015, 2019; Schleyer and Jiao, 1996; Schleyer et al., 1996; Biegler Konig et al., 2001; Hammett, 1935; Zhu et al., 2003; Zhikol et al., 2005; Quinonero et al., 2008). It is defined as the negative value of the absolute shielding computed at a ring center.
The wave functions at the M05-2X/6–311++G** level were used to examine the electron density within the atoms in molecules (AIM) methodology by using the AIM2000 program (Biegler Konig et al., 2001).
3 Results and discussion:
We computationally demonstrated the ability of para-X-phenylboronic acid (para-X-ba) to assemble trans-1,2-bis(4-pyridyl)ethylene (bpe) into π-π stacks via –(B)O − H···N– hydrogen bonds in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes to react to afford rctt-tetrakis(4-pyridyl)cyclobutane (tpcb), (para-X-ba):::tpcb:::(para-X-ba) complexes (Scheme 1). We demonstrated para-X-ba in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes to act as a bifunctional hydrogen-bond-donor template by assembling bpe by way of –(B)O − H···N– hydrogen bonds in a discrete four-component supramolecular assembly to undergo an intermolecular [2 + 2] photodimerization. This photocycloaddition reaction generates (para-X-ba):::tpcb:::(para-X-ba) complexes. The components of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes as discrete four-component molecular assemblies sustained by four –(B)O − H···N– hydrogen bonds and π-π stacks of bpe have enabled the C = C bonds to be stacked in a suitable geometry for a [2 + 2] photodimerization (Scheme 1).
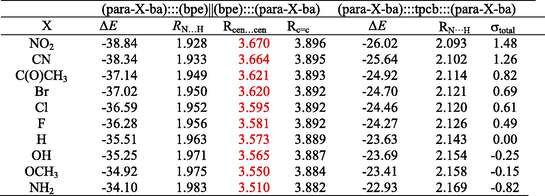
The results summarized in Table 1 allow us to analyze the electronic effects of substituents in terms of the Hammett constant (Hammett, 1935). Hammett constants of substituents demonstrated that electron-donating or electron-withdrawing substituent (X) influence π-π stacking the hydrogen bond interactions in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes. The electron-donating or electron-withdrawing capabilities of substituents which are including were describes using Hammett constant and the trend in electron withdrawing strength was found to be: NH2 < OH < OCH3 < H < F < Cl < Br < C(O)CH3 < CN < NO2.
The corresponding binding energies of complexes were evaluated using Eq. (1) and are also tabulated in Table 1.
The increase in ΔE arises in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes with the electron withdrawing substituents (see Table 1). Whereas ΔE is in the range from −38.84 to −34.09 kcal/mol for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and from −26.01 to –22.92 kcal/mol for (para-X-ba):::tpcb:::(para-X-ba) complexes. The binding energy from NH2 to NO2 is in the following order: NH2 < OCH3 < OH < H < F < Cl < Br < C(O)CH3 < CN < NO2. The binding energy is more as manifested in the largest ΔE values in –NO2 substituent compared to other substituents.
The lowest values ΔE are found for–NH2 substituent that is equal to −34.10 and –22.92 kcalmol−1 to (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively.
The constants σpara or σmeta are not effective parameters to describe intermolecular interactions in these complexes. Therefore, both para and meta positions, that are, σpara and σmeta, certainly have an effect on the binding energies so new parameter σtotal (σtotal = (σpara + σmeta)) was applied to describe these interactions (see Table 1) (Ghafari and Gholipour, 2015, 2019; Zhu et al., 2003; Ebrahimi et al., 2009). Increasing binding energies with electron withdrawing character of substituents show a meaningful correlation between the Hammett constant of substituents and the interaction energies of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes. We demonstrated the correlation between the Hammett’s parameter and the value of ΔE (Fig. 1).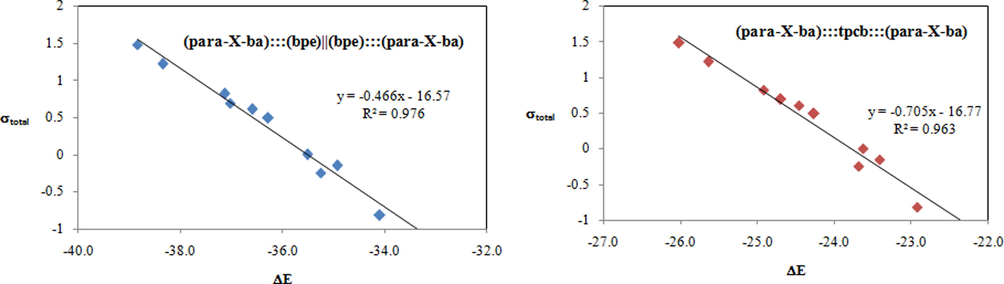
Correlation between ΔE and Hammett constant σtotal for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) (left) and (para-X-ba):::tpcb:::(para-X-ba) (right) complexes.
