

Translate this page into:
Partial hydrogenation of 1,3-butadiene over nickel with alumina and niobium supported catalysts
⁎Corresponding author. kausiva@uaeu.ac.ae (Kaushik Sivaramakrishnan),
⁎⁎Corresponding author. mn.Altarawneh@uaeu.ac.ae (Mohammednoor Altarawneh)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
The hydrogenation of 1,3 butadiene (BD) is an important industrial process that is generally used to generate butenes and aromatics, which are important petrochemicals and constituents of transportation fuels. Ni-based catalysts have gained increasing interest in the academic community and industry because of their superior hydrogenation activity, ability to exist in multiple oxidation states and most importantly, affordability compared to the less-abundant precious metal catalysts. In this work, we explore the ability of alumina (Al2O3) and niobia (Nb2O5) as catalytic supports for Ni as the active metal since they have shown excellent potential for hydrogenation of alkenes. Specifically, we compare the metal-support interactions, which disperse Ni effectively throughout the catalyst surface, textural and pore parameters, Ni crystallite size, surface morphology, elemental composition, coke formation and tendencies of both catalysts through extensive characterization techniques such as H2-temperature programmed reduction (H2-TPR), N2-adsorption–desorption, X-ray diffraction (XRD), scanning electron microscopy combined with electron-dispersive spectra (SEM-EDS), and thermal analysis of the spent catalysts. Limited reports on the use of niobium oxides and alumina as supports for Ni in the partial hydrogenation of BD prompted us to explore this literature gap. The catalytic reactions were conducted using a quartz tube reactor between a temperature range of 50 and 400 °C at regular intervals of 50 °C under operating pressures of 1 atm. A comprehensive analysis of the products from the reaction was carried out using gas-chromatography with mass spectrometry (GC–MS) detector and corroborated these results with those from infrared spectroscopy (FTIR). Reaction mechanisms of aliphatic and aromatic product formation were proposed and verified with literature and our own characterization results. The tests on the spent catalysts also helped to reveal the extent of deactivation of the catalysts due to carbon deposition. Mechanisms of catalyst deactivation and nature of carbonaceous deposits were explored and elaborate scientific reasoning was provided as to why Ni/alumina showed better performance in terms of product yields and BD conversion. Ways to mitigate the harmful effects of coke formation and its deposition are also discussed. The novelty of our work is that a comprehensive comparison of the performance of Ni/alumina and Ni/niobia for hydrogenation of BD along with extensive discussions on coke formation, its chemical nature and proposed reaction chemistry based on obtained characterization results of the catalysts and the products of BD hydrogenation has not been done before for this system. In this way, we show the low-temperature conversion of BD represents an economically efficient and relatively simple method to convert toxic, carcinogenic and environmentally hazardous BD into high-value products using low-cost and chemically efficient catalysts.
Keywords
1,3-butadiene
Infrared spectroscopy
Heterogeneous catalysis
catalyst characterization
metal-support interactions
partial hydrogenation

1 Introduction
With an annual production of ∼19 million tons in 2022, which is 3 times the production in 2007, and an average annual growth rate of > 5 % in the next 5 years, 1,3-butadiene is an important bulk chemical in the petrochemical industry (Berndt and Böge, 2007; Butadiene et al., 2023). Being the simplest conjugated diene, 1,3-butadiene (BD) is an important component in the manufacture of rubber, plastics, resins and other simple and complex polymers (Bonnevide et al., 2020). It has been reported in petroleum literature that ∼95 % of BD is acquired as a side product of steam cracking of kerosene-type hydrocarbons while automobile exhausts contribute to ∼80 % of BD emission in USA (Ghosh et al., 2010). BD can be formed as an undesirable chemical by-product with harmful effects that promote catalyst poisoning due to its polymerization ability (White, 2007). Furthermore, open burning contributes to ∼20 % BD emissions while industrial emissions result in atmospheric concentrations up to 10 ppb. (Vimal et al., 2008) BD poses a number of threats to human beings as well as the environment such as: (i) being a known carcinogen and mutagen due to its epoxide metabolites (Kramp and Paulson, 2000); (ii) 30 times higher risk factor than benzene, (Duffy and Nelson, 1997) and (ii) being classified as a Hazardous Air Pollutant by the Environmental Protection Agency (EPA) continuously under the Clean Air Act for the past few decades and last updated in December 2022 (Initial List of Hazardous Air Pollutants with Modifications, 2023). Even the storage of BD can become dangerous because of its high reactivity and tendency to undergo uncontrolled polymerization reactions, resulting in explosions when exposed to air for long periods of time. Frequent temperature fluctuations can also affect this process (Liang et al., 2022). On the academic and industrial scale, oligomerization and polymerization of BD poisons catalysts that are used with olefin conversions. Hence, there was a need to find an alternative way to convert toxic BD to value-added products to reduce its exposure to humans and the environment and reduce its impact on catalyst poisoning. This formed the motivation for our work in this paper and we decided to hydrogenate BD to obtain more stable and useful products.
The partial hydrogenation of butadiene is a chemical reaction of BD with hydrogen (H2) to mainly produce both linear and branched butenes. Aromatics show up as additional by-products due to various mechanisms such as cyclization, aromatization, hydrogen transfer and carbocation formation in acidic medium. Butadiene is especially reactive to cyclization and aromatization reactions due to two highly reactive conjugated double bonds present. Benzene, toluene and xylene (BTX) aromatics obtained from this process can be blended into gasoline to meet specific performance requirements (Poddar et al., 2013). Furthermore, Maki-Arvela et al. (Mäki-Arvela et al., 2021) recommend a minimum aromatics content of 8 %wt. in aviation jet fuel protects its leakage through seals, small holes and gaskets. it a minimum concentration of 8 %wt. However, the aromatic content in any fuel should not exceed 25 %wt. (according to ASTM regulations) due to the possibility of sooty material in the ignition chambers, which eventually affects important chemical properties associated with their combustion such as energy density and heating value. These sooty particles are known to have long-term effects on the engine by blocking essential fuel pipes and damaging the rotating turbines (Ali et al., 2023; Kuttiyathil et al., 2023). On the other hand, aliphatic products from BD hydrogenation such as butenes and isobutenes also have important industrial uses such as in the manufacture of polyethylene and poly(but-1-ene) (Pomalaza et al., 2020; Shao et al., 2018). Polyethylene is a well-known petrochemical used in the manufacture of household and everyday-use items such as plastic bottles, food containers and synthetic rubber. Hydrogenated polybutenes find a variety of uses such as lipsticks, lipgloss, repellents for birds and squirrels, and as the sticky agent in mouse traps (Cohen, 2023). 1-butene in its monomeric form finds its use mainly in packaging materials and construction industry for pipes and adhesives due to its high tensile strength (Arnd Schade, n.d.). Hence, there needs to be a balance achieved between improving the aromatics yield and extent of hydrogenation that gives the required aliphatics.
Noble metals such as Pt, Pd, Ru, Rh are highly active and selective in olefin hydrogenation (Maier et al., 2020). However, their rare occurrence in the earth’s crust resulting in low availability combined with high cost has prompted academic researchers and industry experts to look at transition metals such as Ni, Fe, Mn, and Co (Méndez et al., 2018; Mooibroek et al., 2013; Reina et al., 2018; Sandl et al., 2019; Docherty et al., 2017; Kallmeier and Kempe, 2018). These metals have some empty 3d orbitals that can coordinate with ligands, accept electrons from other species, participate in charge transfer reactions, compatibility with various supports and ability to show high dispersion in them. Most importantly, they are readily available naturally, more affordable, and less toxic. The dissociation of H2 is an essential step in hydrogenation and Ni shows a great affinity to aid in this dissociation and creating active hydrogen on the catalyst surface at room temperature and atmospheric pressure itself (Mooibroek et al., 2013; Shittu et al., 2022). Heterolytic dissociation of H2 is also naturally facilitated by the enzyme called hydrogenase that is commonly found in certain microorganisms (Cammack and van Vliet, 1998). These natural enzymes have been the inspiration for homogeneous hydrogenation studies of simple olefins that were catalyzed by Ni2+ with diarylphosphane ligands (Mooibroek et al., 2013). Due to the favorable hydrogenation-activity of Ni, it has found applications in a variety of selective hydrogenation studies in different forms such as zero-valent well-dispersed Ni nanoparticles in glycerol using Ni-cycloocta-1,5-diene as the precursor, (Reina et al., 2018; Zaramello et al., 2017) synthesis of stereoselective amino acid derivatives through enantioselective hydrogenation of acylamino-acrylates, (Li et al., 2017) asymmetric hydrogenation of acylamino nitroolefins, (Gao et al., 2017) hydrogenation of sterically hindered and bulky alkenes, (Shittu et al., 2022; Léonard and Chirik, 2018) hydrodesulfurization (HDS), hydrodeoxygenation (HDO) and hydrodenitrogenation (HDN) reactions, (Rocha et al., 2007; Méndez et al., 2020; Xu et al., 2022) hydrogenation of ; -unsaturated —C⚌O compounds, (Hu et al., 2010) and upgrading of biomass-derived compounds, (Qin et al., 2023; Jiang et al., 2021) and reforming of alkanes (Bao et al., 2015; Olafsen et al., 2006).
Selective hydrogenation of BD has come into attention quite frequently in the past decade and carried out over various catalyst-support combinations. Metal-support interactions (MSIs) are important to channel the selectivity of the catalysts and the work by Lee et al. (Lee et al., 2003) showed that modifying Pd/SiO2 with TiO2 resulted in an improved selectivity for 1-butene through a beneficial effect of MSI. Here, the Pd particles were interacting with the partially reduced TiOx and this was evident from the shift in the XPS peak of Pd (3d) signal to lower binding energy. They reasoned that this was due to charge transfer of Ti to Pd, thereby helping 1-butene to desorb effectively. Other precious metals such as Au, Pd and their combinations have also successfully been used for BD hydrogenation. Okumura et al. (Okumura et al., 2002) showed that Au/Al2O3 had excellent hydrogenation performance in terms of BD conversion and product selectivity and not dependent on the size of Au particles on the support. Hugon et al. (Hugon et al., 2010) suggested that bimetallic catalysts such as Au-Pd on alumina support also showed good performance but monometallic Au/Al2O3 matched the butene selectivity from the bimetallic study. Hence, we focused our study on monometallic catalysts with a suitable support. However, we chose to work with nickel due to its abundance, low cost and excellent hydrogenation activity with a variety of supports and feedstocks as discussed before. However, there have been some reports questioning the resistance of Ni-based catalysts towards coking and sintering and the tendency to agglomerate due to its low Taman temperature (Xiao et al., 2021). We have attempted to address these concerns in this paper as well.
Catalytic hydrogenation is affected by a variety of factors such as the metal loading, nature of the support as well as the preparation method. However, it was seen than catalyst supports played an important role in the overall performance of olefin hydrogenation. Co-precipitated Ni/alumina catalyst was shown to be more efficient towards selective deoxygenation involving hydrogenation of waste cooking oil and sunflower oil since it possessed a higher surface area, enabled better Ni dispersion, and reduced coke formation (Nikolopoulos et al., 2023). Al2O3 supports typically have high surface areas, which provide a large number of active sites for catalysis, making Ni/Al2O3 effective for hydrogenation reactions. In addition, Al2O3 is thermally stable, making it suitable for high-temperature hydrogenation reactions, and can be used for a broad range of hydrogenation reactions, including the reduction of various functional groups like alkenes, alkynes, and carbonyl compounds. The beneficial effect of Ni/alumina alone was shown by Kang et al. (Kang et al., 2002) where they observed agglomeration of Ni species when Cu was added to the catalyst and this decreased the conversion rate of BD for its hydrogenation. Modification of classical catalysts by using a strong reducing agent quickened the preparation process and reduced the need for pretreatment (Yu et al., 2010). However, Tan et al. (Tan et al., 2016) found that classical catalysts of Ni/Al2O3 performed much better towards olefin hydrogenation and H2 uptake than non-classical catalysts of the same due to higher stability, lower aggregation tendencies, higher surface area and because of the additional calcination step in the classical catalyst preparation that removes any impurities that may facilitate deactivation. Mendez et al. (Méndez et al., 2016) showed that in the gas phase, bimetallic Ni-Pd on alumina support exhibited good selectivity towards butene in BD hydrogenation and this was further enhanced by adding CeO2 to alumina support in the liquid phase. Ceria has gained enormous interest as excellent hydrogenation catalysts due to the presence of multiple oxidation states of cerium. (Razmgar et al., 2022) Additionally, when associated with multivalent cations, oxygen vacancies are created which aid in dissociating H2. Niobium pentoxide (Nb2O5) or niobia support displays similar properties due to the ability to switch between multiple oxidation states of Nb (Nb5+, Nb4+, Nb2+, Nb0) (Shinzato et al., 2021). Before reduction, the highest oxidized form of Nb is known to stabilize the heterogeneous catalysis process. The advantage of niobia is that it is niobium being smaller in size can interact very well with the active metal species, show enhanced activity towards redox reactions, and created additional Bronsted sites on the support surface through Nb-O bonds in NbOx species, that help in cyclization and aromatization for improved product yields in olefin hydrogenation (Rodrigues et al., 2012; Razmgar et al., 2022). Moreover, the 4 d-electrons in Nb are known to help in heterolytically cleave H2 molecule effectively (Shinzato et al., 2021). The reduction of the pentoxide to NbO form plays a critical role in H2 sorption/desorption kinetics and for this, amorphous Nb2O5 in its metastable state is more active than the crystalline form. (Shinzato et al., 2021) Moreover, since our reaction involved 2 —C⚌C in BD, the acidity of the catalyst support was significant in determining the product distribution. Hydrotreating activity was seen to improve with Nb addition to Ni/alumina catalyst due to the enhanced acidity of the combined catalyst (Rocha et al., 2007). Beneficial interactions between Nb and H2 with the formation of -NbHx single phase species have been shown by Gabriel et al. (Gabriel et al., 2011) as well. However, in some cases of catalytic hydrogenation, the synergic bond between Ni and alumina was found to be better than between Ni and niobia (Farojr and Dossantos, 2006).
Having conducted an extensive review of pertinent catalytic hydrogenation literature, we noticed that no previous work compared the performances of Ni/alumina with Ni/niobia for BD hydrogenation at low temperatures. Hence, our current work investigates hydrogenation of BD over two Ni-supported catalysts: namely, Ni/Al2O3 and Ni/Nb2O5.The key contributions of this work are two-fold: (i) comparison of the performances of the two catalysts used in terms of BD conversion and nature and yields of products produced; (ii) deduction of reaction mechanisms based on the product distribution and their respective yields at different temperatures and insights into the reaction chemistry of BD hydrogenation involving cyclization, aromatization, and hydrogen transfer steps; (iii) discussion on effect of metal support interactions (MSIs) of both catalysts on the product yields; and (iv) proposing plausible mechanisms of coke formation through ring closure that lead to catalyst deactivation through characterization of spent catalysts. Therefore, the objective of our work in this paper is three-fold: (i) to investigate the utility of Ni-based catalysts on alumina and niobia supports separately for hydrogenation of butadiene; (ii) compare the product distribution, conversion of BD at different temperatures < 400 °C and levels of metal-support interactions present; and finally (iii) inspect the incumbent reaction chemistry and mechanisms involved in the formation of aromatic products and coke. We have utilized various characterization methods for analyzing the morphology, chemical environment and reduction state of the catalysts during and after the reaction as well as to know the quantitative and chemical composition of the products after hydrogenation.
2 Methodologies
2.1 Materials and equipment
The partial hydrogenation of BD reaction was carried out using a quartz tube reactor that held the catalysts in the isothermal region of the reactor. Gas cylinders used for this experiment were butadiene in helium (99 % He, 1 % BD) and hydrogen in helium (95 % He, 5 % H2) which were obtained from Air Gulf LLC. The precursor for nickel was nickel nitrate hexahydrate (Ni(NO3)2·6H2O), while aluminium oxide (Al2O3) powder and niobium pentoxide (Nb2O5) powder served as the two supports for our work. All of these were obtained from Sigma-Aldrich with >99 % purity (Monroy et al., 2010).
2.2 Catalyst preparation
In a typical preparation, both catalysts were prepared using the incipient wetness impregnation method (Kang et al., 2002). Here, it was interesting to note that though Ni(NO3)2·6H2O was the more popular precursor for Ni, the chloride version (NiCl2·6H2O) has also been used previously. (Shittu et al., 2022; Yu et al., 2018) However, Yu et al. (Yu et al., 2018) observed that there were significant unwanted chemical interactions between Cl- and the alumina support. Furthermore, the chloride species remained in the samples even after calcination and increased the possibility of sintering for the Ni/Al2O3 catalysts. Hence, we decided to employ the nitrate precursor for Ni in this work. Preparation of Ni/Al2O3 and Ni/Nb2O5 began with adding masses of alumina and niobia to de-ionized water followed by adding the nickel precursor to form a slurry. This mixture was stirred on a heating plate at 70 rpm for 1 hr, first at room temperature and then pre-drying at 150 °C to evaporate the remaining water from the mixture. Next, the solid-type mixture was left in a hot air oven for 2 hrs at 100 °C for complete drying. Following this, the dried solid mixture of both catalysts were each grounded into small-sized powder so as to obtain maximum pore size and increase the surface area. Finally, both catalysts were calcined by passing air at a heating ramp of 10 °C min−1 (increased from room temperature steadily) for 5 hrs at 500 °C, to remove all the volatile components from the catalysts. The prepared catalysts were identified as 5 % Ni/Al2O3 and 5 % Ni/Nb2O5 for the entire domain of this paper. This meant that 5 % of Nickel was used in combination with 95 % of the respective supports.
2.3 Catalyst characterization
Characterization of calcined and reduced Ni/Al2O3 and Ni/Nb2O5 catalysts in this hydrogenation reaction was reported. Different characterization techniques were utilized to understand and analyse the chemical/physical properties of both catalysts, and to link these – when possible with catalyst reactivity (Katano et al., 2008). Powder X-ray diffraction (Malvern Panalytical) tool was used (10–80 2ϴ range, scanning rate of 2 degrees min−1) to analyze the composition of crystalline phases, crystalline size and strain, phase concentrations and amorphous content. Fourier transform infrared (FTIR, Jasco FT/IR-4700, USA) connected with an ATR accessory (16 cm−1 resolution) analysis was done to analyze each peak area and functional group for both catalysts. Scanning electron microscopy with Energy dispersive spectroscope (SEM-EDS, JEOL JSM 6010 PLUS/LA), were used to study the morphology and the elemental composition of the catalyst (10–100 µm resolution). Hydrogen-temperature programmed reduction was performed to test the reducibility (reduction temperature) of both catalysts (Quantachrome, ChemBET-TPR/TPD 151, chemisorption) with a temperature range of 0–900 °C with a heating rate of 10 °C min−1. N2 adsorption–desorption experiments were performed using a Quatachrome instrument (NOVAtouch NT 2LX-1, USA) to estimate the Brunauer-Emmett-Teller (BET) surface areas for both catalysts. The region of relative pressures ( ) between 0.1 and 0.4 in the adsorption branch was used for the BET method since this corresponded to the linear region of the plot. The pore volume was calculated using the amount of N2 adsorbed at a relative pressure of 0.99 and expressed in cm3/g of the catalyst. The Barrett-Joyner-Halenda (BJH) method was used to calculate the pore size from the desorption part of the isotherms between relative pressures of 0.8 and 0.6 (Méndez et al., 2016, 2017). Thermogravimetric analysis (TGA Q50 V20.10 Build 36 analyzer) was performed on the spent catalysts to analyze the weight loss occurred to the catalyst after the reaction (in air with a heating rate of 10 °C min−1), along with SEM-EDS analysis to identify the carbon mass percentage.
