

Translate this page into:
Recent advances in synthesis of ketenimines
⁎Corresponding author. m.bayat@sci.ikiu.ac.ir (Mohammad Bayat)
-
Received: ,
Accepted: ,
This article was originally published by Elsevier and was migrated to Scientific Scholar after the change of Publisher.
Peer review under responsibility of King Saud University.
Abstract
Ketenimines are a kind of reactive species that can be used as synthetic intermediates. In the last two decades, there has been a surge of interest in this class of building blocks and their applications, which has led to extensive research on ketenimine derivatives such as fluorine ketenimine, metal complexes of ketenimines, and the various methods of their preparation. Ketenimines have been prepared by a variety of methods, including photolysis, elimination, or rearrangement reactions. As well as, ketenimines can be prepared using a variety of useful reagents, including isocyanates, copper acetylide, amides, organometallic compounds, and metal complexes. An overview of these achievements is presented here.
Keywords
Ketenimine
Rearrangement
Organometallic
Transition metal-catalyzed

1 Introduction
In more than 180 years, chemists have made significant advances in the development of chemical reactions and methods for the selective and efficient conversion of organic compounds. Thanks to these discoveries, many of the most complicated products can now be produced by organic synthesis (Corey and Cheng, 1995; Nicolaou and Sorensen, 1996; Nicolaou and Snyder, 2003; Nicolaou et al., 2000). Yet, it was not so long ago that the possibilities in this field were not well developed. For example, chemical research has shown that the fluorine atom and fluorine-containing motifs have a significant impact on the composition, reactivity, and function of organic and inorganic molecules (Kirsch, 2004; Groult et al., 2017; Haufe and Leroux, 2019). Since ketenimines are extremely versatile intermediates and can be used in a wide range of reactions, including nucleophilic additions and pericyclic reactions (Krow, 1971; Gambaryan, 1976; Aumann, 1988a,b). However, fluorinated ketenimines are understudied, possibly because there are few synthetic methods for their preparation. On the other hand, ketenimine complexes are used as versatile synthetic building blocks, but due to the lack of suitable methods for their preparation, they have received less attention. These shortcomings have challenged the synthesis initiatives of chemists and stimulated innovative methods for their synthesis, constantly updating the creativity of chemists involved in the Synthesize topologically complex molecules. Literature survey shows that most of the papers about the chemistry of ketenimines were published in the Journals of the American Chemical Society (www.scopus.com). In the last decade, scientists have developed a variety of methods for the Synthesize ketenimines, such as the Wittig reagent, alkane nitriles, amides, metal complexes as “non-classical” building blocks, cleavage of heterocyclic compounds, elimination reactions, isocyanide and rearrangement reactions, and pericyclic processes. Although three reviews on the chemistry of ketenimines have been published, none of them provides a comprehensive evaluation of the synthetic methods for these compounds (Lu et al., 2012; Dodd and Cariou, 2018; Alajarin et al., 2012). Therefore, in this review paper, we have detailed and explained various methods for the preparation of ketenimines, which serve as important intermediates for [2+2]-cycloaddition reactions (Alajarin et al., 1996) or for the formation of five-or six-membered ring systems (Aumann, 1988a,b). The review paper is roughly chronological in its development.
2 Background
2.1 Chemistry
In chemistry, ketenimines (IUPAC name: 1-alkenylideneamines) are considered a main category of compounds as valuable synthetic intermediates or reactive species. The ketenimine system contains an axial dissymmetry that is parallel to the allenes. Due to their special properties, such as a C—C double bond of the ethylene type and a C—N double bond of the azomethine type, they can be chiral. Nevertheless, the ability to perceive axial disharmony depends on the structural constancy of ketenimine, which is likely to reverse its formation either by turning around the imine or inversion of the nitrogen lone pair. The calculated barrier to interconversion is
[Simon et al., 1968], which is consistent with available experimental data. Another reason that causes ketenimines to be chiral is that they have a linear nitrilium resonance form that leads to very fast epimerization with all carbon-substituted groups, and this barrier is only
[Moss et al., 1995]. For ketenimines, resonance structures
are the most crucial ones. As shown in Scheme 1, describing ketenimine as a structure
may in fact oversimplify the situation. Other potential canonical forms include a zwitterionic form 1′, which emphasizes the nucleophilicity of the
-carbon atom, and an alternative zwitterion 1″ which emphasizes the electrophilicity of the central carbon atom.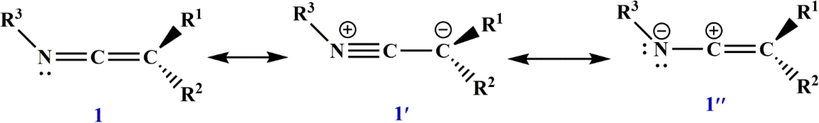
Resonance structures of ketenimines.
Ketenimines' structure is almost linear (
) and has a triad CCN bond, according to single-crystal
-ray diffraction studies, but there are a few examples (“e.g., with (R1 = R2 = MeSO2, R3 = Me”) that prove that with a strong electron-withdrawing sulfone group on the ketene portion and a methyl group on the imine portion of ketenimine 1 the CH3-N-C bond angle is
. The large contribution of the resonance structure 1′ can account for the linearity in the crystalline state. However, when the N-methyl group is replaced by an N-ethyl group, the CH2-N-C bond angle is
. The development of the ketenimine was confirmed by observing the absorption band of the C⚌C⚌N in FT-IR spectra, which showed a very robust absorption around
. The strong shielding of the terminal allenic carbon (
for various methyl- and phenyl- substituted ketenimines) also suggests the conjugative interaction between the C⚌C bond and the lone nitrogen pair implicit in the canonical form 1′, and the up field 15N chemical shift support this conclusion. An attempt to observe the effects of the barrier to interconversion of ketenimines on the chemical shifts of the diastereotopic methylene hydrogens in ketenimines
to
, shown in Table 1, was unsuccessful. The protons
,
or
,
appeared magnetically equivalent over a temperature range of
to +80 °C (Krow, 1971).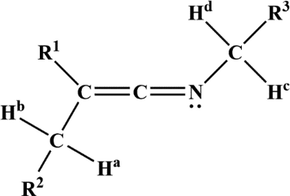
R1
R2
R3
I
Ph
CH3
tert-C4H9
II
Ph
Ph
tert-C4H9
III
Ph
CH3
Ph
IV
H
C(CH3)2Cl
tert-C4H9
Table 1. Ketenimines were used in an attempt to observe diastereotopic hydrogens.
Jochims et al. (Jochims and Anet, 1970) used the NMR method to determine the barriers to racemization of 3-methyl-N,2-diphenylbut-1-en-1-imine
and 2,3-dimethyl-N-phenylbut-1-en-1-imine
(Scheme 2). At −113 °C, the methyl protons of the isopropyl group of 3-methyl-N,2-diphenylbut-1-en-1-imine
separated into a double pair with a
chemical shift difference, corresponding to a free activation energy for racemization of
. The barrier in
was raised to
.
Structure of ketenimines.
In general, ketenimines possess a higher stability level than ketenes and can reach even higher stability through bulky groups or heteroatoms such as phosphorus and silicon, and likewise through mesmeric groups (Kim et al., 2011). Their C⚌C⚌N cumulenic system contributes a high potential for reactions to the members of this class of organic complexes (Kirsch, 2004; Groult et al., 2017; Haufe and Leroux, 2019; Borrmann, 1968), which have the potential to undergo a wide variety of modifications which are particularly significant in the structure of nitrogen heterocycles (Molina et al., 1991; Alajarin et al., 2000). It is believed that ketenimines, as tautomers of aliphatic nitriles, can contribute as intermediates in some important chemical reactions. In modern organic synthesis, ketenimines with high stability levels are absorbed elements (Kaneti and Nguyen, 1982).
2.2 History
The first stable member of the group was described by Staudinger et al. in 1920 (Staudinger and Meyer, 1919). Although no yield was given and no attempts were made to extend the synthesis to other ketenimines, they discovered that the reaction of (Z)-2-choro-N,2,2-triphenylacetimidoyl chloride 2 with zinc leads to the preparation of ketenimine 3 (Scheme 3).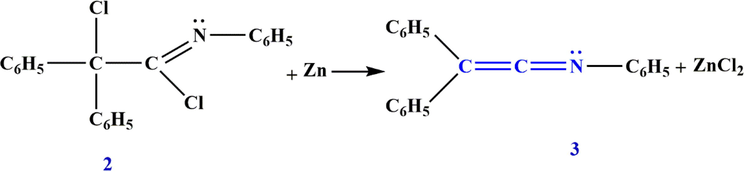
Structure of the first ketenimine 3.
Their results showed that the C⚌C⚌N unit is very susceptible to decomposition by hydrolysis, dimerization, polymerization, and other reactions. They actually developed a new class of highly reactive compounds containing C—C and C—N double bonds. Another successful method for the preparation of ketenimine was proposed by Stevens et al. in 1953 (Stevens and French, 1953). They showed that 2-chloro-2,2-diphenyl-N-(p-tolyl)acetimidoyl chloride 4 was readily dehalogenated with sodium iodide, resulting in the formation of 2,2-diphenyl-N-(p-tolyl)ethen-1-imine 5 as a ketenimine derivative (Scheme 4).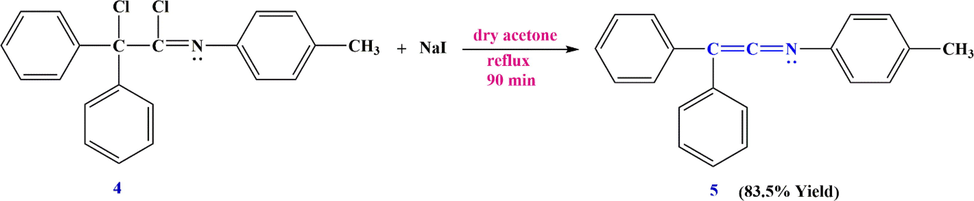
Synthesizing 2,2-diphenyl-N-(p-tolyl)ethen-1-imine 5.
Continuing the work of other researchers, Dijkstra et al. (Dijkstra and Backer, 1954) have shown that the reaction between diazomethane 7 and bis-methylsulphonylacetonitrile 6 can produce N,2-dimethylprop-1-en-1-imine-sulfur (
) oxide 8 in a medium yield (Scheme 5).
Synthesize N,2-dimethylprop-1-en-1-imine-sulfur (
) oxide 8.
Due to the unique properties and high reactivity of ketenimines, the chemistry of these compounds was first studied by Krow (Corey and Cheng, 1995). By 1971, the methods for the preparation of ketenimines had been further developed, and despite substitution reactions in ketenes, isocyanates, and carbodiimides, other addition reactions of isocyanides with carbenes or cyclopropenones, alkynes, as well as elimination and rearrangement reactions, were proposed by other scientists. But the methods he proposed for the synthesis of ketenimine were very limited. In 1976, Gambaryan (Nicolaou and Sorensen, 1996) dealt with the subject of fluorinated ketenimines in a specialized review paper and investigated the methods used for obtaining imines of fluorinated ketenes as well as their dimerization and nucleophilic addition. Other scientists have shown that metal complexes of ketenimines are of particular importance for synthesis. Namely, they have shown how the reactivity of these compounds changes upon complexation and how ketenimine complexes can be used as versatile synthetic building blocks. To further evaluate the generality and scope of the reaction, Ariyaratne et al. (Ariyaratne and Green, 1963) indicated that the metal-
complex of ketenimin-iron 10 can be synthesized by proton-catalyzed isomerization of the
-complex 9 (Scheme 6). The un-complexed imine function can be identified by the characteristic infrared bands at
.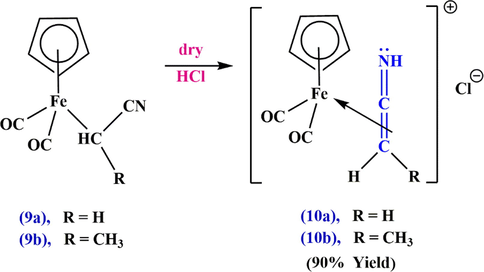
Synthesize the ketenimin-iron complex 10.
Beck et al. (Beck et al., 1966; Beck et al., 1967) showed another method for the preparation of metal complexes ketenimine using a nitrogen transition metal compound. Using X-ray experiments, they demonstrated that the iridium-tetracyanoethylene complex should be the ((2,3,3-tricyanoprop-1-en-1-ylidene) amino) iridium-tetracyanoethylene complex 11, and that the iridium N—C bond angle of 162° is a good indicator of the relative importance of the resonance structure 12 (Scheme 7).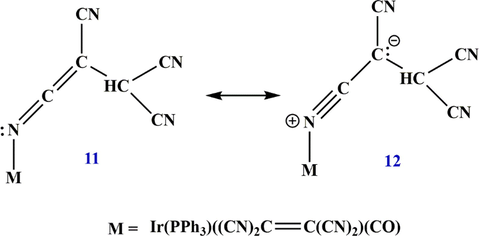
Resonance structures of the metal complex ketenimine.
Also in 1988, Aumann (Aumann, 1988a,b) showed that metal complexes of ketenimines are of particular importance in synthesis. As a matter of fact, he showed that ketenimine complexes 15 are formed by the insertion of a 1,2-bond N⚌C to the bond M⚌C carbon 13, the complexes being bonded to isocyanide 14 (Scheme 8). The insertion of isocyanides into M⚌C bonds is not specific to any particular metal, and complexes 15 with M⚌V, Cr, Fe, Ni, Th, Ir, Mg, Li, U, Au, Cu, Co, and Pd have been crystallographically characterized. It is possible to build up palladium-ketenimine complexes catalytically from isocyanides 14, allyl bromides 16, and triethylamine. Catalysis involves the N-substituted alkenimidoyl complexes 18 as intermediates (18, R2⚌CH3, H, has been isolated) which are probably formed by the insertion of R1-NC into a Pd⚌C
bond. However, coupling via Pd⚌C bonds cannot be excluded. Coupling reactions of carbenes (dichloride-, diphenyl-, dimethoxycarbonylcarbene) or with isocyanides to form ketenimines are also known (Scheme 9).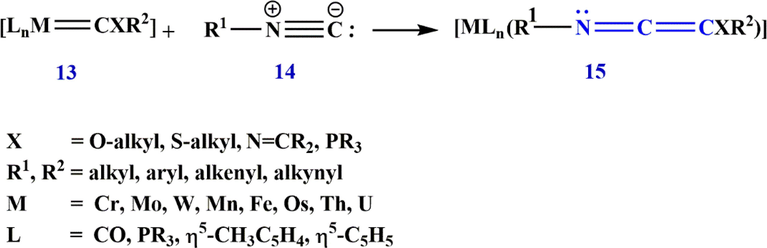
The reaction between carbon complex and isocyanide.
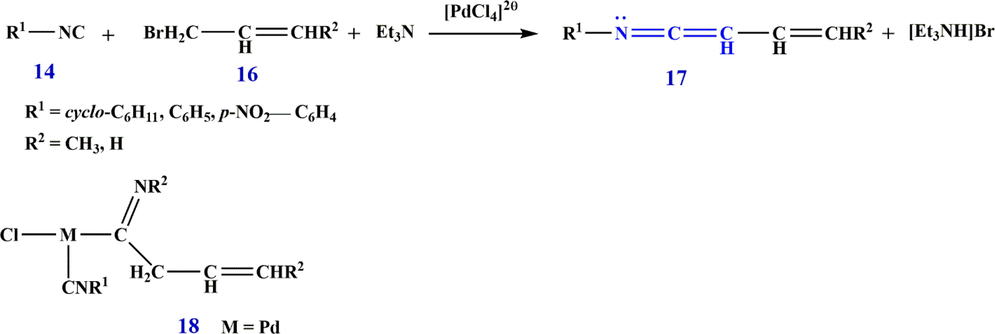
Synthesize ketenimine 17.
2.3 Mechanism
The reaction to produce ketenimines was developed by Staudinger et al., but they did not describe the mechanism of the reaction. It appears that organometallic reagents, including zinc, tend to act as strong reducing agents in the preparation of ketenimines. In fact, the reactions leading to the preparation of ketenimines must follow one of the proposed mechanisms, such as: substitution in heterocumulene, elimination reactions, rearrangement reactions, elimination-rearrangement, metal complexes, or transition metal complexes.
