

Translate this page into:
Photochemical synthesis and antimicrobial studies of new chromen-4-one based vinyl ethers
⁎Corresponding author. Tel.: +91 175 3046409; fax: +91 175 2283073. yusuf_sah04@yahoo.co.in (Mohamad Yusuf)
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.

Abstract
The chromen-4-one based vinyl ethers 3a–3e have been synthesized from the photochemical reaction of cyclobutylmethoxychromen-4-ones 2a–2e in dry MeOH. The compounds 2a–2e were easily obtained from the O-alkylation of different hydroxychromen-4-ones 1a–1e with bromomethyl-cyclobutane in the presence of anhydrous K2CO3/dry acetone and Bu4N+I− (PTC). The structure of the these compounds have been characterized from the rigorous analysis of their IR, 1H-NMR, 13C-NMR, ESI-MS and elemental analysis. The antibacterial and antifungal activities of newly prepared compounds were also evaluated against selected pathogens which include Staphylococcus aureus, Bacillus subtilis, Escherichia coli, Pseudomonas aeruginosa, Pseudomonas florescens, Staphylococcus pyrogens, Klebsiella pneumoniae and Aspergillus janus, Aspergillus niger, Fusarium oxysporium, Aspergillus sclerotium, Penicillium glabrum respectively. Some of the studied compounds exhibited significant activity against the tested microorganisms.
Keywords
Vinyl ether
Photochemical reaction
Chromen-4-ones
Antibacterial and antifungal activity

1 Introduction
Vinyl ethers are the valuable intermediates which are used in a wide range of chemical transformations in organic synthesis (Trost and Fleming, 1991; Crousse et al., 1998; Kerr et al., 2001; Malaga and Mannucci, 2001). These compounds are utilized for the synthesis of 1,4-dicarbonyl substrates and also act as two carbon synthons (Muthusamy and Srinivasan, 2006) for the preparation of thienyl, pyrrolyl, furyl heterocycles and terpenoids (Christopel and Miller, 1986; Freeman and Kim, 1992; Misaki et al., 2005; Biava et al., 2005). Many methods are reported for the synthesis of vinyl ethers such as acetylation of alcohols, mercury catalyzed trans vinylation of alcohols, carbometallation of alkynic ethers or ester methylation promoted by metal complexes etc (Okimoto and Sakaguchi, 2002). However most of these methods involved various difficulties such as harsh reaction conditions, laborious manipulations and overall low yields. Another method for the synthesis of vinyl ether involves the photochemical hydrogen abstraction from various allyl ethers (Gupta et al., 2002). The cyclobutylmethyl derivatives are easily accessible by a number of reliable methods that are known to give high yields (De Meijere, 1997). The inherent ring strain in the cyclobutane ring is responsible for its ring opening reactions (Wong et al., 1986) and the selective cleavage of a cyclobutane bond has been found to be a facile process. Cyclobutane derivatives can be used as starting material for the synthesis of acyclic and cyclic compounds. Apparently the broad synthetic range of applications is one of the important reasons that the chemistry of cyclobutane derivatives has been extensively studied during the last two decades. Photochemical reactions have been utilized as an exciting synthetic tool by organic chemists in the past decades (Horspool, 1986; Wagner, 1989; Sumathi and Balasubramaniam, 1992; Kraus and Wu, 1992; Sumathi and Balasubramaniam, 1992; Park et al., 2001; Scheffer and Wang, 2001; Griesbeck and Heckroth, 2002; Wessig et al., 2004; Pedrosa et al., 2005; Kumar and Yusuf, 2006). These reactions often yield such products which are inaccessible by the thermal routes and proceed along the excited state pathways (Steven, 1961; Froster, 1969). So by keeping these aspects in mind, we are aiming hereby to synthesize new vinyl ethers 3a–3e from the photolysis of 3-cyclobutylmethoxy-chromen-4-ones. The major interest behind this study was to develop a simple method for the synthesis of chromen-4-one based vinyl ethers and to investigate their antimicrobial behaviors.
2 Experimental
Melting points reported are uncorrected. IR spectra were scanned in KBr pellets on a Perkin Elmer RXIFT Infrared spectrophotometer. 1H-NMR spectra were recorded on a 400 MHz Bruker spectrometer using TMS as the internal standard. The mass spectra have been scanned on the Waters Micromass Q-T of a Micro (ESI) spectrometer. TLC plates were coated with silica gel suspended in MeOH–CHCl3 and iodine vapors were used as the visualizing agent.
2.1 Synthesis of 3-(cyclobutylmethoxy)-2-phenyl-4H-chromen-4-one 2a
A suspension of 1a (2.00 g, 0.0089 mol), bromomethyl-cyclobutane (1.32 g, 0.0089 mol) and tetrabutylammonium iodide (1.0 g) in dry acetone (25 ml) was refluxed for 6 h with continuous stirring. The progress of the reaction was monitored by TLC. After the completion of the reaction, the reaction mixture turned into a colorless mass which was poured into iced-HCl to obtain a solid substance. The crude product thus obtained was crystallized with MeOH to yield a pure compound 2a.
