

Translate this page into:
Potential reaction initiation points of polycyclic aromatic hydrocarbons
⁎Corresponding author at: Institute of Chemistry, University of Miskolc, Miskolc-Egyetemváros, H-3515 Miskolc, Hungary. kemfiser@uni-miskolc.hu (Béla Fiser)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Reaction initiation points of the 16 priority polycyclic aromatic hydrocarbons (PAHs) have been determined by calculating all the different C-H bond dissociation enthalpy (BDE) values. Six density functional theory methods (B3LYP, B3LYP-D3, B97D3, M06-LD3, M06-2X-D3, and ωB97X-D) in combination with 4 basis sets (6-31G(d), 6-31+G(d,p), 6-311++G(d,p), def2-TZVP) have been applied and the most feasible combination has been selected. The BDE values and the corresponding bond lengths have been used to determine potential attack points on the structures. The studied molecules have been categorized structurally as well, within which the position of the hydrogen atoms is considered. Results show that most of the hydrogens are in zig-zag positions, and the BDE and bond length values for the 16 priority PAHs are in a range between 342.0 and 485.6 kJ/mol and 1.0817–1.952 Å, respectively. Most of the initiation points are represented by armchair and peak hydrogens. The lowest and highest BDE and shortest and longest bond length values belong to fluorene and acenaphthylene where the hydrogens were aliphatic and in peak position, respectively.
Keywords
PAH
Bond dissociation enthalpy
BDE
DFT
C-H bond

1 Introduction
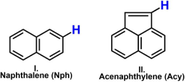
The investigation of polycyclic aromatic hydrocarbons (PAHs) has relevance in numerous research fields due to their harmful properties and their omnipresence in the environment (Abdel-Shafy and Mansour, 2016). PAHs are formed under incomplete combustion processes of organic materials, attributing a human-induced origin to most of them. The most common anthropogenic sources of PAH pollution come from the emissions of coal-fired power plants (Yang et al., 2019; Liu et al., 2019), domestic heating (Wang, 2016; Venturini et al., 2018), coke production (Inceoğlu et al., 2019), waste incineration (Li et al., 2019), tobacco smoke (Dobaradaran, 2019), petroleum spills (Wagner and Barker, 2019), and especially the emission of various means of transportation powered by gasoline, biodiesel, kerosene (Zielinska, 2004). However, natural processes like lightning-caused forest fires or volcanic activities contribute to PAH emissions as well (Siddens, 2012; Moorthy et al., 2015). Based on the enumerated sources of polycyclic aromatic hydrocarbons, their emission can be considered permanent which raises constant concern. The carcinogenicity of PAHs has been widely proven by experiments, therefore their biological relevance is self-explanatory (Siddens, 2012; Moorthy et al., 2015). By intermolecular reactions, strong interaction can be formed between PAH radical cations and the nucleophilic centers of DNA, which can result in cancer cell formations (Phillips and Sims, 2019; Kriek and Westra, 2019). The monitoring of these molecules in the environment started by the creation of a list of 16 priority PAHs (also called parent PAHs, Fig. 1) done by the U.S. Environmental Protection Agency (EPA) (Liu et al., 2019; Wang, 2016).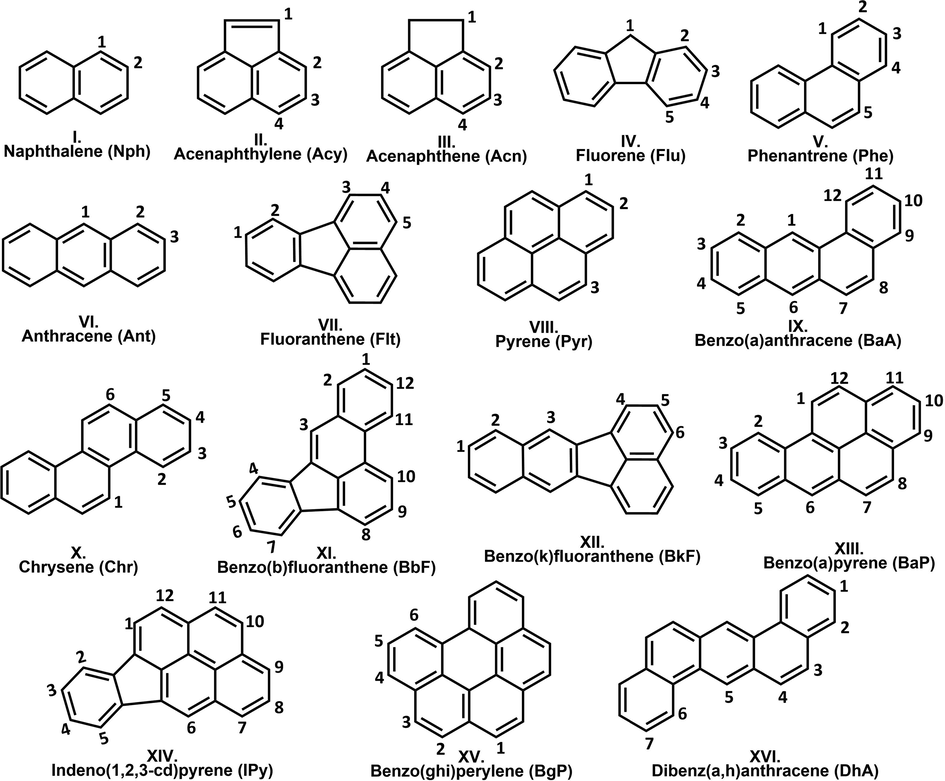
2D structures of the priority PAHs. C-H bonds for which bond dissociation enthalpies (BDEs) have been computed are indicated and marked by numbers.