2.4 Catalytic run and experimental setup
Catalytic hydrogenation of BD was carried out in a quartz tube reactor, placed in an electrical heated chamber (SAFTherm China) controlled using a PID temperature controller, at atmospheric pressure (1 atm) with a temperature range of 50–400 °C at a ramp rate of 50 °C. The uncertainties in pressure were of the order of ± 0.5 atm, which arose due to the change in pressure inside the quartz tube reactor caused by minor fluctuations in the flowrate of the reactants and the small size of the quartz tube (20 mm outer diameter). On the other hand, the temperature reading during the collection of samples in the tedlar bags after the reactions fluctuated slightly due to the surrounding temperature in the laboratory and the micro-surroundings of the bag itself. Furthermore, the exact furnace reactor temperature might not be 100 % accurate when it changes from one temperature to the next and the flowrate of the reactants (BD and H2) from the cylinders into the reactor could further affect the temperature. Moreover, our reactor had a heating zone length of 200 mm, so the temperature inside the heated chamber could be different from the actual number shown in the digital screen of the temperature controller. All these factors resulted in a total uncertainty of ± 5 °C as an averaged value over all the temperature conditions carried out in this work. A mass flow controller was used to control the flow of the gases such as BD (with a flowrate of 100 ml min−1) and H2 (flowrate of 150 ml min−1), both in helium by maintaining a stoichiometric ratio of 10:2 respectively. Prior to the reaction, an experimental run was conducted without catalyst to compare the conversion values obtained with catalyst. The catalysts were initially reduced in H2 at a ramp temperature change of 10 °C min−1 for 6 hrs from room temperature to 600 °C, before starting the reaction. About 0.5 g of each catalyst was placed in the middle of the reactor and quartz wool was used on either side to hold the catalyst.
The yields of the various products were obtained for analysis using GCMS (8890 GC/5977B MSD, Agilent Technologies, US), equipped with a HP-5MS-UI column. Helium was the carrier gas with a flowrate of 1 ml min−1. Fourier transform infrared spectroscopy (FTIR - INVENIO S, Bruker Technologies, US, optical length 10 cm) was performed to identify the functional groups present in the gaseous reaction products and corroborate with the GC–MS results (Iffländer et al., 2020; Shittu and Altarawneh, 2021). The gaseous products collected in the tedlar bags after the reaction were fed into the FTIR as shown in Fig. 1, which depicts the entire experimental setup and reaction steps for this process. As the sample is exposed to IR radiation, some amount is absorbed due to the vibrational frequencies of the covalent bonds in the samples, and the light which is transmitted is detected and analyzed for the presence of specific functional groups. A gas cell with a volume of 25 cm3 and 10 cm optical length is attached to the instrument and filled with N2 before each measurement to minimize interference and reduce noise from previous remaining samples. The range of wavenumber channels used in the FTIR analysis was 4000 – 400 cm−1. However, below 500 cm−1, the noise levels in the transmitted frequencies were high and hence omitted from the analysis.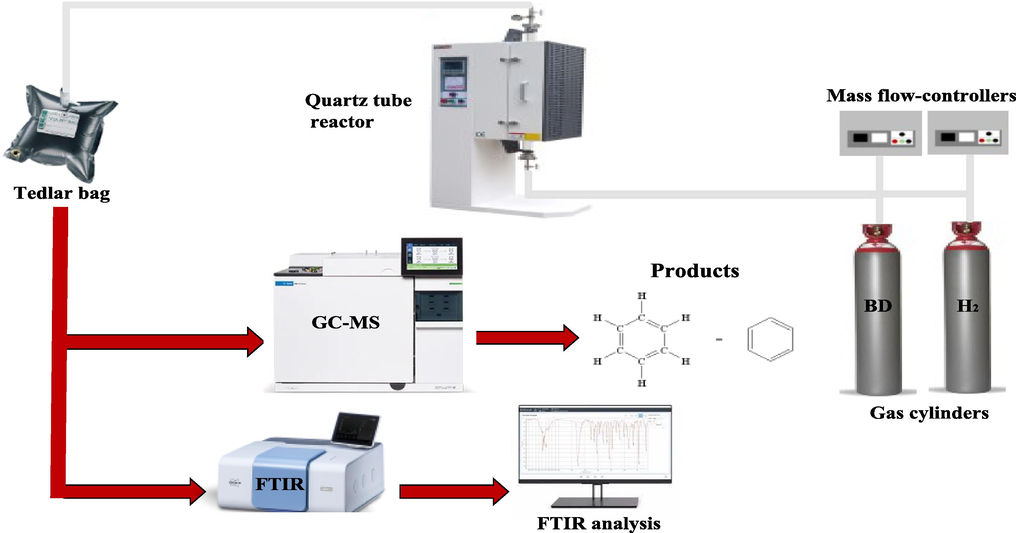
Schematic of the entire experimental procedure following in this work.
3 Results and discussion
3.1 Catalyst characterization
3.1.1 XRD analysis
Fig. 2 represents the XRD patterns of both catalysts (5 % load of Ni) after calcination. In the case of 5 % Ni/Al2O3 and 5 % Ni/Nb2O5, XRD identified the phase present in the sample and determined their crystal structure. The patterns show nickel peaks with low intensity due to the 5 % load of alumina catalyst at 44.5°, 66.2° and 76.4° which correspond to the (1 1 1), (2 0 0) and (2 2 0) crystal phases of nickel. Alumina diffraction peaks corresponding to 4 crystal planes of (3 1 1), (2 2 2), (4 0 0) and (5 2 2) were identified to have 2
values at 37.9°, 39.7°, 45.8°, and 67.2°, respectively. These are indicated in the blue spectra as filled black circles in Fig. 2. The sharpness of the peaks, especially at 67.2° indicate that the crystallinity is preserved even after doping with Ni (Ye et al., 2020). Interestingly, diffraction peaks at 19.1°, 31.6°, and 60.2° could be attributed to the crystal planes (1 1 1), (2 2 0) and (5 1 1) of a nickel-spinel phase (NiAl2O4). The formation of this phase indicated strong metal-support interactions between Ni and alumina and this would be a potential reason for the better performance of this catalyst as compared to Ni/niobia. The effects of metal-support interactions are elaborately discussed in section 3.1.4 in the Results and Discussion section. The challenge for the Ni/alumina catalyst is in distinguishing between the peaks of Ni, NiO, the spinel phase and alumina itself. This becomes especially tough for 2
values between 37° and 39° since they can correspond to NiO in addition to alumina. The most important peak for NiO species is indicated by a hexagon in Fig. 2 and this occurs at 37.2°. This occurs at the same position for both catalysts. It is important to note that it is very difficult to spot the peak for metallic Ni0 species in the XRD spectra of Ni combined with supports due to its low crystallite size in the calcined catalytic samples before reduction. Shittu et al. (Shittu et al., 2022) and Ali et al. (Ali et al., 2023) reported two main peaks for Ni0 species at 44° and 52° only in the reduced Ni/CeO2 samples. We have tried to locate these peaks in our XRD spectra for Ni/alumina and Ni/niobia catalysts but even comparing them with the spectra of unloaded carriers, the 44° peak falls on the shoulder/slope of the rising peak for crystalline Al2O3 and Nb2O5. The peak at 52° was very low in intensity for both catalysts, signifying that the concentration of reduced Ni0 species was very low to be detected for this crystal plane. Even in the works by Shittu et al. (Shittu et al., 2022) and Ali et al. (Ali et al., 2023), there was a very minute peak of Ni in the calcined samples, which spiked when the samples were reduced. The XRD spectra for the unloaded carrier supports were taken from the works by Rodrigues et al. (Rodrigues et al., 2012) and Shittu et al. (Shittu et al., 2022) and compared with our results. It was found that undoped calcined Nb2O5 had characteristic XRD peaks at 2
values of 22°, 28.7°, 36.7°, 46°, 51°, 55.4°, and 71° correspond to the (0 0 1), (1 0 0), ( −1 1 0), (2 0 1), (2 0 2), (2 1 0), (−3 1 2) and (−3 1 3) crystal planes of orthorhombic Nb2O5 (Shittu et al., 2022). However, among these, the peaks at 22° and 28.4° were seen to be most intense and as the 2
values increased, the intensity of the peaks declined significantly (Rodrigues et al., 2012). Interestingly, the diffraction peaks for Ni/niobia indicated a shift to the left by 1-2° for the Nb2O5 species and were of lower intensities than that of the unloaded support. This indicated that dispersion of Ni on niobia affected its XRD patterns due to lower metal-support interactions as compared to Ni/alumina and also the possibility of the agglomeration of Nb2O5 species with the catalytic framework. This was clearly evident from Mendez et al., (Méndez et al., 2017) who also reported similar behaviour for Nb2O5 when the active metals such as Ni and Co were added and reported a decreased surface area of niobia. These issues with Ni/niobia were further confirmed in our other results such as H2-TPR, SEM-EDS of the fresh and spent catalysts, N2-adsorption/desorption, increased coke deposition through thermal analysis, Raman spectroscopy and stability analysis.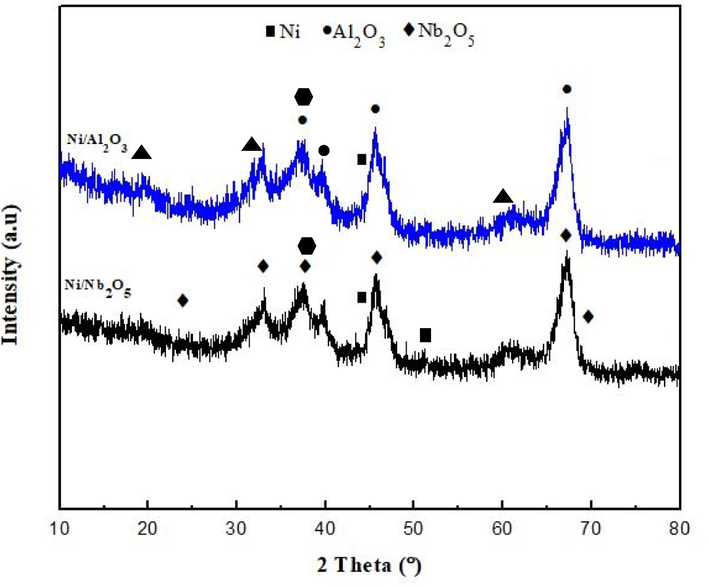
XRD patterns of both catalysts: Ni/alumina and Ni/niobia.
3.1.2 SEM-EDS analysis
SEM-EDS analysis was done for both catalysts to provide information about the elemental composition, distribution, and surface morphology of calcined alumina and niobium catalysts respectively. SEM images and EDS spectra are shown in Fig. 3. As observed, the morphology of the catalysts will gradually change with increasing the load of the catalysts (Echeverría and Jover, 2022). For example, when the load of nickel metal increases on alumina or niobium supports, the average particle size of nickel increases, which leads to decreased dispersion and altered metal-support interactions (Jasik et al., 2005; Jeon et al., 2018). In our case we used a 5 % load for both catalysts to obtain better conversion of butadiene at a low temperature (Chen et al., 2016).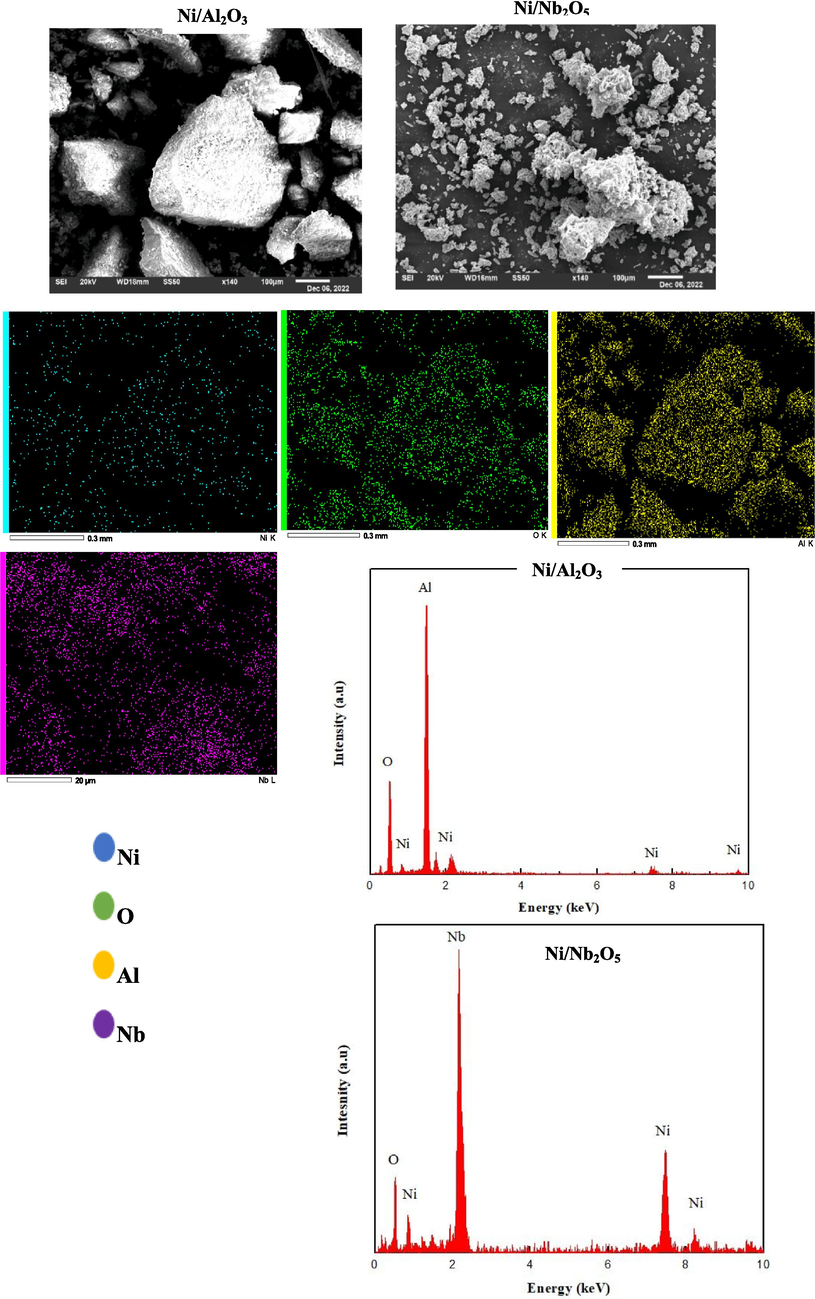
SEM images with EDS spectra and mapping for both Ni/alumina and Ni/niobia catalysts.
The elemental mapping composition analysis for the calcined alumina catalyst shows the presence of 43.91 % of O, 46.71 % of Al and 9.93 % Ni mass ratio respectively as this area of EDS shows a higher nickel content around 10 % compared to 5 % wt.% of Ni/Al2O3. This mass ratio is due to the loading present of the fresh catalyst as adding more load to the catalyst for instant, 10–30 % will affect the crystal structure of the catalyst and the surface will be more compact for higher loadings (Razmgar et al., 2022).
In addition, the mapping of alumina catalyst indicates the atoms distribution on the surface of the catalyst, as observed there are more atoms of O and Al than Ni metal which is expected as the support has a high percentage ratio of 95 %. Niobium catalyst represents elemental mapping composition of 37.94 % O, 53.80 % Nb and 8.26 % Ni mass ratio respectively, as seen this EDS area has more nickel content as well. Due to the high load of the support (95 %), there is an increased mass ratio of Nb and O which indicates the mapping distribution of atoms on the surface of the catalyst. As noted, the Nb and O shows more than Ni on the surface with active sites for niobia catalyst.
EDS analysis also determines the presence of Ni, Al and Nb species in the catalyst matrix. According to the spectra of alumina catalyst, the peaks of Ni are shown in a (1.2, 7.8 and 8.2) KeV respectively, while the peaks of O and Al show in a (0.9 and 1.8) Kev as well. Moreover, spectra of niobia catalyst point to (0.9, 7.5 and 8.3) KeV of Ni, and (0.5 and 2.2) KeV of O and Nb similarly.
3.1.3 N2 adsorption–desorption: Textural characteristics of the catalysts
Fig. 4 provides the adsorption–desorption isotherms of both prepared catalysts while Table 1 gives their textural properties. Both the isotherms given in Fig. 4 fall under the Type IV category according to IUPAC classification of adsorption isotherms and show H1-type hysteresis loop between
values of 0.7 and 0.9 for both the Ni-impregnated catalysts. The type of hysteresis shown in the adsorption–desorption process is directly related to the textural properties such as size and shape of the pores and whether the catalyst particles are agglomerated or not. There is sufficient evidence from catalysis literature that pure oxides of alumina and niobia display H3-type hysteresis loop for N2 adsorption, but when a transition metal is introduced into the matrix, they tend to shift to H1-loop. (ALOthman, 2012; Rocha et al., 2007; Xu et al., 2022) In our work, Ni/Al2O3 showed a steep increase (Fig. 4a) in the N2 adsorption in the region of
between 0.75 and 0.95 while this rate of N2 uptake decreased for Ni/Nb2O5 as seen from Fig. 4b. It can be stated that adsorption occurred was initially limited to a mono-molecular layer in Nb2O5 supported Ni due to the steady increase in N2 uptake whereas multi-molecular layer adsorption was prevalent at all times in Ni/Al2O3. (Xu et al., 2020) The type IV isotherms that exhibit hysteresis clearly indicate the presence of mesopores and the average pore size determined by the BJH method confirm this observation (Table 1). Furthermore, according to IUPAC, materials showing H1-type hysteresis consist of either cylindrical-shaped pores or spheres that agglomerate or a combination of both these shapes. (ALOthman, 2012) This classification was developed on a previous indication given by de Boer (Broekhoff, 1979; Sing, 1985). Shittu et al. (Shittu et al., 2022) reported that Nb2O5 consisted of slit-like pores due to H3-type hysteresis loop for N2 adsorption, but again this changed to H1-type hysteresis on adding Ni into the matrix.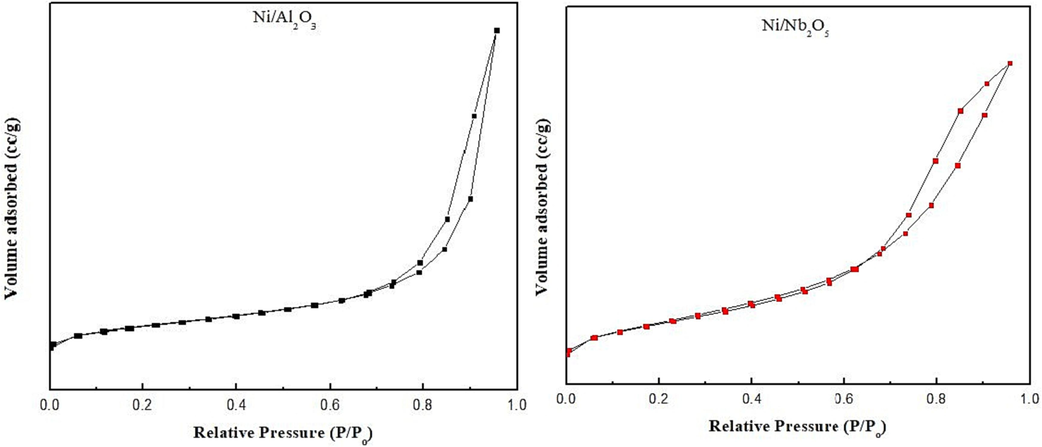
N2 adsorption–desorption isotherms for: (a) Ni/Al2O3 and (b) Ni/Nb2O5 catalysts.
