

Translate this page into:
One-pot synthesis, X-ray crystal structure, and identification of potential molecules against COVID-19 main protease through structure-guided modeling and simulation approach
⁎Corresponding authors. yns.elbakri@gmail.com (Youness El Bakri), sosaid@alfaisal.edu (Souraya Goumri-Said)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Although antimicrobial resistance before the Covid-19 pandemic is a top priority for global public health, research is already ongoing on novel organic compounds with antimicrobial and antiviral properties in changing medical environments in connection with Covid 19. Thanks to the Biginelli reaction, which allows the synthesis of pyrimidine compounds, blockers of calcium channels, antibodies, antiviral, antimicrobial, anti-inflammatory, or antioxidant therapeutic compounds were investigated. In this paper, we aim to present Biginelli's synthesis, its therapeutic properties, and the structural–functional relationship in the test compounds that allows the synthesis of antimicrobial compounds. Both the DFT and TD-DFT computations of spectral data, molecular orbitals (HOMO, LUMO) analysis, and electrostatic potential (MEP) surfaces are carried out as an add-on to synthetic research. Hirshfeld surface analysis was also used to segregate the different intermolecular hydrogen bonds involved in the molecular packing strength. Natural Bond Orbital (NBO) investigation endorses the existence of intermolecular interactions mediated by lone pair, bonding, and anti-bonding orbitals. The dipole moment, linear polarizability, and first hyperpolarizabilities have been explored as molecular parameters. All findings based on DFT exhibit the best consistency with experimental findings, implying that synthesized molecules are highly stable. To better understand the binding mechanism of the SARS-CoV-2 Mpro, we performed molecular docking, molecular dynamics (MD) simulations, and binding free energy calculations.
Keywords
Dihydropyrimidinones
SARS-CoV-2 Mpro
Hirshfeld surface analysis
NBO analysis
Molecular docking

1 Introduction
In 1843, Pietro Biginelli was discovered. The reaction is based on the condensation of aldehydes, urea, and 1,3-dicarbonyl compounds in an acidic medium. As a result of the synthesis, heterocyclic systems of dihydropyrimidinones are formed, which play an important role in the construction of biomolecules such as DNA and RNA. In addition, in the fields of application, the synthesis and development of materials with optical properties, the design of polymers, dyes, and adhesives are also included. The most widely used area of biological impact is medicine. The multicomponent nature of the Biginelli reaction provides large product diversity bearing different pharmacophoric groups in the structure of dihydropyrimidines, which ensure optimal supramolecular interactions with a specific biological target (molecular recognition) and triggers or blocks its biological response. Investigations through molecular manipulations reveal that this class of compounds demonstrate antiviral, anti-filarial, antifungal, analgesic, anti-leishmanial, antiproliferative, antitumor, anti-convulsant, antibacterial, anti-inflammatory, anti-hypertensive, melanin-concentrating hormone 1 receptor antagonist, anti-HIV, antiepileptic, antidiabetic, anti-SARS, anti-malarial, pages-1 inhibitors, anti-hyperglycemic, antitubercular, TRPA1 antagonist, miscellaneous, potassium and calcium channels, and a1a adrenergic antagonists. Similarly, the structural core of quinoline is frequently associated with medicinal applications such as anticancer, antimicrobial, HIV-1 integrase inhibition, HIV protease inhibitors, antileishmanial activity, NK-3 receptor antagonists, PLT antagonists, and antimalarial activity. As a result of these studies, the use of various types of drugs such as riboflavin, idoxuridine, aminophylline, emivirine, 5-fluorouracil, methylthiouracil, batzelladine A, and B based on dihydropyrimidines has allowed widespread use in medicine. Due to the utmost importance of dihydropyrimidines in pharmaceutics, robust, efficient, cost-effective, and “green” chemical synthesis and transformations of dihydropyrimidine scaffolds were developed (Kaur et al., 2015; Nagarajaiah et al., 2016; Terracciano et al., 2015; Yadlapalli et al., 2012; Zhu et al., 2010).(See Scheme 1 Scheme 2.).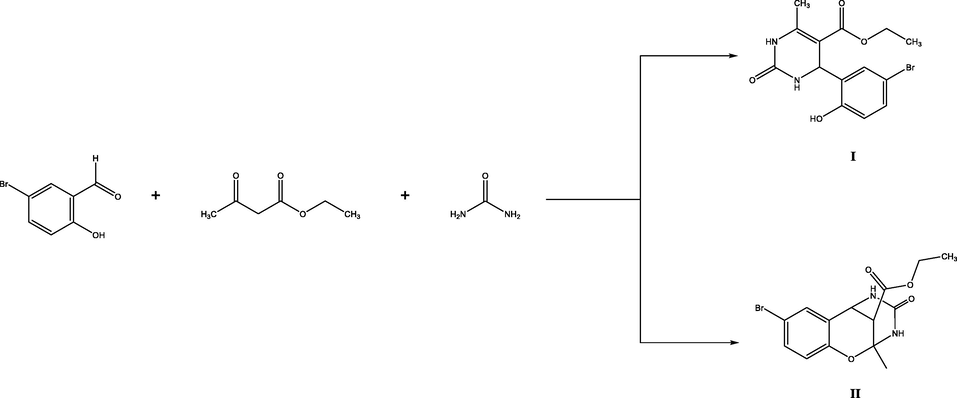
Synthesis of compounds (I) and (II).
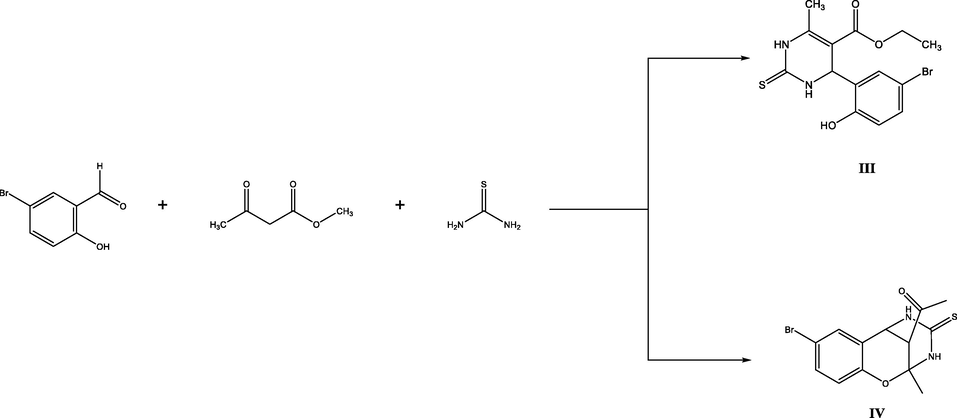
Synthesis of compounds (III) and (IV).
In the biological aspects of the motif, fixed molecules are needed that can easily be obtained for evaluation/study, and in 1930, the woolly-containing activity of these molecules was patented. These molecules bore similarities to the clinical spectrum of nifedipine. Biginelli researched by analogy with their analogs, and later it turned out that they showed the similarity of natural sea alkaloids with batzelladine B. All three building blocks are aldehyde, active methylene, and urea and are caused by the molecular diversity of dihydropyrimidines, which have a multicomponent reaction. The potent pyrimidine molecules produced by the Bignelli reaction have been successfully used and experimented with in stress and pollution-induced diseases. The following are selective molecules having significant activity, and they are examined with clinically used drugs in vivo/in vitro, establishing QSAR (Ashok et al., 2007; Chiang et al., 2009; Chitra et al., 2010; Deshmukh et al., 2009; Kidwai et al., 2005; Rajanarendar et al., 2010). There are several reasons that Biginelli's reaction has been studied in recent decades (Bais et al., 2020; de Fátima et al., 2015; Gireesh et al., 2013; Ismaili et al., 2008; Kaur et al., 2017; Li et al., 2020; Liu et al., 2019; Mokale et al., 2010; Rani et al., 2016; Sawant and Sarode, 2011; Silva et al., 2015).
As well as the inhibitory potential of antimicrobial and antibacterial pyrimidine derivatives has been evaluated against SARS-CoV-2 (Khan et al., 2021). Considering the importance of this class of compound, herein, two compounds are synthesized, characterized, and used in different computational approaches to unveil their anti-SARS-CoV-2 activity. Computational drug designing has been a good platform for predicting the binding capacity of compounds to different therapeutic targets and thus saves time and cost that are usually required in experimental testing. In this study, the docking studies are revalidated by more sophisticated molecular dynamics simulation and binding free energy analysis. In a nutshell, the findings obtained in this study could be useful for experimentalists to test the compounds against SARS-CoV-2 and in particular to be tested in enzymatic assay against the SARS-CoV-2 main protease enzyme.
2 Materials and methods
2.1 Instrumentation
The NMR spectra were recorded at 25 °C from solutions in DMSO‑d6 on a Bruker-300 spectrometer operating at 300 MHz. The IR spectra were obtained on a Specord 75IR instrument from samples dispersed in mineral oil. The progress of reactions and the purity of products were monitored by TLC.
