

Translate this page into:
Physically crosslinked poly(vinyl alcohol)-hydroxyethyl starch blend hydrogel membranes: Synthesis and characterization for biomedical applications
*Corresponding author. Tel.: +20 1283320302; fax: +20 34593414 badawykamoun@yahoo.com (Elbadawy A. Kamoun)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Available online 7 June 2013
Peer review under responsibility of King Saud University.

Abstract
Poly(vinyl alcohol), PVA is a polymer of great importance because of its many appealing characteristics specifically for various pharmaceutical and biomedical applications. Physically crosslinked hydrogel membranes composed of different amounts of hydroxyethyl starch (HES) in (PVA) and ampicillin were prepared by applying freeze–thawing method. This freezing–thawing cycle was repeated for three consecutive cycles. Physicochemical properties of PVA–HES membrane gel such as gel fraction, swelling, morphology, elongation, tensile strength, and protein adsorption were investigated. Introducing HES into freeze–thawed PVA structure affected crystal size distribution of PVA; and hence physicochemical properties and morphological structure have been affected. Increased HES concentration decreased the gel fraction %, maximum strength and break elongation. Indeed it resulted into a significant incrementing of the swelling ability, amount of protein adsorption, broader pore size, and pore distribution of membrane morphological structure. Furthermore, an increase in HES concentration resulted in better and still lower thermal stability compared to virgin PVA and freeze–thawed PVA. The maximum weight loss of PVA–HES hydrogel membranes ranged between 18% and 60% according to HES content, after two days of degradation in phosphate buffer saline (PBS), which indicates they are biodegradable. Thus, PVA–HES hydrogel membranes containing ampicillin could be a novel approach for biomedical application e.g. wound dressing purposes.
Keywords
Poly(vinyl alcohol)
Hydroxyethyl starch
Hydrogel membranes
Freeze–thaw method
Physicochemical properties
Thermal properties

1 Introduction
PVA hydrogels have been previously used for numerous biomedical and pharmaceutical applications (Tanigami et al., 1995). PVA hydrogels have several advantages that make them proper candidates for biomaterials. Some of these advantages include their non-toxic, non-carcinogenic, biocompatible, bio-adhesive characteristics, excellent film-forming, excellent transparency, and additionally their ease of processing. PVA has a simple chemical structure and its chemical modification is possible using simple reaction as well. Meanwhile, PVA gels possess a high degree of swelling in water or biological fluids and an elastic or rubbery nature structure (Chan, 1999). Because of latter advantages, PVA is capable of simulating natural tissues and can be completely accepted into the body. PVA gels have been applied in different biomedical application sites such as contact lenses, the lining for artificial hearts, wound dressing, and drug delivery applications. Peppas and Merrill (1977a,b) have revealed in their earliest work in considering PVA hydrogels as biomaterials. Generally, hydrogels were achieved by crosslinking process of polymers, which may be done by a chemical reaction (e.g. radical polymerization, chemical reaction of complementary groups, using high energy irradiation, or enzymatic reaction) or by physical reaction (e.g. ionic interaction, crystallization of the polymeric chain, hydrogen bond between chains, protein interaction, or design of amphiphilic block and graft copolymers) (Hennink and Nostrum, 2002).
In recent decades, the need of physical crosslinked gels has been potentially increased, (Van Tomme et al., 2005) to avoid the use of chemical crosslinking agents and reagents. These agents are not only often toxic compounds which can be removed or extracted from prepared gels before application, but also can affect the integrity of the substances when entrapped (e.g. proteins, drugs, and cells). Therefore, the physical crosslinking method has been chosen and preferred comparable with the chemical crosslinking method for most crosslinked polymers’ preparation. Several attempts have been done to prepare crosslinked PVA-based hydrogels including radiation crosslinking, (Park and Chang, 2003) chemical reaction with glyoxal, (Teramoto et al., 2001) bifunctional reagents with glutaraldehyde, (Dai and Barbari, 1999) or reaction with borates (Korsmeyer and Peppas, 1981).
Although, an aqueous solution of PVA can form low strength of hydrogel upon exposure to very long storage time at room temperature, but this method did not meet any application requirements, where the mechanical properties are the most important character in hydrogel properties. The earliest attempt for crosslinking of PVA using freezing–thawing method has been pioneered by Peppas (1975). Semi-crystalline PVA gels were prepared by exposing PVA aqueous solution to repetitive freezing–thawing cycles which induced crystallization and result in a network structure, which act as physical crosslinking sites in the network. The freezing–thawing method is regarded the best and the preferred method for obtaining physically crosslinked PVA hydrogel without using any traditional toxic chemical crosslinking agent (Yokoyama et al., 1986). While, the obtained mechanical properties of physically crosslinked PVA hydrogel are tunable structure and can be adjusted by the molecular weight and concentration of PVA or the cycle number of freeze–thaw method. Many polymers have been previously blended to PVA to meet such clinical demands or sometimes to develop a polymeric system suitable for specific biomedical applications, such as drug delivery application, tissue engineering or wound dressing. The blended polymers with PVA are like PVP, (Park and Chang, 2003) chitosan, (Kim et al., 2003a) poly (N-isopropylacrylamide), (Kim et al., 2003b) carboxymethyl chitosan, (Zhao et al., 2003) alginate, (Kim et al., 2008) and dextran (Cascone et al., 1999; Fathi et al., 2011).