Catalyst
BET surface area (m2/g)
Average pore diameter – BJH (nm)
Pore volume (cm3 g−1)
Ni/Al2O3
78.25
1.23
22.52
0.61
1.92
0.08
Ni/Nb2O5
34.19
1.04
23.08
0.69
1.45
0.07
It was interesting to note that the BET surface area of the niobia-supported catalyst was found to be lower than that of the alumina-supported catalyst by more than half (Table 1). There can be many reasons attributed to this result. First of all, let us consider the catalyst preparation process. Niobia is less-soluble in water than alumina and this could be further due to the larger atomic size of niobium (atomic number = 41) as compared to aluminium (atomic number = 13). This could affect the dispersion of niobia in the catalytic matrix and was evident from the EDS spectra given in the previous section (Fig. 3). Rocha et al. (Rocha et al., 2007) also observed lower surface areas for niobia as compared to alumina-supported NiMoS catalysts for hydrodesulfurization and hydrodenitrogenation reactions. In their work, the BET surface area of pure niobia samples was 7 times lower than that of pure alumina and Ni-added niobia showed a BET surface area that was 6 times lower than that of Ni-alumina catalysts. In general, addition of metals to pure oxide supports decreased the surface area due to increase in the catalyst density and blockage of the pores in the support materials. However, it was reasoned that this pore blockage was lower in the case of Ni/alumina than for Ni/niobia due to higher dispersion of Ni in alumina and resulting in higher surface area. Furthermore, the acidity of niobia is higher than that of alumina, as shown by a number of previous studies (Pham et al., 2011; Sobczak et al., 2010; Ziolek et al., 2011). Rodrigues et al. (Rodrigues et al., 2012) clearly demonstrated the creation of additional Bronsted acid sites on the catalyst support surface as the loading of Nb was increased, which was associated with the formation of Nb-O bonds through NbO7, NbO8 species. Rocha et al. (Rocha et al., 2007) suggested that this acidic medium could cause redissolution and reprecipitation of niobia support during the preparation process while impregnating the active metal, thus decreasing the BET surface area. Another important reason for the decrease in the surface area of niobia can be attributed to the agglomeration of the Nb2O5 species, as was also reported by Mendez et al. (Méndez et al., 2017). This was also evident from the SEM images of the spent catalysts in our work (Figs. 12 and 13). They also attributed the laboratory conditions such as humidity in the air, exposure to water for long periods during the preparation stage can significantly affect mesoporous materials such as niobia. They also followed the same impregnation method as our work and reported lower surface area. This resulted in an increased average pore diameter as was observed in our case (Table 1). Along this line of enquiry, possible damage of pore walls of the niobia support during the impregnation of metal precursors in their aqueous solutions have been reported before and can decrease the BET surface area (Kumar Rana and Viswanathan, 1998; Méndez et al., 2017). Furthermore, agglomeration of nickel on the niobia support can plug the pores and reduce the effective surface area available. All these reasons not only affected the surface area but also decreased the pore volume, which is another textural characteristic of the catalysts.
On the other hand, there was only one work to the best of our knowledge that reported alumina to have lower surface area than niobia support (Rodrigues et al., 2012). They noticed that alumina exhibited Type II isotherm without any hysteresis for N2 adsorption, which was characteristic of a non-porous material and the entire adsorption–desorption process being reversible (ALOthman, 2012). They noticed a whole 2-orders of magnitude difference between the surface area of alumina (0.48 m2/g) and niobia (185 m2/g) and increase of hysteresis as the loading of niobia was increased on alumina support. No particular reason was provided for these observations except the presence of -phase of Al2O3, which decreased its surface area due to its crystalline nature. Interestingly, the specific surface area of niobia-supported CoMo catalysts was only found to decrease as the % loading of Nb increased in the matrix during the investigation of hydrodeoxygenation of guaiacol by Xu et al. (Xu et al., 2020) Hence, the observation of decreased surface area for alumina in the study by Rodrigues et al. (Rodrigues et al., 2012) can be considered an anomaly and can also depend on the precursor material taken during catalyst preparation since they synthesized niobia from niobium oxalate, whereas niobium pentoxide was directly used in our work.
3.1.4 A comprehensive analysis of metal-support interactions (MSIs) through comparison of H2-TPR and XRD results and correlating with X-ray photoelectron spectroscopy (XPS) literature
H2-TPR analysis was performed for both catalysts to obtain information about the reducibility of Ni-containing species and analyze the nature of the metal-support interactions (MSIs) present by identifying the potential form of nickel species present in alumina and niobia. The curve shown in Fig. 5 was obtained by plotting the amount of hydrogen consumed as a function of temperature. This section also correlates the results of TPR with our XRD spectra and verify the corresponding analysis with XPS results from literature to confirm the relative amounts of MSIs present in each catalyst. It is well-established in literature that nickel exists in the form of its oxide, NiO, within the catalyst matrix and this is clearly seen in the XRD patterns (Fig. 2) as well. (Ewbank et al., 2015; He et al., 2021; Zieliski, 1982) There was also evidence of the presence of a ‘surface-nickel species’ called nickel aluminate (NiAl2O4) in the XRD patterns, which was a result of the strong interaction between nickel and alumina. This species is formed when NiO is ‘fixed’ through the chemical reaction between the oxide and alumina and it was found to have a spinel structure (Li et al., 2017). Hence, we will also refer to this stable nickel aluminate as ‘the spinel phase’ and Li et al. (Li et al., 2017) found that the Ni/alumina catalysts containing the spinel phase also yielded small Ni0 active species that increased the catalytic performance and in addition, were thermally stable as well. It was determined that a calcination temperature > 400 °C was required to form this spinel phase. Depending on the type of NiO and the strength of the MSIs, the peaks occur in different temperature regions in the H2-TPR curve of Ni-based catalysts. A low temperature peak (∼300 – 500 °C) usually implies weak Ni-alumina interaction and corresponds to easily reduceable
-type nickel oxide. As the TPR peak shifts towards the higher temperatures, the MSI becomes stronger and indicates the presence of
- and
-type NiO species. The peak shown by the Ni/alumina catalyst in our work is around the 600 – 800 °C range, thus indicating the formation of the nickel aluminate spinel and absence of
-type NiO. This is also corroborating with our XRD results (diffraction peaks indicated by a triangle in Fig. 2) and the existence of the spinel-type species of non-stoichiometric nickel aluminate was strongly supported by evidence from a number of studies in literature as well. (Ewbank et al., 2015; Smoláková et al., 2016; Wang et al., 2020; Zieliski, 1982) In an interesting work, He et al. (He et al., 2021) calculated the degree of reduction and %dispersion of Ni on the alumina catalyst at different calcination temperatures based on the amount of H2 consumed. They found that as the reduction TPR peak shifted to higher temperatures such as in our work, the degree of reduction decreased and in turn, the dispersion of Ni increased. This meant that there was a strong interaction between Ni and alumina support, and the presence of the stable nickel aluminate caused nickel to remain in its Ni2+ state, with minimal reduction to Ni0 state. An advantage of this spinel phase of nickel aluminate is that it prevents further diffusion and migration of Ni2+ species over the catalyst surface, thereby preventing sintering and contributing to the thermal stability of Ni/alumina. This leads to higher conversion of BD and lower deactivation for Ni/alumina as compared to Ni/niobia, as observed in this work. Wang et al. (Wang et al., 2011) further elaborated that MSI allowed the Ni2+ species to migrate into tetrahedral vacancies of the
-alumina. This could also be a factor for the lesser coking observed with alumina as compared to niobia (as shown by the TGA of the spent catalysts in Fig. 14), thereby forming both aliphatic butenes and BTX aromatics in the process.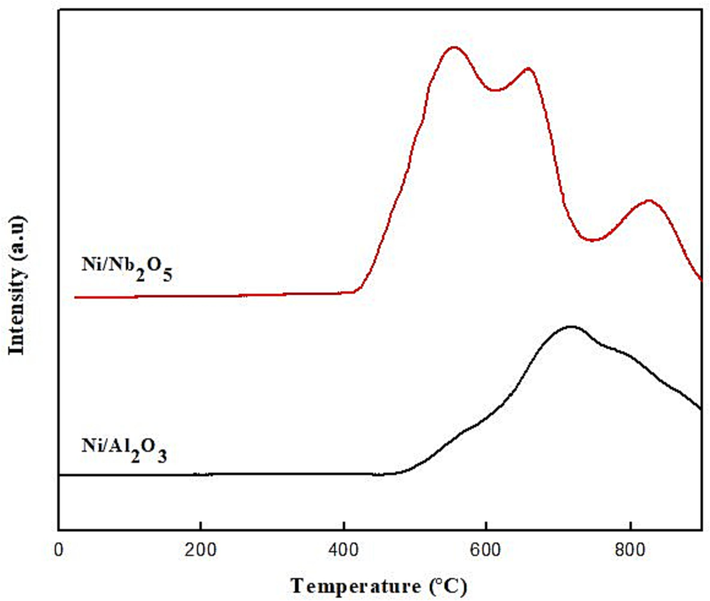
H2-TPR profiles for Ni/alumina and Ni/niobia catalysts before participation in the reaction.
Since X-ray photoelectron spectroscopy (XPS) could not be performed in our work due to unavailability of the instrument, we confirmed the presence of NiO and NiAl2O4 from multiple studies on Ni/alumina used for hydrogenation and dry reforming in the catalysis literature. The XPS spectra for Ni-doped catalysts show Ni(2p3/2) signals, which are asymmetric and the different electronic states of Ni can be interpreted by deconvoluting the signals. The binding energies for the two types of Ni2+ species in the matrix, namely, NiO and NiAl2O4, occur at 854.6 eV and 856.5 eV, respectively. (Ewbank et al., 2015; He et al., 2021) More easily reduceable NiO formed metallic Ni0 species, which shows a peak at a lower binding energy of 852.6 eV as seen in ceria-supported Ni catalysts. (Ali et al., 2023; Shittu et al., 2022) The higher the binding energy in XPS, the higher the metal-support interaction. Thus, it was expected that XPS would show peaks at 854.6 and 856.5 eV for our Ni/alumina catalysts, indicating strong MSI through the presence of NiO, non-reduceable spinel species and low amounts of metallic Ni0. These claims and observations are all in agreement with XRD and TPR results in our work (Fig. 2 and Fig. 5). Moreover, the lower consumption of H2 by the alumina catalyst (1072 µmol g−1) as compared to 2055 µmol g−1 for niobia, could explain the potentially expected lower amounts/absence of metallic Ni0 species. It needs to be kept in mind that achieving higher stability through increasing MSI and confining the active metal species in meso and micropores might also result in lower activity, selectivity and overall performance of the catalyst. (Gu et al., 2021) Hence, tuning of the MSI should be done with caution.
XRD patterns for the calcined Ni/Nb2O5 catalyst (Fig. 2b) are in agreement with those of previous studies and show moderate peaks for face-centered cubic NiO species and strong peaks for hexagonal-structured Nb2O5. Fresh uncalcined niobia is known to exist in the less crystalline TT phase and is amorphous in nature. Introduction of Ni into niobia did not change the hexagonal structure of the support. Interestingly, the H2-TPR analysis of Ni/niobia gave 2 clear sets of peaks with the first peak corresponding to the reduction of Ni2+ to Ni0 (between 450 and 650 °C) and the second peak associated with the reduction of the niobia species itself (>800 °C). TPR of pure niobia shows a major peak at the high temperature of > 1050 K (800 °C) and a small shoulder at a low temperature of ∼250 °C (Chary et al., 2004; Wojcieszak et al., 2006). These peaks are traditionally attributed to reduction of Nb2O5 present in the bulk and some excess NbOx species present on the surface. The peak corresponding to the reduction of Nb2O5 species at >800 K in our work was supported by interesting evidence from literature where Wojcieszak et al. (Wojcieszak et al., 2006) observed that this peak was unchanged when 5 % Ni was added to niobia but shifted by 100 °C to a lower temperature when 1 % Ni was added. They also observed that the intensity of the peak between 450 and 650 °C that corresponded to Ni reduction was far more for the lower loaded (1 %wt.) catalyst as compared to higher loaded Ni (5 %wt) in the niobia support. It was reasoned that as the loading of nickel increases on the support, the metal-support interaction decreases and more free Ni2+ species are available, which reduce easily to Ni0. In our work, the peak for the Ni2+ reduction in the TPR plot was shifted towards the left, i.e., to a lower temperature for Ni/niobia as compared to Ni/alumina as can be seen from Fig. 5. This clearly implied a weaker MSI between Ni and niobia as compared to Ni alumina and that more Ni2+ species present as NiO are susceptible to be reduced to Ni0. This was also confirmed from the work by Chary et al. (Chary et al., 2004) where they observed that too high Ni loadings in niobia shifted the peak maximums in the TPR to the left and this indicated weaker metal-support interactions. Metallic Ni0 species are generally very hard to be detected in the XRD spectra even after subjecting the catalysts to hydrogenation reaction, but were observed by Gnanakumar et al. after CO2 hydrogenation to form methane. Interestingly, in our work, we found mild peaks at 2 values of 74° from the XRD patterns of Ni/Nb2O5 catalyst (Fig. 2), which correspond to metallic Ni0 species. The fact that these were not found in Ni/alumina catalyst clearly indicates the stronger MSI for alumina as compared to niobia.
Gnanakumar et al. (Gnanakumar et al., 2019) further reported that MSIs between nickel and niobia increase only when the pretreatment temperature of Nb2O5 increases. The pretreatment temperature was not changed in our work and hence, stronger MSIs were not expected for Ni/niobia. The Ni(2P3/2) signals in the XPS spectra of unreacted Ni/niobia shows 3 peaks as follows: (i) 856.2 eV, 855.2 eV corresponding to Ni2+ existing as NiO and (ii) a broad satellite peak at 862 eV corresponding to Ni3+ species existing as Ni2O3. Reduced Ni0 species was not seen in the XPS spectra for Ni/niobia before its participation in the reaction in any of the literature works inspected. (Chary et al., 2004; Gnanakumar et al., 2019; Hernández Mejía et al., 2020; Wojcieszak et al., 2006) This was attributed to the difficulty in detecting metallic Ni in the XPS. However, when the XPS of fresh and spent Ni/niobia catalysts were compared for Ni (3p) signals in Gnanakumar et al., (Gnanakumar et al., 2019) there was a minute shift in the binding energy of the spent catalyst by about 1 eV to the lower side. This implied that the hydrogenation environment was conducive to the reduction of Ni2+ species in the catalyst. This meant that a shift to a lower binding energy was directly correlated with a lower MSI and Ni/niobia is prone to this behavior. On related lines, Wosciesak et al. (Wojcieszak et al., 2006) also suggested that the lower the nickel species on the catalyst, the higher the interaction between niobia and nickel, and in our case, the opposite was true since the potential of Ni0 existing on the surface of Ni/niobia during the reaction was very high as indicated by TPR results. Furthermore, Nb 3d5/2 signals occurred at 207.1 and 209.9 eV, which corresponded to multiple oxidation states of Nb such as NbO2 (Nb4+) and Nb2O5 (Nb5+). The reduced states of Nb such as Nb0 and Nb2+ as in NbO were not seen in the unreduced or reduced samples. However, interpretation of Nb (3d) spectra must be carried out with caution due to the lower number of d electrons causing difficulty in observing changes at the electronic level in response to XPS characterization. Overall, it is important to note that though MSIs are highly prominent in both Ni-based catalysts explored in this work, we observed that relative to Ni/niobia, Ni/alumina displayed a higher MSI.
3.1.5 FTIR analysis
The FTIR spectrum analysis performed of both catalysts is shown in Fig. 6. This spectrum analysis depends on the specific preparation method and conditions used to synthesize both catalysts and to understand the nature of the catalyst (Iffländer et al., 2020). For alumina catalyst, the presence of Ni in the sample is detected by the peak of 580 cm−1 which indicates the Ni—O stretching vibration. In addition, the peak at 1020 cm−1 represents the Al—O stretching vibration as Al2O3 was expected to show at this range due to the stretching vibration of the Al—O bond (Pattamakomsan et al., 2011). The peak at 3538 cm−1 illustrates the surface hydroxyl groups of Al2O3, as it’s detected by a broad peak in that range due to the O—H stretching vibration. On the other hand, niobia catalyst, indicates a peak at 590 cm−1 Ni—O stretching vibration with the existence of Ni, another peak is at 745 cm−1 due to the Nb⚌O stretching vibration in the spectrum of Nb2O5. This peak also provides information regarding the oxidation state and direction location of the Nb species (Shittu et al., 2022). Moreover, the peak at 1080 cm−1 denotes the Nb—O stretching vibration. The last peak at 3640 cm−1 signifies the O—H stretching vibration. Nb2O5 support has a surface hydroxyl group detected by a broad peak at this range.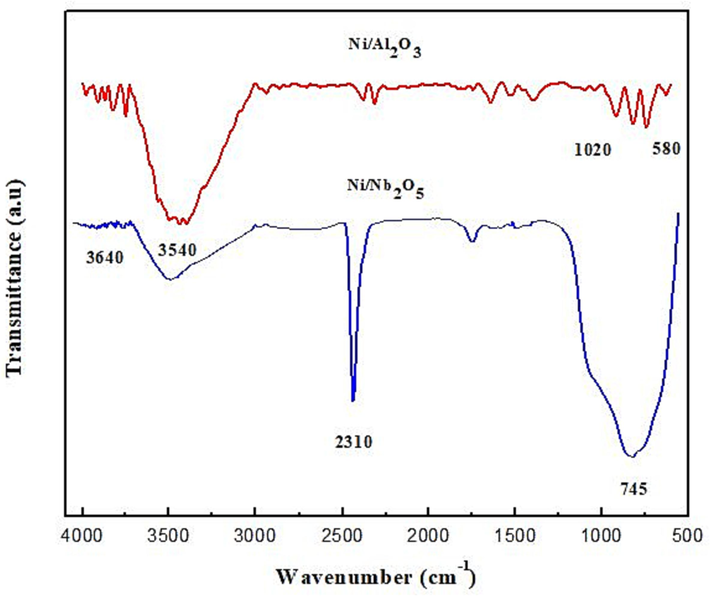
FTIR spectra for both calcined catalysts.