2.2 X-ray diffraction study
Single crystals of compounds I and IV suitable for X-ray analysis were obtained by double recrystallization from ethanol (Kurbanova, 2010). The X-ray diffraction data were acquired on a Bruker APEX II CCD diffractometer (100 k), λMoKα irradiation, graphite monochromator, φ- and ω-scanning, 20max = 56°). The structures were solved by the direct method and refined by the least-squares procedure in an anisotropic approximation for non-hydrogen atoms. Hydrogen atoms in the hydroxy and amino groups were localized by the Fourier difference syntheses, and their positions were refined in an isotropic approximation with fixed positional and thermal parameters. The positions of the other hydrogen atoms were calculated based on geometry considerations and were refined in isotropic approximation with fixed positional and thermal parameters. All calculations were performed using the SHELXTL PLUS and SADABS software packages (Sheldrick, 2003, n.d.). The complete sets of crystallographic data for compounds I and IV were deposited at the Cambridge Crystallographic Data Centre (entry nos. CCDC 694,407 and CCDC 707349).
2.3 Computational studies
Computational studies of the compounds were performed to evaluate their binding affinity for different anti-SARS-CoV-2 targets. These targets include the SARS-CoV-2 main protease enzyme, RNA-dependent RNA polymerase (RdRp), and envelop (E) protein. The crystal structures of these targets were retrieved from protein data banks and underwent preprocessing in UCSF Chimera 1.15 (Pettersen et al., 2004). The PDB IDs used for retrieval of the proteins are the main protease enzyme (6y2e), RdRp (7D4F), and E (7M4R). During preprocessing, all co-crystalized ligands were removed, and the structures were relaxed using the steepest descent and conjugate gradient algorithms using default parameters.
2.4 Molecular docking and dynamics simulation studies
Molecular docking of the compounds with the targeted enzymes was conducted through AutoDock Vina software (Trott and Olson, 2010). Before that, the compound structure was drawn in ChemDraw 12.0 (Cousins, 2011) and processed into.pdb format. The proteins were also imported into the docking software. The grid box was set around the active site of the enzyme along a 15 Å on XYZ axis. The number of compounds docked was set at 100. Each compound conformation is associated with binding energy, and the one with the lowest score was complexed with the best-docked enzyme. Visualization of complexes was done in UCSF Chimera 1.15 (Kaliappan and Bombay, 2016) and Discovery studio v2021(Biovia, 2017). Further, the best-docked complex for both compounds was subjected to molecular dynamics simulation for 300 ns. Simulations were performed via AMBER20 software (Case et al., 2020). In the simulation, the protein parameters were prepared using ff14SB (Maier et al., 2015), while the compounds were parameterized using GAFF (He et al., 2020). Energy minimization of complexes was accomplished via steepest descent and conjugate gradient steps, each of which was run for 1000 cycles. Heating was achieved at 310 K for 100 ps. Equilibration was done for 200 ps. SHAKE and Langevin were used to constrain hydrogen bonds and maintain temperature during a production run, respectively (Izaguirre et al., 2001; Kräutler et al., 2001). CPPTRAJ was applied to analyze simulation trajectories (Roe and Cheatham III, 2013). MMPBSA.py module of AMBER was considered for MMGBSA and MMPBSA binding free energies estimation on 100 frames of simulation trajectories (Genheden and Ryde, 2015; Miller et al., 2012).
3 Results and discussion
3.1 Chemical synthesis
The Biginelli reaction of salicylaldehyde derivatives can lead to the formation of 2 types of dihydropyrimidines (Kurbanova, 2010). The reaction of 5-bromosalicylaldehyde with ethyl acetoacetate and urea in the presence of trichloroacetic acid gave, depending on the conditions, ethyl 4-(5-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (I) or ethyl 8-bromo-2-methyl-4-oxo-3,4,5,6-tetrahydro-2H-2,6-methanobenzo[g][1,3,5].
oxadiazocine-11-carboxylate (II). Ester, I was formed when the reactants were heated for 2–4 h in boiling ethanol, whereas bicyclic compound II was isolated when the reaction mixture was heated for 7–9 h at 40 °C.
A mixture of 0.025 mol of 5-bromosalicylaldehyde, 0.038 mol of ethyl acetoacetate, 0.025 mol of urea, and 25 mg of trichloroacetic acid in 10 ml of ethanol was stirred for 2–4 h on heating under reflux. The progress of the reaction was monitored by TLC. When the reaction was complete, the mixture was cooled to room temperature, and the precipitate was filtered off, washed with ethanol, dried, and recrystallized from aqueous ethanol.
The structure of compound I was proved by IR and NMR spectroscopy and X-ray diffraction data.
Ethyl 4-(5-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (I). Yield 75 %, mp = 190–191 °C. IR spectrum, v, cm−1: 3325, 1745, 1660. 1H NMR spectrum, v, ppm: 1.30 t (3H, CH2CH3), 2.57 s (3H, CH3), 4.25 q (2H, CH2O), 5.70 s (1H, CH), 7.70–7.00 m (4H, Harom), 8.00 s (1H, NH), 9.41 s (1H, NH), 10.11 s (1H, OH).(See Fig. 1).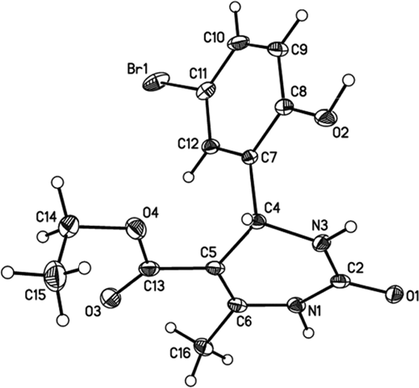
Structure of I according to the X-ray diffraction data.
Ethyl 8-bromo-2-methyl-4-oxo-3,4,5,6-tetrahydro-2H-2,6-methanobenzo[g][1,3,5]
oxadiazocine-11-carboxylate (II) Yield 55 %, mp = 197–199 °C. IR spectrum, v, cm−1: 3350, 1740, 1680. 1H NMR spectrum, v, ppm: 1.27 t (3H, CH2CH3), 2.35 s (3H, CH3), 3.27 d (1H, CH), 4.16 q (2H, CH2O), 4.46 d (1H, CH), 6.77–7.21 m (3H, Harom), 8.36 s (1H, NH), 8.41 s (1H, NH).
X‐ray crystallographic analysis was performed by double crystallization of compound I in ethyl alcohol (Kurbanova et al., 2009). The monoclinic, yellow crystals, with sizes 0.20 × 0.20 × 0.20 mm3, one striped: a = 9.3207(2), b = 16.841(2), c = 10.0908(13) Å, β = 116.678(1)˚, V = 1415.3(3) Å3, space group P21/n, Z = 4, ds = 1.667 q/sm3, μ = 2.922 mm−1 were obtained.
For comparison, three-component condensation of 5-bromosalicylaldehyde with acetylacetone and thiourea was examined. Under analogous conditions, the products were l-[4-(5-bromo-2-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl]-ethanone (Ill) and 1-(8-bromo-2-methyl-4-thioxo-3,4,5,6-tetrahydro-2H-2,6-methanobenzo[g][1,3,5].
oxadiazocin-11-yl)ethan-1-one (IV).
A mixture of 0.025 mol of 5-bromosalicylaldehyde, 0.038 mol of acetylacetone, 0.025 mol of thiourea, and 25 mg of trichloroacetic acid in 10 ml of ethanol was stirred for 2–4 h on heating under reflux. The progress of the reaction was monitored by TLC. When the reaction was completed, the mixture was cooled to room temperature, and the precipitate was filtered off, washed with ethanol, dried, and recrystallized from aqueous ethanol.
Compound IV was isolated as a crystalline substance and its structure was proved by IR and NMR spectroscopy and confirmed by X-ray analysis.
1-[4-(5-Bromo-2-hydroxypheny1)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl[ethanone (III).
Yield 75 %, mp = 175 °C. IR spectrum, v, cm−1: 3345, 1665. 1H NMR spectrum, o, ppm: 1.90 s (3H, CH3), 2.47 s (3H, CH3), 5.57 s (1H, CH), 7.10 s (1H, NH), 7.36–7.45 m (4H, Harom), 8.14 s (1H, NH), 9.50 s (1H, OH).
1-(8-bromo-2-methyl-4-thioxo-3,4,5,6-tetrahydro-2H-2,6-methanobenzo[g][1,3,5]
3.2 oxadiazocin-11-yl)ethan-1-one (IV).
Yield 55 %, mp = 187–188 °C. IR spectrum, v, cm−1: 3350, 1670. 1H NMR spectrum, o, ppm: 1.79 s (3H, CH3), 2.31 s (3H, CH3), 3.33 d (I H, CH), 4.59 d (IH, CH), 6.84–7.20 m (3H, Harom), 8.97 s (1H, NH), 9.12 s (1H, NH).