These molecules have been considered by researchers as being representatives for all the PAHs, but the extension of the list was suggested several times (Venturini et al., 2018; Inceoğlu et al., 2019; Li et al., 2019). In addition, PAHs are considered as the precursors of soot (Keller et al., 2019), which initiated several combustion chemistry studies to build a model to predict their formation (Slavinskaya et al., 2012; Appel et al., 2000; Raj, 2009; Wang et al., 2007; Frenklach and Wang, 1991). Numerous growth mechanisms have been already proposed and a significant amount of data of elementary reactions were produced (Narayanaswamy et al., 2014; Zhu et al., 2017; Wang et al., 2013; Lin et al., 2016; Liu et al., 2016; Mao et al., 2017; Yuan et al., 2019; Liu et al., 2018; Goldaniga et al., 2000; Whitesides and Frenklach, 2010). Despite the continuous progress in the description of PAH formation, the current situation is still far from being complete. The detailed description of formation mechanisms can contribute to the in-depth understanding of the synthesis of various nano-scale graphene materials (such as graphene nanoribbons or graphene quantum dots) (Wang et al., 2019) and the atomic description of carbon vapor deposition (CVD) and carbon vapor infiltration (CVI) processes as well (May et al., 2000). PAHs also have astrochemical significance as they are among the largest and most stable ubiquitous molecules detected in the interstellar medium (ISM) (Zhang et al., 2019). Moreover, PAHs are around 10–20% of all carbon materials in ISM (Hoover, 2014; Zhen et al., 2018). Several PAH growth mechanisms start with the abstraction of a hydrogen atom from the PAH skeleton and thus, a polycyclic aromatic hydrocarbon radical is forming. Enough to mention the HACA (hydrogen abstraction acetylene addition) (Frenklach et al., 2019), MAC (methyl addition cyclization) (Reizer et al., 2019), and PAC (phenyl addition cyclization) (Shukla et al., 2008) mechanisms, within which a hydrogen abstraction is the first step, to achieve a reactive PAH radical structure, and in some cases, this step occurs more than ones during the reaction. This can be achieved by hydrogen abstraction or addition of a radical, which in the case of PAHs mostly occurs via H•, •OH and •CH3. In different environments (i.e. atmosphere, combustion) the concentrations of the radicals are also different. In the atmosphere the radical sites on PAHs are generated often with •OH (i.e nitro-PAHs), then the process continues further with the involvement of O2 (Arey et al., 1986; Lee et al., 2015). Furthermore, •OH, O3, NO3 radicals play an important role also in the atmospheric degradation of toxic PAHs (Ding et al., 2021). In addition, the reaction of PAHs with •OH radical is traditionally considered as crucial evidence in determining their atmospheric lifetimes (Ding et al., 2019). Taking into account the previously mentioned facts, it can be stated that the strengths of the carbon-hydrogen bonds (C-H bonds) are crucial parameters and could define the starting points of reactions. Nevertheless, the occurrence of a given reaction mechanism is strongly influenced by the corresponding barrier height of the rate-determining step. Through the determination of the bond dissociation enthalpy (BDE) values, the strength of bonds can be assessed. Furthermore, BDE calculations represent a key tool for the investigation of antioxidant related mechanisms, and for the study of chemical degradations with environmental relevance (Santos et al., 2020; Filho and De Souza, 2020). It is interesting to point out that the correlation between BDE and bond length has been studied before. For instance, it is found that in the case of different para- and meta- substituted phenol structures the BDE showed a better correlation to the C-O than to the O-H bond length (Klein and Lukeš, 2006). The importance of radical addition reactions is well known in PAH growth, but in case of several major above-mechanisms such as MAC, HACA, and HAERA, the first step is a hydrogen abstraction (Reizer et al., 2019; Reizer et al., 2021), and thus, to determine potential reaction initiation points the strength of C-H bonds has to be known. The purpose of this study is to calculate and compare the bond strength of the carbon-hydrogen bonds for the priority or parent PAHs (Fig. 1). By using these, potential reaction initiation points will be determined, and geometrical and energetical features can be associated with the prevalence of formation mechanisms. Furthermore, a method test will also be carried out, to select an appropriate density functional theory method and basis set combination for future studies.
2 Material and methods
Bond dissociation enthalpy (BDE) is an appropriate thermodynamic property to measure bond strength. It determines the enthalpy change associated with bond breakings and is equivalent to the difference between the sum of the enthalpy of the formed radicals (in this case a PAH radical (
) and atomic hydrogen (
)) and the initial PAH structure (
) (Eq. (1)).
The BDE of a few polycyclic aromatic hydrocarbons has already been determined (Luo, 2007), but for many PAHs, this data is missing (Fujiwara et al., 1996; Meot-Ner, 1980; Bauschlicher and Langhoff, 1999). In this work, the calculations have been carried out by using the Gaussian 09 program package (Frisch, 2004). Calculations are performed with six selected density functional theory (DFT) methods (B3LYP, B3LYP-D3, B97D3, M06-LD3, M06-2X-D3, and ωB97X-D) combined with four basis sets (6-31G(d), 6-31+G(d,p), 6-311++G(d,p), def2-TZVP) in gas phase at 298.15 K and 1 atm. Dispersion corrections have been applied as they were implemented in the Gaussian 09 program package for the corresponding methods. To calculate the BDE for each studied carbon-hydrogen bonds (BDEC-H) of the 16 priority PAH (Fig. 1), 104 radicals, 16 molecules, and 1 hydrogen atom have been calculated with each method, and thus, in total 2904 calculations have been performed [(104 radical + 16 reactant + 1 hydrogen)•24] (Table S1, Table S4). In the selection of the studied C-H bonds, the symmetry of the structures is considered and thus, positions that lead to identical radicals are excluded from the study (Fig. 1). Method test has also been performed within which the applied DFT methods combined with the above-mentioned basis sets have been compared to experimental values to select the best DFT/basis set combination for future studies.