Hydroxyethyl starch, (HES) is a synthetic polymer prepared by reacting naturally occurring amylopectin with ethylene oxide resulting in hydroxyethyl groups being added to oxygen at different carbon positions at glucopyranose unit C2, C3, or C6 to be in the final form of α-1,4-linked d-glucopyranose residues (Kalhorn et al., 1984). HES has valuable medical applications e.g. as blood plasma volume expander polymers (Deitrich, 2001). Leukapheresis agent, as cryo-preservative (Kalhorn et al., 1984), as polymer drug delivery, (Kamoun and Menzel, 2012) and as blood isotonic electrolyte solutions, which further evidenced its non toxicity, biodegradability, and biocompatibility with the human body.(Dorothee et al., 1998) Thus, HES has been chosen to incorporate with PVA membranes due to its unique biomedical characteristics mentioned earlier, additionally its appealing intrinsic properties e.g. high hydrophilicity, abundance natural sources, and low cost polymers compared to other polysaccharides e.g. sodium alginate and dextran, which have been previously blended with PVA membranes.
In the light of such contributions, the blended HES with PVA hydrogel has not been previously reported yet, and in this work the results of PVA–HES blend membrane based hydrogels are explained in detail for the first time in the literature. PVA–HES blend gel membranes were prepared and entanglement physically using freeze–thaw cycle method at high concentrations of PVA (10%, w/w) and high HES contents (0%, 25%, 33%, 50%, 65%, and 75%, w/w). The PVA–HES blend gel membranes were characterized by Fourier transformer infrared (FT-IR), scanning electron microscope (SEM), differential scanning calorimetry (DSC), and thermal gravimetric analysis (TGA). In addition, the physicochemical properties of gel membranes e.g. gel fraction, swelling behavior, maximum tensile strength, protein adsorption, and protein release profile have been assessed for wound dressing polymeric membrane materials.
2 Experimental
2.1 Materials
PVA (typically average Mw = 72,000 g/mol; 98.9% hydrolyzed) was obtained from Biochemica, Germany. HES (average Mw = 130,000 g/mol as determined by GPC, and DS = 0.5), albumin from bovine serum (BSA, fraction V, minimum 96% electrophoresis, nitrogen content 16.2%), and ampicillin sodium salt were purchased from Sigma–Aldrich Chemie GmbH, Steinheim, Germany. Folin & Ciocalteu,s phenol reagent (FC, 2 N with respect to acid), was exported from Park Scientific Limited, Northampton, UK. Distilled water was used throughout this research. All other chemicals were used without any further purification.
2.2 Instrumental analysis and measurements
2.2.1 Preparation of PVA–HES hydrogels
PVA–HES hydrogel membranes were prepared by freezing–thawing (F–T) cycle according to the reported procedure of (Peppas and Stauffer, 1991). Briefly, aqueous solution containing 10% (w/v) PVA and 1.5% (w/v) HES and 20 mg of ampicillin sodium salt were carefully dissolved in deionized water. Different proportions of PVA and HES contents (0%, 25%, 33%, 50%, 65%, and 75%) solutions were mixed, sonicated, and vortexed for 3 h. Proper amounts of this mixture were poured in standard disposable polypropylene Petri dishes (inside dimension is 84 mm diameter × 12 mm H), followed by freezing at −20 °C for 18 h and thawing for 6 h at 25 °C for three continuous cycles, to provide mechanically acceptable hydrogel properties for further experiments. The obtained PVA–HES hydrogel membranes were frozen in liquid nitrogen for 10 min before being lyophilized fractures for SEM investigations. All samples were left in deionized water for 72 h to extract leachable sol fraction or unconnected HES from polymer matrix for further characterizations.
2.2.2 Gel fraction
The obtained PVA–HES hydrogel membranes were dried first in a laminar airflow chamber under sterile conditions for 24 h, then dried again at 50 °C in an oven for 24 h and weighted (W0). The dried xerogel samples were soaked in distilled water for 24 h up to an equilibrium swelling weight (Ws) for removing the leachable or soluble HES parts from membrane. The gel membrane then dried directly at 50 °C in an oven and weighted again (We). The gel fraction (GF %) was calculated by the following equation (Yang et al., 2008).
2.2.3 Swelling behavior
In order to measuring the swelling degree of PVA–HES hydrogel membranes, membrane samples were cut into 2 × 2 cm pieces and dried at 50 °C in an oven for 24 h, the weight of dried sample was determined (We). The dried samples were soaked in distilled water, maintained and incubated at 37 °C, then weighted (Ws) at specific interval times. The water uptake of PVA–HES hydrogel membranes was determined using the following formula (Yang et al., 2008).
2.2.4 Protein adsorption study
The amount of adsorbed bovine serum albumin (BSA) was detected by UV–visible spectrophotometer (Type: Ultrospec 2000, Pharmacia Biotech., Cambridge, England). In order to establish the relationship between the visible absorbance of BSA at 630 nm and the concentration of BSA, a calibration curve was drawn for standard solution of BSA ranging from 3–60 mg/ml. All standard solutions were prepared with distilled water. From the calibration curve a study was made restricting the curve to the linear part that followed Beer’s law where A is the absorbance, c is the concentration, a is a proportionality constant, and L is the path-length which is constant.(Queiroz et al., 2001) Pieces of PVA hydrogel membranes cut into 1 × 1 cm were immersed in 10 ml phosphate buffer saline (pH 7.4), and incubated at 37 °C for 24 h until reaching equilibrium swelling weight. The swollen hydrogel pieces were transferred to buffer solution containing BSA (30 mg/ml) and shacked for 4 h at 37 °C. After protein adsorption, the hydrogel pieces were gently removed. The protein adsorption of the each sample was calculated by the difference between protein concentrations before and after immersing hydrogel pieces in protein/phosphate buffer solution using albumin reagent kit (absorbance range at 630 nm), this procedure has been adapted and modified from the procedure of Lin et al. (2006)
2.2.5 Hydrolytic degradation
PVA–HES membranes contacting ampicillin have been dried under vacuum at 50 °C for 24 h. Dried membrane samples with size of 15 × 8 mm, were weighted and immersed in 3 ml phosphate buffer saline (0.1 M, pH 7.4, at 37 °C). The samples were removed at timed intervals and then wiped gently with soft paper to remove surface water. The samples were dried again under the same mentioned drying conditions above and finally weighted. All experiments were done in duplicate.