X‐ray crystallographic analysis was performed by double crystallization of compound IV in ethyl alcohol. The monoclinic, yellow crystals, with sizes 0.20 × 0.20 × 0.30 mm3, one striped: a = 16.845(3), b = 11.374(2), c = 14.680(3) Å, β = 103.822(2)˚, V = 2731.2(9) Å, space group C2/c, Z = 8, dc = 1.660 q/sm3, μ = 3.162 mm−1 were obtained.
3.3 Hirshfeld surface analysis and 2D fingerprint plots
With the Hirshfeld surface, it is possible to witness intermolecular interactions and fashions in crystal packing (Maity et al., 2018). The compounds I and IV were subjected to the Hirshfeld surface mapping across the normalized contact distance dnorm shown in Fig. 4. The color schemes shown in the map are red, blue, and white. The red (dnorm is negative) and blue (dnorm is positive) colored regions on the surface are from shorter and longer contacts than van der Waals radii respectively, whereas the white areas indicate the van der Waals radii of the contacts, or dnorm = 0 (McKinnon et al., 2007). In the brighter red zone, hydrogen bonding is responsible for the strong interactions, but electron density in the blue zone represents the weaker interactions. Qualitative measures of morphological structure, such as the “shape index,” can be sensitive to even the tiniest changes in surface curvature (or curvedness). The concave region with red triangles in the shape index of atoms of a molecule represents the C—H…π and π…π stacking interactions, while convex regions with blue triangles represent aromatic ring atoms inside the surface. For compounds I and IV, massive green flat areas with blue outlines on the relatively curved surfaces were observed due to the π…π stacking interactions as shown in Fig. 3.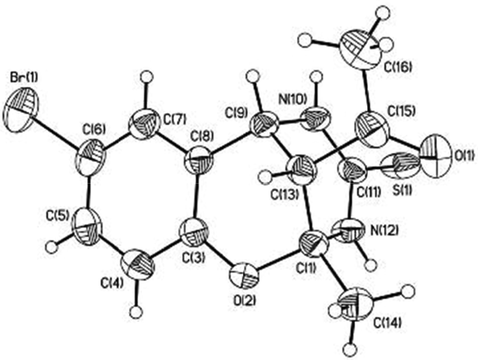
Structure of IV according to the X-ray diffraction data.
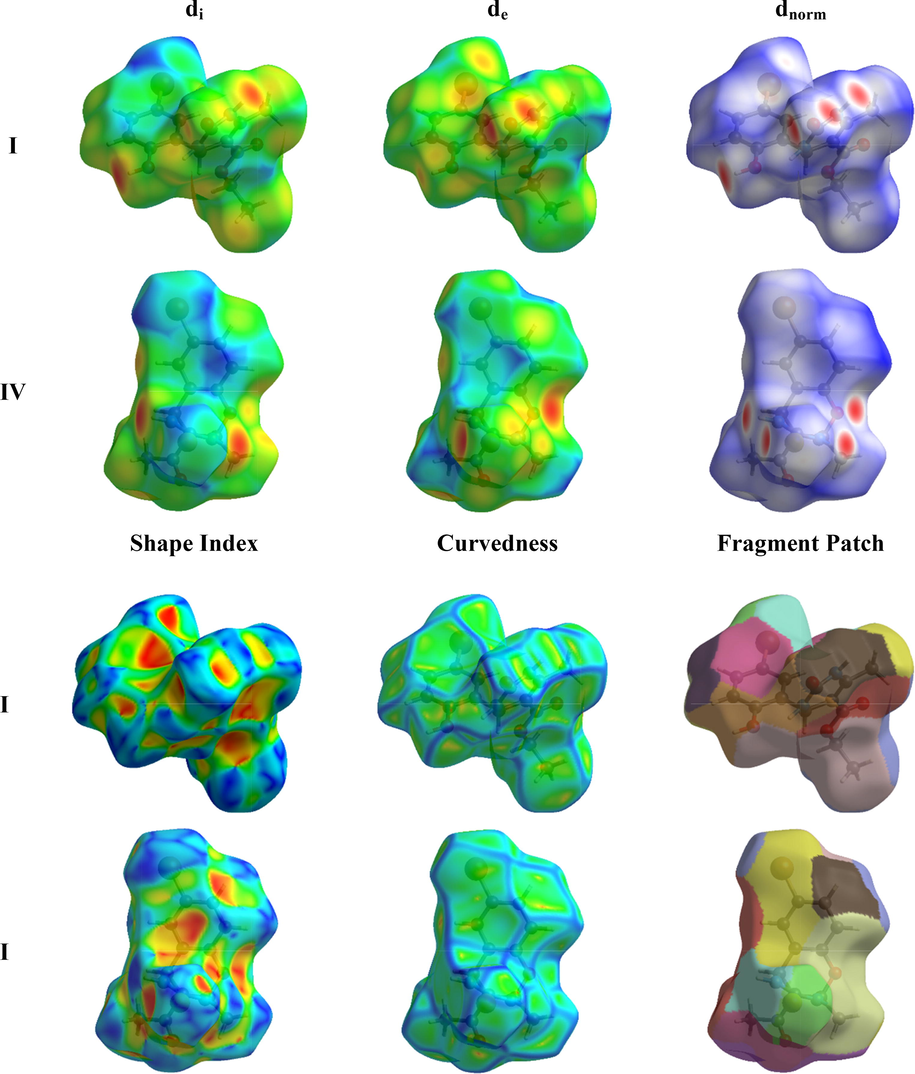
Hirshfeld surface mapped of di, de, dnorm, curvedness, shape index, and fragment patch of compounds I and IV.
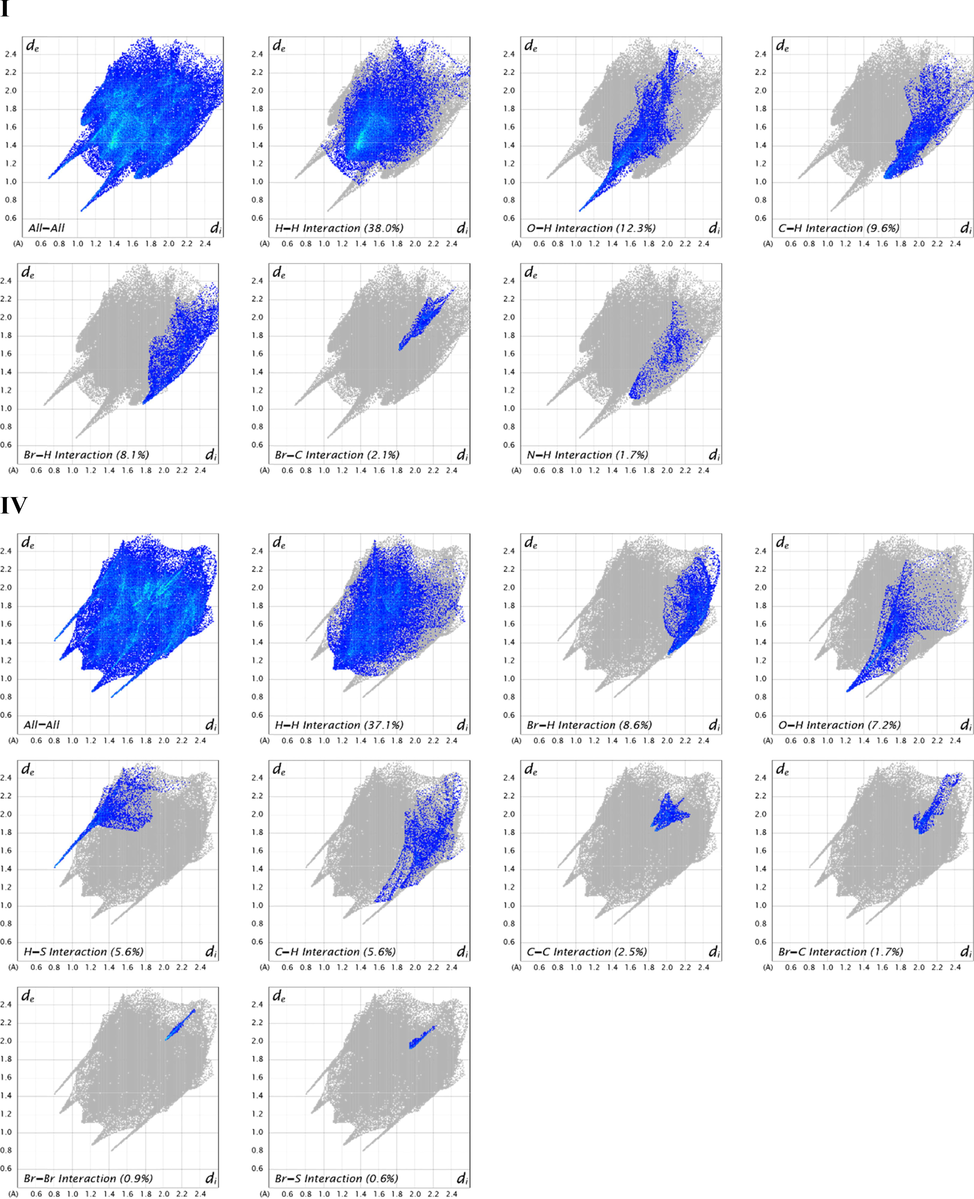
2D fingerprint plots of I and IV displaying contacts for specific pairs of atoms contributing majorly.