2a: Light brown solid; Yield 58%; m.p.: 168–170 °C; IR (KBr) cm−1: 1642 (C⚌O), 1616 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.27 (1H, dd, Jm,o = 2.0, 7.2 Hz, H-5), 8.16 (2H, dd, Jm,o = 2.8, 8.6 Hz, H-2′, 6′), 7.70 (1H, dd, Jm,o = 2.1, 8.0 Hz, H-7), 7.51 (4H, m, H-6, 3′, 4′, 5′), 7.40 (1H, d, Jo = 8.0 Hz, H-8), 4.05 (2H, d, Jvic = 6.8 Hz, OCH2), 2.69 (1H, quintet, Jvic = 6.8 Hz, H-1″), 2.17 (2H, m, H-3″), 1.90 (4H, m, H-2″, 4″); 13C-NMR (100 MHz, CDCl3): δ 172.64 (C-4), 156.21 (C-8a), 138.43 (C-3), 137.68 (C-2), 134.90 (C-7), 132.72 (C-1′), 131.15 (C-5), 128.64 (C-3′, 5′), 127.23 (C-2′, 4′, 6′), 120.79 (C-6), 120.48 (C-4a), 115.79 (C-8), 67.42 (OCH2), 34.90 (C-1″), 26.36 (C-2″, 4″), 18.67 (C-3″); MS (ESI): m/z (M)+ 306; Anal. Calc. For C20H18O3: C, 78.43; H, 5.88; Found C, 78.14; H, 5.90%.
2.2 Synthesis of 3-(cyclobutylmethoxy)-2-p-tolyl-4H-chromen-4-one 2b
The compound 2b was obtained from the reaction of 1b (2.00 g, 0.0079 mol) with bromomethyl-cyclobutane (1.17 g, 0.0079 mol) under similar conditions as described above for 2a.
2b: Light brown solid; Yield 58%; m.p.: 202–205 °C; IR (KBr) cm−1: 1638 (C⚌O), 1604 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.29 (1H, dd, Jm,o = 2.0, 7.8 Hz, H-5), 7.90 (2H, d, Jo = 7.8 Hz, H-2′, 6′), 7.62 (1H, dd, Jm,o = 2.2, 7.8 Hz, H-7), 7.48 (1H, d, Jo = 8.0 Hz, H-6), 7.42 (1H, d, Jo = 8.0 Hz, H-8), 7.26 (2H, d, Jo = 8.0 Hz, H-3′, 5′), 4.12 (2H, d, Jvic = 6.7 Hz, OCH2), 2.60 (1H, quintet, Jvic = 6.7 Hz, H-1″), 2.44 (3H, s, 4′-CH3), 2.14 (2H, m, H-3″), 2.01 (4H, m, H-2″, 4″); 13C-NMR (100 MHz, CDCl3): δ 174.53 (C-4), 155.28 (C-2), 156.02 (C-8a), 137.86 (C-3), 133.70 (C-7), 131.65 (C-5), 129.82 (C-4′), 124.40 (C-1′), 126.90 (C-2′, 6′), 123.04 (C-6), 122.28 (C-4a), 118.10 (C-8), 113.64 (C-3′, 5′), 70.25 (OCH2), 31.56 (C-1″), 24.93 (C-2″, 4″), 23.38 (4′-CH3), 18.72 (C-3″); MS (ESI): m/z (M)+ 320; Anal. Calc. For C21H20O3: C, 78.75; H, 6.25; Found C, 78.56; H, 6.22%.
2.3 3-(Cyclobutylmethoxy)-2-(4-methoxyphenyl)-4H-chromen-4-one 2c
The compound 2c was synthesized from the reaction of 1c (2.00 g, 0.0074 mol) with bromomethyl-cyclobutane (1.10 g, 0.0074 mol) under similar conditions as described above for 2a.
2c: Light brown solid; Yield 58%; m.p.: 185–187 °C; IR (KBr) cm−1: 1630 (C⚌O), 1612 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.26 (1H, d, Jm,o = 2.1, 7.6 Hz, H-5), 8.12 (2H, d, Jo = 8.8 Hz, H-2′, 6′), 7.68 (1H, td, Jm,o = 2.3, 8.6 Hz, H-7), 7.52 (1H, d, Jo = 8.1 Hz, H-6), 7.39 (1H, t, Jo = 7.8 Hz, H-8), 7.00 (2H, dd, Jm,o = 2.3, 7.9 Hz, H-3′, 5′), 3.92 (3H, s, 4′-OCH3), 4.04 (2H, d, Jvic = 6.9 Hz, OCH2), 2.82 (1H, m, H-1″), 1.98 (2H, m, H-3″), 1.80 (4H, m, H-2″, 4″); 13C-NMR (100 MHz, CDCl3): δ 175.80 (C-4), 156.43 (C-4′), 155.68 (C-2), 154.29 (C-8a), 137.24 (C-3), 135.40 (C-7), 132.89 (C-5), 130.21 (C-2′, 6′), 128.71 (C-1′), 123.66 (C-6), 121.78 (C-4a), 115.24 (C-8), 72.39 (OCH2), 54.91 (4′-OCH3), 32.36 (C-1″), 25.54 (C-2″, 4″), 20.06 (C-3″); MS (ESI): m/z (M)+ 336; Anal. Calc. For C21H20O4: C, 75.00; H, 5.95; Found C, 75.30; H, 5.98%.