2.2.6 Operating procedure of in vitro drug release profiles of the hydrogel
15 ml of FC reagent was added into a series of 50 ml conical flasks containing ampicillin (0.2–0.8 mg). The contents were completely mixed and kept into a thermostated water bath at 95 °C for 30 min. The flasks were taken out cooled at room temperature at ∼25 °C, then transferred into 25 ml slandered volumetric flask and diluted up to the mark with distilled water. The absorbance of the resulting blue color of dye was measured against a reagent blank at 750 nm using spectrophotometer as previously discussed (Queiroz et al., 2001; Lin et al., 2006; Ahmad et al., 2004).
Two pieces of PVA–HES hydrogel membranes contacting ampicillin (the obtained casted membrane from the Petri dish, each piece with surface area is almost 55 cm2 and membrane thickness is between 0.06 and 0.1 mm), were immersed in phosphate buffer (pH 8, at 37 °C) and kept in continuous shaking. 200 μL of last solution was withdrawn at timed intervals each 15 min and was added to 3 ml FC reagent. The last mixture was heated at 95 °C for 30 min, and then taken out for cooling at room temperature; 1.8 ml of FC reagent was added to the cooled mixture and carefully vortexed before measuring. The absorbance of ampicillin released from prepared samples was detected by spectrophotometer at 750 nm. All experiments were done in triplicate.
2.2.7 Characterizations
-
●
FT-IR
Vacuumed and dried samples of freeze–thawed PVA, HES, and freeze–thawed PVA–HES xerogels were analyzed by FT-IR on an EQUINOX 55 instrument (BRUKER, Germany). Translucent KBr-disks were prepared by grinding the dried sample materials together with infrared grade KBr and then pressing. The FTIR spectra were obtained by recording 64 scans between 4000 and 400 cm−1 with a resolution of 2 cm−1. All samples were freeze-dried using liquid nitrogen, crushed to a fine powder (KBr: sample = 140 mg: 2 mg), and pressed by applying a force 105 N into a transparent disk (maximum disk weight = 145 mg) with a diameter of 13 mm. All samples were measured in absorbance mode.
-
●
Thermal properties
The thermal characterization of vacuumed-dried PVA–HES xerogels, has been accomplished using TGA and DSC thermograms. The thermo-gravimetric analysis (TGA) was performed on a 204 Phoenix TGA instrument (NETZSCH, Germany) from 50 to 600 °C at a heating rate of 10 °C/min. The onset temperature (Tonset) was determined by TGA thermograms. Tonset is defined as the temperature at the intersection of the baseline mass and tangent drawn to the mass curve at the inflection point or point of greatest rate of mass loss % (Kamoun and Menzel, 2012).
Glass transition temperatures (Tg) of dried PVA–HES xerogels were determined using a differential scanning calorimeter, DSC (model: 204 Phoenix DSC instrument), (NETSCH, Germany). All measurements were made at a heating rate of 5 °C min−1 from 25 to 500 °C under nitrogen. The Tg, Tonset, the melting temperature (Tm), and the heat of fusion or enthalpy (ΔHm) were measured from DSC thermograms. Tg values were determined as mid-point in the thermograms, as measured from the extensions of the pre-and post-transition baselines. Whereas, the degree of crystallization of PVA was also determined using the following equation,
Xc = (ΔHm/ΔHom) × 100, where PVA crystallization degree has been discussed elsewhere (Hassan and Peppas, 2000).
-
●
Mechanical properties
The maximum tensile strength and the elongation degree to break PVA–HES blend hydrogel membranes have been conducted using a tensile test machine (model: AG-I/50–10KN, Japan). PVA–HES membranes were cut into specific a dog-bone shape (6 cm long, 2 cm wide at the ends, and 1 cm at the middle). The analysis was performed at a stretching rate of 20 mm/min with pre-load of 0.5 N to determine load for each sample (Alencar et al., 2003). The thickness of membrane samples was measured with a digimatic caliper before examination.
-
●
Scanning electron microscope
The surface and internal structure of the xerogel membrane samples were investigated by Analytical-SEM (type: JEOL, JSM-6360LA, Japan) with 15 kV voltage for secondary electron imaging. The xerogel membranes were dehydrated by freeze-dryer and coated with Au using an ion sputter coater in (model: 11430, USA, combined with vacuum base unit or SPi module control, model: 11425, USA).