The intermolecular percent contribution of all kinds of contacts to the overall Hirshfeld surface area was calculated using 2D fingerprint plots (Seth, 2018). Fig. 4 shows the 2D fingerprint of all the contacts. The major contribution of both I and IV is from H—H contacts, 38.0 %, and 37.1 % respectively, to the total Hirshfeld surface area. The remaining contributions of C1 are from O—H, C—H, Br-H, Br-C, and N—H contacts, which contribute about 12.3 %, 9.6 %, 8.1 %, 2.1 %, 1.7 % respectively to the Hirshfeld surface area. The remaining contributions of C2 are from Br-H, O—H, H—S, C—H, C—C, Br-C, Br-Br, and Br-S contacts, which contribute about 8.6 %, 7.2 %, 5.6 %, 5.6 %, 2.7 %, 1.7 %, 0.9 %, 0.6 % respectively to the total Hirshfeld surface area. Spikes arise in the fingerprint plots showing greater intermolecular interactions in the crystal packing of both I and IV.
3.4 3D energy frameworks analysis
Energy framework analysis reveals the quantitative study of interaction energies and supramolecular design of molecules in a crystal. A cluster of molecules within 3.8 A˚ of the radius was built around a single I and IV compound. The scale factors used for developing the energy framework for B3LYP/6-31G(d,p) electron densities are k_ele = 1.057, k_pol = 0.740, k_disp = 0.871, k_rep = 0.618 (Edwards et al., 2017). Table 1 summarizeso the computed interaction energies in kJ/mol and the symmetry operations for eight molecules of I. The blue-colored molecule positioned at 10.44 A˚ has the greatest total interaction energy (-74.7 kJ/mol), whereas the green-colored one positioned at 10.09 A˚ has the lowest total interaction energy (0.9 kJ/mol). Similarly, as shown in Table 1, the computed energy interactions and symmetry operations for seven IV molecules are also given in kJ/mol.
I
N
Symmetry Operation
R
Electron Density
Eele
Epol
Edis
Erep
Etot
1
x, y, z
9.32
B3LYP/6-31G(d,p)
−4.3
−1.0
−20.1
16.4
−12.7
0
-x + 1/2, y + 1/2, -z + 1/2
8.49
B3LYP/6-31G(d,p)
−6.9
−1.5
−26.8
18.4
−20.5
0
-x, -y, -z
12.42
B3LYP/6-31G(d,p)
−0.4
−0.2
−7.2
0.0
−6.9
0
x, y, z
10.09
B3LYP/6-31G(d,p)
6.0
−1.3
−6.8
2.5
0.9
1
x + 1/2, -y + 1/2, z + 1/2
8.60
B3LYP/6-31G(d,p)
−12.8
−2.6
−25.7
30.7
−18.8
0
-x, -y, -z
10.44
B3LYP/6-31G(d,p)
−77.2
−20.0
−17.6
59.9
−74.7
1
x + 1/2, -y + 1/2, z + 1/2
5.63
B3LYP/6-31G(d,p)
−13.2
−3.4
−60.8
46.1
−40.9
0
-x, -y, -z
10.13
B3LYP/6-31G(d,p)
−2.2
−1.0
−8.1
2.6
−8.5
IV
1
-x, -y, -z
6.85
B3LYP/6-31G(d,p)
−40.8
−10.5
−54.1
69.8
−54.8
1
x, -y, z + 1/2
9.4
B3LYP/6-31G(d,p)
−15.9
−5.1
−13.3
15.6
–22.5
1
x, y, z
11.37
B3LYP/6-31G(d,p)
−0.3
−0.1
−4.4
2.2
−2.9
1
x + 1/2, y + 1/2, z
10.16
B3LYP/6-31G(d,p)
−2.5
−2
−12
4.8
−11.6
1
-x + 1/2, y + 1/2, -z + 1/2
8.73
B3LYP/6-31G(d,p)
−10.7
−4.4
−20.5
14
–23.7
1
-x, y, -z + 1/2
4.89
B3LYP/6-31G(d,p)
−9.7
−5.7
−69
46.6
−45.8
1
-x, -y, -z
6.53
B3LYP/6-31G(d,p)
−10.7
−2.1
−35.3
29
−25.7
1
-x + 1/2, -y + 1/2, -z
10.24
B3LYP/6-31G(d,p)
0.5
−0.2
−2.4
0.1
−1.7
0
-x + 1/2, -y + 1/2, -z
7.87
B3LYP/6-31G(d,p)
−105.8
–22.3
−19.1
116
−73.4
1
x + 1/2, -y + 1/2, z + 1/2
9.76
B3LYP/6-31G(d,p)
1.5
−0.7
−5.7
1.3
−3.1
S-axis is shown in Fig. 5. The Coulomb interactions in I look to be comparable in strength as R values are closer for both. The calculated interaction energies for electrostatic, polarization, dispersion, and repulsion of I are −111.0, −31.0, −173.1, and 176.6 kJ/mol, respectively. The calculated interaction energies for electrostatic, polarization, dispersion, and repulsion of C2 are −194.4, −53.1, −235.8, and 299.4 kJ/mol, respectively. The total energies of I and IV are −182.1 and −265.2 kJ/mol respectively. The dispersion energy has the highest value among all interaction energies in I and IV, i.e., the dispersion interaction energy dominates over the electrostatic Coulomb interaction energy. This is because of the presence of the bromo group, which has a large electron cloud in each compound. The more electrons an atom or molecule has (the most polarizable), the stronger dispersion forces are. The scale factors for benchmarked energies used for the construction of energy models were taken from Mackenzie et al. (Mackenzie et al., 2017).
Molecular pairs involved in the calculation of interaction energies of I and IV along the b-axis.
The visualization of different interaction energies like Coulomb interaction energy, dispersion energy, and total interaction energy is represented by red, green, and blue colors, respectively, for the compounds, I and IV along different axes as shown in Fig. 6. The cylinders in the energy framework represent the relative strengths of molecular packing in several directions. An overall scale factor is used to contract or expand the size of the cylinders in the framework (Turner et al., 2015). There is an absence of cylinders in a particular direction due to the exclusion of a few interactions under certain threshold energy. These weaker interactions have been ignored only to make the figures less crowded.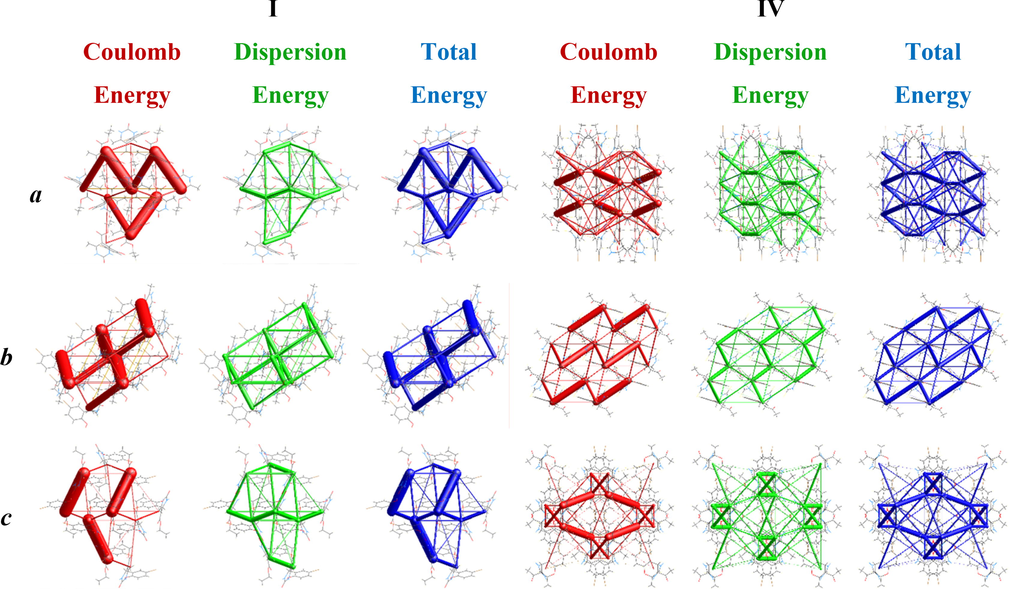
The graphical representation of electrostatic interactions: Coulomb interaction energy (red), dispersion energy (green), and total interaction energy (blue) of I and IV along a, b, and c axes.