3.2 Catalytic performance
3.2.1 Product yields through GC–MS analysis: Reaction chemistry of product formation
As mentioned in section 2.4, the products obtained from the hydrogenation of BD at five different temperatures between 200 and 400 °C were analyzed using both chromatography (GC–MS) and spectroscopy (FTIR) techniques. In this section we will be discussing the results from both catalysts and comparing them qualitatively and quantitatively to shed light into the effect of the deployed support. We will also propose some reaction sequences that lead to the different products observed from GC–MS and FTIR. The distribution of products from BD hydrogenation using both catalysts are provided in Fig. 7 and Fig. 8 for Ni/Al2O3 and Ni/Nb2O5, respectively.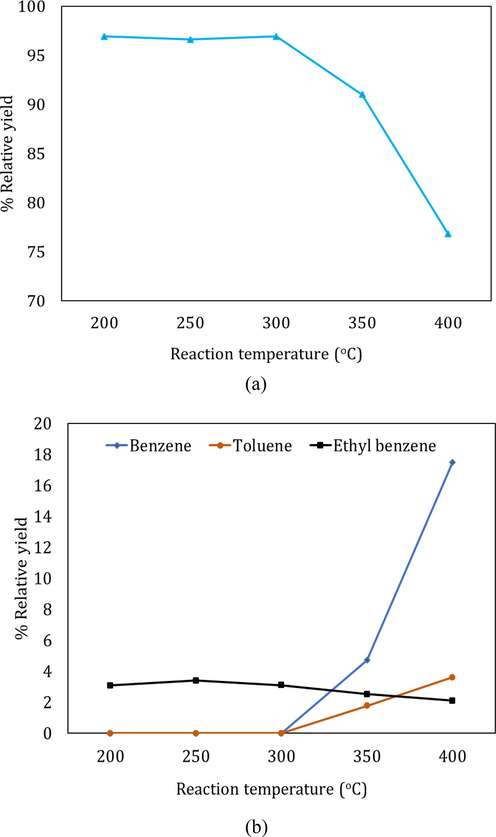
Product yields corresponding to (a) Isobutene, 1-butene and 2-butene as aliphatics, (b) benzene, toluene and ethyl benzene as aromatics, from hydrogenation of BD using Ni/alumina catalyst.
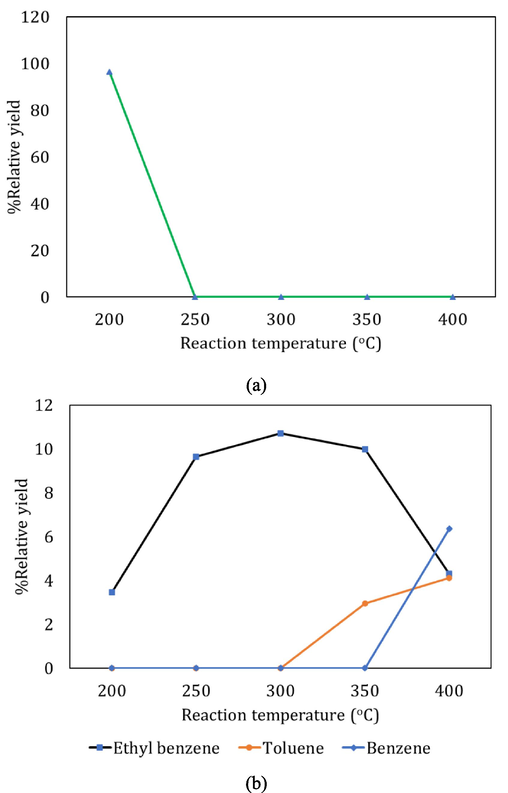
Product yields corresponding to (a) Isobutene, 1-butene and 2-butene as aliphatics, (b) benzene, toluene and ethyl benzene as aromatics, from hydrogenation of BD using Ni/niobia catalyst.
In the case of Ni/Al2O3, the yield of aliphatic compounds consisting of the butenes and isobutene was the highest at all temperatures as compared to the other products as seen from Fig. 7a and 7b. However, it can also be seen from Fig. 7a that the yield of aliphatic butenes (both linear and branched) decreased from 95 % at 200 °C to ∼77 % at the highest temperature of 400 °C. In this same temperature range, the yield of aromatics, especially benzene and toluene increased from 0 to 17.5 % and 0 to 3.6 %, respectively. Interestingly, the yield of ethyl benzene showed a slight decrease from 3 to 2 % from 200 to 400 °C. On this note, it was interesting to note that ethyl benzene was formed at all temperatures but toluene and benzene started forming only at temperatures >300 °C. Fig. 9 presents the plausible reaction chemistry of formation of ethyl benzene from BD.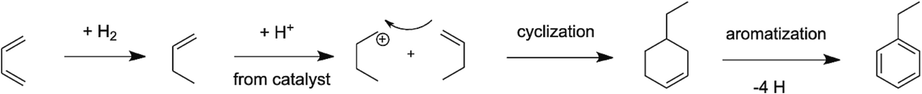
Reaction sequences leading to the formation of ethyl benzene from BD and 1-butene.
Aromatization and cyclization of n-alkanes and alkenes are not uncommon and are supported by sufficient evidence from literature using Ni-based as well as Zn, Zeolite, Alumina composites (Ishihara et al., 2020). These types of mechanisms are also seen extensively in petroleum feedstocks and biomass during their thermal and catalytic conversions (Ali et al., 2023; Kuttiyathil et al., 2023; Puliyanda et al., 2020; Sattari et al., 2020; Sivaramakrishnan et al., 2018, 2021, 2019; Sivaramakrishnan, 2019). Further confidence of dimerization, oligomerization, cyclization, isomerization and aromatization steps occurring in this conversion of 1-butene to aromatics were seen from the work by Sivaramakrishnan et al., (Sivaramakrishnan et al., 2018). Starting with propylene (C3), they obtained a range of products from C1 – C10 hydrocarbons including linear and branched aliphatics, dimers, aromatics with side chains and benzene, which were all observed from GC–MS results. This range of products and the observed reaction chemistry was mainly attributed to the acidic nature of the catalyst used and was observed in other oligomerization reactions with simple alkenes as well (Fuchs et al., 2023; Liu et al., 2019; Quann et al., 1988). The formation of 1-butene is a result of simple hydrogenation of one of the 2 double bonds present in BD. The only possibility to form C6 or greater numbered hydrocarbon compounds from a lower numbered hydrocarbon such as butadiene or 1-butene is carbon addition combined with cracking. An acidic medium is a requirement for activating the double bond of olefins and in our case, the lower acidity of alumina was enough to activate 1-butene, which then forms a primary carbocation. The Bronsted acidity of the alumina and niobia cataysts has already been discussed in section 3.1. This carbocation is attacked by another molecule of 1-butene since the carbocation is electron deficient and electron rich double bond in 1-butene is in its proximity in the reaction medium. This will lead to the formation of a secondary C8 carbocation, which will cyclize to form the ethyl cyclohexene. Next, four hydrogens are lost from the naphthenic ring through hydrogen transfer to form the more stable and resonance-stabilized aromatic, ethyl benzene as shown in Fig. 9. It is important to note that ethyl benzene was formed at all temperatures and hence, this mechanism has a high probability to occur.
Toluene is seen to appear only at 300 °C as shown in Fig. 7b. This suggests more cracking of the side chains as the temperature increases and it is easier to form toluene from ethyl benzene since the bond dissociation energy of the benzyl C-terminal C of the ethyl group in ethyl benzene is lesser than a normal aliphatic C—C bond (Sivaramakrishnan et al., 2021; Wang et al., 2014; Sivaramakrishnan et al., 2019). Cracking follows a free-radical mechanism, which causes the benzyl carbon to bear the free radical from the homolytic cleavage of C—C that requires a high temperature as observed in our work. This benzyl free radical is further stabilized by resonance with the aromatic ring. Further breakage of the Aryl C-benzyl C bond is difficult since the bond dissociation energy of this bond is 433 kJ/mol and this is equivalent to abstracting a hydrogen radical from methane, which is equally difficult (Blanksby and Ellison, 2003). But since benzene forms only at 350 °C and 400 °C, these higher temperatures seem to be sufficient to overcome this barrier of Aryl C-benzyl C bond scission. There is evidence in literature for the breakage of this bond to form benzene at these exact temperatures during thermal cracking of n-hexadecane in aromatic solvents (Khorasheh and Gray, 1993). Sequences of cracking mechanisms of the pathway from ethyl benzene to toluene to benzene, along with the bond dissociation energies of the cracked bonds are provided in Fig. 10. It is interesting to see that the bond dissociation energy (BDE) of a C—C aliphatic bond decreases to 320 kJ/mol in a benzyl carbon from a normal C—C sigma bond, where the BDE is 365 kJ/mol due to resonance stabilization of the free radical formed (Blanksby and Ellison, 2003). Furthermore, the Aryl C-benzyl C is the toughest to break since the BDE increases to 433 kJ/mol since breakage of this bond disturbs the aromatic sextet and leads to instability (Zachariah and De Klerk, 2016).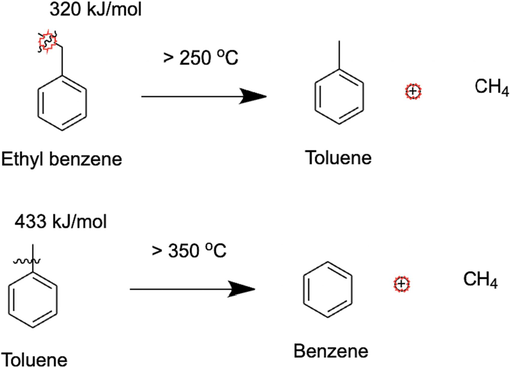
Cracking pathway from ethyl benzene to toluene to benzene at higher temperatures.
Similar trends were seen in the yields of aromatics when Ni/niobia was used as the catalyst for BD hydrogenation. Ethyl benzene formed at all temperatures while toluene appeared only at temperatures >300 °C. Due to the difficulty of breaking the aryl C-benzyl C bond, benzene is seen to form only at the highest temperature of BD hydrogenation carried out in this work. Moreover, the yields of ethyl benzene and toluene were higher when niobia was used as the support and this can be attributed to the higher Bronsted acid sites in niobia as compared to Ni/alumina (Sobczak et al., 2010). Ni is more selective towards aromatic formation than aliphatic compounds as seen from previous studies (Qin et al., 2023; Tercenio and Alexanian, 2019; Veses et al., 2015). Moreover, effective dissociation of H2 was not possible in niobia as compared to alumina due to lower MSI and lower BET surface area as extensively discussed in previous sections 3.1.3 and 3.1.4. This resulted in lower hydrogenation of the double bonds since aliphatic butenes were observed only at the lowest temperature of 200 °C (Fig. 8a). As the temperature increased, aromatization preferably occurred through protonation of the double bonds in the butenes and addition of this created carbocation to other butene molecules to form the cyclic naphthene. Hydrogen transfer from these cyclohexenes led to ethyl benzene and subsequent cracking of the side chains formed toluene. The pathway from toluene to benzene is more difficult and this is shown in Fig. 10. On the other hand, the formation of 2-butene from BD was possible through isomerization of the butyl carbocation shown in Fig. 9. Through this isomerization, the positive charge will shift to the second carbon, making it a secondary carbocation, which is more stable than the primary carbocation due to inductive effects satisfying the electron deficiency at the carbon. On hydrogen abstraction from this carbocation, 2-butene will be formed.
For the niobia-supported Ni, the conversion of BD was found to be very low apart from the lowest temperature of 200 °C. On analyzing of the spent catalysts through SEM-EDS, TGA and Raman spectroscopy, we found more carbon deposits on the niobia catalysts as compared to alumina and these results and a detailed discussion on the nature of carbon deposits and mechanisms of deactivation are provided in section 3.3 in this work.
3.2.2 FTIR analysis of products from BD hydrogenation
Fig. 11a and b show the FTIR spectra of the products from the hydrogenation reactions on butadiene using both catalysts separately (Ni/Al2O3 and Ni/Nb2O5) respectively. FTIR is a strong instrument to identify the functional groups that are present in the products and the inferences can be correlated with those from the GC–MS analysis described in the previous section (Lankmayr et al., 2004; Shittu et al., 2022; Shittu and Altarawneh, 2021; Sivaramakrishnan et al., 2018, 2021; Söyler and Ceylan, 2021; Sun et al., 2022; Sivaramakrishnan et al., 2019; Sivaramakrishnan, 2019). The first observation from the FTIR spectra of the hydrogenation products obtained from both catalysts is that of butadiene at the lowest temperatures of 50 and 100 °C, which indicates no conversion of BD at these temperatures. The characteristic peaks for BD are indicated in Fig. 11a and assigned to the respective entities as follows: sp2—C—H bends at 900–1000 cm−1 since BD can be considered as a mono-substituted alkene on both double bonds, the C⚌C stretch at 1600 cm−1, sp2—C—H symmetric and asymmetric stretches at ∼3000 and ∼3050 cm−1, respectively. As the temperature of the reaction increases above 150 °C, we notice the gradual occurrence of the peak at 680 cm−1, that corresponds to bending vibration of the aromatic ring.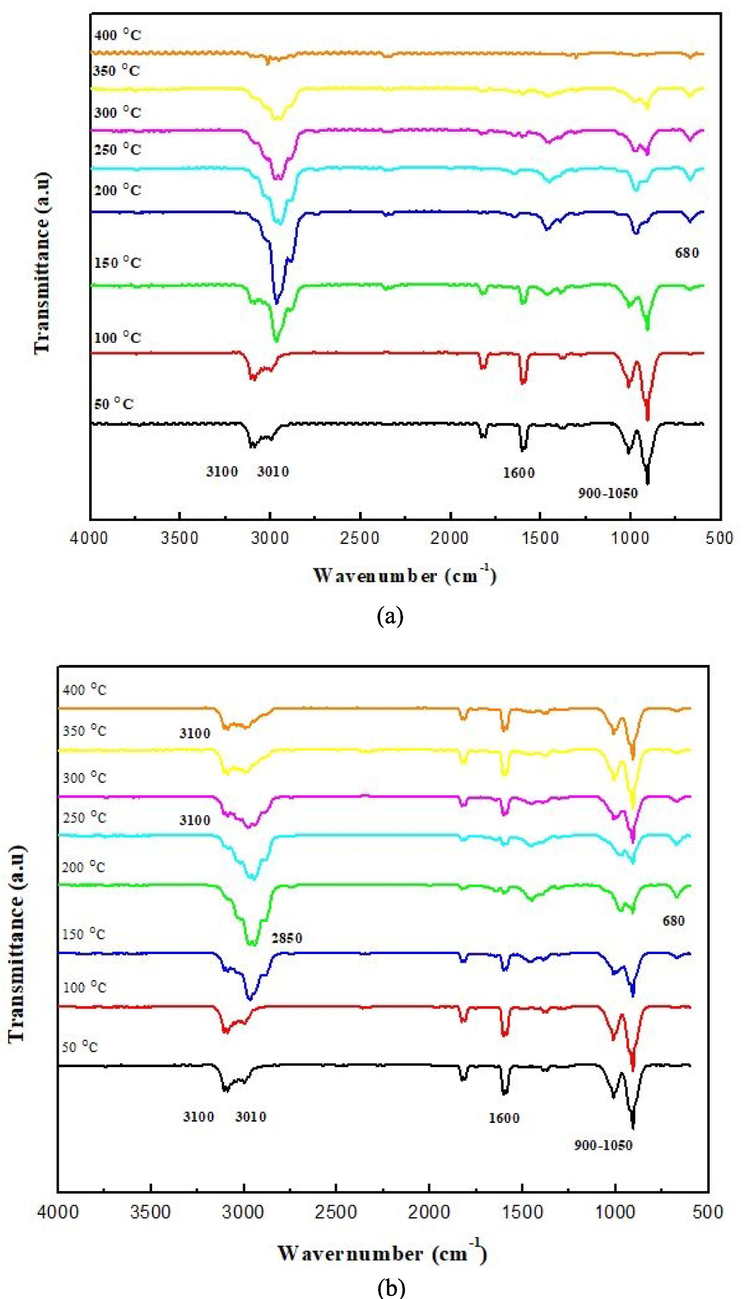
FTIR spectra of the gaseous products from hydrogenation of butadiene using: (a) Ni/Alumina (Ni/Al2O3) and (b) Ni/niobia (Ni/Nb2O5) catalyst.
This indicates the presence of aromatic rings in the products of hydrogenation after 200 °C for both catalysts. Through the hydrogenation of BD, the C⚌C bond is broken and replaced by the C—C sigma and sp3—C—H bonds, resulting the intensity of the C⚌C stretching vibrations to decrease, and consequentially the increase in the intensity of sp3-C—H stretching vibrations at 2850 and 2950 cm−1. Specifically, the 2850 cm−1 corresponds to the methylene group (—CH2—) in 1-butene and the stretch at 2920 and 2950 cm−1 corresponds to the terminal methyl groups in iso-butene, 1-butene and 2-butene. It is to be noted that the methylene group is present only in 1-butene as well as in ethyl benzene, but not in 2-butene. However, the terminal methyl groups are present in the linear butenes, isobutene as well as in toluene and ethyl benzene. This distribution of the aliphatic C—H stretches between 2850 and 2950 cm−1 is summarized in Table 2 below. All these observations corroborate with the GC–MS results in the previous section and the proposed mechanisms for the reaction chemistry can be verified as well.
–CH3 (2920, 2950 cm−1): Terminal methyl
–CH2- (2850 cm−1): In-chain methylene
Iso-butene (2)
1-butene (1)
1-butene (1)
Ethyl benzene (1)
2-butene (2)
Toluene (1)
Ethyl benzene (1)
Total = 7
Total = 2
The numbers in the brackets for each compound indicate the number of that respective functional group present. It can be clearly seen from Table 2 that the number of terminal methyl groups are far more than the number of methylene groups in the products. Since the FTIR spectra is taken on the mixture of the products from hydrogenation of BD, we can see that the peaks for the terminal methyl groups at 2920 and 2950 cm−1 are higher than those of the methylene groups at 2850 cm−1 in Fig. 11 for both the catalysts.
The yields of aromatics increased with an increase in temperature as seen in the previous section. This was also reflected in the FTIR spectra of the products as the appearance of the aromatic ring bend at 680 cm−1 is paired with the peak at 3100 cm−1 for sp2—C—H stretch with the carbon attached to an aromatic ring (indicated in Fig. 11b). Aromatic compounds such as toluene and ethyl benzene are mono-substituted and possess 5 hydrogen atoms attached to sp2—C in the aromatic ring. The bending of this sp2—C—H bond is characterized by wavenumbers in the 750–900 cm−1 but since the bending vibrations from the alkenes are also present in this same range, overlap occurs, and we cannot clearly identify whether the bending vibrations come from the alkenes or aromatics. A similar explanation can be given for the C⚌C stretch at 1610 cm−1 since it can be from both the aromatic ring and from alkenes. Nevertheless, there is sufficient evidence from GC–MS and FTIR that the yield of aromatics increases as the temperature increases for both catalysts. The FTIR spectra of the products obtained at 400 °C using Ni/Al2O3 catalyst was not clear as it appears that all the peaks are close to the baseline and hence, cannot be interpreted.