2.4 Synthesis of 3-(cyclobutylmethoxy)-2-(thiophen-2-yl)-4H-chromen-4-one 2d
The compound 2d was prepared from the reaction of 1d (2.00 g, 0.0087 mol) with bromomethyl-cyclobutane (1.29 g, 0.0087 mol) under similar conditions as described above for 2a.
2d: Off white solid; Yield 62%; m.p.: 174–176 °C; IR (KBr) cm−1: 1632 (C⚌O), 1609 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.24 (1H, dd, Jm,o = 2.1, 7.9 Hz, H-5), 7.98 (1H, d, J3′,4′ = 3.7 Hz, H-3′), 7.80 (1H, td, Jm,o = 2.3, 7.9 Hz, H-7), 7.67 (1H, d, J5′,4′ = 5.0 Hz, H-5′), 7.56 (1H, d, Jo = 8.2 Hz, H-6), 7.41 (1H, d, Jo = 7.9 Hz, H-8), 7.22 (1H, d, J4′,5′ = 5.0 Hz, H-4′), 4.42 (2H, d, Jvic = 6.6 Hz, OCH2), 2.72 (1H, m, H-1″), 2.12 (2H, m, H-3″), 1.98 (4H, m, H-2″, 4″); 13C-NMR (100 MHz, CDCl3): δ 176.48 (C-4), 160.02 (C-2), 154.53 (C-8a), 138.72 (C-3), 132.37 (C-7), 130.23 (C-5), 129.96 (C-5′), 129.74 (C-2′), 127.39 (C-4′), 126.27 (C-3′), 122.78 (C-6), 121.78 (C-4a), 116.26 (C-8), 69.62 (OCH2), 32.80 (C-1″), 23.91 (C-2″, 4″), 19.84 (C-3″); MS (ESI): m/z (M)+ 312; Anal. Calc. For C18H16O3S: C, 69.23; H, 5.12; S, 10.25; Found C, 69.50; H, 5.09; S, 10.21%.
2.5 Synthesis of 3-(cyclobutylmethoxy)-2-(furan-2-yl)-4H-chromen-4-one 2e
The compound 2e was obtained from the reaction of 1c (2.00 g, 0.0082 mol) with bromomethyl-cyclobutane (1.22 g, 0.0041 mol) under similar conditions as described above for 2a.
2e: Off white solid; Yield 62%; m.p.: 190–192 °C; IR (KBr) cm−1: 1640 (C⚌O), 1602 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.14 (1H, dd, Jm,o = 2.0, 7.9 Hz, H-5), 7.83 (1H, d, J3′,4′ = 1.2 Hz, H-3′), 7.74 (1H, td, Jm,o = 2.2, 7.8 Hz, H-7), 7.60 (1H, d, Jo = 8.4 Hz, H-6), 7.43 (1H, td, Jm,o = 2.0, 7.9 Hz, H-8), 7.33 (1H, d, J5′,4′ = 3.2 Hz, H-5′), 6.69 (1H, dd, J4′,3′ = 1.2 Hz, J4′,5′ = 3.5 Hz, H-4′), 4.20 (2H, d, Jvic = 6.6 Hz, OCH2), 2.79 (1H, m, H-1″), 2.08 (2H, m, H-3″), 1.94 (4H, m, H-2″, 4″); 13C-NMR (100 MHz, CDCl3): δ 186.65 (C-4), 162.44 (C-2), 151.19 (C-8a), 145.25 (C-3), 130.37 (C-5), 130.26 (C-7), 130.21 (C-5′), 129.69 (C-2′), 129.52 (C-3′), 118.61 (C-4′), 115.96 (C-4a), 114.18 (C-6), 113.94 (C-8), 67.36 (OCH2), 26.13 (C-1″), 25.23 (C-2″, 4″), 25.18 (C-3″); MS (ESI): m/z (M)+ 296; Anal. Calc. For C18H16O4: C, 72.97; H, 5.40; Found C, 73.38; H, 5.37%.
2.6 Synthesis of (E)-3-(pent-1-enyloxy)-2-p-tolyl-4H-chromen-4-one 3a
A deoxygenated solution of 2a (100 mg, 0.00032 mol) was photolyzed in dry MeOH with light from a 125 Hg arc lamp under nitrogen atmosphere in a Pyrex vessel for 5 h. The progress of the reaction was monitored by TLC. The solvent was distilled out under reduced pressure and the photolysate thus obtained was subjected to extensive column chromatography (100–200 mesh), packed in petroleum ether-benzene (1:1). The elution of the column with benzene-EtOAc (3:1) furnished starting compound 2a (14%, IR, co-tlc and mmp) and a new compound 3a.