3.5 Structure optimization
The synthesized compounds (I and IV) are optimized at the B3LYP/6-31G(d,p) level of theory using Gaussian 16 (Frisch et al., 2016) together with GuassView 6.0 (Dennington et al., 2016) and their fully optimized geometries are presented in Fig. 7.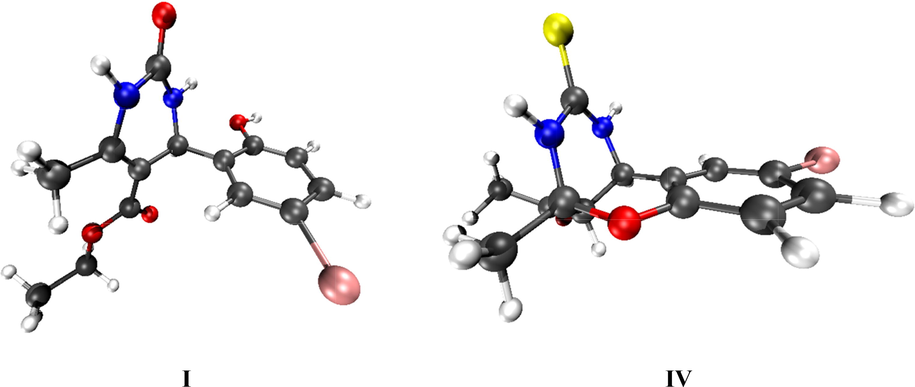
Optimized geometries of I and IV compounds at B3LYP/6-31G(d,p) level of theory.
3.6 Frontier molecular orbital (FMO) analysis
The FMO’s of all compounds calculated at B3LYP/6-31G (d,p) DFT level are given in Fig. 8 along with values in Table 2.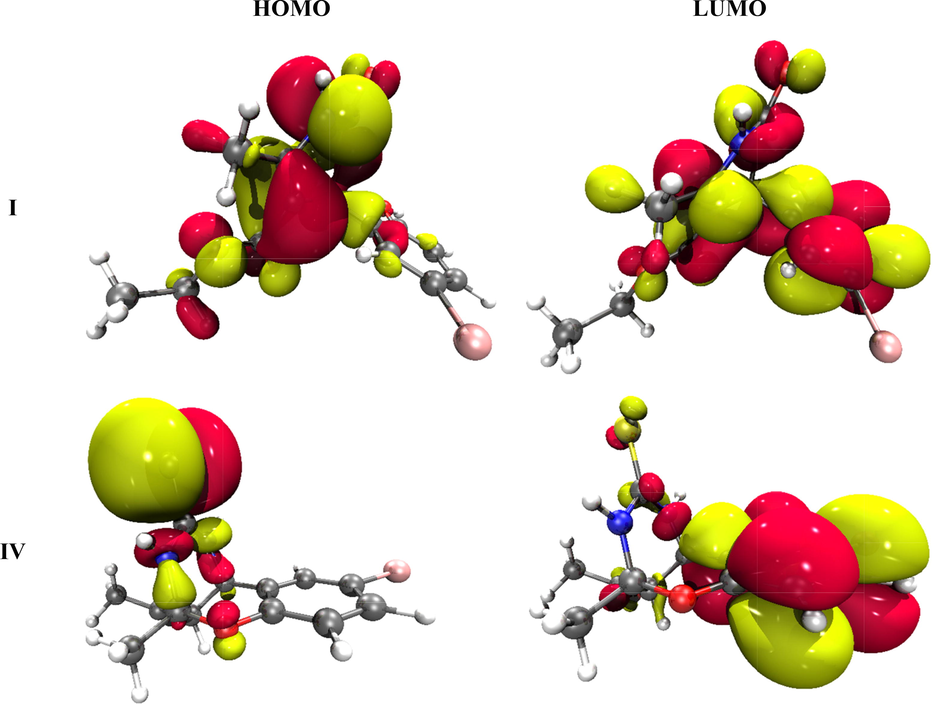
The HOMO and LUMO pictorial representation of I and IV synthesized compounds.
Compounds
EHOMO (eV)
ELUMO (eV)
ΔE = ELUMO – EHOMO (eV)
I
−6.095
−1.045
5.050
IV
−5.806
−1.137
4.668
Compounds
Ionization Potential
Electron Affinity
Electronegativity
Hardness
Chemical Potential
Electrophilicity
e- donor capability
e- acceptor capability
softness
I (eV)
A (eV)
χ (eV)
η (eV)
μ (eV)
ω (eV)
ω- (eV)
ω+ (eV)
S (eV)
I
7.674
−0.578
3.548
4.126
−3.548
1.526
3.815
0.267
0.121
IV
7.592
−0.488
3.552
4.040
−3.552
1.561
3.842
0.290
0.124
The values of energy gaps for compounds I and IV are very close with a minor difference (5.050, 4.668 eV) as shown in Table 2. The compound IV shows a smaller ΔE value of 4.668 eV due to the greater π-conjugation than the I compound. Contrarily, the highest ΔE value of 5.050 eV is seen in I. Thus, the HOMO-LUMO energy gap shows the following increasing order for all compounds: IV < I. The graphical diagrams are shown in Fig. 8 where red and yellow colors indicate the negative and positive phases of molecular orbitals, respectively.
3.7 Global reactivity descriptors
The ionization potential and electron affinity values in Table 3 illustrate that compound, I, show greater ionization potential than electron affinity revealing greater electron donating ability in comparison to the accepting nature. Overall, the electron affinity values are positive for both the compounds and a good gesture for their use in charge transfer reactions. The global electrophilicity values reveal that the electron donor capability (ω-) of these compounds are larger than the electron accepting capability (ω + ). Similarly, the compound IV shows greater softness value followed by I with a minor difference. The electronegativity decreasing order observed among I and IV compounds is as IV > I. The synthesized compounds are harder due to large values of chemical hardness than the lower values of global softness.
These findings reveal a high degree of chemical stability and little reactivity. With the increase in the chemical potential (μ), chemical stability increases while reactivity decreases. The decrease in chemical potential for both the compounds is in the order: [IV (μ = -3.552) > I (μ = -3.548)]. This order indicates the lowest chemical potential (μ) for the compound I making it kinetically less stable and more reactive. Similarly, the molecules with smaller softness are less stable and more reactive.
The results of the global reactivity parameters show both compounds have a higher donating potential, more stability, and a poorer accepting ability and thus are appropriate to participate in charge transfer reactions. Moreover, the purpose of performing DFT computation of these compounds is to endorse experimental studies, cross-check experimental findings, as well as to evaluate stability, reactivity, intramolecular or intermolecular charge transfer, and other parameters are suitable for predicting potential photovoltaic characteristics/biological activities (Pardasani et al., 2003; Pasha et al., 2007). Table 3 presents the global reactivity descriptors and accepting and donating aptitude of all compounds and endorses the relation with photovoltaic characteristics.
3.8 UV–visible study
To observe the electronic transitions, UV–vis spectroscopy is a powerful method, which also estimates the charge transfer probability in a compound and assignments of molecular orbitals corresponding to the transition (Siddique et al., 2021). Both, experimental and theoretical tools are used to study the UV–Visible transitions. The time-dependent DFT (TD-DFT) is performed at B3LYP functional in conjunction with the 6-31G(d,p) basis set.
Table 4 represents the absorption maxima, oscillator strength (f), and molecular assignments. Both experimental and theoretical absorption values are close to each other with minute differences. The compound I shows maximum absorption at 267.3 nm with an oscillator strength of 0.217 which is nearly equal to the experimental λmax of 267.4 nm. Compounds IV shows absorption maxima at 247.8 nm bearing a very small difference with oscillator strengths of 0.210. Overall, the computed UV–vis studies are in fine contract with experimental absorption values. The HOMO to LUMO transition is the characteristic of photovoltaic materials, ΔE gap and chemical hardness indicate molecules are a little bit harder requiring a large amount of energy for reactivity which is an indication of kinetic stability. The biological charge transfer and photovoltaic capability of C1 and C2 are in the order IV > I making them potential candidates.
DFT λmax (nm)
Exp. λmax (nm)
f (oscillator strength)
MO contributions
267.3
267.4
0.217
H-1 → L (86 %) H-2 → L (4 %), H-1 → L + 1 (3 %), H → L (2 %)
247.8
248.9
0.210
H-2 → L + 1 (53 %), H-1 → L + 2 (12 %), H → L + 2 (27 %)
3.9 Molecular electrostatic potential (MEP) analysis
The 3D representation of electron density on all compounds is explored via MEP analysis following equation (1).
Here, V(r) is molecular electrostatic potential, ZA is charge density over the nucleus, A placed at RA, p(r’) defines the electronic density function, and r’ is the integration variable (Muthu and Prabhakaran, 2014; Okulik and Jubert, 2005).