3.2.3 A comparison with catalytic literature of Ni-based catalysts used for hydrogenation
As cited in previous sections in this paper’s discussion, Ni-based catalysts have been used extensively for the hydrogenation of olefins, dry and steam reforming of hydrocarbons and alcohols, methanation and hydrogenation of CO and CO2 as well as biomass upgrading. We have conducted a thorough literature review of these reactions and provided a summary of the information including the type of catalyst preparation method, reaction conditions, conversions of reactants and yields of major products in Table 3.
Catalyst
usedMethod of catalyst preparation
Conversion/yields
(%)Major Products
Feed
References
Pd/SiO2 with TiO2
500 °C and 1 atm
95
cis-2-butene, trans-2-butene and 1-butene
BD in air and H2 in helium
Lee et al. (Lee et al., 2003)
Au/Al2O3
Au/SiO2
Au/TiO2
190 °C and 10 atm
98
1-butene, trans-2-butene and cis-2-butene
BD in H2
Okumura et al. (Okumura et al., 2002)
Pd and Ps-Sn over Al2O3
80 °C and 1 atm
80
1-butene and butane
BD and H2 in helium
Pattamakomsan et al. (Pattamakomsan et al., 2011)
Au-Pd/Al2O3
Au-Al2O3
150 °C and 1 atm
100
but-1-ene but-2-ene trans-but-2-ene
BD and H2 in helium
Hugon et al. (Hugon et al., 2010)
Present work (5 % Ni/Al2O3 and 5 % Ni/Nb2O5)
200 °C and 1 atm, Incipient wetness impregnation
100
Iso-butene 1-butene 2-butene and aromatic compounds
BD and H2 in helium
4 % Ni, 5 % Nb-Ce
150 – 300 °C and 1 atm,
Incipient wetness impregnation71 % conversion
96 % selectivity to styreneStyrene and
ethyl benzenePhenyl acetylene and H2
Shittu et al. (Shittu et al., 2022)
Ni2P/Al2O3
100 °C and 0.3 MPa
In-situ reduction of NiAl-LDH>98 % conversion 88.2 % selectivity to styrene
Styrene and
ethyl benzenePhenyl acetylene and H2
Chen et al. (Chen et al., 2015)
60 % Ni/Al2O3
310 – 350 °C
Co-precipitation and wet impregnation100 % conversion 97 % yield of n-alkanes by co-precipitation
C15 – C18
n-alkanes, acids and estersSunflower oil and Waste Cooking oil separately and H2
Nikolopoulos et al. (Nikolopoulos et al., 2023)
Ni-Co/HZSM-5
400 °C and 1 atm
Impregnation83 % conversion of toluene
33 % aromatics, 22 % alcohols, 15 % aliphaticsAromatics, Aliphatics, alcohols, ketones, furans, esters, phenols
Toluene, cyclopentanone, acetic acid, furfural, guaiacol
Qin et al. (Qin et al., 2023)
Ni/La2O3, Ni/Al2O3
600 °C and 0.1 MPa
Citric acid-assisted sol–gel94 % ethanol conversion, 7000
mol/min H2 production, 90 % methane conversion
H2 and CO (syngas)
Ethanol steam reforming (C2H5OH + H2O),
methane dry reforming
(CH4 + CO2)Xiao et al. (Xiao et al., 2023)
Dilithium nickelate
25 °C and 5 bar
99 % conversion and 99 % yield of product
Triphenyl ethane
Sterically hindered Triphenyl ethylene and H2
Maier et al. (Maier et al., 2020)
Ni/TiO2
200 °C and 1 atm
4 % CO conversion
51 % yield of CH4
C1, C2, C3, C4, C5 paraffins
CO and H2
Vannice and Garten
From the above table, it is apparent that different types of supports have been used along with Ni as the active metal for the hydrogenation of a number of feedstocks such as CO, sterically hindered triphenyl ethylene, model bio-oils, and phenyl acetylene. Ni-based catalysts have also been frequently utilized for the reforming of ethanol and methane. Moreover, having compared our experimental results in this work with a few previous investigations on BD hydrogenation, we observed that none of the previous works utilized Ni/niobia or alumina as the catalyst in the gas phase and neither have they reported the production of BTX aromatic compounds as part of their products. Razmgar et al. (Razmgar et al., 2022) investigated the pathways of hydrogenation of BD through density functional theory (DFT) studies and suggested that hydrogen transfer was a crucial step in the mechanism. Aromatization occurs majorly through hydrogen transfer and this not only validates our mechanisms discussed for aromatics production with moderate yields but also differentiates our work from previous studies that have worked on BD hydrogenation as shown in Table 3.
3.3 Spent catalysts analysis
After performing the hydrogenation reaction, the spent catalysts were analyzed for examining their physical and chemical properties to check their current state and extent of deactivation. There are various reasons for deactivation such as, poisoning by impurities, cocking, sintering and attrition (Olafsen et al., 2006; Zhang and Huang, 2009). In this work, two techniques were performed on the spent catalyst, namely, SEM-EDS and TGA. The former provided information on the catalyst’s structure, surface morphology, homogeneity/heterogeneity and elemental composition while the latter helped us evaluate the effect of the hydrogenation reaction on the thermal stability of the samples.
3.3.1 SEM-EDS
Fig. 12 and Fig. 13 show the SEM images and EDS mapping of both spent catalysts after their use in the hydrogenation reaction. In the case of both spent catalysts, their SEM images revealed changes in surface homogeneity, % composition of each atom, metal dispersion and indicated the formation of new species on the surface. These changes definitely affected the catalytic performance of Ni/Nb2O5 and resulted in a significant deactivation of the catalyst during the reaction. In Fig. 3, the surfaces of the calcined samples were porous, thus providing space for more adsorption of reactant molecules and their interaction. (Shittu et al., 2022) It could be seen that the Nb-supported Ni catalyst displayed a finer and smoother surface than the Al-supported catalyst. However, after reaction, the morphology of the Nb2O5 catalyst changed and showed more cluster-type moeities (Fig. 13) than the unreacted samples. These clusters represent aggregation on the surface, as confirmed by Jiang et al. (Jiang et al., 2021) Shittu et al. (Shittu et al., 2022) observed that this aggregation decreased as the loading of the active metal decreased, and hence, we adopted a constant loading of 5 % in our work. It should be mentioned that there were regions of dot intensities for the uncalcined samples (Fig. 3) but these were removed on calcination. The calcined samples do not show aggregated clusters and hence, the changes in the spent catalysts can be attributed to the carbon deposits on the surface from the reaction.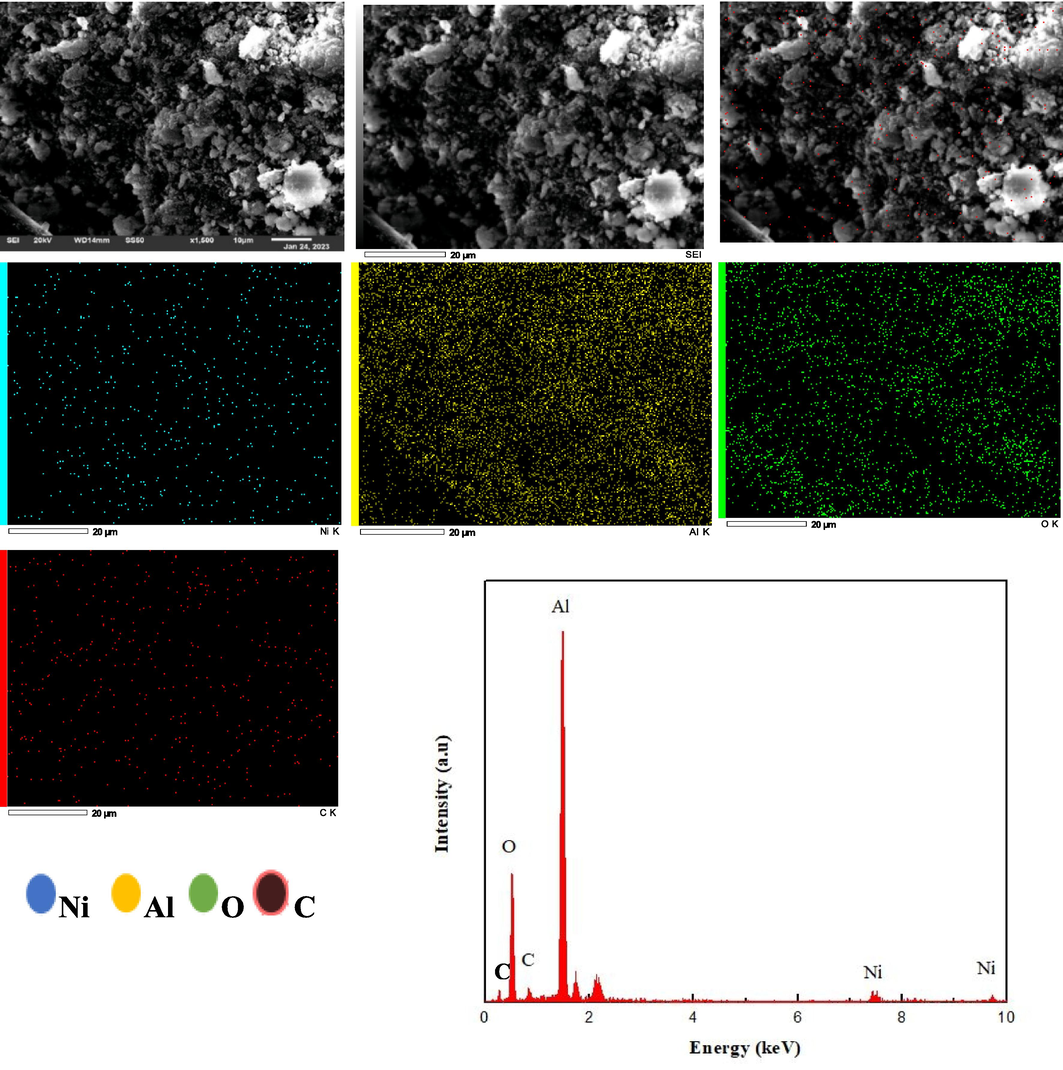
SEM images of the spent catalyst (5% Ni/Al2O3) along with its EDS spectra and mapping.
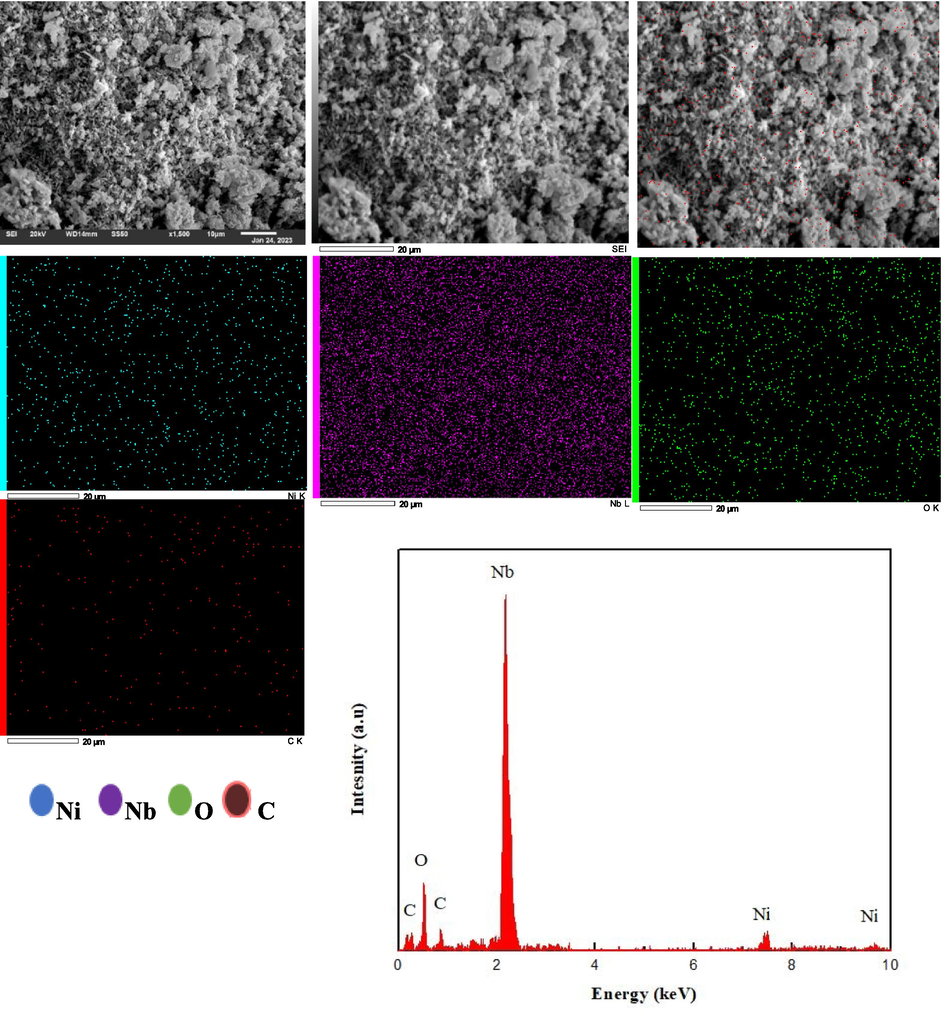
SEM images of the spent catalyst (5% Ni/Nb2O5) along with its EDS spectra and mapping.
The distribution of the elements in both spent catalyst after the reaction is clearly mapped out in the EDS as shown in the other parts of Figs. 12 and 13. We had already seen that the elements were uniformly distributed for the unreacted catalyst (Fig. 3) and this distribution was not disturbed by much in the spent catalysts. The only difference was that carbon was the additional element present in the spent catalyst, as indicated by the pink dots in Figs. 12 and 13 for alumina and niobia catalysts, respectively. Wang et al. (Wang et al., 2020) have discussed the uniform distribution of the catalyst support in order to enable effective metal dispersion, which in turn paved the way for increased catalytic performance. In our work, regions of dot intensity were clearly seen for Al in the EDS spectra of the uncalcined samples but these regions disappear in the spent catalyst. There were some regions of dot intensity seen in the spent catalyst spectra for Ni/Nb2O5 and these confirm the aggregates formed as seen in the SEM images. Interestingly, the EDS spectra is generally not known to capture all the aggregates in the catalyst surface (Ali et al., 2023; Shittu et al., 2022). Furthermore, the elemental mapping composition showed a percentage mass ratio of 40.67 1.78 % for O, 35.48 0.94 % for Al, 6.45 0.11 % for Ni and 17.31 0.77 % for C. The presence of carbon mostly arises from coke deposits, which caused a partial deactivation of the catalyst (Ronda-Lloret et al., 2020; Shittu et al., 2022). However, this did not affect the performance of the Ni/Al2O3 as much as it affected Ni/Nb2O5, as seen before. EDS analysis determined the presence of Ni, Al, O and C species in the catalyst matrix and a point analysis of the samples are presented as the intensity vs. energy plots in Figs. 12 and 13 for alumina and niobia, respectively. Corresponding to the spectra of alumina catalyst peaks of Ni are seen at 1.1, 7.6 and 8.1 KeV, while peaks of O and Al occur at 0.8 and 1.6 KeV, respectively. The peaks of C are seen to occur at 0.4 and 0.8 KeV and all these peaks in the EDS spectra occurred at their respective characteristic values, as verified from literature. (Roy et al., 2018; Wang et al., 2017).
For niobia catalyst, the percentage mass ratio of Ni, Nb, O and C were calculated to be 5.9 0.09 %, 56.91 1.34 %, 23.01 1.08 % and 14.18 0.59 % one-to-one, respectively. In addition, the atomic percentage ratio came out to be 34 1.10 % of carbon, 44 1.18 % of oxygen, 3.1 0.03 % of nickel and 18.1 0.81 % of niobium. The higher % of carbon in the niobia catalyst was also reflected in the EDS plots since the peaks of C at 0.4 and 0.8 KeV were seen to have a higher intensity (Fig. 13) than for the alumina catalyst (Fig. 12). As mentioned before the carbon sites on the surface affected the active catalyst surface and led to a partial deactivation of the catalyst during the reaction. The presence of Ni, Nb, O and C species were located in the EDS spectra of niobia catalyst at similar positions as that of alumina catalyst as: Ni at (1.1, 7.4 and 8.3) KeV, Nb at (0.3, 1.9 and 2.2) KeV, O at (0.7) Kev and C at (0.4 and 0.9) KeV. It is postulated that the carbonaceous coke occupied the vacant active sites of the catalyst and decreased the activity of the niobium catalyst towards BD hydrogenation.
3.3.2 TGA analysis: Nature of carbonaceous deposits
TGA analysis of both the spent catalysts was performed in air with a ramp change of 10 °C min−1 to measure the mass loss of the samples as a function of temperature as well as to analyze thermal stability and any coke deposition. The TGA curves shown in Fig. 14 present a clear picture of the carbonaceous deposits in each catalyst.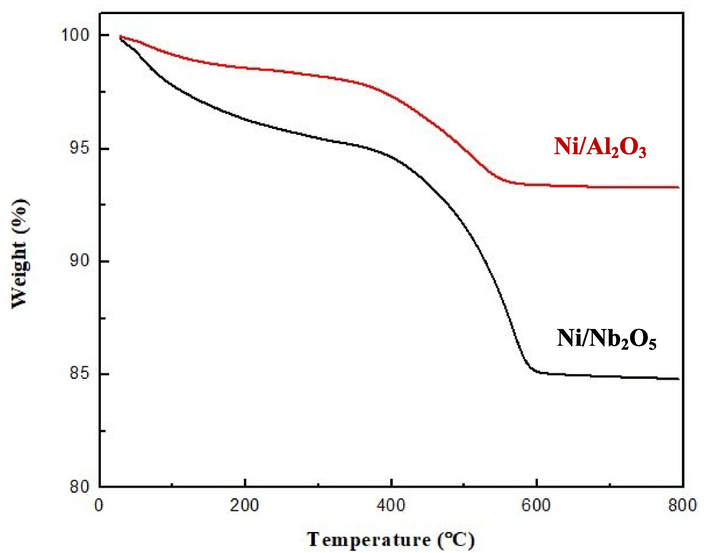
TGA analysis for both the spent catalysts used for BD hydrogenation in thiswork.