3a: Light yellow solid; Yield 48%; m.p.: 210–212 °C; IR (KBr) cm−1: 1648 (C⚌O), 1608 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.24 (1H, dd, Jm,o = 2.3, 8.0 Hz, H-5), 7.98 (2H, m, H-2′, 6′), 7.63 (1H, d, Jm,o = 2.2, 7.0 Hz, H-7), 7.56 (1H, td, Jm,o = 2.1, 8.0 Hz, H-6), 7.49 (3H, m, H-3′, 4′, 5′), 7.20 (1H, d, Jo = 8.0 Hz, H-8), 6.52 (1H, d, J1″,2″ = 12.2 Hz, H-1″), 5.19 (1H, dt, J2″,3″ = 7.0 Hz, J2″,1″ = 12.3 Hz, H-2″), 2.98 (2H, m, H-3″), 1.96 (2H, m, H-4″), 1.76 (3H, t, Jvic = 6.4 Hz, H-5″); 13C-NMR (100 MHz, CDCl3): δ 177.68 (C-4), 155.74 (C-8a), 141.38 (C-1″), 138.36 (C-3), 134.85 (C-7), 132.21 (C-2), 130.24 (C-1′), 127.98 (C-3′, 5′), 127.02 (C-2′, 4′, 6′); 125.64 (C-5), 122.57 (C-6), 120.69 (C-4a), 114.48 (C-8), 102.30 (C-2″), 29.78 (C-3″), 22.49 (C-4″), 14.32 (C-5″); MS (ESI): m/z (M)+ 306 (100%), 186 (40.8%), 120 (22.3%), 92 (10.7%); Anal. Calc. For C20H18O3: C, 78.43; H, 5.88; Found C, 78.68; H, 5.86%.
2.7 Synthesis of (E)-3-(pent-1-enyloxy)-2-p-tolyl-4H-chromen-4-one 3b
The photoirradiation of 2b (100 mg, 0.00031 mol) was carried out under similar conditions as described above for 2a. The resulting reaction mixture upon chromatographic work up yielded a new photoproduct 3b.
3b: Light yellow solid; Yield 55%; m.p.: 222–225 °C; IR (KBr) cm−1: 1628 (C⚌O), 1612 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.32 (1H, dd, Jm,o = 2.0, 7.9 Hz, H-5), 7.90 (2H, m, H-2′, 6′), 7.76 (1H, d, Jm,o = 2.3, 7.2 Hz, H-7), 7.47 (1H, td, Jm,o = 2.1, 8.0 Hz, H-6), 7.32 (1H, d, Jo = 8.0 Hz, H-8), 7.24 (2H, m, H-3′, 5′), 6.47 (1H, d, J1″,2″ = 12.2 Hz, H-1″), 5.23 (1H, dt, J2″,3″ = 7.2 Hz, J2″,1″ = 12.1 Hz, H-2″), 3.02 (2H, m, H-3″), 2.40 (3H, s, 4′-CH3), 1.92 (2H, m, H-4″), 1.63 (3H, t, Jvic = 6.4 Hz, H-5″); 13C-NMR (100 MHz, CDCl3): δ 174.24 (C-4), 154.69 (C-8a), 140.20 (C-1″), 138.01 (C-3), 134.92 (C-7), 135.24 (C-2), 129.28 (C-1′), 127.76 (C-3′, 5′), 126.94 (C-2′, 4′, 6′); 124.98 (C-5), 122.79 (C-6), 120.34 (C-4a), 113.36 (C-8), 104.64 (C-2″), 29.45 (C-3″), 22.18 (C-4″), 14.72 (C-5″); MS (ESI): m/z (M)+ 320 (62.8%), 200 (100%), 120 (35.4%), 92 (28.3%); Anal. Calc. For C21H20O3: C, 78.75; H, 6.25; Found C, 79.02; H, 6.28%.
2.8 Synthesis of (E)-2-(4-methoxyphenyl)-3-(pent-1-enyloxy)-4H-chromen-4-one 3c
The photolysis of 2c (100 mg, 0.00029 mol) was carried out under similar condition as described above for 2a. The chromatographic separations of the photolysate provided new product 3c.
3c: Light yellow solid; Yield 52%; m.p.: 238–240 °C; IR (KBr) cm−1: 1632 (C⚌O), 1602 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.29 (1H, dd, Jm,o = 2.2, 7.9 Hz, H-5), 8.06 (2H, m, H-2′, 6′), 7.68 (1H, d, Jm,o = 2.0, 7.2 Hz, H-7), 7.50 (1H, td, Jm,o = 2.0, 8.0 Hz, H-6), 7.24 (1H, d, Jo = 8.0 Hz, H-8), 6.98 (2H, m, H-3′, 5′), 6.56 (1H, d, J1″,2″ = 12.2 Hz, H-1″), 5.30 (1H, dt, J2″,3″ = 7.0 Hz, J2″,1″ = 12.4 Hz, H-2″), 4.97 (3H, s, 4′-OCH3), 3.07 (2H, m, H-3″), 1.83 (2H, m, H-4″), 1.52 (3H, t, Jvic = 6.4 Hz, H-5″); 13C-NMR (100 MHz, CDCl3): δ 175.79 (C-4), 152.82 (C-8a), 143.64 (C-1″), 137.84 (C-3), 135.13 (C-7), 134.28 (C-2), 129.00 (C-1′), 127.64 (C-3′, 5′), 125.82 (C-2′, 4′, 6′); 124.47 (C-5), 121.65 (C-6), 120.44 (C-4a), 114.22 (C-8), 103.78 (C-2″), 29.32 (C-3″), 21.98 (C-4″), 14.56 (C-5″); MS (ESI): m/z (M)+ 336 (100%), 216 (60.2%), 120 (12.5%), 92 (22.6%); Anal. Calc. For C21H20O4: C, 75.00; H, 5.95; Found C, 75.29; H, 5.92%.