3 Results and discussion
3.1 Validation
BDE values of Nph-2 (second hydrogen of naphthalene, Fig. 1) and Acy-1 (first hydrogen of acenaphthylene, Fig. 1) were calculated with every combination of methods and basis sets and compared to experimental data (Table 1). It can be seen that in the case of naphthalene (Nph-1) each ΔBDEs are below 4 kJ/mol (within chemical accuracy). However, regarding acenaphthylene (Acy-1) the difference between the calculated and experimental BDE values is > 4 kJ/mol except in the case of the ωB97X-D/6-311++G(d,p) level of theory (Luo, 2007). Taking into consideration the experimentally measured (BDEEXP) and the calculated (BDECALC) values, it can be concluded that the best agreement is obtained with the ωB97X-D/6-311++G(d,p) level of theory, as the deviations (BDEEXP-BDECALC) were only 0.1 kJ/mol and 3.9 kJ/mol in the case of Naph-2 and Acy-1, respectively (Luo, 2007).
ΔBDE = BDEEXP-BDECALC/kJ/mol
Basis set
6-31G(d)
6-31+G(d,p)
6-311++G(d,p)
def2-TZVP
Method
Nph-1
Acy-1
Nph-1
Acy-1
Nph-1
Acy-1
Nph-1
Acy-1
B3LYP
4.7
8.2
1.7
5.1
4.4
7.6
5.9
9.2
B3LYPD3
2.5
5.8
1.6
4.4
1.6
4.5
3.8
6.7
B97D3
5.7
8.6
2.5
5.01
1.8
4.4
8.1
10.4
M06L-D3
22.8
27.4
18.0
22.4
17.6
22.1
21.1
25.4
M06-2X-D3
0.2
6.6
1.5
4.4
2.6
8.4
2.8
7.9
ωB97X-D
0.3
4.3
20.1
18.8
0.1
3.9
2.4
6.3
In addition, the extensive work of Mardirossian and Head-Gordon made on the analysis of 200 density functionals showed that ωB97X-D is the best hybrid GGA functional (Mardirossian and Head-Gordon, 2017). The results of ωB97X-D/6-311++G(d,p) are compared with the other DFT/basis set combinations (Figure S1; Figure S2). If the data of ωB97X-D/6-311++G(d,p) is considered as a reference (red line on Figure S2), other DFT values usually underestimate these. The deviation is the highest (<20.3 kJ/mol on average) between the M06-LD3 and ωB97X-D/6-311++G(d,p) values. Not surprisingly the ωB97X-D/6-31+G(d) data is very close to the reference. The difference between them is just 3.8 kJ/mol on average, which value is well below the chemical accuracy (4 kJ/mol), and can be attributed to the difference in the diffuse and polarization functions (+, ++; (d), (d,p)) involved in the basis sets (Figure S1). In addition, among the values obtained with different methods, the M06-2X-D3 values are the closest to the ωB97X-D/6-311++G(d,p) values as, the deviation is just 2.5 kJ/mol (Figure S2, b). Thus, the ωB97X-D/6-31+G(d) and ωB97X-D/6-311++G(d,p) level of theories are both suitable for similar studies, but as the latter is slightly better it was selected and applied in the analysis.
Furthermore, experimentally determined bond length values of chrysene and benzo(a)pyrene were compared to the calculated ones obtained by the selected ωB97X-D/6-311++G(d,p) level of theory and the computed and measured values were also in good agreement with each other (Table S2–S3; Figure S4).
3.2 Structural and energetics considerations based on the results of the ωB97X-D/6-311++G(d,p) level of theory
The studied hydrogen atoms have been categorized based on their position or chemical properties (Fig. 3).