Ni/niobia showed a 15 %wt. loss while Ni/alumina showed a 8 %wt. loss and this thermal decomposition occurred till ∼590 °C for Ni/Nb2O5 but only till 490 °C for Ni/Al2O3. Apart from the removal of any adsorbed water and volatileorganic compounds generated during the reaction, this weight loss was primarily attributed to the decomposition and oxidation of carbonaceous deposits on the surface of the catalysts (Riley et al., 2018; Xiao et al., 2023). For clarity purposes, we will term these carbonaceous deposits formed on the catalysts after reaction as coke. Coke is essentially a mixture of high carbon number compounds that are heavily deficient in hydrogen as compared to the corresponding reactant used in that reaction. They can be both aliphatic or aromatic in nature or a combination of both with long-chain aliphatics and polyaromatic rings fused together, or just amorphous carbon, and contain heteroatoms such as S, O, N. It is evident that coke has a much larger molecular weight than the corresponding feedstock and it can stick to the catalytic surface and pores, causing deactivation. We have already seen that Ni/alumina displayed a stronger metal-support interaction (elaborately discussed in section 3.1.4 from our H2-TPR results and XPS studies in literature), and consisted of smaller Ni-crystallites on its surface (which were not detected by XRD but were detected for Ni/niobia). The lower binding energy shift in the XPS of the reduced Ni/niobia catalyst also indicated a weaker MSI for Ni/niobia compared to Ni/alumina. All this reflected in the catalytic performance and the distribution of products obtained from these two catalysts as previously discussed in section 3.2.1 and 3.2.2. It was also seen that Ni/niobia was more prone to sintering during the reaction due to agglomeration of the niobia support species and Ni-Ni particles as discussed in section 3.1.3, which was also evident from SEM images of the spent catalysts (Figs. 12 and 13). (Méndez et al., 2017) This section also provided a number of possible reasons for the lower surface area found for Ni/niobia as compared to Ni/alumina and this combined with more carbonaceous deposits can easily plug the active sites and pores, causing lower yields and conversion, as observed in our work. Xiao et al. (Xiao et al., 2023) attributed the lower carbon deposition in Ni/La2O3 directly to the stronger MSI and smaller size of Ni particles dispersed on the catalyst surface during their investigation of ethanol and methane reforming using Ni/La2O3 and alumina. The temperature corresponding to the oxidation of encapsulated carbon on the catalyst was ∼594.4 °C and as the amount of carbon deposits decreased, this temperature of mass loss in the TGA of spent catalyst decreased, very similar to our work. They directly correlated the activity, performance and stability of the catalyst to the carbon deposits on the surface of nickel and hence, we can attribute the higher conversion of BD observed in the Ni/Alumina catalyst to its lower coking tendency. All these observations were also confirmed with high confidence by Zhang et al., (Zhang et al., 2018) where Ni/Al2O3 showed lower coking tendency than Ni/SiO2 and they attributed this to stronger MSI and lower crystallite sizes of Ni particles.
Furthermore, it was commonly observed that the more the acidic sites on the catalyst, the higher the probability of coke formation and vice versa for basic sites. This is related to the reaction mechanism as a higher acidity through Bronsted sites in Ni/niobia (this was discussed in section 3.1.3) promotes protonation of the BD molecules and promotes the dimerization and aromatization reactions as already depicted in Fig. 7. These observations were also confirmed by Schmal et al. (Schmal et al., 2000) It was already discussed in section 3.2 that Ni/niobia only yielded aromatics at higher temperatures whereas Ni/alumina yielded more aliphatics than aromatics at all temperatures. The cyclization and aromatization reactions increased at higher temperature and lead to eventual formation of coke by development of fused aromatic rings. Wauters and Martin (Wauters and Marin, 2001) provided a sequence of steps based on free-radical mechanism, for the fusion of aromatic rings into denser cores. Due to the absence of free-radicals in our reaction sytem, these steps were slightly modified as a carbocation mechanism and applied as for BD conversion in our work and shown in Fig. 15. These are summarized as follows: (i) creation of a carbocation by adsorption of BD to a Bronsted acid site in Ni/niobia to form butene carbocation (compound 1). It is important to note that these Bronsted sites can be created by dissociation of H2 on the Ni surface as well; (ii) electrophilic attack of the electron-deficient carbocation on the electron rich aromatic ring in benzene/toluene. It is important to note that the aromatic ring in toluene is more active towards this type of electrophilic substitution at the ortho and para positions due to the positive inductive effect of the –CH3 group in toluene. If this is the case, there will be side chains attached to the fused aromatic core as shown in compound 2; (iii) ring closure by C = C attack on the proximate positive charge formed in the previous step to form compound 3; (iv) loss of hydrogens from the cyclic ring through hydrogen transfer/abstraction to form a fused aromatic (methyl naphthalene – compound 4). It is important to note that once a layer of initial coke is formed, this sequence of steps will continue to happen on the surface of the coke with electrophilic attack of the carbocations from the gas-phase to increase the coke deposits. These types of reactions are very common in petroleum feedstocks and pyrolysis of biomass as well (Qi et al., 2019; Sivaramakrishnan et al., 2021; Yañez Jaramillo and De Klerk, 2018; Zachariah and De Klerk, 2016; Sivaramakrishnan et al., 2019). Schmal et al. (Schmal et al., 2000) refers to this type of coke formation as the ‘poliene’ route for olefins reacting on catalysts with the presence of acid sites. Thus, we can confidently say that the higher acidity of Ni/niobia, along with weaker MSI, larger crystallite size of Ni, agglomeration of Nb2O5 species, increased electron density of Ni were all contributing factors for the higher carbonaceous coke deposits on Ni/niobia as compared to Ni/alumina.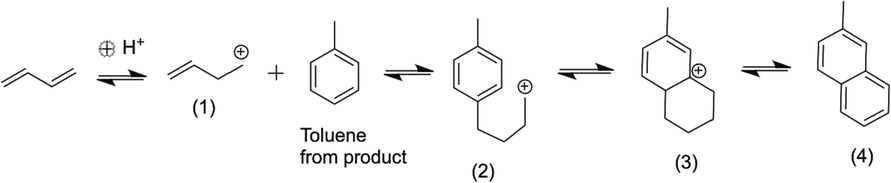
Schematic depicting formation of aromatic coke with fused rings.
Vogt et al. (Vogt et al., 2023) have provided a comprehensive review of the mechanism and nature of carbon deposits responsible for catalyst deactivation in different chemical conversion reactions. Interestingly, apart from concurring with our proposed mechanism in Fig. 15, they also suggested that deactivation could be initiated by trapping of branched aliphatics in the cavities, which can hinder diffusion of reactants and promote accumulation of products in the active sites. But Qian et al. (Qian et al., 2014) confirmed that the probability of polyaromatics causing the deactivation was much higher than lower carbon-numbered aliphatics. With respect to alkene hydrogenation/de-hydrogenation processes, the type of coke was also found to depend on the nature of the active metal employed as the catalyst. For example, when Pt-Sn bimetallic catalyst was used for propane dehydrogenation, 2/3rds of the coke was reported to be aliphatic in nature (Sattler et al., 2014). The reminder was graphitic and aromatic in nature and deposited on Pt. However, as the reaction progressed, most of the aliphatic deposits converted into fused aromatics and the formed propene had a similar reaction with the aromatics in the presence of acid sites as shown in Fig. 15. Alkenes are very reactive and sometimes do not need an acid site to promote coke formation as shown in Li et al. (Li et al., 2011) and Lian et al. (Lian et al., 2021) They noted the formation of ‘soft coke’ composed of oligomers of propene and alkene monomers with < C5 carbon number. However, as mentioned before, this low-carbon route for coke formation was thought to be less-probable for niobia-based catalysts in literature as well. (Schmal et al., 2000) This eventually gives way to ‘hard coke’ through isomerization, cyclization, aromatization and ring closure reactions and this was the traditional coke-forming process in a number of other reactions such as thermal cracking, pyrolysis hydrotreating, dehydro-aromatization, and other conversions involving hydrocarbons and alkenes (Vogt et al., 2023). Evidence of hard coke was apparent from Raman spectra of the Ni/niobia catalyst and readers are referred to section 3.3.4 for a discussion on the different bands of carbon found here. Hence, these points further enhance the confidence in the mechanism proposed in Fig. 15 for coke formation and suggest that the nature of carbonaceous deposits will mostly be aromatic for BD hydrogenation. When zeolites are involved, coke formation was proposed to occur via a carbenium-ion-based mechanism (Ono et al., 1987).
Since Ni-based catalysts were also used extensively for reforming, it was interesting to note the type of carbon deposits on Ni/SiO2 and Ni/Mg(Al)O, a hydrotalcite-derived catalyst in the reforming of methane and propane conducted by Olafsen et al. (Olafsen et al., 2006) They conducted magnetic measurements to inspect the state of Ni after reaction and found that a decrease of ferromagnetism of Ni was directly proportional to propensity of coke formation. They noted the formation of 2 kinds of coke deposits: (i) low temperature-carbon that started accumulating initially but could be removed by hydrogenation at < 500 °C; (ii) high temperature-carbon that formed later on but remained stable for long time. The low temperature carbon was a carbide form of Ni as NiC, Ni2C, Ni3C and depended on the Ni-loading, reaction conditions, and nature of the support. On the other hand, the high temperature carbon was largely graphitic in nature similar to that formed in alkene hydrogenation/dehydrogenation discussed in the previous paragraph. Other forms of low temperature carbons are amorphous polymeric films or filaments referred to as , that forms from atomic C (Bartholomew, 2001). On long exposure to the high temperature, they can eventually be in the form of carbon nanotubes formed by carbon extrusion from dense planes. These nanotubes can deposit on the catalyst surface, thereby limiting the access of the reacting gases to the active sites and eventual deactivation. Hence, the forms of carbon deposits in BD hydrogenation can take the forms of fused aromatic rings, carbides, nanotubes, and encapsulating deactivating carbon. Evidence for the presence of carbon was clear from SEM-EDS analysis in section 3.3.1 as well, where the intensity of elemental carbon in the EDS of Ni/niobia (Fig. 13) was much higher than that for Ni/alumina (Fig. 12). Furthermore, Olafsen et al. (Olafsen et al., 2006) also noted that strong MSI hindered the formation of carbon nanotubes and this led to the formation of in-situ easily removable low-temperature soft-coke for these catalysts. This clearly corroborated with our observations and discussion for Ni/alumina catalyst and further validated the type of carbonaceous deposits on each catalyst and the reasoning behind this phenomenon. Lastly, it can also be reasoned that O-availability was much more in Ni/alumina than in Ni/niobia, and this could have limited the formation of hard-coke in the former. This was because of better particle dispersion and strong interaction of Ni with the support and forming the spinel phase in the Ni/alumina catalyst. Similar observations were made in literature comparing different Ni-based catalysts as well (Olafsen et al., 2006).
3.3.3 Mechanisms of catalyst deactivation and ways of mitigation
Bartholomew (Bartholomew, 2001) classified the mechanisms of catalyst deactivation into the following types: (i) fouling, (ii) poisoning, (iii) attrition/crushing, (iv) sintering due to thermal degradation, (v) vapor compounds formation, (vi) chemical reactions involving vapor-vapor, solid–solid phases. This is a serious problem that can cost millions of dollars to industries if not dealt with properly. The types of deactivation that is relevant to BD hydrogenation in Ni-based catalysts are poisoning and fouling. Poisoning is related to the chemisorption of unwanted molecules or impurities on the active sites of the catalyst. They can either block the sites that interact with the reactants for their adsorption or change the chemical and geometrical environment of the active and support species, making it incompatible to the reactants. Unsaturated hydrocarbons can turn into poisons when they undergo chemisorption on the metal sites through multiple bonds and the empty 3d orbitals in Ni accommodate the -electrons. Arsenic and sulfur compounds are also typical poisons for hydrogenation catalysts. The poisoning extent also depends on the number of molecules chemisorbed, the relative sizes of the molecule compared to the active metal and the strength of the bond. (Satyanarayana et al., 2016) A large poison such as arsenic or sulfur can block more than one active site in Ni and can also affect the electronic environment of its adjacent atoms, thereby affecting their adsorption energy. Reactant molecules that are adsorbed on the metal surface need to diffuse in order to react with each other. Poisoning also affects this phenomenon in BD hydrogenation, hindering the diffusion of BD molecules within the catalyst surface. Interestingly, the degree of toxicity of the poison decreases with increasing positive charge or decreasing electron density on the atom acting as the poison. For example, SO42- (oxidation state of S is + 6) is a less potential poison than H2S (oxidation state of S is −2). In fact, H2S is the most common catalytic poison in hydrocarbon hydrogenation processes due to the sources of the feedstocks being sour in nature (Bartholomew, 2001; Satyanarayana et al., 2016). Sometimes the poison selectively deposits on the more active sites than the less active sites and this is called selective poisoning. For example, Ni surfaces are prone to poisoning by H2S since H2S has a great affinity for Ni and undergoes dissociative adsorption (Bartholomew et al., 1982). This interaction is thermodynamically favorable and is not easily reversible (Bartholomew, 2001). S is dangerous to catalysts as it inhibits any other molecules from adsorbing on the Ni sites and results in significant reduction in catalytic activity. An effective way to deal with sulfur poisoning is doping the catalyst with Mo or Boron since they selectively adsorb S and sulfur-containing molecules (Erekson and Bartholomrw, 1983). On the other hand, if the atomic or molecular size of the poison increases, its potential to deactivate the catalyst increases. Acidic catalysts such as alumina and niobia are greatly affected by the presence of bases such as ammonia, and organic basis such as amines in the reaction media. Hence, we took great caution not to increase the pH of the reaction mixture during BD hydrogenation and no product was formed such that it created a basic medium. Studies have shown that surface coverage of S on Ni depends on the adsorption stoichiometry. The surface coverage remains below 0.25 if the adsorbed S is bonded to 4 atoms of a single Ni crystal and this increases to 0.5 if S is bonded to only 2 Ni atoms.
Fouling involves deposition of mechanically hard species such as carbon in its different forms and carbon-containing compounds such as coke on the surface of the catalyst. This blocks important pores and active sites and eventually damages the catalyst by disintegration. We have already discussed the mechanism of coke formation with respect to BD hydrogenation in the previous section. The location of the coke deposition is important in determining how they deactivate the catalyst. On the active metal, coke or carbon may adsorb physically or chemically similar to a poison, thereby preventing or reducing the adsorption of reactant molecules. Since their molecular sizes are relatively large, they can plug the mesoporous supports and this blocks reactants from diffusing into these pores. Coke can also deposit completely around the Ni particles and this will result in full deactivation. This separates the Ni active sites from the reactant molecules, reduces the MSI between Ni and the supports, and drives the Ni particles to agglomerate since they migrate to the outer surface of the support due to low MSI (Ma et al., 2020). Disintegration of the catalyst can result from long-term continuous deposition of coke in the pores, leading to fracturing of the support. For acidic supports such as alumina and niobia, once the initial layer of coke forms, it strongly adsorbs onto the acidic sites and decreases the activity. This can be an example of selective poisoning of the acidic sites by coke similar to S on Ni as discussed previously. In the case of soft coke leading to hard coke (section 3.3.2), the former forms on the metal first and then migrates to the support and accumulate there for the rest of the reaction time. In this way, retention of fused aromatic rings and clusters in the junctions of 2 pores are a main source for deactivation. Interestingly, if the rate of coke formation is lower and reaction temperature is high, it affects acid sites more. On the other hand, if the rate of coke formation is high and the reaction temperature is low, blockage of pores results in a faster deactivation than acid site neutralization (Satyanarayana et al., 2016). Hence, it would make sense to believe that Ni/niobia deactivated through blockage of pores rather than acid sites poisoning since the reaction temperature was low and rate of coke formation was relatively higher than with Ni/alumina in our work.
Creation of oxygen vacancies in the NiAl2O4 spinel phase (discussed in section 3.1.4) promotes the adsorption and fixation of CO2 molecules, which in turn yield O radicals (Zhang et al., 2021). Oxygen vacancies can be created by doping the support with multivalent cations (Razmgar et al., 2022). Carbonaceous deposits in the metal sites and supports can be oxidized by these oxygen radicals and result in in-situ coke removal. Based on this concept of oxidation of the deposited carbon, Al-Mubaddel et al. (Al-Mubaddel et al., 2021) found that the chemical adsorption of CO2 and its subsequent dissociation to form O radicals is favored when La2O3 was added to Al2O3 supports in Ni-based catalysts. Li et al. (Li et al., 2017) recently proposed an interesting mechanism occurring when lanthanum oxide was added to alumina and the carbon deposits were oxidized to CO2. During the chemisorption of CO2 on La2O3, it can lead to a stable carbonate phase (La2O2CO3), which in turn is reduced by more C species on the catalyst surface, forming CO2 and in this process, removing the carbonaceous deposits. La2O3 can have similar effects for niobia catalysts as well and hence can be added to both catalysts for mitigating carbon deposition in BD hydrogenation. The carbonate phase reduces the acidic site concentrations as well, thereby limiting the aromatization reactions as well (Gao et al., 2021). On the whole, enhancing the basicity or reducing the acidity of the support improves the affinity for CO2 chemisorption, which in turn oxidizes the formed carbon deposits and coke. However, it should be noted that any further activation of CO2 molecules on the surface of the catalyst can affect the chemical environment of Ni particles as well.
Mesoporous alumina has been the focus of catalytic hydrogenation for the past 3 decades due to its larger surface area, ordered pore channels and potential for higher metal dispersion as compared to non-porous alumina (Gao et al., 2021). Confinement of Ni0 species within the mesoporous structure provided greater surface area, higher dispersion, limiting the harmful effects of coke deposition as well as improving resistance to sintering (Gholizadeh et al., 2021). The imposed confinement of Ni0 species in the support limited their agglomerating tendencies as well. Overall, these modifications improved the conversions of CO2 in its hydrogenation reactions as reported by multiple studies (Gholizadeh et al., 2021; Jabbour et al., 2019). Sulfiding of niobia catalysts that are susceptible to hard coke formation reduces the rate of aromatization, ring closure and polymerization of olefins and mitigates coke deposition, thereby prolonging the life of the catalyst (Bartholomew, 2001; Satyanarayana et al., 2016). Another way to deal with the loss of active sites on metal active sites of Ni is to form alloys with noble and other transition metals such as Rh, Ru, Pt, Fe, Co, Sn, which are all compatible with Ni (Gao et al., 2021). This will stabilize the structure, provide more active sites for reactant adsorption and chemisorption, and increase the potential for MSIs. Alloys help in decreasing the metallic crystal size and this will also inhibit carbon deposition and enable oxidation of deposited carbon in addition to providing enhanced dispersion on the support (Yusuf et al., 2021). Sometimes adding an extra metal will enhance the oxygen mobility by creating more surface defects, and this improves the oxygen storage capability and helps in coke mitigation. Ceria is especially active towards improving the oxygen mobility due to its redox cycle between Ce4+ and Ce3+ transitions and their favorable interactions with empty d-orbitals of Ni. CeO2 oxidizes the deposited carbon to get reduced to Ce2O3 and alleviating the carbon deposits. In the dry CO2 reforming methane, Chen et al. (Chen et al., 2015) observed that the concentration of carbon deposits and graphitized carbon decreased significantly as the Ce content increased in the Ni/alumina support. Hence, it is seen that both the active metal and the support can be activated and modified to mitigate and reduce carbon deposition by adding another active metal species or doping the support with other metal oxides.
3.3.4 Raman spectra of the spent catalysts
Raman spectroscopy, which has demonstrated to be an effective method for evaluating the structure of carbon deposits, was also used to study the nature and characteristics of carbon deposits. Raman spectroscopy analysis was conducted on the representative spent Ni/Al2O3 and Ni/Nb2O5 catalysts after catalytic evaluation of BD hydrogenation reaction at 200 °C to study the type of carbonaceous deposition on the catalytic surface during the hydrogenation reaction. Fig. 16 represents the Raman spectra of both Ni-based catalysts employed in this work. Two typical peaks can be seen with hight intensity located at high Raman band positions of ∼1350 and 1600 cm−1 for Ni/Al2O3 and of ∼1290 and ∼1570 cm−1 for Ni/Nb2O5, demonstrating the D- and G-bands of carbon respectively. In addition, the D-band is formed by sp3 bonding defects of amorphous carbon or filaments of carbon deposited on the catalyst surface, while the G-band corresponds to the stretching mode of carbon's sp2 bonds, indicative of the emergence of structured, i.e., graphitic, carbon (Méndez et al., 2017). The occurrence of both D and G bands suggests that the carbonaceous deposition on the catalyst surface post the hydrogenation reaction was heterogeneous and featured filamentous and graphitic carbons (Charisiou et al., 2020). But, the intensities of the bands were higher for niobia and this clearly confirmed the higher coke deposition as compared to alumina.