2.4 Precursors
The reaction of isocyanides with electron-deficient groups such as acetylenic ester, acetylenedicarboxylate, acid chlorides, and diethyl (bromo (phenyl) methyl) phosphonate are the most commonly used reagents for the preparation of ketenimines. These ketenimines can also be synthesized by various reagents, such as isocyanates, copper acetylides, amides, or organometallic or metal complexes of ketenimines by a variety of methods, including cleavage of compounds, photolysis, or elimination or rearrangement reactions. However, as shown in Fig. 1, there are several other ketenimine precursors. All methods for the preparation of ketenimines have been thoroughly investigated in this review paper. As we know, ketenimines are very reactive, and only sterically hindered or inductively stabilized ketenimines can be isolated. Therefore, ketenimines are almost always prepared in situ and trapped with species already in solution. The following sections show how ketenimines are used in organic synthesis to speed up the process of making molecules with many different parts.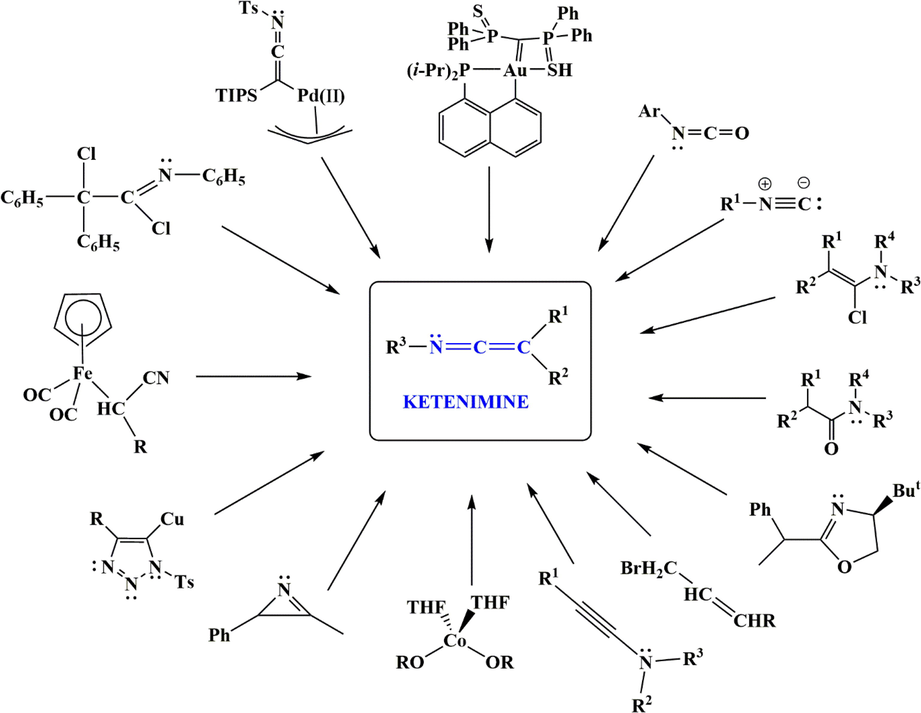
All precursors for the preparation of ketenimines-based intermediates.
3 Ketenimines preparation
Ketenimines are flexible intermediates capable of going through different organic reactions, and therefore their construction and potential for reactions have been the subject of extensive research (Jochims and Anet, 1970). Ketenimines belong to the group of heterocumulenes and are considered as imine equivalents of ketenes (Yoo and Chang, 2009). No wonder that they are mostly used as starting materials in chemical reactions for the synthesis of open-chain and heterocyclic compounds (Denmark and Wilson, 2012; Barker and Rosamond, 1972; Kaufman, 1970) and also peptides (Stevens and Munk, 1958). This is due to their high reactivity. However, this high reactivity and the difficult reaction conditions required for their building can sometimes cause problems in the preparation of functionalized ketenimines. Ketenimines stabilized by heteroatoms such as silicon, phosphorus, and sulfur, as well as those conjugated with vinyl, aryl, and carbonyl functional groups, can be synthesized and separated as active intermediates.
3.1 Ketenimine preparation via coupling reactions
The Wittig reaction is a commonly used method for the preparation of C⚌C or C⚌N bonds of ketenimines (Guan et al., 2019; Xiong et al., 2021). Aryl isocyanate 19 and methyl 2-(triphenylphosphoranylidene) propanoate 20 can be utilized to prepare methyl 3-(arylimino)-2-methylacrylate 21, a highly electrophilic ketenimine (Cheng et al., 2009). In such a reaction, the Wittig reagent serves as an equivalent to the nucleophilic attack of the electron-deficient isocyanates. Since the ester group at the C-terminus stabilizes the ketenimines, they can be isolated (Scheme 10).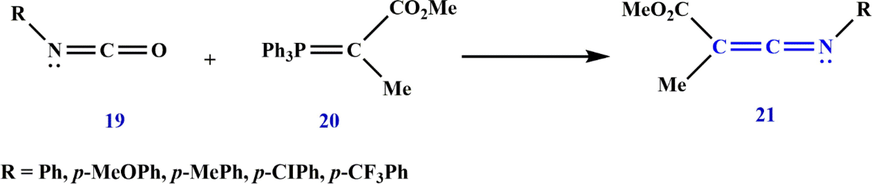
Synthesize ketenimine 21.
Alternatively, the coupling reaction between the Wittig reagent 24 and diphenylketene can be carried out in anhydrous ortho-xylene at room temperature to give the 1,3-oxathiane ketenimine 25, which under the reaction conditions converts to the 3',3'-diphenylspiro[1,3-oxathiane-2,4'(3'H)-quinoline] 26 and spiroquinolines 28 in cis and trans (via 1,3-oxathia-neketenimines [1,5]-H-shift followed by 6-electrocyclisation) in moderate yield (Scheme 11) (Alajarin et al., 2012).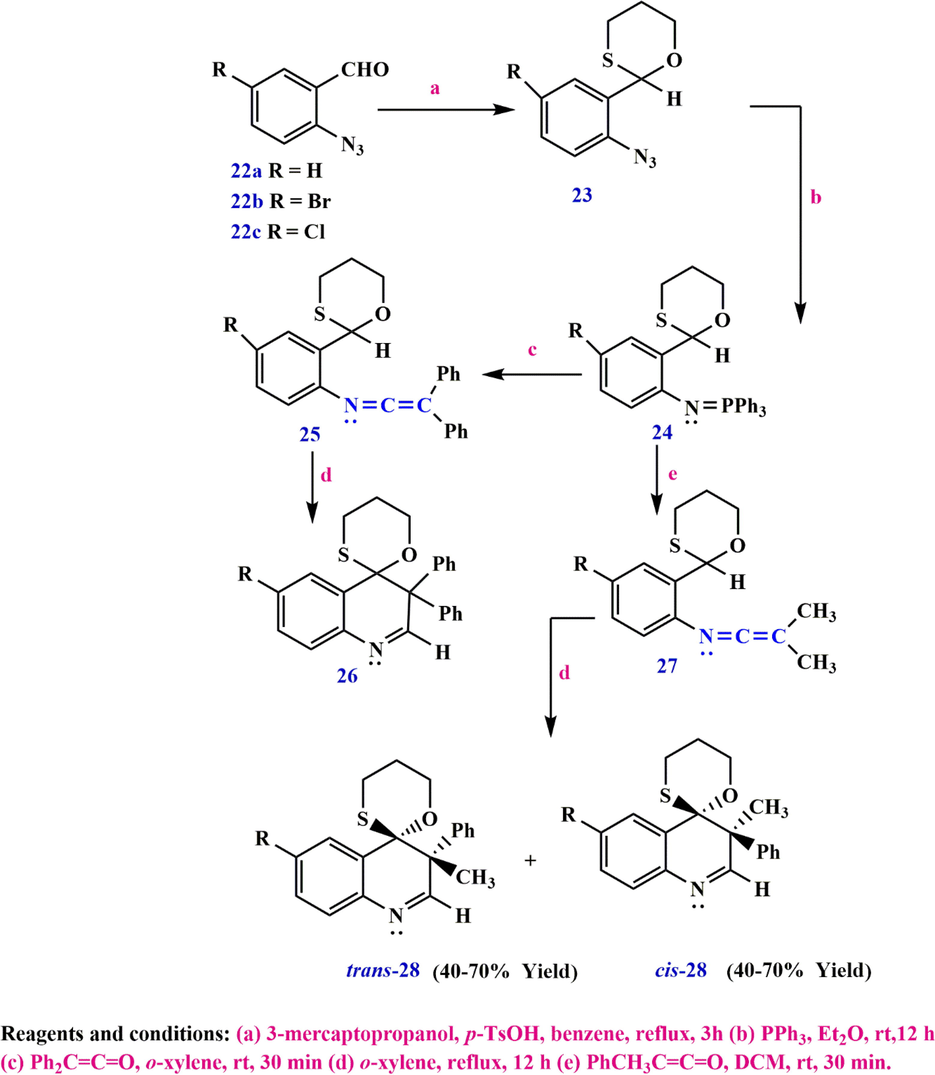
Synthesize spiroquinolines 28 via ketenimine.
3.2 Ketenimine preparation via isocyanide-based multicomponent reactions
Many methods based on multicomponent reactions (MCRs) have gained increasing attention (Zhu and Bienaymé, 2005; Domling, 2006; Orru and Greef, 2003). Imidoyl chloride 29 can be prepared in situ via the Nef reaction between isocyanide and acid chloride. When trialkyl phosphites are used to trap the
-keto imidoyl chloride, chloride through a phosphite, a Perkow-type reaction can take place to form ethyl 3-(substitutedimino)-2-((dialkoxyphosphoryl)oxy) acrylate 30 (Scheme 12) (Coffinier et al., 2011).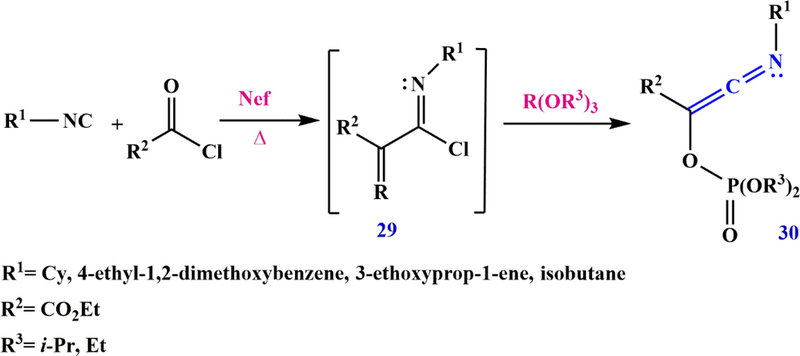
Synthesize ketenimines via a Nef/Perkow sequence.
A parallel reaction model is also detected in the reaction of cyclohexyl isocyanide 31 with dialkyl acetylenedicarboxylate 32 in the presence of aryl sulfonamide 33, which proceeds with a soft reaction in dichloromethane at room temperature to obtain dialkyl 2-cyclohexyliminomethylene-3-arylsulfonylamino succinates 34 in good yield. Despite the lack of a feasible mechanism for the reaction between isocyanides and acetylene esters in the presence of aryl sulfonamide, a plausible mechanism has been presented (Scheme 13). A logical assumption, according to the deep-rooted chemistry of isocyanides, is that the functionalized ketenimine 34 obviously comes from the primary addition of the cyclohexyl isocyanide 31 to the acetylenic ester 32 and the subsequent protonation of the 1:1 adduct 35 by aryl sulfonamide 33. Then the positively charged ion 36 is attacked by anion 37, and finally, product 34 is formed (Anaraki-Ardakani et al., 2011).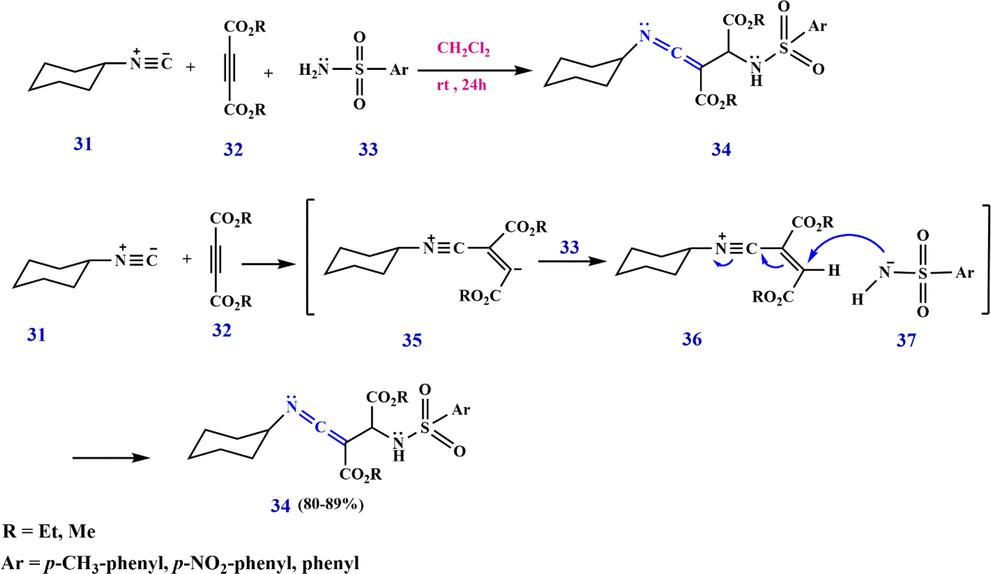
Synthesize ketenimines 34 by cyclohexyl isocyanide.
In 2013, Asghari et al. (Asghari et al., 2013) described a three-component reaction for the synthesis of ketenimines. The reaction starts with the Michael addition of tert-butyl isocyanide to the electron-deficient acetylenic ester, and finally the cationic intermediate, (Z)-N-(4-methoxy-2-(methoxycarbonyl)-4-oxobut-2-en-1-ylidyne)-2-methylpropan-2-aminium 41 is formed (Scheme 14). Subsequently, 2,4-dioxothiazolidin-3-ide, the negatively charged ion 42 can attack the cationic intermediate 41 via routes A and B, and dialkyl-2-((tert-butylimino) methylene)-3-(2,4-dioxothiazolidin-3-yl) succinate 38 is produced by the Michael addition of the intermediate 42 to the cationic intermediate 41.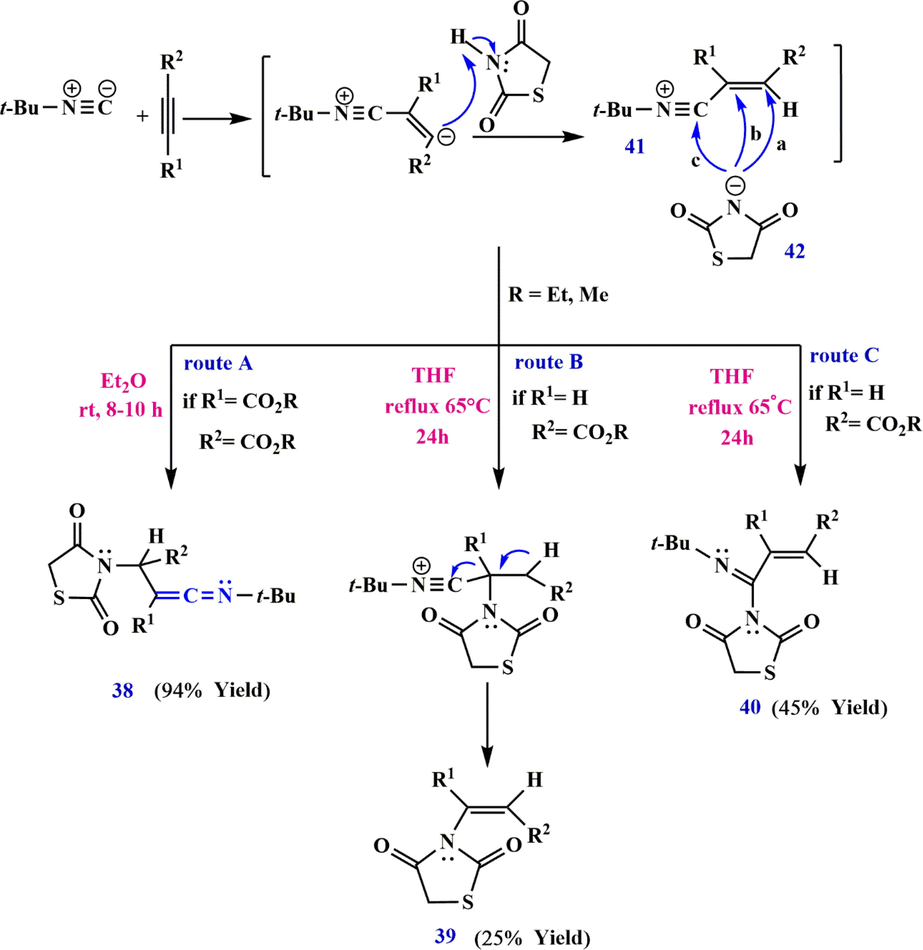
Synthesize ketenimines 38.
Jalli et al.)Jalli et al., 2015) reported the reaction performed using pentyl isocyanide 43, diethyl acetylene dicarboxylate 44, morpholine 45 (or) thiomorpholine and 4-chloro-3-formyl coumarin 46 to give the corresponding ketenimine furyl coumarin 47 and 48, respectively (Scheme 15).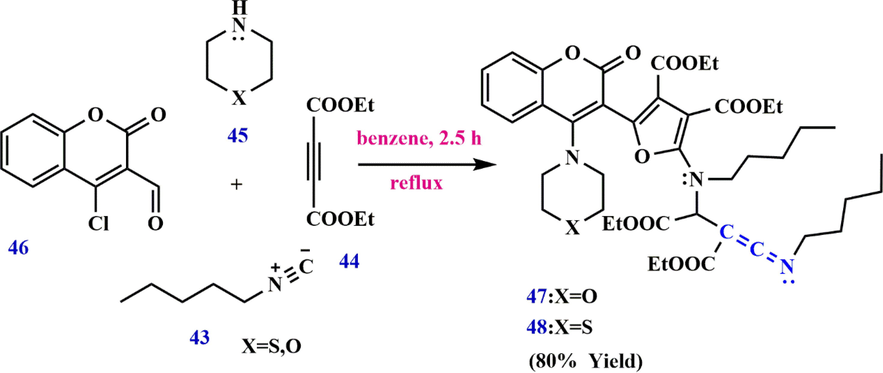
Synthesize ketenimine furyl coumarin 47, 48.