Seven categories are identified, and all of the hydrogens are assigned to one of the following groups taking into account the symmetry of the molecular structures as well: zig-zag (noted with an orange triangle), peak (marked with a green square), armchair (red circle), penta-bay (blue star) anthracene type (purple square), aliphatic (blue rhombus) and next-to-aliphatic (yellow triangle) (Fig. 2). Hydrogens in zig-zag positions are most easily represented on naphthalene, as the hydrogens attached to a connected section of 4 carbon atoms (for example C7–C8–C9–C1 zig-zag chain, Fig. 2, I. Naphthalene (Nph)) between the C2 - C3 and C7 – C6 midpoints constituent symmetry plane. The next category is the peak, where the hydrogen is attached to a carbon and the neighboring C atoms also hold H atoms (Figure 2, C3, C4, C10, BaP). The hydrogen is in an armchair position it belongs to a carbon around a bay region (between C1 and C10 carbons of phenanthrene, a prototype of a “bay region”, Fig. 2). Numerous PAHs have this structural feature (DhA, Chr, BaA, BaP), with additional fused aromatic rings. This type of arrangement has significance in cancer research since the bay region is preserved within the most carcinogenic PAHs (benzo(a)anthracene, benzo(a)pyrene) and is considered as a critical structural feature for the formation of carcinogenic and mutagenic cells (Lehr, 1985). Hydrogens in the penta-bay category are in similar positions to the hydrogens in the armchair group, but here the two benzene rings at the edge of the structural unit are fused with a five-membered ring in the middle (fluorene, Fig. 2). The anthracene-type hydrogens are found on the middle ring of three linearly connected benzene rings (BaP, Fig. 2). The last two categories are related to the aliphatic structural moieties of the PAHs. Aliphatic hydrogens are on the aliphatic unit of the molecules (Acn-1, Flu-1, Fig. 2), while hydrogens which are on an aromatic six-membered ring connected to the aliphatic part of the species (Acn-2, Flu-2) belong to the next-to-aliphatic group. The computed BDE and the corresponding bond length values have been plotted and analyzed (Fig. 2). By comparing all the obtained values, it can be seen that the minimum and maximum values belong to two molecules. Flu-1 has both the lowest BDE value and the longest bond length value at the same time with 342.0 kJ/mol and 1.0952 Å respectively. However, Acy-1 (the first hydrogen on acenaphthylene, Fig. 1) has the shortest bond length value with 1.0817 Å and the highest BDE value with 485.6 kJ/mol. Plotting the BDEs as a function of the corresponding bond lengths, three subgroups were observed (marked with roman numbers, Fig. 3, a). The results of Acy-1 (the first hydrogen of acenaphthylene) define a special case due to the highest BDE value (485.6 kJ/mol) and to the relatively short bond length value (1.0817 Å). Consequently, Acy-1 stays separately from the first and third groups and basically would represent alone the first subgroup of the BDE vs bond length diagram (Fig. 3, a, I.). This can be explained by its special position since it is bound to a carbon atom involved in a five-membered ring which is fused with two six-member rings. Two data points, Flu-1 and Acn-1 represent the third major group (Fig. 3, a, III.), with 342.0 kJ/mol and 348.7 kJ/mol BDE values, respectively. These two points have a BDE value well below the average (466.6 kJ/mol) which is due to the fact that the C-H bond is in α-position to the carbon atom involved in the aromatic system containing alternating single and double bonds (Lengyel et al., 2012; Medina et al., 2015). These C-H bonds could be also the reaction centers of possible oxidative attacks (Lengyel et al., 2012; Medina et al., 2015). Furthermore, if all the data are being compared it can be seen that the longest C-H bond values belong to these with 1.0931 Å and 1.0952 Å. Most of the data points (101 from 104) can be found in the second group (Fig. 3, a, I.) with a bond length and BDE value range between 1.0817 Å (Chr-1) – 1.0863 Å (Ant-1) and 461.2 kJ/mol (Chr-1) – 472.9 kJ/mol (BaP-6), respectively. At first sight, one can suspect a linear correlation within the II. group between the BDE and bond length values. In order to verify this, the data of the II. group were selected and analyzed by using linear regression (Figure S3) and a reasonable correlation was found (R2 > 0.75). By applying the equation of the linear fit, with some uncertainty the bond dissociation enthalpy can be predicted for a C-H bond if the corresponding bond length is known and vice versa. It can be seen that the BDE values increase together with the bond length values, so a proportional trend exist between them. It is interesting to mention that in the work of Klein and Lukes the BDE values were inversely proportional to C–O bonds (Klein and Lukeš, 2006). The positions of the hydrogens are also noted according to the categorization mentioned earlier (Fig. 3). Thirty-eight hydrogens are in zig-zag position (within II.) so this type of arrangement is the most prevalent. In this group, the average BDE value is 469.9 kJ/mol, in a range between 468.0 kJ/mol (Acy-2) to 471.5 kJ/mol (Ipy-9). Regarding the bond lengths, the average is 1.0846 Å with a minimum of 1.0850 Å (Phe-3) and a maximum of 1.0855 Å (BkF-2). 33 hydrogens can be identified as peak (within II.), with a 468.4 kJ/mol BDE and 1.0843 Å bond length, on average.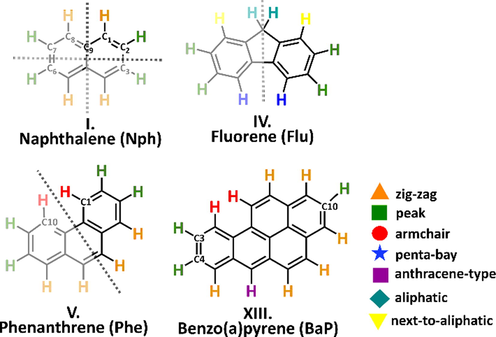
Categorization of the H atoms of the studied PAHs introduced by using specific examples.