Raman spectra of the spent catalysts after BD hydrogenation.
Conversely, the other Raman active modes (Eg, T2g and A1g) were not detectable for both spent catalysts, more likely due to the deposited carbon that was covering the catalytic surface. The outcomes indicated that graphitic carbon species had been deposited on the catalyst during the reaction. The presence of the G and D signals reveals an array of the carbon species produced by the reaction. Both catalysts D band intensities were nearly equal to their G band, indicating that the carbon structures of the used catalysts were similar. Based on these results, the nature of support strongly influenced the stability of the catalysts during hydrogenation reaction and besides contributing to producing different aliphatic products, promotes the presence of carbon precursors deposited on nickel surface during the reaction.
3.4 Stability tests for the catalysts
Catalytic stability analysis is an important aspect of evaluating the performance and durability of catalyst materials. We have performed catalytic stability tests for both the catalysts used in this work: Ni/Al2O3 and Ni/Nb2O5 in order to test the longevity of the catalysts, i.e., the maximum time for which the catalysts could resist coke deposition and subsequent deactivation at a particular temperature. These types of tests were also conducted previously in different studies to study the rate of deactivation and time required to show a decrease in catalytic activity. The concept of these tests is that if the conversion of each reactant and the yields of each product are relatively constant over a long period of time, the catalyst is said to be stable since the catalytic activity is not being inhibited by deactivation due to coke formation.
In this analysis, time-on-stream experiments for the Ni/Al2O3 and Ni/Nb2O5 were carried out up to a reaction time of 12 hrs at the lowest conversion temperature of 200 °C. It was important to note that both aromatics and aliphatic products were formed at this temperature using both catalysts but at higher temperatures, Ni/niobia was seen to produce only aromatic products. The reasons for these observations have already been discussed in previous sections and coke deposition was seen to be a major reason for the absence of aliphatic products when Ni/niobia was used at temperatures > 200 °C. In this stability test, the yields of the different products at different reaction times at a constant temperature of 200 °C have been shown in Fig. 17a and Fig. 17b for Ni/alumina and Ni/niobia, respectively. Prior to testing, the fresh catalytic samples were calcined in air for 5 hrs at 500 °C then reduced at 500 °C in H2 for a further 5 hrs. As observed, the expected products from the hydrogenation at this low temperature were obtained: isobutene, 1-butene, 2-butene and ethylbenzene. No BTX aromatics were observed at this temperature since it was already shown that the formation of aromatics occurred only at higher temperatures due to cyclization, aromatization through loss of hydrogen owing to hydrogen transfer reactions. The findings of this study show that Ni/Al2O3 is more stable than Ni/niobia since the yields of the products were in the small range ∼70–76 % for isobutene, ∼23–28 % for 2-butene and <1 % of ethylbenzene (Fig. 17a). The only exceptions were at 7 and 8 h, where isobutene decreased to 40 % and 2-butene increased to ∼60 % at 7 h while high amounts of 1-butene (75 %) was formed at 8 h with the remaining being isobutene.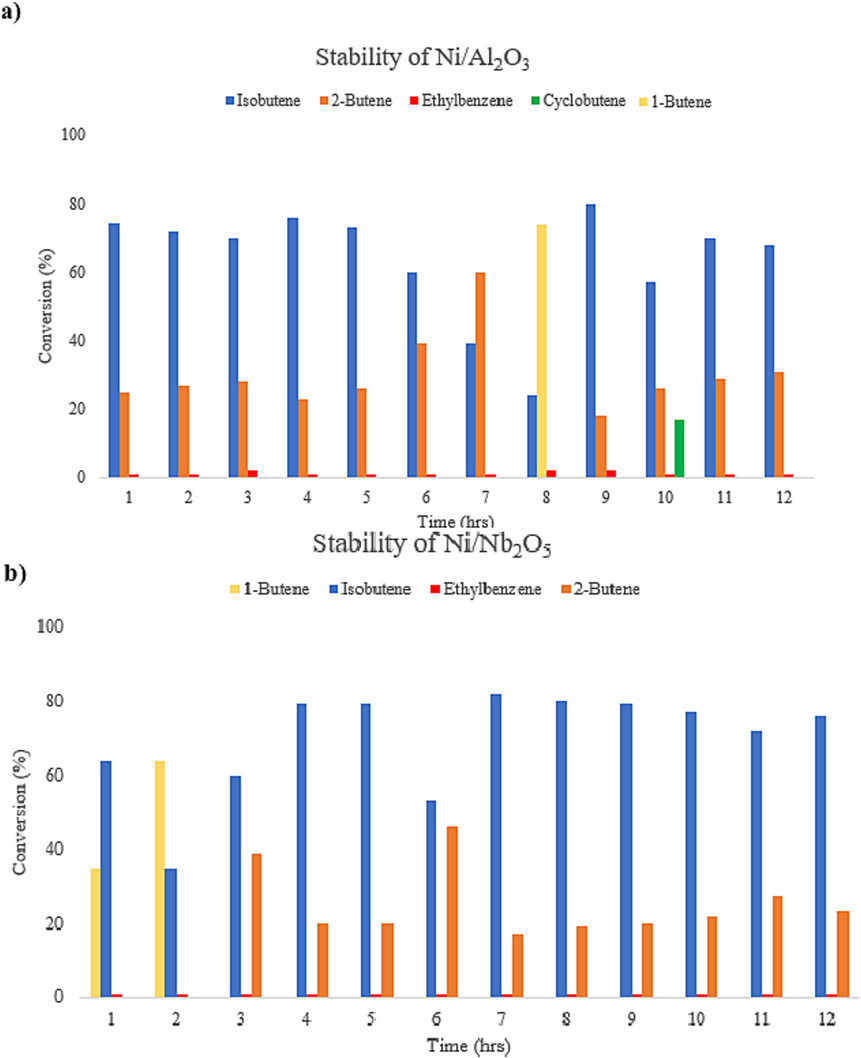
Stability analysis results for a) Ni/Al2O3 and b) Ni/Nb2O5. To be noted that the sum of the conversions for all products in each hour aggregates to 100%.
In contrast, the product distribution for Ni/Nb2O5 was more random with increase in time with much higher variance and spread in the %wts. of each product than that noticed with its alumina counterpart. We noticed the formation of 1-butene for the first 2 h at yields of 35 and 64 %wt. respectively and it disappeared completely afterwards (Fig. 17b). The maximum and minimum yields of 2-butene were 42 %wt. and 20 %wt. with a spread of 22 % whereas the spread in Ni/alumina was only 5 %. Similarly, the maximum and minimum yields of isobutene were 37 %wt. and 82 %wt. with a spread of 45 %, which was way higher than the 6 % observed when Ni/alumina was used over the same reaction times at 200 °C. Furthermore, the conversion of BD remained constant at 100 % at all reaction times when Ni/alumina was used but it showed a decreasing trend after 4 h when Ni/niobia was used (plot not shown here). However, Ni/niobia showed less-varying yields of the products after 6 hrs of reaction time with the range of isobutene and 2-butene as ∼70–82 %wt. and ∼17–27 %wt., respectively. Interestingly, both catalysts were more selective towards isobutene and 2-butene for majority of the reaction times at this lowest temperature of BD hydrogenation. Thus, the stability analysis of Ni/Al2O3 and Ni/Nb2O5 confirms the higher activity and lower coking resistance of Ni/alumina as compared to Ni/niobia and these observations are concurrent with previous inferences of stronger MSIs, more surface area, lesser carbonaceous deposits, low Ni-crystallite size, higher Ni dispersion and higher conversion of BD observed for the former catalyst.
4 Conclusions and future work
This work investigated the hydrogenation of butadiene (BD) over 2 Ni-based catalysts with different supports (Al2O3 and Nb2O5) at low temperatures between 200 and 400 °C with intervals of 50 °C and at 1 atm pressure. Ni/alumina catalyst showed better overall performance considering higher BD conversion at all temperatures and the yields of aliphatic and aromatic products. The yields of aliphatic products such as 1-butene, 2-butene and isobutene were much higher in the case of Ni/alumina than that from Ni/niobia, where no aliphatics were observed at temperatures >200 °C. Aromatics such as benzene, toluene, and ethyl benzene were produced when both catalysts were used but the yields of benzene and toluene were higher for Ni/alumina than that of Ni/niobia. Ethyl benzene was produced at all temperatures, signifying the role of cyclization, aromatization, and hydrogen transfer mechanisms in the reaction chemistry of BD hydrogenation. Furthermore, coke deposition was higher for Ni/niobia and this was attributed to lower metal-support interactions, lower BET surface area, lower Ni dispersion, and agglomeration of Nb2O5 species. All these observations were verified through extensive characterization methods for both catalysts before and after hydrogenation reaction. Mechanisms of coke formation, its deposition and ways of deactivation were also discussed, showing that graphitic/hard coke was more prevalent in Ni/niobia and soft, oligomeric coke and low-temperature carbon was deposited in Ni/alumina, confirmed by Raman spectroscopy as well. The higher acidity of niobia played a role in its higher coke formation tendency. Stability tests indicated that Ni/alumina was more stable than Ni/niobia and produced consistent product yields and BD conversions over 12 h of reaction time. In conclusion, we can confidently say that Ni/alumina was the better catalyst for BD hydrogenation and our work has significant implications in the olefin hydrogenation industry.
Our future plan related to this work is as follows:
-
To investigate nickel-based catalysts with the same supports but with changing the content of based nickel and the supports (10/90 %, 15/85 % and 20/80 %)
-
The benefits of adding niobia to alumina have been investigated previously for other types of reactions such as HDS, HDN and hydrogenolysis but not for hydrogenation of olefins (Rocha et al., 2007; Rodrigues et al., 2012). Hence, this is a definite area for future work since niobia was found to be well-dispersed in alumina, causing increasing in specific surface area and catalytic activity and performance.
-
Other supports such as ceria have shown beneficial effects of metal-support interactions due to their multiple oxidation states and switching ability between them. The combination of Ni with ceria will be explored for BD hydrogenation.
-
Since acidity played a role in olefin hydrogenation reactions, zeolites such as ZSM-5 and metal–organic frameworks will also be a pathway for future investigations.
-
Active metals apart from Ni such as Co and Cu will also be explored in combination with these aforementioned supports.
-
Incorporating density functional theory (DFT) studies to this hydrogenation reaction will enable us to develop a molecular insight.
Acknowledgments
This research was funded by a start-up grant from United Arab Emirates University, awarded to Dr. Kaushik Sivaramakrishnan (#12N104).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Catalytic upgrading of bio-oil from halophyte seeds into transportation fuels. J. Bioresources Bioproducts 2023
- [CrossRef] [Google Scholar]
- Optimizing acido-basic profile of support in Ni supported La2O3+Al2O3 catalyst for dry reforming of methane. Int. J. Hydrogen Energy. 2021;46(27):14225-14235.
- [CrossRef] [Google Scholar]
- A Review: fundamental aspects of silicate mesoporous materials. Materials. 2012;5(12):2874-2902.
- [CrossRef] [Google Scholar]
- Arnd Schade. BUTENE-1. Evonik - Leading Beyond Chemistry.
- Highly active and stable ni-based bimodal pore catalyst for dry reforming of methane. Appl. Catal. A. 2015;491:116-126.
- [CrossRef] [Google Scholar]
- Mechanisms of catalyst deactivation. Appl. Catal. A. 2001;212(1–2):17-60.
- [CrossRef] [Google Scholar]
- Bartholomew, C.H., Agrawal, P.K., Katzer, J.R., 1982. Sulfur Poisoning of Metals, pp 135–242. https://doi.org/10.1016/S0360-0564(08)60454-X.
- Atmospheric reaction of OH radicals with 1,3-Butadiene and 4-Hydroxy-2-Butenal. Chem. A Eur. J.. 2007;111(48):12099-12105.
- [CrossRef] [Google Scholar]
- Synthesis of polyisoprene, polybutadiene and styrene butadiene rubber grafted silica nanoparticles by nitroxide-mediated polymerization. Polymer (guildf). 2020;190:122190
- [CrossRef] [Google Scholar]
- Mesopore determination from nitrogen sorption isotherms: fundamentals, scope. Limitations 1979:663-684.
- [CrossRef] [Google Scholar]
- Butadiene Industry Installed Capacity and Capital Expenditure Forecasts Including Active and Planned Plants to 2027; London, 2023.
- Catalysis by Nickel in Biological Systems. New York: Marcel Dekker; 1998.
- Charisiou, N.D., Siakavelas, G. I., Sebastian, V., Hinder, S.J., Baker, M.A., Papadakis, V.G., Wang, W., 2020. Polychronopoulou, K.; Goula, M. A. Structural Investigation of the Carbon Deposits on Ni/Al2O3 Catalyst Modified by CaO-MgO for the Biogas Dry Reforming Reaction. In: The 1st International Electronic Conference on Catalysis Sciences; MDPI: Basel Switzerland, p. 15. https://doi.org/10.3390/ECCS2020-07569.
- Characterization and catalytic properties of niobia supported nickel catalysts in the hydrodechlorination of 1,2,4-Trichlorobenzene. J. Mol. Catal. A Chem.. 2004;223(1–2):353-361.
- [CrossRef] [Google Scholar]
- Effect of Co-Doping NiO and Nb2O5 on phase stability and mechanical properties of Y2O3-stabilized ZrO2/Al2O3 composites. Adv. Powder Technol.. 2016;27(3):877-881.
- [CrossRef] [Google Scholar]
- Metal phosphides derived from hydrotalcite precursors toward the selective hydrogenation of phenylacetylene. ACS Catal.. 2015;5(10):5756-5765.
- [CrossRef] [Google Scholar]
- Products—but What Does It Do? 2023
- Activation and discovery of earth-abundant metal catalysts using sodium tert-butoxide. Nat. Chem.. 2017;9(6):595-600.
- [CrossRef] [Google Scholar]
- Exposure toEmissions of 1,3-Butadiene and benzene in the cabins of moving motor vehicles and buses in Sydney, Australia. Atmos. Environ.. 1997;31(23):3877-3885.
- [CrossRef] [Google Scholar]
- From simple alkenes and CO2 to fluorinated carboxylic acids: computational studies and predictions. European J Org Chem. 2022;2022(2)
- [CrossRef] [Google Scholar]
- Sulfur poisoning of nickel methanation catalysts. Appl. Catal.. 1983;5(3):323-336.
- [CrossRef] [Google Scholar]
- Effect of metal-support interactions in Ni/Al2O3 catalysts with low metal loading for methane dry reforming. Appl. Catal. A. 2015;494:57-67.
- [CrossRef] [Google Scholar]
- Cumene hydrocracking and thiophene HDS on niobia-supported Ni, Mo and Ni–Mo catalysts. Catal. Today. 2006;118(3–4):402-409.
- [CrossRef] [Google Scholar]
- (Co-)Oligomerization of olefins to hydrocarbon fuels: influence of feed composition and pressure. Chem. Ing. Tech.. 2023;95(5):651-657.
- [CrossRef] [Google Scholar]
- Niobium hydrogenation process: effect of temperature and cooling rate from the hydrogenation temperature. Int. J. Refract Metals Hard Mater.. 2011;29(1):134-137.
- [CrossRef] [Google Scholar]
- Anti-Coking and anti-sintering Ni/Al2O3 catalysts in the dry reforming of methane: recent progress and prospects. Catalysts. 2021;11(8):1003.
- [CrossRef] [Google Scholar]
- Nickel-Catalyzed asymmetric hydrogenation of β-Acylamino nitroolefins: an efficient approach to chiral amines. Chem. Sci.. 2017;8(9):6419-6422.
- [CrossRef] [Google Scholar]
- Catalytic performance of cubic ordered mesoporous alumina supported nickel catalysts in dry reforming of methane. Microporous Mesoporous Mater.. 2021;310:110616
- [CrossRef] [Google Scholar]
- OH Radical initiated oxidation of 1,3-Butadiene: isomeric selective study of the dominant addition channel. Chem. A Eur. J.. 2010;114(16):5299-5305.
- [CrossRef] [Google Scholar]
- Highly efficient nickel-niobia composite catalysts for hydrogenation of CO2 to methane. Chem. Eng. Sci.. 2019;194:2-9.
- [CrossRef] [Google Scholar]
- Synergizing metal-support interactions and spatial confinement boosts dynamics of atomic nickel for hydrogenations. Nat. Nanotechnol.. 2021;16(10):1141-1149.
- [CrossRef] [Google Scholar]
- Tuning metal-support interactions on Ni/Al2O3 catalysts to improve catalytic activity and stability for dry reforming of methane. Processes. 2021;9(4):706.
- [CrossRef] [Google Scholar]
- Stable niobia-supported nickel catalysts for the hydrogenation of carbon monoxide to hydrocarbons. Catal. Today. 2020;343:56-62.
- [CrossRef] [Google Scholar]
- Ionic liquid immobilized Nickel(0) nanoparticles as stable and highly efficient catalysts for selective hydrogenation in the aqueous phase. Chem. Asian J.. 2010;5(5):1178-1184.
- [CrossRef] [Google Scholar]
- Selective hydrogenation of 1,3-Butadiene in the presence of an excess of alkenes over supported bimetallic gold−palladium catalysts. J. Phys. Chem. C. 2010;114(24):10823-10835.
- [CrossRef] [Google Scholar]
- Dehydrogenation of 1-Butene with CO2 over VOx supported catalysts. Appl. Catal. A. 2020;602:117648
- [CrossRef] [Google Scholar]
- Initial List of Hazardous Air Pollutants with Modifications. United States Environmental Protection Agency. https://www.epa.gov/haps/initial-list-hazardous-air-pollutants-modifications#mods (accessed 2023-07-01).
- Effects of a matrix on formation of aromatic compounds by dehydrocyclization of n -Pentane Using ZnZSM-5–Al2O3 composite catalysts. ACS Omega. 2020;5(19):11160-11166.
- [CrossRef] [Google Scholar]
- Ordered mesoporous Fe-Al2O3 based-catalysts synthesized via a direct “one-pot” method for the dry reforming of a model biogas mixture. Int. J. Hydrogen Energy. 2019;44(29):14889-14907.
- [CrossRef] [Google Scholar]
- Study of nickel catalysts supported on Al2O3, SiO2 or Nb2O5 oxides. J. Mol. Catal. A Chem.. 2005;242(1–2):81-90.
- [CrossRef] [Google Scholar]
- Synergistic effects of Nb2O5 promoter on Ru/Al2O3 for an aqueous-phase hydrodeoxygenation of glycerol to hydrocarbons. Appl. Catal. A. 2018;551:49-62.
- [CrossRef] [Google Scholar]
- Selective cleavage of lignin-derived diphenyl ether C-O bond over weakly acidic Ni/Nb2O5 catalyst. Fuel. 2021;295:120635
- [CrossRef] [Google Scholar]
- Mangankomplexe in Der (De)Hydrier-Katalyse – Ein Vergleich Mit Cobalt- Und Eisenkatalysatoren. Angew. Chem.. 2018;130(1):48-63.