Santos et al.)Dos Santos et al., 2017) have shown that imidoyl chloride 49 causes the formation of 1-(4-chlorophenyl)-2-(cyclohexylimino) vinyl dimethyl phosphate 50 in the Nef-Perkow sequence, whereupon the intermediate imidate ester undergoes a Mumm rearrangement, leading to imido phosphate 51(Scheme 16).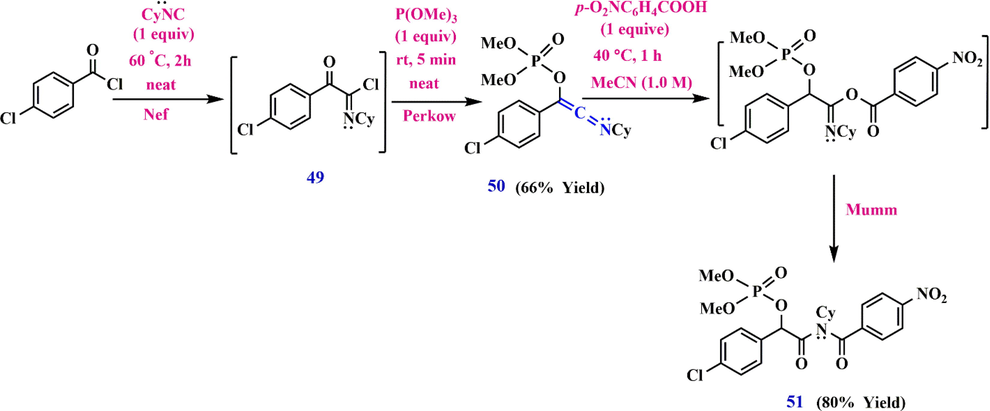
Nef-Perkow-Mumm rearrangement cascade towards imido phosphate 51.
Bayat et al.)Bayat et al., 2008) described a modern and effective approach to the synthesis of ketenimines 55 using alkyl isocyanide 52 with dibenzoylacetylene or dialkyl acetylenedicarboxylate 53 in the presence of NH-acid 54, prepared at ambient temperature in dry diethyl ether or ethyl acetate (Scheme 17).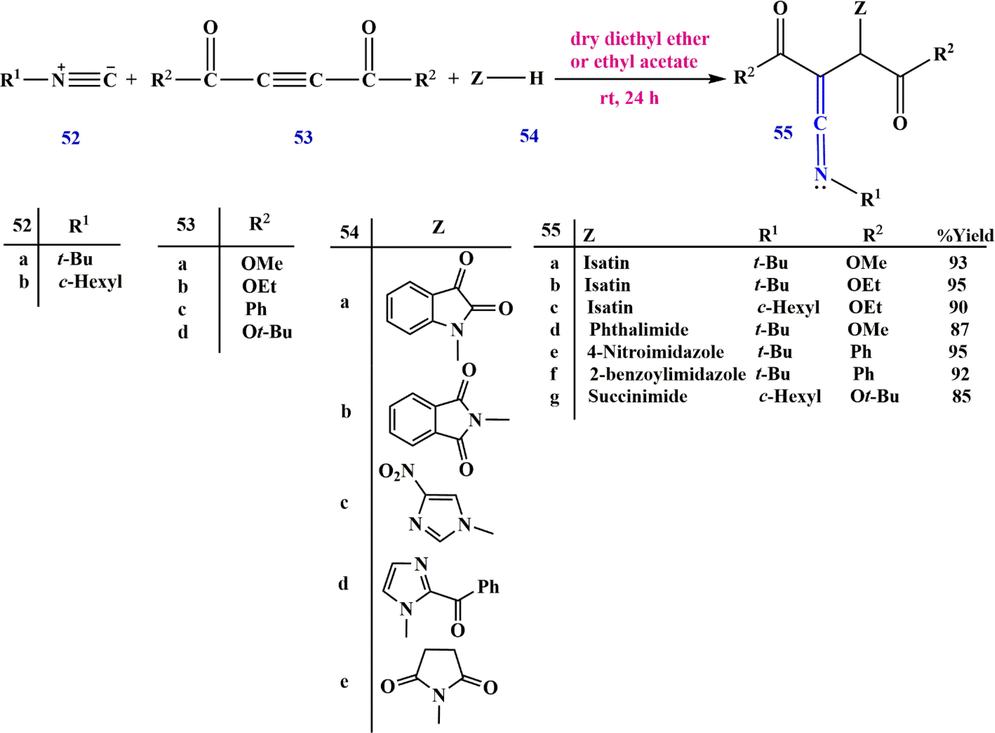
Synthesize ketenimine 55.
Recently, Zhang et al. (Zhang et al., 2019) studied the reaction of difluorocarbene with isocyanide to produce difluoroketenimine 58. They showed that the addition of fluorine substituents at the C-terminus leads to the activation of the ketenimines (Scheme 18).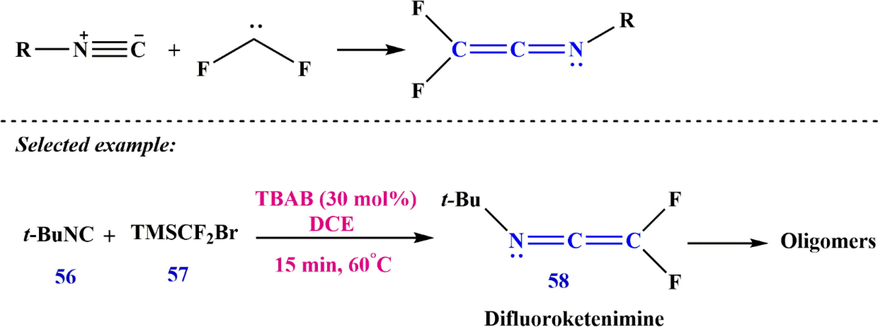
Synthesize difluoroketenimine 58.
3.3 Ketenimine preparation via -chloroenamine and tertiary amide
In 1972, Ghosez (Marchand-Brynaert and Ghosez, 1972) reported that ‘keto’-keteniminium salts 60 (R1, R2
H) can be prepared from the corresponding
-chloroenamine 59, route A in (Scheme 19), or tertiary amide 61 (Falmagne et al., 1981), route B in (Scheme 19). Since the reactivity of the aldo-keteneiminium salts 60 (R1
H) is very high, they are only prepared via route B and lead to the formation of cyclobutenimine salt 62 by the reaction [2 + 2] of the keto-keteneiminium salts 60 (R1, R2 = alkyl, c-alkyl). The iminium salt 60 was then used as dienophiles in Diels–Alder reactions with various functionalized dienes, giving compound 64 (Scheme 20).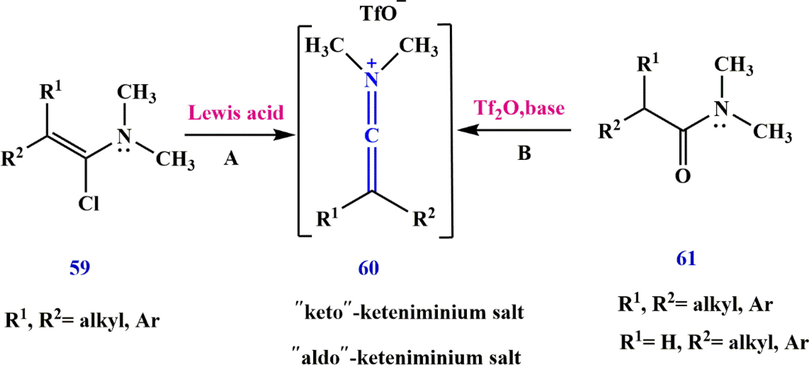
Synthesize keteniminium salt 60.
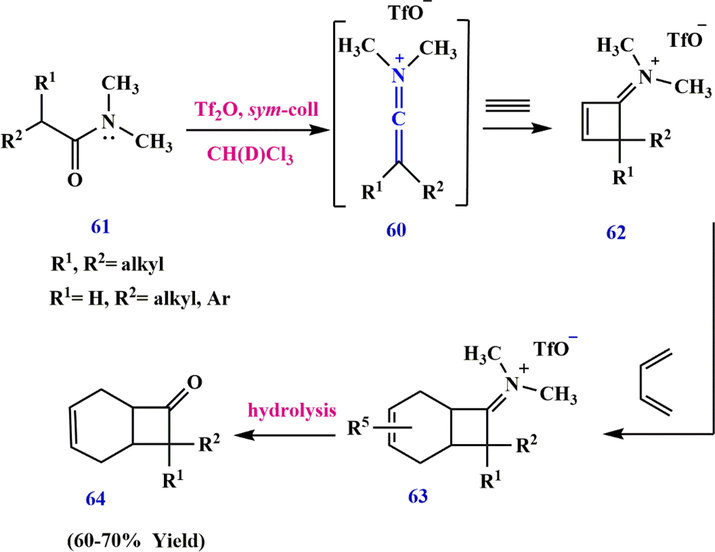
Synthesize cyclobuteniminium salt 62 from keteniminium salt.
3.4 Ketenimine preparation via cleavage of heterocyclic compounds
3.4.1 Cleavage of azirines
Irradiation with a short wavelength of 3-methyl-2-phenyl-2H-azirine in an argon matrix leads to the ylide, while irradiation with a longer wavelength forms the ketenimine (Scheme 21) (Zhang et al., 2014; Weragoda et al., 2017). Irradiation of 2-azidovinylbenzene 65 under reaction conditions leads to 2-phenyl-2H-azirine 66 as the main product, some ketenimine 68, and a small amount of cis-65, while irradiation of vinyl azide 65 under reaction conditions produces only2-phenyl-2H-azirine 66 and 2-phenylethen-1-imine 68 (Scheme 22). Osisioma et al. (Osisioma et al., 2018) showed that irradiation of vinyl azide 69 led to the simultaneous preparation of the parallel azirine (methyl 2-benzoyl-2-chloro-2H-azirine-3-carboxylate) and ketenimine (methyl (2-chloro-3-oxo-3-phenylprop-1-en-1-ylidene) carbamate) derivatives (Scheme 23).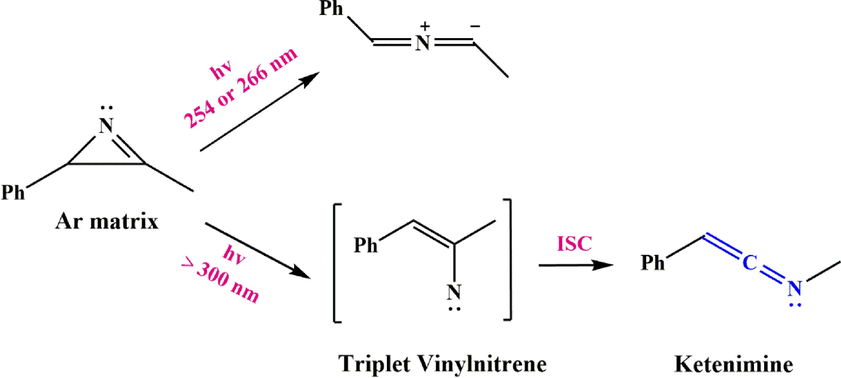
Cleavage of 3-methyl-2-phenyl-2H-azirine.
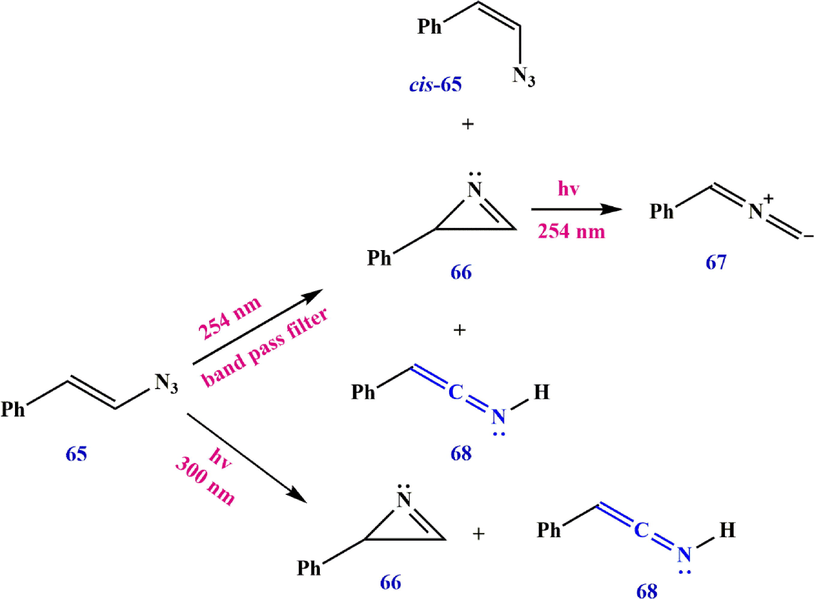
Cleavage of azirine 66.
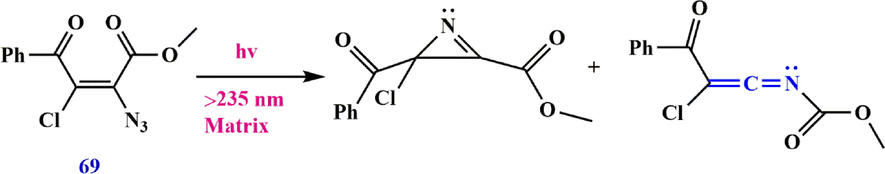
Cleavage of vinyl azide.
Another method of producing ketenimines reported by Inui et al. (Inui and Murata, 2001). In their method, matrix isolation/IR at low temperature leads to the formation of the diradical (2-aryl-3-methyl-2H-azirine), which is used as an intermediate for the synthesis of 2-aryl-N-methylethen-1-imine 70 (Scheme 24).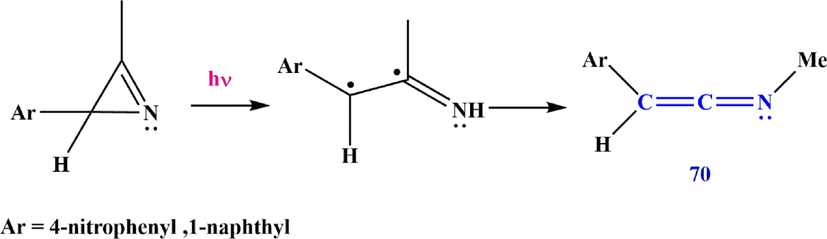
Cleavage of 2H-azirines 70.
3.4.2 Cleavage of oxazolines and isoxazolium salts
The ring cleavage observed in achiral oxazolines during lithiation has now been extended to chiral oxazolines. For example, treatment of oxazoline 71 with BuLi gives lithium (S,E)-4-(tert-butyl)-2-(1-phenylethylidene)oxazolidin-3-ide 72, which is mainly in the ketenimine form 73 (Scheme 25). This ketenimine can be trapped with TMSCl to give 3-(((3R)-4,4-dimethyl-3-((2-phenylprop-1-en-1-ylidene) amino) pentyl) dimethylsilyl) oxazolidin-2-one 74, but it is more useful to react with second equivalents of BuLi and a benzyl bromide to give chiral ketones such as 2-methyl-1,2-diphenylheptan-3-one 75 in 50–70% yield and 50–87% ee after work-up (Dwyer et al., 1999).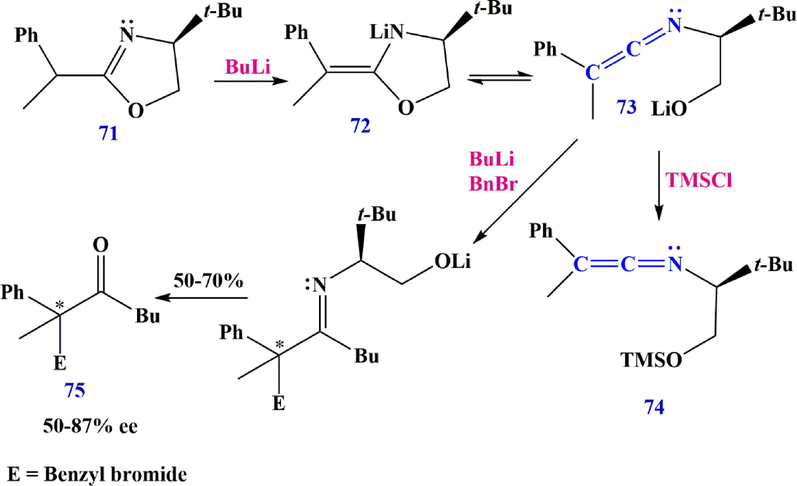
Synthesize ketenimine 74.