2.9 Synthesis of (E)-3-(pent-1-enyloxy)-2-(thiophen-2-yl)-4H-chromen-4-one 3d
The photolysis of 2d (100 mg, 0.00032 mol) under similar conditions as used for 2a yielded a new compound 3d.
3d: Light yellow solid; Yield 54%; m.p.: 248–250 °C; IR (KBr) cm−1: 1640 (C⚌O), 1606 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.40 (1H, dd, Jm,o = 2.4, 7.6 Hz, H-5), 7.95 (2H, d, J3′,4′ = 3,7 Hz, H-3′), 7.84 (1H, d, Jm,o = 2.2, 7.0 Hz, H-7), 7.63 (2H, d, J5′,4′ = 4.8 Hz, H-5′), 7.45 (1H, td, Jm,o = 2.0, 7.6 Hz, H-6), 7.27 (1H, d, Jo = 8.0 Hz, H-8), 7.24 (1H, dd, J4′,3′ = 3.7 Hz, J4′,5′ = 4.8 Hz, H-4′), 6.40 (1H, d, J1″,2″ = 12.2 Hz, H-1″), 5.27 (1H, dt, J2″,3″ = 6.9 Hz, J2″,1″ = 12.3 Hz, H-2″), 2.95 (2H, m, H-3″), 1.85 (2H, m, H-4″), 1.59 (3H, t, Jvic = 6.4 Hz, H-5″); 13C-NMR (100 MHz, CDCl3): δ 177.28 (C-4), 153.12 (C-8a), 140.85 (C-1″), 136.78 (C-3), 133.35 (C-7), 130.48 (C-2), 130.24 (C-5′), 129.54 (C-2′), 128.14 (C-4′), 127.64 (C-3′), 123.87 (C-5), 121.27 (C-6), 120.98 (C-4a), 114.96 (C-8), 103.59 (C-2″), 28.00 (C-3″), 21.46 (C-4″), 14.02 (C-5″); MS (ESI): m/z (M)+ 312 (32.5%), 192 (100%), 120 (23.7%), 92 (11.8%); Anal. Calc. For C18H16O3S: C, 69.23; H, 5.12; S, 10.25; Found C, 69.47; H, 5.15; S, 10.21%.
2.10 Synthesis of (E)-2-(furan-2-yl)-3-(pent-1-enyloxy)-4H-chromen-4-one 3e
The photoirradiation of compound 2e (100 mg, 0.00033 mol) under similar conditions as used earlier for 2a provided a new product 3e.
3e Light yellow solid; Yield 51%; m.p.: 216–218 °C; IR (KBr) cm−1: 1623 (C⚌O), 1590 (C⚌C); 1H-NMR (400 MHz, CDCl3): δ 8.20 (1H, dd, Jm,o = 2.3, 7.8 Hz, H-5), 7.79 (2H, d, J3′,4′ = 1.2 Hz, H-3′), 7.72 (1H, d, Jm,o = 2.2, 7.6 Hz, H-7), 7.59 (1H, td, Jm,o = 2.0, 7.6 Hz, H-6), 7.38 (1H, d, Jo = 8.0 Hz, H-8), 7.31 (1H, d, J5′,4′ = 3.8 Hz, H-5′), 6.63 (1H, dd, J4′,3′ = 1.2 Hz, J4′,5′ = 3.6 Hz, H-4′), 6.52 (1H, d, J1″,2″ = 12.4 Hz, H-1″), 5.35 (1H, dt, J2″,3″ = 6.9 Hz, J2″,1″ = 12.2 Hz, H-2″), 2.90 (2H, m, H-3″), 1.80 (2H, m, H-4″), 1.47 (3H, t, Jvic = 6.4 Hz, H-5″); 13C-NMR (100 MHz, CDCl3): δ 176.04 (C-4), 154.29 (C-8a), 140.85 (C-1″), 136.78 (C-3), 133.35 (C-7), 133.24 (C-2), 129.94 (C-5′), 129.15 (C-2′), 128.75 (C-4′), 125.89 (C-3′), 125.64 (C-5), 122.57 (C-6), 120.69 (C-4a), 114.48 (C-8), 102.30 (C-2″), 29.78 (C-3″), 22.49 (C-4″), 14.32 (C-5″); MS (ESI): m/z (M)+ 296 (100%), 176 (23.8%), 120 (16.4%), 92 (30.9%); Anal. Calc. For C18H16O4: C, 72.97; H, 5.40; Found C, 72.63; H, 5.37%.
3 Results and Discussion
The compounds 2a–2e required for this study were obtained from the O-alkylation of 3-hydroxychromen-4-ones(Yusuf and M., 2012; Gupta et al., 1995; Gupta et al., 1999; Yadav et al., 1990) 1a–1e by using bromomethyl-cyclobutane in the presence of anhydrous K2CO3 and Bu4N+I− (PTC) in dry acetone (Scheme 1). In the absence of PTC, the reactions involved long reaction times and also provided poor yields of alkoxy chromones. The structures of 2a–2e were determined from the rigorous analysis of their IR, 1H-NMR, 13C-NMR and ESI-MS spectral data. The elemental analysis also confirmed the purity of these products.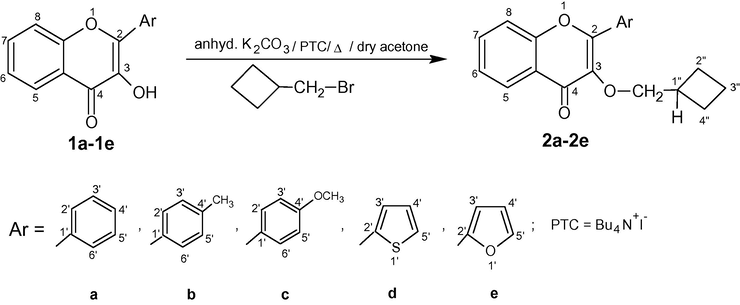
Synthesis of 3-cyclobutylmethoxy-chromones 2a–2e.