The blue and red colors are the indications of nucleophilic and electrophilic centers, respectively. The magnitude of electrostatic potential is increased in order of red < white < blue (Mahalakshmi and Balachandran, 2015). The electrophilic attack is easy on the red site while the blue site facilitates the nucleophilic attack. The 3D-MEP analysis is performed at B3LYP/6-31G(d,p) functional of all synthesized compounds and pictorial representation is given in Fig. 9. The oxygen, nitrogen, and sulfur atoms show negative potential (blue color), while mostly hydrogen and partially carbon and bromine atoms exhibit positive potential (red color). The white color indicates the mean potential being between two boundaries. The red and blue colors represent the different reaction sites.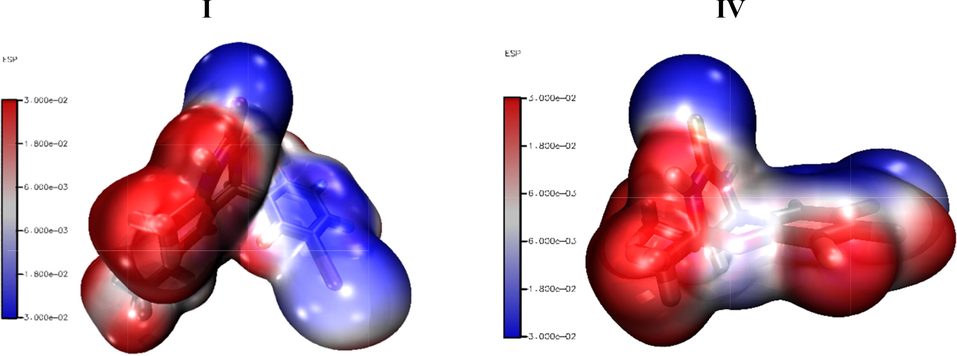
The 3D MEP representation of I and IV at B3LYP/6-31G(d,p) level of DFT.
3.10 NBO analysis
The charge transfer interactions within the bonds are studied using the natural bond orbital (NBO) analysis. The estimate of the off-diagonal NBO Fock matrix elements F(i,j) is given by this interaction energy. This could be calculated using the second-order perturbation method (Foster and Weinhold, 1980; Glendening et al., 1998). The one-center lone pairs and two-center bonds from NBO analysis show a precise representation of chemical bonding for a stable molecular species, which corresponds to a single Lewis structure. The non-Lewis set involves unoccupied valence nonbonding (LP∗) and extra-valence-shell Rydberg (RY∗) orbitals also with the valence anti-bonds (BD∗). The shortfall of the Lewis-type NBOs (bonds and lone pairs) denoting the density matrix can be computed with the occupancy of these NBOs. The delocalization energy, Eij, is calculated as:
Where E(2) is the energy of hyper conjugative interactions, qi is the occupancy of the contributing (Lewis type) orbital, εi and εj are the energies of the donating and accepting orbitals, and Fij is the off-diagonal element of the Fock matrix in the NBO basis (Frisch et al., 2000; Kleinman, 1962). An NBO analysis has been carried out to define the interior of the intramolecular hybridization and delocalization of electron density within the molecule.
Intermolecular hyper conjugative interactions are formed by overlapping the orbitals between the bonds of I (C—H), (C—O), (N—H), (C—C) and IV (C—H), (C—O), (C—N), (C—S), (C—C) and anti-bonding of I (C—C), (C—O), (C—N), (C–Br), (N—C) and IV (C—C), (C—S), (C—N), (C—O), (N—C), (C—H) orbitals, which result in intramolecular charge transfer (ICT) stabilizing the molecular system and leading to strong delocalization. The strong intramolecular hyper conjugative interactions of the electrons of I (C—H), (C—O), (N—H), (C—C), and IV (C—H), (C—O), (C—N), (C—S), (C—C) and anti-bonding of I (C—C), (C—O), (C—N), (C–Br), (N—C) and IV (C—C), (C—S), (C—N), (C—O), (N—C), (C—H) bonds of the ring lead to stabilization of some part of the ring as given in Table 5. For example, in I, the intermolecular hyper conjugative interaction of σ (N2–H22) distributes to σ* (C1–N6) and in IV σ (C15–S16) distributes to σ* (C15–S16), leading to stabilization of 21.129 KJ/mol and 10.91 KJ/mol, respectively.
I
Donor (i)
Acceptor (j)
E(2) kJ/mol
E(2) kcal/mol
E(j)-E(i) a.u.
F(i,j) a.u.
π* C13 - C14
π* C12 - C17
845.796
202.15
0.02
0.083
LP (2) O9
π* C7 - O8
189.703
45.34
0.33
0.112
LP (1) N2
π* C1 - O10
182.632
43.65
0.33
0.109
LP (2) O8
σ* C7 - O9
139.369
33.31
0.62
0.13
π C4 - C5
π* C7 - O8
98.742
23.6
0.29
0.077
π* C1 - O10
σ* C1 - O10
56.2748
13.45
0.49
0.167
LP (3) Br19
π* C15 - C16
38.618
9.23
0.3
0.052
σ N2 - H22
σ* C1 - N6
21.129
5.05
1.05
0.066
σ* C7 - O9
σ* C3 - C4
20.83632
4.98
0.04
0.05
σ C14 - C15
σ* C16 -Br19
20.502
4.9
0.79
0.056
σ C11 - H25
π* C4 - C5
20.334
4.86
0.55
0.049
σ N6 - H24
σ* C1 - N2
15.397
3.68
1.14
0.059
π C1 - O10
π* C1 - O10
8.619
2.06
0.48
0.031
π* C12 - C17
σ* N2 - C3
8.28432
1.98
0.31
0.052
σ C20 - H33
RY*(2) O9
2.803
0.67
1.63
0.029
IV
π* C1 - C6
π* C2 - C3
204.94
0.01
0.08
857.469
LP (1) N14
σ* C15 - S16
52.70
0.25
0.107
220.497
LP (2) O8
π* C4 - C5
28.53
0.34
0.094
119.370
π C4 - C5
π* C1 - C6
23.24
0.28
0.073
97.236
σ* C15 - S16
π* C15 - S16
14.47
0.27
0.109
60.542
LP (2) S16
σ* N13 - C15
11.09
0.62
0.075
46.401
σ C15 - S16
σ* C15 - S16
10.91
0.29
0.057
45.647
LP (3) Br7
π* C1 - C6
9.53
0.3
0.052
39.874
σ C19 - H32
π* C17 - O18
6.66
0.52
0.053
27.865
σ C11 - N14
π* C15 - S16
3.35
1
0.052
14.016
π* C4 - C5
σ* C11 - N14
3.31
0.3
0.059
13.849
σ C4 - O8
RY*(1) C9
2.7
1.84
0.063
11.297
π* C2 - C3
RY*(5) C2
1.79
1.14
0.1
7.489
π C17 - O18
σ* C19 - H32
1.66
0.81
0.033
6.945
σ C17 - C19
RY*(1) C10
1.41
1.63
0.043
5.899
This enhanced further conjugation with an anti-bonding orbital of I π (C4–C5) and IV π (C4–C5) which led to a strong delocalization of 98.742 KJ/mol and 23.24 KJ/mol with π∗ (C7–O8) in C1 and π* (C1–C6) in IV, respectively. The most important highest energy, related to molecule I, is electron-donating from π* (C13–C14) to the anti-bonding acceptor π* (C12–C17) and for C2, electron-donating from π* (C1–C6) to the anti-bonding acceptor π* (C2–C3) with stabilization energies of 845.796 and 204.94 KJ/mol, respectively. The magnitude of charge transferred in C1 from LP (2) O9 to the anti-bonding π* (C7–O8) and LP (1) N2 to the anti-bonding π* (C1–O10) shows that the stabilization energy is 6.38 KJ/mol and 10.89 KJ/mol, respectively. Similarly, in C2, the charge transferred from LP (1) N14 to the anti-bonding σ* (C15–S16) and LP (2) O8 to the anti-bonding π* (C4–C5) with stabilization energies of 52.70 and 28.53 KJ/mol, respectively.
3.11 Optical properties
Nonlinear optical materials may find use in optoelectronic devices, networking, signal manipulation, telecommunications, and other fields. The strength of the donor–acceptor group, as well as the sequence in which they are placed, affects the amount of nonlinear optical efficiency. To put it another way, organic NLO materials have a push–pull conjugated structure because of the π-electron conjugated system they contain, bearing electron donor and acceptor groups on different ends. The present work has been extended to explore the optical properties of the I and IV complexes for their remarkable utilization in optoelectronic. The optoelectronics properties include dipole moment (µtot), polarizability (αtot), and first-order hyperpolarizability (βtot) (Kurtz et al., 1990) and are determined by the following equations:
The first hyperpolarizability is a third rank tensor with a 3 × 3 × 3 matrix, and the 27 components of the 3D matrix are reduced to 10 components due to the Kleinman symmetry. The dipole moment, linear polarizability, and nonlinear first hyperpolarizability of the I and IV are listed in Table 6. The dipole moments of I and IV are 1.543 D and 4.504 D, respectively. The polarizabilities of I and IV are 188.2 au and 186.1 au, respectively. The results clearly show that the largest component of polarizability is along the axial direction, whereas perpendicular components contribute minimally, indicating that the molecule is optically active in the X-direction. The first-order hyperpolarizabilities of I and IV are 526.5 au and 427.6 au. The greater hyperpolarizability of I than IV is mainly attributed to the greater intramolecular charge transfer between different atoms of I. Besides this, the NLO response of I and IV is larger than the reference molecule urea (Kanis et al., 1994), which has a total value of 43 au, indicating that these complexes can be potential NLO candidates for optoelectronic applications.