Flash vacuum pyrolysis (FVP) of 3-aryl-4-[(dimethylamino(alkyl(aryl)amino methylene] isoxazol-5(4H)-ones 76 gives 1-(3-aryl-2H-azirin-2-ylidene)-N-phenylmethanimine 77 and bisiminopropadiene ArN⚌C⚌C⚌C⚌NR 78 with the elimination of dimethylamine and CO2 and rearrangement of the 3-aryl-4-((phenylimino) methylene) isoxazol-5(4H)-one (Scheme 26) (Wolf et al., 1996).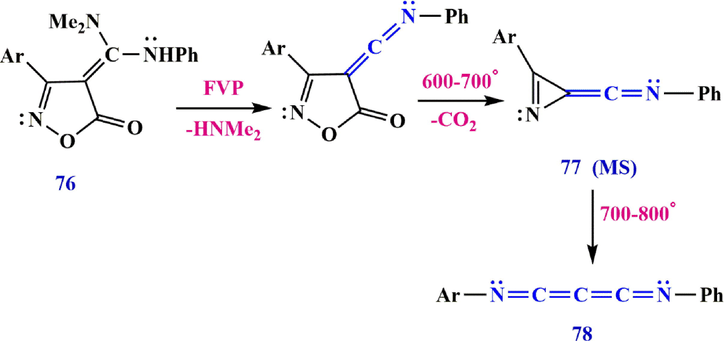
Cleavage of isoxazolone
Woodward's reagent K79 was introduced as a prototype for a class of peptide synthesis reagents in 1961. (Woodward and Olofson, 1961). They showed that the initial deprotonation at the unsubstituted 3-position has been attributed to the reactivity of these isoxazolium salts, accompanied by ring cleavage to form C-acyl ketenimine 80, which was intercepted by a carboxylate to afford enol esters suitable for further attack (Scheme 27). Spectroscopic evidence for acyl intermediates of ketenimine was provided by Woodward et al.in later studies. This route is limited to relatively stable compounds such as N-(t-butyl)-benzoylketenimine and the related acetyl compound, which are prepared in 60% and 70–80% yields from isoxazolium perchlorates and triethylamine, respectively, for preparative purposes. 4,5-disubstituted isoxazolium perchlorates or tetrafluoroborates have also been used to isolate C-acyl ketenimines 80-84. By reacting the salt with triethylamine in an acetonitrile solution, the comparatively unstable C-acyl ketenimine 80 can be prepared from reagent K itself. With the addition of cuprate, other susceptible acyl ketenimines can be intercepted (Scheme 28). Several isoxazolium salts with heteroatomic substituents were also found to produce ketenimines when treated with a base. The 5-aminoisoxazolium precursor 85 of ketenimine-amide 86 and the 4-azido isoxazolium salt 87 were used to prepare the special C-azido ketenimine 88, which is stable only below −60 °C.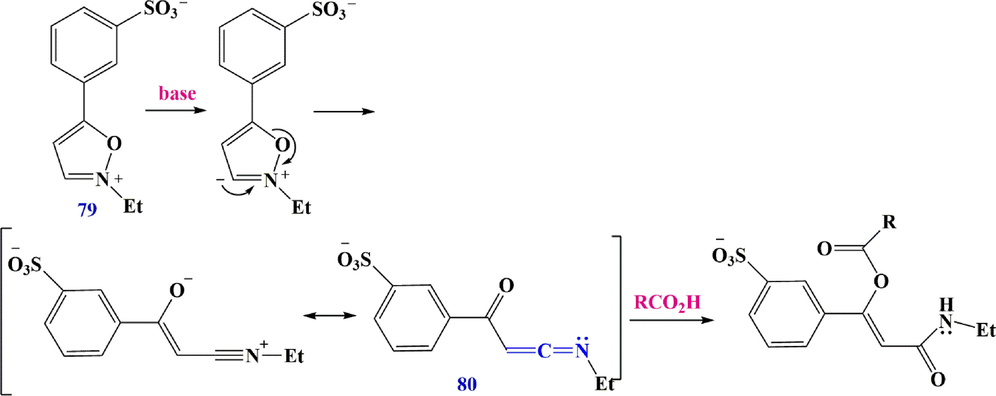
Synthesize ketenimines with Woodward's reagent.
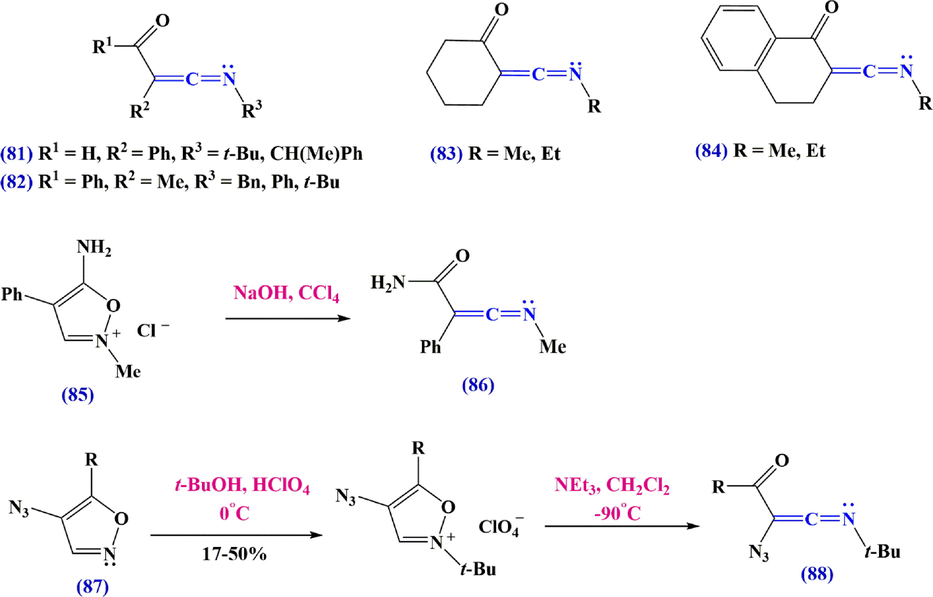
Synthesize ketenimines from 4-azido isoxazolium salts.
The K reagents were developed in another study by Peng et al. via a solution-phase nucleophilic addition reaction accompanied by a gas-phase elimination reaction. The reaction pathway for reagent ion preparation which involves the irreversible conversion of the wrk 89 to a reactive N-ethyl keto-ketenimine (Ki-Et) 90 by proton abstraction from the 3 isoxa-zole positions (Scheme 29). The intermediate 91, termed amidine, has the potential to be converted by tautomerism to compound 92, which in turn reacts with a primary amine of an amino acid. The C—N bonds of both amidines, 91 and 92, are easily cleavable upon collisional activation in the gas phase, which either generates the N-amino acid keto-ketenimine (Ki-X-OPG) 93 with the loss of an ethylamine, or reproduces the Ki-Et 90 at the loss of the NH2-X-OPG (Peng and McLuckey, 2015).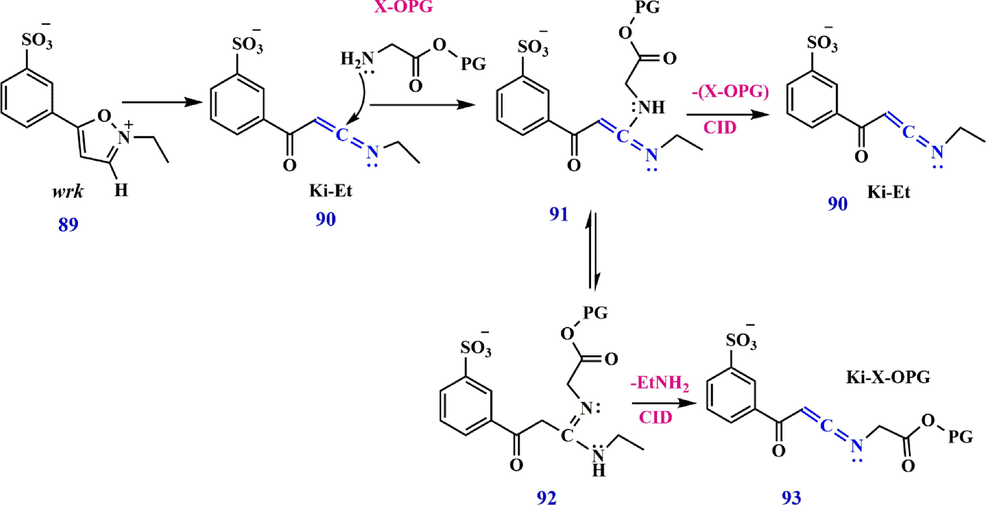
Reaction of amidine synthesis between keto-ketenimine (Ki-Et) and Woodward’s reagent K.
3.5 Transition Metal-Catalyzed Synthesis of Ketenimine
3.5.1 Pd -Catalyzed synthesis of ketenimine
For the first time in the history of chemistry, scientists have developed an effective method for the synthesis of C-phosphonoketenimines using palladium-catalyzed migratory insertion of isocyanides (Qian et al., 2016). The peculiarity of this method is that it involves a large number of functional groups and has good atomic economy. In this method, tert-butylisocyanide 94 was reacted with diethyl(bromo(phenyl)methyl) phosphonate 95 to synthesis of diethyl (2-(tert-butylimino)-1-phenylvinyl) phosphonate 96 (Scheme 30).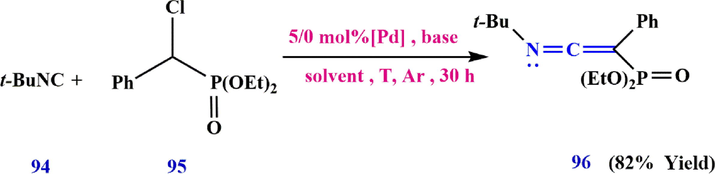
Synthesis diethyl (2-(tert-butylimino)-1-phenylvinyl) phosphonate 96.
Interestingly, ketenimines can be prepared very rapidly using rearrangement of N-allocynamides and a functional group-tolerant Pd catalyst under neutral conditions. Hsung et al. (Zhang et al., 2009; De Korver et al., 2010; De Korver et al., 2011) previously described the heat and palladium catalyzed rearrangement of N-allyl-N-tosyl ynamides to obtain 4-methyl-N-(2-(triisopropylsilyl)penta-1,4-dien-1-ylidene)benzenesulfonamide 97 as ketenimine, which can be trapped by an exogenous nucleophile such as an amine or an alcohol (Scheme 31).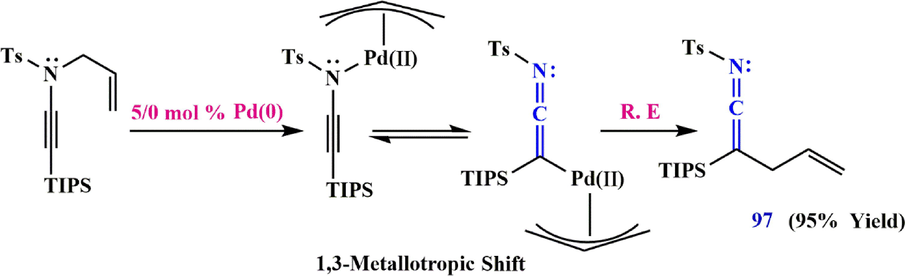
Recommended mechanism to the synthesis of ketenimine 97.
Another method for the preparation of ketenimine in the presence of Pd is the substitution reaction (Hiroi and Sato, 1985; Alexander and Cook, 2017). Although various heteroatom nucleophiles have been used to prepare amidine and imidate compounds, the use of carbon nucleophiles is much less common. The imine 102 with a fully substituted
-carbon results from the reaction of 2-allyl-N-arylhex-1-en-1-imine 99 with sulfur (Scheme 32).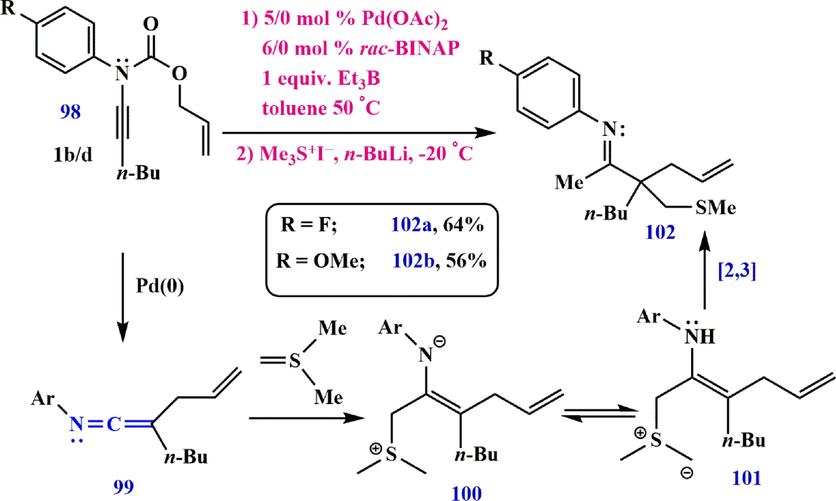
Synthesize ketenimine 99.
Hydrolysis of ketenimine 105 results in the formation of an
,
-unsaturated carboxamide 106, which is produced by the reaction of allyl ethyl carbonate 103 with isocyanide 104 in the presence of Pd (OAc)2 as a catalyst (route A in Scheme 33). Moreover, a three-component synthesis of 1,5-disubstituted tetrazole 107 can be achieved by [3+2]-cycloaddition with hydrazoic acid (HN3) or TMSN3 in situ trapping of ketenimine 105 (route B in Scheme 33). The oxidative addition of an allyl carbonate 108 to
give an
-allyl Pd complex C that is symmetric to the
-allyl Pd species D (Qiu et al., 2016). Migratory insertion of D would produce an imidoylpalladium intermediate E, which upon
-hydride elimination would furnish a ketenimine 105 with the concurrent rearrangement of the
catalyst (Scheme 34).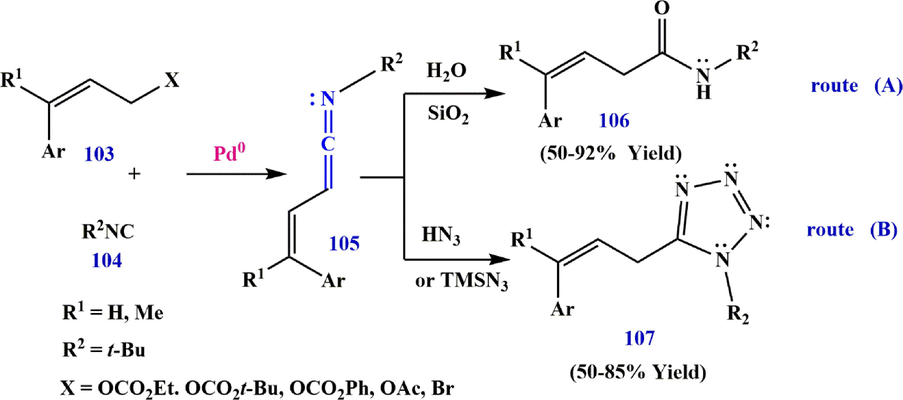
Synthesize ketenimine 105.
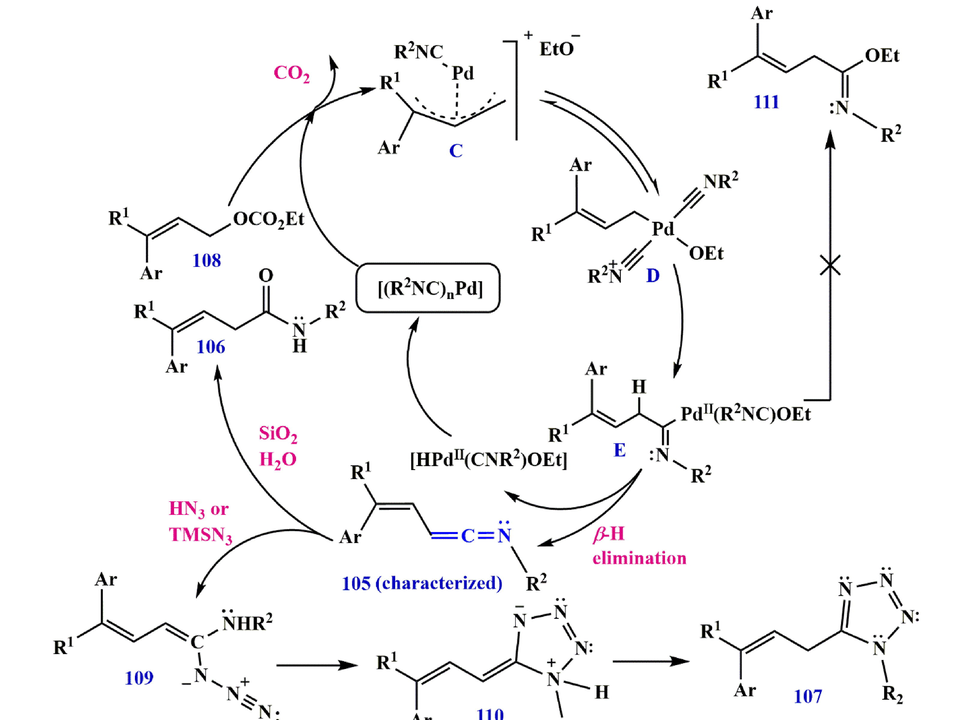
Recommended mechanism for the synthesis of ketenimine 105.