The presence of conjugated C⚌O and C⚌C groups in 2a–2e was confirmed by the appearance of strong IR bands in the region of 1642–1630 and 1616–1602 cm−1 respectively. The major feature of their 1H-NMR (400 MHz, CDCl3) spectra was the presence of a doublet at δ 4.42–4.05 (Jvic = 6.9–6.6 Hz) and a multiplet at δ 2.82–2.60 which could be assigned to OCH2 and H-1″ respectively. In addition to these signals in the aliphatic region, two multiplets were observed at δ 2.17–1.98 (2H, H-3″) and 2.01–1.80 (4H, H-2″, 4″). The benzenoid ring protons H-5, H-7, H-6 and H-8 produced suitable resonances at δ 8.27–8.14 (1H, dd, Jm,o = 2.1–2.0, 7.9–7.6 Hz), 7.80–7.43 (1H, td, Jm,o = 2.3–2.1, 8.6–7.9 Hz), 7.59–7.48 (1H, d, Jo = 8.4–8.0 Hz) and 7.49–7.39 (1H, d, Jo = 8.0–7.8 Hz) respectively. The hydrogens of 2-aryl ring in 2a–2c were found to be present at δ 8.16–7.00 and two additional singlets also appeared at δ 2.44 and 3.92 due to 4′-CH3 and 4′-OCH3 in 2b and 2c respectively. But in the compounds 2d and 2e, three protons of 2-thienyl and 2-furanyl ring were resonating at δ 7.98 (1H, d, J3′,4′ = 3.7 Hz, H-3′), 7.67 (1H, d, J5′,4′ = 5.0 Hz, H-5′), 7.22 (1H, dd, J4′,3′ = 3.7 Hz, J4′,5′ = 5.0 Hz, H-4′) and 7.83 (1H, d, J3′,4′ = 1.2 Hz, H-3′), 7.33 (1H, d, J5′,4′ = 3.2 Hz, H-5′), 6.69 (1H, dd, J4′,3′ = 1.2 Hz, J4′,5′ = 3.5 Hz, H-4′) respectively. 13C-NMR (100 MHz, CDCl3) spectra of 2a–2e revealed most downfield resonance at δ 186.65–172.64 (C⚌O) and other signals present at δ 162.44–154.48 (C-2), 156.21–151.19 (C-8a) and 140.79–137.24 (C-3) appeared downfield due to the direct linkage of these carbons to the electronegative oxygen atom. The remaining noticeable signals in the aromatic region were found to be placed at δ 132.89–129.86 (C-5), 135.49–130.26 (C-7), 118.16–113.94 (C-8) and 123.66–114.18 (C-6). The carbon atoms of 2-phenyl/thienyl/furanyl rings were also resonating at appropriate positions in the aromatic region (vide experimental). Suitable resonances were also observed in the aliphatic region at δ 72.39–67.36 (OCH2) and 34.90–18.51 (C-1″, 2″, 3″, 4″) respectively.
The photochemical reactions of 3-cyclobutylmethoxy-chromones 2a–2e were carried out under inert atmosphere in dry MseOH with Pyrex filtered light from a 125 W Hg arc lamp. The progress of the photoreactions was monitored by TLC and after about 5–6 h most of the starting compounds were transformed into new products (Scheme 2). The column chromatographic separation of the reaction mixtures yielded 3a–3e (48–55%).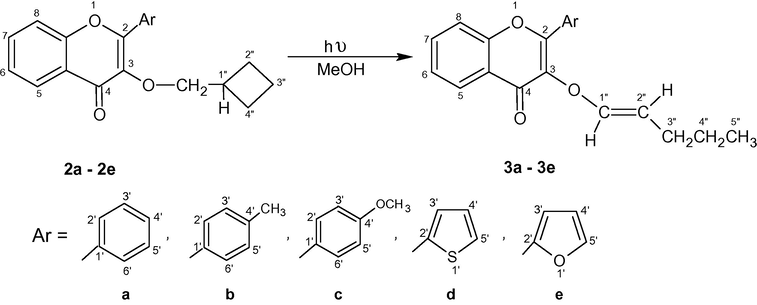
Photolysis of 3-cyclobutylmethoxy-chromones 2a–2e.