4 Geometrical parameters
The geometrical data of all the optimized complexes included in this study were gathered in Table 1 at the M05-2X/6–311++G** level of theory which is contained RN···H, Rcen···cen and RC=C geometrical parameters (see Scheme 1). Selected geometrical parameters for the related complexes are represented in Scheme 1. The RN···H, Rcen···cen and RC=C geometrical parameters are selected to derive the general trend of the relative strength of these interaction. RN…H is the -(B)O − H···N- intermolecular hydrogen bond distance between para-X-ba and bpe molecule. Rcen···cen is the distance between the center of pyridine rings in two bpe molecules;. RC=C is the distance between the C = C bonds stacked in two bpe molecules.
As it can be seen from Scheme 1, para-X-ba are connected to two bpe molecules through –(B)O − H···N– hydrogen bonds. Substitutions of the studied complexes result in essentially change in the RN…H hydrogen bond lengths (see Table 1). The four RN…H hydrogen bond distances of each substitute have almost the same length. The RN…H distances found range from 1.928 to 1.983 Å (mean value) for complexes (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and from 2.093 to 2.169 Å (mean value) for (para-X-ba):::tpcb:::(para-X-ba) complexes. As can be seen in Table 1, the RN···H for these complexes increases with increasing electron-donating character of substituents.
An electron-withdrawing substituent tends to decrease the RN…H in complexes in comparison with other substituents and this quantity increases through the incorporation of an electron- donating substituent. As depicted in Table 1, this observation signifies that RN…H for (X = NH2) complex is greater than any other complexes and this quantity in (X = NO2) complex is shorter than those of the corresponding complexes. The RN…H falls in the order NH2 > OCH3 > OH > H > F > Cl > Br > C(O)CH3 > CN > NO2. We demonstrated the correlation between the RN…H and the value of ΔE (Fig. 2).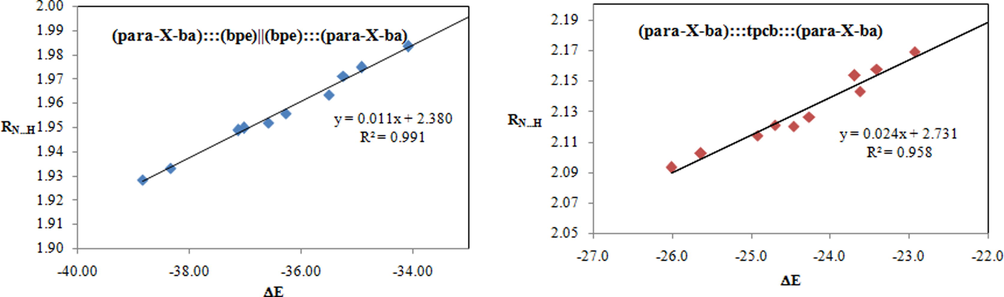
Correlation between ΔE and RN…H for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) (left) and (para-X-ba):::tpcb:::(para-X-ba) (right) complexes.
The information on the geometry (i.e. RN…H, Rcen···cen and RC=C) were important in the complexes, thus, Rcen···cen and RC=C of bpe molecules are suitable geometry parameters for strength of π-π stacking interaction. The π-π stacking of the bpe molecule would place the C = C bonds in positions to undergo intermolecular [2 + 2] photodimerizations in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes.