3.5.2 Cu -Catalyzed synthesis of ketenimine
Ketenimine 116 can be prepared via the coupling reaction between copper acetylide 113, formed from phenylacetylene 112 and CuI (undergoes a 1,3-dipolar cycloaddition) reaction with sulfonyl azide 114, to give the triazole derivative 115 (Yoo et al., 2007). This intermediate can be converted to the ketenimine derivative 116, which is attacked by cyanoguanidine 117 to afford 118. This intermediate undergoes intramolecular cyclization and tautomerization to diaminopyrimidine derivatives 120 (Yavari et al., 2012) (Scheme 35).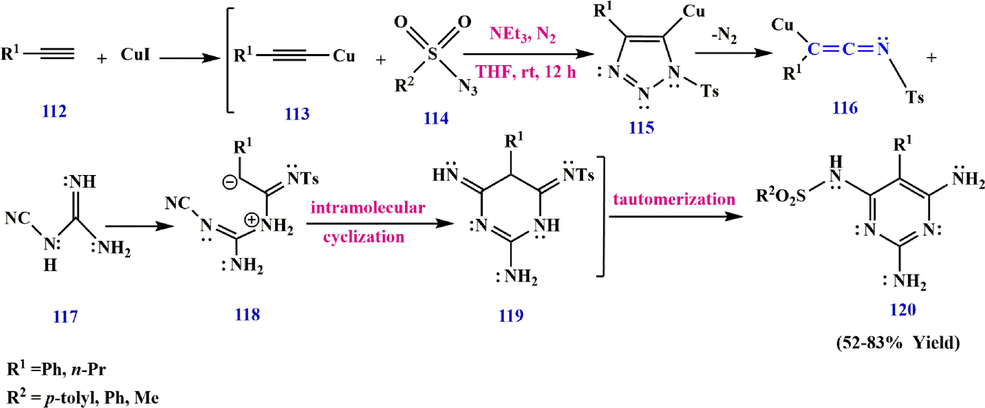
Recommended mechanism for the synthesis of diaminopyrimidine derivatives 120.
Similarly, copper acetylide 122 undergoes 1,3-dipolar cycloaddition with sulfonyl azide 123 to form triazole 124. This intermediate is converted to the ketenimine derivative 125, which is then cycloaddition with the nitrile imine 126, which is prepared from hydrazonoyl chloride and Et3N, to afford 127. Finally, the intermediate 127 is converted to the product tetrasubstituted pyrazole 128 via a 1,3-H shift (Yavari et al., 2012) (Scheme 36).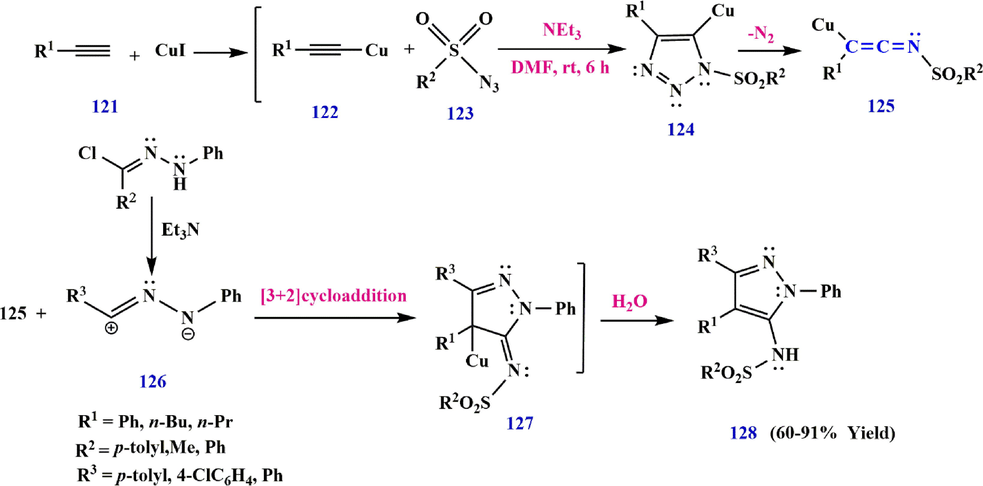
A possible mechanism for the synthesis of pyrazole derivatives 128.
Based on the above results, a reaction pathway for the synthesis of ketenimine III was developed leading to the preparation of 3-trifluoromethyl-substituted 1,2,4-triazinone 131 (Scheme 37). First, ring-opening of II leads to the formation of ketenimine III, which can be converted to IV by nucleophilic addition with CF3CO2H. Subsequently, the amine moiety is cyclized with the ketone C⚌O bond to form V, which then undergoes condensation and aromatization to produce the 3-trifluoromethyl-substituted 1,2,4-triazinone as the major product (Scheme 38) (Wu et al., 2017).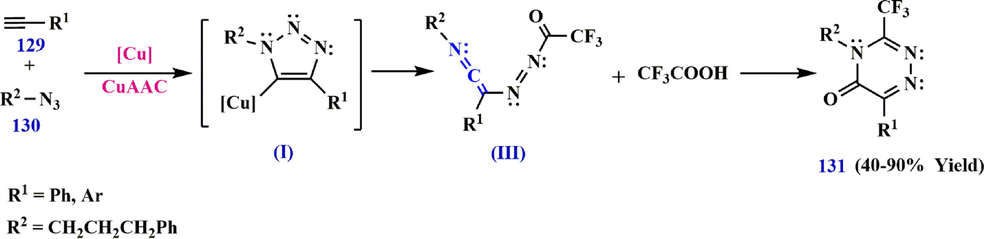
Synthesize 3-trifluoromethyl-substituted 1,2,4-triazinone 131.
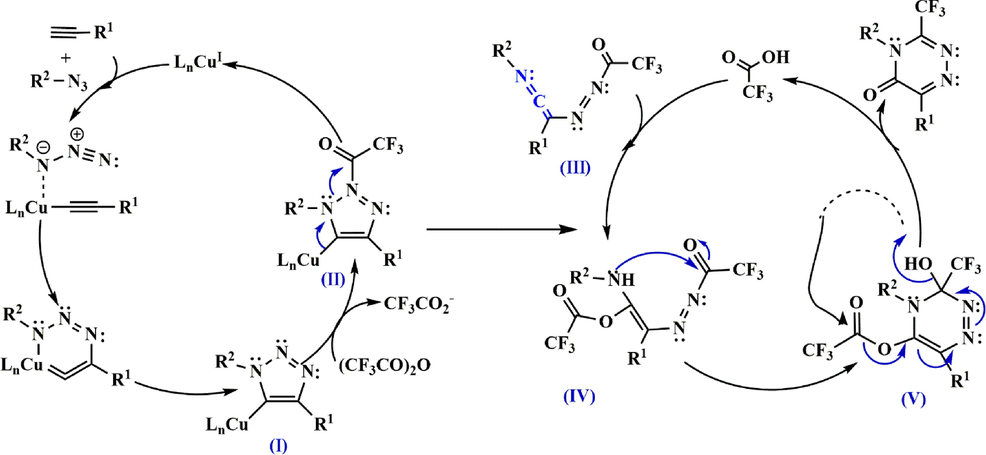
Recommended mechanism for the synthesis of 3-trifluoromethyl-substituted 1,2,4-triazinone 131.
3.5.3 Co -Catalyzed synthesis of ketenimine
The high-valent cobalt carbene Co(OR)2(⚌CPh2) (OR⚌OCtBu2Ph) reacts with various isocyanides CNR′ to form ketenimine. While spectroscopic and theoretical studies reveal a significant radical characteristic in carbene functionality, only sluggish cyclopropanation reactivity with styrene was observed. The carbene complex, (diphenylmethylene)bis((2,2,4,4-tetramethyl-3-phenylpentan-3-yl)oxy)cobalt 134 is obtained by treating the cobalt bis (alkoxide) complex 132 with diphenyldiazomethane 133. Subsequent, 1H-NMR investigation revealed that treatment of 134 with 132 in the presence of 2,6-dimethylphenyl isocyanide 133 (CN(2,6-Me2Ph) resulted in the formation of ketenimine 135 in around 30% yield. The preparation of ketenimine 135 was confirmed by GC-MS, and the results, unlike previous reports (Grass et al., 2019), showed that the process produced ketenimine 135 without photolysis or heating (Scheme 39).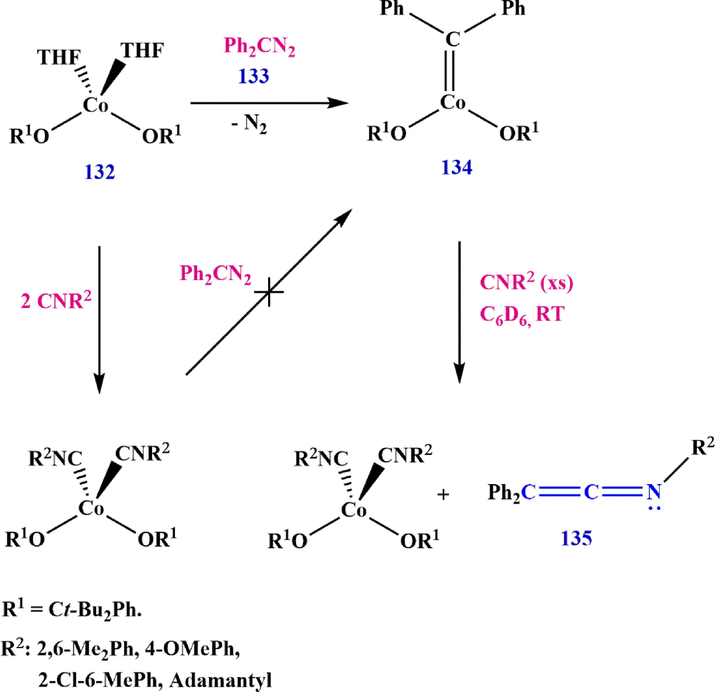
Synthesize ketenimine 135.
According to a possible mechanism, carbene complex 134, formed from complex 132 and diazoalkane, undergoes an intramolecular reaction with an isocyanide, resulting in the formation of ketenimine. In the presence of excess isocyanide, the isocyanides replace the ketenimine to form Co(OR1)2(CNR2)2Co(OR1)2(CNR2)2, which then reacts with the diazoesters to restore carbene functionality, releasing two equivalents of isocyanide and dinitrogen molecules (Scheme 40).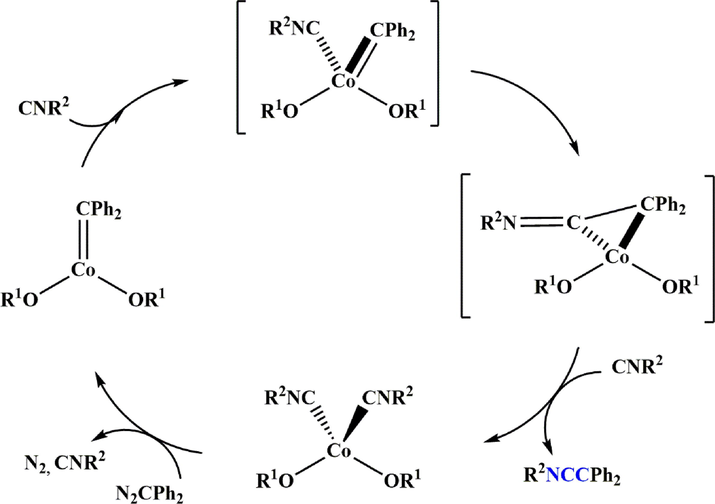
Recommended mechanism for the synthesis of ketenimines 135.
3.6 Ketenimine preparation via organometallic
3.6.1 Ketenimine preparation via Ir complexes
The use of Ir-OH or Ir-OTF complexes, which was introduced by Bissember et al. (Bissember et al., 2018) in 2018, is another method for the preparation of ketenimines. They showed that the carboxamide product 136 is produced exclusively when R is an aromatic group. However, when R
methyl, a mixture of hydration product 136 and
-deprotonated product 137 is formed, and when R
CHPh2, ketenimine complex 138 was the only product (Scheme 41).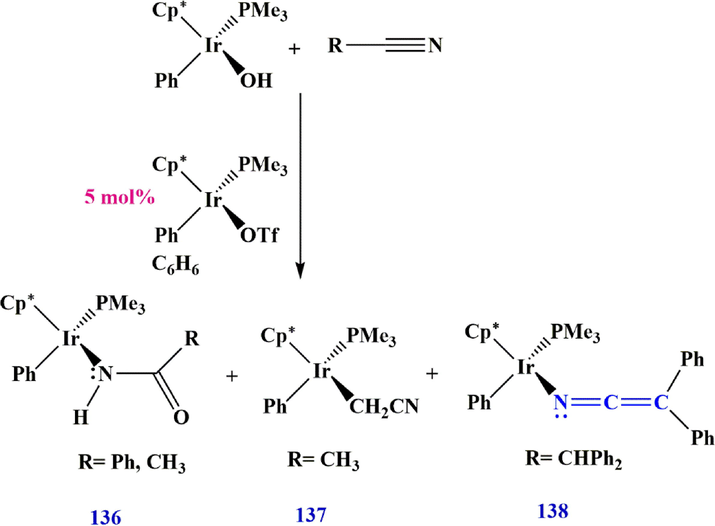
Synthesize the Ir-ketenimine complex 138.
3.6.2 Ketenimine preparation via Mg complexes
More basic ligands, such as alkyl ligands, can generate
-cyanocarbanion complexes when they react with a suitable nitrile. Indeed, an Mg complex with methyl-bridged ligands was reacted with diphenyl acetonitrile to produce the desired ketenimine complex (Fedushkin et al., 2009) with loss of methane (Scheme 42).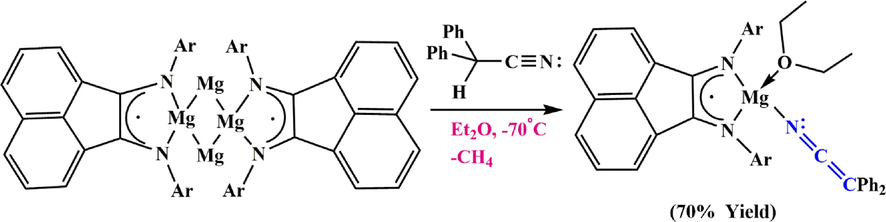
Synthesize Mg-ketenimine complex.
3.6.3 Ketenimine preparation via Li complexes
Iravani et al. (Iravani and Neumüller, 2003) showed that lithium ketenimine salts were prepared by reacting diphenyl acetonitrile with
-butyl lithium then salt metathesis with indium (
) and gallium (
) halide complexes (Scheme 43).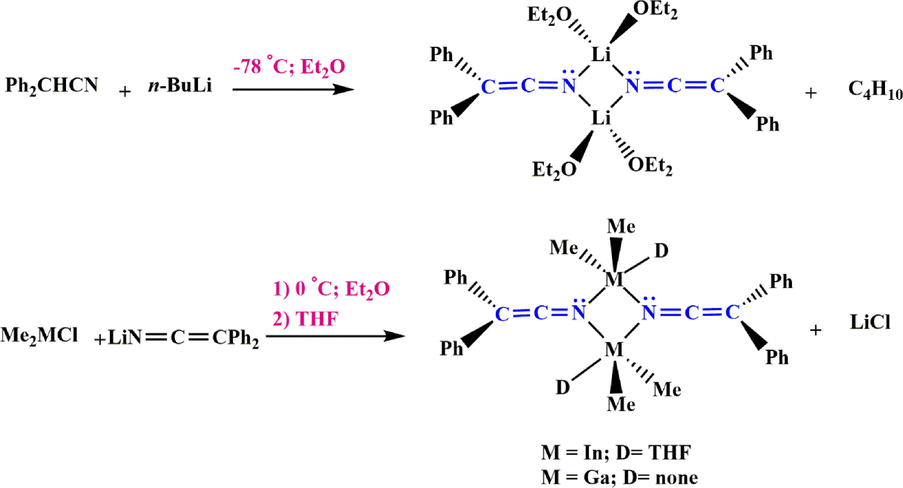
Synthesize Li, In and Ga-ketenimine complex.
3.6.4 Ketenimine preparation via Au complexes
The first gold (
) carbene complex was generated by the reaction of a geminal di-anion with a (P, C) cyclometallated gold (
) precursor that can react with a variety of electrophiles. Heterocumulenes have been shown to be the reagent of choice to test the nucleophilicity of carbene complexes, while another method was proposed by Pujol et al. (Pujol et al., 2017) via isocyanate. They showed that complex 139 reacts with PhNCS to form (2-(phenylimino)ethene-1,1-diyl)bis(diphenylphosphine sulfide) 140 and the monomeric “Au⚌S” complex A, which is formed by an Aura-Wittig reaction (Scheme 44).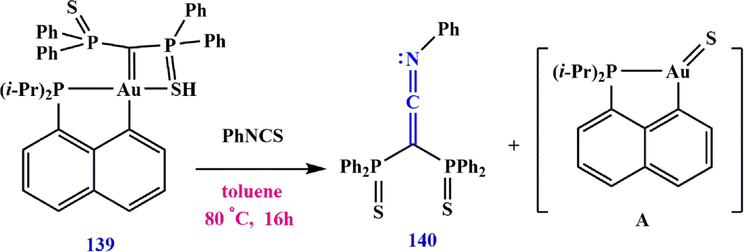
Synthesize (2-(phenylimino) ethene-1,1-diyl) bis (diphenylphosphine sulfide) 140.