3 Results and discussion
Poly(vinyl alcohol)-hydroxyethyl starch blend hydrogel membranes were synthesized using the freeze–thawing technique, while the crosslinking was accomplished physically by crystallization step. In Fig. 1, FTIR spectra of pure freeze–thawed PVA, pure HES as blend materials, and freeze–thawed PVA–HES blend polymer membranes are shown. It clearly reveals the main peaks associated with freeze–thawed PVA. For example, it can be easily observed that C–H broad alkyl stretching band (ν = 2850 cm−1) and the typical strong –OH group bands for free unreacted alcohol (non-bonded –OH stretching band at ν = 3650–3590 cm−1) and hydrogen bonded bands (bonded –OH stretching bands at ν = 3600–3200 cm−1). The hydrogen bonding between –OH groups can occur among PVA chains due to high hydrophilic forces (Mansur et al., 2004). Also, presence of sharp absorption peak was noted at ν = 1150 cm−1. This band has been used as an indicator for PVA structure, because it is a semi-crystalline synthetic polymer able to form some domains depending on several process parameters such as the F–T cycle number, the molecular weight and concentration of used PVA (Mansur et al., 2004). Additionally, it was found that a notable stretching band at ν = 1569–1460 cm−1 of –CH2 groups which are regarded as feature groups for chemical structure of PVA and PVA–HES blend polymer. All last mentioned stretching peaks, have been detected in structure of both PVA and PVA–HES blend polymer. Furthermore, all vibration peaks of PVA and HES have been verified in IR-spectrum of PVA–HES.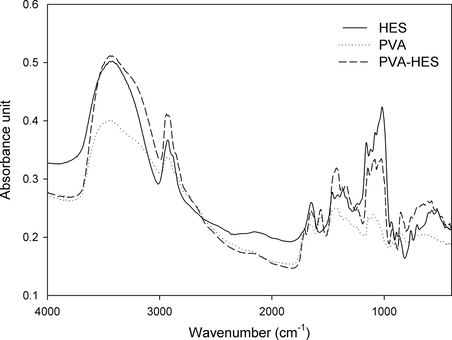
FTIR spectra of pure freeze–thawed PVA membrane, pure HES, and freeze–thawed PVA–HES blend polymer.
The consecutive F–T cycles formed entangled PVA–HES polymer hydrogel membranes. The influence of HES contents (0%, 25%, 33%, 50%, 65%, and 75%) and drug introduction on the gel fraction percentage (GF %), is displayed in Fig. 2. Generally, the lower gel fraction was the weakest mechanical stability and less flexibility of gel was. In the absence of HES and drug (0% HES content and without drug), the gel fraction increased to the maximum value which was about 86% and relatively high, suggesting that the PVA was almost crystallized in the highest degree and consequently crosslinked. This result is consistent with the obtained results by Yokoyama et al. (1986). While, GF% monotonically decreased with increasing HES contents or addition of drug in PVA hydrogel and decreased drastically to less than 40% at 75% of HES content in the PVA hydrogel. This behavior can be attributed to HES content and addition of drug in PVA hydrogel may reduce the crosslinking reaction and consequently the gelation process is clearly reduced.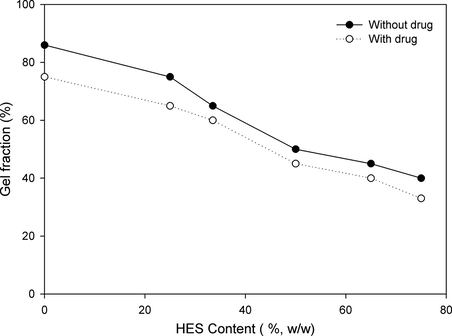
Effect of HES content in PVA hydrogel membranes on gel fraction.
Fig. 3 presents the water uptake percent of PVA–HES hydrogel membranes versus HES contents. In the light of our preliminary swelling study, when PVA–HES hydrogel membrane was immersed in distilled water for 20 min, small amounts of blended HES were dissolved in swelling medium. The dissolved amount of HES is sharply depending on the initial blended HES in PVA hydrogel. Moreover, the dissolved amount of HES significantly affected the swelling test. As seen in Fig. 3, the maximum swelling ability increases with increasing the HES content in PVA hydrogel up to a certain limit of huge swelling, the hydrogel structure was then destructed. This is due to HES does not crosslink and has high ability to solubilize in water of swelling medium. While, in the absence of HES (0%, HES content); high crosslinked structure for PVA hydrogel has been obtained and this structure could not retain water amount within which result in low swelling ability with a water uptake % of about 1500. When HES content increased to 75%, the water uptake % progressively increases to 2700%, after this content of HES the swelling ability decrease again. This is due to, the high content of HES in PVA film increases the wettability and hydrophilicity characters of hydrogel which somewhat results in partial or complete destruction of hydrogel structure. These results are compatible with the reported results of Balakrishnan et al. (2005) and Choi et al. (1999)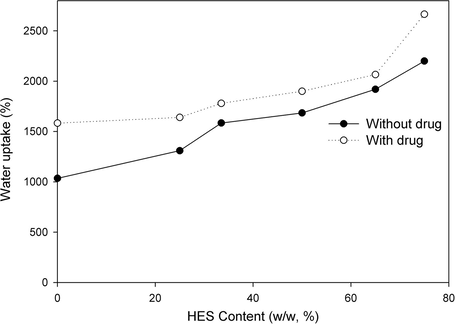
Water uptake (%) of PVA–HES hydrogel membranes as a function of HES contents in hydrogel membranes, swelled and incubated in distilled water at 37 °C.