The Rcen···cen distances found range from 3.670 to 3.609 Å (mean value) (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes. Inspection of the data in Table 1 shows that the distant between center of pyridine rings in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes (Rcen···cen) is affected by the substituents. Noteworthy, Rcen···cen of the π-π stacking interaction can be reduced by the electron donating substituents, the lowest Rcen-cen is observed for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes (X = NH2) (see Table 1), also in agreement with the energy analysis, the trend in the Rcen···cen is: NH2 < C(O)CH3 < H < Br < F < Cl < OCH3 < OH < CN < NO2. Table 1 shows that (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes have shorter Rcen-cen which increases with increasing electron- withdrawing character of the substituents, and a reverse behavior is shown for electron- donating substituents. It is largest in (para-NO2-ba):::(bpe)||(bpe):::(para-NO2-ba) complex and smallest in (para-NH2-ba):::(bpe)||(bpe):::(para-NH2-ba) complex.
The RC=C distances found range from 3.896 to 3.882 Å (mean value) (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes. Inspection of the data in Table 1 shows that the distant between center of pyridine rings in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes (RC=C) is affected by the substituents. The lowest Rcen-cen is observed for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes (X = NH2) (see Table 1), also in agreement with the energy analysis, the trend in the RC=C is: NH2 < C(O)CH3 < H < Br < F < Cl < OCH3 < OH < CN < NO2. Table 1 shows that (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes have shorter RC=C which increases with increasing electron- withdrawing character of the substituents, and a reverse behavior is shown for electron- donating substituents. It is largest in (para-NO2-ba):::(bpe)||(bpe):::(para-NO2-ba) complex and smallest in (para-NH2-ba):::(bpe)||(bpe):::(para-NH2-ba) complex.
5 NMR spectroscopy
The NMR analysis will be carried out to understand chemical shielding of δN-H, δHa, δHb δHc, δHa′, δHb′ and δHf proton that gathered in Table 2 for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes with electron withdrawing and donating substituents at M05-2X/6–311++G** level of theory. The strength of the hydrogen bonding interactions was explained when setting the electron withdrawing and donating substituents by NMR data. For direct comparison of the 1H shielding values, we calculated the same for H atoms of tetramethyl silane (TMS) at the same level of theory (δH = σTMS - σH). For TMS protons, NMR shielding appears at 31.62 ppm.
X
(para-X-ba):::(bpe)||(bpe):::(para-X-ba)
(para-X-ba):::tpcb:::(para-X-ba)
δN-H
δHa
δHb
δHc
NICS(1)
δN-H
δHa′
δHb′
δHf
NICS(1)
NO2
8.77
7.90
7.46
7.68
−11.01
7.07
8.32
6.69
4.56
−10.82
CN
8.63
7.99
7.60
7.78
−10.75
6.76
8.35
6.66
4.53
−10.63
C(O)CH3
8.32
8.02
7.56
7.86
−10.21
6.43
8.38
6.62
4.56
−10.36
Br
8.26
8.05
7.54
7.73
−10.28
6.19
8.39
6.62
4.52
−10.14
Cl
8.26
8.08
7.56
7.76
−10.37
6.26
8.40
6.63
4.51
−10.29
F
8.12
8.91
7.46
7.66
−10.08
6.11
8.42
6.63
4.53
−10.02
H
8.13
9.46
7.83
7.75
−10.11
5.88
9.37
7.94
4.57
−9.84
OH
7.81
9.51
7.80
7.75
−9.61
5.63
8.52
8.00
4.52
−10.01
OCH3
7.94
9.51
7.78
7.75
−10.00
5.51
9.32
6.72
4.52
−9.93
NH2
7.68
8.97
7.40
7.63
−9.38
5.29
8.58
6.74
4.55
−9.63
The δN-H values are presented in Table 2 that have found range from 7.68 to 8.77 ppm (mean value) for complexes (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and 5.29–7.07 ppm (mean value) for (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively. The variation of δN-H can be characterized as the effect of the substituents by influencing –(B)O − H···N– bond that results in stabilization of hydrogen bonding (Table 2).
In order to study the influence of the substituents on the H shielding, the value of H shielding in substituted complexes has been calculated. This hydrogen bonding in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes manifests itself by a shielded N-H proton signal. The influence of the electron withdrawing and the electron-donating substituents on the δN-H is observed and presented in Table 2. The (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes with electron withdrawing substituents have a larger δN-H. In particular, the δN-H decreases for these complexes which bear an electron-donation substituent (Table 2). The largest magnitude of δN-H correspond to –NO2 electron donating substituent that is equal to 8.77 and 7.07 ppm in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively. We see from Table 2 that in fact δN-H is lowest for NH2 substituent with the values of 7.68 and 5.29 ppm for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively. The reduction of –(B)O − H···N– hydrogen bonds distances is also accompanied by increasing H shielding in complextion. The minimum value of H shielding is accompanied by the highest RN…H hydrogen bond lengths.