3.6.5 Ketenimine preparation via Ni complexes
Nickel complexes are also used for the preparation of ketenimines. In this reaction, the Ni2(CPh2) complex 141 reacted with
-BuNC, leading to the formation of isonitrile 142.When the Ni2(
-CPh2)(CN
-Bu) complex 142 is subsequently dissolved in C6H6, N-(tert-butyl)-2,2-diphenylethen-1-imine 143 is formed (Maity et al., 2018) (Scheme 45).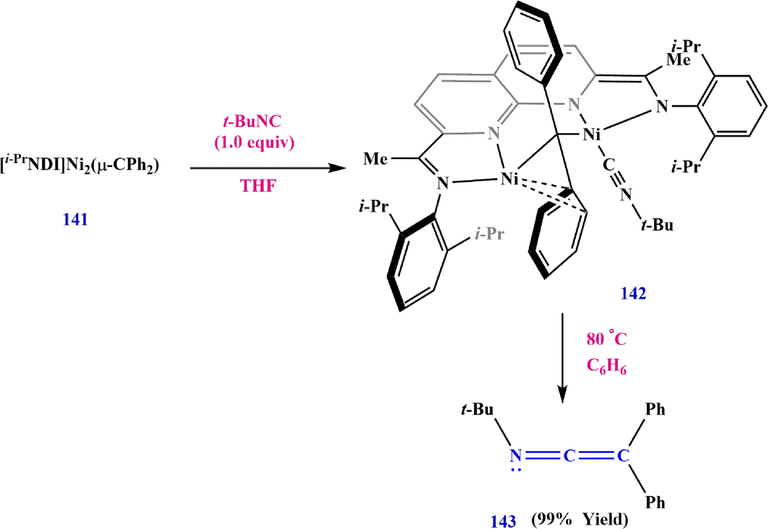
Synthesize ketenimine 143.
3.7 Ketenimine preparation via elimination reactions
Ketenimine A, formed from N, N-disulfonyl ynamide 141 and
-PrOLi reacts with aldehyde 142 to form oxetene C. Ring-opening of oxetene C gives amide D, which is protonated to produce the
,
-unsaturated 143 after work-up (route A in Scheme 46). On the other hand, N, N-disulfonyl ynamide 141 may be reacted with salicylaldehyde 144 in the presence of Et3N to produce the ketenimine intermediate E. Subsequently, after addition of the second molecule 144, followed by intramolecular nucleophilic addition and dehydration, iminocoumarin 145 is generated (route B in Scheme 46) (Yu and Cao, 2014).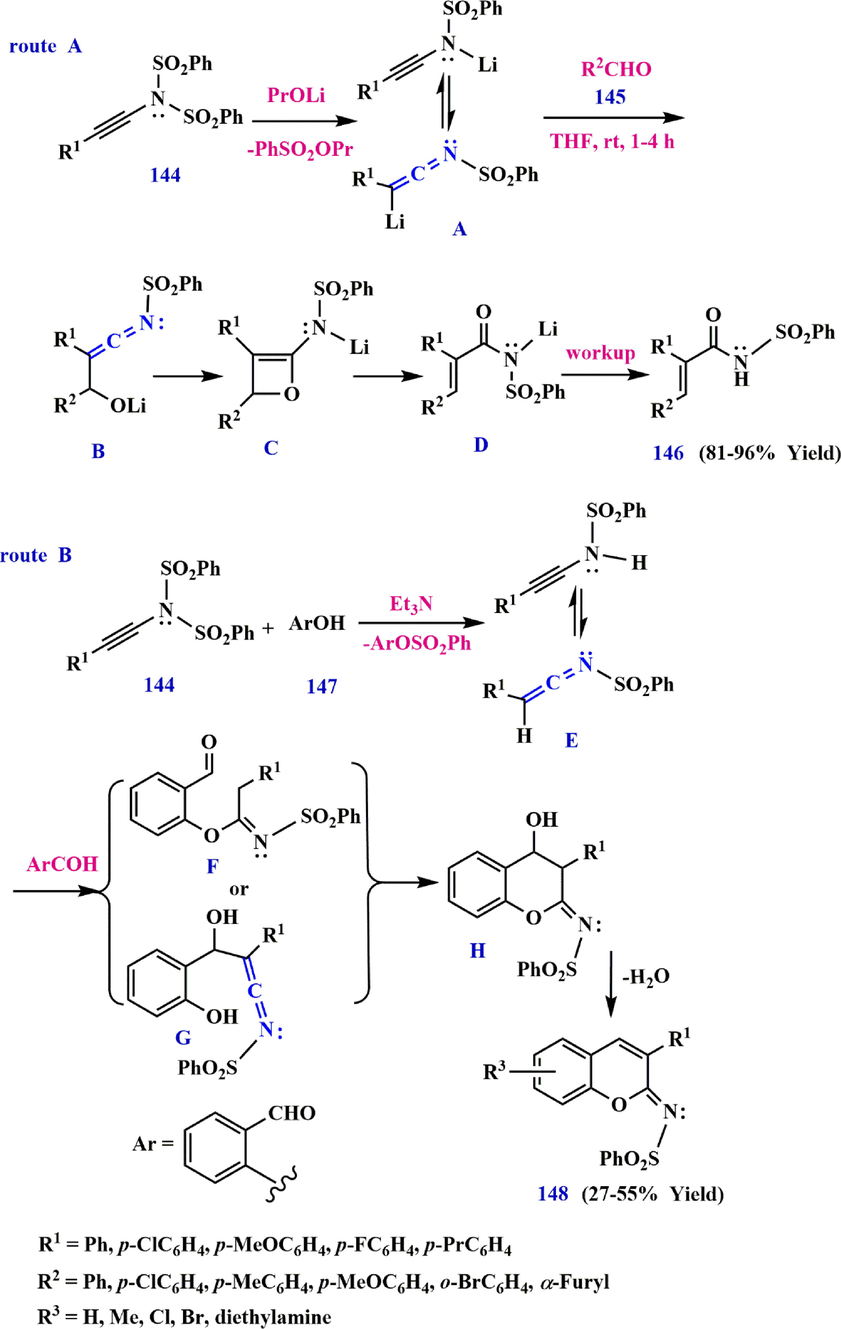
Synthesize ketenimine from N, N-disulfonyl ynamide.
Jin et al. have described another method for the synthesis of ketenimines using amides. The C-diaromatic ketenimine derivatives of 150a-c are prepared by the elimination of water from secondary amides 149 using
of anhydrous pyridine or triethylamine. Route B also shows that C-dialiphatic ketenimines 154d-f can be readily prepared by chlorination of secondary amides 152 with phosphoryl chloride via established procedures to give imidoyl chloride 153, followed by their dehydrochlorination with an excess of trimethylamine (route A in Scheme 47) (Stevens and Singhal, 1964; Jin et al., 2015).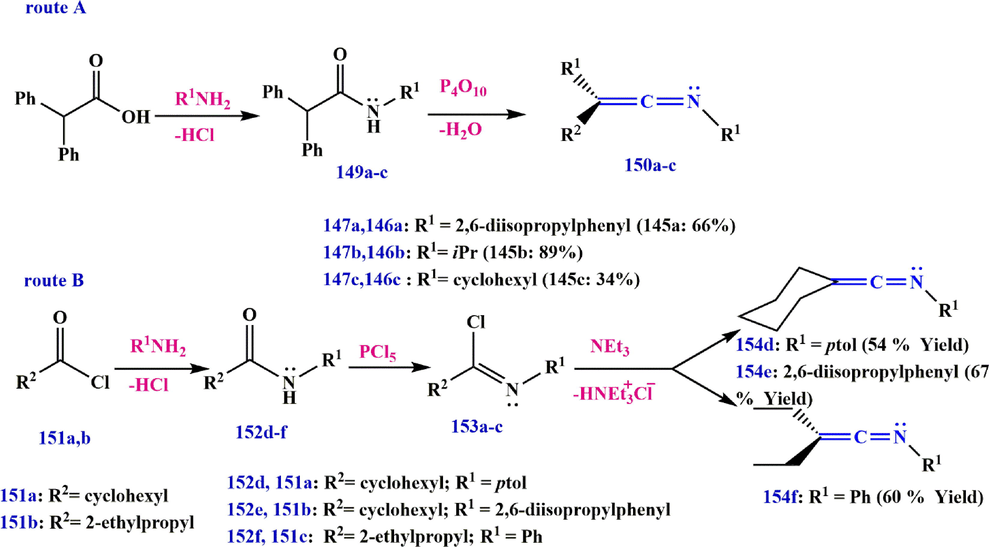
Synthesize ketenimines 150 and 154.
In 2009, Katagiri et al. (Katagiri et al., 2009) reported the first example of the effective synthesis of 2-trifluoromethylketenimines. In fact, imidyl chlorides 157 play a significant role in this synthesis because they engage in the elimination process, which is catalyzed by a triethylamine chloride, and leads to the generation of trifluoromethylketenimine (N- polymethylpentene -3,3,3-trifluoroprop-1-en-1-imine) 158 (Scheme 48).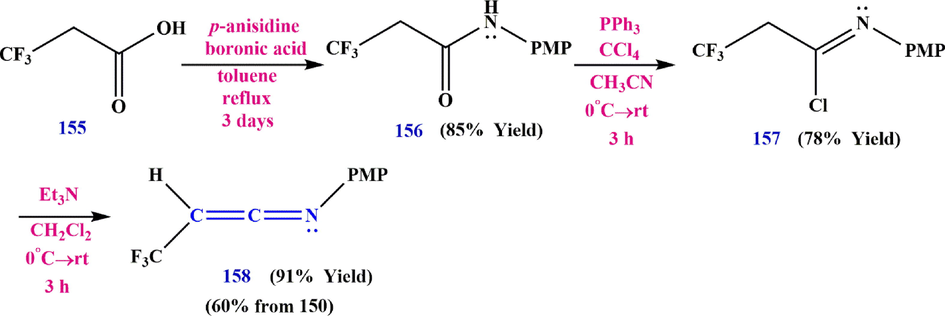
Synthesis trifluoromethylketenimine (N- polymethylpentene -3,3,3-trifluoroprop-1-en-1-imine) 158.
3.8 Ketenimine via rearrangement reactions
3.8.1 Rearrangement of enolizable nitriles
Due to the strong electron-withdrawing ability of the nitrile group, the reactivity of the ynimines under anionic conditions investigated by examining the result of the reaction between non-enolizable benzophenone-derived ynimines 159a and 159b and organolithium reagents. The addition of the organolithium reagent to the starting ynimine 159 produces a lithiated ynamine A which is in equilibrium with the lithiated ketenimine form. Using the electrophile to trap this intermediate would lead to the formation of a transient, unstable ketenimine C, which then rearranges, as seen previously with comparable ketenimines (Clarke et al., 1992; Alajarin et al., 2004; Khlebnikov et al., 2003). Subsequently, rapid rearrangement of C leads to the formation of a stable benzhydryl radical D, which is then reacted with radicals D and E/E′ to form nitrile 160 (Bendikov et al., 2005; Laouiti et al., 2014) (Scheme 49).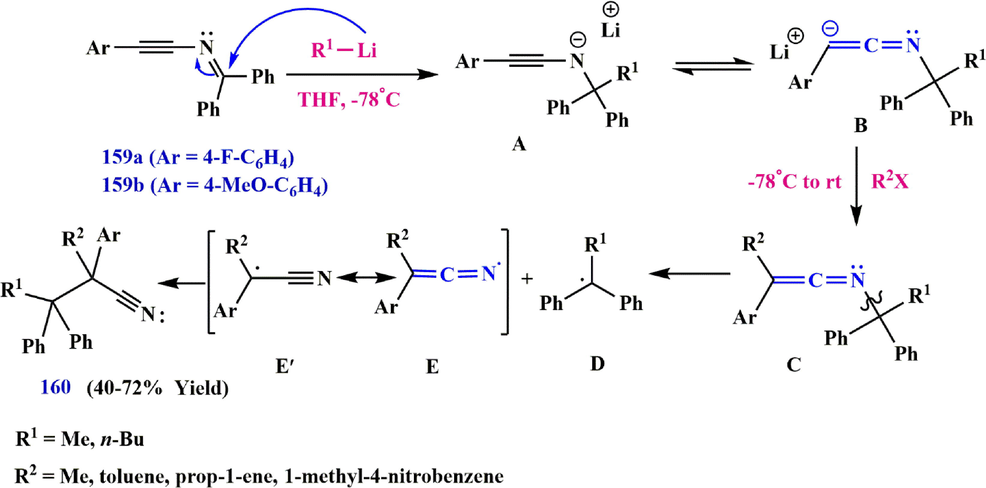
Recommended mechanism for the transformation of ynimine to alkanenitril.
Based on the synthesis of TMS-ketenimine 162 by the reaction of diphenylacetonitrile 161 with LDA, leading to the formation of compounds 164 and 165 (Long et al., 2013), the Li-stabilized ketenimine 161a also exhibits a parallel reaction pattern (Scheme 50).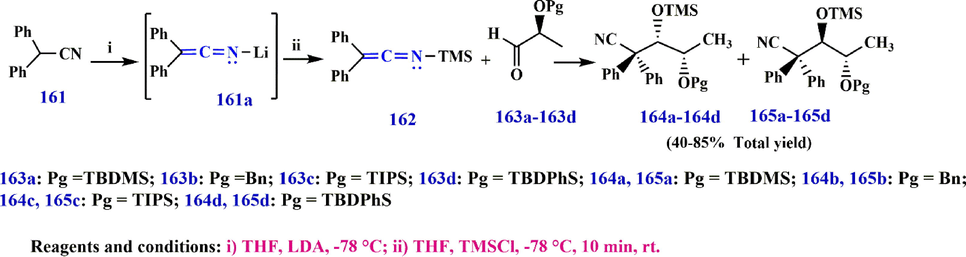
Synthesize N-TBDMS- keteneimine 162.
3.8.2 [1,3]-Brook rearrangement
In 2014, Evano et al. (Laouiti et al., 2014) investigated the possibility of trapping intermediate lithiated-silylketenimine 167 with aromatic aldehydes. They showed that the nucleophilic addition of 167 to these aldehydes should allow the formation of alkoxysilylketenimine 168, which could be formed via a [1,3]-Brook rearrangement, a nucleophilic addition of the alkoxide to the central carbon atom of the ketenimine moiety, or a Peterson-type olefination. Finally, [3]-azacumulene hydrolysis (E)-3-aryl-N-(1,1-diphenylethyl)propa-1,2-dien-1-imine 169 leads to the formation of the
,
-unsaturated amide 170 (Doney et al., 1983) (Scheme 51).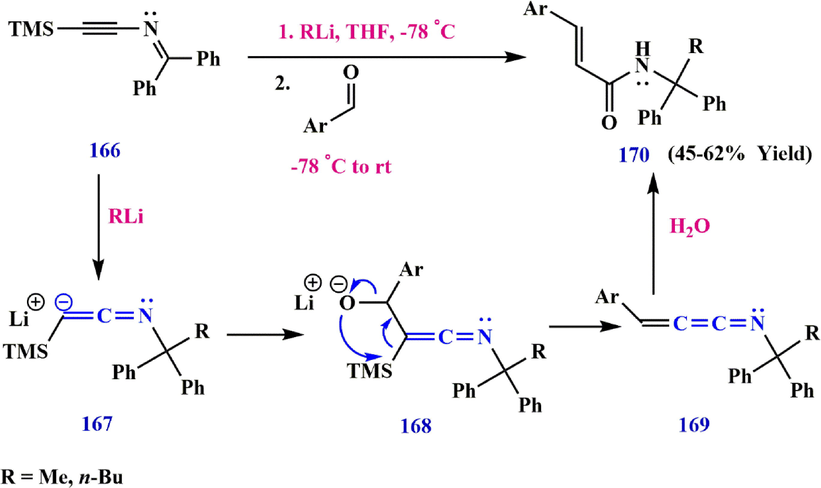
Synthesize ketenimine 169.
3.8.3 Aza-Claisen rearrangement of ynamides derivatives
The first thermal conversion of an ynamide to a ketenimine was reported by Bendikov et al. (Bendikov et al., 2005). They showed that the
-tosyl group of compounds 171 migrates from N to C under reaction conditions, leading to the formation of ethyl 3-((4-methoxybenzyl)imino)-2-tosylacrylate 172 (Scheme 52).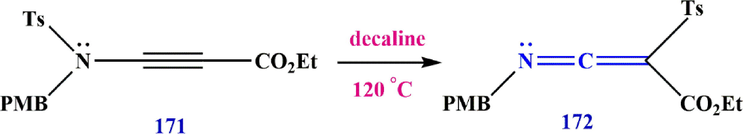
Synthesize ketenimines 172.