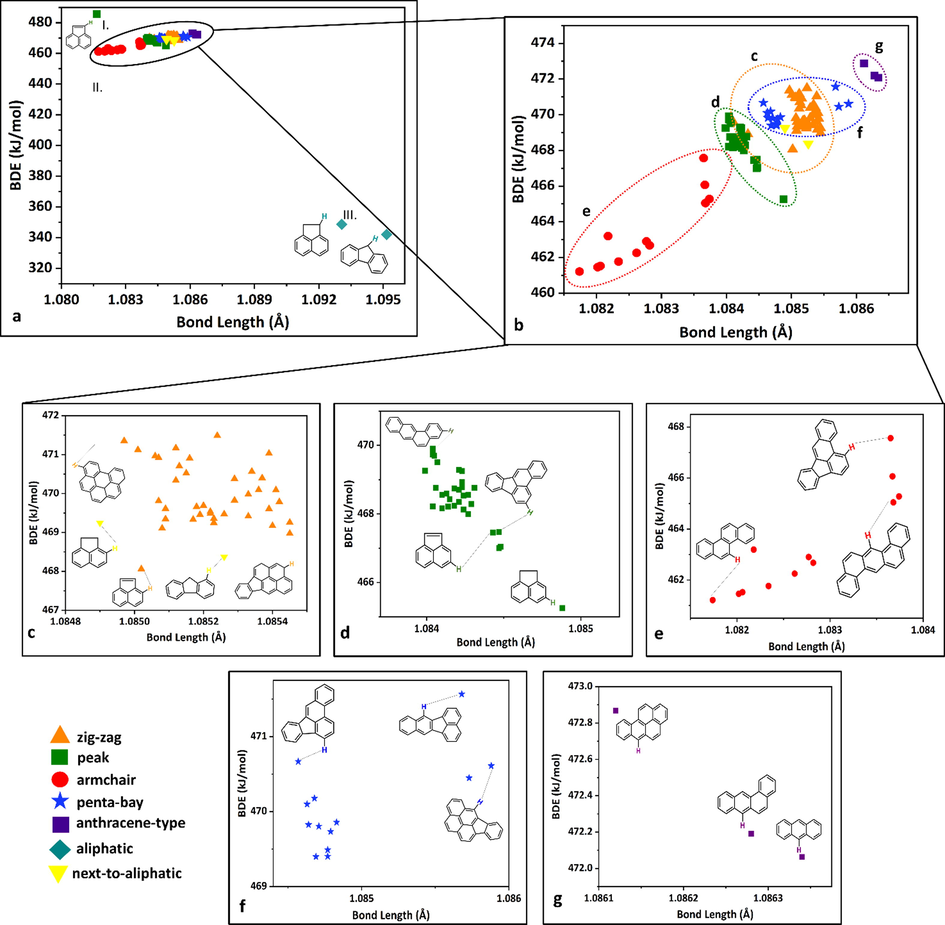
Bond dissociation enthalpy (BDE, in kJ/mol) values as a function of the corresponding C-H bond lengths calculated at the ωB97X-D/6-311++G(d,p) level of theory and the categories of the H atoms.
The maximum BDE value appeared on the tenth hydrogen of benzo(a)anthracene (BaA-10) with 469.9 kJ/mol and the minimum is obtained on Acn-3 with 465.3 kJ/mol. The longest C-H bond has been located on the same molecule (Acn-3) within the peak category with 1.0849 Å. However, the minimum bond length is obtained on the first hydrogen of benzo(b)fluoranthene (BbF-1) with 1.0840 Å. 12 hydrogens within the II. group are in armchair arrangement (Fig. 3). Interestingly both the minimum BDE and bond length values belong to Chr-1 with 461.2 kJ/mol and 1.082 Å, respectively. The maximum BDE value in armchair position is attributed to BbF-10 (benzo(b)fluoranthene) with 467.6 kJ/mol. The longest bond length belongs to BaA-1 with 1.0837 Å. There are 13 hydrogens in the penta-bay group. Among them, indeno(1,2-cd)pyrene represents the minimum BDE and the longest bond length values with 469.4 kJ/mol (Ipy-1) and 1.0859 Å (Ipy-6), respectively. The highest BDE is attributed to the third hydrogen of benzo(k)fluoranthene (Bkf-3), and the shortest bond length to benzo(b)fluoranthene. The fifth category in the second group is anthracene-type which includes only three hydrogens (Ant-1, BaP-6, and BaA-6). Based on the results it can be observed that the BDE and bond length values are changing inversely proportional with the growth of benzene rings. Ant-1 being the simplest prototype in this category has the longest bond length value (1.0863 Å) with the lowest BDE value (472.1 kJ/mol). The sixth hydrogen on benzo(a)pyrene (BaP-6) consists of two additional benzene rings than anthracene, has the highest BDE value (472.9 kJ/mol), and the shortest bond length value (1.0861 Å) in this category. Being in concordance with previous observations the results of BaA-6 are between the results of Ant-1 and BaP-6, obtaining a 472.2 kJ/mol and a 1.0863 Å of BDE and bond length values, respectively. Among the 13 hydrogens located in the penta-bay category the first and third hydrogens on indeno(1,2-cd)pyrene represent the minimum bond length and maximum BDE value with 1.086 Å and 469.4 kJ/mol, respectively. The maximum BDE is found at BkF-3 with 471.6 kJ/mol, and the minimum bond length corresponds to BbF-8 with 1.0846 Å. The two hydrogens of Acn-2 and Flu-2 represent the last group that bears the next-to-aliphatic name. The BDE values here are 469.3 kJ/mol (Acn-2) and 468.4 kJ/mol (Flu-2), while the bond length values are ∼1.08 Å. Based on the obtained BDEs, potential reaction initiation points can be determined on each molecule (Fig. 4). For 7 molecules (Phe, BaA, Chr, BaP, BbF, DhA, BgP) from the 16 parent PAHs, the most reactive hydrogens are in an armchair position with a BDE range of 461.2–466.1 kJ/mol, and the most reactive molecule in this respect is chrysene (Chr) due to its slightly lower BDE value (461.2 kJ/mol) than the others. The hydrogens in peak positions represent the second most reactive subgroup (Fig. 4). Among them, the most reactive molecule is acenaphthylene (Acy) which has a 468.7 kJ/mol BDE value. Thus, under a given mechanism the further growth of PAHs probably will start with the formation of the PAH radicals by the removal of hydrogens from these positions.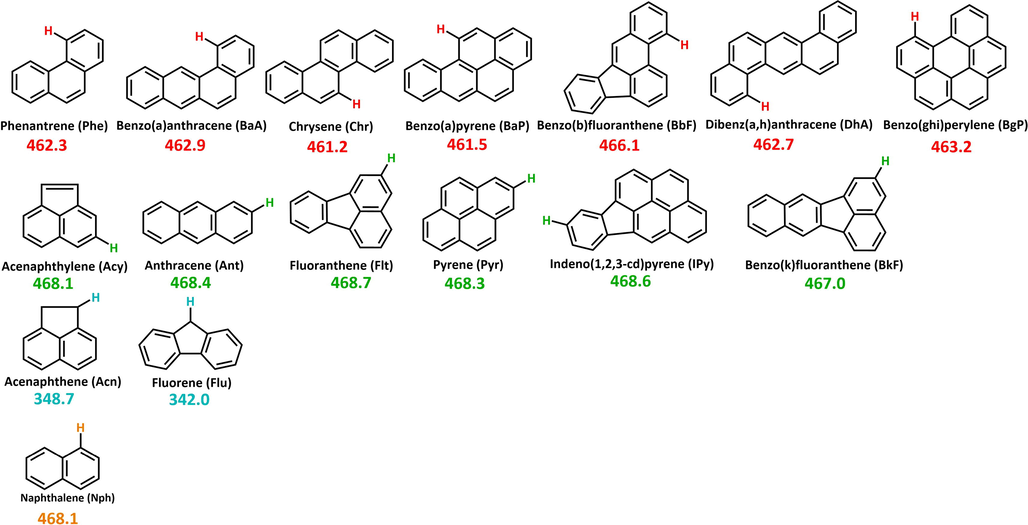
Reaction initiation points of the 16 priority PAHs along with the corresponding bond dissociation enthalpy (BDE, in kJ/mol) values calculated at the ωB97X-D/6-311++G(d,p) level of theory. Hydrogens are colored according to their respective positions and categories: armchair – red, peak – green, aliphatic – blue, and zig-zag – orange. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