The δHa, δHb δHc, δHa′, δHb′ and δHf of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes were calculated by ab initio method at the M05-2X/6–311++G** level of theory (see Scheme 2), which shows features in reasonable agreement with experimental data (Alvarado et al., 2018). The δHa;δ Hb δHc, δHa′, δHb′ and δHf are also changed due to influence of the electron withdrawing and donating substituents that summarized in Table 2.
The δN-H, δHa, δHb δHc, δHa′, δHb′ and δHf of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes.
The nucleus independent chemical shift (NICS) index is one of the most widely employed indicators of aromaticity, which was proposed by Schleyer and co-workers (Chen et al.; Schleyer and Jiao, 1996; Schleyer et al., 1996). It is defined as the negative value of the absolute shielding computed at a ring center.
We have also studied the aromatic character of para-X-ba by means of NICS calculations. the aromaticity of the para-X-ba acid in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes changes when para-X-ba participate in hydrogen bonding interactions. There are observed changes in aromaticity of these complexes by predictions of NICS due to electron withdrawing and donating substituents. NICS(1) (1 Å above the plane of the ring) essentially reflects π effects. The electron withdrawing and donating effect of the substituent manifests itself by increasing and decreasing the aromaticity of para-X-ba ring in the (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes, it is apparent that X = NO2 in these complexes has the largest NICS(1). If we compare the NICS(1) of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes with different substituents, we can see that the –NH2 substituent have lowest one.
The NICS values are presented in Table 2 that have found range from −9.38 to −11.01 ppm for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes and from −9.63 to −10.82 ppm for (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively. We demonstrated the correlation between the NICS and the value of δN-H. (Fig. 3).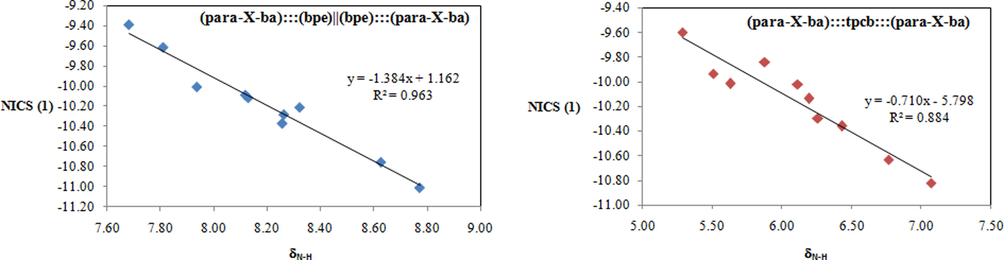
Correlation between δN-H and NICS(1) for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) (left) and (para-X-ba):::tpcb:::(para-X-ba) (right) complexes.
6 AIM analysis
To rationalize the observed trends, the electron density at bond critical points (ρBCP) and at cage critical points (ρCCP) of computed (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes were performed using the AIM analysis at M05-2X/6–311++G** level together with QTAIM that reported in Table 3. The effect of substituents on the hydrogen bonds was completely surveyed by the electron density at bond critical points ρBCP (Table 3). The ρBCP values are presented in Table 3 that have found range from 5.128 to 5.947 a.u (mean value) for complexes (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and from 2.799 to 3.445 a.u (mean value) for (para-X-ba):::tpcb:::(para-X-ba) complexes, respectively.
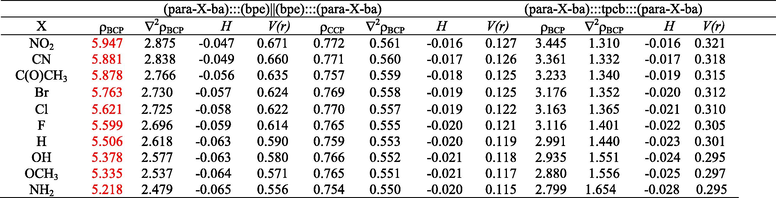
Comparison of ρBCP indicates that the largest magnitude of ρBCP occurred in the presence of –NO2 substituent and the lowest value of ρBCP is related to –NH2 complex, as given in Table 3. The ρBCP varies in the order: NH2 < OCH3 < OH < H < F < Cl < Br < C(O)CH3 < CN < NO2. This trend can be expected because the strength decreases from electron withdrawing to electron donating substituents.