A comparison of 1H-NMR spectra of 3a–3e and 2a–2e showed that signals placed at δ 4.42–4.05 in latter were found to be missing altogether in the former which suggest the involvement of the OCH2 group hydrogen in the photoreaction. The appearance of a doublet and a doublet of triplet at δ 6.61–6.40 (1H, J1″,2″ = 12.4–12.2 Hz, H-1″) and 5.35–5.19 (1H, J2″,3″ = 7.2–6.9 Hz, J2″,1″ = 12.4–12.2 Hz, H-2″) describes the formation of trans double bond between C-1″ and C-2″; downfield occurrence of the former as compared to latter can be ascribed to its direct attachment to the electronegative oxygen atom. The remaining aromatic protons in 3a–3c (H-5, 6, 7, 8 and H-2′, 3′, 4′, 5′, 6′) were easily resonating at δ 8.40–7.24. The 2-thienyl and 2-furanyl ring protons (H-3′, H-4′ and H-5′) in 3d and 3e were found to be placed at expected positions in the aromatic region (vide experimental).
The 13C-NMR (100 MHz, CDCl3) spectra of 3a–3e were also very helpful to corroborate the proposed structures. The downfield signals at δ 177.68–174.24 were observed due to carbonyl group of the pyrone ring. The resonances present at δ 155.74–153.12, 143.64–140.20 and 138.01–136.78 may be assigned to C-8a, C-1″ and C-3 due to their direct linkage to the electronegative atom. The signals due to C-2″ and propyl chain (C-3″, 4″ and 5″) were placed at δ 104.64–102.30 and 29.78–13.64 respectively.
The structures of the prepared compounds 2a–2e and 3a–3e were also confirmed by their ESI-MS spectra (vide experimental).
Mechanistically, phototransformation of 2a-2e may be occurring through H-abstraction by the photoexcited C⚌O group from C3–OCH2 group which lead to the formation of 1,4-biradical 2a'–2e'. The latter causes the cleavage of the cyclobutane ring due to its inherent ring strain to give 1,8-biradical 2a''–2e'' which finally captures H. from the solvent (MeOH) to provide vinyl ethers 3a–3e as the end products (Scheme 3).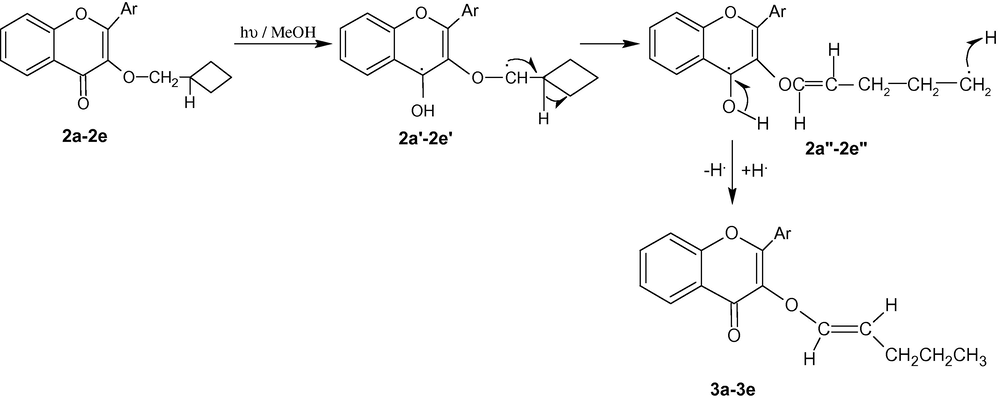
Mechanism showing the phototransformations of 2a–2e.
4 Antimicrobial Activity
The antimicrobial activity of the newly prepared compounds was screened in vitro against seven bacterial and fungal strains namely Staphylococcus aureus (MTCC 96), Bacillus subtilis (MTCC 441), Escherichia coli (MTCC 443), Pseudomonas aeruginosa (MTCC 424), Pseudomonas florescens (MTCC 103), Staphylococcus pyrogens (MTCC 442) and Klebsiella pneumoniae (MTCC 3384) and Aspergillus janus (2751), Aspergillus niger (MTCC 281), Fusarium oxysporium (MTCC 2480), Aspergillus sclerotium (MTCC 1008) and Penicillium glabrum (4951). The in vitro MIC of these compounds were evaluated by using the serial tube dilution method(Pandey and Khan, 2008) at concentrations of 128, 64, 32, 16, 8 and 4 μg/mL against the above said microorganisms. The bacterial pathogens were subcultured on nutrient agar medium whereas fungal strains were subcultured on the malt extract medium. The bacterial and fungal strains were incubated at 37°C and 28°C respectively. DMSO was used as the negative control while Amoxicillin and Fluconozole were used as reference drugs for comparison. The minimum inhibitory concentrations (MIC) of the studied compounds were noted by the appearance of turbidity in test tubes after incubation. The results of the MIC determinations have been presented in Table 1 (2a–2e) and Table 2 (3a–3e).
Compound No.
Gram (−ve) bacteria
Gram (+ve) bacteria
Fungi
E. coli
K. pneumoniae
P. Aeruginosa
P. florescens
S. Aureus
B. subtilis
S. pyrogens
A. janus
P. glabrum
A. niger
F. oxysporium
A. sclerotiorum
2a
16
8
16
16
16
16
16
16
16
32
16
8
2b
32
16
16
16
32
16
32
32
32
16
32
32
2c
32
16
8
8
32
8
16
16
16
32
32
16
2d
16
8
8
8
16
8
16
16
32
32
16
16
2e
32
8
16
16
32
16
16
16
16
16
16
8
Amoxicillin
4
4
4
4
2
2
4
–
–
–
–
–
Fluconozole
–
–
–
–
–
–
–
2
2
2
2
2
Compound No.