4 Conclusions
Bond dissociation enthalpies (BDEs) of the 16 priority PAHs were calculated for each unique C-H bond. The calculations have been carried out by using six density functional theory methods (B3LYP, B3LYP-D3, B97D3, M06-LD3, M06-2X-D3, and ωB97X-D) in combination with 4 basis sets (6-31G(d), 6-31+G(d,p), 6-311++G(d,p), def2-TZVP). 104 radicals, 16 molecules, and 1 hydrogen atom have been studied with each method, and thus, in total 2904 calculations were performed. The results compared with experimental values and the best method/basis set combination, the ωB97X-D/6-311++G(d,p) level of theory, has been selected. The correlation between the computed BDE values and the corresponding computed C-H bond lengths were also examined. By plotting the BDEs against the corresponding bond lengths, three clusters can be identified. The first and third groups contained only three structures, acenaphthylene, Flu-1 and Acn-1. Acenaphthylene has a relatively high BDE 485.6 kJ/mol and short bond length value (1.0817 Å), while Flu-1 and Acn-1 are outliers because of their small BDEs, 342.0 kJ/mol and 348.7 kJ/mol, respectively. The largest cluster includes 101 points, where the bond lengths are in a range between 1.0817 Å and 1.0863 Å, while the corresponding BDE values are between 461.2 kJ/mol and 472.9 kJ/mol. Linear correlation was found between the BDE and bond length values in this cluster (R2 > 0.75) and it can be seen that the BDE values increase together with the bond length values, so a proportional trend exist between them. By using the equation of the linear fit, the BDE of a given C-H bond can be predicted with some uncertainty if the corresponding bond length is known and vice versa. Furthermore, all the studied hydrogens have been categorized by using structural and chemical properties. Seven categories have been defined, zig-zag, peak, armchair, penta-bay, anthracene type, aliphatic, and next-to-aliphatic. Most of the H atoms are in zig-zag positions. The BDE and bond length values for the C-H bonds within the 16 priority PAH were in a range between 342.0 and 485.6 kJ/mol and 1.0817–1.0952 Å, respectively. The minimum and maximum BDE and bond length values belong to fluorene and acenaphthylene, where the corresponding hydrogens are in the aliphatic and peak subgroup. The BDE values have been used to determine potential reaction initiation points on each molecule. Most of these initiation points are hydrogens in an armchair (7 of them) or peak position (6 of them). Further growth of the structures would start with the removal of hydrogens from these points. These results can be used to assess the feasibility of potential growth mechanisms.
Acknowledgments
This research is supported by the European Union and the Hungarian State, co-financed by the European Regional Development Fund in the framework of the GINOP-2.3.4-15-2016-00004 project, which aimed to promote the cooperation between higher education and the industry. Further support has been provided by the National Research, development and Innovation Fund (Hungary) within the TKP2021-NVA-14 project. The GITDA (Governmental Information-Technology Development Agency, Hungary) is gratefully acknowledged for allocating computing resources used in this work.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egypt. J. Pet.. 2016;25(1):107-123.
- [CrossRef] [Google Scholar]
- Kinetic modeling of soot formation with detailed chemistry and physics: laminar premixed flames of C2 hydrocarbons. Combust. Flame. 2000;121(1–2):122-136.
- [CrossRef] [Google Scholar]
- The formation of nitro-PAH from the gas-phase reactions of fluoranthene and pyrene with the OH radical in the presence of NOx. Atmos. Environ.. 1986;20(12):2339-2345.