I
IV
Dipole moment (Debye)
Hyperpolarizability (au)
Dipole moment (Debye)
Hyperpolarizability (au)
Parameter
Value
Parameter
Value
Parameter
Value
Parameter
Value
µx
1.543
βxxx
15.49
µx
0.000
βxxx
73.29
µy
0.000
βyyy
29.25
µy
0.000
βyyy
24.19
µz
0.000
βzzz
479.7
µz
4.504
βzzz
133.1
µtot
1.543
βxyy
50.63
µtot
4.504
βxyy
32.06
Polarizability (au)
βxxy
87.27
Polarizability (au)
βxxy
−114.8
αxx
191.6
βxxz
−17.42
αxx
226.9
βxxz
106.1
αxy
7.690
βxzz
229.7
αxy
−20.52
βxzz
77.00
αxz
−15.34
βyzz
−69.17
αxz
−15.14
βyzz
−94.06
αyy
202.2
βyyz
−29.24
αyy
161.1
βyyz
100.6
αyz
−8.241
βxyz
66.00
αyz
11.33
βxyz
−53.14
αzz
170.7
βtot
526.5
αzz
170.3
βtot
427.6
αtot
188.2
αtot
186.1
3.12 Molecular docking study
The docking was conducted to examine the binding affinity of compounds with different targets of the SARS-CoV-2 virus. Of the different targets used, it was found the main protease enzyme was the best-docked enzyme for the compounds used, as shown in Table 7. Compound I had binding energy of −12.67 kcal/mol for the SARS-CoV-2 main protease enzyme, while compound IV had binding energy of −11.24 kcal/mol. Both the compounds dock well inside the binding cavity of the enzyme and are engaged by hydrophilic and hydrophobic interactions. The compound I formed hydrogen bonds with Asn142, Ser144, and Gly143. Similarly, compound IV formed hydrogen bonds with Phe 140, Gly143, and Cys145. The binding mode and chemical interactions of the compounds with the enzyme are given in Fig. 10.
Compound
Main Protease Enzyme
RdRp Enzyme
E Protein
Compound I
−12.67
−6.35
−7.65
Compound IV
−11.24
−5.65
−6.65
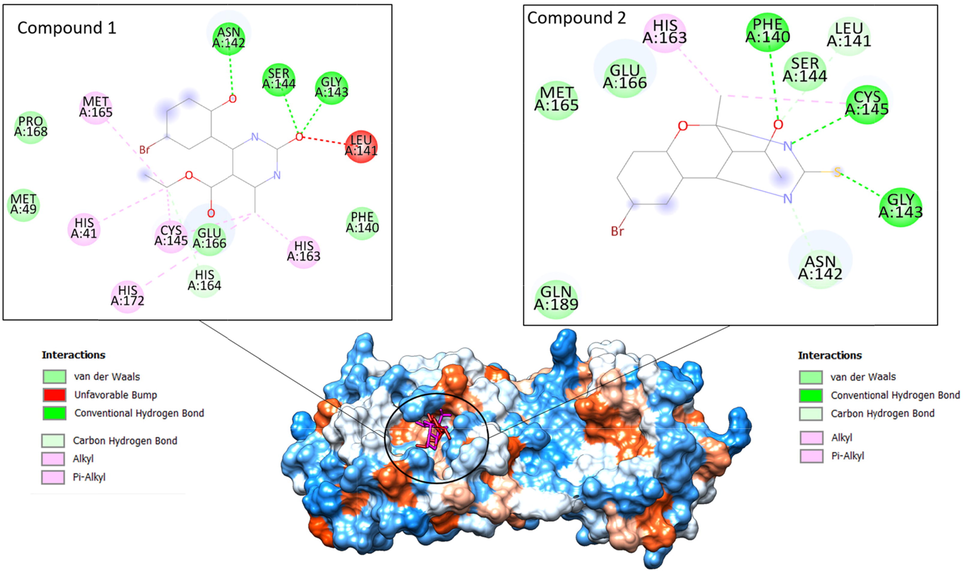
Binding of the compounds at substrate binding cavity of SARS-CoV-2 main protease enzyme. Compound I is shown by a red stick while compound IV is pink. The enzyme is on hydrophobicity surface. The binding interactions of compounds are also given.
3.13 Molecular dynamics simulation
As the compounds showed stronger binding affinity with the SARS-CoV-2 main protease enzyme compared to others, only main protease enzyme compounds best docked complexes were subjected to molecular dynamics simulation analysis. According to the molecular dynamic simulations, compound I is more dynamically stable compared to compound IV. The root means square deviation (RMSD) reached a maximum level of 2 Å. The compound IV, on the other hand, showed significant deviation (up to 7 Å) in the first 100 ns. Followed that, the complex was found very stable towards the end of the simulation. The residue-wise fluctuations were measured via the root mean square fluctuation (RMSF), which indicated that in the presence of compound I, the enzyme remained very stable compared to compound IV. The RMSD and RMSF of compounds are given in Fig. 11.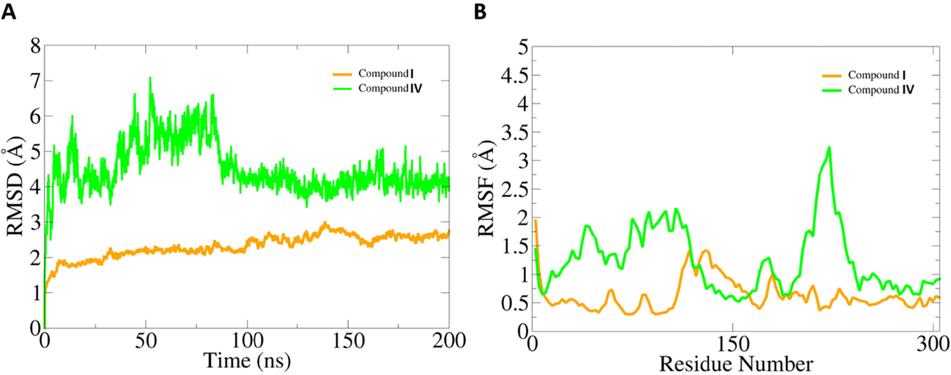
Dynamics investigation of compounds with SARS-CoV-2 main protease enzyme. A. RMSD and B. RMSF. Both values are measured in Å.
3.14 MMPB\GBSA analysis
Different binding free energies of the compounds with the enzyme active site residues were also estimated. Again, compound I was ranked as the best-docked molecule with the main protease enzyme. The binding free energies of compounds are tabulated in Table 8. The net binding free energy of compound I is −59 kcal/mol in MMGBSA and −59.553 kcal/mol in MMPBSA. The energy value of compound I is better than that of compound IV, which has a net energy of −46.69 kcal/mol in MMGBSA and net energy of −41.03 kcal/mol in MMPBSA.
Parameter
Compound I Complex
Compound IV Complex
MMGBSA
Van der Waals Energy
−55.36
−43.88
Electrostatic Energy
−25.41
−19.18
Polar Solvation Energy
35.13
29.03
Non-Polar Solvation Energy
−13.36
−12.66
Net Energy
−59
−46.69
MMPBSA
Van der Waals Energy
−55.36
−43.88
Electrostatic Energy
−25.41
−19.18
Polar Solvation Energy
33.497
32.04
Non-Polar Solvation Energy
−12.28
−10.04
Net Energy
−59.553
−41.03
4 Conclusions
In this paper, we carried out several computation analyses to treat test dihydropyrimidines due to the possible benefits that can hit today’s prominent pandemic problem.
Furthermore, from the DFT theoretical calculations, the optimized geometrical parameters are analyzed and are in good agreement with the XRD results. We found that the HOMO-LUMO energy gap affects the stability of compounds. Similarly, NBO and natural atomic charge analyses were performed to explore the stabilization energy of different inter- and intramolecular interactions within the systems and the net charge distribution, respectively. Insights from the DFT study of these compounds reveal their good stability by using other analyses like global indices of reactivity and molecular electrostatic potential analysis. Molecular docking and MD simulations were used to assess the binding affinity and stability of these drugs to the binding site of the SARS-CoV-2 receptor. Through molecular docking studies with various types of interactions, substantial binding affinity for both examined compounds was established. Following the computational techniques, the substrates were subjected to MD simulations to measure the rigidity of the protein structure, fluctuations caused by interactions, and overall structural stability by calculating RMSD.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Convenient one-pot synthesis of some novel derivatives of thiazolo [2, 3-b] dihydropyrimidinone possessing 4-methylthiophenyl moiety and evaluation of their antibacterial and antifungal activities. Eur. J. Med. Chem.. 2007;42:380-385.
- [Google Scholar]
- One pot synthesis of micromolar BACE-1 inhibitors based on the dihydropyrimidinone scaffold and their thia and imino analogues. Molecules. 2020;25:4152.
- [Google Scholar]
- Discovery studio visualizer. CA, USA: San Diego; 2017.
- Case, D.A., Belfon, K., Ben-Shalom, I., Brozell, S.R., Cerutti, D., Cheatham, T., Cruzeiro, V.W.D., Darden, T., Duke, R.E., Giambasu, G., others, 2020. Amber 2020.
- Select pyrimidinones inhibit the propagation of the malarial parasite, Plasmodium falciparum. Bioorg. Med. Chem.. 2009;17:1527-1533.