Dekorver et al. (Dekorver et al., 2010) obtained similar results for the thermal rearrangement of N-allylsulfonylynamide 173, although in this case the allyl group first migrates through an Aza-Claisen rearrangement. Their results showed that the nature of the R group has a significant effect on the outcome of the process and the phenyl group 173a or a protected primary alcohol 173b, the intermediate ketenimine 174, eventually migrates to the corresponding tertiary nitrile 175 by a 1,3-sulfonyl shift (route A in Scheme 53). On the other hand, the substituent a TIPS brings sufficient stabilization to allow isolation of the ketenimine 174 or its trapping by nucleophilic addition with pyrrolidine to give the amidine 176. Prolonged heating, results in desilylation of the ketenimine and triggers its rearrangement into the 2-tosylpent-4-enenitrile (secondary nitrile) 175d (route B in Scheme 53).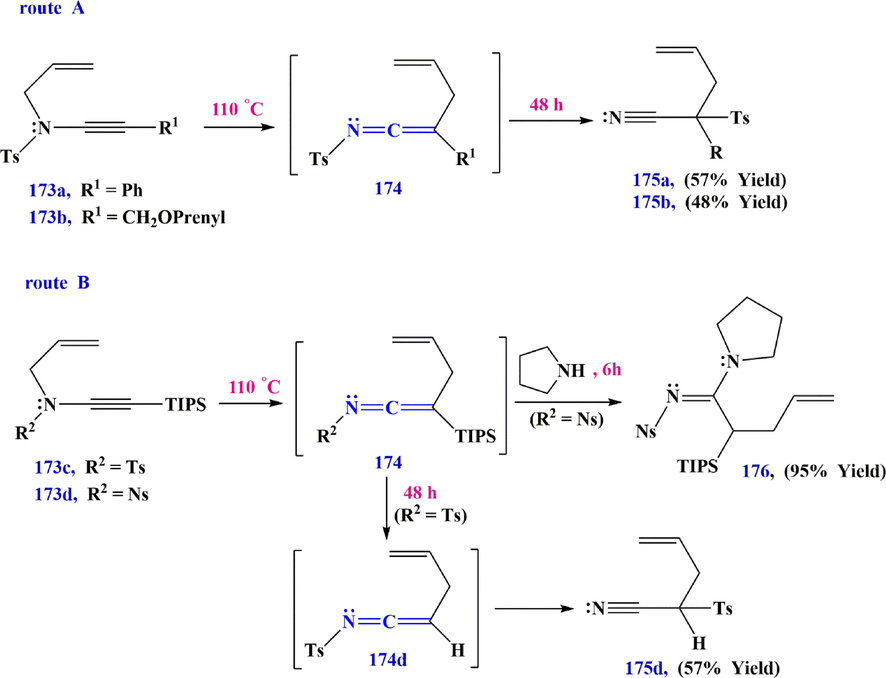
Synthesize compounds 175 and 176 via Aza-Claisen rearrangement.
In addition, thermal Aza-Claisen rearrangement of N-phosphoryl ynamide 177 can generate allyl ketenimines (5,5-dimethyl-2-((2-phenylpenta-1,4-dien-1-ylidene)amino)-1,3,2-dioxaphosphinane 2-oxide) 180, which subsequently undergo a 1,3-phosphoryl shift from nitrogen to carbon to generate nitriles 178 or cyclopentenimine 179. On the other hand, heating N-phosphoryl ynamide 177 in toluene in the absence of a competing 1,3-phosphoryl shift leads to the formation of cyclopentenimine 179 in an isolated yield of 50%. (Dekorver et al., 2010) (Scheme 54).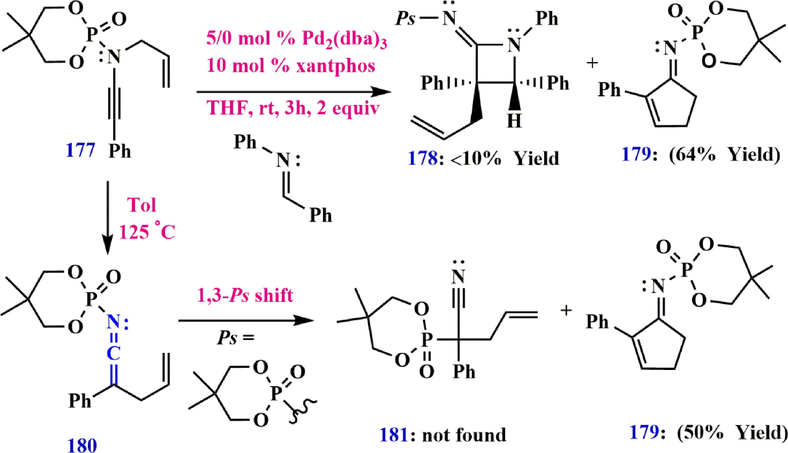
Synthesize N-phosphoryl ynamide from allyl ketenimines.
In 2012,continuing their studies, Dekorver et al. demonstrated that N-phosphorylyl ynamide 182e leads to the formation of compounds 183e and TIPS-ketenimine 184e under reaction conditions. They showed that in the presence of Pd as a catalyst, N-phosphoryl ynamide 185 can undergo the Rautenstrauch rearrangement, yielding Pd-π-allyl ketenimines 186b as an intermediate (Rautenstrauch, 1984; Shi et al., 2005), leading to the formation of compound 187. On the other hand, the carbocyclization of N-phosphoryl ynamide 188 by ketenimine 189 appears to be accelerated under both palladium-catalyzed and thermal conditions in the absence of a competing 1,3-phosphoryl shift. Subsequently, carbocyclization of ketenimine 189 (Hanessian et al., 2005; Sosa et al., 2008) gives an intermediate 190, which undergoes a 1,2-H shift to provide cyclopentenimine 191. Carbocyclization of ketenimine 193 without the use of a palladium catalyst leads to the formation of 194a− 194c, avoiding possible scrambling (Scheme 55). Also, the possibility of trapping intermediates with bound nucleophiles to produce bicyclic scaffolds such as 195a−195c is predictable (Scheme 56). Furthermore, when ynamide 196 is heated to 135 °C in toluene with a bound benzene ring, only cyclopentenemine 200 is formed, resulting from a 1,2-H shift in 199. In contrast, the
-methoxy group, which is an electron-donating group, can enrich the benzene ring in yanmide 197 with electrons, and 2-((7-methoxy-3a-methyl-3-phenyl-3a,4,5,9b-tetrahydro-1H-cyclopenta[α]naphthalen-2-yl) amino)-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide 202 is immediately synthesized in 85% yield in a combination of cis and trans isomers via the Aza-Claisen/Friedel-Craft electrophilic aromatic substitution process (Dekorver et al., 2012; Wang et al., 2013) (Scheme 57).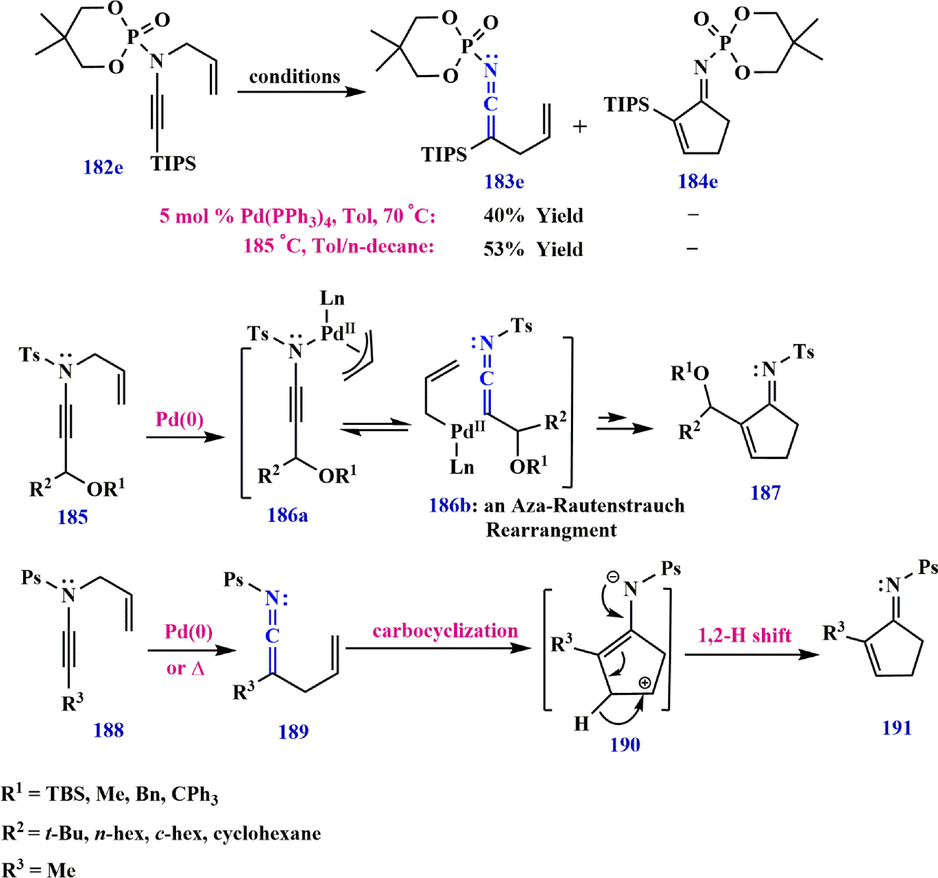
Synthesize ketenimine from ynamide.
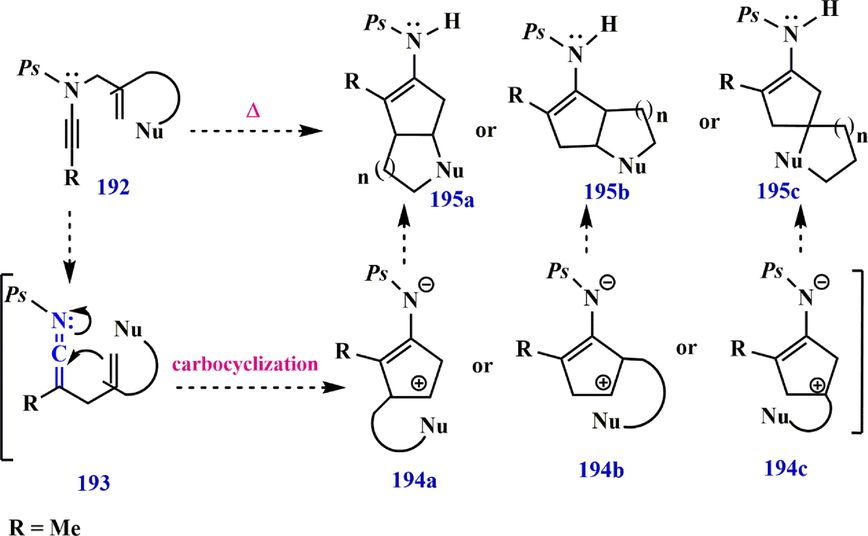
Various pathways of N-phosphoryl ynamide synthesis.
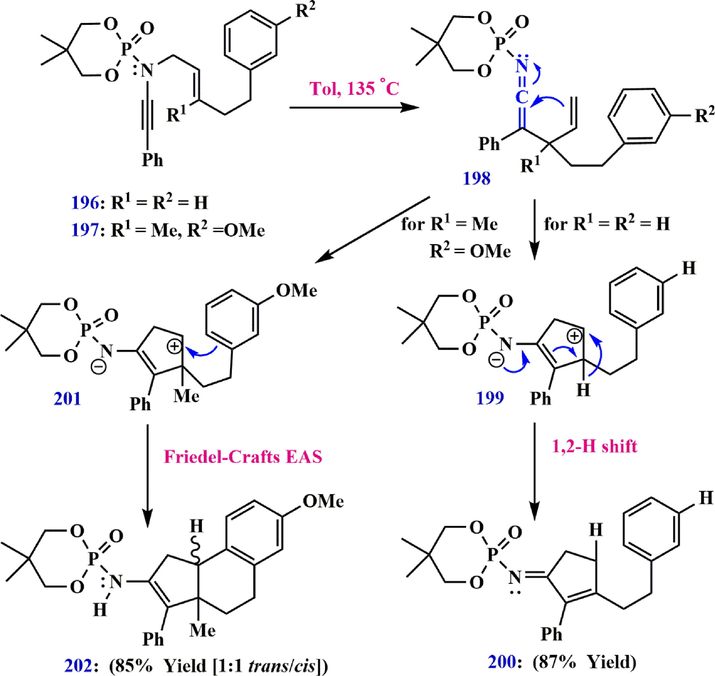
Synthesis routes of compounds 200 and 202.
3.8.4 Beckmann rearrangement of oxime derivatives
The SCF3 substituent has attracted much attention in recent years, and various methods for its direct introduction using electrophilic, nucleophilic, and radical sources have been investigated. (Tlili and Billard, 2013; Toulgoat et al., 2014). The Beckmann rearrangement of ketoxime 204 is one of the simplest methods to obtain fluorinated ketenimines. Treatment of (Z)-1-phenyl-2-((trifluoromethyl)thio)ethan-1-one oxime 203 under the reaction conditions led to the preparation of ketoxime 204. Monitoring of, GC-MS showed that a solution of 204 was stable under argon for several days (Scheme 58).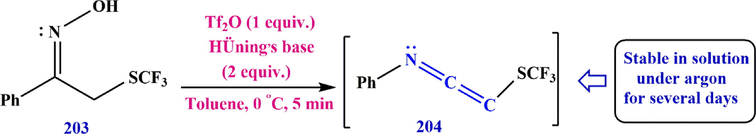
Synthesize ketoxime 204.
According to the plausible mechanism, the oxime is first converted to its triflic ether derivative I, as (Z)-1-phenyl-2-((trifluoromethyl)thio)ethan-1-one O-((trifluoromethyl)sulfonyl) oxime (Guérin et al., 2020), then the phenyl group migrates to the nitrogen as the triflate anion leaves, resulting in the synthesis of intermadiate II, a mesomeric form of nitrilium III. The remainder of the base deprotonates II or III to generate ketoxime 204 (Scheme 59).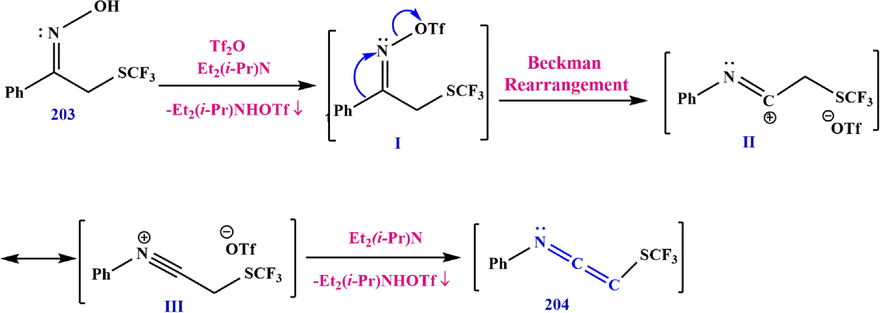
Synthesize ketenimine 204.
4 Summary and outlook
The chemistry of ketenimines is interesting. Because of their excellent lability, ketenimines are often prepared in situ as reactive intermediates and used in one-pot processes. In the last decade, stabilizing substituents have also been used to create a variety of isolable ketenimines. In this review, we have presented a summary of the latest developments in the chemistry of ketenimines, which are a significant category of structural units with a significant application in biological synthesis. Despite the tremendous progress that has been made in this area, developing new methods for the generation of ketenimines and applying them in modern organic synthesis continues to be a time-consuming task. It's worth noting that the chirality transfers and preparation of chiral ketenimines have yet to be reported. However, since the chemistry of ketenimines is much more complex than that summarized in this study, this is most likely just the tip of the iceberg. The application of asymmetric variations of these transformations, a problem that has yet to be tackled, would most likely be of special importance for future research in the area. We hope that this study will encourage chemists to examine the flourishing field of ketenimine chemistry.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ketenimine Complexes from Carbene Complexes and Isocyanides: Versatile Building Blocks for Carbocycles and N-Heterocycles [New Synthetic Methods. Angew. Chem. Int. Ed. Engl.. 1988;27:1456-1467.
- [Google Scholar]
- Intramolecular [2 + 2] cycloaddition of ketenimines with imines. Tetrahedron Lett.. 1996;37:8945-8948.
- [Google Scholar]
- Keteniminkomplexe aus Carbenkomplexen und Isocyaniden – vielseitige Bausteine für Carbocyclen und N-Heterocyclen. Angew Chem.. 1988;100:1512-1524.
- [Google Scholar]
- Periselective intramolecular [4+2] cycloadditions of ketenimines: synthesis of pyrido[1,2-a] benzimidazoles. Tetrahedron Lett.. 2000;41:7029-7032.
- [CrossRef] [Google Scholar]
- Tandem [1,5]-H shift/6p-electrocyclizations of ketenimines bearing 1,3-oxathiane units. Computational assessment of the experimental diastereoselection. Tetrahedron Lett.. 2012;68:4672-4681.