Gram (−ve) bacteria
Gram (+ve) bacteria
Fungi
E. coli
K. pneumoniae
P. aeruginosa
P. florescens
S. aureus
B. subtilis
S. pyrogens
A. janus
P. glabrum
A. niger
F. oxysporium
A. sclerotiorum
3a
16
8
8
16
8
16
16
16
8
32
16
8
3b
32
16
8
16
32
8
32
32
32
16
32
8
3c
32
16
16
16
32
8
8
16
16
32
32
16
3d
32
8
16
8
32
16
16
16
8
16
16
8
3e
16
8
8
8
16
8
8
16
32
32
16
16
Amoxicillin
4
4
4
4
2
2
4
–
–
–
–
–
Fluconozole
–
–
–
–
–
–
–
2
2
2
2
2
It is evident from Table 1 that compounds 2a, 2d and 2e showed significant activity (MIC-8 μg/mL) against Klebsiella pneumoniae whereas 2c and 2d were having similar MIC against Pseudomonas aeruginosa, Bacillius subtilis and Pseudomonas florescens. The compound 2a also had MIC of 8 μg/mL against Aspergillius janus.
Table 2 describes that most of the compounds showed significant activities (MIC-8 μg/mL) against tested microorganisms except Aspergillus niger, Fusarium oxysporium and Aspergillus janus. Compounds 3b, 3c and 3d exhibited noticeable activities (MIC-8 μg/mL) against Bacillius subtilis and 3c also had similar activity against Staphylococcus pyrogens. The compounds 3a and 3d were found to be active against Aspergillus sclerotium, Penicillium glabrum, Klebsiella pneumoniae, Pseudomonas aeruginosa and Aspergillus sclerotium, Penicillium glabrum, Klebsiella pneumoniae, Pseudomonas florescens and Pseudomonas aeruginosa respectively. Compounds 3b and 3e could provide significant MIC (8 μg/mL) against Pseudomonas aeruginosa and Klebsiella pneumoniae, Pseudomonas florescens respectively.
The above study clearly suggests that compounds 2a, 2d, 3a, 3c and 3d exhibited significant antimicrobial results against most of the tested microorganisms. It was also found that chromen-4-one substituted vinyl ethers 3a–3e seem to be better antimicrobial agents than the corresponding cyclobutylmethoxy derivatives 2a–2e. The importance of this research work lies in the possibility that a thorough investigation regarding the structure–activity relationship, toxicity and biological effects of these compounds could be helpful in designing more potent antibacterial agents for therapeutic uses.
5 Conclusion
This study provides a simple photochemical method for the synthesis of new chromen-4-one based vinyl ethers. The significance of this method lies in the fact that here vinyl ethers are obtained under mild conditions without using any toxic and specific reagents and some of the resultant compounds also exhibited significant antimicrobial behaviors.
Acknowledgement
Authors are extremely thankful to the CSIR, New Delhi for providing financial assistance for this research work.
References
- Bioorg. Med. Chem.. 2005;13:1221-1230.
- J. Org. Chem.. 1986;51:4169-4175.
- Tetrahedron Lett.. 1998;39:5765-5768.
- De Meijere, A. (Ed.), 1997. Carbocyclic of four membered Ring Compounds, vol. 17 e/f. Houben-Weyl Methods Org. Chem., Thieme, Stuttgart.
- J. Org. Chem.. 1992;57:550-552.
- Chem. Int. Ed.. 1969;8:333-343.
- J. Am. Chem. Soc.. 2002;124:396-403.
- J. Chem. Soc. Perkin Transaction. 1995;1(2):177-184.
- J. Chem. Soc. Perkin Transaction. 1999;1(1):2391-2395.
- Tetrahedron Lett.. 2002;43:6875-6877.
- Photochemistry in Organic Synthesis. In: Coyle J.D., ed. Carbonyl Compounds H-abstraction. London: The Royal Society of Chemistry; 1986. p. :61-79.
- [Google Scholar]
- J. Organomet. Chem.. 2001;630:104-117.
- J. Am. Chem. Soc.. 1992;114:8705-8707.
- Arkivoc. 2006;9:239-264.
- Tetrahedron Lett.. 2001;42:2023-2025.
- J. Am. Chem. Soc.. 2005;127:2854-2855.
- Tetrahedron Lett.. 2006;47:6297-6300.
- J. Am. Chem. Soc.. 2002;124:1590-1591.
- Archiv der Pharmazie. 2008;341(7):418-423.
- J. Org. Chem.. 2001;66:6800-6802.
- J. Org. Chem.. 2005;70:1408-1416.
- Synthesis 2001:1253-1257.
- Nature. 1961;192:725-727.
- Tetrahedron Lett.. 1992;31:3775-3778.
- Tetrahedron Lett.. 1992;33:2213-2216.
- Comprehensive Organic Synthesis. 1991;9
- Acc. Chem. Res.. 1989;22:83-91.
- J. Org. Chem.. 2004;69:7582-7591.
- Topic Curr. Chem.. 1986;446
- J. Ind. Chem. Soc.. 1990;67(9):770-772.
- The Scientific World Journal Article ID 954934 2012:8.