The ρBCP was well correlated with the ΔE, in summary, as the electron density ρBCP increases, there is seen a significant enhancement in the binding energies ΔE. The ρBCP and interaction energy of these molecules also reveal the same trend (see Tables 1 and 3).
The ρBCP is at a maximum in NO2 substituent that presents the shortest -(B)O − H···N- hydrogen bonds distances, confirming that the ρBCP at the bond critical points is a good indicator of the strength of the interaction. In other words, the -(B)O − H···N- hydrogen bonds weakening can be reflected from a decrease in ρBCP.
The electron density at cage critical points (ρCCP) is in good correlation with strength of π-π stacking interaction which was previously reported (Ebrahimi et al., 2009; Zhikol et al., 2005; Quinonero et al., 2008). To explain the strength of π-π stacking interaction, the ρCCP was applied to give reliable results about strength and stabilization of this interaction. The increase in ρCCP is a direct consequence of the presence of electron withdrawing substituents, which predicts that electron withdrawing substituents have reinforced effects on (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes (Table 3). The lowest value of ρCCP is attributed to the NH2 and NO2 substituent exhibit largest values of ρCCP than the corresponding ρCCP for other substituents. The results also indicate that the NO2 substituent apparently enhances the π-π stacking interaction.
The results obtained for the Laplacian (∇2ρ), potential energy density, V(r) and the energy density (H) at the critical points (CPs) are evaluated by means of the AIM approach at the M05-2X/6–311++G** level for the studied complexes.
Moreover, we have found that the sign of the Laplacian of the electron density at the bond critical point (∇2ρ) and that of the energy density (H) could characterize the strength of hydrogen bonding interaction. If the classification of Rozas et al. (Rozas et al., 2000) has been corrected for individual interactions in hydrogen bonding interactions. Thus, weak hydrogen bonding show both ∇2ρBCP and HBCP > 0, and medium hydrogen bonding show ∇2ρBCP > 0 and HBCP < 0, while strong hydrogen bonding show both ∇2ρBCP and HBCP < 0. In these interactions, with regard to ∇2ρBCP > 0 and HBCP < 0 at BCPs in the complexes, they will be classified as medium hydrogen bonding interaction (see Table 3).
7 Conclusion
The computational work that was done in this study complements and extends the general research on the prediction of binding energy (Alvarado et al., 2018); chemical shielding and electron density of (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes.
It was found that the hydrogen bonds and π-π stacking in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes with electron-withdrawing substituents were generally observed better activities than the one with electron-donating substituents. Complexes involving electron withdrawing substituents obtained more favorable binding energy values than those involving electron donating substituents. (para-NO2-ba):::(bpe)||(bpe):::(para-NO2-ba) Complex involving NO2 obtained the largest interaction energy value of the study (-38.84 kcal/mol). On the contrary, (para-NH2-ba):::(bpe)||(bpe):::(para-NH2-ba) complex involving the NH2 substituent obtained the lowest interaction energy value of the study (−34.10 kcal/mol).