To investigate the additional influence of HES on the mechanical properties of PVA hydrogel membranes, their maximum tensile strength and elongation at break have been evaluated and shown in Fig. 4. As shown, the maximum tensile strength and elongation at break of PVA–HES hydrogel membranes, sharply decreased with increasing HES contents. Proportionally, the maximum tensile strength at break possessed the same pattern behavior to elongation at break of hydrogel membranes. These results can be ascribed to the addition of HES into PVA hydrogels that may accelerate and destabilize the break elongation of hydrogel resulting in decreasing and deconstructing of the maximum tensile strength. These results are consistent with the obtained results of Rosiak et al. (2001). They have referred that the maximum tensile strength of PVA hydrogel decreased with increasing blend materials due to decreased crosslinking density. Similarly, our results are completely consistent with the reported results by Hwang et al. (2010). They have demonstrated that the maximum tensile strength of PVA hydrogel has sharply decreased with increasing dextran portions in the hydrogel.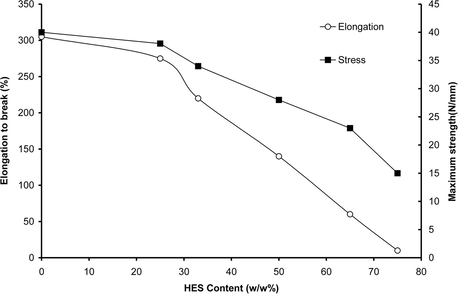
Effect of HES content in PVA hydrogel membranes on maximum strength and elongation to break.
The morphology structure of PVA–HES hydrogel membranes was investigated by SEM. The SEM micrographs of the surface and interior structures of PVA hydrogel membranes versus HES portions are shown in Fig. 5. According to SEM micrographs, the absence of HES presents a very smooth, uniform, and non pore shape surface structure. However, addition of HES in PVA hydrogel in different potions 50% and 75% provides very tiny pores at the surface; these pores notably increase with increasing HES contents. Furthermore, the number of pores and pore distribution area formation are significantly noticed at the surface of PVA hydrogel, when high HES contents were incorporated, while the pore diameter size have not been determined. This morphological change can be attributed to the extraction of HES particles in different agglomeration numbers and this explanation has been previously verified in swelling study. Although, excess amount of HES in PVA hydrogel can perturb the formation of PVA crystallites because of partial miscibility with PVA, but these parts of excess HES can be dissolved again and transferred from the hydrogel network into swelling solution. Also, it can be speculated that morphological changes are due to the presence of big difference in the homogeneity or miscibility degrees between two components of membrane (i.e. PVA and HES), which resulted in ordered-crystalline phase and uniform shape structure in case 0% HES, due to high entanglement of PVA and disordered-crystalline phase for PVA hydrogel blended with high portions of HES. The morphological results and our speculation are compatible with obtained results of Cascone et al. (1999) and Fathi et al. (2011) On the other hand, layered and channeling cross-section-shape structures were observed at 0% HES in PVA hydrogel. In addition, pores, cracks, and somewhat caves have appeared after incorporation of different portions of HES in cross-section structure of PVA hydrogel.
SEM images depicting the surface and the cross-section morphology distinctions of different HES contents in PVA hydrogel membranes, (original magnification was ×2500).
The protein adsorption onto PVA–HES blend hydrogel membranes has been conducted via in vitro experiments, and has been shown in Fig. 6. As shown in Fig. 6, the adsorption of BSA increased linearly from 0.25 to 1.125 mg/cm2 as the amounts of HES increased in the PVA hydrogels from 0 to 75 (%, w/w). Most importantly, the PVA hydrogels with both 25% and 75% HES showed the significant highest adsorption of BSA, while 33–65% of HES the differences in adsorption amounts were much closed, indicating that HES content affected the protein adsorption behavior onto the surface of PVA hydrogel. These results are consistent with the reported results by Kim et al. (2008) who revealed that the adsorption of protein increased with increasing blended alginate in PVA hydrogels. Moreover, the mechanism of protein adsorption on the surface of PVA–HES membranes can be attributed to various types of interaction forces between protein molecules and the membrane surface, such as weak bonding (van der Waal interactions, ionic bonding, hydrogen bonding or hydrophobic interactions) or strong chemical bonding due to chemically surface modified for membranes. Thus, in our study the clearest values of protein adsorption on PVA–HES surface have been detected with the highest values of hydrophilic surface interaction due to the addition of HES portions.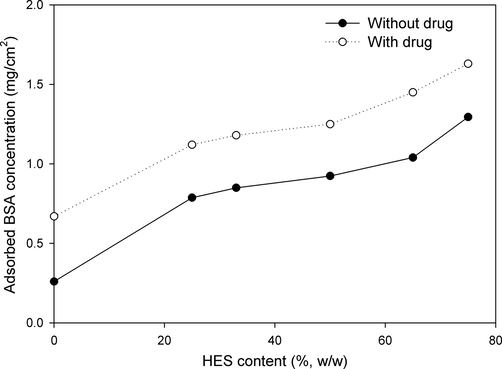
Effect of HES content in PVA hydrogel membranes on protein adsorption.