- [CrossRef] [Google Scholar]
- Bond dissociation energies for substituted polycyclic aromatic hydrocarbons and their cations. Mol. Phys.. 1999;96(4):471-476.
- [CrossRef] [Google Scholar]
- Theoretical investigation on atmospheric oxidation of fluorene initiated by OH radical. Sci. Total Environ.. 2019;669:920-929.
- [Google Scholar]
- Atmospheric degradation of chrysene initiated by OH radical: A quantum chemical investigation. Chemosphere. 2021;263:128267
- [CrossRef] [Google Scholar]
- Cigarette butts: An overlooked source of PAHs in the environment? Environ. Pollut.. 2019;249:932-939.
- [CrossRef] [Google Scholar]
- Examining the degradation of environmentally-daunting per- and poly-fluoroalkyl substances from a fundamental chemical perspective. Phys. Chem. Chem. Phys.. 2020;22(31):17659-17667.
- [CrossRef] [Google Scholar]
- Detailed modeling of soot particle nucleation and growth. Symp. Combust.. 1991;23(1):1559-1566.
- [CrossRef] [Google Scholar]
- On the low-temperature limit of HACA. Proc. Combust. Inst.. 2019;37(1):969-976.
- [CrossRef] [Google Scholar]
- Gaussian 03, Revision C.02. Wallingford CT: Gaussian Inc.; 2004.
- CH bond dissociation energies of polycyclic aromatic hydrocarbon molecular cations: Theoretical interpretation of the (M-1)+ peak in the mass spectra. J. Mass Spectrom.. 1996;31(11):1216-1220.
- [CrossRef] [Google Scholar]
- The kinetic modeling of soot precursors in a butadiene flame. Combust. Flame. 2000;122(3):350-358.
- [CrossRef] [Google Scholar]
- Hoover, R. 2014. Need to Track Organic Nano-Particles Across the Universe? NASA’s Got an App for That, 2014, Accessed: Mar. 22, 2019. [Online]. https://www.nasa.gov/ames/need-to-track-organic-nano-particles-across-the-universe-nasas-got-an-app-for-that/#.VlgyCSs2W09
- “VOC and PAH characterization of petroleum coke at maximum thermal decomposition temperature. Energy Sources Part A Recover. Util. Environ. Eff.. 2019;41(11):1305-1314.
- [CrossRef] [Google Scholar]
- Reactive intermediates via Fourier transform mass spectrometry. J. Phys. Org. Chem.. 2002;15(8):461-468.
- [CrossRef] [Google Scholar]
- A Theoretical multiscale approach to study the initial steps involved in the chemical reactivity of soot precursors. Energy Fuels. 2019;33(10):10255-10266.
- [CrossRef] [Google Scholar]
- DFT/B3LYP study of O-H bond dissociation enthalpies of para and meta substituted phenols: Correlation with the phenolic C-O bond length. J. Mol. Struct. Theochem. 2006;767(1–3):43-50.
- [CrossRef] [Google Scholar]
- Metabolic activation of aromatic amines and amides and interactions with nucleic acids. In: Chemical Carcinogens and DNA. CRC Press; 2019. p. :1-28.
- [Google Scholar]
- Formation of polyaromatic hydrocarbon (PAH)-quinones during the gas phase reactions of PAHs with the OH radical in the atmosphere. Environ. Chem.. 2015;12(3):307-315.
- [CrossRef] [Google Scholar]
- Bay region theory of polycyclic aromatic hydrocarbon carcinogenesis. ACS Symp. Ser. 1985:63-84.
- [CrossRef] [Google Scholar]
- Oxidation of sterols: Energetics of C-H and O-H bond cleavage. Food Chem.. 2012;133(4):1435-1440.
- [CrossRef] [Google Scholar]
- Emission characteristics of parent and halogenated PAHs in simulated municipal solid waste incineration. Sci. Total Environ.. 2019;665:11-17.
- [CrossRef] [Google Scholar]
- Formation of the first aromatic ring through the self-recombination of but-1-ene-3-yne with H-assistance in combustion. Int. J. Hydrogen Energy. 2016;41(31):13736-13746.
- [CrossRef] [Google Scholar]
- The growth of PAHs and soot in the post-flame region. Proc. Combust. Inst.. 2018;37(1):977-984.
- [CrossRef] [Google Scholar]
- Development of a reduced toluene reference fuel (TRF)-2,5-dimethylfuran-polycyclic aromatic hydrocarbon (PAH) mechanism for engine applications. Combust. Flame. 2016;165:453-465.
- [CrossRef] [Google Scholar]
- Distribution of organic compounds in coal-fired power plant emissions. Energy Fuels. 2019;33(6):5430-5437.
- [CrossRef] [Google Scholar]
- Comprehensive Handbook of Chemical Bond Energies, ISBN-13:978-0-8493-7366-4. CRC Press Taylor & Francis Group; 2007.
- Formation of incipient soot particles from polycyclic aromatic hydrocarbons: A ReaxFF molecular dynamics study. Carbon N. Y.. 2017;121:380-388.
- [CrossRef] [Google Scholar]
- Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys.. 2017;115(19):2315-2372.
- [CrossRef] [Google Scholar]
- Structures and C–H bond energies of hydrogenated polycyclic aromatic hydrocarbons. Phys. Chem. Chem. Phys. 2000;2(22):5089-5092.
- [CrossRef] [Google Scholar]
- Theoretical study on the oxidative damage to cholesterol induced by peroxyl radicals. J. Phys. Org. Chem.. 2015;28(7):504-508.