- [CrossRef] [Google Scholar]
- γ-Alumina supported Cu-Ni bimetallic catalysts: characterization and selective hydrogenation of 1,3-Butadiene. Can. J. Chem. Eng.. 2002;80(1):63-70.
- [CrossRef] [Google Scholar]
- Partial Hydrogenation of 1,3-Butadiene on Hydrogen-Precovered Pd(110) in the Balance of π-Bonded C4 Hydrocarbon Reactions. J. Phys. Chem. C. 2008;112(44):17219-17224.
- [CrossRef] [Google Scholar]
- High-Pressure Thermal Cracking of n-Hexadecane in Aromatic Solvents. Ind. Eng. Chem. Res.. 1993;32:1864-1876.
- [Google Scholar]
- The gas phase reaction of ozone with 1,3-Butadiene: formation yields of some toxic products. Atmos. Environ.. 2000;34(1):35-43.
- [CrossRef] [Google Scholar]
- Catalytic upgrading of pyrolytic bio-oil from salicornia bigelovii seeds for use as jet fuels: exploring the ex-situ deoxygenation capabilities of Ni/Ze catalyst. Bioresour Technol Rep. 2023;22:101437
- [CrossRef] [Google Scholar]
- Chemometrical classification of pumpkin seedoils using UV–Vis, NIR and FTIR spectra. J. Biochem. Bioph. Methods. 2004;61:95-106.
- [Google Scholar]
- Selective hydrogenation of 1,3-Butadiene on TiO2-modified Pd/SiO2 catalysts. Appl. Catal. A. 2003;244(1):83-91.
- [CrossRef] [Google Scholar]
- Air-Stable α-Diimine nickel precatalysts for the hydrogenation of hindered, unactivated alkenes. ACS Catal.. 2018;8(1):342-348.
- [CrossRef] [Google Scholar]
- Ni/Al2O3 catalysts derived from spinel NiAl2O4 for low-temperature hydrogenation of maleic anhydride to succinic anhydride. Chin. J. Catal.. 2017;38(7):1166-1173.
- [CrossRef] [Google Scholar]
- Coke formation on Pt–Sn/Al2O3 catalyst in propane dehydrogenation: coke characterization and kinetic study. Top. Catal.. 2011;54(13–15):888-896.
- [CrossRef] [Google Scholar]
- Nickel-Catalyzed enantioselective hydrogenation of β-(Acylamino)Acrylates: synthesis of chiral β-amino acid derivatives. Org. Lett.. 2017;19(19):5130-5133.
- [CrossRef] [Google Scholar]
- Coke deposition on Pt-based catalysts in propane direct dehydrogenation: kinetics, suppression, and elimination. ACS Catal.. 2021;11(15):9279-9292.
- [CrossRef] [Google Scholar]
- Oxidation reaction and thermal stability of 1,3-butadiene under oxygen and initiator. Arab. J. Chem.. 2022;15(11):104289
- [CrossRef] [Google Scholar]
- Oligomerization of light olefins catalyzed by Brønsted-acidic metal-organic framework-808. J. Am. Chem. Soc.. 2019;141(29):11557-11564.
- [CrossRef] [Google Scholar]
- Stabilizing Ni on bimodal mesoporous-macroporous alumina with enhanced coke tolerance in dry reforming of methane to syngas. J. CO2 Util.. 2020;35:288-297.
- [CrossRef] [Google Scholar]
- Heterogeneous olefin hydrogenation enabled by a highly-reduced Nickel(−II) catalyst precursor. Chem. – A Eur. J.. 2020;26(28):6113-6117.
- [CrossRef] [Google Scholar]
- Hydroconversion of fatty acids and vegetable oils for production of jet fuels. Fuel. 2021;306:121673
- [CrossRef] [Google Scholar]
- Selective Hydrogenation of 1,3-Butadiene in Presence of 1-Butene under liquid phase conditions with NiPd/Al2O3 catalysts. Appl. Petrochem. Res.. 2016;6(4):379-387.
- [CrossRef] [Google Scholar]
- Dibenzothiophene Hydrodesulfurization with NiMo and CoMo Catalysts Supported on Niobium-Modified MCM-41. Appl Catal B. 2017;219:479-491.
- [CrossRef] [Google Scholar]
- Promoting Effect of Ceria on the Performance of NiPd/CeO2 –Al2O3 Catalysts for the Selective Hydrogenation of 1,3-Butadiene in the Presence of 1-Butene. New J. Chem.. 2018;42(13):11165-11173.
- [CrossRef] [Google Scholar]
- NiMo Catalysts Supported on Al, Nb, Ti or Zr-Containing MCM-41 for Dibenzothiophene Hydrodesulfurization. Catal. Today. 2020;349:217-227.
- [CrossRef] [Google Scholar]
- Monroy, T.G., Abella, L.C., Gallardo, S.M., Hinode, H., 2010. Catalytic Dry Reforming of Methane Using Ni/MgO-ZrO2 Catalyst. In: Proceedings of the 2nd Annual Gas Processing Symposium; Elsevier, pp 145–152. https://doi.org/10.1016/S1876-0147(10)02017-3.
- Homogeneous hydrogenation and isomerization of 1-octene catalyzed by Nickel(II) complexes with bidentate diarylphosphane ligands. Inorg. Chem.. 2013;52(14):8190-8201.
- [CrossRef] [Google Scholar]
- Nickel—Alumina catalysts for the transformation of vegetable oils into green diesel: the role of preparation method, activation temperature, and reaction conditions. Nanomaterials. 2023;13(3):616.
- [CrossRef] [Google Scholar]
- Hydrogenation of 1,3-Butadiene and of crotonaldehyde over highly dispersed Au catalysts. Catal. Today. 2002;74(3–4):265-269.
- [CrossRef] [Google Scholar]
- Light alkanes CO2 reforming to synthesis gas over Ni based catalysts. Catal. Today. 2006;115(1–4):179-185.
- [CrossRef] [Google Scholar]
- Transformation of But-1-Ene into aromatic hydrocarbons over ZSM-5 zeolites. J. Chem. Soc. Faraday Trans.: Phys. Chem. Condensed Phases. 1987;83(9):2913.
- [CrossRef] [Google Scholar]
- Selective hydrogenation of 1,3-Butadiene over Pd and Pd–Sn catalysts supported on different phases of alumina. Catal. Today. 2011;164(1):28-33.
- [CrossRef] [Google Scholar]
- Improved hydrothermal stability of Niobia-supported Pd catalysts. Appl. Catal. A. 2011;397(1–2):153-162.
- [CrossRef] [Google Scholar]
- Polycyclic aromatic hydrocarbons from the co-pyrolysis of 1,3-Butadiene and propyne. Proc. Combust. Inst.. 2013;34(1):1775-1783.
- [CrossRef] [Google Scholar]
- Ethanol-to-Butadiene: the reaction and its catalysts. Cat. Sci. Technol.. 2020;10(15):4860-4911.
- [CrossRef] [Google Scholar]
- Data fusion by joint non-negative matrix factorization for hypothesizing pseudo-chemistry using bayesian networks. React. Chem. Eng.. 2020;5(9):1719-1737.
- [CrossRef] [Google Scholar]
- Synthesis of 1,3-Butadiene and Its 2-Substituted monomers for synthetic rubbers. Catalysts. 2019;9(1):97.
- [CrossRef] [Google Scholar]
- Combined operando UV/Vis/IR spectroscopy reveals the role of methoxy and aromatic species during the methanol-to-olefins reaction over H-SAPO-34. ChemCatChem. 2014;6(12):3396-3408.
- [CrossRef] [Google Scholar]
- Catalytic performance of Ni–Co/HZSM-5 catalysts for aromatic compound promotion in simulated bio-oil upgrading. RSC Adv.. 2023;13(11):7694-7702.
- [CrossRef] [Google Scholar]
- Chemistry of olefin oligomerization over ZSM-5 catalyst. Ind. Eng. Chem. Res.. 1988;27(4):565-570.
- [CrossRef] [Google Scholar]
- Selective hydrogenation of 1,3-Butadiene over ceria catalyst: a molecular insight. Mol. Catal.. 2022;524:112331
- [CrossRef] [Google Scholar]
- Ceria-supported niobium oxide catalyst for low-temperature oxidation of 1,3-butadiene. Mol. Catal.. 2022;518:112083
- [CrossRef] [Google Scholar]
- Stable zero-valent nickel nanoparticles in glycerol: synthesis and applications in selective hydrogenations. Adv. Synth. Catal.. 2018;360(18):3544-3552.
- [CrossRef] [Google Scholar]
- Stable zero-valent nickel nanoparticles in glycerol: synthesis and applications in selective hydrogenations. Adv. Synth. Catal.. 2018;360(18):3544-3552.
- [CrossRef] [Google Scholar]
- Design of effective catalysts for selective alkyne hydrogenation by doping of ceria with a single-atom promotor. J. Am. Chem. Soc.. 2018;140(40):12964-12973.
- [CrossRef] [Google Scholar]
- Alumina-, niobia-, and niobia/alumina-supported NiMoS catalysts: surface properties and activities in the hydrodesulfurization of thiophene and hydrodenitrogenation of 2,6-Dimethylaniline. J. Catal.. 2007;252(2):321-334.
- [CrossRef] [Google Scholar]
- Effect of niobia and alumina as support for Pt catalysts in the hydrogenolysis of glycerol. Chem. Eng. J.. 2012;198–199:457-467.
- [CrossRef] [Google Scholar]
- Butane dry reforming catalyzed by cobalt oxide supported on Ti 2 AlC MAXphase. ChemSusChem. 2020;13(23):6401-6408.
- [CrossRef] [Google Scholar]
- Influence of sintering temperature on microstructure and electrical properties of Er2O3 Added ZnO-V2O5-MnO2-Nb2O5 varistor ceramics. J. Alloy. Compd.. 2018;749:687-696.
- [CrossRef] [Google Scholar]
- Cobalt-catalyzed hydrogenations via olefin cobaltate and hydride intermediates. ACS Catal.. 2019;9(8):7596-7606.
- [CrossRef] [Google Scholar]
- Chemoinformatic investigation of the chemistry of cellulose and lignin derivatives in hydrous pyrolysis. Ind. Eng. Chem. Res.. 2020;59(25):11582-11595.
- [CrossRef] [Google Scholar]
- Real-time quantitative operando raman spectroscopy of a CrOx/Al2O3 propane dehydrogenation catalyst in a pilot-scale reactor. ChemCatChem. 2014;6(11):3139-3145.
- [CrossRef] [Google Scholar]
- Satyanarayana, C.V.; Srikant, D., Gurav, H.R. Catalyst Deactivation and Regeneration. In: Industrial Catalytic Processes for Fine and Specialty Chemicals; Elsevier, 2016; pp 187–219. https://doi.org/10.1016/B978-0-12-801457-8.00005-7.
- A study of the promoting effect of noble metal addition on niobia and niobia alumina catalysts. Catal. Today. 2000;57(3–4):169-176.
- [CrossRef] [Google Scholar]
- Synthesis of spherical polyethylene/poly(1-butene) reactor blends with two-stage sequence polymerization technology. Polymer(guildf). 2018;144:72-79.
- [CrossRef] [Google Scholar]
- Catalytic activities of various niobium oxides for hydrogen absorption/desorption reactions of magnesium. ACS Omega. 2021;6(36):23564-23569.
- [CrossRef] [Google Scholar]
- Investigative properties of CeO2 doped with niobium: a combined characterization and DFT studies. Nanotechnol. Rev.. 2021;11:191-203.
- [CrossRef] [Google Scholar]
- Functionalized ceria-niobium supported nickel catalysts for gas phase semi-hydrogenation of phenylacetylene to styrene. Cat. Sci. Technol.. 2022;12(23):7133-7150.
- [CrossRef] [Google Scholar]
- Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984) Pure Appl. Chem.. 1985;57(4):603-619.
- [CrossRef] [Google Scholar]
- Sivaramakrishnan, K., De Klerk, A., Prasad, V., 2019. Viscosity of Canadian Oilsands Bitumen and Its Modification by Thermal Conversion. In: Chemistry Solutions to Challenges in the Petroleum Industry (In Press); American Chemical Society, pp 115–199.
- Least squares-support vector regression for determining product concentrations in acid-catalyzed propylene oligomerization. Ind. Eng. Chem. Res.. 2018;57:13156-13176.
- [Google Scholar]
- A data-driven approach to generate pseudo-reaction sequences for the thermal conversion of athabasca bitumen. React. Chem. Eng.. 2021;6:505-537.
- [CrossRef] [Google Scholar]
- Sivaramakrishnan, K., 2019. Application of Chemometric and Experimental Tools for Monitoring Processes of Industrial Importance. PhD, University of Alberta.
- Nickel catalyst with outstanding activity in the DRM reaction prepared by high temperature calcination treatment. Int. J. Hydrogen Energy. 2016;41(20):8459-8469.
- [CrossRef] [Google Scholar]
- Glycerol oxidation on gold catalysts supported on group five metal oxides—A comparative study with other metal oxides and carbon based catalysts. Catal. Today. 2010;158(1–2):121-129.
- [CrossRef] [Google Scholar]
- Thermokinetic analysis and product characterization of waste tire-hazelnut shell co-pyrolysis: TG-FTIR and fixed bed reactor study. J. Environ. Chem. Eng.. 2021;9(5):106165
- [CrossRef] [Google Scholar]
- Experimental study of the co-pyrolysis of sewage sludge and wet waste via TG-FTIR-GC and artificial neural network model: synergistic effect, pyrolysis kinetics and gas products. Renew. Energy. 2022;184:1-14.
- [CrossRef] [Google Scholar]
- Hydrogenation of liquid styrene by alumina supported nickel catalysts: comparison between classical and non-classical methods. IOP Conf. Ser. Mater. Sci. Eng.. 2016;133:012017
- [CrossRef] [Google Scholar]
- Nickel-catalyzed, ring-forming aromatic C-H alkylations with unactivated alkyl halides. Tetrahedron. 2019;75(31):4143-4149.
- [CrossRef] [Google Scholar]
- Catalytic upgrading of biomass derived pyrolysis vapors over metal-loaded ZSM-5 zeolites: effect of different metal cations on the bio-oil final properties. Microporous Mesoporous Mater.. 2015;209:189-196.
- [CrossRef] [Google Scholar]
- Experimental and Ab initio dynamical investigations of the kinetics and intramolecular energy transfer mechanisms for the OH + 1,3-Butadiene reaction between 263 and 423 K at low pressure. Chem. A Eur. J.. 2008;112(31):7227-7237.
- [CrossRef] [Google Scholar]
- Carbon deposit analysis in catalyst deactivation, regeneration, and rejuvenation. Angew. Chem. Int. Ed.. 2023;62(29)
- [CrossRef] [Google Scholar]
- Effect of metal-support interaction on the catalytic performance of Ni/Al2O3 for selective hydrogenation of isoprene. J. Mol. Catal. A Chem.. 2011;344(1–2):122-127.
- [CrossRef] [Google Scholar]
- Conversion of Ethanol to n -Butanol over NiCeO2 based catalysts: effects of metal dispersion and niceinteractions. Ind. Eng. Chem. Res.. 2020;59(51):22057-22067.
- [CrossRef] [Google Scholar]
- Characterizations of Ni-CeO2 nanocomposite coating by interlaced jet electrodeposition. J. Alloy. Compd.. 2017;727:269-277.
- [CrossRef] [Google Scholar]
- Visbreaking oilsands-derived bitumen in the temperature range of 340–400 °C. Energy Fuel. 2014;28:5014-5022.
- [Google Scholar]
- Computer generation of a network of elementary steps for coke formation during the thermal cracking of hydrocarbons. Chem. Eng. J.. 2001;82(1–3):267-279.
- [CrossRef] [Google Scholar]
- White, Wm. C. Butadiene Production Process Overview. Chem Biol Interact 2007, 166 (1–3), 10–14. https://doi.org/10.1016/j.cbi.2007.01.009.
- Nickel niobia interaction in non-classical Ni/Nb2O5 catalysts. J. Mol. Catal. A Chem.. 2006;256(1–2):225-233.
- [CrossRef] [Google Scholar]
- Methane dry reforming by Ni–Cu nanoalloys anchored on periclase-Phase MgAlOx nanosheets for enhanced syngas production. ACS Appl. Mater. Interfaces. 2021;13(41):48838-48854.
- [CrossRef] [Google Scholar]
- Effects of metal-support interactions and interfaces on catalytic performance over M2O3- (,Al-) supported Ni catalysts. Int. J. Energy Res.. 2023;2023:1-14.
- [CrossRef] [Google Scholar]
- The role of Nb2O5 in controlling metal-acid sites of CoMoS/γ-Al2O3 catalyst for the enhanced hydrodeoxygenation of guaiacol into hydrocarbons. J. Catal.. 2022;407:19-28.
- [CrossRef] [Google Scholar]
- Pore structure and fractal characteristics of different shale lithofacies in the dalong formation in the western area of the lower Yangtze platform. Minerals. 2020;10(1):72.
- [CrossRef] [Google Scholar]
- Partial upgrading of bitumen by thermal conversion at 150–300 °C. Energy Fuels. 2018;32:3299-3311.
- [Google Scholar]
- High-performance of nanostructured Ni/CeO2 catalyst on CO2 methanation. Appl Catal B. 2020;268:118474
- [CrossRef] [Google Scholar]
- Steam reforming of acetic acid over nickel-based catalysts: the intrinsic effects of nickel precursors on behaviors of nickel catalysts. Appl Catal B. 2018;237:538-553.
- [CrossRef] [Google Scholar]
- A Remarkable enhancement of catalytic activity for KBH4 treating the carbothermal reduced Ni/AC catalyst in glycerol hydrogenolysis. Catal. Commun.. 2010;11(5):493-497.
- [CrossRef] [Google Scholar]
- Contemporary trends in composite Ni-based catalysts for CO2 reforming of methane. Chem. Eng. Sci.. 2021;229:116072
- [CrossRef] [Google Scholar]
- Kinetic investigation into the chemoselective hydrogenation of α, β-unsaturated carbonyl compounds catalyzed by Ni(0) nanoparticles. Dalton Trans.. 2017;46(15):5082-5090.
- [CrossRef] [Google Scholar]
- Study on the preparation conditions of Ni/Al2O3 catalysts to selective hydrogenation of butadiene in FCC C4 fraction. Pet. Sci. Technol.. 2009;27(3):291-301.
- [CrossRef] [Google Scholar]
- A review of CH4CO2 reforming to synthesis gas over Ni-based catalysts in recent years (2010–2017) Int. J. Hydrogen Energy. 2018;43(32):15030-15054.
- [CrossRef] [Google Scholar]
- NixAl1O2-δ mesoporous catalysts for dry reforming of methane: the special role of NiAl2O4 spinel phase and its reaction mechanism. Appl Catal B. 2021;291:120074
- [CrossRef] [Google Scholar]
- Catalytic performance of niobium species in crystalline and amorphous solids—gas and liquid phase oxidation. Appl. Catal. A. 2011;391(1–2):194-204.
- [CrossRef] [Google Scholar]