DSC thermograms showing distinctions in the glass transition temperatures for PVA and PVA–HES blend xerogels are depicted in Table 1. The changes in thermal properties due to incorporation of HES in different portions have been summarized in Table 1. According to a freeze–thawing procedure which has been used to entangle PVA polymer, this procedure shows a significant reduction in Tg from 87 °C for virgin PVA to 45 °C for physically crosslinked PVA and refers to nearly rubbery xerogels in approximately ambient conditions. The significant reduction in Tg value can be attributed to increasing in the free volume throughout amorphous regions which resulted from the physical crosslinking or crystallization process of PVA or decreasing in hydrogel bonding in the same amorphous network. Incorporation of a small amount of HES (30%, w/w), shows a prominent increase in Tg value from 45 to 63 °C. Consequently, addition of HES in a high amount results in increasing in Tg from 45 °C for crosslinked PVA (0% HES) to 72 °C for PVA–HES xerogel (75%, HES) (Table 1). The notable increase in Tg values due to blending HES, can be ascribed to higher Tg values of HES itself, its higher miscibility and its good homogeneity with PVA. Accordingly, the onset temperature (Tonset) values increase progressively with increasing the content of HES. The presented data are consistent with the reported data of Tg for chitosan/PVA hydrogels (Cascone et al., 1999). Cascone et al. (1999) have demonstrated that the Tg of the first thermogram peak for chitosan/PVA increased with increasing chitosan content until certain concentration, then it remained constant. Also, the current results of Tg, are consistent with results of Fathi et al. (2011) who revealed that the addition of dextran to PVA increased significantly the Tg values of xerogels in the first decomposition thermogram peak. The melting point (Tm) values were determined from the third relaxation in PVA–HES xerogels, during the melting of the crystallization region upon decomposition at temperature above 140 °C. Although, the determination of the melting temperature of PVA-based materials, is very difficult at temperature above 130 °C, (Marten et al., 1985) but according to the popular estimated method for the equilibrium Tm, has been used (Hassan and Peppas, 2000). As shown in Table 1, relative changes in the melting temperatures have been observed after the addition of HES up to 50%. While, Tm significantly decreased from 220 °C for pure crosslinked PVA to 204 °C for PVA–HES blend xerogel (75%, HES content). The depicted data in Table 1 related to Tm are consistent with those of crystallization degree (Xc). The heat of fusion and the shoulder temperature are increased significantly after freeze–thawing process, and the Xc therefore decreased significantly after the addition of HES content from 74% (for 0%, HES content) to 49% (for 75%, HES content), this is due to the reduction of crystallization or entanglement process and increase of HES content which does not have the ability for crosslinking and perturbing the crystallization process. –: Do not determine.
Sample No.
HES content (%, w/w)
DSC results
TGA results
Tg, °C
Tonset, °C
Tm, °C
Xc,%
Tonset, °C
Td region, °C
PVA
87
–
217
–
–
–
HES
233
–
–
–
–
–
KE0
0
45
200
220
74
261
228–318
KE30
33
63
202
219
72
262
235–338
KE50
50
68
203
218
63
263
238–348
KE75
75
72
205
204
49
250
220–345
The thermal degradation of PVA–HES xerogels was conducted using TGA as drawn in Fig. 7, and the concluded thermogram data have been complied in Table 1. The thermal onset decomposition temperature (Tonset) and the sharp decomposition temperature (Td) in the second decomposition stage have been studied. As HES increased, the residual weight loss after complete volatilization and the onset temperatures slightly increase after the addition of HES up to 50%, then it decreased significantly from 261 (for 0% HES content) to 250 °C (for 75% HES content), (Fig. 7, and Table 1). Thus, addition of HES improved the thermal stability of PVA–HES xerogels, because the hydroxyl groups of HES might form hydrogen bonding with those of PVA. Additionally, the notable reduction of thermal onset temperature at high HES content, can be also tentatively attributed to a high HES content acts as an insulator and mass-transport barrier to the volatile products generated during thermal decomposition and this heat barrier acts in a reverse mode, that is the accumulation heat by the decomposed materials could be released, acting as a new heat source causing an acceleration on the decomposition process at the same time with the outside heat source. The presented data of thermal degradation in Table 1, are consistent with reported data by Hwang et al. (2010) who revealed that the addition of dextran to PVA improved thermal properties of hydrogels. The first degradation step at 26–85 °C, can be ascribed to the removal of traces of water or solvent vapor. The second degradation step between 220 and 500 °C results in the highest residual weight loss and this is due to the decomposition and volatilization of organic components of polymer. The thermal decomposition temperature (Td) range of PVA–HES blend xerogels shifted significantly toward temperature ranges higher than those of pure crosslinked PVA xerogels (0% HES content) until certain HES contents (Table 1). The third decomposition step is after approximately 550 °C, the curves all become flat, and mainly the organic residues are completely volatilized, (Fig. 7).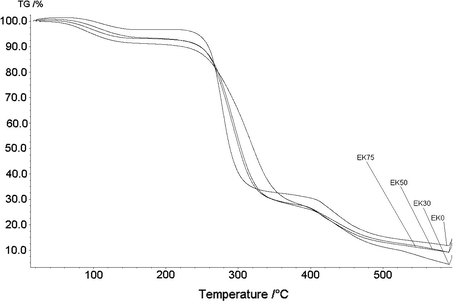
TGA thermograms of PVA based xerogel membranes containing various amounts of HES showing changes in thermal degradation profile as loss weight percent.
Gravimetric determination was used to study the hydrolytic degradation of PVA–HES hydrogel membranes (Gan et al., 1999). Due to the hydrolytic cleavage of hydrogen bonding among –OH groups of PVA chains, apparently weight loss values can be observed in Fig. 8. Also, this weight loss was previously noticed and expected during experiments in Fig. 3, due to the high water solubility of HES as a blend material and high hydrophilic forces between PVA chains. As shown in Fig. 8, it was found that the maximum weight loss of pure freeze–thawed PVA membrane (0% HES) is only 18%, while for PVA–HES membrane (75% HES content) reaches to 60%. These results refer to the weight loss of PVA–HES hydrogels which dramatically increased with increasing HES contents. This phenomenon can be ascribed to the degradation of PVA–HES hydrogel membranes that are predominantly the cleavage of entanglement segments of PVA and is consistent with the fact that the degradation of PVA is quite limited, whereas the degradation of PVA–HES is quite high. In addition, as PVA and HES are nontoxic materials, (Kamoun and Menzel, 2012; Xiao and Zhou, 2003) the PVA–HES hydrogel membranes and their degradation by-products might be expected to be nontoxic too.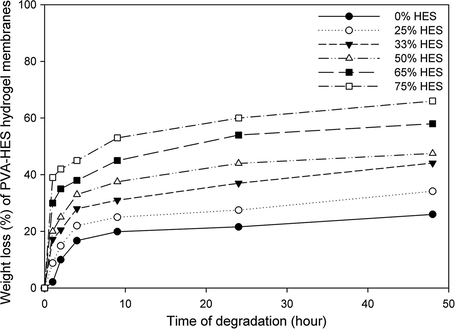
Effect of HES contents on weight loss of the PVA–HES hydrogel membranes after different degrading times in phosphate buffer saline (PBS) (0.1 M, pH 7.4, at 37 °C).