- [CrossRef] [Google Scholar]
- Dimer cations of polycyclic aromatics. Experimental bonding energies and resonance stabilization. J. Phys. Chem.. 1980;84(21):2724-2728.
- [CrossRef] [Google Scholar]
- Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol. Sci.. 2015;145(1):5-15.
- [CrossRef] [Google Scholar]
- A chemical mechanism for low to high temperature oxidation of n-dodecane as a component of transportation fuel surrogates. Combust. Flame. 2014;161(4):866-884.
- [CrossRef] [Google Scholar]
- Polycyclic aromatic hydrocarbon metabolites: their reactions with nucleic acids. In: Chemical Carcinogens and DNA. CRC Press; 2019. p. :29-58.
- [Google Scholar]
- A statistical approach to develop a detailed soot growth model using PAH characteristics. Combust. Flame. 2009;156(4):896-913.
- [CrossRef] [Google Scholar]
- Reed, D.R., Kass, S.R. 2000. Experimental determination of the a and b C-H bond dissociation energies in naphthalene.
- Formation Mechanism of Benzo(a)pyrene: One of the Most Carcinogenic Polycyclic Aromatic Hydrocarbons (PAH) Molecules. 2019;24(6):1040.
- [CrossRef] [Google Scholar]
- Formation mechanism of benzo(a)pyrene: One of the most carcinogenic polycyclic aromatic hydrocarbons (PAH) Molecules. 2019;24(6)
- [CrossRef] [Google Scholar]
- Formation and growth mechanisms of polycyclic aromatic hydrocarbons: a mini-review. Chemosphere 2021:132793.
- [CrossRef] [Google Scholar]
- Probing structural properties and antioxidant activity mechanisms for eleocarpanthraquinone. J. Mol. Model.. 2020;26(9):1-8.
- [CrossRef] [Google Scholar]
- Role of phenyl radicals in the growth of polycyclic aromatic hydrocarbons. J. Phys. Chem. A. 2008;111:9532-9543.
- [CrossRef] [Google Scholar]
- Polycyclic aromatic hydrocarbons as skin carcinogens: comparison of benzo[a]pyrene, dibenzo[def, p]chrysene and three environmental mixtures in the FVB/N mouse. Toxicol. Appl. Pharmacol.. 2012;264(3):377-386.
- [CrossRef] [Google Scholar]
- Detailed numerical modeling of PAH formation and growth in non-premixed ethylene and ethane flames. Combust. Flame. 2012;159(3):979-995.
- [CrossRef] [Google Scholar]
- Effect of fuel quality classes on the emissions of a residential wood pellet stove. Fuel. 2018;211:269-277.
- [CrossRef] [Google Scholar]
- Distribution of polycyclic aromatic hydrocarbons (PAHs) from legacy spills at an Alaskan Arctic site underlain by permafrost. Cold Reg. Sci. Technol.. 2019;158:154-165.
- [CrossRef] [Google Scholar]
- Influence of different types of coals and stoves on the emissions of parent and oxygenated PAHs from residential coal combustion in China. Environ. Pollut.. 2016;212:1-8.
- [CrossRef] [Google Scholar]
- Wang, H., You, X., Joshi, A.V., Davis, S.G., Laskin, A., Law, F.E.C.K. 2007. USC Mech Version II. High-Temperature Combustion Reaction Model of H2/CO/C1-C4 Compounds”. [Online]. Available: http://ignis.usc.edu/USC_Mech_II.htm, May 2007.
- Development of an n-heptane/toluene/polyaromatic hydrocarbon mechanism and its application for combustion and soot prediction. Int. J. Engine Res.. 2013;14(5):434-451.
- [CrossRef] [Google Scholar]
- Polycyclic aromatic hydrocarbons in the graphene era. Sci. China Chem.. 2019;62(9):1099-1144.
- [CrossRef] [Google Scholar]
- Detailed Kinetic Monte Carlo Simulations of Graphene-Edge Growth. J. Phys. Chem. A. 2010;114(2):689-703.
- [CrossRef] [Google Scholar]
- PAHs accumulations in plant leaves around coal-fired power plant and identification of their potential use as bioindicators. Arch. Environ. Contam. Toxicol.. 2019;76(2):346-355.
- [CrossRef] [Google Scholar]
- Study on soot nucleation and growth from PAHs and some reactive species at flame temperatures by ReaxFF molecular dynamics. Chem. Eng. Sci.. 2019;195:748-757.
- [CrossRef] [Google Scholar]
- Laboratory photochemistry of covalently bonded fluorene clusters: observation of an interesting PAH bowl-forming mechanism. Astrophys. J.. 2019;872(1):38.
- [CrossRef] [Google Scholar]
- Laboratory gas-phase infrared spectra of two astronomically relevant PAH cations: diindenoperylene, and dicoronylene. Astrophys. J.. 2018;854(1):27.
- [CrossRef] [Google Scholar]
- The growth mechanism of polycyclic aromatic hydrocarbons from the reactions of anthracene and phenanthrene with cyclopentadienyl and indenyl. Chemosphere. 2017;189:265-276.
- [CrossRef] [Google Scholar]
- Phase and size distribution of polycyclic aromatic hydrocarbons in diesel and gasoline vehicle emissions. Environ. Sci. Technol.. 2004;38(9):2557-2567.
- [CrossRef] [Google Scholar]
Appendix A
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2022.103839.
Appendix A
Supplementary material
The following are the Supplementary data to this article:Supplementary data 1
Supplementary data 1