- [CrossRef] [Google Scholar]
- Three-component synthesis of dialkyl 2-(cyclohexyliminomethylene)-3-arylsulfonylamino succinate. J. Chem. Res.. 2011;35:98-100.
- [CrossRef] [Google Scholar]
- One-pot synthesis of N-substituted 2,4-thiazolidinediones and computational investigation of the products. Monatsh. Chem.. 2013;144:337-343.
- [CrossRef] [Google Scholar]
- Formation of Ketenimines via the Palladium-Catalyzed Decarboxylative π–Allylic Rearrangement of N-Alloc Ynamides. Org. Lett.. 2017;19:5822-5825.
- [CrossRef] [Google Scholar]
- Imino-Ketenimines on an Ortho-Benzylic Scaffold. Nitrogen to Carbon [1,3] Shift of an Ortho-Functionalized Benzyl Group. Lett. Org. Chem.. 2004;1:340-342.
- [CrossRef] [Google Scholar]
- Methoden der Organischen Chemie. (Houben-Weyl); 1968.
- Über Metall-Stickoxid-Komplexe. XXI. Tricyanomethanido–Nitrosyl-Komplexe von Kobalt und Nickel. Z. Anorg. Allg. Chem.. 1966;344:285-291.
- [CrossRef] [Google Scholar]
- Pseudohalogeno—Metallverbindungen XVIII. Anionische (Tricyanomethanido)pentacarbonyl-Komplexe von Chrom, Molybdän und Wolfram. J. Organometal. Chem.. 1967;8:547-550.
- [CrossRef] [Google Scholar]
- Heterocycles from ketenimines. V. 2-Iminoazetidines through thermolysis. J. Heterocyclic Chem.. 1972;9:1147-1148.
- [CrossRef] [Google Scholar]
- Synthetic Communications: An International Journal for Rapid Communication of Synthetic Organic Chemistry. Synth. Commun.. 2008;38:2567-2574.
- [CrossRef] [Google Scholar]
- α-Cyanocarbanion complexes and their application in synthesis. J. Organomet. Chem.. 2018;869:213-226.
- [CrossRef] [Google Scholar]
- An Unexpected Two-Group Migration Involving a Sulfonynamide to Nitrile Rearrangement. Mechanistic Studies of a Thermal N → C Tosyl Rearrangement. Org. Lett. 2005;7:783-786.
- [CrossRef] [Google Scholar]
- The Logic of Chemical Synthesis. John Wiley & Sons; 1995.
- An Unprecedented Chemospecific and Stereoselective Tandem Nucleophilic Addition/Cycloaddition Reaction of Nucleophilic Carbenes with Ketenimines. J. Org. Chem.. 2009;74:850-855.
- [CrossRef] [Google Scholar]
- A new multicomponent reaction for the synthesis of pyridines via cycloaddition of azadienes and ketenimines. Tetrahedron Lett.. 2011;52:3023-3025.
- [CrossRef] [Google Scholar]
- Relatively Stable N-Benzhydryl- and N-Benzyldiarylketene Imines and Their Conversion to Cyanodiarylmet hanes via an Isolable Radical. Org. Chem.. 1992;57:362-366.
- [CrossRef] [Google Scholar]
- Ketenimines Generated from Ynamides: Versatile Building Blocks for Nitrogen-Containing Scaffolds. Chem. Eur. J.. 2018;24:2297-2304.
- [Google Scholar]
- Imenes derived from methylsulphonyl-acetonitrile: (Properties of the sulphonyl group XLIII) Recl. Trav. Chim. Pays-Bas.. 1954;73:575-581.
- [CrossRef] [Google Scholar]
- Silyl Ketene Imines: Highly Versatile Nucleophiles for Catalytic, Asymmetric Synthesis. Angew. Chem. Int. Ed.. 2012;51:9980-9992.
- [CrossRef] [Google Scholar]
- Recent Developments in Isocyanide Based Multicomponent Reactions in Applied Chemistry. Chem. Rev.. 2006;106:17-89.
- [CrossRef] [Google Scholar]
- Nef–Perkow–Mumm Cascade towards Imido Phosphate Derivatives. Synlett. 2017;28:2637-2641.
- [CrossRef] [Google Scholar]
- An asymmetric oxazoline ketenimine rearrangement. Construction of chiral o -quaternary carbon ketones. Tetrahedron Lett.. 1999;40:4765-4768.
- [CrossRef] [Google Scholar]
- A Divergent Mechanistic Course of Pd(0)-Catalyzed Aza-Claisen Rearrangement and Aza-Rautenstrauch-Type Cyclization of N-Allyl Ynamides. Org. Lett.. 2010;12:1840-1843.
- [CrossRef] [Google Scholar]
- N-Allyl-N-sulfonyl Ynamides as Synthetic Precursors to Amidines and Vinylogous Amidines. An Unexpected N-to-C 1,3-Sulfonyl Shift in Nitrile Synthesis. J. Org. Chem. 2011;76:5092-5103.
- [CrossRef] [Google Scholar]
- New Donors with Two-Electron Oxidation. Synthesis and Electrochemical Properties of Highly Conjugated Bis(4H-pyrans), Bis(4H-thiopyrans), and Bis(flavenes) J. Org. Chem.. 1983;4:2757-2761.
- [CrossRef] [Google Scholar]
- A Divergent Mechanistic Course of Pd (0)-Catalyzed Aza-Claisen Rearrangement and Aza-Rautenstrauch-Type Cyclization of N-Allyl Ynamides. Org. Lett.. 2010;12:1840-1843.
- [CrossRef] [Google Scholar]
- Carbocyclization Cascades of Allyl Ketenimines via Aza-Claisen Rearrangements of N-Phosphoryl-N-allyl-ynamide. Org. Lett.. 2012;14:1768-1771.
- [CrossRef] [Google Scholar]
- Cyclobutanone and Cyclobutenone Derivatives by Reaction of Tertiary Amides with Alkenes or Alkynes. Angew. Chem. Int.. 1981;20:879-880.
- [CrossRef] [Google Scholar]
- Magnesium (II) Complexes of the dpp-BIAN Radical-Anion: Synthesis, Molecular Structure, and Catalytic Activity in Lactide Polymerization. Eur. J. Inorg. Chem.. 2009;2009:4995-5003.
- [CrossRef] [Google Scholar]
- Modern Synthesis Processes and Reactivity of Fluorinated Compounds. Elsevier Science; 2017.
- One-Pot Three-Component Synthesis of Pyrrolidin-2-ones via a Sequential Wittig/Nucleophilic Addition/Cyclization Reaction. Synth.. 2019;51:2402-2408.
- [CrossRef] [Google Scholar]
- Ketenimine Formation Catalyzed by a High-Valent Cobalt Carbene in Bulky Alkoxide Ligand Environment. J. Organomet. Chem.. 2019;38:962-972.
- [CrossRef] [Google Scholar]
- Synthesis and use of trifluoromethylthiolated ketenimines. Chem. Eur. J.. 2020;26:14852-14855.
- [CrossRef] [Google Scholar]
- Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals. Academic Press; 2019.
- Asymmetric Induction Reactions. I. Asymmetric [2, 3] Sigmatropic Rearrangements of Sulfur Ylides Derived from Chiral Ketenimines and Trimethylsulfonium YlideChem. Pharm. Bull.. 1985;33:2331-2338.
- [CrossRef] [Google Scholar]
- Synthetic Studies in the Intramolecular Carbocyclization of N-Acyloxyiminium Ions. Stereoelectronic and Steric Implications of Nucleophilic Alkene, Alkyne, and Allene Tethers. Org. Chem.. 2005;70:5070-5085.
- [CrossRef] [Google Scholar]
- Control of C-C and C–N Bond Cleavage of 2H-Azirine by Means of the Excitation Wavelength: Studies in Matrices and in Solutions. Chem. Commun.. 2001;2001:1036-1037.
- [CrossRef] [Google Scholar]
- Trimerization of Phenylacetonitrile. InMe3 as a Base for C-H Acidic Nitriles. Organometallics. 2003;22:4129-4135.
- [CrossRef] [Google Scholar]
- Ketenimines. Geometry and barriers to racemization. J. Am. Chem. Soc. 1970;92:5524-5525.
- [CrossRef] [Google Scholar]
- One-pot four component synthesis of novel 3-furyl coumarin derivatives. J. Chem. Sci.. 2015;128:217-226.
- [CrossRef] [Google Scholar]
- Hydroalumination of Ketenimines and Subsequent Reactions with Heterocumulenes: Synthesis of Unsaturated Amide Derivatives and 1,3-Diimines. J. Org. Chem.. 2015;80:6062-6075.
- [CrossRef] [Google Scholar]
- Modern Fluoroorganic Chemistry. Wiley-VCH; 2004.
- Sulfonyl and Phosphoryl Azides: Going Further Beyond the Click Realm of Alkyl and Aryl Azides. Chem. Asian. J.. 2011;6:2618-2634.
- [CrossRef] [Google Scholar]
- Theoretical study of ketenimine: Geometry, electronic properties, force constants and barriers to inversion and rotation. J. Mol. Struct.. 1982;87:205-210.
- [CrossRef] [Google Scholar]
- Reaction of ethyl azidoformate with dimethyl- and diethylketen-N-(p-tolyl) imine. J. Org. Chem.. 1970;35:4244-4245.
- [CrossRef] [Google Scholar]
- Preparations and reactions of 2-trifluoromethylketenimines. J. Fluor. Chem. 2009;130:714-717.
- [CrossRef] [Google Scholar]
- Cascade Transformations of (2,2-Diaryl-3,3-dichloroaziridin-1-yl) acetates. Russ. J. Org. Chem. 2003;39:559-573.
- [CrossRef] [Google Scholar]
- The thriving chemistry of ketenimines. Chem. Soc. Rev. 41,5687–5705. Chem. Soc. Rev.. 2012;41:5687-5705.
- [Google Scholar]
- Exploring the Anionic Reactivity of Ynimines, Useful Precursors of Metalated Ketenimines. Org. Lett.. 2014;16:2252-2255.
- [CrossRef] [Google Scholar]
- Efficient Aldol-Type Reaction of O-Protected α-Hydroxy Aldehydes and N-Trimethylsilyl Ketene Imines: Synthesis of β, γ-Dihydroxy-Nitriles. Eur. J. Org. Chem. 2013;2013:5127-5142.
- [CrossRef] [Google Scholar]
- Glossary of class names of organic compounds and reactivity intermediates based on structure (IUPAC Recommendations 1995) Pure. Appl. Chem.. 1995;67:1307-1375.
- [CrossRef] [Google Scholar]
- Domino reactions. One-pot preparation of fluoreno[2,3,4-ij] isoquinoline derivatives from conjugated ketene imines. J. Org. Chem.. 1991;56:4008-4016.
- [CrossRef] [Google Scholar]
- Cycloadditions of keteneimmonium cations to olefins and dienes. New synthesis of four-membered rings. J. Am. Chem. Soc.. 1972;94:2870-2872.
- [CrossRef] [Google Scholar]
- Carbene Formation and Transfer at a Dinickel Active Site. J. Organomet. Chem.. 2018;37:2437-2441.
- [CrossRef] [Google Scholar]
- Classics in Total Synthesis. Wiley-VCH; 1996.
- Classics in Total Synthesis II. Wiley-VCH; 2003.
- The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century. Angew. Chem. Int. Ed.. 2000;39:44-122.
- [Google Scholar]
- Recent Advances in Solution-Phase Multicomponent Methodology for the Synthesis of Heterocyclic Compounds. Synth.. 2003;10:1471-1499.
- [CrossRef] [Google Scholar]
- Wavelength-dependent photochemistry of 2-azidovinylbenzene and 2-phenyl-2H-azirine. J. Mol. Struct.. 2018;1172:94-101.
- [CrossRef] [Google Scholar]
- C-terminal peptide extension via gas-phase ion/ion reactions. Int. J. Mass Spectrom.. 2015;391:17-23.
- [CrossRef] [Google Scholar]
- A Nucleophilic Gold (III) Carbene Complex. Angew. Chem. Int. Ed.. 2017;56:12264-12267.
- [CrossRef] [Google Scholar]
- Palladium-Catalyzed Migratory Insertion of Isocyanides for Synthesis of C-Phosphonoketenimines. ACS Catal. 2016;6:4715-4719.
- [CrossRef] [Google Scholar]
- Ketenimines from Isocyanides and Allyl Carbonates: PalladiumCatalyzed Synthesis of β, γ-Unsaturated Amides and Tetrazoles. Angew. Chem.. 2016;128:15603-15607.
- [CrossRef] [Google Scholar]
- 2-Cyclopentenones from 1-ethynyl-2-propenyl acetates Org. Chem.. 1984;49:950-952.
- [CrossRef] [Google Scholar]
- Uber neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta.. 1919;2:635-646.
- [CrossRef] [Google Scholar]
- Nitrogen Analogs of Ketenes. A New Method of Preparation. J. Am. Chem. Soc.. 1953;75:657-660.
- [CrossRef] [Google Scholar]
- Nitrogen Analogs of Ketenes. V. Formation of the Peptide Bond. J. Am. Chem. Soc.. 1958;80:4065-4071.
- [CrossRef] [Google Scholar]
- Nitrogen Analogs of Ketenes. VI.1 Dehydration of Amides. Org. Chem.. 1964;29:34-37.
- [CrossRef] [Google Scholar]
- Synthesis of 2-Cyclopentenones by Gold(I)-Catalyzed Rautenstrauch Rearrangement. J. Am. Chem. Soc.. 2005;127:5802-5803.
- [CrossRef] [Google Scholar]
- Synthesis of Alkynyl Ethers and Low-Temperature Sigmatropic Rearrangement of Allyl and Benzyl Alkynyl Ethers. Org. Lett.. 2008;10:5091-5094.
- [CrossRef] [Google Scholar]
- Formation of C-SCF3 Bonds through Direct TrifluoromethylthiolationAngew. Chem. Int.. 2013;52:6818-6819.
- [CrossRef] [Google Scholar]
- Direct Trifluoromethylthiolation Reactions: The “Renaissance” of an Old Concept. Eur. J. Org. Chem.. 2014;2014:2415-2428.
- [CrossRef] [Google Scholar]
- Singlet Photoreactivity of 3-Methyl-2-phenyl-2H-azirine. J. Chem.. 2017;70:413-420.
- [CrossRef] [Google Scholar]
- Novel Heterocumulenes: Bisiminopropadienes and Linear Ketenimines. Chem. Eur. J.. 1996;2:1318-1329.
- [CrossRef] [Google Scholar]
- Trifluoroacetic Anhydride-Promoted Copper(I)-Catalyzed Interrupted Click Reaction: From 1,2,3-Triazoles to 3- Trifluoromethyl-Substituted 1,2,4-Triazinones. Angew. Chem Int.. 2017;56:10476-10480.
- [CrossRef] [Google Scholar]
- J. Org. Chem.. 2013;78:6233-6244.
- [CrossRef]
- One-Pot Synthesis of Polysubstituted Pyrroles via Sequential Ketenimine Formation/Ag(I)-Catalyzed Alkyne Cycloisomerisation Starting from Ylide Adducts. Chin. Chem. Lett.. 2021;39:1553-1557.
- [CrossRef] [Google Scholar]
- Copper-Catalyzed Multicomponent Reactions: Securing a Catalytic Route to Ketenimine Intermediates and their Reactivities. Curr. Org. Chem.. 2009;13:1766-1776.
- [CrossRef] [Google Scholar]
- Copper-Catalyzed Synthesis of N-Sulfonyl-1,2,3-triazoles: Controlling SelectivityAngew. Chem. Int.. 2007;46:1730-1733.
- [CrossRef] [Google Scholar]
- One-pot synthesis of 2,6-diamino-4-sulfonamidopyrimidines from sulfonyl azides, terminal alkynes and cyanoguanidine. Tetrahedron Lett.. 2012;53:942-943.
- [CrossRef] [Google Scholar]
- Copper-catalyzed one-pot synthesis of tetrasubstituted pyrazoles from sulfonyl azides, terminal alkynes, and hydrazonoyl chloride. Tetrahedron Lett.. 2012;53:1889-1890.
- [CrossRef] [Google Scholar]
- Synthesis of α, β-unsaturated amides and iminocoumarins from N, N-disulfonyl ynamides with aldehydes via the ketenimine intermediate. Org Biomol. Chem.. 2014;12:3986-3990.
- [CrossRef] [Google Scholar]
- Zhu, J., Bienaymé, H., 2005. Multicomponent Reactions. France: Eds. Wiley: Weinheim.
- The Generation of Difluoroketenimine and Its Application in the Synthesis of α, α-Difluoro-β-amino Amides. Angew. Chem.. 2019;131:5800-5804.
- [CrossRef] [Google Scholar]
- Comparison of the Photochemistry of 3–Methyl-2-phenyl–2H–azirine and 2–Methyl-3-phenyl–2H–azirine. J. Org. Chem.. 2014;79:653-663.
- [CrossRef] [Google Scholar]
- Synthesis of Amidines Using N-Allyl Ynamides. A Palladium-Catalyzed Allyl Transfer through an Ynamido-π-Allyl Complex. Org. Lett.. 2009;11:899-902.
- [CrossRef] [Google Scholar]