Fig. 9 shows the cumulative percentage of ampicillin release profile from PVA–HES hydrogel membranes with different HES contents in phosphate buffer (pH 7.5, at 37 °C). The initial release rate of ampicillin from PVA–HES hydrogels was rapid, particularly after the first 15 min of release profile, but it increases slightly after several hours. In addition, the cumulative percentage of ampicillin was notably increased with increasing HES contents in PVA–HES hydrogel membranes. This initial burst release may be attributed to the rapid diffusion of ampicillin that was loaded close to the surface of the hydrogel membrane due to the de-swelling of the ampicillin-loaded hydrogel in the buffer solution. Later on, ampicillin was released more slowly from the hydrogel as compared to the initial release to the total ampicillin release after 6 h. The increase of ampicillin release profile as HES content increases can be ascribed to the formation of spongy and porous shape-structure for PVA–HES hydrogels containing high HES contents, as was confirmed in SEM investigations (Fig. 5). Moreover, high HES contents in hydrogels facilitate the diffusion of loaded ampicillin due to high water uptake probability as discussed in Fig. 3. This means the release of ampicillin from PVA–HES hydrogel membranes is considered to be mostly controlled by diffusion during high water uptake and porous structure of carrier hydrogels.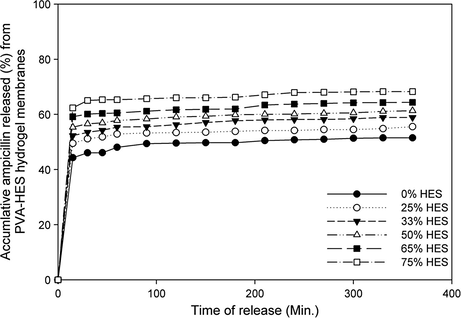
In vitro cumulative release profile of released ampicillin percentage from PVA–HES hydrogel membranes in phosphate buffer solution (at 37 °C in pH 7.5–8.0), as a function of different HES contents in membranes.
4 Conclusion
In summary, the PVA–HES-ampicillin blend hydrogel membranes have been developed using freezing–thawing technique as a physical crosslinking method. The PVA–HES hydrogels have been characterized on many levels, examined for numerous purposes and incredible potential applications in medicine and pharmacy. FTIR results indicate that absorption peak is related to hydrogen bonding between –OH groups of HES and PVA. Addition of HES in the physically crosslinked PVA network significantly influenced its molecular structure, thermal, mechanical, and morphological properties. SEM results showed that morphology structures of PVA hydrogels were strongly dependent on HES contents, where pore size and pore area distribution obviously related to the introduction of high HES contents. Furthermore, physically crosslinked PVA–HES hydrogel gave more swellable, flexible, elastic, and higher protein adsorbent compared to that with only PVA. Additionally, HES incorporation to PVA hydrogel improved the thermal stability. The pure PVA xerogels exhibited lower Tg values in comparison to virgin PVA or blended PVA with HES up to certain content. Moreover, the overall thermal stability was notably improved by introduction of HES as blend materials. However, the crystallization degree displayed a significant reduction with HES incorporation. Both hydrolytic degradation of PVA–HES hydrogel membranes and release profile of loaded-ampicillin, have apparently increased with increasing HES contents in hydrogels. Finally, it was concluded from our results that the physicochemical, morphological, mechanical, thermal properties, degradation, and release profile study showed that the addition of HES–PVA hydrogels is expected to improve utility as hydrogel membrane for biomedical applications, specifically for wound dressing application mildly.
Acknowledgments
Authors gratefully acknowledge Prof. Dr. Henning Menzel, Institute for Technical Chemistry, Braunschweig University of Technology, Braunschweig, Germany; for conducting FTIR, and thermal characterizations by Dr. E.A. Kamoun. Also, E.A. Kamoun acknowledges DAAD Cairo-Office through (WAP-2012) program for financial support and grant.
References
- Spectrophotometric determination of ampicillin, amoxycillin, and carbenicillin using Folin-Ciocalteu phenol reagent. J. Anal. Chem.. 2004;59(2):119-123.
- [Google Scholar]
- Development of new hydroactive dressings based on chitosan membranes: characterizations and in vivo behavior. J. Biomed. Mater. Res.. 2003;64A:147-154.
- [Google Scholar]
- Evaluation of an in situ forming hydrogel wound dressing based on oxidized alginate and gelatin. Biomaterials. 2005;26:6335-6342.
- [Google Scholar]
- Effect of chitosan and dextran on the properties of poly(vinyl alcohol) hydrogels. J. Mater. Sci. Mater. Med.. 1999;10:431-435.
- [Google Scholar]
- Evaluation of permeability and mechanical properties of composite polyvinyl alcohol films. Chem. Pharm. Bull.. 1999;47(10):1412-1416.
- [Google Scholar]
- Study on gelatin-containing artificial skin. Part I. preparation and characteristics of novel gelatin–alginate sponge. Biomaterials. 1999;20:409-417.