The hydrogen bond interaction was further proven by 1H NMR. A clear upfield shift (from δN-H = 7.68 to 8.77 ppm in (para-X-ba):::(bpe)||(bpe):::(para-X-ba) complexes) and (from δN-H = 5.29 to 7.07 ppm in (para-X-ba):::tpcb:::(para-X-ba) complexes) of the δN-H proton signal was observed after complextion with electron-withdrawing, indicating these substituents is making the hydrogen bond stronger. The system bearing the electron withdrawing substituents yields an increase of the corresponding δN-H. It was demonstrated that for these complexes with electron donating, the calculated shielding of the proton engaged in the hydrogen bonding δN-H was reduced. The ρBCP increases in the complexes having electron-donating or withdrawing substituents and the calculated ρBCP values suggest that –NO2 substituent has the maximum ρBCP for both (para-X-ba):::(bpe)||(bpe):::(para-X-ba) and (para-X-ba):::tpcb:::(para-X-ba) complexes, it is clear that this substituent is making the hydrogen bond stronger. The maximum value of H shielding is accompanied by the highest ρBCP hydrogen bond lengths (see Fig. 4).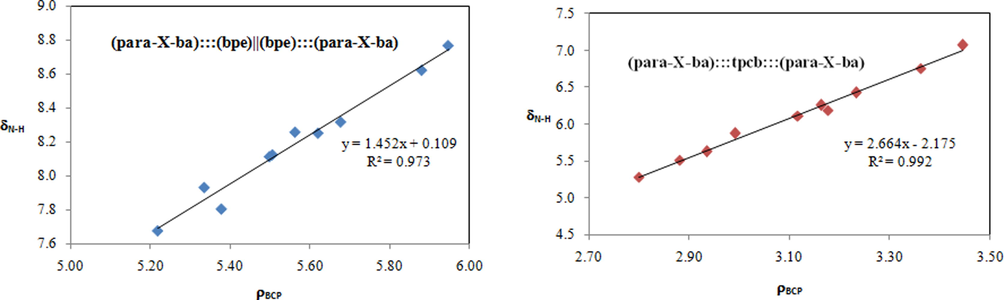
Correlation between δN-H and ρBCP for (para-X-ba):::(bpe)||(bpe):::(para-X-ba) (left) and (para-X-ba):::tpcb:::(para-X-ba) (right) complexes.
References
- Org. Lett.. 2018;20:5490-5492.
- J. Comput. Chem.. 2001;22:545-559.
- Mol. Phys.. 1970;19:553-566.
- Science. 1985;229:23.
- Cryst. Growth Des.. 2010;10:3182-3190.
- Arabian J. Chem.. 2020;13:2982-2994.
- Chen, Z., Wannere, C.S., Corminboeuf, C., Puchta, R., Schleyer, P.V.R., Chem. Rev.
- Chem. Rev.. 2006;106:3652-3711.
- Chem.- Eur. J.. 2016;22:4723-4726.
- J. Phys. Org. Chem.. 2008;21:472.
- Org. Lett.. 2017;19:5794-5797.
- J. Org. Chem.. 2019;84:1126-1138.
- Phys. Chem. Chem. Phys.. 2009;11:11424-11431.
- Theor. Chem. Acc.. 2009;124:115-122.
- Chem. Asian J.. 2008;3:1076-1091.
- J. Mol. Model.. 2015;21:253-259.
- Chem. Phys. Lett.. 2019;721:91-98.
- Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine. John Wiley & Sons; 2006.
- Hall, D.G., 2005. In Boronic Acids-Preparation and Applications in Organic Synthesis and Medicine. Hall, D.G., (Ed.), Wiley-VCH:Weinheim, pp. 1–26.
- Chem. Rev.. 1935;17:125-136.
- Curr. Opin. Struct. Biol.. 2006;16:514.
- Phys. Chem. Chem. Phys.. 2019;21:5796-5802.
- Beilstein J. Org. Chem.. 2016;12:391-405.
- Chem. Commun.. 2015;51:2005-2020.
- Science. 2017;356:7355-7362.
- Angew. Chem. Int. Ed.. 2003;42:1210.
- Chem. Commun.. 2011;47:1124-1150.
- Nucleic Acids Res.. 2010;38:7179.
- J. Elguero Chem. Phys. Lett.. 2008;460:406-410.
- Can. J. Chem.. 1977;55:3071.
- J. Am. Chem. Soc.. 2000;122:11154-11161.
- Arab. J. Chem.. 2019;12:800-815.
- Pure Appl. Chem.. 1996;68:209-218.
- J. Am. Chem. Soc.. 1996;118:6317-6318.
- J. Comput. Chem.. 1993;14:1347-1363.
- Nat Chem.. 2015;7:389-393.
- Phys. Chem. Chem. Phys.. 2016;18:14168-14171.
- Theor Chem Account. 2008;120:215-241.
- Zheng, H., McDonald, R., Hall, D.G., Chem. Eur. J. 16 (2010) 5454−5460.
- Angew. Chem. Int. Ed.. 2012;51:6187-6190.
- J. Chem. Phys.. 2005;122 144104-1–144104-144111
- J. Phys. Chem. A. 2003;107:2296-2303.