- [Google Scholar]
- Synthesis, Spectral, Anti-Liver Cancer and Free Radical Scavenging Activity of New Azabicyclic Thienoyl Hydrazone Derivatives. Eur. J. Med. Chem. 2010;45:367-371.
- [Google Scholar]
- Cousins, K.R., 2011. Computer review of ChemDraw ultra 12.0.
- de Fátima, Â., Braga, T.C., Neto, L. da S., Terra, B.S., Oliveira, B.G.F., da Silva, D.L., Modolo, L. V, 2015. A mini-review on Biginelli adducts with notable pharmacological properties. J. Adv. Res. 6, 363–373
- Dennington, R., Keith, T.A., Millam, J.M., 2016. GaussView 6.0. 16. Semichem Inc. Shawnee Mission. KS, USA.
- A novel and efficient one step synthesis of 2-amino-5-cyano-6-hydroxy-4-aryl pyrimidines and their anti-bacterial activity. Eur. J. Med. Chem.. 2009;44:2651-2654.
- [Google Scholar]
- Intermolecular interactions in molecular crystals: what’s in a name? Faraday Discuss.. 2017;203:93-112.
- [Google Scholar]
- Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Petersson, G.A., Nakatsuji, H., 2016. Gaussian 16.
- Gaussview user manual. Pittsburgh, PA: Gaussian Inc.; 2000. p. :556.
- The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov.. 2015;10:449-461.
- [CrossRef] [Google Scholar]
- Synthesis of sydnone substituted Biginelli derivatives as hyaluronidase inhibitors. Arch. Pharm. (Weinheim). 2013;346:645-653.
- [Google Scholar]
- Glendening, E.D., Reed, A.E., Carpenter, J.E., Weinhold, F., 1998. NBO Version 3.1. Google Sch. There is no Corresp. Rec. this Ref.
- Fast, accurate, and reliable protocols for routine calculations of protein–ligand binding affinities in drug design projects using AMBER GPU-TI with ff14SB/GAFF. ACS Omega. 2020;5:4611-4619.
- [Google Scholar]
- Synthesis and antioxidant activity evaluation of new hexahydropyrimido [5, 4-c] quinoline-2, 5-diones and 2-thioxohexahydropyrimido [5, 4-c] quinoline-5-ones obtained by Biginelli reaction in two steps. Eur. J. Med. Chem.. 2008;43:1270-1275.
- [Google Scholar]
- Kaliappan, S., Bombay, I.I.T., 2016. UCSF Chimera-Superimposing and Morphing.
- Design and construction of molecular assemblies with large second-order optical nonlinearities. Quant. Chem. Asp. Chem. Rev.. 1994;94:195-242.
- [Google Scholar]
- Recent synthetic and medicinal perspectives of dihydropyrimidinones: a review. Eur. J. Med. Chem.. 2017;132:108-134.
- [Google Scholar]
- Primaquine–pyrimidine hybrids: synthesis and dual-stage antiplasmodial activity. Eur. J. Med. Chem.. 2015;101:266-273.
- [Google Scholar]
- Novel pyrimidine-benzimidazole hybrids with antibacterial and antifungal properties and potential inhibition of SARS-CoV-2 main protease and spike glycoprotein. Digit. Chinese Med.. 2021;4:102-119.
- [Google Scholar]
- Synthesis of 4-aryl-7, 7-dimethyl-1, 2, 3, 4, 5, 6, 7, 8-octahydroquinazoline-2-one/thione-5-one derivatives and evaluation as antibacterials. Eur. J. Med. Chem.. 2005;40:816-819.
- [Google Scholar]
- A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem.. 2001;22:501-508.
- [Google Scholar]
- Bicyclic compounds obtained by the Biginelli reaction. Russ. J. Org. Chem.. 2010;46:599-601.
- [Google Scholar]
- Crystal structures of ethyl-4-(5-bromo-2-hydroxyphenyl)-6-methyl-2-oxo-1, 2, 3, 4-tetrahydropyrimidine-5-carboxylate and ethyl-1-methyl-15-oxo-2-oxa-14, 16-diazatetracyclo [11.3. 1.03. 12.06. 11] heptadeca-3, 5, 7, 9, 11-pentaene-17-carboxylate. J. Struct. Chem.. 2009;50:505-509.
- [Google Scholar]
- Calculation of the nonlinear optical properties of molecules. J. Comput. Chem.. 1990;11:82-87.
- [Google Scholar]
- Synthesis, characterization, and anticancer activities evaluation of compounds derived from 3, 4-dihydropyrimidin-2 (1H)-one. Molecules. 2019;24:891.
- [Google Scholar]
- CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ. 2017;4:575-587.
- [Google Scholar]
- NBO, HOMO, LUMO analysis and vibrational spectra (FTIR and FT Raman) of 1-Amino 4-methylpiperazine using ab initio HF and DFT methods. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2015;135:321-334.
- [Google Scholar]
- ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput.. 2015;11:3696-3713.
- [Google Scholar]
- Quantifying conventional C-H⋯ π (aryl) and unconventional C–H⋯ π (chelate) interactions in dinuclear Cu (II) complexes: experimental observations, Hirshfeld surface and theoretical DFT study. New J. Chem.. 2018;42:10202-10213.
- [Google Scholar]
- Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007:3814-3816.
- [Google Scholar]
- MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput.. 2012;8:3314-3321.
- [CrossRef] [Google Scholar]
- Synthesis and anti-inflammatory activity of some 3-(4, 6-disubtituted-2-thioxo-1, 2, 3, 4-tetrahydropyrimidin-5-yl) propanoic acid derivatives. Bioorg. Med. Chem. Lett.. 2010;20:4424-4426.
- [Google Scholar]
- Vibrational spectroscopic study and NBO analysis on tranexamic acid using DFT method. Spectrochim. Acta Part A Mol. Biomol. Spectrosc.. 2014;129:184-192.
- [Google Scholar]
- Theoretical analysis of the reactive sites of non-steroidal anti-inflammatory drugs. Internet Electron. J. Mol. Des.. 2005;4:17-30.
- [Google Scholar]
- Theoretical and synthetic approach to novel spiroheterocycles derived from isatin derivatives and L-proline via 1, 3-dipolar cycloaddition. Heteroat. Chem. An Int. J. Main Gr. Elem.. 2003;14:36-41.
- [Google Scholar]
- Quantitative structure activity relationship (QSAR) study of estrogen derivatives based on descriptors of energy and softness. Chem. Biol. Drug Des.. 2007;70:520-529.
- [Google Scholar]
- UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem.. 2004;25:1605-1612.
- [Google Scholar]
- Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3, 6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2, 3, 6, 10b-tetrahydro-1H-pyrimido [5, 4-c] quinolin-5-ones. Bioorg. Med. Chem. Lett.. 2010;20:6052-6055.
- [Google Scholar]
- Biological potential of pyrimidine derivatives in a new era. Res. Chem. Intermed.. 2016;42:6777-6804.
- [Google Scholar]
- PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput.. 2013;9:3084-3095.
- [Google Scholar]
- Synthesis, spectral characterization and analgesic activity of 2-methylthio-1, 4-dihydropyrimidines. Iran. J. Pharm. Res. IJPR. 2011;10:733.
- [Google Scholar]
- Structural characterization and Hirshfeld surface analysis of a CoII complex with imidazo [1, 2-a] pyridine. Acta Crystallogr. Sect. E Crystallogr. Commun.. 2018;74:600-606.
- [Google Scholar]
- SHELXTL. Version 6.14. Structure Determination Software Suite. Madison (WI, USA): Bruker AXS; 2003.
- Sheldrick, G.M., n.d. SHELXTL, v. 6.12, Structure Determination Software Suite,(2001) Bruker AXS. Madison, Wisconsin, USA
- Efficient tuning of zinc phthalocyanine-based dyes for dye-sensitized solar cells: a detailed DFT study. RSC Adv.. 2021;11:27570-27582.
- [Google Scholar]
- The Biginelli reaction under batch and continuous flow conditions: catalysis, mechanism and antitumoral activity. RSC Adv.. 2015;5:48506-48515.
- [Google Scholar]
- Structural insights for the optimization of dihydropyrimidin-2 (1 H)-one based mPGES-1 inhibitors. ACS Med. Chem. Lett.. 2015;6:187-191.
- [Google Scholar]
- AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem.. 2010;31:455-461.
- [Google Scholar]
- Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun.. 2015;51:3735-3738.
- [Google Scholar]
- Synthesis and in vitro anticancer and antitubercular activity of diarylpyrazole ligated dihydropyrimidines possessing lipophilic carbamoyl group. Bioorg. Med. Chem. Lett.. 2012;22:2708-2711.
- [Google Scholar]
- 2, 4-Diaryl-4, 6, 7, 8-tetrahydroquinazolin-5 (1H)-one derivatives as anti-HBV agents targeting at capsid assembly. Bioorg. Med. Chem. Lett.. 2010;20:299-301.
- [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.104230.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1
Supplementary data 2
Supplementary data 2