- [Google Scholar]
- Hydrogel membranes with mesh size asymmetry based on the gradient crosslinking of poly(vinyl alcohol) J. Membr. Sci.. 1999;156:67-79.
- [Google Scholar]
- Characterization of acetyl starch by means of NMR spectroscopy and SEC/MALLS in comparison with hydroxyethyl starch. Starch – Stärke. 1998;50(10):431-437.
- [Google Scholar]
- Physically crosslinked polyvinyl alcohol–dextran blend xerogels: morphology and thermal behavior. Carbohydr. Polym.. 2011;84:145-152.
- [Google Scholar]
- Enzymatic degradation of poly(ε-caprolactone)/poly(dl-lactide) blends in phosphate buffer solution. Polymer. 1999;40(10):2859-2862.
- [Google Scholar]
- Structure and applications of poly (vinyl alcohol) hydrogels produced by conventional crosslinking or by freezing/thawing methods. Adv. Polym. Sci.. 2000;153:37-65.
- [Google Scholar]
- Novel crosslinking methods to design hydrogels. Adv. Drug Del. Rev.. 2002;54:13-36.
- [Google Scholar]
- Gentamicin-loaded wound dressing with polyvinyl alcohol/dextran hydrogel: gel characterization and in vivo healing evaluation. AAPS Pharm. Sci. Technol.. 2010;11(3):1092-1103.
- [Google Scholar]
- Biliary excretion of hydroxyethyl starch in man. Biomed. Mass Spectrom.. 1984;11(4):164-166.
- [Google Scholar]
- HES-HEMA nanocomposite polymer hydrogel: swelling behavior and characterization. J. Polym. Res.. 2012;19:9851-9865.
- [Google Scholar]
- Swelling behavior of interpenetrating polymer network hydrogels composed of poly(vinyl alcohol) and chitosan. React. Funct. Polym.. 2003;55:53-59.
- [Google Scholar]
- Synthesis and characteristics of interpenetrating polymer network hydrogels composed of poly(vinyl alcohol) and poly(nisopropylacrylamide) React. Funct. Polym.. 2003;55:61-67.
- [Google Scholar]
- Development of polyvinyl alcohol–sodium alginate gel-matrix-based wound dressing system containing nitrofurazone. Inter. J. Pharm. 2008;359:79-86.
- [Google Scholar]
- Effect of the morphology of hydrophilic polymeric matrices on the diffusion and release of water soluble drugs. J. Membr. Sci.. 1981;9:211-227.
- [Google Scholar]
- Blood compatibility of novel PGA (poly glutamic acid)/poly vinyl alcohol hydrogels. Colloids Surf. B Biointer.. 2006;47:43-49.
- [Google Scholar]
- Characterization of poly(vinyl alcohol)/poly(ethylene glycol) hydrogels and PVA-derived hybrids by small-angle X-ray scattering and FTIR spectroscopy. Polymer. 2004;45(21):7193-7202.
- [Google Scholar]
- Encyclopedia of Polymer Science and Engineering. New York: John Wiley and Sons; 1985. 17, 167
- Synthesis of PVA/PVP hydrogels having two-layer by radiation and their physical properties. Radiat. Phys. Chem.. 2003;67:361-365.
- [Google Scholar]
- Turbidimetric studies of aqueous poly(vinyl alcohol) solutions. Macro. Chem.. 1975;176:3433-3440.
- [Google Scholar]
- Development of semicrystalline Poly (vinyl alcohol) hydrogels for biomedical applications. J. Biomed. Mater. Res.. 1977;11:423-434.
- [Google Scholar]
- Crosslinked poly(vinyl alcohol) hydrogels as swollen elastic networks. J. Appl. Polym. Sci.. 1977;21:1763-1770.
- [Google Scholar]
- Reinforced uncrosslinked poly(vinyl alcohol) gels produced by cyclic freezing–thawing processes: a short review. J. Control. Rel.. 1991;16:305-310.
- [Google Scholar]
- Adsorption and release studies of sodium ampicillin from hydroxyapatite and glass-reinforced hydroxyapatite composites. Biomaterials. 2001;22(11):1393-1400.
- [Google Scholar]
- Irradiation of polyvinyl alcohol and polyvinyl pyrrolidone mixed hydrogel for wound dressing. Radiat. Phys. Chem.. 2001;62:107-113.
- [Google Scholar]
- Anomalous swelling of poly(vinyl alcohol) film in mixed solvents of dimethylsulfoxide and water. Polymer. 1995;36(15):2941-2946.
- [Google Scholar]
- Morphology and mechanical properties of pullulan/poly(vinyl alcohol) blends crosslinked with glyoxal. J. Appl. Polym. Sci.. 2001;82:2273-2280.
- [Google Scholar]
- Self-gelling hydrogels based on oppositely charged dextran microsphere. Biomaterials. 2005;26:2129-2135.
- [Google Scholar]
- Synthesis and properties of degradable poly(vinyl alcohol) hydrogel. Polym. Degrad. Stab.. 2003;81:297-301.
- [Google Scholar]
- Investigation of PVA/ws-chitosan hydrogels prepared by combined gama-irradiation and freeze–thawing. Carbohydr. Polym.. 2008;73:401-408.
- [Google Scholar]
- Morphology and structure of highly elastic poly vinyl alcohol) hydrogel prepared by repeated freezing-and melting. Colloid Polym. Sci.. 1986;264:595-601.
- [Google Scholar]
- Synthesis of antibacterial PVA/CM-chitosan blend hydrogels with electron beam irradiation. Carbohydr. Polym.. 2003;53:439-446.
- [Google Scholar]




